 Ricardo A. L. Sousa1
Ricardo A. L. Sousa1 Asmaa Yehia
Asmaa Yehia Osama A. Abulseoud
Osama A. Abulseoud
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurosci., 24 July 2023
Sec. Neuropharmacology
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1237153
This article is part of the Research TopicExploring the Key Targets and Compounds That Manipulate Brain Neurocircuits Against Mental Disorders and PsychiatricView all 15 articles
Coronavirus disease-19 (COVID-19), caused by severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2), is associated with the persistence of pre-existing or the emergence of new neurological and psychiatric manifestations as a part of a multi-system affection known collectively as “post-COVID syndrome.” Cognitive decline is the most prominent feature among these manifestations. The underlying neurobiological mechanisms remain under intense investigation. Ferroptosis is a form of cell death that results from the excessive accumulation of intracellular reactive iron, which mediates lipid peroxidation. The accumulation of lipid-based reactive oxygen species (ROS) and the impairment of glutathione peroxidase 4 (GPX4) activity trigger ferroptosis. The COVID-19-associated cytokine storm enhances the levels of circulating pro-inflammatory cytokines and causes immune-cell hyper-activation that is tightly linked to iron dysregulation. Severe COVID-19 presents with iron overload as one of the main features of its pathogenesis. Iron overload promotes a state of inflammation and immune dysfunction. This is well demonstrated by the strong association between COVID-19 severity and high levels of ferritin, which is a well-known inflammatory and iron overload biomarker. The dysregulation of iron, the high levels of lipid peroxidation biomarkers, and the inactivation of GPX4 in COVID-19 patients make a strong case for ferroptosis as a potential mechanism behind post-COVID neuropsychiatric deficits. Therefore, here we review the characteristics of iron and the attenuation of ferroptosis as a potential therapeutic target for neuropsychiatric post-COVID syndrome.
Ferroptosis is defined as a controlled form of cell death driven by excess intracellular labile iron and loss of the anti-oxidant enzyme glutathione peroxidase 4 (GPX4) activity, with consequent accumulation of lipid-based reactive oxygen species (ROS), especially lipid hydroperoxides (Yang and Stockwell, 2016). Interestingly, a knockout of GPX4 in mice led to lethality before embryonic day 9 (E9), which indicates a vital role for GPX4 in mouse development (Matsui, 1996; Yant et al., 2003). In the absence of GPX4, lipid peroxidation in vivo has a lethal nature, especially in neurons (Seiler et al., 2008). Alongside the unfavorable effect of GPX4 activity loss, iron overload contributes to the pathogenesis of coronavirus disease-19 (COVID-19), inciting inflammation, hypercoagulation, and immune dysfunction. Iron overload fosters an environment with free, unbound reactive iron, which triggers ROS generation (Habib et al., 2021). Ferritin, an iron storage protein, is a well-known inflammatory and iron overload biomarker, and is considered a direct mediator of the immune system in COVID-19 (Kappert et al., 2020; Kaushal et al., 2022; Lee et al., 2022). Iron overload is strongly suggested to contribute to the development of post-COVID neurological deficits (Fratta Pasini et al., 2021; Pandey et al., 2021; Zhang et al., 2022).
The post-COVID progressive and intense neurological clinical deterioration seems to occur due to the cytokine storm in COVID-19 patients (Para et al., 2022), which could create a vicious cycle with ferroptosis. Patients with COVID-19 presenting with high serum ferritin levels are usually in a severe condition (Abulseoud et al., 2022; Para et al., 2022). In addition, COVID-19 patients with comorbidities such as severe acute liver injury, diabetes, thrombotic complications, and cancer present with significantly higher levels of ferritin than those without (Cheng et al., 2020; Li et al., 2021).
A recent study analyzed COVID-19 effects over a 2-year retrospective cohort of 1,248,437 patients and revealed that cognitive decline, brain fog, and dementia are increasing over a 2-year follow-up period (Taquet et al., 2022). Another recent study by Wang et al. analyzed 6,245,282 older adults (over 65 years old), and the authors showed that older people infected by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) were at significantly higher risk for a new diagnosis of Alzheimer’s disease (Wang et al., 2022). Post-COVID neurological consequences are tightly linked to other systems, including changes in the cardiovascular and immune systems, as well as higher levels of stress, anxiety, and depression (Campos et al., 2019; Salari et al., 2020; Zubair et al., 2020; Improta-Caria et al., 2021; Júnior et al., 2021; Sousa et al., 2021). The enigmatic nature of the post-COVID syndrome’s underlying mechanism necessitates intense investigation in order to achieve effective management strategy. Uncovering the role of iron as a potential therapeutic target is a critical step in pursuing better management of the post-COVID neurological consequences. Here, we review the characteristics of iron, and the attenuation of ferroptosis as a potential therapeutic target for neurological post-COVID syndrome.
Iron is the 26th element in the periodic table and is located in the transition metals group. It can exist in different oxidation states and possess catalytic properties (Neyens and Baeyens, 2003). Ferrous (Fe+2) and ferric (Fe+3) irons are the two most common iron states in biological systems (Cabantchik, 2014). From an atomic orbital energy standpoint, a ferrous iron atom has a total of 26 electrons distributed in the following manner: two electrons in 1 s (the least energy orbit), 2 electrons in 2 s, 6 electrons in 2p, 2 electrons in 3 s, 6 electrons in 3p, 2 electrons in 4 s, and 6 electrons in the 4d orbit (Fe: 1s22s22p63s23p63d64s2). The position energy in the 3rd orbit (3d) is slightly higher than the position energy in the 4th orbit (4 s). This means that electrons will fill the 4 s position first before filling the 3d position, and also that the 4 s electrons will be lost first before the 3d electrons (Sherry and Fürstner, 2008). That is why ferrous iron [(Fe2+): 1s22s22p63s23p63d6] has lost two electrons from the 4 s position, while ferric iron [(Fe3+): 1s22s22p63s23p63d5] has lost a total of three electrons (two from the 4 s and one from the 3d position). As such, ferric iron (Fe3+) is relatively more stable than ferrous iron (Fe2+). Stability means the balance between positive and negative charges in the atom. Changing the number of electrons disturbs this balance. The atom holds electrons through electron binding energy, which is the minimum energy required to remove an electron from an atom. This energy is directly proportional to the atomic number (heavier atoms have more energy) and inversely proportional to the distance from the nucleus (electrons in outer orbits require less energy to be removed from the atom). As stated, ferrous iron has lost two electrons from 4 s, leaving six electrons in 3d, while ferric iron has lost two electrons from 4 s and one from 3d leaving five electrons in 3d. Electrons in partially filled orbits require more energy to remove from the orbit compared to electrons in fully filled orbits, which is why ferric iron is more stable compared to ferrous iron. The ability of ferric iron (Fe3+) to accept an electron and become ferrous iron (Fe2+) is called reduction, and the ability of ferrous iron (Fe2+) to donate an electron and become ferric iron (Fe3+) is called oxidation (Figure 1).
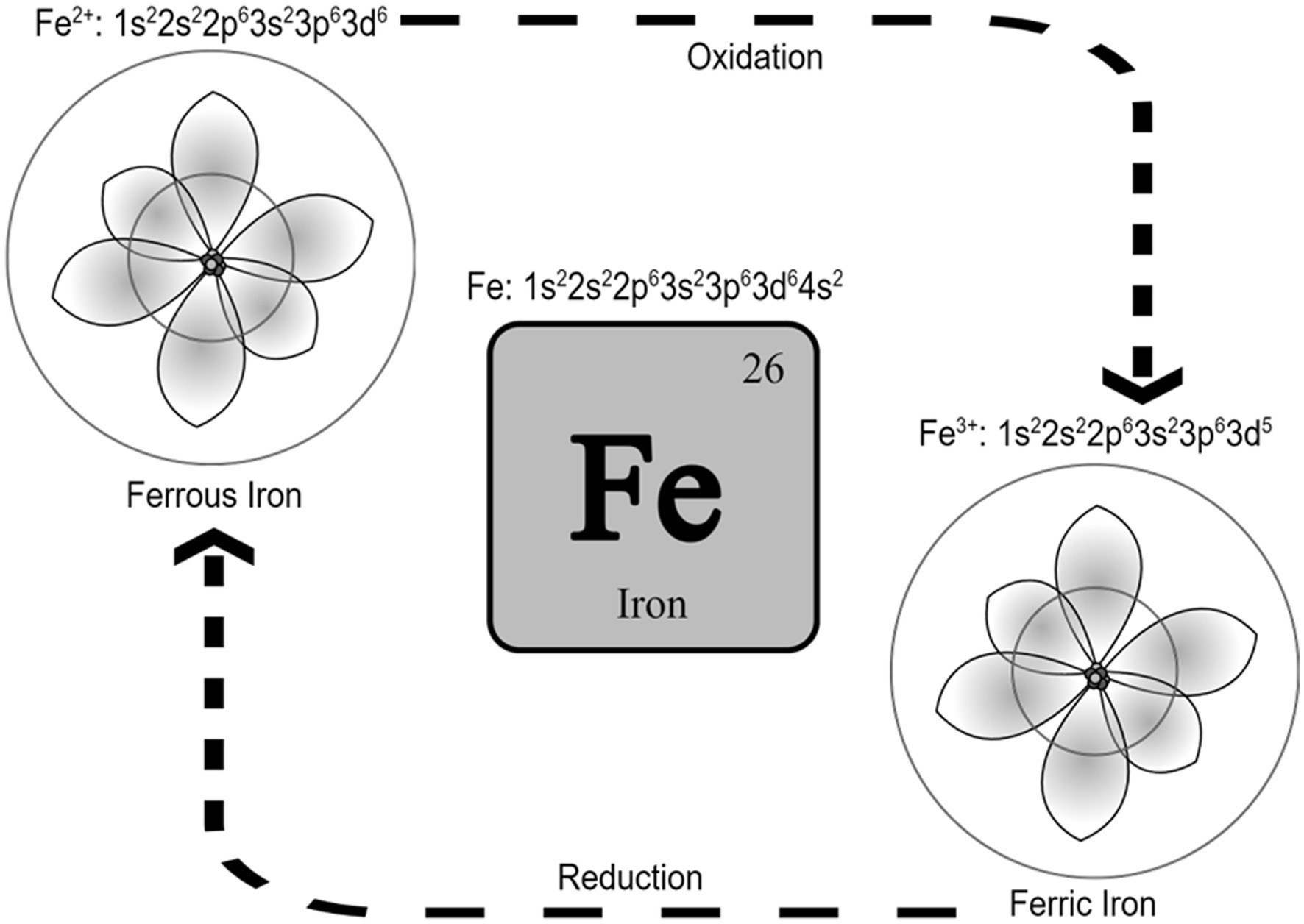
Figure 1. Iron and the two most common iron states in biological systems. It is called reduction when Fe3+ accepts an electron and becomes Fe2+, and it is called oxidation when Fe2+ donates an electron and becomes Fe3+. Schrodinger atom model is represented in the figures for ferrous and ferric iron.
This reduction–oxidation or Redox, is what makes iron a catalyst for reactions that require electron transfer (Hosseinzadeh and Lu, 2016). In the Fenton reaction, for example, iron catalyzes the decomposition of hydrogen peroxide (H2O2) to produce hydroxyl radicals (●OH) at an acidic PH with high oxidizing properties (Neyens and Baeyens, 2003) [see the reaction below. The unpaired electron of a free radical is represented with a dot (●)].
Numerous key enzymes rely on iron redox properties such as the mitochondrial respiratory chain enzymes (Gille and Reichmann, 2011), the aconitase enzyme in the tri-carboxylic acid cycle, which facilitates the conversion of citrate to iso-citrate via cis-aconitate (Kennedy et al., 1983; Robbins and Stout, 1989), and tyrosine hydroxylase, which is the rate-limiting enzyme in catecholamine synthesis (Ramsey et al., 1996; Daubner et al., 2011). Moreover, the serotonergic system may require ferrous iron (Fe2+) for binding serotonin to serotonin-binding proteins (Tamir and Liu, 1982). As such, iron is beneficial in regulating energy production and neurotransmitter synthesis, such as glutamate (McGahan et al., 2005; Lall et al., 2008) and dopamine (Ramsey et al., 1996). However, the same inherent “pro-oxidant” ability of ferrous iron (Fe2+) to donate an electron and catalyze enzymatic reactions also causes hydrogen peroxide (H2O2) to breakdown into hydroxyl radicals (●OH) causing lipid peroxidation and oxidative damage to proteins, deoxyribonucleic acid (DNA), and ribonucleic acid (RNA) (Graham et al., 2007). For this reason, the oxidative property of ferrous iron (Fe2+) must be managed by transforming it into a more stable ferric iron (Fe3+) and shielding this ferric iron (Fe2+) from water by transporting ferric iron into protein transporters.
The liver is the major organ involved in iron homeostasis. The liver produces iron transporters, including transferrin and divalent metal transporter-1 (DMT1). Transferrin transports ferric iron in the intestinal lumen and plasma, while DMT1 transports ferrous iron from the interior of the endosome to the cytosol. The liver also produces ferritin and hepcidin, which serve as the iron storage protein, and the only iron hormonal regulator, respectively. Furthermore, it produces transferrin receptor 1 (TfR1) and the iron responsive element binding proteins, which act as iron sensors and regulate the mRNA iron responsive element to enhance transferrin receptor translation and to down-regulate ferritin translation (Winterbourn, 1995). Moreover, the liver controls the oxidative state of iron by producing ferrireductase and ferroxidase enzymes. Ferrireductase reduces endosomal ferric iron, at low PH, within endocytosis vesicles into ferrous iron before it is released from the transferrin and reduces stored ferric iron into ferrous iron before release from ferritin, while ferroxidase oxidizes ferrous iron into ferric iron to be stored in ferritin (Graham et al., 2007).
Dietary ferric iron uptake by the intestinal mucosa depends on hepatic transferrin, Transferrin Receptor 1 (TfR1), and iron content within enterocytes. Transferrin is synthesized by hepatocytes and excreted through the biliary system into the intestinal lumen. Transferrin binds two atoms of iron per protein molecule and brings them into the cells by endocytosis (Bezkorovainy, 1989b). Duodenal and upper jejunal (low PH), but not ileal (Huebers et al., 1983), enterocyte crypts express TfR1 at the basolateral border (Barisani and Conte, 2002). These cells uptake iron from plasma transferrin by receptor-mediated endocytosis (Morgan and Oates, 2002). TfR1 is involved in sensing body iron stores. TfR1 expression increases with iron deficiency and decreases with iron overload (Barisani and Conte, 2002). Within endosomes, transferrin, TfR1, and ferric iron (Fe3+) are subject to low PH to separate ferric iron (Fe3+) from transferrin and TfR1. Transferrin and TfR1 are cycled back to the cell surface or plasma, while ferric iron (Fe3+) is reduced by the ferrireductase enzyme into ferrous iron (Fe2+). DMT1 transports ferrous iron (Fe2+) out of the endosome and into the cytoplasm, where it enters a transient pool of metabolically active iron known as the labile iron pool (LIP). LIP iron can be utilized for cellular processes such as DNA synthesis, repair, and cell cycling. Alternatively, excess LIP iron can be stored in ferritin (Paul et al., 2017) or exit the cell through ferroportin. Iron storage as ferric iron (Fe3+) within ferritin protein may occur (Ponka et al., 1998; Zandman-Goddard and Shoenfeld, 2007; Finazzi and Arosio, 2014). However, iron must be in the ferrous state (Fe2+) to enter and exit the ferritin molecule (Crichton, 1973; Bezkorovainy, 1989a; Hintze and Theil, 2006). The enzymes ferroxidase and ferrireductase change the state of iron back and forth between ferric (Fe3+) and ferrous (Fe2+). Ferritin synthesis is up regulated by several factors, including high toxic oxygen radical or cytokine concentrations, typically seen during infections. High ferritin production reduces the bioavailability of iron which leads to less reactive oxygen radical production (Koorts and Viljoen, 2007; Zandman-Goddard and Shoenfeld, 2007).
On the other hand, rapid degradation of ferritin could be toxic due to the uncontrolled release of free reactive iron. However, degradation within membrane-encapsulated “secondary lysosomes’ may avoid this problem through the formation of hemosiderin, which is another form of iron storage protein (Harrison and Arosio, 1996). Iron exit depends on the iron export channel ferroportin expression and the hepatic hormone hepcidin concentration. Ferroportin is an iron transporter on the surface of absorptive enterocytes, hepatocytes, and other cells. The main function of ferroportin is to export ferrous iron from iron-containing cells into plasma transferrin as ferric iron (Fe3+). The ferroxidase enzyme oxidizes ferrous iron into ferric iron (Nemeth et al., 2004; Ganz, 2005, 2006, 2007; Drakesmith et al., 2015). Hepcidin is an iron-regulatory hormone synthesized in hepatocytes. Hepcidin binds, internalizes, and degrades the cellular iron exporter ferroportin and thereby decreases iron efflux into plasma. Hepcidin synthesis is stimulated by high plasma iron and iron stores and inhibited by erythropoietic activity (Ganz, 2007). Hepcidin deficiency causes iron overload in hereditary hemochromatosis and ineffective erythropoiesis (Ganz and Nemeth, 2012; Ginzburg, 2019).
Blood transferrin binds to transferrin receptors on epithelial cells of the choroid plexus and oligodendrocytes. Among glial cells, oligodendrocytes synthesize 90% of brain transferrin since iron plays a significant role in their development and in myelin formation (Todorich et al., 2009). Neurons and glial cells take up iron released into the brain interstitium, and apo-transferrin is recycled back to the blood (Bloch et al., 1987; Crowe and Morgan, 1992; Moos, 2002). This process increases during the period of rapid brain growth and iron deficiency and declines with age (Taylor and Morgan, 1990). It also can be reversed (from the brain interstitium back to the blood) during brain iron overload (Broadwell, 1989). Most iron entering the brain across the capillary endothelium finally leaves the system with the bulk outflow of the cerebrospinal fluid (CSF) through the arachnoid villi and other channels (Bradbury, 1997). Interestingly, approximately half of the transferrin in the CSF is derived from the choroid plexus, while the other half comes from the blood in the adult brain (Crowe and Morgan, 1992). Neuronal function is iron-dependent because of the high energy demand, oxidative metabolism, and cytochrome participation in the respiratory chain. The function of oligodendrocytes is also iron-dependent since iron is involved in lipid synthesis needed for myelin synthesis (Connor and Menzies, 1996). Microglial iron is essential for the inflammatory release of hydrolytic enzymes and free radicals via the oxidation of ferrous iron. Microglial iron also participates in the formation of nitric oxide, where iron acts as a co-factor for the nitric oxide synthase enzyme that catalyzes the formation of nitric oxide from the amino acid L-arginine (Moos, 2002). Several iron-related molecular pathways have been reported to be involved in COVID-19 (Farahani et al., 2022).
Uncovering the molecular mechanisms involved in SARS-CoV-2 infection is crucial for a better understanding and management of COVID-19’s consequences. The angiotensin converting enzyme 2 (ACE2)/Angiotensin 1–7 Mas receptor pathway is an important part of the renin-angiotensin system (RAS), which converts angiotensin II into a heptapeptide (Angiotensin 1–7) and angiotensin I into a nonapeptide (Angiotensin 1–9). ACE2 works as a cell surface receptor through which SARS-CoV-2 can enter the cell (De Sousa et al., 2021; Farahani et al., 2022). Viral brain invasion occurs through the olfactory nerve, infection of the vascular endothelium, or migrating leukocytes crossing the blood–brain barrier (Zubair et al., 2020). SARS-CoV-2 infection leads to higher levels of ROS that will cause harmful effects on proteins, lipids, and DNA, creating a similar state to cell necrosis (Kouhpayeh et al., 2021). Ferroptosis is considered a novel type of cell death that shares some aspects with cell necrosis (Anthonymuthu et al., 2021). The excess of iron in the plasma and body organs is tightly related to COVID-19 (Liu P. et al., 2020; Habib et al., 2021; Li et al., 2021; Zhang et al., 2022).
Among the COVID-19 related molecular pathways, there are 22 pathways identified (RAS, NF-kappa B, mTOR, Notch, HIF-1, MAPK, JAK–STAT, TNF signaling pathway, autophagy, apoptosis, necroptosis, B cell receptor signaling pathway, chemokine signaling pathway, IL-17 signaling pathway, natural killer cell mediated cytotoxicity, NOD-like receptor signaling pathway, T cell receptor signaling pathway, Th1 and Th2 cell differentiation, Th17 cell differentiation, toll-like receptor signaling pathway, complement and coagulation cascades, and cytokine-cytokine receptor interaction pathway) with non-cross-talk genes and cross-talk genes making up 561 genes. The cytokine-cytokine receptor interaction pathway is the most significant pathway, presenting 197 crosstalk genes of the 561 total genes (Farahani et al., 2022). ADAM17 is also identified as an important mediator of the major signaling pathways involved in the deleterious consequences of COVID-19 since it processes various substrates, like membrane-anchored cytokines, growth factors, cell adhesion molecules, receptors, and other proteins. The reported damage to body organs and brain regions in COVID-19 results mainly from the cytokine storm, one of the main SARS-CoV-2 infection harmful consequences (Li et al., 2020).
Greater levels of inflammatory cytokines combined with a hypoxic state resulting from pulmonary dysfunction can lead to a reduction in blood flow and oxygen supply (Fratta Pasini et al., 2021). The cytokine storm is a prominent feature of the SARS-CoV-2 infection, instigating systemic flooding with pro-inflammatory cytokines such as interleukin-6 (IL-6), IL-1β, IL-8, interferon-γ (IFN-γ), tumor necrosis factor-alpha (TNF-α), monocyte chemo-attractant protein-1 (MCP-1), and macrophage inflammatory protein-1A (MIP-1A) (Fara et al., 2020; Kempuraj et al., 2020). Moreover, the cytokine storm co-exists with a massive increase in coagulopathies and acute phase reactants such as C-reactive protein (CRP) and serum ferritin which correlate with the severity of the disease (Cheng et al., 2020; Lino et al., 2021; Savla et al., 2021). High levels of peripheral pro-inflammatory cytokines compromise the blood brain barrier (BBB) integrity, cross over to the brain vicinity, and activate its resident immune cells, causing microglial activation which in turn creates a medium of neuroinflammation (Almutairi et al., 2021). Interleukin-6 (IL-6) stimulates the synthesis of ferritin and hepcidin in a cytokine storm (Daher et al., 2017; Bessman et al., 2020). Hepcidin and hepcidin-like proteins bind to ferroportin, the cellular iron exporter, which prevents iron outflow and contributes to enhanced LIP, posing the risk of the Fenton reaction and ferroptosis when GPX4 does not eliminate the excess lipid ROS (Frazer and Anderson, 2014; Ganz, 2018). Hoarding iron into the cell as in cases of iron overload could be detrimental since SARS-CoV-2 replication requires iron (Liu W. et al., 2020). Furthermore, SARS-CoV-2 attacks hemoglobin, leading to iron release into the circulation (Zhang et al., 2022). Therefore, the interaction between the cytokine storm and iron dysregulation, with potential subsequent ferroptosis in COVID-19, could activate molecular mechanisms that result in brain damage. In that case, brain damage could heavily rely on higher hepcidin levels, excessive iron influx through transferrin receptors, and the release of free iron into the circulation due to infection. In addition, during ferroptosis, mitochondria stop elongating, condense, and reduce in size and number. Microglia get activated, engulf synapses, and are polarized to a pro-inflammatory phenotype, flooding the brain with pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), leading to changes in cognition and behavior (Figure 2; Zhang et al., 2022).
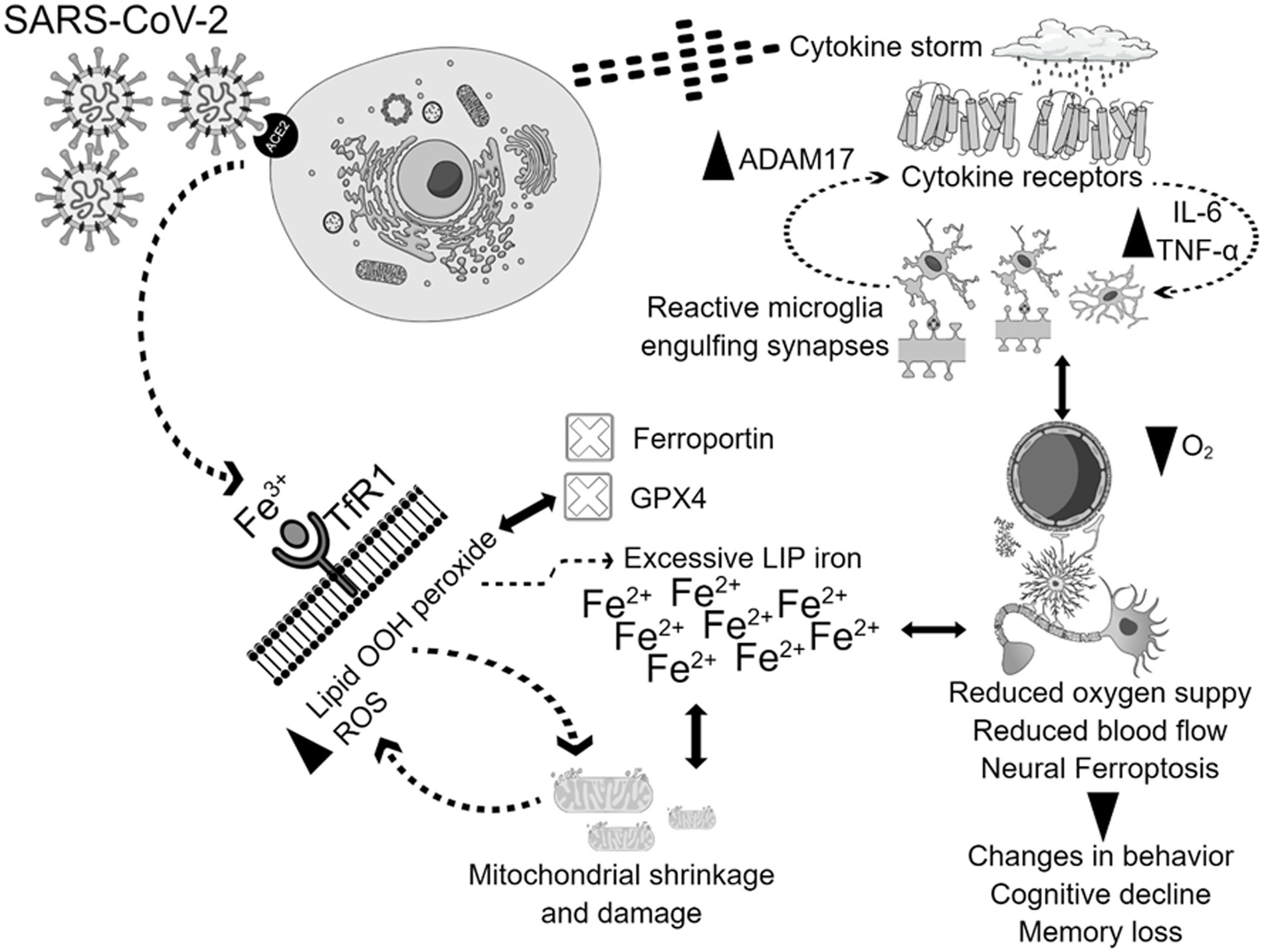
Figure 2. Potential mechanisms of physiological and neurological ferroptosis-associated changes due to SARS-CoV-2 infection. SARS-CoV-2 virus enters the cells through the ACE2 receptor, and triggers cytokine storm, and ferroptosis, which together contributes to mitochondrial damage, and microglial activation. The activation of all these mechanisms together contribute to the enhancement of ADAM17, TNF-, IL-6, with simultaneous inhibition of GPX4, ferroportin, reduction of oxygen supply, and blood flow. These physiological and neurological changes will lead to behavioral disturbances, cognitive decline, and memory loss.
Ferroptosis can be classified as a new type of cell death that is dependent on lipid peroxidation and characterized by mitochondrial shrinkage (Liu P. et al., 2020). Ferroptosis contributes to the development of several pathologic processes, including inflammation and neurodegenerative diseases (Zhang et al., 2022). Multiple neurodegenerative diseases present with iron accumulation and lipid peroxidation in the brain (Farahani et al., 2022). Hambright et al. tested the efficacy of tamoxifen-induced deletion of forebrain neuronal GPx4 gene (Gpx4BIKO mice) as a model of ferroptosis. Gpx4BIKO mice exhibited significant deficits in spatial learning and memory function associated with lipid peroxidation and hippocampal neurodegeneration. Treatment with the ferroptosis inhibitor liproxstatin-1 ameliorated neurodegeneration (Hambright et al., 2017). Recent studies have also documented the role of ferroptosis in mediating cognitive dysfunction in animal models of Alzheimer’s disease (AZ). Using the 5xFAD mouse model (has low GPx4 and cognitive impairment), Chen et al. generated a 5xFAD mice that overexpress Gpx4 (5xFAD/GPX4). These mice with overexpressed GPx4 performed significantly better in memory and learning tasks compared to the control 5xFAD mice and had reduced neurodegeneration (Chen et al., 2022). Bao et al. showed that selective genetic deletion of ferroportin 1 led to ferroptosis, hippocampal atrophy and memory deficits, while restoring ferroportin 1 ameliorated ferroptosis and memory impairment in the APPswe/PS1dE9 mouse model of AZ (Bao et al., 2021). Along the same lines, Hao et al. showed that cognitive dysfunction in the streptozotocin rat model of type 1 diabetes is related to hippocampal iron overload and ferroptosis mediated by down regulation of ferroportin 1 gene (Hao et al., 2021). A growing body of evidence suggests ferroptosis as a plausible mechanism behind the SARS-CoV-2-associated neuropsychiatric symptoms, cognitive decline, and memory loss (Zhang et al., 2022). In an ischemic stroke model, Acyl-coenzyme A synthase long-chain family member 4 (ACSL4), which is crucial to ferroptosis-related lipid peroxidation, promoted ferroptosis-induced brain injury and neuroinflammation with similar findings to neuro-COVID-19 events, such as infarct size increase, reduced neurological function, microglial activation, and increased pro-inflammatory cytokines (Cui et al., 2021). In the context of COVID 19, we can see glimpses of potential iron perturbation along with neuroinflammation in several forms. High serum ferritin (Cheng et al., 2020) and hepcidin levels (Hortová-Kohoutková et al., 2023), low serum iron levels (Gaiatto et al., 2023), and low transferrin saturation (Claise et al., 2022) have been significantly correlated with COVID-19 severity, hospitalization, and mortality (Zhou et al., 2020; Kaushal et al., 2022; Suriawinata and Mehta, 2022). In COVID-19 patients, altered iron metabolism, depletion of glutathione (GSH) (Kumar et al., 2022), inactivation of GPX4 (Muhammad et al., 2021), and up regulation of lipid peroxidation biomarkers strongly propose ferroptosis as a plausible mechanism for COVID-19 multi-organ affection, including neuropsychiatric sequelae (Yang and Lai, 2020).
It is repeatedly reported that the use of multiple iron chelators such as deferoxamine prevents the formation of ROS by averting electron donation from iron to oxygen, which could minimize ferroptosis (Ren et al., 2020; Anthonymuthu et al., 2021). To prevent ferroptosis, ferrostatin-1 plays a role as a lipid ROS scavenger (Yang and Stockwell, 2016). Ferrostatin-1 alleviates angiotensin II-induced inflammation and ferroptosis by inhibiting the enhancement of ROS levels in astrocytes and the subsequent reactive gliosis (Li et al., 2021). Another recent study showed that ferrostatin-1 diminishes the levels of ROS and malondialdehyde and enhances superoxide dismutase activity in HT-22 cells, revealing a protective effect of this ferroptosis inhibitor (Chu et al., 2020). Curiously, a recent study reported that ferrostatin-1, in the presence of reduced iron levels, eliminates lipid hydroperoxides, presenting a similar effect as GPX4 (Miotto et al., 2020). The use of deferoxamine as a ferroptosis inhibitor showed success in reducing inflammation and improving memory in different models of neurodegenerative diseases (Xue et al., 2016; Fine et al., 2020; Lee et al., 2021). These results propose ferroptosis inhibition as a plausible approach to managing the post-COVID neurological disturbances (Table 1).
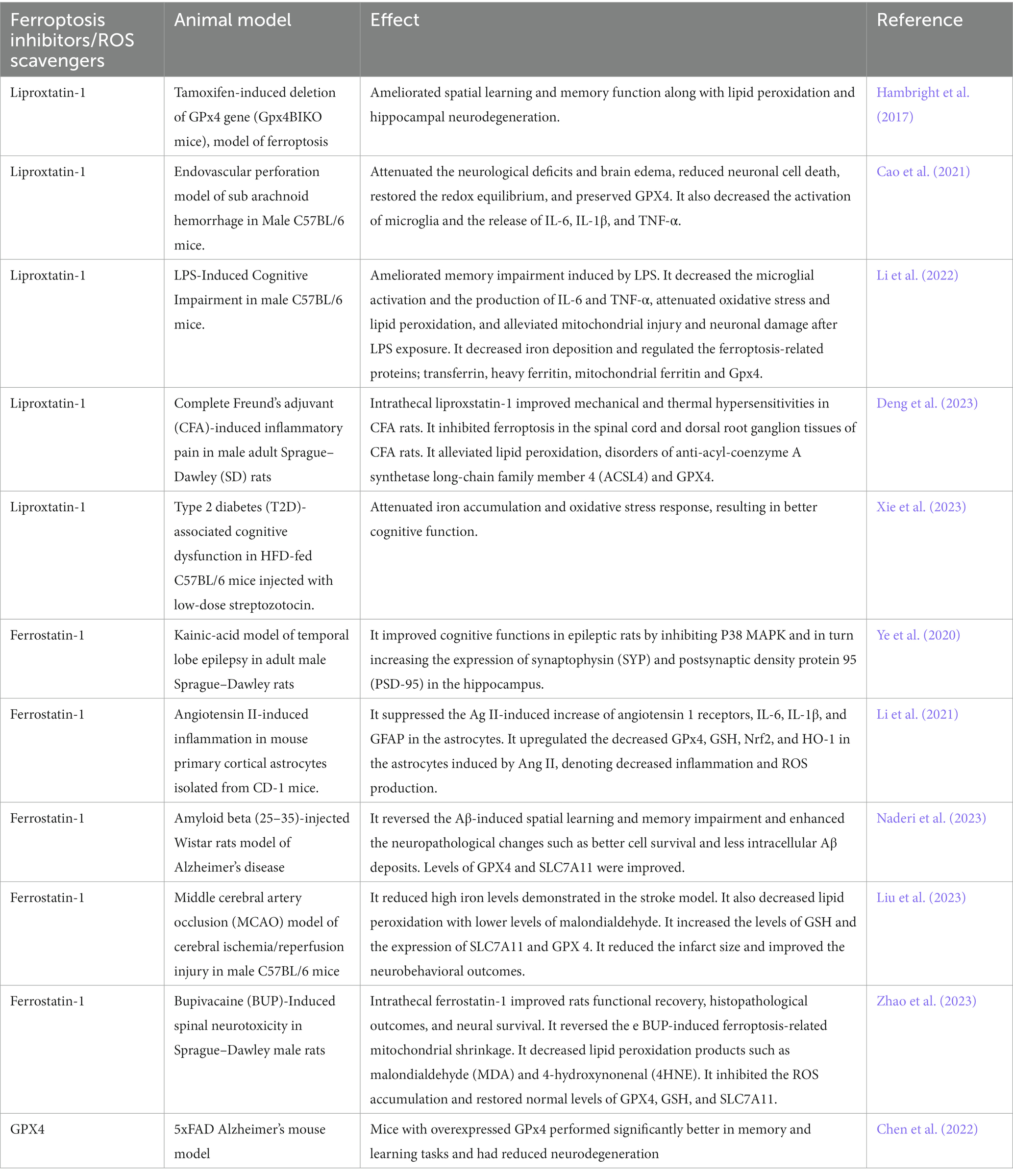
Table 1. Ferroptosis inhibitors/ROS scavengers.
The attenuation of ferroptosis as a potential therapeutic target for neurological post-COVID syndrome is not yet fully established. However, inactivation of GPX4 and up regulation of lipid peroxidation and ROS are constitutive components of both SARS-CoV-2 infection and ferroptosis, suggesting a potentially major role for ferroptosis inhibitors. Identifying the possible beneficial molecular changes in the brain caused by these inhibitors in the context of COVID-19 would provide a great insight into managing post-COVID neuropsychiatric manifestations.
OA: concept and design, drafting of the manuscript, and supervision. All authors: critical revision of the manuscript for important intellectual content.
This work was funded by the Department of Psychiatry and Psychology at the Mayo Clinic Arizona.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abulseoud, O. A., Yehia, A., Egol, C. J., Nettey, V. N., Aly, M., Qu, Y., et al. (2022). Attenuated initial serum ferritin concentration in critically ill coronavirus disease 2019 geriatric patients with comorbid psychiatric conditions. Front. Psych. 13:1035986. doi: 10.3389/fpsyt.2022.1035986
Almutairi, M. M., Sivandzade, F., Albekairi, T. H., Alqahtani, F., and Cucullo, L. (2021). Neuroinflammation and its impact on the pathogenesis of COVID-19. Front. Med. 8:745789. doi: 10.3389/fmed.2021.745789
Anthonymuthu, T. S., Tyurina, Y. Y., Sun, W. Y., Mikulska-Ruminska, K., Shrivastava, I. H., Tyurin, V. A., et al. (2021). Resolving the paradox of ferroptotic cell death: Ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 38:101744. doi: 10.1016/j.redox.2020.101744
Bao, W.-D., Pang, P., Zhou, X.-T., Fan, H., Xiong, W., Chen, K., et al. (2021). Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Different. 28, 1548–1562. doi: 10.1038/s41418-020-00685-9
Barisani, D., and Conte, D. (2002). Transferrin receptor 1 (TfR1) and putative stimulator of Fe transport (SFT) expression in iron deficiency and overload: an overview. Blood Cells Mol. Dis. 29, 498–505. doi: 10.1006/bcmd.2002.0588
Bessman, N. J., Mathieu, J. R. R., Renassia, C., Zhou, L., Fung, T. C., Fernandez, K. C., et al. (2020). Dendritic cell-derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science 368, 186–189. doi: 10.1126/science.aau6481
Bezkorovainy, A. (1989a). Biochemistry of nonheme iron in man. I. Iron proteins and cellular iron metabolism. Clin. Physiol. Biochem. 7, 1–17.
Bezkorovainy, A. (1989b). Biochemistry of nonheme iron in man. II. Absorption of iron. Clin. Physiol. Biochem. 7, 53–69.
Bloch, B., Popovici, T., Chouham, S., Levin, M. J., Tuil, D., and Kahn, A. (1987). Transferrin gene expression in choroid plexus of the adult rat brain. Brain Res. Bull. 18, 573–576. doi: 10.1016/0361-9230(87)90122-5
Bradbury, M. W. (1997). Transport of iron in the blood-brain-cerebrospinal fluid system. J. Neurochem. 69, 443–454.
Broadwell, R. D. (1989). Transcytosis of macromolecules through the blood-brain barrier: a cell biological perspective and critical appraisal. Acta Neuropathol. 79, 117–128. doi: 10.1007/BF00294368
Cabantchik, Z. I. (2014). Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Front. Pharmacol. 5:45. doi: 10.3389/fphar.2014.00045
Campos, B. P. e. S., dos Santos Gomes, G. D., de Sousa Braz, A., and de Vilela, A. T. (2019). Cardiovascular risk factors and risk measurement in patients with psoriatic arthritis in a university hospital. Int. J. Cardiovasc. Sci. 33, 112–118.
Cao, Y., Li, Y., He, C., Yan, F., Li, J.-R., Hang-Zhe, X., et al. (2021). Selective ferroptosis inhibitor liproxstatin-1 attenuates neurological deficits and neuroinflammation after subarachnoid hemorrhage. Neurosci. Bull. 37, 535–549. doi: 10.1007/s12264-020-00620-5
Chen, L., Dar, N. J., Na, R., McLane, K. D., Yoo, K., Han, X., et al. (2022). Enhanced defense against ferroptosis ameliorates cognitive impairment and reduces neurodegeneration in 5xFAD mice. Free Radic. Biol. Med. 180, 1–12. doi: 10.1016/j.freeradbiomed.2022.01.002
Cheng, L., Li, H., Li, L., Liu, C., Yan, S., Chen, H., et al. (2020). Ferritin in the coronavirus disease 2019 (COVID-19): a systematic review and meta-analysis. J. Clin. Lab. Anal. 34:e23618. doi: 10.1002/jcla.23618
Chu, J., Liu, C. X., Song, R., and Li, Q. L. (2020). Ferrostatin-1 protects HT-22 cells from oxidative toxicity. Neural Regen. Res. 15, 528–536. doi: 10.4103/1673-5374.266060
Claise, C., Saleh, J., Rezek, M., Vaulont, S., Peyssonnaux, C., and Edeas, M. (2022). Low transferrin levels predict heightened inflammation in patients with COVID-19: new insights. Int. J. Infect. Dis. 116, 74–79. doi: 10.1016/j.ijid.2021.12.340
Connor, J. R., and Menzies, S. L. (1996). Relationship of iron to oligodendrocytes and myelination. Glia 17, 83–93. doi: 10.1002/(SICI)1098-1136(199606)17:2<83::AID-GLIA1>3.0.CO;2-7
Crichton, R. R. (1973). Structure and function of ferritin. Angew. Chem. Int. Ed. Engl. 12, 57–65. doi: 10.1002/anie.197300571
Crowe, A., and Morgan, E. H. (1992). Iron and transferrin uptake by brain and cerebrospinal fluid in the rat. Brain Res. 592, 8–16. doi: 10.1016/0006-8993(92)91652-U
Cui, Y., Zhang, Y., Zhao, X., Shao, L., Liu, G., Sun, C., et al. (2021). ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 93, 312–321. doi: 10.1016/j.bbi.2021.01.003
Daher, R., Manceau, H., and Karim, Z. (2017). Iron metabolism and the role of the iron-regulating hormone hepcidin in health and disease. Presse Med. 46, e272–e278. doi: 10.1016/j.lpm.2017.10.006
Daubner, S. C., Le, T., and Wang, S. (2011). Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 508, 1–12. doi: 10.1016/j.abb.2010.12.017
Deng, Y.-F., Xiang, P., Jing-Yi, D., Liang, J.-F., and Li, X. (2023). Intrathecal liproxstatin-1 delivery inhibits ferroptosis and attenuates mechanical and thermal hypersensitivities in rats with complete Freund’s adjuvant-induced inflammatory pain. Neural Regen. Res. 18, 456–462. doi: 10.4103/1673-5374.346547
Drakesmith, H., Nemeth, E., and Ganz, T. (2015). Ironing out Ferroportin. Cell Metab. 22, 777–787. doi: 10.1016/j.cmet.2015.09.006
Fara, A., Mitrev, Z., Rosalia, R. A., and Assas, B. M. (2020). Cytokine storm and COVID-19: a chronicle of pro-inflammatory cytokines. Open Biol. 10:200160. doi: 10.1098/rsob.200160
Farahani, M., Niknam, Z., Mohammadi Amirabad, L., Amiri-Dashatan, N., Koushki, M., Nemati, M., et al. (2022). Molecular pathways involved in COVID-19 and potential pathway-based therapeutic targets. Biomed. Pharmacother. 145:112420. doi: 10.1016/j.biopha.2021.112420
Finazzi, D., and Arosio, P. (2014). Biology of ferritin in mammals: an update on iron storage, oxidative damage and neurodegeneration. Arch. Toxicol. 88, 1787–1802. doi: 10.1007/s00204-014-1329-0
Fine, J. M., Kosyakovsky, J., Baillargeon, A. M., Tokarev, J. V., Cooner, J. M., Svitak, A. L., et al. (2020). Intranasal deferoxamine can improve memory in healthy C57 mice, suggesting a partially non-disease-specific pathway of functional neurologic improvement. Brain Behav. 10:e01536. doi: 10.1002/brb3.1536
Fratta Pasini, A. M., Stranieri, C., Girelli, D., Busti, F., and Cominacini, L. (2021). Is Ferroptosis a key component of the process leading to multiorgan damage in COVID-19? Antioxidants 10:1677. doi: 10.3390/antiox10111677
Frazer, D. M., and Anderson, G. J. (2014). The regulation of iron transport. Biofactors 40, 206–214. doi: 10.1002/biof.1148
Gaiatto, A. C., Macedo, T. A., Bibo, N. D. G. M., Raimundo, J. R. S., da Costa Aguiar Alves, B., Gascón, T., et al. (2023). COVID-19 compromises iron homeostasis: transferrin as a target of investigation. J. Trace Elem. Med. Biol. 76:127109. doi: 10.1016/j.jtemb.2022.127109
Ganz, T. (2005). Hepcidin--a regulator of intestinal iron absorption and iron recycling by macrophages. Best Pract. Res. Clin. Haematol. 18, 171–182. doi: 10.1016/j.beha.2004.08.020
Ganz, T. (2006). Hepcidin--a peptide hormone at the interface of innate immunity and iron metabolism. Curr. Top. Microbiol. Immunol. 306, 183–198. doi: 10.1007/3-540-29916-5_7
Ganz, T. (2007). Molecular control of iron transport. J. Am. Soc. Nephrol. 18, 394–400. doi: 10.1681/ASN.2006070802
Ganz, T., and Nemeth, E. (2012). Hepcidin and iron homeostasis. Biochim. Biophys. Acta 1823, 1434–1443. doi: 10.1016/j.bbamcr.2012.01.014
Gille, G., and Reichmann, H. (2011). Iron-dependent functions of mitochondria--relation to neurodegeneration. J. Neural Transm. (Vienna) 118, 349–359. doi: 10.1007/s00702-010-0503-7
Ginzburg, Y. Z. (2019). Hepcidin-ferroportin axis in health and disease. Vitam. Horm. 110, 17–45. doi: 10.1016/bs.vh.2019.01.002
Graham, R. M., Chua, A. C., Herbison, C. E., Olynyk, J. K., and Trinder, D. (2007). Liver iron transport. World J. Gastroenterol. 13, 4725–4736. doi: 10.3748/wjg.v13.i35.4725
Habib, H. M., Ibrahim, S., Zaim, A., and Ibrahim, W. H. (2021). The role of iron in the pathogenesis of COVID-19 and possible treatment with lactoferrin and other iron chelators. Biomed. Pharmacother. 136:111228. doi: 10.1016/j.biopha.2021.111228
Hambright, W. S., Fonseca, R. S., Chen, L., Na, R., and Ran, Q. (2017). Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17. doi: 10.1016/j.redox.2017.01.021
Hao, L., Mi, J., Song, L., Guo, Y., Li, Y., Yin, Y., et al. (2021). SLC40A1 mediates ferroptosis and cognitive dysfunction in type 1 diabetes. Neuroscience 463, 216–226. doi: 10.1016/j.neuroscience.2021.03.009
Harrison, P. M., and Arosio, P. (1996). The ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1275, 161–203. doi: 10.1016/0005-2728(96)00022-9
Hintze, K. J., and Theil, E. C. (2006). Cellular regulation and molecular interactions of the ferritins. Cell. Mol. Life Sci. 63, 591–600. doi: 10.1007/s00018-005-5285-y
Hortová-Kohoutková, M., Skotáková, M., Onyango, I. G., Slezáková, M., Panovský, R., Opatřil, L., et al. (2023). Hepcidin and ferritin levels as markers of immune cell activation during septic shock, severe COVID-19 and sterile inflammation. Front. Immunol. 14:1110540. doi: 10.3389/fimmu.2023.1110540
Hosseinzadeh, P., and Lu, Y. (2016). Design and fine-tuning redox potentials of metalloproteins involved in electron transfer in bioenergetics. Biochim. Biophys. Acta 1857, 557–581. doi: 10.1016/j.bbabio.2015.08.006
Huebers, H. A., Huebers, E., Csiba, E., Rummel, W., and Finch, C. A. (1983). The significance of transferrin for intestinal iron absorption. Blood 61, 283–290. doi: 10.1182/blood.V61.2.283.283
Improta-Caria, A. C., Soci, Ú. P. R., Pinho, C. S., Júnior, R. A., De Sousa, R. A. L., and Bessa, T. C. B. (2021). Physical exercise and immune system: perspectives on the COVID-19 pandemic. Rev. Assoc. Med. Bras. 67, 102–107. doi: 10.1590/1806-9282.67.suppl1.20200673
Júnior, R. A., Durães, A., Roever, L., Macedo, C., Aras, M. G., Nascimento, L., et al. (2021). The impact of COVID-19 on the cardiovascular system. Rev. Assoc. Med. Bras. 67, 163–167. doi: 10.1590/1806-9282.67.suppl1.20201063
Kappert, K., Jahic, A., and Tauber, R. (2020). Assessment of serum ferritin as a biomarker in COVID-19: bystander or participant? Insights by comparison with other infectious and non-infectious diseases. Biomarkers 25, 616–625. doi: 10.1080/1354750X.2020.1797880
Kaushal, K., Kaur, H., Sarma, P., Bhattacharyya, A., Sharma, D. J., Prajapat, M., et al. (2022). Serum ferritin as a predictive biomarker in COVID-19. A systematic review, meta-analysis and meta-regression analysis. J. Crit. Care 67, 172–181. doi: 10.1016/j.jcrc.2021.09.023
Kempuraj, D., Selvakumar, G. P., Ahmed, M. E., Raikwar, S. P., Thangavel, R., Khan, A., et al. (2020). COVID-19, mast cells, cytokine storm, psychological stress, and neuroinflammation. Neuroscientist 26, 402–414. doi: 10.1177/1073858420941476
Kennedy, M. C., Emptage, M. H., Dreyer, J. L., and Beinert, H. (1983). The role of iron in the activation-inactivation of aconitase. J. Biol. Chem. 258, 11098–11105. doi: 10.1016/S0021-9258(17)44390-0
Koorts, A. M., and Viljoen, M. (2007). Ferritin and ferritin isoforms II: protection against uncontrolled cellular proliferation, oxidative damage and inflammatory processes. Arch. Physiol. Biochem. 113, 55–64. doi: 10.1080/13813450701422575
Kouhpayeh, S., Shariati, L., Boshtam, M., Rahimmanesh, I., Mirian, M., Esmaeili, Y., et al. (2021). The molecular basis of COVID-19 pathogenesis, conventional and nanomedicine therapy. Int. J. Mol. Sci. 22:5438. doi: 10.3390/ijms22115438
Kumar, P., Osahon, O., Vides, D. B., Hanania, N., Minard, C. G., and Sekhar, R. V. (2022). Severe glutathione deficiency, oxidative stress and oxidant damage in adults hospitalized with COVID-19: implications for GlyNAC (glycine and N-acetylcysteine) supplementation. Antioxidants 11:50. doi: 10.3390/antiox11010050
Lall, M. M., Ferrell, J., Nagar, S., Fleisher, L. N., and McGahan, M. C. (2008). Iron regulates L-cystine uptake and glutathione levels in lens epithelial and retinal pigment epithelial cells by its effect on cytosolic aconitase. Invest. Ophthalmol. Vis. Sci. 49, 310–319. doi: 10.1167/iovs.07-1041
Lee, J. X., Chieng, W. K., Abdul Jalal, M. I., Tan, C. E., and Lau, S. C. D. (2022). Role of serum ferritin in predicting outcomes of COVID-19 infection among sickle cell disease patients: a systematic review and meta-analysis. Front. Med. 9:919159. doi: 10.3389/fmed.2022.919159
Lee, K. E., Mo, S., Lee, H. S., Jeon, M., Song, J. S., Choi, H. J., et al. (2021). Deferoxamine reduces inflammation and osteoclastogenesis in avulsed teeth. Int. J. Mol. Sci. 22:8225. doi: 10.3390/ijms22158225
Li, Z., Liu, T., Yang, N., Han, D., Mi, X., Li, Y., et al. (2020). Neurological manifestations of patients with COVID-19: potential routes of SARS-CoV-2 neuroinvasion from the periphery to the brain. Front. Med. 14, 533–541. doi: 10.1007/s11684-020-0786-5
Li, Y., Sun, M., Fuyang Cao, Y., Chen, L. Z., Li, H., Cao, J., et al. (2022). The ferroptosis inhibitor liproxstatin-1 ameliorates LPS-induced cognitive impairment in mice. Nutrients 14:4599. doi: 10.3390/nu14214599
Li, S., Zhou, C., Zhu, Y., Chao, Z., Sheng, Z., Zhang, Y., et al. (2021). Ferrostatin-1 alleviates angiotensin II (Ang II)- induced inflammation and ferroptosis in astrocytes. Int. Immunopharmacol. 90:107179. doi: 10.1016/j.intimp.2020.107179
Lino, K., Guimarães, G. M. C., Alves, L. S., Oliveira, A. C., Faustino, R., Fernandes, C. S., et al. (2021). Serum ferritin at admission in hospitalized COVID-19 patients as a predictor of mortality. Braz. J. Infect. Dis. 25:101569. doi: 10.1016/j.bjid.2021.101569
Liu, P., Feng, Y., Li, H., Chen, X., Wang, G., Xu, S., et al. (2020). Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell. Mol. Biol. Lett. 25:10. doi: 10.1186/s11658-020-00205-0
Liu, X., Yue, D., Liu, J., Cheng, L., He, W., and Zhang, W. (2023). Ferrostatin-1 alleviates cerebral ischemia/reperfusion injury through activation of the AKT/GSK3β signaling pathway. Brain Res. Bull. 193, 146–157. doi: 10.1016/j.brainresbull.2022.12.009
Liu, W., Zhang, S., Nekhai, S., and Liu, S. (2020). Depriving Iron supply to the virus represents a promising adjuvant therapeutic against viral survival. Curr. Clin. Microbiol. Rep. 7, 13–19. doi: 10.1007/s40588-020-00140-w
Matsui, M. (1996). Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev. Biol. 178, 179–185. doi: 10.1006/dbio.1996.0208
McGahan, M. C., Harned, J., Mukunnemkeril, M., Goralska, M., Fleisher, L., and Ferrell, J. B. (2005). Iron alters glutamate secretion by regulating cytosolic aconitase activity. Am. J. Physiol. Cell Physiol. 288, C1117–C1124. doi: 10.1152/ajpcell.00444.2004
Miotto, G., Rossetto, M., Di Paolo, M. L., Orian, L., Venerando, R., Roveri, A., et al. (2020). Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 28:101328. doi: 10.1016/j.redox.2019.101328
Morgan, E. H., and Oates, P. S. (2002). Mechanisms and regulation of intestinal iron absorption. Blood Cells Mol. Dis. 29, 384–399. doi: 10.1006/bcmd.2002.0578
Muhammad, Y., Kani, Y. A., Iliya, S., Muhammad, J. B., Binji, A., Ahmad, A. E.-F., et al. (2021). Deficiency of antioxidants and increased oxidative stress in COVID-19 patients: a cross-sectional comparative study in Jigawa, Northwestern Nigeria. SAGE Open Med. 9:205031212199124. doi: 10.1177/2050312121991246
Naderi, S., Khodagholi, F., Pourbadie, H. G., Naderi, N., Rafiei, S., Janahmadi, M., et al. (2023). Role of amyloid beta (25− 35) neurotoxicity in the ferroptosis and necroptosis as modalities of regulated cell death in Alzheimer’s disease. Neurotoxicology 94, 71–86. doi: 10.1016/j.neuro.2022.11.003
Nemeth, E., Tuttle, M. S., Powelson, J., Vaughn, M. B., Donovan, A., Ward, D. M., et al. (2004). Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093. doi: 10.1126/science.1104742
Neyens, E., and Baeyens, J. (2003). A review of classic Fenton’s peroxidation as an advanced oxidation technique. J. Hazard. Mater. 98, 33–50. doi: 10.1016/S0304-3894(02)00282-0
Pandey, K., Thurman, M., Johnson, S. D., Acharya, A., Johnston, M., Klug, E. A., et al. (2021). Mental health issues during and after COVID-19 vaccine era. Brain Res. Bull. 176, 161–173. doi: 10.1016/j.brainresbull.2021.08.012
Para, O., Caruso, L., Pestelli, G., Tangianu, F., Carrara, D., Maddaluni, L., et al. (2022). Ferritin as prognostic marker in COVID-19: the FerVid study. Postgrad. Med. 134, 58–63. doi: 10.1080/00325481.2021.1990091
Paul, B. T., Manz, D. H., Torti, F. M., and Torti, S. V. (2017). Mitochondria and Iron: current questions. Expert. Rev. Hematol. 10, 65–79. doi: 10.1080/17474086.2016.1268047
Ponka, P., Beaumont, C., and Richardson, D. R. (1998). Function and regulation of transferrin and ferritin. Semin. Hematol. 35, 35–54.
Ramsey, A. J., Hillas, P. J., and Fitzpatrick, P. F. (1996). Characterization of the active site iron in tyrosine hydroxylase. Redox states of the iron. J. Biol. Chem. 271, 24395–24400.
Ren, J. X., Sun, X., Yan, X. L., Guo, Z. N., and Yang, Y. (2020). Ferroptosis in neurological diseases. Front. Cell. Neurosci. 14:218. doi: 10.3389/fncel.2020.00218
Robbins, A. H., and Stout, C. D. (1989). The structure of aconitase. Proteins 5, 289–312. doi: 10.1002/prot.340050406
Salari, N., Hosseinian-Far, A., Jalali, R., Vaisi-Raygani, A., Rasoulpoor, S., Mohammadi, M., et al. (2020). Prevalence of stress, anxiety, depression among the general population during the COVID-19 pandemic: a systematic review and meta-analysis. Glob. Health 16, 1–11. doi: 10.1186/s12992-020-00589-w
Savla, S. R., Prabhavalkar, K. S., and Bhatt, L. K. (2021). Cytokine storm associated coagulation complications in COVID-19 patients: pathogenesis and management. Expert Rev. Anti-Infect. Ther. 19, 1397–1413. doi: 10.1080/14787210.2021.1915129
Seiler, A., Schneider, M., Forster, H., Roth, S., Wirth, E. K., Culmsee, C., et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8, 237–248. doi: 10.1016/j.cmet.2008.07.005
Sherry, B. D., and Fürstner, A. (2008). The promise and challenge of Iron-catalyzed cross coupling. Acc. Chem. Res. 41, 1500–1511. doi: 10.1021/ar800039x
Sousa, D., Leoni, R. A., Improta-caria, A. C., Aras-júnior, R., De Oliveira, E. M., Soci, Ú. P. R., et al. (2021). Physical exercise effects on the brain during COVID-19 pandemic: links between mental and cardiovascular health. Neurol. Sci. 42, 1325–1334. doi: 10.1007/s10072-021-05082-9
Suriawinata, E., and Mehta, K. J. (2022). Iron and iron-related proteins in COVID-19. Clin. Exp. Med. 1591–9528. doi: 10.1007/s10238-022-00851-y
Tamir, H., and Liu, K.-P. (1982). On the nature of the interaction between serotonin and serotonin binding protein: effect of nucleotides, ions, and sulfhydryl reagents. J. Neurochem. 38, 135–141. doi: 10.1111/j.1471-4159.1982.tb10864.x
Taquet, M., Sillett, R., Zhu, L., Mendel, J., Camplisson, I., Dercon, Q., et al. (2022). Neurological and psychiatric risk trajectories after SARS-CoV-2 infection: an analysis of 2-year retrospective cohort studies including 1 284 437 patients. Lancet Psychiatry 9, 815–827. doi: 10.1016/S2215-0366(22)00260-7
Taylor, E. M., and Morgan, E. H. (1990). Developmental changes in transferrin and iron uptake by the brain in the rat. Brain Res. Dev. Brain Res. 55, 35–42. doi: 10.1016/0165-3806(90)90103-6
Todorich, B., Pasquini, J. M., Garcia, C. I., Paez, P. M., and Connor, J. R. (2009). Oligodendrocytes and myelination: the role of iron. Glia 57, 467–478. doi: 10.1002/glia.20784
Wang, L., Davis, P. B., Volkow, N. D., Berger, N. A., Kaelber, D. C., and Xu, R. (2022). Association of COVID-19 with new-onset Alzheimer’s disease. J. Alzheimers Dis. 89, 411–414. doi: 10.3233/JAD-220717
Winterbourn, C. C. (1995). Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol. Lett. 82-83, 969–974. doi: 10.1016/0378-4274(95)03532-X
Xie, Z., Wang, X., Luo, X., Yan, J., Zhang, J., Sun, R., et al. (2023). Activated AMPK mitigates diabetes-related cognitive dysfunction by inhibiting hippocampal ferroptosis. Biochem. Pharmacol. 207:115374. doi: 10.1016/j.bcp.2022.115374
Xue, H., Chen, D., Zhong, Y. K., Zhou, Z. D., Fang, S. X., Li, M. Y., et al. (2016). Deferoxamine ameliorates hepatosteatosis via several mechanisms in Ob/Ob mice. Ann. N. Y. Acad. Sci. 1375, 52–65. doi: 10.1111/nyas.13174
Yang, M., and Lai, C. L. (2020). SARS-CoV-2 infection: can ferroptosis be a potential treatment target for multiple organ involvement? Cell Death Discovery 6:130. doi: 10.1038/s41420-020-00369-w
Yang, W. S., and Stockwell, B. R. (2016). Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 26, 165–176. doi: 10.1016/j.tcb.2015.10.014
Yant, L. J., Ran, Q., Rao, L., Van Remmen, H., Shibatani, T., Belter, J. G., et al. (2003). The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 34, 496–502. doi: 10.1016/S0891-5849(02)01360-6
Ye, Q., Zeng, C., Luo, C., and Yuan, W. (2020). Ferrostatin-1 mitigates cognitive impairment of epileptic rats by inhibiting P38 MAPK activation. Epilepsy Behav. 103:106670. doi: 10.1016/j.yebeh.2019.106670
Zandman-Goddard, G., and Shoenfeld, Y. (2007). Ferritin in autoimmune diseases. Autoimmun. Rev. 6, 457–463. doi: 10.1016/j.autrev.2007.01.016
Zhang, R., Sun, C., Chen, X., Han, Y., Zang, W., Jiang, C., et al. (2022). COVID-19-related brain injury: the potential role of ferroptosis. J. Inflamm. Res. 15, 2181–2198. doi: 10.2147/JIR.S353467
Zhao, Y., Luo, Y., Liu, Z., Chen, Y., Wei, L., Luo, X., et al. (2023). Ferrostatin-1 ameliorates bupivacaine-induced spinal neurotoxicity in rats by inhibiting ferroptosis. Neurosci. Lett. 809:137308. doi: 10.1016/j.neulet.2023.137308
Zhou, C., Chen, Y., Ji, Y., He, X., and Xue, D. (2020). Increased serum levels of hepcidin and ferritin are associated with severity of COVID-19. Med. Sci. Monit. 26:e926178. doi: 10.12659/MSM.926178
Keywords: ferroptosis, post-COVID syndrome, SARS-CoV-2, therapeutic target, long COVID
Citation: Sousa RAL, Yehia A and Abulseoud OA (2023) Attenuation of ferroptosis as a potential therapeutic target for neuropsychiatric manifestations of post-COVID syndrome. Front. Neurosci. 17:1237153. doi: 10.3389/fnins.2023.1237153
Edited by:
Song Zhang, Shanghai Jiao Tong University, ChinaReviewed by:
Barnali Ray Basu, University of Calcutta, IndiaCopyright © 2023 Sousa, Yehia and Abulseoud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Osama A. Abulseoud, QWJ1bHNlb3VkLm9zYW1hQE1heW8uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.