 Osama Hamadelseed
Osama Hamadelseed Mike K. S. Chan2,3
Mike K. S. Chan2,3 Thomas Skutella
Thomas Skutella- 1Department of Neuroanatomy, Institute of Anatomy and Cell Biology, University of Heidelberg, Heidelberg, Germany
- 2EW European Wellness Academy GmbH, Edenkoben, Germany
- 3Baden R&D Laboratories GmbH, Edenkoben, Germany
- 4Stellar Biomolecular Research GmbH, Edenkoben, Germany
Objectives: We critically review research findings on the unique changes in brain structure and cognitive function characteristic of Down syndrome (DS) and summarize the similarities and differences with other neurodevelopmental disorders such as Williams syndrome, 22q11.2 deletion syndrome, and fragile X syndrome.
Methods: We conducted a meta-analysis and systematic literature review of 84 studies identified by searching PubMed, Google Scholar, and Web of Science from 1977 to October 2022. This review focuses on the following issues: (1) specific neuroanatomic and histopathological features of DS as revealed by autopsy and modern neuroimaging modalities, (2) language and memory deficits in DS, (3) the relationships between these neuroanatomical and neuropsychological features, and (4) neuroanatomic and neuropsychological differences between DS and related neurodevelopmental syndromes.
Results: Numerous post-mortem and morphometric neuroimaging investigations of individuals with DS have reported complex changes in regional brain volumes, most notably in the hippocampal formation, temporal lobe, frontal lobe, parietal lobe, and cerebellum. Moreover, neuropsychological assessments have revealed deficits in language development, emotional regulation, and memory that reflect these structural changes and are more severe than expected from general cognitive dysfunction. Individuals with DS also show relative preservation of multiple cognitive, linguistic, and social domains compared to normally developed controls and individuals with other neurodevelopmental disorders. However, all these neurodevelopment disorders exhibit substantial heterogeneity among individuals.
Conclusion: People with Down syndrome demonstrate unique neurodevelopmental abnormalities but cannot be regarded as a homogenous group. A comprehensive evaluation of individual intellectual skills is essential for all individuals with neurodevelopment disorders to develop personalized care programs.
1. Introduction
1.1. The development of Down syndrome
Down syndrome (DS) is the most common chromosomal abnormality associated with intellectual disability (Nadel, 2003). Individuals with DS harbor an extra (third) copy of chromosome 21 in all somatic cells, so DS is also referred to as trisomy 21. The extra copy results from a fertilization event involving an egg or sperm carrying two copies of chromosome 21 due to “non-disjunction” or failure of chromosome separation during meiosis. The cause of non-disjunction is unknown, although studies show it occurs more frequently with maternal age. There are no known environmental or behavioral factors that directly cause non-disjunction.1 The DS phenotype is also the same whether the extra chromosome comes from the egg or sperm. Further, there is no evidence that nationality, ethnicity, diet, medication use, illness history, or upbringing influences DS risk. Because trisomy 21 is present from conception, maternal behavior during pregnancy does not affect the disease course. In contrast to maternal age, paternal age appears to have little or no impact on DS risk. Two theories have been proposed to explain the influence of maternal age. According to one theory, all women carry a few eggs with an extra chromosome, which are more likely to be released at the end of reproductive life. Alternatively, the second theory posits that the rate of trisomic conception is the same in all maternal age groups but that impaired pregnancies are more likely to continue in older women (i.e., are less likely to end in miscarriage) due to some mechanism that protects later pregnancies.2
1.2. Clinical and pathological features of Down syndrome
The clinical characteristics of DS include physical abnormalities, intellectual disabilities, and deficits in emotional regulation. Physical features indicative of DS include a small chin, slanted eyes, poor muscle tone, a flat nasal bridge, a single crease on the palm, a large protruding tongue, and a relatively small mouth. Emotional abnormalities such as depression increase in number and severity with age and may be comorbid with neurological sequela, including epilepsy. For instance, Evenhuis reported generalized tonic–clonic seizures in 6 of 12 moderately intellectually impaired (IQ 35–55) individuals with DS and in all five severely restricted (IQ 25–35) individuals (Evenhuis, 1990). Lai and Williams (1989) also reported markedly elevated incidences of epilepsy (84%) and Parkinsonism (20%) in DS, while Ropper and Williams (1980) reported significantly increased incidences of depression and dementia. Individuals with DS also demonstrate substantial delays and limitations in speech development (Chapman and Hesketh, 2000). Many pathological changes observed in young people with DS are absent in utero and emerge during early postnatal development. The proportion of DS individuals with cognitive impairment increases progressively with age, a trend resembling Alzheimer’s disease (AD). These age-related impairments usually begin with slowly progressive memory loss, leading to pervasive cognitive decline accompanied by dementia and emotional changes (Schapiro et al., 1987; Lai and Williams, 1989; Alexander et al., 1997; Nelson et al., 2001). Moreover, senile plaques and neurofibrillary tangles resembling those of the AD brain accumulate in the brains of aging individuals with DS. Further, the regional and temporal patterns of neuropathology are strikingly similar to AD. Therefore, studying the pathogenesis of age-related cognitive dysfunction in DS could help reveal the mechanisms underlying AD and vice versa (Teipel et al., 2003; Hamadelseed et al., 2022). Postmortem studies also show that neurocytological changes in DS are similar to those observed in AD, including multiple neurodegenerative foci. Although AD neuropathology is not an essential aspect of normal aging, there is speculation that DS is associated with accelerated aging (Lott, 1982; Raz et al., 1995).
1.3. Neuroanatomical and neuropsychological characteristics of Down syndrome
Down syndrome is the most prevalent hereditary cause of developmental delay, affecting around one out of every 700 live births (Parker et al., 2010). Intellectual disability is the most extensively studied feature of the DS cognitive-behavioral phenotype (Gibson, 1978). However, children with DS have relative strengths and weaknesses that distinguish them from younger, typically developing children and same-age peers with other forms of intellectual disability (Fidler et al., 2008). Children with DS usually display linguistic deficits that surpass general cognitive limits. Furthermore, explicit memory deficits exceed overall intellectual dysfunction (Jarrold et al., 2007; Hamner et al., 2018). However, the precise neuroanatomic correlates of these childhood deficits (and preserved functions) are unclear due to the limited number of post-mortem and neuroimaging studies in infants with DS as well as the small study samples, narrow age ranges, and low spatial resolutions achieved in most previous studies (Pinter et al., 2001a). This research gap is due partly to the challenges associated with properly implementing developmental neuroimaging in this group, such as suppression of movement (Hamner et al., 2018). Nonetheless, neuroimaging research employing sophisticated acquisition and analysis techniques and best practice standards are necessary to achieve progress in this field (Raschle et al., 2012). The goals of this review are to critically evaluate the existing literature on structural neuroimaging of individuals with DS (magnetic resonance imaging [MRI], computed tomography [CT], and (or) voxel-based morphometry [VBM]), particularly studies combining neuroimaging with cognitive–behavioral assessments or comparing cognitive–behavioral phenotypes between DS and other neurodevelopment disorders like fragile X syndrome (FXS), Williams syndrome (WS), and 22q11.2 deletion syndrome (DS22q11.2). It is hoped that synthesis of existing knowledge will provide clues or generate new perspectives on the mechanisms underlying DS-associated brain maldevelopment, the associated intellectual deficits in childhood, adolescence, and adulthood, and age-related AD-like neurodegeneration (Zigman and Lott, 2007; Lee et al., 2015).
2. Materials and methods
This study followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines. PubMed, Google Scholar, and Web of Science databases were used to identify relevant peer-reviewed studies of DS published in English and with adequate methodological and statistical information to allow comprehensive critical analysis and replication. We excluded duplicate publications and studies of animal models except for the background. Regarding the excluded 119 articles on “neuroanatomy-brain changes” here, we used the full and restricted exclusion criteria that included only relevant peer-reviewed studies of DS published in English and with adequate methodological and statistical information to allow comprehensive critical analysis and replication and the studies related to this topic represent the greatest number of studies, so the excluded number is expected to be more significant. This review focuses primarily on the following topics: (1) specific brain changes in DS revealed by structural neuroimaging, (2) language and memory deficits as well as preserved functions in DS, (3) the relationship between neuroanatomical and neuropsychological features, and (4) similarities and differences in brain structure and intellectual function between DS and other related neurodevelopmental syndromes such as FXS, WS, and DS22q11.2. These discussions are not restricted by age or gender.
First, we searched for articles with “Brain changes in Down syndrome,” “Neuroanatomy of Down syndrome,” or similar in the title using the keywords “brain changes,” “hippocampus,” “temporal lobe,” “parietal lobe,” “neuroanatomy,” and “Down syndrome.” In addition, we search for articles with “Cognitive profile of Down syndrome,” “Neuropsychology of Down syndrome,” or similar using the keywords “cognitive profile,” “neuropsychology,” “language,” “memory,” and “Down syndrome.” We selected functional and structural neuroimaging studies using CT, MRI, and (or) VBM that also examined the associations of structural changes, especially in the parietal lobe, temporal lobe, and hippocampus, with memory and language function. We also searched for studies with “Comparing brain changes in Down syndrome and Fragile X syndrome,” “Comparing the neuroanatomy of Down syndrome and Fragile X syndrome,” “Comparing brain changes in Down syndrome and Williams syndrome,” “Comparing the neuroanatomy of Down syndrome and Williams syndrome,” “Comparing brain changes in Down syndrome and 22q11.2 deletion syndrome,” “Comparing neuroanatomy of Down syndrome and 22q11.2 deletion syndrome,” or similar in the title using the keywords “comparison,” “brain changes,” “neuroanatomy,” “hippocampus,” “temporal lobe,” “parietal lobe,” “Down syndrome,” “fragile X syndrome,” “Williams syndrome,” and “22q11.2 deletion syndrome.” In addition, we analyzed articles with the titles: “Comparing the cognitive profile of Down syndrome and Fragile X syndrome,” “Comparing neuropsychology of Down syndrome and Fragile X syndrome,” “Comparing the cognitive profile of Down syndrome and Williams syndrome,” “Comparing neuropsychology of Down syndrome and Williams syndrome,” “Comparing the cognitive profile of Down syndrome and 22q11.2 deletion syndrome” “Comparing neuropsychology of Down syndrome and 22q11.2 deletion syndrome” or similar in the title using the keywords “comparison, cognitive profile,” “neuropsychology,” “language,” “memory,” “Down syndrome,” “fragile X syndrome,” “Williams syndrome,” and “22q11.2 deletion syndrome.”
The meta-analysis was performed on 41 brain structure measurements from 6 studies with sufficient data (e.g., large sample size and broad age range of Down syndrome participants) for the statistical meta-analysis framework. DerSimonian and Laird’s tau estimation was calculated as a standardized mean difference metric for scaling variables, and Cohen’s d was calculated as an estimate of variance among studies (see Table 1). The systematic review is supplemented with descriptive tables, while pooled results of the meta-analysis are presented using forest plots. Funnel plots and Egger’s tests are used to evaluate study publication bias. Eighty four studies were selected in total after using the above-mentioned inclusion and exclusion criteria. A PRISMA flow chart of study inclusion and exclusion is illustrated in Figure 1.
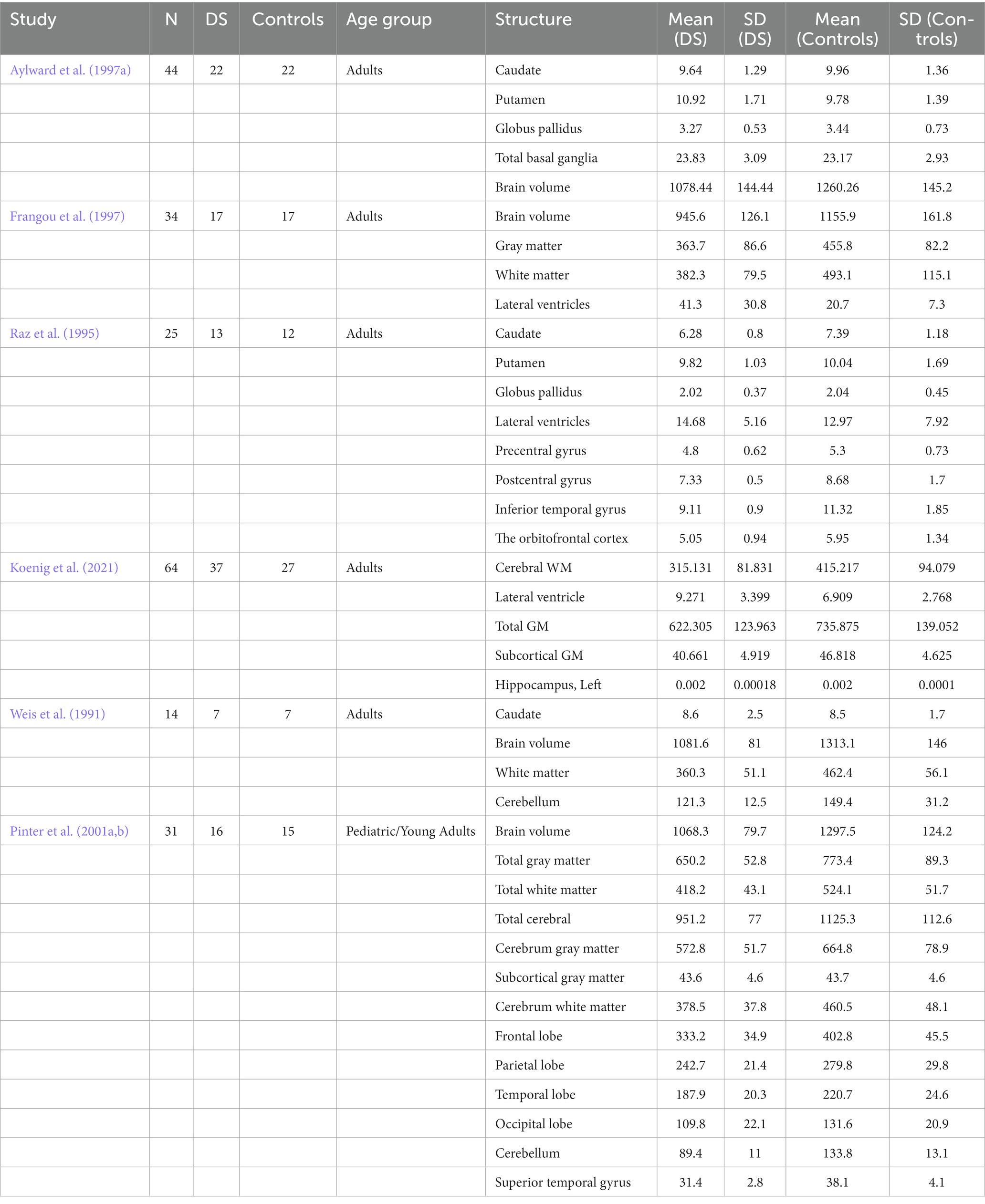
Table 1. Descriptive statistics used to express and analyze regional brain volume differences between DS and Control groups in individual studies.
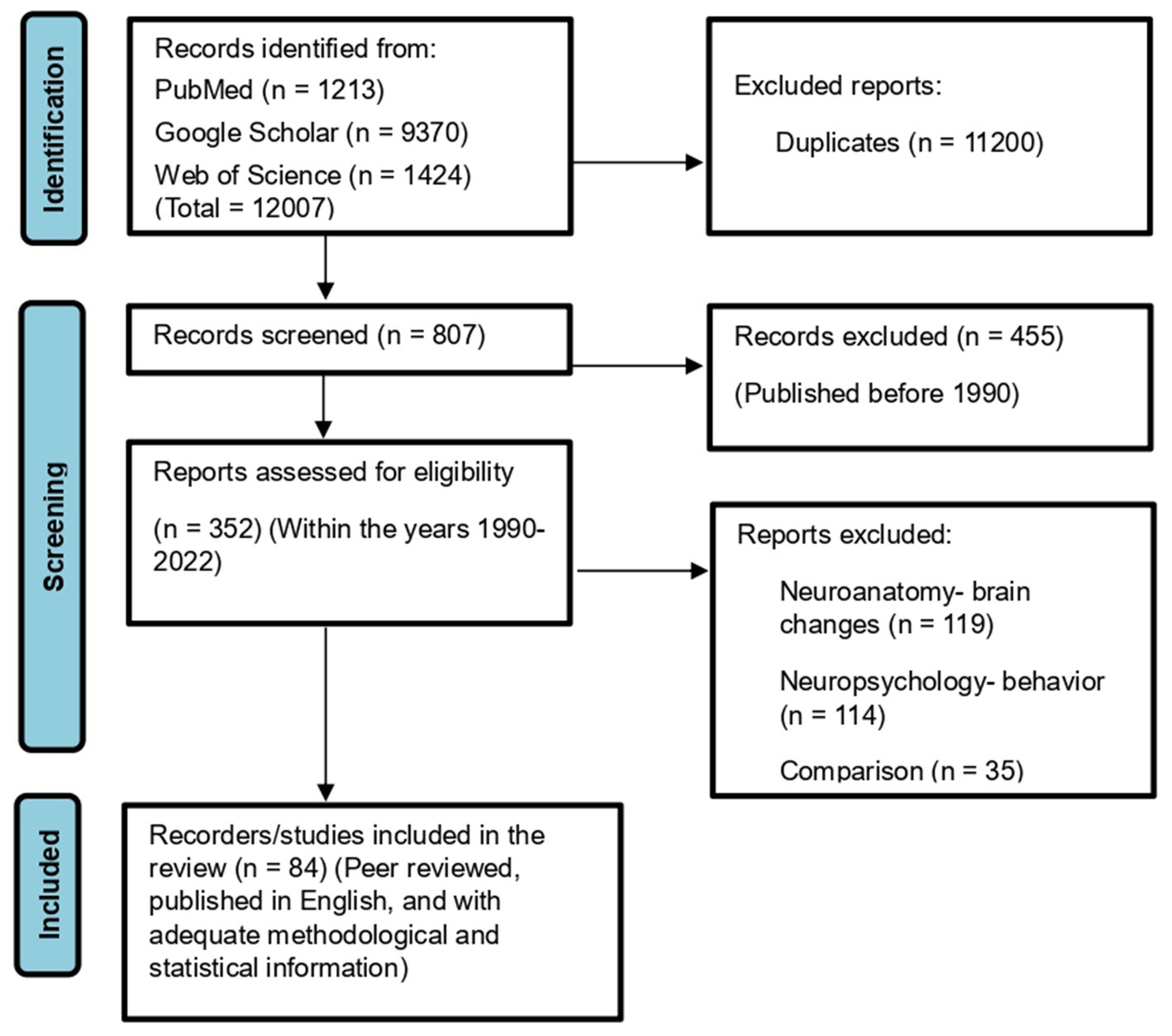
Figure 1. PRISMA 2020 flowchart showing the selection of studies analyzed in this systematic review and meta-analysis.
3. Results
3.1. Neuroanatomical changes in people with Down syndrome
The neuroanatomical changes characteristic of DS can be divided into two categories: (1) those arising from maldevelopment and directly related to the DS genotype and (2) progressive brain atrophy superimposed on neurodevelopmental abnormalities that contribute to AD-like pathology. In recent years, postmortem data on DS neuroanatomy have been supplemented by a growing number of in vivo structural neuroimaging studies utilizing CT and MRI (Teipel and Hampel, 2006). We have identified six studies reporting neuroanatomical changes in the brains of people with DS based on neuropathological evidence (see Table 2). Postmortem studies have identified several consistent abnormal features of the DS brain, including reduced total weight, frontal and temporal lobe atrophy, widespread gyral atrophy, cerebral ventricle enlargement, a smaller brain stem, and cerebellar atrophy, particularly in the vermis (Zellweger, 1977; Vogt et al., 1991; Teipel and Hampel, 2006). However, two major factors hamper the interpretation of postmortem studies. First, agonal condition and the long-term pathological changes that lead to death can alter brain anatomy. Second, the lack of information about the patient’s cognitive and developmental status makes selecting a suitable control group difficult. Non-invasive in vivo imaging techniques have largely eliminated these limitations. Kesslak et al. (1994) found reduced hippocampal formation volume in a DS group but unexpectedly reported enlargement of the parahippocampal gyrus, possibly reflecting anomalies in neurogenesis throughout development, while differences were found in the volumes of lateral ventricles, combined subcortical nuclear structures, whole neocortex, or temporal gyri between DS and matched control groups. Pinter, Eliez, et al. (2001b) found large volume reductions in frontal and occipital lobes, planum temporale, and superior temporal gyrus. Still, the absolute volume reductions in whole temporal and parietal lobes were no longer significant after adjustment for total brain volume. The relative preservation of temporal lobe volume was associated with greater white matter (WM) volume. According to the authors, this may be due to a specific aberration in WM maturation. However, their technique depended on a semi-automated parcellation of the brain using a normal pediatric template, while individuals with DS may have differently shaped brains, so there may have been methodological challenges in detecting the posterior margins of the temporal lobes on MRI images (Pinter et al., 2001b). Both Jernigan et al. (1993) and Pinter et al. (2001b) found that subcortical gray matter (GM) structures (basal ganglia and thalamus) were normal in DS. Although DS is associated with prenatal defects in neurogenesis and synaptogenesis, the gross brain pathology observed in young individuals is not present in utero but occurs during early postnatal development (Zellweger, 1977; Wisniewski, 1990; Vogt et al., 1991). In addition to morphometric changes arising from maldevelopment, evidence from postmortem studies suggests that other morphological changes reflect AD-like neuropathology (Ball et al., 1986; Mann, 1988). Lott postulated that DS is associated with premature aging (Lott, 1982). If so, relatively young people with DS should show neuroanatomical changes characteristic of older normal adults or AD patients. In particular, people with DS, like ordinary older adults and AD patients, would be expected to exhibit some loss of brain tissue, accompanied by pronounced shrinkage of tertiary association areas such as the dorsolateral prefrontal cortex and lower parietal lobule (Haug, 1985; Raz et al., 1993; Kemper, 1994). In addition, persons with DS demonstrated premature shrinkage of the caudate nucleus, mammillary bodies, lobules VI to VIII of the cerebellar vermis, and especially of the hippocampus, all of which have been observed in normal aging, while hippocampal shrinkage is a hallmark of AD (Raz et al., 1992; Kemper, 1994). Raz et al. (1995) investigated these changes in detail by examining neuroanatomical abnormalities of DS and correlations with cognitive changes, while Fernandez-Alcaraz and Carvajal-Molina (2014) reviewed the latest neuroanatomic studies of DS. The reviewed studies indicated that most individuals with DS exhibit a variety of anatomical abnormalities, including brachycephaly and reduced regional brain volumes, particularly in the cerebellum, frontal and temporal lobes, and brainstem. A considerable increase in the volume of the cerebral ventricles was also noted, as well as a smaller hippocampus and amygdala, a narrower superior temporal gyrus, and a decrease in the number and depth of cortical sulci and convolutions (Fernandez-Alcaraz and Carvajal-Molina, 2014). Similarly, distinctive neuropathological signs of AD in the fourth decade of life were reported in the earliest related studies (Wisniewski et al., 1985; de la Monte and Hedley-Whyte, 1990). In addition, the studies reviewed by Fernandez-Alcaraz and Carvajal-Molina (2014) indicated that these anatomical changes occur in similar locations of both hemispheres, including bilateral hippocampus, amygdala, temporal and frontal lobes, and cerebellum, in addition to the brainstem and corpus callosum.

Table 2. Summary of studies reporting neuroanatomical changes in the brains of people with DS based on neuropathological evidence.
3.2. Neuroimaging findings in people with Down syndrome
Until recently, our understanding of the structural brain abnormalities in DS depended on autopsy studies (Fernandez-Alcaraz and Carvajal-Molina, 2014). These studies consistently showed a lower total brain weight, brachycephaly, reduced cerebellar volume, smaller frontal and temporal lobes, simplified sulcal morphology, and a narrower superior temporal gyrus compared to matched controls (Coyle et al., 1986; Wisniewski, 1990; Becker et al., 1991; Vogt et al., 1991). However, post-mortem studies cannot reveal the developmental and pathological mechanisms leading to these differences or control for agonal brain structure changes independent of DS. For these reasons, post-mortem investigations have been largely replaced by in vivo neuroimaging (Kates et al., 1997b; Fernandez-Alcaraz and Carvajal-Molina, 2014). There are at least 14 published structural MRI studies on the DS brain, but most have included only subjects older than five years (Jernigan and Bellugi, 1990; Pearlson et al., 1990; Weis et al., 1991; Jernigan et al., 1993; Kesslak et al., 1994; Raz et al., 1995; Aylward et al., 1997a, 1999; Krasuski et al., 2002; White et al., 2003; Beacher et al., 2010; Mullins et al., 2013; Hamner et al., 2018; Koenig et al., 2021). Therefore, the early developmental abnormalities in DS are still largely unknown. Nonetheless, recent improvements in MRI acquisition and processing techniques have enabled higher-resolution quantitative investigations of brain structure in individuals with DS (McCann et al., 2021). We have shown 31 neuroimaging studies reporting anatomical abnormalities in DS in Table 3. In accord with early post-mortem studies, volumetric neuroimaging studies of adults with DS have consistently shown reduced total brain volume and a disproportionately smaller cerebellum, brainstem, frontal lobe, and hippocampal formation (Weis et al., 1991; Kesslak et al., 1994; Raz et al., 1995; Aylward et al., 1997a, 1999; Koenig et al., 2021). In contrast to some post-mortem studies, volumetric MRI studies have reported typical basal ganglion volumes in DS (Raz et al., 1995; Aylward et al., 1997b). Surprisingly, few MRI studies have investigated young children despite the high prevalence of DS among neurodevelopment disorders (Pinter et al., 2001b; Śmigielska-Kuzia et al., 2011; Carducci et al., 2013; Rodrigues et al., 2019). Among the few studies investigating the neuroanatomy of infants and toddlers with DS (Fujii et al., 2017; Gunbey et al., 2017; Levman et al., 2019), a volumetric MRI study by Jernigan et al. (1993) also reported a smaller total brain volume and disproportionately smaller frontal, temporal, and cerebellar regions in six children with DS. Pinter et al. (2001b) assessed brain volumes and tissue composition in DS from early childhood to young adulthood and reported (1) reduced overall brain volume and reduced GM and WM volumes, (2) a disproportionately lower cerebellar volume, and (3) no significant subcortical and cortical volume changes, including in parietal GM and temporal WM, after correction for reduced total GM and WM. These findings are generally consistent with previous pathological and imaging studies (Coyle et al., 1986; Becker et al., 1991; Jernigan et al., 1993; Aylward et al., 1997a). In contrast to studies reporting lower cerebellar and frontal lobe volumes, but consistent with findings from previous studies in adults and children with DS (Jernigan et al., 1993; Raz et al., 1995; Aylward et al., 1997b; Pinter et al., 2001b) found remarkable preservation of subcortical structures, possible due to compensatory subcortical GM adjustment. In addition to increased subcortical GM volumes (Pinter et al., 2001b), others have reported larger lateral ventricles (Pearlson et al., 1998). Automated MRI brain analysis technologies can evaluate cortical thicknesses in different brain regions (Fischl, 2012). To provide a comprehensive evaluation of morphological changes in the DS brain, White et al. (2003) used a fully automated VBM technique on 19 persons with DS living without dementia and 11 age-matched controls. The cerebellum, cingulate gyrus, left medial frontal lobe, right middle/superior temporal gyrus, and left CA2/CA3 area of the hippocampus all showed significant reductions in GM volume (p < 0.05, adjusted for multiple comparisons) while WM volume was significantly reduced across the inferior brainstem. In contrast, a superior/caudal section of the brainstem and left parahippocampal gyrus showed statistically significant GM expansion (p < 0.05, adjusted for multiple comparisons), while the bilateral parahippocampal gyrus showed significantly increased WM volume. The DS group also showed substantial cerebral spinal fluid (CSF) volume increases in (larger) lateral ventricles. These findings are consistent with the previous region of interest (ROI)-based imaging investigations of people with DS living without dementia and further contribute to our knowledge of the three-dimensional topography of the DS brain.
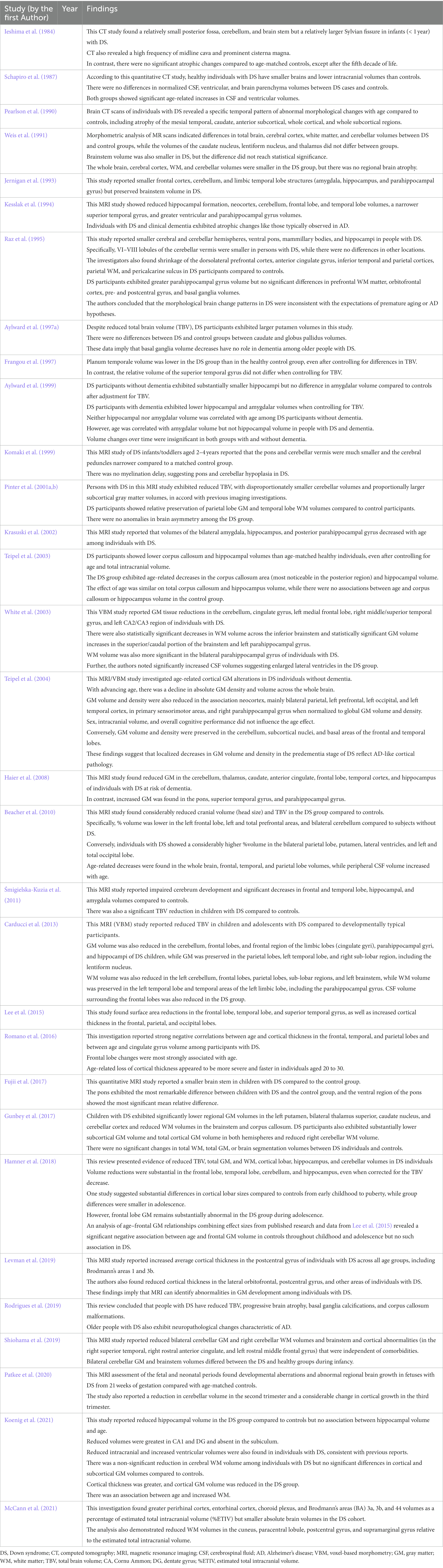
Table 3. Summary of neuroimaging studies reporting anatomical abnormalities in DS.
In general, VBM has provided high-resolution quantitative measures of neuroanatomic abnormalities in DS that are generally consistent with prior imaging studies (White et al., 2003). Menghini et al. (2011) identified numerous brain anomalies in a homogenous sample of 12 teenagers with DS compared to healthy controls using VBM, including significantly reduced GM density in the left cerebellum (posterior), right inferior temporal gyrus/medial temporal lobe (fusiform gyrus and hippocampus), and left medial temporal lobe, but greater GM density in the left cerebellum (anterior), right medial temporal lobe (fusiform gyrus), bilateral putamen and caudate nucleus, bilateral insula, bilateral superior frontal gyri, right superior and middle temporal gyri, and bilateral inferior frontal gyri. A similar study by Carducci et al. (2013) of 21 children and adolescents with DS and 27 age-matched controls using MRI-based VBM revealed a smaller whole-brain volume, reduced GM volumes in the cerebellum, frontal lobes, frontal region of the limbic lobe, parahippocampal gyri, and hippocampi, and reduced WM volume in the cerebellum, frontal and parietal lobes, sub-lobar regions, and brainstem of the DS group compared to the control group. In contrast, the GM volumes of the parietal lobes, temporal lobe, and sub-lobar areas, as well as the WM volumes of the temporal lobe and limbic lobe temporal regions, were preserved in DS. Carducci et al. (2013) also reported reduced CSF volume in the frontal lobes. A recent retrospective MRI investigation of individuals with DS aged 0–22 and a cohort of neurotypical participants (aged 0–32) by McCann et al. (2021) reported decreased whole brain, cerebral, cerebellar brainstem, and hippocampal volumes, and increased parahippocampal gyrus volume. Levman et al. (2019) quantitatively assessed the brain morphology of newborns and toddlers with DS using structural MRI and found smaller WM volumes in the right cerebellum, reduced GM volumes in the brainstem and bilateral cerebellum, and morphological abnormalities in the right superior temporal cortex, right rostral anterior cingulate cortex, and left rostral middle frontal cortex. An in vivo fetal and neonatal MRI assessment by Patkee et al. (2020) also reported early alterations in cortical and cerebellar growth in DS from 21 weeks of gestation associated with later global declines in whole brain and cortical volumes. Thus, this study detected early biomarkers that may predict the degree of cognitive deficits in DS.
Previous research has focused mainly on children and adults rather than infants with DS, so it is difficult to distinguish between direct pathophysiological effects on the brain and secondary or compensatory processes that occur through living with the disability. However, Patkee et al. (2020) reported that trisomy directly impacts the early in vivo growth of the brain and cerebellum, so it may be possible to anticipate the ultimate severity of cognitive deficits for genetic counseling or treatment planning (Patkee et al., 2020). A recent study by Koenig et al. (2021) using a high-resolution 7-T MRI to assess the structure and function of the hippocampus in young adults with DS (mean age 24.5 ± 6.5 years) found reduced whole hippocampal volume, reduced Cornu Ammon (CA) field 1 (CA1) and dentate gyrus volumes, but not significant difference subicular volume. Further, these regional anatomic abnormalities were associated with cognitive deficits (see next section).
In contrast to VMB findings, studies examining regional cortical thickness abnormalities in DS have produced inconsistent results. Lee et al. (2015) and Levman et al. (2019) reported increased cortical thickness in some brain regions of individuals with DS, but Romano et al. (2016) reported decreased cortical thickness. Lee et al. (2015) found that the primary regions affected were the frontal, parietal, and occipital lobes. This study also concluded that people with DS have reduced cortical surface areas in the frontal lobe, temporal lobe, and superior temporal gyrus. Romano et al. (2016) compared the CT scans of older DS participants to those of neurotypical participants as part of a separate study at another imaging center (Levman et al., 2019) and found significant enlargement of CSF spaces, such as in the chiasmatic cistern (Ieshima et al., 1984; Pearlson et al., 1990). Alternatively, Schapiro et al. (1987) found no evidence of age-related ventriculomegaly between DS and normal controls (Schapiro et al., 1987). CT and MRI studies have primarily replicated previously found lower cerebral parenchyma volume (Schapiro et al., 1987). Two MRI studies (Weis et al., 1991; Jernigan et al., 1993) reported that the cerebellum of people with DS was significantly smaller than that of age-matched normal controls, and one (Jernigan et al., 1993) observed global and regional differences in cerebral cortex volume in a small sample of young people with DS compared to normal controls. These two MRI studies (Weis et al., 1991, Jernigan et al., 1993) also showed a preserved average gross volume of the basal ganglia. Fujii et al. (2017) reported a smaller brainstem in children with DS, particularly in the pons, consistent with previous studies (Raz et al., 1995; Komaki et al., 1999). Gunbey et al. (2017) assessed WM integrity and associated WM and GM volumetric changes in early childhood. Haier et al. (2008) reported less GM in brain areas such as the cerebellum, anterior cingulate, frontal lobe, and temporal lobe, including part of the hippocampus. They also reported greater GM in other brain areas such as the pons, superior temporal gyrus, and parahippocampus (Haier et al., 2008).
Figure 2 reveals that overall regional brain volume is significantly lower among individuals with DS compared to matched controls (p < 0.05) with moderate effect size (overall Cohen’s d = −0.59, 95% CI, −0.93−−0.24) and significant heterogeneity (88%, p < 0.01). According to a random effects model, individual brain volumes are shown as gray boxes, and the overall difference is as a diamond. The red bar indicates the 95% Cl of the overall effect. The error bars represent the confidence intervals for individual measures.
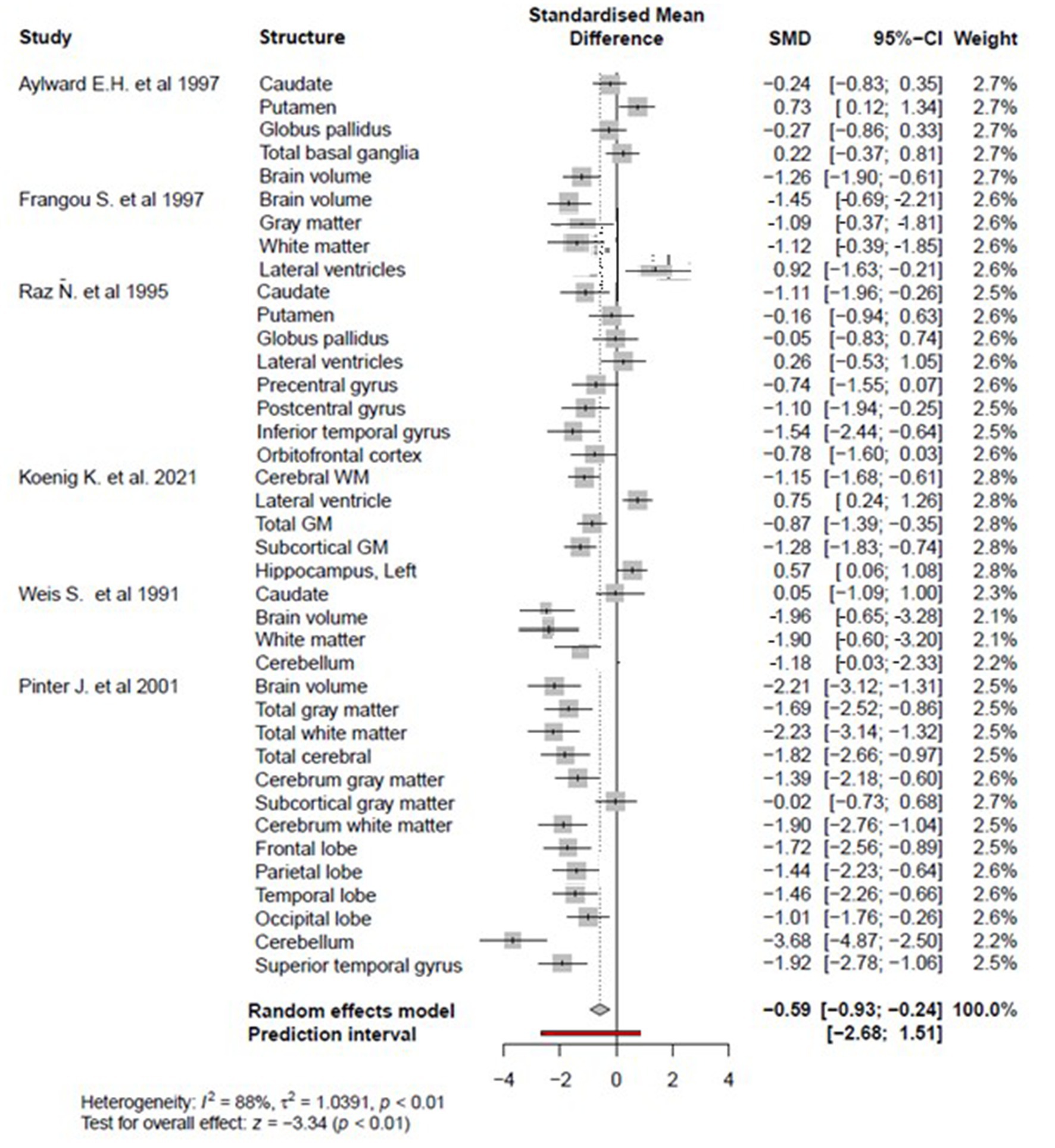
Figure 2. The forest plot for the meta-analysis compares regional brain volumes between individuals with DS and healthy matched controls.
There is no or slight publication bias, as evidenced by plot symmetry. Egger’s publication bias test was also non-significant (t = −1.23, df = 38, p = 0.22) (see Figure 3).

Figure 3. Funnel plot for analysis of publication bias.
3.3. Neuroanatomical changes in the hippocampus of people with Down syndrome
The hippocampus is essential for forming and consolidating explicit memories, including memories for life episodes and semantic information (Squire, 1992), as hippocampal lesions and degeneration lead to catastrophic memory impairments (Isaacs et al., 2003; Savage et al., 2004). The hippocampus changes markedly in size and structure during childhood, with volume peaking between 9 and 11 years of age and decreasing progressively during adolescence (Uematsu et al., 2012). Youth with DS exhibit substantial impairments in hippocampus-dependent memory tasks (Pennington et al., 2003; Clark et al., 2017). Hippocampal atrophy is also a hallmark of AD pathology and a reliable neuroimaging biomarker for the disease (Blanken et al., 2017). Given the AD-like pathology observed in DS, it is surprising that there is little research on hippocampal changes in adolescents with DS (Hamner et al., 2018). Nonetheless, the limited research suggests no significant differences in hippocampal volume between infants with DS and younger typically developing peers (Gunbey et al., 2017). However, children with DS exhibited an absolute reduction in hippocampal volume compared to controls (Śmigielska-Kuzia et al., 2011) and a relative decrease when adjusted for TBV (Pinter et al., 2001a). In addition, VBM studies indicated significant reductions in specific hippocampal subregions in DS during childhood (Menghini et al., 2011; Carducci et al., 2013; Hamner et al., 2018). Carducci et al. (2013) observed a decrease in hippocampal GM volume, especially on the right side, consistent with previous MRI studies of young people with DS (Jernigan et al., 1993; Pinter et al., 2001a) and adults with DS (Raz et al., 1995; Aylward et al., 1997a,b; Pearlson et al., 1998; Pinter et al., 2001b; White et al., 2003). These studies suggest that hippocampal volume reduction in DS begins in childhood and persists throughout adulthood as a direct result of the trisomic genotype (Carducci et al., 2013). Aylward et al. (1999) reported that hippocampus volumes were disproportionately smaller in DS adults with eventual dementia even before dementia onset, while hippocampal volumes did not decrease with age among DS individuals without dementia (Aylward et al., 1999). In contrast, Teipel et al. (2003) found an age-related decrease in hippocampal volumes among DS adults without dementia. Koenig et al. (2021) measured hippocampal subfield volumes and functional connectivity in a group of age-matched normal controls and adolescents and young adults with DS using high-resolution MRI and found significantly reduced total hippocampal volume as well as reduced CA1 and DG volumes, but no change in subicular volume, in the DS group. However, in accord with Aylward et al. (1999), there was no association between age and hippocampal volume. Given the essential function of the subiculum in memory and AD pathology, the lack of volume decrease is unexpected (Koenig et al., 2021). Collectively these observations suggest hippocampal subfield volume reductions in more severely afflicted individuals with DS (i.e., those ultimately developing early onset AD).
3.4. Neuroanatomical changes in the temporal and parietal lobes of people with Down syndrome
The parietal and lateral temporal lobes are involved in linguistic and visuomotor skills (Pinter et al., 2001b), both impaired in DS. Kates et al. (2002) reported reduced total parietal and temporal lobe volumes in individuals with DS, while and Pinter et al. (2001b) found an increase in total volumes. Alternatively, a voxel-based whole-brain morphometry study of children and adolescents with DS by Carducci et al. (2013) found symmetrical GM volume maintenance in the right postcentral gyrus, left precuneus, and upper lobule of the parietal lobe, consistent with previous MRI studies on young adults with DS (Jernigan et al., 1993; Pinter et al., 2001b; Kates et al., 2002). However, a few studies of adults with DS have reported reduced parietal lobe volume with aging (Teipel et al., 2004). Conversely, White et al. (2003) found no significant volumetric changes in this region, suggesting that damage to the parietal lobe occurs much later in life. Carducci et al. (2013) also observed a relative decrease in WM volume in the parietal lobes. Moreover, Pinter et al. (2001b) suggested that the deficits in speech and visual processing observed in people with DS are partly due to excessive cell numbers (i.e., insufficient developmental pruning) and concomitant dysfunction of the parietal lobes. The same study by Carducci et al. (2013) also reported preservation of GM volume in the left temporal lobe, while Menghini et al. (2011) found GM loss in the temporal lobe, in accord with previous MRI studies on adults with DS (Kesslak et al., 1994; Raz et al., 1995; White et al., 2003). In addition, postmortem studies by Wisniewski (1990) and Posner and Dehaene (1994) supported late damage to this structure. Thus, early deficits in linguistic and visuomotor skills are not associated with detectable volumetric changes in the parietal and lateral temporal lobes. Rather, these deficits likely reflect disorganization or dysfunction of language circuits within or projecting to these regions.
3.5. Hippocampus dysfunction and memory impairment in Down syndrome
Several studies examining hippocampal function in DS have found deficits in both verbal and nonverbal explicit memory (Pennington et al., 2003; Godfrey and Lee, 2018). Impaired verbal short-term memory may significantly impede the development of language skills, particularly vocabulary (Jarrold et al., 2008), and verbal short-term memory is consistently impaired in DS (Pennington et al., 2003). Persons with DS also appear to have impaired explicit long-term memory for verbal information and some deficits in explicit long-term memory for visual object associations (Godfrey and Lee, 2018). In contrast, implicit memory may be less affected in DS, suggesting that neural substrates for this form of memory are relatively well preserved. Moreover, intact implicit memory may provide strategies for improving language development in DS. Impairments in explicit verbal and non-verbal memory support morphometric studies that the hippocampus and functionally associated structures, such as the pre-frontal cortex, are malformed in DS (Jarrold et al., 2008). Pennington et al. (2003) studied hippocampal functions in a sample of 28 school-age children with DS using tasks first developed in animal models and found substantial deficits in hippocampus-dependent memory.
The neurocellular basis of these deficits has been examined in DS mouse models such as Ts65Dn. Hyde and Crnic (2001) reported that hippocampal dysfunction in Ts65Dn mice was influenced by age and associated with reduced numbers of cholinergic hippocampal neurons. In addition, studies of the Ts65Dn mouse model and individuals with DS have suggested that impaired neurogenesis in the molecular layer of the hippocampal dentate gyrus (DG) may contribute to long-term memory deficits (Contestabile et al., 2007). However, the specific contributions of DG neurogenesis to the DS phenotype are still not widely studied (Jarrold et al., 2008).
Given the crucial role of the hippocampus in explicit memory, changes in hippocampal structure and function are a major focus of research investigating biomarkers for AD (Pujol et al., 2018). Neuroimaging studies have shown reduced hippocampal volume associated with memory deficits in the predementia stage of AD among adults with DS (Krasuski et al., 2002; Beacher et al., 2010; Koenig et al., 2021; Hamadelseed et al., 2022). Memory loss is one of the first symptoms of AD (Jahn, 2013) and the temporal order of regional neurodegeneration determines the clinical course and severity of the disease (Hoehne, 2007). Histopathological studies have confirmed that the hippocampus is one of the first and most severely damaged regions by AD (Nagy et al., 1996). Therefore, more detailed structural and functional neuroimaging of the hippocampus may allow for both better prognosis of memory and language impairments in DS and identify the underlying neural substrates, especially in the preclinical stage of DS-associated AD (Godfrey and Lee, 2018; Pujol et al., 2018).
3.6. Dysfunction of temporoparietal junction and occipitotemporal structures as markers of deficient language performance in Down syndrome
As discussed earlier, Pinter et al. (2001b) identified morphological changes in multiple temporal lobe structures implicated in speech, although such findings have not been reported consistently across studies. Further, they did not find superior temporal gyrus (STG) volume reduction in DS compared to normal adults with speech deficits, as expected if STG atrophy is involved in DS-associated language dysfunction. They also found no volumetric evidence of an irregular pattern of brain asymmetry (including the temporal lobe) in DS. In contrast, psychological tests examining selective attention and lateralization of brain function within the auditory system suggest lateralized speech dysfunction in DS. While this finding requires additional support from structural and functional neuroimaging studies (Hartley, 1981, 1985; Giencke and Lewandowski, 1989; Pinter et al., 2001b), it appears that language deficits in DS cannot be explained simply by temporal lobe atrophy.
Language function also depends on a supportive network of neural connections that provide related information (Friederici and Gierhan, 2013). Two of these centers are located in the temporal and lower frontal lobes containing Wernicke’s and Broca’s areas (Hamner et al., 2018). Other neural language network structures are located within the temporoparietal junction, occipital lobe (e.g., fusiform gyrus), and superior temporal gyrus (Hickok and Poeppel, 2004; Friederici and Gierhan, 2013; Hamner et al., 2018). The temporoparietal junction structures serve as an auditory–motor interface that transmits heard language to premotor and motor representations in Broca’s area that drive speech output. This hypothesis is consistent with functional imaging evidence that connects perceptual learning to the auditory-motor pathways (Scott and Johnsrude, 2003; Hickok and Poeppel, 2004; Rauschecker and Scott, 2009). High-resolution fMRI and functional connectivity studies are required to identify if any of these pathways are disrupted in DS.
3.7. Differences in neuroimaging findings between Down syndrome and fragile X syndrome, Williams syndrome, and chromosome 22q11.2 deletion syndrome (DS22q11.2)
We identified four studies comparing neuroanatomic abnormalities in DS with FXS, WS, and chromosome 22q11.2 deletion syndrome (DS22q11.2) based on imaging data (see Table 4). Previous neuroimaging studies have found an age-related increase in hippocampal volume among fragile X syndrome (SXF) patients compared to a DS group (Reiss et al., 1994; Kates et al., 1997a). In addition, these studies provided evidence of an age-related volume reduction in the superior temporal gyrus, which is essential for speech processing (Reiss et al., 1994). Kates et al. (2002) confirmed these results by studying volumetric changes at the cerebral and lobar levels in young males with FXS (age range: 2–7 years). In addition, they compared persons with the full mutation (FM) and mosaic (Mos) FXS to age-matched controls, individuals with developmental language delay (DLD), and individuals with DS. They found a relative reduction in temporal lobe volume (in the essential GM) but relative preservation or increased parietal lobe WM volume among young individuals with FM FXS (Kates et al., 2002). While the temporal lobe volume reduction was not specific to FXS, as it was also observed in DLD and DS, the parietal lobe preservation/enlargement was specific to FXS.
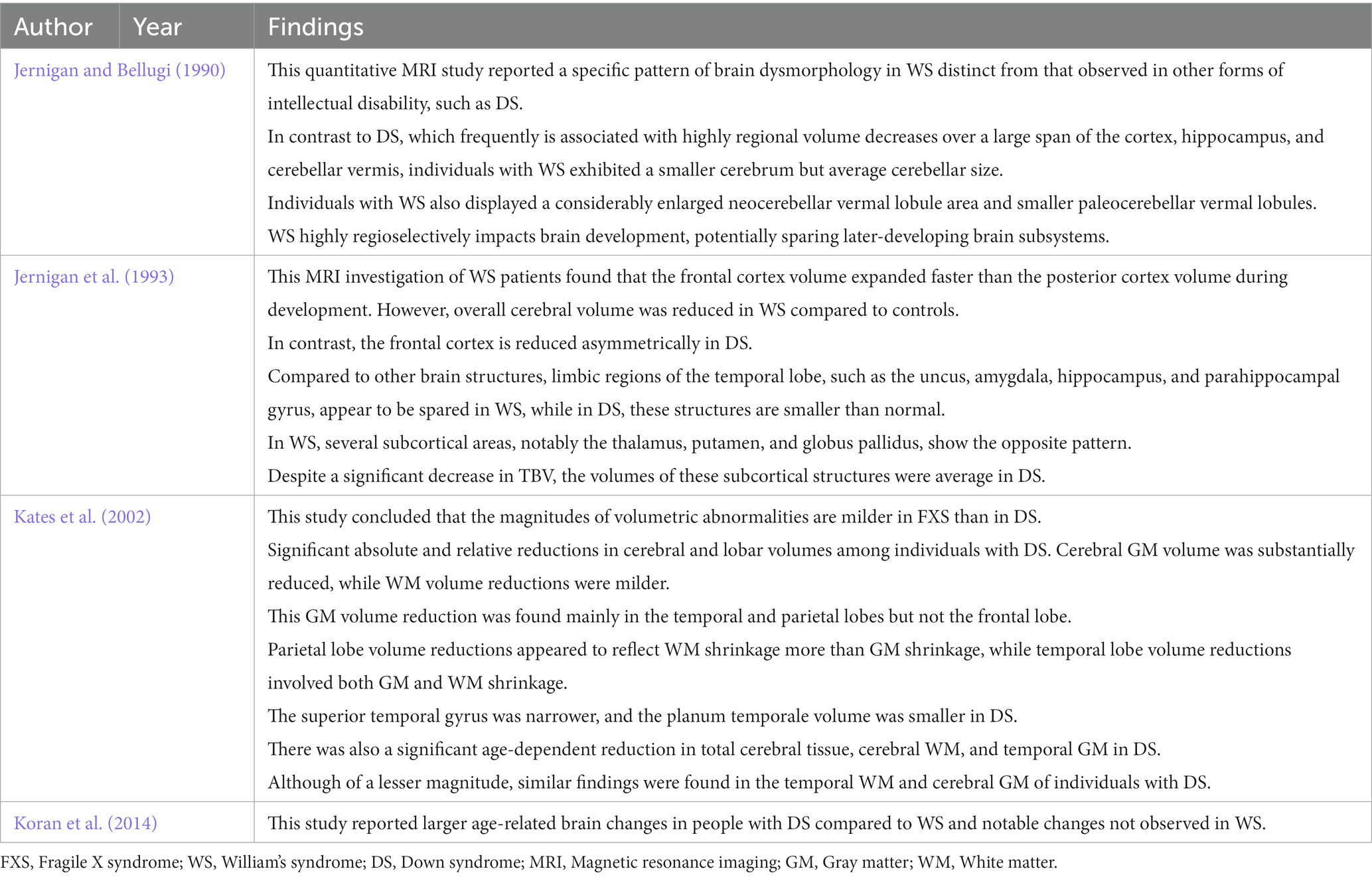
Table 4. Summary of studies comparing neuroanatomic abnormalities in DS with FXS, WS, and chromosome 22q11.2 deletion syndrome (DS22q11.2) based on imaging data.
Neuroimaging studies of adolescents and adults with Williams syndrome have consistently reported reduced TBV with more significant effects on WM than GM (Jernigan and Bellugi, 1990; Jernigan et al., 1993; Reiss et al., 2000, 2004; Chiang et al., 2007; Koran et al., 2014). In contrast to DS, Meyer-Lindenberg et al. (2005) found largely preserved hippocampal size but subtle changes in shape among adolescents and adults with WS, while a tensor-based morphometry study by Chiang et al. (2007) found reduced parietal lobe volume near the temporoparietal junction and relative preservation of the superior temporal gyrus (Chiang et al., 2007). In contrast, an MRI study by Sampaio et al. (2008) found reduced superior temporal gyrus volume in WS. Further, Campbell et al. (2009) found increased GM volume in the left temporal lobe of children with WS.
Many studies have also examined the brain structure–behavior relations in chromosome 22q11.2 deletion syndrome (DS22q11.2) (Debbané et al., 2006; Kates et al., 2006; DeBoer et al., 2007). Such studies have reported several neuroanatomic abnormalities, including GM and WM changes in the temporal lobe (DeBoer et al., 2007). Eliez et al. (2001) found significant overall brain, superior temporal gyrus, and hippocampal volume reductions in children with DS22q11.2 and schizophrenia and a smaller middle temporal lobe (Eliez et al., 2001). Further, the latter effect was correlated with age. Chromosome 22q11.2 deletion syndrome is considered by some as a genetic subtype of schizophrenia (Zinkstok and van Amelsvoort, 2005). DeBoer et al. (2007) reported a reduction in hippocampal volume in children with DS22q11.2 associated with cognitive impairment, specifically lower Intelligence Quotient (IQ). Zinkstok and van Amelsvoort (2005) also found reduced parietal lobe volume in children and adults with DS22q11.2. Eliez et al. (2000) found loss of normal parietal lobe tissue symmetry due to a significant reduction in left parietal lobe GM among children with DS22q11.2. In addition, Chow et al. (2002) found reduced GM in the left parietal lobe of DS22q11.2 adults associated with schizophrenia symptoms. In contrast, Kates et al. (2002) reported a reduction in parietal WM in DS22q11.2. Finally, Simon et al. (2005) found reduced TBV among children with DS22q11.2 that were associated with a more significant decrease in WM volume than GM volume, consistent with several previous morphometric studies.
In summary, various neuroanatomic abnormalities observed in FXS, WS, and DS22q11.2 are unique to each disorder and distinct from those of DS. However, as with DS, there are few simple direct associations with disease course or symptoms.
3.8. Cognitive profile of Down syndrome
We identified 27 studies analyzing the cognitive profile in patients with Down syndrome (see Table 5). Children and adults with DS have numerous intellectual deficits as well as domains of relative preservation or strength, and the profile of strengths and weaknesses can differ markedly among individuals (Wang, 1996; Pulina et al., 2019; Channell et al., 2021; Onnivello et al., 2022). Certain language domains are frequently among the most severely impaired, especially domains related to language expression, while language processing (receptive language function) tends to be less impaired (Abbeduto et al., 2001; Lanfranchi et al., 2009; Pulina et al., 2019). Verbal short-term memory, explicit long-term memory, and executive functions such as working memory, all of which are essential for language development, are also generally impaired in DS (Abbeduto et al., 2001; Chapman R., 2003; Chapman, 2006; Jarrold et al., 2008; Grieco et al., 2015), while visual–spatial short-term memory, implicit long-term memory, perception, and social cognition are preserved or even relative strengths among individuals with DS (Fidler, 2005; Vicari, 2006; Lanfranchi et al., 2009; Martin et al., 2009; Yang et al., 2014; Pulina et al., 2019). These memory deficits are apparent in children with DS compared to developmentally matched controls and include poor immediate verbal memory, simultaneous processing and storage, delayed memory and learning, and gist recall. In contrast, immediate visuospatial memory, semantic information recall, and phonological information retrieval are generally preserved. As a result, children with DS struggle when applying semantic/conceptual information for narrative recall, which is essential for reading development, but do better in phonological information retrieval and semantic/conceptual information retrieval (Conners et al., 2011). The cognitive profile of DS can be assessed using tasks such as the Peabody Picture Vocabulary Test (PPVT), Kaufman Assessment Battery for Children (K-ABC), and Stanford-Binet Intelligence Scale (Chapman, 2006; Pulina et al., 2019).
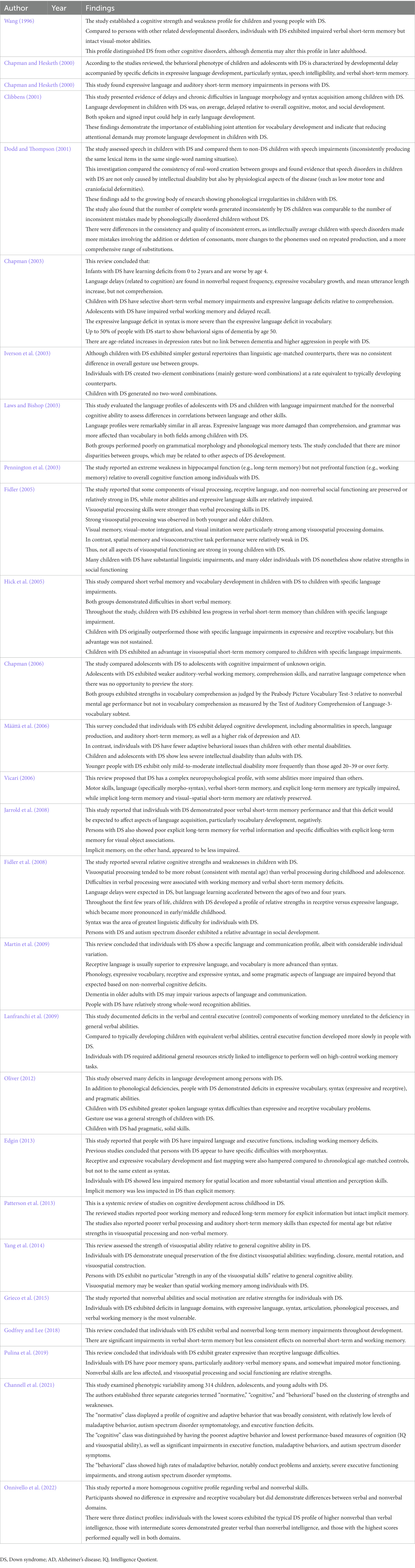
Table 5. Summary of studies analyzing the cognitive profile of Down syndrome.
3.9. Cognitive development in Down syndrome
General intelligence deficits in DS range from moderate to severe (IQ = 25–70) (Pulina et al., 2019). However, a few reports have indicated that some children with DS are in the average IQ range (Chapman and Hesketh, 2001; Vicari, 2006; Vicari et al., 2007; Martin et al., 2009). The IQ scores in DS change with age and are positively or negatively influenced by many genetic and environmental factors influencing typically developing children (Pennington et al., 2003). In addition, AD pathology may be the predominant influence on IQ scores in later life (Bush and Beail, 2004). Longitudinal studies have shown that cognitive development slows during early childhood, and IQ may decrease (Patterson et al., 2013; Sarimski, 2014). Studies monitoring IQ with advancing chronological age have generally found that intellectual disability is mild in children and adolescents compared to adults (Määttä et al., 2006; Vicari, 2006). Couzens et al. (2011) tracked the developmental courses of individuals with DS from childhood to early adulthood using the Stanford Binet Test (4th edition) and found distinct developmental patterns, with crystallized skills (vocabulary, understanding, and quantitative subtests) declining after about 20 years of age. The same trend was also observed for short-term memory. In contrast, fluid abilities (such as pattern analysis) increased rapidly in the first few years of life, followed by constant growth and no decline for at least 30 years (Couzens et al., 2011). A recent literature review by Godfrey and Lee (2018) concluded that long-term memory is impaired from an early age and that verbal short-term memory ability decreases from childhood to adulthood (although there is a lack of IQ studies on young children). Nonverbal short-term and working memory may show a similar trend, although limited data are available.
3.10. Language development in Down syndrome
Language and communication skills follow a characteristic pattern in DS. Receptive language is typically better preserved than expressive language, and vocabulary is stronger than syntax, albeit with considerable individual variability (Martin et al., 2009). During development, the language domains most delayed in children with DS are vocabulary, syntax, and morphology, including grammar, verb form, word order, word roots, suffixes, and prefixes (Dodd and Thompson, 2001; Laws and Bishop, 2003; Roberts et al., 2007). Children with DS usually show good pragmatic skills and build a large and varied vocabulary. Typical children with DS also have good social interaction skills and can communicate successfully through language, gestures, and facial expressions (Martin et al., 2009). The use of gestures by children with DS is an important communication skill in the early stages of language development (Iverson et al., 2003). Many studies have reported the effectiveness of gestures and suggested that such use should be promoted to improve social interactions and communication between children with DS and other groups (Launonen, 1994; Miller, 1999; Clibbens, 2001). While there are characteristic patterns of language development, there are certain areas of individual variation. For instance, some children with DS speak their first words at 9 months, whereas others may not speak until several years after birth. Also, word combinations can begin at 18 months in some children with DS but not until school-age in others (Fowler, 1995; Aktas, 2004). According to the National DS Society,3 speech problems can also result from difficulties articulating specific sounds, poor mouth or facial muscle tone, sensory processing deficits, phonological processes, and (or) deficits in motor planning for speech. Otitis media, oral anatomy, and atypical facial muscles are all problems that can affect speech development in children with DS (Price et al., 2007; Oliver, 2012). There are also many interventions proposed to improve language skills during the development of children with DS (Hixson, 1993; McWilliam et al., 1996; Chapman R. S., 2003; Dodge, 2004; Fidler, 2005; Hick et al., 2005; McCauley et al., 2006; Paul, 2007; Chapman, 2008). These interventions promote early communication, speaking skills, more complex language and literacy skills, and communication methods such as sign language, images, and symbols (Martin et al., 2009).
3.11. Correlations between region-specific hippocampal atrophy and cognitive status in Down syndrome
According to previous neuropathological and neuroimaging studies, hippocampal volumes are disproportionately smaller in DS even before the major declines in cognition observed during development (Wisniewski et al., 1985; Kesslak et al., 1994; Raz et al., 1995; Aylward et al., 1999). Moreover, hippocampal volume decline significantly correlates with the deterioration in memory function, even after controlling for changes in total cognitive score and age (Krasuski et al., 2002; Teipel and Hampel, 2006; Hamadelseed et al., 2022). Two studies also identified atrophy of the hippocampus and parahippocampal gyrus before the onset of dementia in DS (Kesslak et al., 1994; Raz et al., 1995). In agreement with other studies (e.g., Jernigan et al., 1993), both studies showed reductions in hippocampal and parahippocampal gyrus volume in older people with DS and dementia compared to younger individuals with DS but without dementia (Raz et al., 1995; Pearlson et al., 1998; Aylward et al., 1999). Many reports have also used hippocampal atrophy as a marker of cortical neuron loss in AD (Bobinski et al., 1996, 1999; Nagy et al., 1996; Schröder and Pantel, 2016; Hamadelseed et al., 2022; Rao et al., 2022). Several neuroimaging investigations of people with DS but without dementia indicated that the volumes of the hippocampus and neighboring medial temporal lobe structures decrease dramatically with age and that these changes are correlated with early allocortical degenerative alterations and memory loss (Kesslak et al., 1994; Lawlor et al., 2001; Krasuski et al., 2002). However, other studies have found no association between hippocampal volume and age in individuals with DS (Raz et al., 1995; Aylward et al., 1999). A study by Mullins et al. (2013) reported a volume reduction in the hippocampus of people with DS and dementia correlated with cognitive decline. This finding is consistent with studies showing that Mini-mental Status Examination (MMSE) performance correlates directly with hippocampal volume (Ball et al., 1986) and that this MMSE–hippocampal volume association reflects actual atrophy and hippocampal dysfunction (Mullins et al., 2013).
3.12. Correlations between region-specific temporal lobe atrophy and language impairment in Down syndrome
Pinter et al. (2001b) found no imaging evidence for disproportionately smaller total temporal lobe volume in DS and found more significant relative (corrected) temporal lobe volume, although the change did not reach statistical significance. Segmenting by tissue type indicated that this relative expansion was due to a significantly larger corrected temporal lobe WM volume. However, it remains unclear if these temporal lobe WM volume changes are associated with cognitive decline in DS, particularly language impairments. Such a relationship may be expected based on functional MRI studies showing significant superior temporal gyrus (STG) activation in healthy adults during auditory and language processing and the prominent language deficits in children with DS (Démonet et al., 1992; Price et al., 1992; Schlosser et al., 1998; Pinter et al., 2001b). Although, Pinter et al. (2001b) did find that corrected STG WM volume was significantly smaller in the DS group, there was no significant difference after adjustment. Krasuski et al. (2002) also reported reduced medial temporal lobe volume and correlations with overall cognitive and memory functions but no correlation with language ability. Menghini et al. (2011) reported reduced regional GM density in the lateral and medial temporal lobes of individuals with DS compared to controls and refined the location of this GM density reduction to the bilateral fusiform gyrus. These results suggest that the right middle temporal gyrus contributes to morphosyntactic ability, and indeed high GM density in this region is positively associated with better morphosyntactic production (Menghini et al., 2011). Finally, Mullins et al. (2013) reported reduced temporal lobe volume in people with DS and dementia and further found a correlation between this volume reduction and cognitive status, including language skills.
3.13. Distinct cognitive-behavioral phenotypes of Down syndrome, fragile X syndrome, Williams syndrome, and microdeletion syndrome 22q11
We identified 20 studies that compared cognitive-behavioral phenotypes of DS with FXS, WS, and chromosome 22q11.2 deletion syndrome (DS22q11.2) (see Table 6). Previous studies on FXS have described delayed language development, relative weaknesses in executive function, visual–spatial working memory, perception, and thinking, visual-motor coordination, and short-term memory, but relative strengths in verbal thinking skills and simultaneous processing tasks (Powell et al., 1997; Roberts et al., 2007; Kogan et al., 2009). Further, some of these studies reported sex differences in FXS-related intellectual impairments (Cornish et al., 2007; Huddleston et al., 2014). Males with FXS demonstrated substantial abnormalities in most verbal and visuospatial domains of working memory (Cornish et al., 2007), as well as in memorizing and recounting stories, abilities crucial for developing reading skills (Conners et al., 2011), while delayed memory was relatively less impaired. Rapid phonological recall and semantic/conceptual memory are also crucial aspects of reading development, particularly for young males, but relatively little is known about such capacities in FXS (Conners et al., 2011). The cognitive profile of DS appears distinct from that of FXS in several respects, including more severe language deficits in DS but better reading skills, visual memory, visual perception, and visuomotor skills than among individuals with FXS.
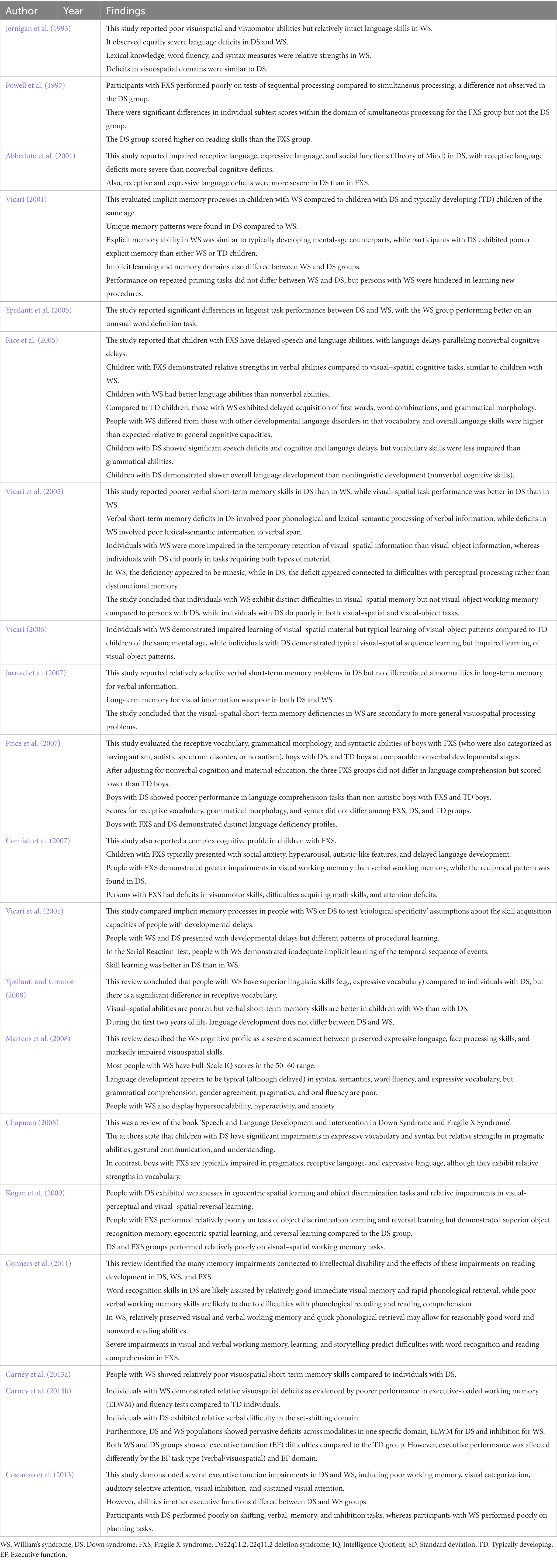
Table 6. Summary of studies comparing cognitive-behavioral phenotypes of DS with FXS, WS, and chromosome 22q11.2 deletion syndrome (DS22q11.2).
Alternatively, Ypsilanti and Grouios (2008) reported that the cognitive profile of DS is more similar to that of WS, although others have not reported such similarities (Carney et al., 2013a). These similarities include strengths in similar language domains and shared deficiences in visuospatial ability (Bellugi et al., 2000; Atkinson et al., 2001; Vicari et al., 2005; Carney et al., 2013b). However, characterizing the cognitive profile of WS and DS in terms of a dissociation between language and visuospatial abilities is an oversimplification as both syndromes exhibit unequal strengths and weaknesses within each cognitive domain (Vicari, 2001; Rice et al., 2005; Menghini et al., 2011; Costanzo et al., 2013). The typical cognitive profile of WS includes relatively intact language and facial processing skills but severely impaired visual–spatial skills (Jernigan et al., 1993), in contrast to DS. Language development in WS appears to be typical in syntax, semantics, word flow, and expressive vocabulary (albeit delayed; Ypsilanti et al., 2005; Ypsilanti and Grouios, 2008). Conversely, deficits were observed in grammatical comprehension, gender matching, pragmatics, and oral fluency (Martens et al., 2008). Several studies have confirmed visual–spatial impairments in WS (Atkinson et al., 2001; Vicari et al., 2005; Jarrold et al., 2007). Individuals with WS also show significant behavioral problems, including hyperactivity, anxiety, and hypersociality (Martens et al., 2008). Immediate spatial recall, verbal and spatial delayed memory and learning, and semantic retrieval requiring proper names are also deficient in WS. However, relative strengths are instantaneous verbal and visual recall, visual-delayed memory and learning, and phonological retrieval. Weaknesses in visual–auditory learning and semantic retrieval, as well as relative strengths in instantaneous recall (both visual and verbal) and rapid phonological retrieval, are particularly significant influences on reading development (Conners et al., 2011).
Individuals with DS22q11.2 also exhibit a complex neurocognitive profile characterized by a variable IQ score, relatively poor language, computational power, visual–spatial processing skills, and visual-object short-term memory, as well as weaknesses in executive function, motor skills, psychosocial function, and working memory (Woodin et al., 2001; Sobin et al., 2005; Persson et al., 2006; Jacobson et al., 2010; Niklasson and Gillberg, 2010; Vicari et al., 2012; Morrison et al., 2020). Sobin et al. (2005) also reported severe visual attention and working memory impairments in DS22q11.2.
4. Discussion
We searched the literature over the past several decades to identify studies investigating regional brain volume abnormalities and cognitive deficits in children and adults with DS and comparing pathological features among DS, FXS, WS, and DS22q11.2. A synthesis of results from the 84 included studies indicates that DS is typically associated with reduced total brain volume and regional volume reductions within the hippocampal formation, frontal, parietal, temporal, and occipital lobes, brain stem, and cerebellum. However, morphometric, and cognitive abnormalities differed across studies, suggesting the influence of factors such as age, sex, and comorbidity profile. While there is no ‘stereotypical’ DS profile, this review identified several brain regions and cognitive domains with relatively stable differences compared to matched TD groups and compared to FXS, WS, and DS22q11.2 groups that may provide clues to disease pathogenesis and guidance for the development of more targeted and individualized therapeutic interventions.
Although DS is a neurodevelopmental condition exhibiting substantial changes in symptom profile with age, most of the reviewed neuroimaging studies, particularly those employing VBM, included adolescents and adults. Moreover, from middle life onwards, individuals with DS are at heightened risk of AD dementia, and even those without dementia exhibit significant age-related changes in regional brain volume (Kesslak et al., 1994; Krasuski et al., 2002; Teipel et al., 2004). Furthermore, according to Teipel and Hampel (2006), the specific effects of age may be obscured by more significant variability in brain morphology. Therefore, larger-scale studies of multiple well-delineated clinical subgroups are required to address the independent impact of aging on the DS brain, cognitive capacity, and behavior.
Despite heterogeneity within and across DS study groups, there were relatively consistent associations between working memory deficits and volume reductions within the parietal lobe (Menghini et al., 2011) and temporal lobe (Galaburda and Schmitt, 2003; Pennington et al., 2003; Vicari, 2006; Menghini et al., 2011). Further, reduced temporal lobe volume was strongly associated with language deficits, although some studies, such as Pinter et al. (2001b), reported a larger corrected temporal lobe volume in DS. Nonetheless, the balance of neuroimaging evidence suggests that a disproportionately smaller temporal lobe contributes to language deficits in DS. Also, Fabbro et al. (2002) proposed that language deficits are partly due to impairments in frontocerebellar structures involved in articulation and verbal working memory. However, we did not include frontocerebellar structures in our review.
There were also relatively stable associations between regional reductions in hippocampal volume and deficits in language and memory (Raz et al., 1995; Krasuski et al., 2002; Pennington et al., 2003). Moreover, the hippocampus is among the most vulnerable regions to damage by AD, including from the premature AD observed in many individuals with DS concomitant with dementia onset (Aylward et al., 1999; Hamadelseed et al., 2022). Less consistent was the association between age and hippocampal volume. Raz et al. (1995) and Aylward et al. (1999) found no correlation between age and hippocampal volume, while Kesslak et al. (1994) found a significant correlation. A decrease in hippocampal volume with age may help explain the learning and memory deficits that start in infancy, continue throughout childhood, and frequently worsen in adulthood (Kates et al., 1997b). We speculate that deficits in hippocampal circuit function and plasticity undetectable by structural neuroimaging contribute to specific language and memory deficits in DS. Similarly, long-term memory impairments may be linked to medial temporal lobe volume reductions or circuit dysfunction. Higher-resolution functional neuroimaging studies are needed to elucidate these mechanisms.
The memory profile observed in DS appears distinct from that of the other cognitive impairment-related hereditary diseases reviewed. For instance, verbal and visual short-term memory is relatively preserved. In contrast, spatial working memory is relatively deficient in WS, and a similar pattern is observed in explicit long-term memory among adolescents with WS. Conversely, individuals with WS demonstrate reduced implicit process learning capacity (Vicari, 2006), while this form of learning is relatively intact among individuals with DS.
A likely reason for the variability observed within and across studies is the small individual sample size and limited statistical power (Deinde et al., 2021). However, recruiting large numbers of individuals with neurodevelopmental disorders, especially children, for comprehensive neuropsychological testing and neuroimaging is challenging. First, many people (regardless of cognitive status) are afraid of brain scans (Enders et al., 2011), while MRI acquisition may be challenging for people with DS as many are obese and thus may experience claustrophobia (Melville et al., 2005; Head et al., 2018). Furthermore, individuals with DS may experience discomfort in the prone position during extended periods due to the disorder’s distinctive neck and facial anatomy (Head et al., 2018). Third, fear of scanning may be further compounded by an inability to understand the purpose of the procedure or follow instructions. Such individuals may find it difficult to remain stationary during the scanning procedure, resulting in movement artifacts (Neale et al., 2018). While sedation may be helpful, it may also influence the neural processes under study (Landt et al., 2011; Head et al., 2018). Allowing the DS participants to visit the scanning site and become familiar with the scanning technique may help alleviate fear and promote relaxation and trust in the operator. Mock scanners and shorter scan wait times may also help set participants at ease.
A general problem limiting the sample size for neuroimaging is the operating expense and equipment availability (Neale et al., 2018). For this reason, MRI should be combined with cognitive and clinical tests to maximize data acquisition and provide an independent data stream for better interpretation. It is also necessary to compare neuroimaging results between people with DS with and without dementia. Such information could help identify regions most vulnerable to AD, predictive biomarkers, and underlying pathogenic mechanisms for effective treatment.
5. Conclusion
This review is one of the few to assess the unique neuroanatomic and neuropsychological characteristics of Down syndrome comprehensively and critically. A synthesis of the included studies suggests that cognitive deficits in DS, especially within language and memory domains, may be associated with abnormalities in regional brain volume. Indeed, DS is characterized by relatively consistent GM and WM loss in regions subserving memory and language, including the hippocampal formation and lateral temporal lobe. Thus, deficits in memory and language are more severe than expected from global intellectual dysfunction. In addition, a reduction in parietal lobe volume may be a significant predictor of cognitive deficits in DS, particularly among individuals with impaired visuospatial processing ability. Neuroimaging is a powerful technique that can help researchers detect early structural, functional, neurochemical, and metabolic alterations associated with DS, including AD development. These neuroimaging discoveries could allow physicians to treat high-risk patients before AD onset, thereby maintaining cognitive function and quality of life. Neuroimaging in DS may also facilitate the development of new therapies and improved treatment response monitoring. Large-scale longitudinal studies will be critical for expanding our understanding of AD pathogenesis in DS.
In summary, this review provides educational psychologists and instructors with crucial information to identify and address the various learning and social challenges experienced by individuals with DS. Understanding the cognitive and behavioral phenotypes of DS will help professionals and parents better understand and manage the condition in daily life, helping people with DS achieve their highest level of independent function.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
OH, MC, MW, and TS performed material preparation, data collection, and analysis. OH wrote the first draft of the manuscript, and all authors commented on previous versions. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This research project was made possible through funding from the Friedrich-Naumann Foundation for Freedom (Scholar-Number: 8683/P611, 2021).
Acknowledgments
We thank our families for their outstanding support and patience during the data collection and analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
1. ^ https://rarediseases.info.nih.gov/diseases/10247/down-syndrome
2. ^ http://www.intellectualdisability.info/diagnosis/articles/the-genetics-of-downs-syndrome
References
Abbeduto, L., Pavetto, M., Kesin, E., Weissman, M. D., Karadottir, S., O’Brien, A., et al. (2001). The linguistic and cognitive profile of Down syndrome: evidence from a comparison with fragile X syndrome. Downs Syndr. Res. Pract. 7, 9–15. doi: 10.3104/reports.109
Aktas, M. (2004). Sprachentwicklungsdiagnostik bei Kindern mit Down-Syndrom: Entwicklung eines diagnostischen Leitfadens zum theoriegeleiteten Einsatz standardisierter Verfahren.
Alexander, G. E., Saunders, A. M., Szczepanik, J., Strassburger, T. L., Pietrini, P., Dani, A., et al. (1997). Relation of age and apolipoprotein E to cognitive function in Down syndrome adults. Neuroreport. 8, 1835–1840. doi: 10.1097/00001756-199705260-00009
Atkinson, J., Anker, S., Braddick, O., Nokes, L., Mason, A., and Braddick, F. (2001). Visual and visuospatial development in young children with Williams syndrome. Dev. Med. Child Neurol. 43, 330–337. doi: 10.1017/S0012162201000615
Aylward, E. H., Habbak, R., Warren, A. C., Pulsifer, M. B., Barta, P. E., Jerram, M., et al. (1997a). Cerebellar volume in adults with Down syndrome. Arch. Neurol. 54, 209–212. doi: 10.1001/archneur.1997.00550140077016
Aylward, E. H., Li, Q., Habbak, Q. R., Warren, A., Pulsifer, M. B., Barta, P. E., et al. (1997b). Basal ganglia volume in adults with Down syndrome. Psychiatry Res. 74, 73–82. doi: 10.1016/S0925-4927(97)00011-5
Aylward, E. H., Li, Q., Honeycutt, N. A., Warren, A. C., Pulsifer, M. B., Barta, P. E., et al. (1999). MRI volumes of the hippocampus and amygdala in adults with Down’s syndrome with and without dementia. Am. J. Psychiatr. 156, 564–568. doi: 10.1176/ajp.156.4.564
Ball, M., Schapiro, M., and Rapoport, S. I. (1986). Neuropathological relationships between Down syndrome and senile dementia Alzheimer type. The Neurobiol. Down Syndrome 45, 58.
Beacher, F., Daly, E., Simmons, A., Prasher, V., Morris, R., Robinson, C., et al. (2010). Brain anatomy and ageing in non-demented adults with Down’s syndrome: an in vivo MRI study. Psychol. Med. 40, 611–619. doi: 10.1017/S0033291709990985
Becker, L., Mito, T., Takashima, S., and Onodera, K. (1991). Growth and development of the brain in Down syndrome. Prog. Clin. Biol. Res. 373, 133–152.
Bellugi, U., Lichtenberger, L., Jones, W., Lai, Z., and George, S. (2000). I. the neurocognitive profile of Williams syndrome: a complex pattern of strengths and weaknesses. J. Cogn. Neurosci. 12, 7–29. doi: 10.1162/089892900561959
Blanken, A. E., Hurtz, S., Zarow, C., Biado, K., Honarpisheh, H., Somme, J., et al. (2017). Associations between hippocampal morphometry and neuropathologic markers of Alzheimer’s disease using 7 T MRI. NeuroImage Clin. 15, 56–61. doi: 10.1016/j.nicl.2017.04.020
Bobinski, M. M. J., de Leon, M. J., Wegiel, J., DeSanti, S., Convit, A., Saint Louis, L. A., et al. (1999). The histological validation of post mortem magnetic resonance imaging-determined hippocampal volume in Alzheimer's disease. Neuroscience 95, 721–725. doi: 10.1016/S0306-4522(99)00476-5
Bobinski, M., Wegiel, J., Wisniewski, H. M., Tarnawski, M., Bobinski, M., Reisberg, B., et al. (1996). Neurofibrillary pathology — correlation with hippocampal formation atrophy in Alzheimer disease. Neurobiol. Aging 17, 909–919. doi: 10.1016/s0197-4580(97)85095-6
Bush, A., and Beail, N. (2004). Risk factors for dementia in people with Down syndrome: issues in assessment and diagnosis. Am. J. Ment.Retard. 109, 83–97. doi: 10.1352/0895-8017(2004)109<83:RFFDIP>2.0.CO;2
Campbell, L. E., Daly, E., Toal, F., Stevens, A., Azuma, R., Karmiloff-Smith, A., et al. (2009). Brain structural differences associated with the behavioural phenotype in children with Williams syndrome. Brain Res. 1258, 96–107. doi: 10.1016/j.brainres.2008.11.101
Carducci, F., Onorati, P., Condoluci, C., Di Gennaro, G., Quarato, P. P., Pierallini, A., et al. (2013). Whole-brain voxel-based morphometry study of children and adolescents with Down syndrome. Funct. Neurol. 28, 19–28.
Carney, D. P. J., Brown, J. H., and Henry, L. A. (2013a). Executive function in Williams and Down syndromes. Res. Dev. Disabil. 34, 46–55. doi: 10.1016/j.ridd.2012.07.013
Carney, D. P. J., Henry, L. A., Messer, D. J., Danielsson, H., Brown, J. H., and Rönnberg, J. (2013b). Using developmental trajectories to examine verbal and visuospatial short-term memory development in children and adolescents with Williams and Down syndromes. Res. Dev. Disabil. 34, 3421–3432. doi: 10.1016/j.ridd.2013.07.012
Channell, M. M., The Down Syndrome Cognition Project, Mattie, L. J., Hamilton, D. R., Capone, G. T., Mahone, E. M., et al. (2021). Capturing cognitive and behavioral variability among individuals with Down syndrome: a latent profile analysis. J. Neurodev. Disord. 13:16. doi: 10.1186/s11689-021-09365-2
Chapman, R. S. (2003). Language and communication in individuals with Down syndrome. Int. Rev. Res. Ment. Retard. 27, 1–34. doi: 10.1016/S0074-7750(03)27001-4
Chapman, R. S. (2006). Language learning in Down syndrome: the speech and language profile compared to adolescents with cognitive impairment of unknown origin. Downs Syndr. Res. Pract. 10, 61–66. doi: 10.3104/reports.306
Chapman, R. S. (2008). Speech and language development and intervention in Down syndrome and fragile X syndrome, Baltimore: Brookes.
Chapman, R. S., and Hesketh, L. J. (2000). Behavioral phenotype of individuals with Down syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 6, 84–95. doi: 10.1002/1098-2779(2000)6:2<84::AID-MRDD2>3.0.CO;2-P
Chapman, R. S., and Hesketh, L. J. (2001). Language, cognition, and short-term memory in individuals with Down syndrome. J. Sarah Duff. Centre 7, 1–7. doi: 10.3104/reviews.108
Chiang, M. C., Reiss, A. L., Lee, A. D., Bellugi, U., Galaburda, A. M., Korenberg, J. R., et al. (2007). 3D pattern of brain abnormalities in Williams syndrome visualized using tensor-based morphometry. NeuroImage 36, 1096–1109. doi: 10.1016/j.neuroimage.2007.04.024
Chow, E. W., Zipursky, R. B., Mikulis, D. J., and Bassett, A. S. (2002). Structural brain abnormalities in patients with schizophrenia and 22q11 deletion syndrome. Biol. Psychiatry 51, 208–215. doi: 10.1016/S0006-3223(01)01246-X
Clark, C. A. C., Fernandez, F., Sakhon, S., Spanò, G., and Edgin, J. O. (2017). The medial temporal memory system in Down syndrome: translating animal models of hippocampal compromise. Hippocampus 27, 683–691. doi: 10.1002/hipo.22724
Clibbens, J. (2001). Signing and lexical development in children with Down syndrome. J. Sarah Duff. Centre 7, 101–105. doi: 10.3104/reviews.119
Conners, F. A., Moore, M. S., Loveall, S. J., and Merrill, E. C. (2011). Memory profiles of down, Williams, and fragile X syndromes: implications for reading development. J. Dev. Behav. Pediat. 32, 405–417. doi: 10.1097/DBP.0b013e3182168f95
Contestabile, A., Fila, T., Ceccarelli, C., Bonasoni, P., Bonapace, L., Santini, D., et al. (2007). Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with Down syndrome and in Ts65Dn mice. Hippocampus 17, 665–678. doi: 10.1002/hipo.20308
Cornish, K. M., Levitas, A., and Sudhalter, V. (2007). “Fragile X syndrome: the journey from genes to behavior” in Neurogenetic developmental disorders: Manifestation and identification in childhood. eds. M. M. Mazzocco and J. Ross (Cambridge, MA: MIT Press), 73–103.
Costanzo, F., Varuzza, C., Menghini, D., Addona, F., Gianesini, T., and Vicari, S. (2013). Executive functions in intellectual disabilities: a comparison between Williams syndrome and Down syndrome. Res. Dev. Disabil. 34, 1770–1780. doi: 10.1016/j.ridd.2013.01.024
Couzens, D., Cuskelly, M., and Haynes, M. (2011). Cognitive development and Down syndrome: age-related change on the Stanford-Binet test (fourth edition). Am. J. Intellect. Dev. Disabil. 116, 181–204. doi: 10.1352/1944-7558-116.3.181
Coyle, J. T., Oster-Granite, M., and Gearhart, J. D. (1986). The neurobiologie consequences of Down syndrome. Brain Res. Bull. 16, 773–787. doi: 10.1016/0361-9230(86)90074-2
de la Monte, S. M., and Hedley-Whyte, E. T. (1990). Small cerebral hemispheres in adults with Down’s syndrome: contributions of developmental arrest and lesions of Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 49, 509–520. doi: 10.1097/00005072-199009000-00006
Debbané, M., Schaer, M., Farhoumand, R., Glaser, B., and Eliez, S. (2006). Hippocampal volume reduction in 22q11. 2 deletion syndrome. Neuropsychologia 44, 2360–2365. doi: 10.1016/j.neuropsychologia.2006.05.006
DeBoer, T., Wu, Z., Lee, A., and Simon, T. J. (2007). Hippocampal volume reduction in children with chromosome 22q11.2 deletion syndrome is associated with cognitive impairment. Behav. Brain Funct. 3:54. doi: 10.1186/1744-9081-3-54
Deinde, F., Kotecha, J., Lau, L. S. L., Bhattacharyya, S., and Velayudhan, L. (2021). A review of functional neuroimaging in people with Down syndrome with and without dementia. Dement. Geriatr. Cogn. Disord. Extra 11, 324–332. doi: 10.1159/000520880
Démonet, J. F., Chollet, F., Ramsay, S., Cardebat, D., Nespoulous, J. L., Wise, R., et al. (1992). The anatomy of phonological and semantic processing in normal subjects. Brain J. Neurol. 115, 1753–1768. doi: 10.1093/brain/115.6.1753
Dodd, B., and Thompson, L. (2001). Speech disorder in children with Down’s syndrome. J. Intellect. Disabil. Res. 45, 308–316. doi: 10.1046/j.1365-2788.2001.00327.x
Dodge, E. P. (2004). Communication skills: the foundation for meaningful group intervention in school-based programs. Top. Lang. Disord. 24, 141–150. doi: 10.1097/00011363-200404000-00007
Edgin, J. O. (2013). Cognition in Down syndrome: a developmental cognitive neuroscience perspective. Wiley Interdiscip. Rev. Cogn. Sci. 4, 307–317. doi: 10.1002/wcs.1221
Eliez, S., Blasey, C. M., Schmitt, E. J., White, C. D., Hu, D., and Reiss, A. L. (2001). Velocardiofacial syndrome: are structural changes in the temporal and mesial temporal regions related to schizophrenia? Am. J. Psychiatr. 158, 447–453. doi: 10.1176/appi.ajp.158.3.447
Eliez, S., Schmitt, J. E., White, C. D., and Reiss, A. L. (2000). Children and adolescents with velocardiofacial syndrome: a volumetric MRI study. Am. J. Psychiatr. 157, 409–415. doi: 10.1176/appi.ajp.157.3.409
Enders, J., Zimmermann, E., Rief, M., Martus, P., Klingebiel, R., Asbach, P., et al. (2011). ‘Reduction of claustrophobia during magnetic resonance imaging: methods and design of the CLAUSTRO’ randomized controlled trial. BMC Med. Imaging 11:4. doi: 10.1186/1471-2342-11-4
Evenhuis, H. M. (1990). The natural history of dementia in Down’s syndrome. Arch. Neurol. 47, 263–267. doi: 10.1001/archneur.1990.00530030029011
Fabbro, F., Alberti, B., Gagliardi, C., and Borgatti, R. (2002). Differences in native and foreign language repetition tasks between subjects with William’s and Down’s syndromes. J. Neurolinguistics 15, 1–10. doi: 10.1016/S0911-6044(00)00016-6
Fernandez-Alcaraz, C., and Carvajal-Molina, F. (2014). Cytogenetic and neurobiological advances in Down syndrome. Anales de Psicología 30, 347–355.
Fidler, D. J. (2005). The emerging Down syndrome behavioral phenotype in early childhood: implications for practice. Infants Young Child. 18, 86–103.
Fidler, D., Most, D., and Philofsky, A. (2008). The Down syndrome behavioural phenotype: taking a developmental approach.
Fowler, A. E. (1995). “Language variability in persons with Down syndrome” in Down syndrome: Living and learning in the community (New York: Wiley-Liss)
Frangou, S., Aylward, E., Warren, A., Sharma, T., Barta, P., and Pearlson, G. (1997). Small planum temporale volume in Down’s syndrome: a volumetric MRI study. Am. J. Psychiatr. 154, 1424–1429. doi: 10.1176/ajp.154.10.1424
Friederici, A. D., and Gierhan, S. M. (2013). The language network. Curr. Opin. Neurobiol. 23, 250–254. doi: 10.1016/j.conb.2012.10.002
Fujii, Y., Aida, N., Niwa, T., Enokizono, M., Nozawa, K., and Inoue, T. (2017). A small pons as a characteristic finding in Down syndrome: a quantitative MRI study. Brain Dev. 39, 298–305. doi: 10.1016/j.braindev.2016.10.016
Galaburda, A., and Schmitt, J. (2003). Neuroanatomical considerations specific to the study of neurogenetics.
Giencke, S., and Lewandowski, L. J. C. (1989). Anomalous dominance in Down syndrome young adults. J. Dev. Study Nerv. Syst. Behav. 25, 93–102. doi: 10.1016/S0010-9452(89)80009-7
Godfrey, M., and Lee, N. R. (2018). Memory profiles in Down syndrome across development: a review of memory abilities through the lifespan. J. Neurodev. Disord. 10:5. doi: 10.1186/s11689-017-9220-y
Grieco, J., Pulsifer, M., Seligsohn, K., Skotko, B., and Schwartz, A. (2015). Down syndrome: cognitive and behavioral functioning across the lifespan. Am. J. Med. Genet. C Semin. Med. Genet. 169, 135–149. doi: 10.1002/ajmg.c.31439
Gunbey, H. P., Bilgici, M. C., Aslan, K., Has, A. C., Ogur, M. G., Alhan, A., et al. (2017). Structural brain alterations of Down’s syndrome in early childhood evaluation by DTI and volumetric analyses. Eur. Radiol. 27, 3013–3021. doi: 10.1007/s00330-016-4626-6
Haier, R. J., Head, K., Head, E., and Lott, I. T. (2008). Neuroimaging of individuals with Down’s syndrome at-risk for dementia: evidence for possible compensatory events. NeuroImage 39, 1324–1332. doi: 10.1016/j.neuroimage.2007.09.064
Hamadelseed, O., Elkhidir, I. H., and Skutella, T. (2022). Psychosocial risk factors for Alzheimer’s disease in patients with Down syndrome and their association with brain changes: a narrative review. Neurol. Ther. 11, 931–953. doi: 10.1007/s40120-022-00361-9
Hamner, T., Udhnani, M. D., Osipowicz, K. Z., and Lee, N. R. (2018). Pediatric brain development in Down syndrome: a field in its infancy. J. Int. Neuropsychol. Soc. 24, 966–976. doi: 10.1017/S1355617718000206
Hartley, X. Y. J. C. (1981). Lateralisation of speech stimuli in young Down’s syndrome children. J. Dev. Stud. Nerv. Syst. Behav. 17, 241–248. doi: 10.1016/S0010-9452(81)80044-5
Hartley, X. Y. J. J. (1985). Receptive language processing and ear advantage of Down’s syndrome children. J. Ment. Defic. Res. 29, 197–205. doi: 10.1111/j.1365-2788.1985.tb00328.x
Haug, H. (1985). Are neurons of the human cerebral cortex really lost during aging? A morphometric examination. Senile dementia of the Alzheimer type Singapore, Springer: 150–163.
Head, E., Powell, D. K., and Schmitt, F. A. (2018). Metabolic and vascular imaging biomarkers in Down syndrome provide unique insights into brain aging and Alzheimer disease pathogenesis. Front. Aging Neurosci. 10:191. doi: 10.3389/fnagi.2018.00191
Hick, R. F., Botting, N., and Conti-Ramsden, G. (2005). Short-term memory and vocabulary development in children with Down syndrome and children with specific language impairment. Dev. Med. Child Neurol. 47, 532–538. doi: 10.1017/S0012162205001040
Hickok, G., and Poeppel, D. (2004). Dorsal and ventral streams: a framework for understanding aspects of the functional anatomy of language. Cognition 92, 67–99. doi: 10.1016/j.cognition.2003.10.011
Hixson, P. K. (1993). An integrated approach to program development. Top. Lang. Disord. 14, 41–57. doi: 10.1097/00011363-199311000-00006
Hoehne, C. (2007). Messungen der regionalen Corpus-callosum- und hippocampus-atrophie bei nicht dementen Erwachsenen mit Down-Syndrom, lmu.
Huddleston, L. B., Visootsak, J., and Sherman, S. L. (2014). Cognitive aspects of fragile X syndrome. Wiley Interdisciplinary Reviews. Cogn. Sci. 5, 501–508. doi: 10.1002/wcs.1296
Hyde, L. A., and Crnic, L. S. (2001). Age-related deficits in context discrimination learning in Ts65Dn mice that model Down syndrome and Alzheimer’s disease. Behav. Neurosci. 115, 1239–1246. doi: 10.1037/0735-7044.115.6.1239
Ieshima, A., Kisa, T., Yoshino, K., Takashima, S., and Takeshita, K. (1984). A morphometric CT study of Down’s syndrome showing small posterior fossa and calcification of basal ganglia. Neuroradiology 26, 493–498. doi: 10.1007/BF00342687
Isaacs, E. B., Vargha-Khadem, F., Watkins, K. E., Lucas, A., Mishkin, M., and Gadian, D. G. (2003). Developmental amnesia and its relationship to degree of hippocampal atrophy. Proc. Natl. Acad. Sci. U. S. A. 100, 13060–13063. doi: 10.1073/pnas.1233825100
Iverson, J. M., Longobardi, E., and Caselli, M. C. (2003). Relationship between gestures and words in children with Down’s syndrome and typically developing children in the early stages of communicative development. Int. J. Lang. Commun. Disord. 38, 179–197. doi: 10.1080/1368282031000062891
Jacobson, C., Shearer, J., Habel, A., Kane, F., Tsakanikos, E., and Kravariti, E. (2010). Core neuropsychological characteristics of children and adolescents with 22q11. 2 deletion. J. Intellect. Disabil. Res. 54, 701–713. doi: 10.1111/j.1365-2788.2010.01298.x
Jahn, H. (2013). Memory loss in Alzheimer’s disease. Dialogues Clin. Neurosci. 15, 445–454. doi: 10.31887/DCNS.2013.15.4/hjahn
Jarrold, C., Baddeley, A. D., and Phillips, C. (2007). Long-term memory for verbal and visual information in Down syndrome and Williams syndrome: performance on the doors and people test. J. Dev. Study Nerv. Syst. Behav. 43, 233–247. doi: 10.1016/S0010-9452(08)70478-7
Jernigan, T. L., and Bellugi, U. (1990). Anomalous brain morphology on magnetic resonance images in Williams syndrome and Down syndrome. Arch. Neurol. 47, 529–533. doi: 10.1001/archneur.1990.00530050049011
Jernigan, T. L., Bellugi, U., Sowell, E., Doherty, S., and Hesselink, J. R. (1993). Cerebral morphologic distinctions between Williams and Down syndromes. Arch. Neurol. 50, 186–191. doi: 10.1001/archneur.1993.00540020062019
Kates, W. R., Abrams, M. T., Kaufmann, W. E., Breiter, S. N., and Reiss, A. L. (1997a). Reliability and validity of MRI measurement of the amygdala and hippocampus in children with fragile X syndrome. Psychiatry Res. 75, 31–48. doi: 10.1016/S0925-4927(97)00019-X
Kates, W. R., Folley, B. S., Lanham, D. C., and Capone, G. T. (2002). Cerebral growth in fragile X syndrome: Review and comparison with Down syndrome. Microsc. Res. Tech. 57, 159–167.
Kates, W. R., Kaufmann, W. E., and Reiss, A. L. (1997b). Neuroimaging of developmental and genetic disorders. Child Adolesc. Psychiatr. Clin. N. Am. 6, 283–304. doi: 10.1016/S1056-4993(18)30305-5
Kates, W. R., Miller, A. M., Abdulsabur, N., Antshel, K. M., Conchelos, J., Fremont, W., et al. (2006). Temporal lobe anatomy and psychiatric symptoms in velocardiofacial syndrome (22q11.2 deletion syndrome). J. Am. Acad. Child Adolesc. Psychiatry 45, 587–595. doi: 10.1097/01.chi.0000205704.33077.4a
Kesslak, J. P., Nagata, S. F., Lott, I., and Nalcioglu, O. (1994). Magnetic resonance imaging analysis of age-related changes in the brains of individuals with Down’s syndrome. Neurology 44, 1039–1045. doi: 10.1212/WNL.44.6.1039
Koenig, K. A., Oh, S. H., Stasko, M. R., Roth, E. C., Taylor, H. G., Ruedrich, S., et al. (2021). High resolution structural and functional MRI of the hippocampus in young adults with Down syndrome. Brain Commun. 3:fcab088. doi: 10.1093/braincomms/fcab088
Kogan, C. S., Boutet, I., Cornish, K., Graham, G. E., Berry-Kravis, E., Drouin, A., et al. (2009). A comparative neuropsychological test battery differentiates cognitive signatures of fragile X and Down syndrome. J. Intellect. Disabil. Res. 53, 125–142. doi: 10.1111/j.1365-2788.2008.01135.x
Komaki, H., Hamaguchi, H., and Hashimoto, T. (1999). Assessment of the brainstem and the cerebellar lesions and myelination using magnetic resonance images in children with Down syndrome. No To Hattatsu 31, 422–427.
Koran, M. E. I., Hohman, T. J., Edwards, C. M., Vega, J. N., Pryweller, J. R., Slosky, L. E., et al. (2014). Differences in age-related effects on brain volume in Down syndrome as compared to Williams syndrome and typical development. J. Neurodev. Disord. 6:8. doi: 10.1186/1866-1955-6-8
Krasuski, J. S., Alexander, G. E., Horwitz, B., Rapoport, S. I., and Schapiro, M. B. (2002). Relation of medial temporal lobe volumes to age and memory function in nondemented adults with Down’s syndrome: implications for the prodromal phase of Alzheimer’s disease. Am. J. Psychiatr. 159, 74–81. doi: 10.1176/appi.ajp.159.1.74
Lai, F., and Williams, R. S. (1989). A prospective study of Alzheimer disease in Down syndromeDown syndrome. Arch. Neurol. 46, 849–853. doi: 10.1001/archneur.1989.00520440031017
Landt, J., d’Abrera, J. C., Holland, A. J., Aigbirhio, F. I., Fryer, T. D., Canales, R., et al. (2011). Using positron emission tomography and carbon 11-labeled Pittsburgh compound B to image brain Fibrillar β-amyloid in adults with Down syndrome: safety, acceptability, and feasibility. Arch. Neurol. 68, 890–896. doi: 10.1001/archneurol.2011.36
Lanfranchi, S., Jerman, O., and Vianello, R. (2009). Working memory and cognitive skills in individuals with Down syndrome. Child Neuropsychol. J. Norm. Abnorm. Dev. Childh. Adolesc. 15, 397–416. doi: 10.1080/09297040902740652
Launonen, K. (1994). Enhauncing communication skills of children with Down syndrome: early use of manual signs. Augment. Altern. Commun. Euro. Perspect. 213–231.
Lawlor, B. A., McCarron, M., Wilson, G., and McLoughlin, M. (2001). Temporal lobe-oriented CT scanning and dementia in Down’s syndrome. Int. J. Geriatr. Psychiatry 16, 427–429. doi: 10.1002/gps.358
Laws, G., and Bishop, D. V. (2003). A comparison of language abilities in adolescents with Down syndrome and children with specific language impairment. J. Speech Lang. Hear. Res. 46, 1324–1339. doi: 10.1044/1092-4388(2003/103)
Lee, N. R., Adeyemi, E. I., Lin, A., Clasen, L. S., Lalonde, F. M., Condon, E., et al. (2015). Dissociations in cortical morphometry in youth with Down syndrome: evidence for reduced surface area but increased thickness. Cereb. Cortex Commun. 26, 2982–2990. doi: 10.1093/cercor/bhv107
Levman, J., MacDonald, A., Baumer, N., MacDonald, P., Stewart, N., Lim, A., et al. (2019). Structural magnetic resonance imaging demonstrates abnormal cortical thickness in down syndrome: newborns to young adults. NeuroImage. Clin. 23:101874. doi: 10.1016/j.nicl.2019.101874
Lott, I. T. J. A. (1982). Down’s syndrome, aging, and Alzheimer’s disease: a clinical review. Ann. N. Y. Acad. Sci. 396, 15–27. doi: 10.1111/j.1749-6632.1982.tb26840.x
Määttä, T., Tervo-Määttä, T., Taanila, A., Kaski, M., and Iivanainen, M. (2006). Mental health, behaviour and intellectual abilities of people with Down syndrome. J. Sarah Duff. Centre 11, 37–43. doi: 10.3104/reports.313
Mann, D. M. A. (1988). Alzheimer’s disease and Down’s syndrome. Histopathology 13, 125–137. doi: 10.1111/j.1365-2559.1988.tb02018.x
Martens, M. A., Wilson, S. J., and Reutens, D. C. (2008). Research review: Williams syndrome: a critical review of the cognitive, behavioral, and neuroanatomical phenotype. J. Child Psychol. Psychiatry 49, 576–608. doi: 10.1111/j.1469-7610.2008.01887.x
Martin, G. E., Klusek, J., Estigarribia, B., and Roberts, J. E. (2009). Language characteristics of individuals with Down syndrome. Top. Lang. Disord. 29, 112–132. doi: 10.1097/TLD.0b013e3181a71fe1
McCann, B., Levman, J., Baumer, N., Lam, M. Y., Shiohama, T., Cogger, L., et al. (2021). Structural magnetic resonance imaging demonstrates volumetric brain abnormalities in down syndrome: newborns to young adults. NeuroImage Clin. 32:102815. doi: 10.1016/j.nicl.2021.102815
McCauley, R. J., Fey, M. E., and Gillam, R. B. (2006). Treatment of language disorders in children, Washington: Paul H. Brookes Pub.
McWilliam, P., Winton, P. J., and Crais, E. R. (1996). Practical strategies for family-centered early intervention, San Diego: Singular.
Melville, C. A., Cooper, S. A., McGrother, C. W., Thorp, C. F., and Collacott, R. (2005). Obesity in adults with Down syndrome: a case-control study. J. Intellect. Disabil. Res. 49, 125–133. doi: 10.1111/j.1365-2788.2004.00616.x
Menghini, D., Costanzo, F., and Vicari, S. (2011). Relationship between brain and cognitive processes in Down syndrome. Behav. Genet. 41, 381–393. doi: 10.1007/s10519-011-9448-3
Meyer-Lindenberg, A., Mervis, C. B., Sarpal, D., Koch, P., Steele, S., Kohn, P., et al. (2005). Das and S. J. T. J. O. C. I. Kippenhan . “Functional, structural, and metabolic abnormalities of the hippocampal formation in Williams syndrome ” 115: 1888–1895.
Miller, J. F. (1999). Profiles of language development in children with Down syndrome. Improving the communication of people with Down syndrome. Genet. Med., 11–39.
Morrison, S., Chawner, S. J. R. A., van Amelsvoort, T. A. M. J., Swillen, A., Vingerhoets, C., Vergaelen, E., et al. (2020). Cognitive deficits in childhood, adolescence and adulthood in 22q11.2 deletion syndrome and association with psychopathology. Transl. Psychiatry 10:53. doi: 10.1038/s41398-020-0736-7
Mullins, D., Daly, E., Simmons, A., Beacher, F., Foy, C. M., Lovestone, S., et al. (2013). Dementia in Down’s syndrome: an MRI comparison with Alzheimer’s disease in the general population. J. Neurodev. Disord. 5:19. doi: 10.1186/1866-1955-5-19
Nadel, L. (2003). Down’s syndrome: a genetic disorder in biobehavioral perspective. Genes Brain Behav. 2, 156–166. doi: 10.1034/j.1601-183X.2003.00026.x
Nagy, Z., Jobst, K. A., Esiri, M. M., Morris, J. H., King, E. M., MacDonald, B., et al. (1996). Hippocampal pathology reflects memory deficit and brain imaging measurements in Alzheimer’s disease: clinicopathologic correlations using three sets of pathologic diagnostic criteria. Dementia 7, 76–81. doi: 10.1159/000106857
Neale, N., Padilla, C., Fonseca, L. M., Holland, T., and Zaman, S. H. (2018). Neuroimaging and other modalities to assess Alzheimer’s disease in Down syndrome☆. NeuroImage Clin. 17, 263–271. doi: 10.1016/j.nicl.2017.10.022
Nelson, L. D., Orme, D., Osann, K., and Lott, I. T. (2001). Neurological changes and emotional functioning in adults with Down Syndrome. J. Intellect. Disabil. Res. 45, 450–456. doi: 10.1046/j.1365-2788.2001.00379.x
Niklasson, L., and Gillberg, C. (2010). The neuropsychology of 22q11 deletion syndrome. A neuropsychiatric study of 100 individuals. Res. Dev. Disabil. 31, 185–194. doi: 10.1016/j.ridd.2009.09.001
Oliver, C. D. (2012). Down syndrome and language development, IL: Southern Illinois University Carbondale.
Onnivello, S., Pulina, F., Locatelli, C., Marcolin, C., Ramacieri, G., Antonaros, F., et al. (2022). Cognitive profiles in children and adolescents with Down syndrome. Sci. Rep. 12:1936. doi: 10.1038/s41598-022-05825-4
Parker, S. E., Mai, C. T., Canfield, M. A., Rickard, R., Wang, Y., Meyer, R. E., et al. (2010). Updated national birth prevalence estimates for selected birth defects in the United States, 2004-2006. Birth Defects Res. A Clin. Mol. Teratol. 88, 1008–1016. doi: 10.1002/bdra.20735
Patkee, P. A., Baburamani, A. A., Kyriakopoulou, V., Davidson, A., Avini, E., et al. (2020). Early alterations in cortical and cerebellar regional brain growth in Down syndrome: an in vivo fetal and neonatal MRI assessment. NeuroImage Clin. 25:102139. doi: 10.1016/j.nicl.2019.102139
Patterson, T., Rapsey, C. M., and Glue, P. (2013). Systematic review of cognitive development across childhood in Down syndrome: implications for treatment interventions. J. Intellect. Disabl. Res. 57, 306–318. doi: 10.1111/j.1365-2788.2012.01536.x
Paul, R. (2007). Language disorders from infancy through adolescence: assessment & intervention, Salt Lake City: Elsevier Health Sciences.
Pearlson, G. D., Breiter, S. N., Warren, A. C., Grygorcewicz, M., and Frangou, S. (1998). MRI brain changes in subjects with Down syndrome with and without dementia. Neurology 40, 326–334.
Pearlson, G. D., Warren, A. C., Starkstein, S. E., Aylward, E. H., Kumar, A. J., Chase, G. A., et al. (1990). Brain atrophy in 18 patients with Down syndrome: a CT study. AJNR Am. J. Neuroradiol. 11, 811–816.
Pennington, B. F., Moon, J., Edgin, J., Stedron, J., and Nadel, L. (2003). The neuropsychology of Down syndrome: evidence for hippocampal dysfunction. Child Dev. 74, 75–93. doi: 10.1111/1467-8624.00522
Persson, C., Niklasson, L., Óskarsdóttir, S., Johansson, S., Jönsson, R., and Söderpalm, E. (2006). Language skills in 5-8-year-old children with 22q11 deletion syndrome. Int. J. Lang. Commun. Disord. 41, 313–333. doi: 10.1080/13682820500361497
Pinter, J. D., Brown, W. E., Eliez, S., Schmitt, J. E., Capone, G. T., and Reiss, A. L. (2001a). Amygdala and hippocampal volumes in children with Down syndrome: a high-resolution MRI study. Neurology 56, 972–974. doi: 10.1212/WNL.56.7.972
Pinter, J. D., Eliez, S., Schmitt, J. E., Capone, G. T., and Reiss, A. L. (2001b). Neuroanatomy of Down’s syndrome: a high-resolution MRI study. Am. J. Psychiatr. 158, 1659–1665. doi: 10.1176/appi.ajp.158.10.1659
Posner, M. I., and Dehaene, S. (1994). Attentional networks. Trends Neurosci. 17, 75–79. doi: 10.1016/0166-2236(94)90078-7
Powell, L., Houghton, S., and Douglas, G. (1997). Comparison of etiology-specific cognitive functioning profiles for individuals with fragile X and individuals with Down syndrome. J. Spec. Educ. 31, 362–376. doi: 10.1177/002246699703100305
Price, J., Roberts, J., Vandergrift, N., and Martin, G. (2007). Language comprehension in boys with fragile X syndrome and boys with Down syndrome. J. Intellect. Disabil. Res. 51, 318–326. doi: 10.1111/j.1365-2788.2006.00881.x
Price, C., Wise, R., Ramsay, S., Friston, K., Howard, D., Patterson, K., et al. (1992). Regional response differences within the human auditory cortex when listening to words. Neurosci. Lett. 146, 179–182. doi: 10.1016/0304-3940(92)90072-F
Pujol, J., Fenoll, R., Ribas-Vidal, N., Martínez-Vilavella, G., Blanco-Hinojo, L., García-Alba, J., et al. (2018). A longitudinal study of brain anatomy changes preceding dementia in Down syndrome. NeuroImage Clin. 18, 160–166. doi: 10.1016/j.nicl.2018.01.024
Pulina, F., Vianello, R., and Lanfranchi, S. (2019). Chapter three. Cognitive profiles in individuals with Down syndrome. International review of research in developmental disabilities. Lanfranchi: Academic Press.
Rao, Y. L., Ganaraja, B., Murlimanju, B. V., Joy, T., Krishnamurthy, A., and Agrawal, A. (2022). Hippocampus and its involvement in Alzheimer’s disease: a review. 3 Biotech 12:55.
Raschle, N., Zuk, J., Ortiz-Mantilla, S., Sliva, D. D., Franceschi, A., Grant, P. E., et al. (2012). Pediatric neuroimaging in early childhood and infancy: challenges and practical guidelines. Ann. N. Y. Acad. Sci. 1252, 43–50. doi: 10.1111/j.1749-6632.2012.06457.x
Rauschecker, J. P., and Scott, S. K. (2009). Maps and streams in the auditory cortex: nonhuman primates illuminate human speech processing. Nat. Neurosci. 12, 718–724. doi: 10.1038/nn.2331
Raz, N., Torres, I. J., Briggs, S. D., Spencer, W. D., Thornton, A. E., Loken, W. J., et al. (1995). Selective neuroanatomic abnormalities in Down’s syndrome and their cognitive correlates: evidence from MRI morphometry. Neurology 45, 356–366. doi: 10.1212/WNL.45.2.356
Raz, N., Torres, I. J., Spencer, W. D., and Acker, J. D. J. P. (1993). Pathoclysis in aging human cerebral cortex: evidence from in vivo MRI morphometry. Psychobiology 21, 151–160. doi: 10.3758/BF03332042
Raz, N., Torres, I. J., Spencer, W. D., White, K., and Acker, J. D. (1992). Age-related regional differences in cerebellar vermis observed in vivo. Arch. Neurol. 49, 412–416. doi: 10.1001/archneur.1992.00530280106030
Reiss, A. L., Eckert, M. A., Rose, F. E., Karchemskiy, A., Kesler, S., Chang, M., et al. (2004). An experiment of nature: brain anatomy parallels cognition and behavior in Williams syndrome. J. Neurosci. 24, 5009–5015. doi: 10.1523/JNEUROSCI.5272-03.2004
Reiss, A. L., Eliez, S., Schmitt, J. E., Straus, E., Lai, Z., Jones, W., et al. (2000). IV. Neuroanatomy of Williams syndrome: a high-resolution MRI study. J. Cogn. Neurosci. 12, 65–73. doi: 10.1162/089892900561986
Reiss, A. L., Lee, J., and Freund, L. (1994). Neuroanatomy of fragile X syndrome: the temporal lobe. Neurology 44, 1317–1324. doi: 10.1212/WNL.44.7.1317
Rice, M. L., Warren, S. F., and Betz, S. K. (2005). Language symptoms of developmental language disorders: an overview of autism, Down syndrome, fragile X, specific language impairment, and Williams syndrome. Appl. Psycholinguist. 26, 7–27. doi: 10.1017/S0142716405050034
Roberts, J. E., Price, J., and Malkin, C. (2007). Language and communication development in Down syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 13, 26–35. doi: 10.1002/mrdd.20136
Rodrigues, M., Nunes, J., Figueiredo, S., Martins de Campos, A., and Geraldo, A. F. (2019). Neuroimaging assessment in Down syndrome: a pictorial review. Insights Imaging 10:52. doi: 10.1186/s13244-019-0729-3
Romano, A., Cornia, R., Moraschi, M., Bozzao, A., Chiacchiararelli, L., Coppola, V., et al. (2016). Age-related cortical thickness reduction in non-demented Down’s syndrome subjects. J. Neuroimag. Off. J. Am. Soc. Neuroimag. 26, 95–102. doi: 10.1111/jon.12259
Ropper, A. H., and Williams, R. S. (1980). Relationship between plaques, tangles, and dementia in Down syndrome. Neurology 30, 639–644. doi: 10.1212/WNL.30.6.639
Sampaio, A., Sousa, N., Férnandez, M., Vasconcelos, C., Shenton, M. E., and Gonçalves, O. F. (2008). MRI assessment of superior temporal gyrus in Williams syndrome. Cogni. Behav. Neurol. Off. J. Soc. Behav. Cogn. Neurol. 21, 150–156. doi: 10.1097/WNN.0b013e31817720e4
Sarimski, K. (2014). Entwicklungspsychologie genetischer Syndrome 4., überarb und erw. Aufl. Göttingen [u.a.], Hogrefe.
Savage, L. M., Buzzetti, R. A., and Ramirez, D. R. (2004). The effects of hippocampal lesions on learning, memory, and reward expectancies. Neurobiol. Learn. Mem. 82, 109–119. doi: 10.1016/j.nlm.2004.05.002
Schapiro, M. B., Creasey, H., Schwartz, M., Haxby, J. V., White, B., Moore, A., et al. (1987). Quantitative CT analysis of brain morphometry in adult Down’s syndrome at different ages. Neurology 37, 1424–1427. doi: 10.1212/WNL.37.8.1424
Schlosser, M. J., Aoyagi, N., Fulbright, R. K., Gore, J. C., and McCarthy, G. (1998). Functional MRI studies of auditory comprehension. Hum. Brain Mapp. 6, 1–13. doi: 10.1002/(SICI)1097-0193(1998)6:1<1::AID-HBM1>3.0.CO;2-7
Schröder, J., and Pantel, J. (2016). Neuroimaging of hippocampal atrophy in early recognition of Alzheimer’s disease – a critical appraisal after two decades of research. Psych. Res. Neuroimag. 247, 71–78. doi: 10.1016/j.pscychresns.2015.08.014
Scott, S. K., and Johnsrude, I. S. (2003). The neuroanatomical and functional organization of speech perception. Trends Neurosci. 26, 100–107. doi: 10.1016/S0166-2236(02)00037-1
Shiohama, T., Levman, J., Baumer, N., and Takahashi, E. (2019). Structural magnetic resonance imaging-based brain morphology study in infants and toddlers with Down syndrome: the effect of comorbidities. Pediatr. Neurol. 100, 67–73. doi: 10.1016/j.pediatrneurol.2019.03.015
Simon, T. J., Ding, L., Bish, J. P., McDonald-McGinn, D. M., Zackai, E. H., and Gee, J. (2005). Volumetric, connective, and morphologic changes in the brains of children with chromosome 22q11.2 deletion syndrome: an integrative study. NeuroImage 25, 169–180. doi: 10.1016/j.neuroimage.2004.11.018
Śmigielska-Kuzia, J., Boćkowski, L., Sobaniec, W., Sendrowski, K., Olchowik, B., Cholewa, M., et al. (2011). A volumetric magnetic resonance imaging study of brain structures in children with Down syndrome. Neurol. Neurochir. Pol. 45, 363–369. doi: 10.1016/S0028-3843(14)60107-9
Sobin, C., Kiley-Brabeck, K., Daniels, S., Khuri, J., Taylor, L., Blundell, M., et al. (2005). Neuropsychological characteristics of children with the 22Q11 deletion syndrome: a descriptive analysis. Child Neuropsychol. J. Norm. Abnorm. Dev. Childh. Adolesc. 11, 39–53. doi: 10.1080/09297040590911167
Squire, L. R. (1992). Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol. Rev. 99, 195–231. doi: 10.1037/0033-295X.99.2.195
Teipel, S. J., Alexander, G. E., Schapiro, M. B., Möller, H. J., Rapoport, S. I., and Hampel, H. J. B. (2004). Age-related cortical grey matter reductions in non-demented Down’s syndrome adults determined by MRI with voxel-based morphometry. Brain J. Neurol. 127, 811–824. doi: 10.1093/brain/awh101
Teipel, S. J., and Hampel, H. (2006). Neuroanatomy of Down syndrome in vivo: a model of preclinical Alzheimer’s disease. Behav. Genet. 36, 405–415. doi: 10.1007/s10519-006-9047-x
Teipel, S. J., Schapiro, M. B., Alexander, G. E., Krasuski, J. S., Horwitz, B., Hoehne, C., et al. (2003). Relation of corpus callosum and hippocampal size to age in nondemented adults with Down’s syndrome. Am. J. Psychiatr. 160, 1870–1878. doi: 10.1176/appi.ajp.160.10.1870
Uematsu, A., Matsui, M., Tanaka, C., Takahashi, T., Noguchi, K., Suzuki, M., et al. (2012). Developmental trajectories of amygdala and hippocampus from infancy to early adulthood in healthy individuals. PLoS One 7:e46970. doi: 10.1371/journal.pone.0046970
Vicari, S. (2001). Implicit versus explicit memory function in children with down and Williams syndrome. Downs Syndr. Res. Pract. 7, 35–40. doi: 10.3104/reports.112
Vicari, S. (2006). Motor development and neuropsychological patterns in persons with Down syndromeDown syndrome. Behav. Genet. 36, 355–364. doi: 10.1007/s10519-006-9057-8
Vicari, S., Bellucci, S., and Carlesimo, G. A. (2005). Visual and spatial long-term memory: differential pattern of impairments in Williams and down syndromes. Dev. Med. Child Neurol. 47, 305–311. doi: 10.1017/S0012162205000599
Vicari, S., Mantovan, M., Addona, F., Costanzo, F., Verucci, L., and Menghini, D. (2012). Neuropsychological profile of Italian children and adolescents with 22q11.2 deletion syndrome with and without intellectual disability. Behav. Genet. 42, 287–298. doi: 10.1007/s10519-011-9499-5
Vicari, S., Verucci, L., and Carlesimo, G. A. (2007). Implicit memory is independent from IQ and age but not from etiology: evidence from down and Williams syndromes. J. Intellect. Disabil. Res. 51, 932–941. doi: 10.1111/j.1365-2788.2007.01003.x
Vogt, B., Peters, A., Jones, E., and Kemper, T. L. (1991). Cerebral cortex: Normal and altered states of function chapter 12: Down syndrome Singapore: Springer.
Wang, P. P. (1996). A neuropsychological profile of Down syndrome: cognitive skills and brain morphology. Ment. Retard. Dev. Disabil. Res. Rev. 2, 102–108. doi: 10.1002/(SICI)1098-2779(1996)2:2<102::AID-MRDD8>3.0.CO;2-V
Weis, S., Weber, G., Neuhold, A., and Rett, A. (1991). Down syndrome: MR quantification of brain structures and comparison with normal control subjects. AJNR Am. J. Neuroradiol. 12, 1207–1211.
White, N. S., Alkire, M. T., and Haier, R. J. J. N. (2003). A voxel-based morphometric study of nondemented adults with Down syndrome. NeuroImage 20, 393–403. doi: 10.1016/S1053-8119(03)00273-8
Wisniewski, K. E. (1990). Down syndrome children often have brain with maturation delay, retardation of growth, and cortical dysgenesis. Am. J. Med. Genet. Suppl. 7, 274–281. doi: 10.1002/ajmg.1320370755
Wisniewski, K. E., Wisniewski, H. M., and Wen, G. Y. (1985). Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann. Neurol. 17, 278–282. doi: 10.1002/ana.410170310
Woodin, M., Wang, P. P., Aleman, D., McDonald-McGinn, D., Zackai, E., and Moss, E. (2001). Neuropsychological profile of children and adolescents with the 22q11.2 microdeletion. Genet. Med. Off. J. Am. Coll. Med. Genet. 3, 34–39. doi: 10.1097/00125817-200101000-00008
Yang, Y., Conners, F. A., and Merrill, E. C. (2014). Visuo-spatial ability in individuals with Down syndrome: is it really a strength? Res. Dev. Disabil. 35, 1473–1500. doi: 10.1016/j.ridd.2014.04.002
Ypsilanti, A., and Grouios, G. (2008). Linguistic profile of individuals with Down syndrome: comparing the linguistic performance of three developmental disorders. Child Neuropsychol. J. Norm. Abnorm. Dev. Childh. Adolesc. 14, 148–170. doi: 10.1080/09297040701632209
Ypsilanti, A., Grouios, G., Alevriadou, A., and Tsapkini, K. (2005). Expressive and receptive vocabulary in children with Williams and Down syndromes. J. Intellect. Disabil. Res. 49, 353–364. doi: 10.1111/j.1365-2788.2005.00654.x
Zigman, W. B., and Lott, I. T. (2007). Alzheimer’s disease in Down syndrome: neurobiology and risk. Ment. Retard. Dev. Disabil. Res. Rev. 13, 237–246. doi: 10.1002/mrdd.20163
Keywords: Down syndrome, neuroimaging, neuroanatomy, neuropsychology, fragile X syndrome, Williams syndrome, 22q11.2 deletion syndrome
Citation: Hamadelseed O, Chan MKS, Wong MBF and Skutella T (2023) Distinct neuroanatomical and neuropsychological features of Down syndrome compared to related neurodevelopmental disorders: a systematic review. Front. Neurosci. 17:1225228. doi: 10.3389/fnins.2023.1225228
Edited by:
Josef P. Kapfhammer, University of Basel, SwitzerlandReviewed by:
Chi Chiu Wang, The Chinese University of Hong Kong, ChinaOliver Schmitt, Medical School Hamburg, Germany
Copyright © 2023 Hamadelseed, Chan, Wong and Skutella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Osama Hamadelseed, T3NhbWEuaGFtYWRlbHNlZWRAdW5pLWhlaWRlbGJlcmcuZGU=