 Judy Ghalayini
Judy Ghalayini Gabrielle L. Boulianne
Gabrielle L. Boulianne
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 11 April 2023
Sec. Neurodegeneration
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1166973
This article is part of the Research Topic Drosophila as a model to study Neurodegenerative diseases View all 11 articles
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder for which there is no cure. Recently, several studies have reported a significant reduction in the incidence and progression of dementia among some patients receiving antihypertensive medications such as angiotensin-converting enzyme inhibitors (ACE-Is) and angiotensin receptor blockers (ARBs). Why these drugs are beneficial in some AD patients and not others is unclear although it has been shown to be independent of their role in regulating blood pressure. Given the enormous and immediate potential of ACE-Is and ARBs for AD therapeutics it is imperative that we understand how they function. Recently, studies have shown that ACE-Is and ARBs, which target the renin angiotensin system in mammals, are also effective in suppressing neuronal cell death and memory defects in Drosophila models of AD despite the fact that this pathway is not conserved in flies. This suggests that the beneficial effects of these drugs may be mediated by distinct and as yet, identified mechanisms. Here, we discuss how the short lifespan and ease of genetic manipulations available in Drosophila provide us with a unique and unparalleled opportunity to rapidly identify the targets of ACE-Is and ARBs and evaluate their therapeutic effectiveness in robust models of AD.
AD is a devastating neurodegenerative disorder that accounts for 70–80% of dementia cases worldwide (Barker et al., 2002). Dementia is a general term used to describe symptoms associated with a decline in cognitive functions including memory, thinking and social abilities that are distinguishable from normal ageing. Currently, around 55 million people have dementia worldwide and as the aging population continues to grow, this number is estimated to triple to 150 million by 2050 (Nichols et al., 2022). In its initial stages, AD is characterized by subtle changes in cognition. However, as the disease progresses, individuals present more severe symptoms including extreme memory loss, impaired spatial and temporal orientation, language disturbances, behavioral changes and motor deficits (Castellani et al., 2010). Ultimately AD renders patients unable to carry out simple day-to-day activities. These symptoms arise from extreme neuronal deterioration mainly in areas of the brain responsible for cognition such as the hippocampus and cortex (Castellani et al., 2010; Holtzman et al., 2011).
AD was first characterized over a century ago, by psychiatrist and neuropathologist Alois Alzheimer who described a 51-year-old patient with memory and language deficits as well as severe disorientation and hallucinations (Graeber et al., 1997; Goedert and Ghetti, 2007). Although these symptoms matched the definition of what was then called dementia, it was atypical for them to be present in someone so young. A post-mortem autopsy revealed various abnormalities in the patient’s brain including extensive atrophy of the cerebral cortex and abnormal protein deposits inside and between nerve cells (Graeber et al., 1997; Goedert and Ghetti, 2007). These protein deposits, known as amyloid plaques and neurofibrillary tangles (NFTs), soon became the pathological hallmarks of AD.
Amyloid plaques in the extracellular matrix of brain tissue are composed of amyloid-β (Aβ) peptides (Glenner and Wong, 1984). These peptides are derived from a precursor transmembrane protein known as APP, found mainly in neurons (Kang et al., 1987; van der Kant and Goldstein, 2015). APP undergoes sequential processing by enzyme complexes, β-secretase and γ-secretase, to produce a C-terminal fragment (C99) followed by an Aβ peptide, respectively (Vassar et al., 1999; Haapasalo and Kovacs, 2011; van der Kant and Goldstein, 2015). C99 may be cleaved at different sites by γ-secretase thus, amyloid-β peptides vary in size ranging from 38 to 43 residues, with Aβ40 and Aβ42 being the most prominent species (Takami et al., 2009). Aβ40 and Aβ42 only differ by two residues however, Aβ42 is more prone to aggregation and is found enriched in amyloid plaques (Miller et al., 1993; Iwatsubo et al., 1994; Mak et al., 1994). Furthermore, Aβ42 has a much higher level of neurotoxicity compared to Aβ40. The ratio of Aβ42/Aβ40 is often used as a reference to AD pathogenesis (Kuperstein et al., 2010). However, Aβ levels and deposition appear to plateau at the onset of clinical symptoms and correlate poorly with the degree of cognitive impairment in the dementia phases of AD (Masters et al., 2015). In contrast, measurements of biomarkers for neurofibrillary tangles (NFTs) strongly correlate with disease progression and severity at the onset of clinical manifestation (Masters et al., 2015).
NFTs are intraneuronal aggregates mainly composed of hyperphosphorylated Tau, a microtubule associated protein that interacts with tubulin to support its assembly into microtubules and to stabilize its structure (Barbier et al., 2019). Tau is also known to promote neurite outgrowth and axonal transport (Barbier et al., 2019). However, in AD, tau proteins are hyperphosphorylated, which causes them to assemble into filamentous bundles (Busche and Hyman, 2020). As a result, the structural integrity of microtubules is damaged, impairing axonal transport and neurite development, which leads to neuronal cell death (Busche and Hyman, 2020). Accordingly, NFTs and phosphorylated tau levels correlate strongly with the degree of neurodegeneration in AD patients and are predictive of disease severity (Masters et al., 2015).
The relationship between Aβ and NFTs is complex. Findings show high Aβ load - either as plaques or non-fibrillar, soluble, oligomeric forms, precede NFT formation and suggest that Aβ initiates AD in part by acting on pathophysiological mechanisms that lead to tau-hyperphosphorylation and aggregation into NFTs (Busche and Hyman, 2020). Consistent with this model, animal studies have demonstrated that Aβ peptides promote tau-hyperphosphorylation resulting in its aggregation into NFTs (Götz et al., 2001; Gomes et al., 2019). This concept is consistent with the amyloid-cascade hypothesis whereby Aβ is the primary initiator of AD (described below).
An understanding of how Aβ peptides lead to senile plaque formation was revealed through familial genetic studies of AD, which identified dominant mutations in APP and presenilin 1 and presenilin 2; the core catalytic components of γ-secretase (Holtzman et al., 2011; van der Kant and Goldstein, 2015). These mutations were shown to profoundly alter APP metabolism, increasing and favoring the production of aggregation prone Aβ species such as Aβ42 (Holtzman et al., 2011; van der Kant and Goldstein, 2015). Ultimately these studies led to the development of the amyloid cascade hypothesis of AD pathogenesis (Hardy and Selkoe, 2002). This long-standing hypothesis states that neurotoxic forms of β-amyloid peptides initiate AD pathology and precede all other disease hallmarks. However, familial AD (FAD), also known as early-onset AD (EOAD), accounts for only 1–5% of all cases (Holtzman et al., 2011). Most cases of AD fall under the “sporadic” late-onset form (LOAD), which lacks clearly defined genetic factors. However, genome-wide association studies (GWAS) have identified certain genetic risk factors including the apolipoprotein E gene (apoE) (Bertram and Tanzi, 2009). Individuals carrying the apoE e4 allele have up to a 12-fold increased risk of developing the disease (Bertram and Tanzi, 2009). As more genetic risk factors are discovered, it becomes increasingly clear that AD is a complex and multifactorial disorder.
Neuroinflammation has also emerged as a significant factor in the pathogenesis of AD, alongside amyloid plaques and NFT (Newcombe et al., 2018). Neuroinflammation refers to an inflammatory response that occurs within the CNS and involves the production of cytokines, chemokines, reactive oxygen species and secondary messengers. Microglial cells are the primary players in neuroinflammation and act as the resident macrophages of the CNS, serving several roles including acting as a first line of immune defence against brain injury or infection (Calsolaro and Edison, 2016). However, it has been suggested that the activation of microglia and the subsequent release of pro-inflammatory factors can cause significant neuronal damage. Although microglial activation can be protective within the CNS, if the stimulus for activation is not resolved, chronic inflammation can develop and contribute to neuronal dysfunction, injury and loss (Calsolaro and Edison, 2016; Fakhoury, 2018). Post-mortem studies of AD patient brains have revealed activated microglia co-localized with amyloid plaques. Amyloid peptides, fibrils and APP have also been shown to activate microglia, triggering an inflammatory response and the release of neurotoxic cytokines (Calsolaro and Edison, 2016; Fakhoury, 2018). PET studies have demonstrated a correlation between microglial activation and amyloid load in AD patients. Moreover, neuroinflammation has been detected before the onset of dementia, suggesting its early occurrence in AD pathology. Finally, GWAS studies have identified a relationship between components of the innate immune system and the incidence of sporadic AD, further supporting a link between the immune system and AD (Newcombe et al., 2018).
Despite distinct etiologies of sporadic and familial forms of AD, both exhibit comparable clinical manifestations, including rates of disease progression and similar biomarker profiles. As a result, genetic factors known in FAD are commonly employed to model the disease in research aimed at understanding its underlying causes. Moreover, these models are widely utilized in preclinical studies to evaluate the effectiveness of potential therapeutics.
Recently, there has been a significant advancement in the treatment of AD with the approval of two monoclonal antibodies, aducanumab and lecanemab, as potential therapies for AD (Tampi et al., 2021; Reardon, 2023). These drugs are designed to target and clear β-amyloid plaques. However, while clinical studies have demonstrated their efficacy in reducing Aβ levels, their impact on cognitive improvement has been limited (Tampi et al., 2021; Reardon, 2023). This suggests that targeting Aβ and tau alone may not be sufficient to effectively treat AD and that a more comprehensive approach that targets multiple pathways may be more effective. Therefore, ongoing research to develop therapies that target other aspects of AD is crucial. One promising avenue of research revolves around the potential benefits of drugs that target the renin angiotensin system (RAS).
Systemic RAS (or peripheral RAS) is a hormonal system that plays a critical role in regulating blood pressure and fluid balance in the body (Yim and Yoo, 2008; Wu et al., 2018). It is made up of several hormones and enzymes, including renin, angiotensinogen, angiotensin (Ang) I, Ang II, and angiotensin converting enzyme (ACE). Renin is produced and released by the kidneys in response to low blood pressure or low blood volume and it acts on angiotensinogen (a pro-peptide produced by the liver) to produce Ang I (Yim and Yoo, 2008; Wu et al., 2018). Ang I is then converted to Ang II by ACE, a zinc- and chloride-dependent metallopeptidase, which is expressed in the lungs and other tissues (Yim and Yoo, 2008; Wu et al., 2018). Ang II is a potent vasoconstrictor, exerting its effects by binding to angiotensin II type 1 receptors (AT1Rs), while binding to angiotensin II type 2 receptors (AT2Rs) induces vasodilation (Yim and Yoo, 2008; Wu et al., 2018). Ongoing research of the RAS has led to the discovery of additional components, such as ACE2, a homolog of ACE. ACE2 cleaves Ang I or Ang II into the heptapeptide angiotensin 1–7 (Ang 1–7). This peptide was later found to bind to the Mas receptor (MasR), resulting in vasodilation (Yim and Yoo, 2008; Wu et al., 2018).
A pivotal discovery relating to the RAS, was the development of captopril, the first ACE inhibitor (ACE-I) drug in 1975 (Zheng et al., 2022). Captopril selectively targets ACE by binding to its active site and preventing the formation of Ang II thereby inhibiting the RAS. It marked a significant milestone in the treatment of hypertension paving the way for the development of additional ACE-Is with improved activity and bioavailability (Zheng et al., 2022). In addition, a new class of drugs known as angiotensin receptor blockers (ARBs), which selectively inhibit Ang II by competitive antagonism of the AT1Rs, were also developed (Barreras and Gurk-Turner, 2003). These drugs have proven to be clinically relevant in the treatment of various cardiovascular conditions, including hypertension, heart failure, diabetic nephropathy and continue to be an important therapeutic option for patients today (Barreras and Gurk-Turner, 2003; Zheng et al., 2022).
Well after its discovery, it became evident that the RAS system is also expressed in numerous organs including the brain (now referred to as local RAS) and possesses additional functionalities revealing its degree of complexity (Paul et al., 2006). Local synthesis of cerebral RAS is necessary due to the blood–brain barrier (BBB) preventing peripheral RAS components from accessing most regions of the brain (Jackson et al., 2018). Astrocytes are the primary source of angiotensinogen, which is constitutively secreted and cleaved into various neuroactive peptides (Jackson et al., 2018). As previously described and illustrated in w, renin converts angiotensinogen to Ang I, which is further processed by ACE to produce Ang II. Ang II is the main effector protein that binds to AT1R or AT2R. Ang II can also be further processed into Ang IV by aminopeptidases (AP-A and AP-N), which binds to AT4R (Jackson et al., 2018). Alternatively, ACE2 converts Ang II to Ang 1–7, which binds to MasRs. Ang 1–7 can also be produced through first processing of Ang I by ACE2 to produce Ang 1–9 and then by ACE (Jackson et al., 2018). AT1R and AT2R are present in neurons, astrocytes, oligodendrocytes and microglia of the cortex, hippocampus and basal ganglia both on the cell surface and intracellularly at mitochondrial and nuclear levels allowing for the regulation of oxidative stress, transcription and trafficking of receptor types (Jackson et al., 2018). Activation of AT1R has been associated with deleterious effects such as promoting neuroinflammation, oxidative stress and neuronal cell death (Jackson et al., 2018; Cosarderelioglu et al., 2020). In contrast, AT2R appears to be neuroprotective, counteracting AT1R’s effects by inhibiting neuroinflammation, reducing oxidative stress and influencing neuronal regeneration. Of note, while Ang II can bind both receptors, ACE upregulation specifically leads to increased AT1R activation (Jackson et al., 2018). MasR also located in these brain regions are expressed by neurons, astrocytes and microglia, and similar to AT2R, have both antioxidant and anti-inflammatory properties and promote cell survival (Jackson et al., 2018). The expression of AT4R is restricted to neurons localized in the cortex, hippocampus and basal ganglia, where it is believed to induce LTP and mediate learning and memory consolidation (Jackson et al., 2018; Cosarderelioglu et al., 2020). The interplay between the receptors and enzymes of RAS in the brain has been suggested to work synergistically and is thought to be crucial in maintaining cognitive balance in a healthy brain. Accordingly, misregulation of RAS has been implicated in pathologies underlying neurodegenerative diseases including AD (Cosarderelioglu et al., 2020; Gouveia et al., 2022). Changes in RAS components have been documented in brains of AD patients compared to control individuals (Savaskan et al., 2001). For example, studies have found increased levels of ACE in the hippocampus, frontal cortex and caudate nucleus of AD patients and its activity is increased and correlates positively with parenchymal Aβ load (Arregui et al., 1982; Miners et al., 2008; MacLachlan et al., 2022). While the expression of ACE appears to be upregulated in AD, the opposite has been reported for ACE2. Researchers observed a significant reduction in ACE2 activity in the mid-frontal cortex of AD patients and this reduction was inversely correlated with total β-amyloid and tau load as well as ACE activity (Kehoe et al., 2016). Similarly, a systematic analysis of ACE2 protein expression in different brain regions revealed a downregulation in the basal nucleus, hippocampus and entorhinal cortex, middle frontal gyrus, visual cortex and amygdala of AD patient brains (Cui et al., 2021). However, contrary to these findings, Ding et al. (2021) reported higher ACE2 protein expression levels in hippocampal tissues of AD patients compared to control subjects. Although, in a more recent study that evaluated ACE and ACE2 protein expression and enzyme activity in the frontal and temporal cortex in early AD stages, authors report that both ACE and ACE2 protein level are unchanged and that only ACE enzyme activity was elevated (MacLachlan et al., 2022). ACE and ACE2 act on different axes of the RAS having either neurotoxic or neuroprotective properties, respectively and an imbalance between these axes may play a role in AD pathogenesis. Taken together, these discrepancies suggest the need for further analysis.
Genetic studies have also implicated ACE as a probable risk factor for AD (Alvarez et al., 1999; Elkins et al., 2004). Recent AD GWAS meta-analyses identified common genetic variants in the Ace locus outside of exonic regions, which are associated with an increased risk of AD (Marioni et al., 2018; Kunkle et al., 2019). In the past, genetic studies of Ace and AD have focused primarily on a common insertion/deletion (I/D) variant that influences ACE serum levels. Individuals with the D/D haplotype have higher serum ACE levels than those with I/I haplotype (Elkins et al., 2004). Most studies, Kehoe et al. (1999), Narain (2000), Kölsch et al. (2005), and Lehmann et al. (2005) although at times contradictory (Chou et al., 2016), have indicated that individuals carrying an insertion allele are at higher risk of AD than those with the D/D haplotype. It is important to note that ACE serum levels do not reflect ACE enzymatic activity levels. In a more recent genetic study, Cuddy et al. (2020) performed whole genome sequencing to identify rare coding variants in the Ace gene associated with AD. They selected one variant (R1297Q) for functional analysis to gain a better understanding of the role that ACE plays in AD. This was achieved by generating knock-in (KI) mice that harbored the cognate mutation, R1297Q, in the murine Ace gene. The authors report that while the mutation had no effect on blood pressure and cerebrovasculature, it did result in increased levels of neuronal ACE protein and activity, memory impairment, neuroinflammation and hippocampal neurodegeneration. Moreover, these reported phenotypes were exacerbated in an AD mouse model of amyloidosis. These findings strongly suggest that increased ACE activity is associated with AD pathogenesis.
Following this apparent genetic link between RAS and AD, epidemiological and clinical studies were performed to examine the effects of RAS targeting drugs including ACE-Is and ARBs on the incidence of AD. A retrospective study conducted by Barthold et al. (2018) assessed the risk of developing AD in patients being treated with either RAS targeting or non-RAS anti-hypertensive medication. The study findings indicated that RAS-acting drugs were more effective in reducing the risk of AD development compared to non-RAS acting drugs. The study also compared the effects between ARBs and ACE-Is and found that ARBs demonstrated superior preventative efficacy against AD than ACE-Is. These findings are in line with a previous prospective cohort study conducted by Li et al. (2010) that reported reduced incidence and progression of AD in participants taking ARBs compared to those taking other cardiovascular drugs and a nested case–control analysis following AD patients who were prescribed different anti-hypertensive drugs including ACE-Is and ARBs (Davies et al., 2011). Specifically, this study found a 53% decrease in AD incidence for ARB use and a 24% decrease in AD incidence from ACE-I use (Davies et al., 2011). Furthermore, ACE-Is and ARBs have also been evaluated for their ability to reduce the rate of cognitive decline and improve cognitive performance in AD patients. From a cross-sectional and retrospective cohort study of an elderly population, Hajjar et al. (2005) reported patients taking ARBs had improved cognitive performances while those taking ACE-Is presented a lower rate of cognitive decline. A follow up double-blind randomized clinical trial reported similar findings whereby the ARB, candesartan, was associated with improvement in cognition and outperformed the ACE-I, lisinopril (Hajjar et al., 2012). While studies continue to support the potential benefits of ARBs in AD (Ouk et al., 2021; Deng et al., 2022), a recent clinical study evaluating the effect of 12-month losartan (an ARB) treatment on brain atrophy in patients diagnosed with mild-to-moderate AD reported no significant reduction in brain volume loss (Kehoe et al., 2021). These findings suggest that further studies regarding ARB treatment duration and time of treatment are needed to determine when potential benefits of ARB use may arise. Regarding ACE-Is, their potential benefits for AD are not as clear. Even though a prospective cohort study by Soto et al. (2013) showed a slower rate of cognitive decline in older adults taking ACE-Is, it appears that ACE-Is as a pharmacological class do not reduce the risk of developing dementia or improve cognition in AD (Sink et al., 2009; Solfrizzi et al., 2013; O’Caoimh et al., 2014). However, a closer examination of subgroups of ACE-Is, such as those that can penetrate the BBB vs. those that cannot, imply potential beneficial effects may arise exclusively from drugs that can penetrate the BBB (Ellul et al., 2006; Gao et al., 2013; O’Caoimh et al., 2014). Indeed, in an observational study, O’Caoimh et al. (2014) found a decrease in the rate of cognitive decline in patients with mild to moderate AD receiving BBB-penetrating ACE-Is compared to those on no drug. Finally, it is important to note that the beneficial effects of ACE-Is and ARBs remain the same even after studies adjusted for blood pressure or hypertension suggesting that the beneficial effects are independent of blood pressure or hypertension regulation (Hajjar et al., 2008). While the precise mechanisms by which ACE-Is and ARBs exert their effects in AD remain unclear, their promise as potential therapeutics has inspired researchers to elucidate their mechanisms of action utilizing in vivo model systems.
Several studies have now examined the effects of ARBs on cognition animal models, as shown in Table 1. While the majority of studies have demonstrated that ARBs can improve learning and memory in AD mouse models (Wang et al., 2007; Takeda et al., 2009; Ongali et al., 2014; Royea et al., 2017; Torika et al., 2017) others have failed to demonstrate any or have shown limited beneficial effects (Papadopoulos et al., 2016; Trigiani et al., 2018). Nonetheless, improvements in memory performance and retrieval, spatial learning, and prevention of cognitive deficits have been documented through the use of different ARBs such as losartan, olmesartan, and telmisartan (Wang et al., 2007; Takeda et al., 2009; Ongali et al., 2014; Royea et al., 2017; Torika et al., 2017). These beneficial effects are suggested to result, in part, by the ability of ARBs to reduce β-amyloid load, neuroinflammation and oxidative stress in the brain (Wang et al., 2007; Takeda et al., 2009; Danielyan et al., 2010; Torika et al., 2017, 2018) all of which are neuropathological hallmarks of AD (Krstic and Knuesel, 2012; Cassidy et al., 2020). With the increasing number of studies in this field, it is evident that ARBs have strong therapeutic potential and efforts into revealing their mechanism of action are underway.
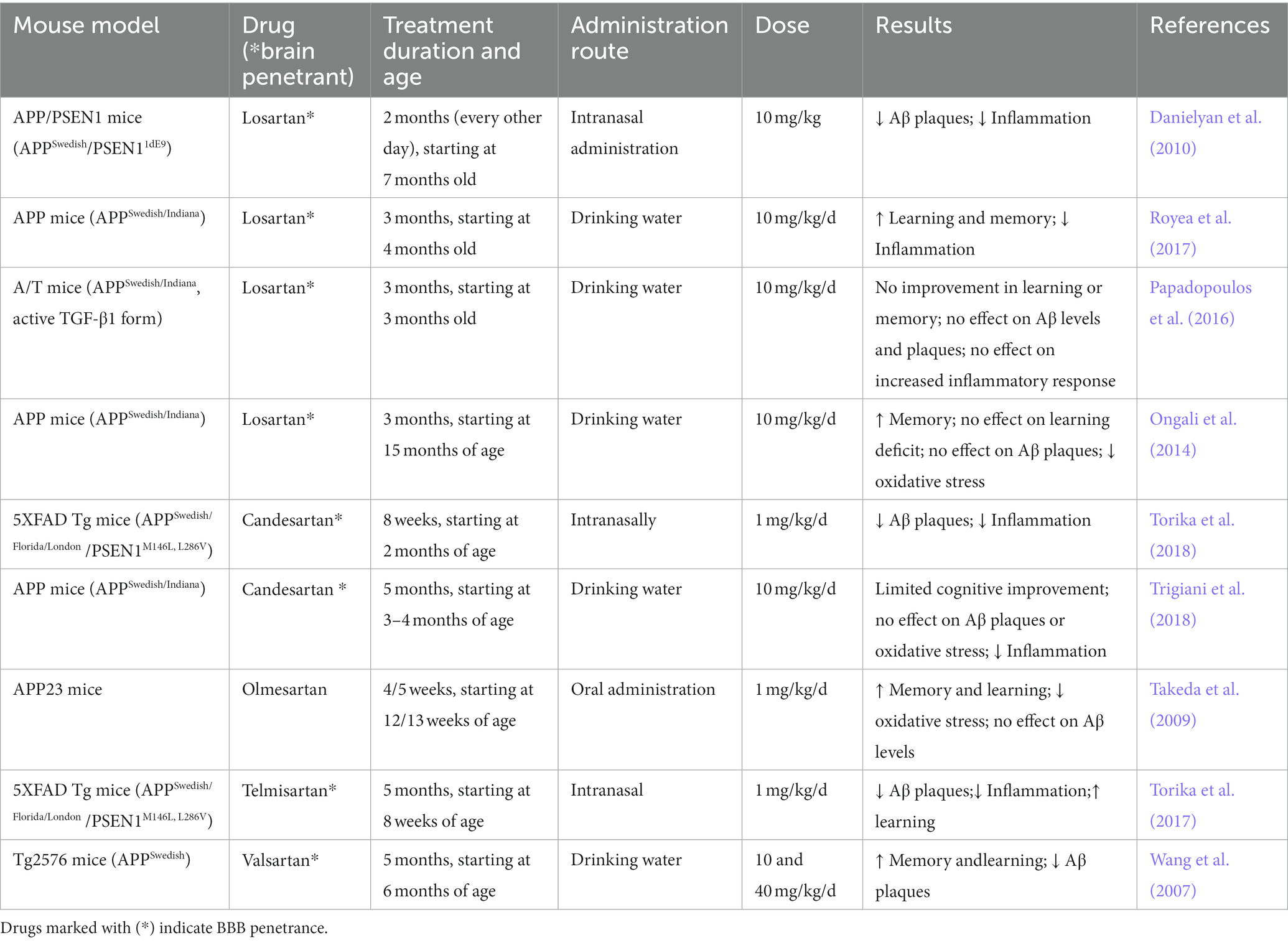
Table 1. Summary of effects of ARBs in mouse models of AD including Aβ levels and deposition, neuroinflammation and oxidative stress and, cognitive deficits.
However, as summarized in Table 2, the effects of ACE-Is in AD are more ambiguous. Some studies suggest that ACE-Is may be detrimental as they led to an increase in Aβ accumulation in AD mice (Zou et al., 2007; Liu et al., 2019). For example, Liu et al. (2019) reported that treating Tg2576 AD mice with captopril, an ACE-I, for 11 months resulted in increased levels of Aβ42 and β-amyloid plaque deposition in the hippocampus and neocortex. In contrast, other studies have shown that captopril treatment reduced Aβ burden (AbdAlla et al., 2013; Torika et al., 2016; Asraf et al., 2018). For example, AbdAlla et al. (2013) demonstrated that treating Tg2576 AD mice with captopril for 6 months led to reduced amyloidogenic processing of full-length APP resulting in slower accumulation of Aβ in the hippocampus. However, other studies reported no effect of ACE inhibition on Aβ (Hemming et al., 2007; Dong et al., 2011). These discrepancies make it difficult to conclude whether ACE inhibition may be beneficial in AD. However, an evaluation of the differences in a number of factors across these studies including, method of administration, drug dose and treatment duration may help explain the contrasting results. For instance, Hemming et al. (2007) evaluated the effect of different concentrations of captopril delivered by oral administration in AD mice over 28 days on its ability to inhibit ACE activity in the brain. They found that only high concentrations of captopril, approximately 10 times relative to the highest amount used in therapeutic doses in humans, led to a significant but modest reduction in ACE activity and there was no change in either cerebral Aβ levels or deposition. In a related study by Liu et al. (2019), captopril was administered orally at similar concentrations resulting in a significant reduction in systolic and diastolic blood pressure likely due to inhibition of peripheral ACE. However, no data was provided as to whether there was a significant effect on inhibiting brain ACE that could account for the increased levels of Aβ42 observed by the authors as a result of captopril treatment. In contrast, studies whereby captopril was administered intranasally reported reduced Aβ42 load in hippocampal and cortical areas (Torika et al., 2016; Asraf et al., 2018). It is worth noting that treatment duration also impacts the outcome of captopril treatment. Torika et al. (2016) found that when AD mice were treated with captopril for 3.5 weeks vs. 7 months, no changes in β-amyloid load were observed. However, this was not the case when they administered perindopril, another ACE-I, for a short duration suggesting that the type of ACE-I used could contribute to discrepancies in the literature. Finally, in addition to altering Aβ levels, ACE-Is appear to alter immune responses in brains of AD mice similar to that observed using ARBs (Dong et al., 2011; Torika et al., 2016; Asraf et al., 2018). For example, studies showed that ACE inhibition reduced the level of CD11b, a marker of activated microglial and reactive oxygen species (ROS) suggesting an overall reduction in inflammation (AbdAlla et al., 2013; Torika et al., 2016; Asraf et al., 2018). Altogether, these studies suggest that while ARBs and ACE-Is may have beneficial effects in AD animal models further studies are needed to decipher both the method of delivery and their mechanism of action.
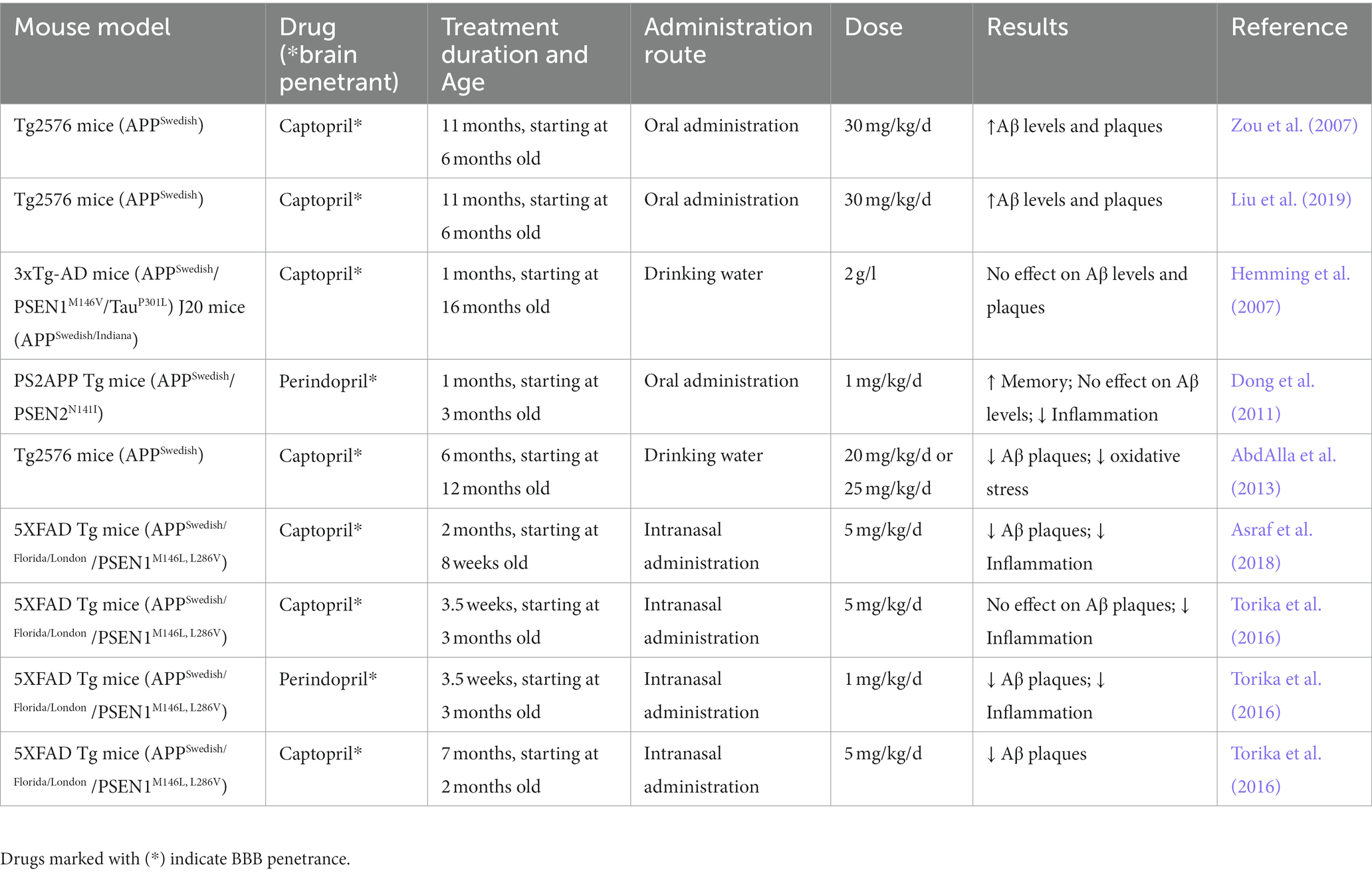
Table 2. Summary of effects of ACE-Is in mouse models of AD including Aβ levels and deposition, neuroinflammation and oxidative stress and, cognitive deficits.
Drosophila has proven to be an excellent model system to study neurodegenerative diseases. The short life span of flies coupled with powerful genetic approaches has made it possible to generate models of disease that faithfully recapitulate many features observed in patients, including age-dependent neurodegeneration and progressive defects in synaptic plasticity and memory. Once a model has been generated it can also be used to perform genetic screens to identify modifiers of known disease-causing genes or drug screens to identify and evaluate novel therapies. Many of the genes associated with neurodegeneration are also conserved in Drosophila including those implicated in AD. For example, Drosophila possess a homolog of APP known as APP-like or APPL (Luo et al., 1992). Flies deficient in this gene exhibit a behavioral defect that can be partially rescued by expressing a human APP transgene suggesting functional homology between APP and APPL (Luo et al., 1992). However, APPL lacks the amyloidogenic Aβ peptide sequence at the C- terminus found in human APP and does not appear to be processed in vivo as is human APP (Prüßing et al., 2013). Similarly, while Presenilin and the γ-secretase complex are well conserved (Periz and Fortini, 2004) there is no clear homolog of β-secretase in flies (Prüßing et al., 2013).
Several transgenic fly lines based on the expression of human and/or Drosophila AD-related genes have been generated and are readily available to study disease processes. One such model is based on co-expression of human β-secretase along with human APP and fly presenilin, both of which possess FAD-linked mutations (Greeve et al., 2004). Co-expression of all three transgenes in the fly eye led to β-amyloid plaque formation and age-dependent neurodegeneration (Greeve et al., 2004) demonstrating that Drosophila could be used as a model for AD. Soon after, additional models were generated, whereby transgenic flies expressed different Aβ transgenes in specific tissues. The transgenes varied in several ways, including whether or not they possessed a signal sequence to allow for expression outside cells, the number of Aβ42 copies expressed in tandem, and whether they contained an FAD mutation (Prüßing et al., 2013). All exhibited similar phenotypic defects when expressed in neuronal tissue, including plaque formation, neurodegeneration, as well as motor and cognitive defects (Prüßing et al., 2013). The main difference between the models was the severity of the phenotypes, which often correlated with the levels of Aβ42 expression and the degree of protein aggregation. Transgenic flies that express Tau in neuronal tissue are also available and similarly show robust phenotypes reminiscent of those observed in AD (Prüßing et al., 2013). Functional genomic studies using various AD fly models have also facilitated our understanding of the role of cellular mechanisms including inflammation, oxidative stress, mitochondrial dysfunction and apoptosis in AD pathogenesis (reviewed by Jeon et al., 2020). Finally, the ability to perform large-scale genetic screens together with the availability of RNA interference for all genes annotated in the fly genome, make it possible to identify novel modifiers of Aβ42 and Tau (Jeon et al., 2020) that not only provide insight into the molecular and cellular pathways implicated in AD but also potential novel therapeutic targets for this devastating disease.
The RAS has been well studied in humans and many mammalian model organisms due to its intricate role in regulating blood pressure. However, components of the RAS have also been found in non-mammalian organisms that lack a closed circulatory system (Fournier et al., 2012). ACE homologs have been identified in several invertebrate organisms, including Drosophila. There are six ACE-like factors in Drosophila, including Ance, Ance-2,-3,-4,-5, and Acer (Cornell et al., 1995; Taylor et al., 1996; Houard et al., 1998). Of these, only Acer and Ance are believed to be active zinc metallopeptidases as they possess an intact conserved active site motif (HExxH) (Coates et al., 2000). Their catalytic activity was demonstrated through biochemical assays that showed their ability to hydrolyze an ACE synthetic substrate, Hip-His-Leu, which mimics the C-terminal sequence of Ang I, to a similar degree as mammalian ACE (Houard et al., 1998; Coates et al., 2000).
ACE-Is have also been shown to be effective in inhibiting Acer and Ance catalytic activity although both enzymes structurally differ from their mammalian counterparts (Houard et al., 1998; Coates et al., 2000; Bingham et al., 2006; Akif et al., 2012). In mammals, the ACE gene is subjected to alternative splicing that gives rise to two distinct enzymes (Hubert et al., 1991). Somatic ACE (sACE), is widely distributed and contains two active sites (N and C-terminal) while germinal ACE (gACE), is expressed exclusively in the testes and only possesses the second active site (C-terminal) (Hubert et al., 1991; Shen et al., 2008). Drosophila Acer and Ance only possess a single catalytic domain (Houard et al., 1998; Coates et al., 2000). The active site of Acer is similar to the N-terminal active site of somatic ACE, whereas the active site of Ance is similar to the C-terminal active site of somatic ACE. Moreover, Acer and Ance lack a C-terminal transmembrane anchor that is found in mammalian ACE and may be proteolytically cleaved to yield soluble enzymes (Houard et al., 1998; Coates et al., 2000). However, both Acer and Ance possess a signal sequence that leads to their secretion outside the cell (Coates et al., 2000; Rylett et al., 2007; Carhan et al., 2011). A comparison between Drosophila and human ACE proteins is illustrated in Figure 2.
The first study to suggest a link between ACE and AD in Drosophila was a modifier screen aimed to identify genes that either enhanced or suppressed phenotypes resulting from over-expression of Psn followed by a secondary screen to determine if any of the Psn modifiers could also suppress phenotypes generated from expression of the truncated form of APP, called C99 (van de Hoef et al., 2009). Numerous candidates that suppressed or enhanced both the Psn and C99 phenotypes were identified, including some that had previously been shown to interact with Psn. More importantly, two ACE-like factors were identified: Acer and Ance-5. Acer and Ance-5 modified Psn-dependent phenotypes while Ance-5 also modified the C99-dependent phenotype (van de Hoef et al., 2009). This suggested that ACE-like factors might be involved in regulating Psn function and that further characterization of the interaction between psn, APP, and ACE might aid our understanding of AD pathogenesis and the potential of ACE-Is for AD therapeutic development. However, while there is increasing evidence that ACE-Is may be beneficial in AD patients, the mechanisms behind the beneficial effects of the drugs remain poorly understood. One reason is the inability to disentangle their effects on blood pressure from their direct effects on local RAS. As such, Drosophila, which does not have a conserved RAS pathway (Salzet et al., 2001; Fournier et al., 2012), provides a unique model to study the relationship between ACE-Is and AD. Toward this goal, Lee et al. (2020) evaluated the effects of ACE-Is and ARBs in fly lines expressing different AD-related transgenes. Using the GAL4/UAS system, fly lines were generated that expressed the following human transgenes: C99WT and C99V717I (a common mutation of APP found in FAD), and Aβ42 in CNS tissue, including the eye and brain. They showed that all of the flies exhibit age-dependent defects, including neuronal cell death and impaired memory, with the mildest defects observed in C99WT flies and the most severe in Aβ42 flies. Importantly, both the cell death and memory defects observed in C99 and Aβ42 flies were suppressed when flies were fed either captopril (an ACE-I) or losartan (an ARB), as summarized in Table 3. Moreover, measurements of Aβ42 levels and plaques in drug treated flies were similar to untreated flies, suggesting that the observed beneficial effects are independent to changes in Aβ42 pathology. This is consistent with findings from some of the mouse studies mentioned previously. Finally, to confirm that ACE-Is work in Drosophila in a similar manner to mammals, Lee et al. (2020) evaluated the effects of an Acer null mutant on various transgenic AD-related lines mentioned above. They found that a complete loss of Acer recapitulated the effects of captopril demonstrating that captopril exerts its effects by specifically inhibiting Acer.
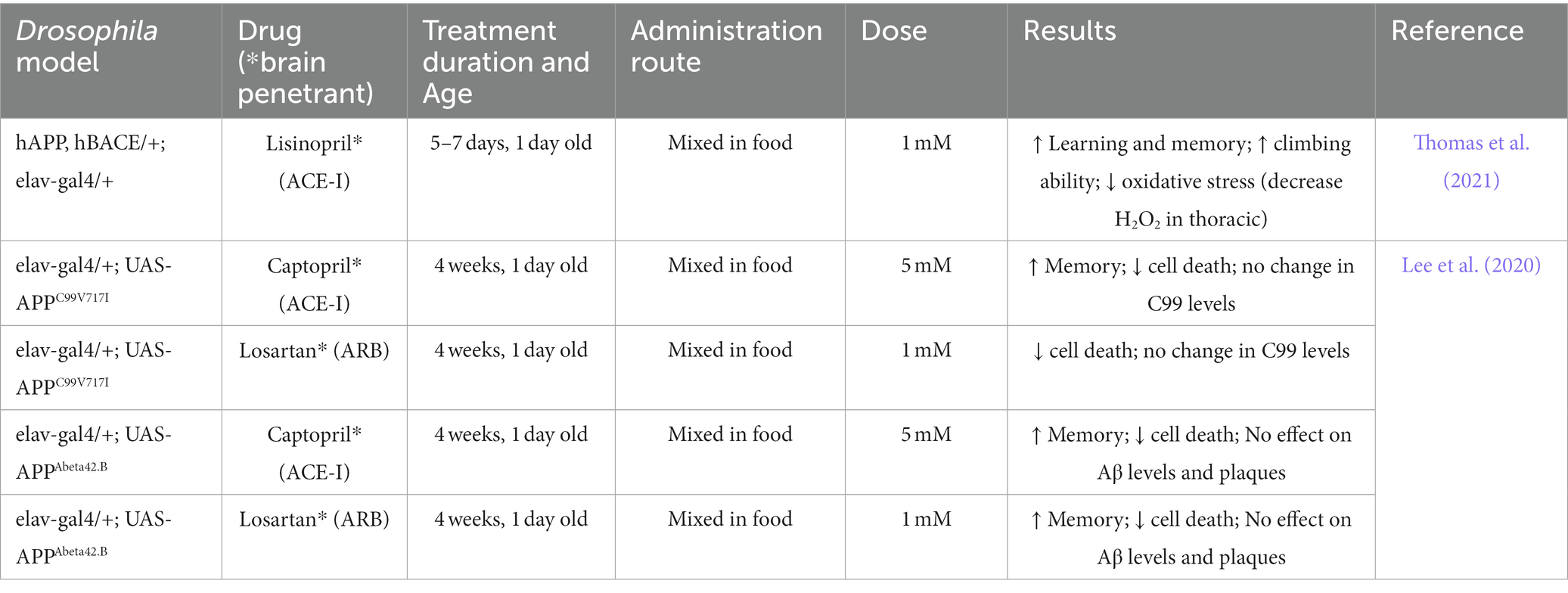
Table 3. Summary of effects of ACE-Is and ARBs in Drosophila models of AD including Aβ levels and deposition, oxidative stress and, cognitive deficits.
In a similar study, Thomas et al. (2021) aimed to determine whether the administration of lisinopril, an ACE-I, would have beneficial effects in a Drosophila model of AD that overexpresses human APP and human β-secretase in CNS tissue. Compared to control flies, these flies displayed deficits in learning and memory as well as in climbing ability, as determined through an aversive phototaxis suppression assay and a negative geotaxis assay, respectively. Upon lisinopril administration, AD flies displayed both improved cognition and climbing ability as well as a significant reduction in oxidative stress levels compared to untreated AD flies, as shown in Table 3.
To date, the mechanisms by which ARBs and ACE-Is function to suppress cell death and memory defects in Drosophila remain unknown. The effects of losartan are surprising given that their known target in mammals, AT1R, is not conserved in flies (Fournier et al., 2012). These findings suggest that losartan functions through an unknown and potentially novel target in Drosophila. This warrants further studies to help elucidate what its target(s) in flies might be. With regards to ACE-Is, Lee et al. (2020) demonstrated that the ability of captopril to suppress AD-related phenotypes in flies could be recapitulated by a null mutation in Acer demonstrating that Acer or a downstream effector of Acer, is the target of captopril. However, as previously mentioned, apart from ACE, RAS substrates do not appear to be conserved in Drosophila (Fournier et al., 2012) suggesting the existence of novel targets for ACE that extend beyond its conventional role in the canonical RAS. Such targets can be readily identified and validated in flies using a combination of biochemical and genetic approaches. Characterization of these targets will not only reveal the cellular pathways on which Acer acts in Drosophila but may also reveal novel roles for mammalian ACE beyond its canonical role in RAS and lead to the development of additional therapies for AD.
Although the molecular targets of Acer in Drosophila are unknown, several studies have begun to elucidate the physiological roles of Acer and a closely related gene, Ance, either through the use of ACE-Is or genetic nulls (Figure 1B). Such studies may provide insight into how inhibition of Acer using captopril or a null mutation, suppresses AD-related phenotypes. Both genes are broadly expressed in a variety of tissues throughout development and adult stages of flies (Chintapalli et al., 2007). However, only Ance is highly expressed in male accessory glands of the fly reproductive system suggesting a role in male fertility similar to that of mammalian gACE. Accordingly, males homozygous for hypomorphic alleles of Ance are infertile (Hurst et al., 2003; Rylett et al., 2007). Not limited to this role, Ance has also been suggested to influence aging in flies. Specifically, Gabrawy et al. (2019) examined the effect of lisinopril (an ACE-I) on Drosophila lifespan and found that lisinopril extended life span due to inhibiting Ance. They demonstrated this by first showing that knockdown of Ance extended lifespan and that treatment of lisinopril failed to enhance this effect.
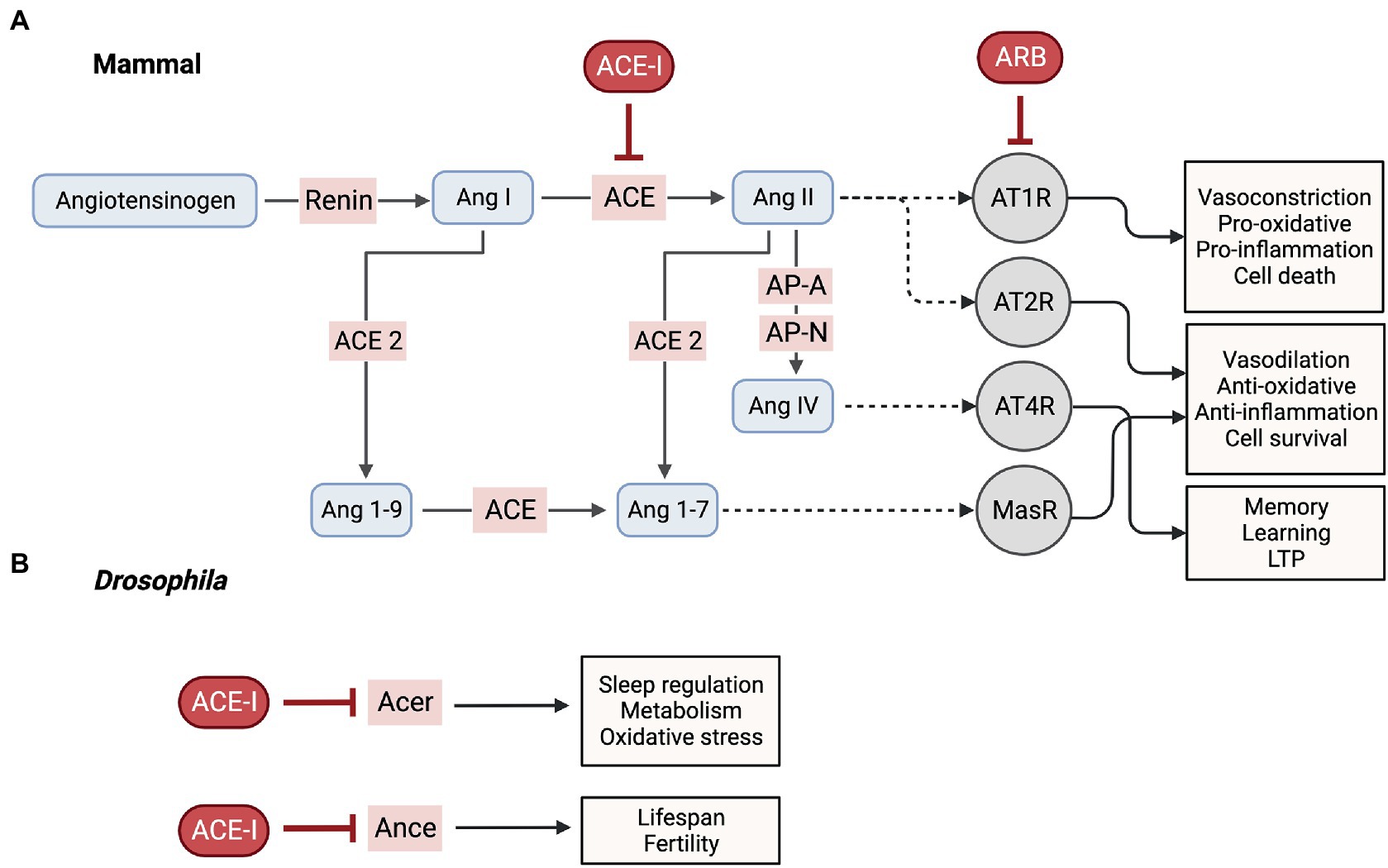
Figure 1. RAS in mammals vs. Drosophila. (A) Mammalian brain RAS pathways and its inhibitors. ACE, angiotensin converting enzyme; ACE-I, angiotensin converting enzyme inhibitor; Ang, angiotensin; AP-A, aminopeptidase A; AP-N, aminopeptidase N; ARB, angiotensin receptor blocker; AT1R, angiotensin II type 1 receptor; AT2R, angiotensin II type 2 receptor; AT4R, angiotensin 4 receptor; LTP, long-term potentiation; MasR, Mas receptor. (B) ACE-I inhibit Acer and Ance, the Drosophila ACE homologs. Acer, angiotensin-converting-enzyme related; Ance, angiotensin converting enzyme. (Created with BioRender.com).
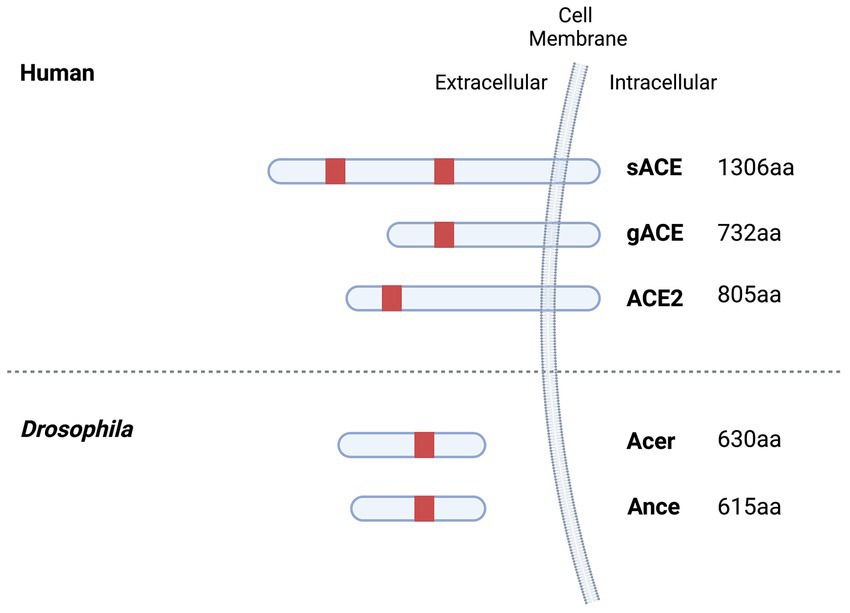
Figure 2. ACE family members in humans and Drosophila. Ance and Acer are homologous to ACE and share 61% (45% identity, 48% coverage) and 58% (41% identify, 45% coverage) amino acid similarity with ACE. Active site domains containing the conserved catalytic consensus zinc-binding motif (HEXXH) are indicated in red. sACE possess two active protein domains (N- and C- domain) whereas gACE, ACE2, Ance and Acer only have one. gACE is identical to that of the C domain of sACE except for its first 36 residues. Human ACE and ACE2 are integral-membrane proteins whereas Drosophila Ance and Acer lack a transmembrane domain. (Created with BioRender.com).
In contrast to Ance, Acer is highly expressed in adult heads, fat body (analogous to mammalian white adipose tissue and liver) and cardiac cells suggesting a role in different physiological processes, including cardiac function and metabolism (Crackower et al., 2002; Carhan et al., 2011). In fact, studies by Crackower et al. (2002) initially showed that an Acer mutation generated by a transposon (P-element) insertion gave rise to defects in heart morphogenesis that resulted in embryonic lethality. In line with these findings, Liao et al. (2013) presented a role for Acer in cardiac function by demonstrating that knock-down of Acer, specifically in adult heart tissue, results in heart defects such as impaired contractile properties. However, a contradictory study by Carhan et al. (2011) showed that flies homozygous for an Acer null mutation developed normally without significant heart defects, implying that previous heart development phenotypes observed by Crackower et al. (2002) may be attributed to a second-site mutation in the transposon-induced mutant line (Liao et al., 2013). Therefore, a role for Acer in Drosophila cardiac function remains to be established. Nevertheless, Carhan et al. (2011) identified a potential role for Acer in sleep regulation. Building upon a study that noted the cyclical expression of Acer in adult heads regulated by the circadian gene (clock) (McDonald and Rosbash, 2001; Carhan et al., 2011) hypothesized a possible role for Acer in circadian behavior. Indeed, they found Acer null flies exhibit a reduction in night-time sleep and greater sleep fragmentation. Moreover, this was also observed using an ACE-I, fosinopril.
Mechanisms underlying Acer’s contribution to defective sleep patterns remain unclear. Though, there is speculation that it may be due to changes in metabolic processes. Acer expression is strong in the fat body, a tissue with various functions, including protein and carbohydrate metabolism, lipid storage, and hormone secretion (Arrese and Soulages, 2010). Recent studies have also indicated a role for the fat body in regulating complex behaviors, including sleep (Yurgel et al., 2018). Therefore, Acer potentially possesses a functional role in metabolic processes in the fat body that, when disrupted, result in sleep defects. Beyond its prominent role in metabolism, the fat body plays an integral role in innate immune response regulation. It is responsible for the humoral response, synthesizing and secreting antimicrobial peptides into the hemolymph. While there is evidence in AD mouse studies, that ACE-Is mitigate neuroinflammatory responses that are known to contribute to AD pathology, it remains to be determined whether Drosophila Acer plays a similar role in immune response regulation.
Recently, a study by Glover et al. (2019) identified a potential role for Acer in metabolism including glycogen storage. Stored levels of lipids and glycogen in Drosophila are known to respond to dietary intake of sugar and yeast. This study found that under certain dietary conditions, Acer null mutant flies exhibit reduced glycogen levels compared to controls. However, just as with its role in sleep modulation, the mechanisms underlying this role for Acer are unknown. Nevertheless, it does pose an interesting avenue for further research to investigate the role of Acer in AD.
A prominent feature of AD is a significant reduction in glucose metabolism that is believed to contribute to disease progression and underlie cognitive dysfunction (Kumar et al., 2022). A decrease in metabolism is suggested to result from poor cerebral uptake of glucose into the brain. Interestingly, a study by Niccoli et al. (2016), showed that increasing glucose uptake in neurons in a Drosophila AD model, alleviated neurodegeneration and extended lifespan. Therefore it is worth exploring whether Acer plays a role in maintaining proper glucose metabolism in the brain of flies that could explain how its inhibition results in increased levels that in turn rescues cell death and memory phenotypes found in AD models.
Looking for new strategies to treat AD is an unmet clinical need. Targeting the RAS system has great potential for AD therapeutics. While many studies in patients and animal models have shown promising beneficial effects from inhibiting this system, it remains unclear what mechanisms underlie these outcomes. The RAS is well studied for its peripheral role in regulating blood pressure, fluid and electrolytes. However, its role in organs such as the brain appears to be more complex with new components having been discovered. For this reason, Drosophila provides a unique opportunity to understand how ACE-Is may function in the context of AD. Given that only ace like factors have been identified in the fly, it is possible to study the role ACE has in AD in isolation without confounding effects from other RAS components. More so, it is evident from human studies that beneficial effects of ACE-Is arise from their ability to penetrate the BBB and act on central RAS. Therefore, their effects appear to be independent of their ability to regulate blood pressure. For that reason, using an invertebrate model such as Drosophila with an open circulatory system provides an advantage of disentangling the effects of ACE-Is from their vascular hemodynamic effects and focusing directly on their effects in the brain.
JG wrote the first draft of the manuscript. JG and GB wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a grant from the Canadian Institutes of Health Research (PJT153063) to GB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AbdAlla, S., Langer, A., Fu, X., and Quitterer, U. (2013). ACE inhibition with captopril retards the development of signs of neurodegeneration in an animal model of alzheimer’s disease. Int. J. Mol. Sci. 14, 16917–16942. doi: 10.3390/ijms140816917
Akif, M., Masuyer, G., Bingham, R. J., Sturrock, E. D., Isaac, R. E., and Acharya, K. R. (2012). Structural basis of peptide recognition by the angiotensin-1 converting enzyme homologue AnCE from Drosophila melanogaster. FEBS J. 279, 4525–4534. doi: 10.1111/febs.12038
Alvarez, R., Alvarez, V., Lahoz, C. H., Martinez, C., Pena, J., Sanchez, J. M., et al. (1999). Angiotensin converting enzyme and endothelial nitric oxide synthase DNA polymorphisms and late onset alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 67, 733–736. doi: 10.1136/jnnp.67.6.733
Arregui, A., Perry, E. K., Rossor, M., and Tomlinson, B. E. (1982). Angiotensin converting enzyme in alzheimer’s disease: increased activity in caudate nucleus and cortical areas. J. Neurochem. 38, 1490–1492. doi: 10.1111/j.1471-4159.1982.tb07930.x
Arrese, E. L., and Soulages, J. L. (2010). Insect fat body: energy, metabolism, and regulation. Annu. Rev. Entomol. 55, 207–225. doi: 10.1146/annurev-ento-112408-085356
Asraf, K., Torika, N., Apte, R. N., and Fleisher-Berkovich, S. (2018). Microglial activation is modulated by captopril: in vitro and in vivo studies. Front. Cell. Neurosci. 12:e00116. doi: 10.3389/fncel.2018.00116
Barbier, P., Zejneli, O., Martinho, M., Lasorsa, A., Belle, V., Smet-Nocca, C., et al. (2019). Role of tau as a microtubule-associated protein: structural and functional aspects. Front. Aging Neurosci. 11:e00204. doi: 10.3389/fnagi.2019.00204
Barker, W. W., Luis, C. A., Kashuba, A., Luis, M., Harwood, D. G., Loewenstein, D., et al. (2002). Relative frequencies of alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the state of Florida brain Bank. Alzheimer Dis. Assoc. Disord. 16, 203–212. doi: 10.1097/00002093-200210000-00001
Barreras, A., and Gurk-Turner, C. (2003). Angiotensin II receptor blockers. Baylor Univ. Med. Center Proc. 16, 123–126. doi: 10.1080/08998280.2003.11927893
Barthold, D., Joyce, G., Wharton, W., Kehoe, P., and Zissimopoulos, J. (2018). The association of multiple anti-hypertensive medication classes with alzheimer’s disease incidence across sex, race, and ethnicity. PLoS One 13:e0206705. doi: 10.1371/journal.pone.0206705
Bertram, L., and Tanzi, R. E. (2009). Genome-wide association studies in alzheimer’s disease. Hum. Mol. Genet. 18, R137–R145. doi: 10.1093/hmg/ddp406
Bingham, R. J., Dive, V., Phillips, S. E., Shirras, A. D., and Isaac, R. E. (2006). Structural diversity of angiotensin-converting enzyme. Insights from structure-activity comparisons of two drosophila enzymes. FEBS J. 273, 362–373. doi: 10.1111/j.1742-4658.2005.05069.x
Busche, M. A., and Hyman, B. T. (2020). Synergy between amyloid-β and tau in alzheimer’s disease. Nat. Neurosci. 23, 1183–1193. doi: 10.1038/s41593-020-0687-6
Calsolaro, V., and Edison, P. (2016). Neuroinflammation in alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 12, 719–732. doi: 10.1016/j.jalz.2016.02.010
Carhan, A., Tang, K., Shirras, C. A., Shirras, A. D., and Isaac, R. E. (2011). Loss of angiotensin-converting enzyme-related (ACER) peptidase disrupts night-time sleep in adult drosophila melanogaster. J. Exp. Biol. 214, 680–686. doi: 10.1242/jeb.049353
Cassidy, L., Fernandez, F., Johnson, J. B., Naiker, M., Owoola, A. G., and Broszczak, D. A. (2020). Oxidative stress in alzheimer’s disease: a review on emergent natural polyphenolic therapeutics. Complement. Ther. Med. 49:102294. doi: 10.1016/j.ctim.2019.102294
Castellani, R. J., Rolston, R. K., and Smith, M. A. (2010). Alzheimer disease. Dis. Mon. 56, 484–546. doi: 10.1016/j.disamonth.2010.06.001
Chintapalli, V. R., Wang, J., and Dow, J. A. (2007). Using FlyAtlas to identify better drosophila melanogaster models of human disease. Nat. Genet. 39, 715–720. doi: 10.1038/ng2049
Chou, P.-S., Wu, S.-J., Kao, Y.-H., Chou, M.-C., Tai, S.-Y., and Yang, Y.-H. (2016). Angiotensin-converting enzyme insertion/deletion polymorphism is associated with cerebral white matter changes in alzheimer’s disease. Geriatr Gerontol Int 17, 945–950. doi: 10.1111/ggi.12815
Coates, D., Isaac, R. E., Cotton, J., Siviter, R., Williams, T. A., Shirras, A., et al. (2000). Functional conservation of the active sites of human and drosophila angiotensin I-converting enzyme. Biochemistry 39, 8963–8969. doi: 10.1021/bi000593q
Cornell, M. J., Williams, T. A., Lamango, N. S., Coates, D., Corvol, P., Soubrier, F., et al. (1995). Cloning and expression of an evolutionary conserved single-domain angiotensin converting enzyme from drosophila melanogaster. J. Biol. Chem. 270, 13613–13619. doi: 10.1074/jbc.270.23.13613
Cosarderelioglu, C., Nidadavolu, L. S., George, C. J., Oh, E. S., Bennett, D. A., et al. (2020). Brain renin–angiotensin system at the intersect of physical and cognitive frailty. Front. Neurosci. 14:586314. doi: 10.3389/fnins.2020.586314
Crackower, M. A., Sarao, R., Oudit, G. Y., Yagil, C., Kozieradzki, I., Scanga, S. E., et al. (2002). Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417, 822–828. doi: 10.1038/nature00786
Cuddy, L. K., Prokopenko, D., Cunningham, E. P., Brimberry, R., Song, P., Kirchner, R., et al. (2020). Aβ-accelerated neurodegeneration caused by Alzheimer’s-associated ace variant R1279Q is rescued by angiotensin system inhibition in mice. Sci. Transl. Med. 12:eaaz2541. doi: 10.1126/scitranslmed.aaz2541
Cui, H., Su, S., Cao, Y., Ma, C., and Qiu, W. (2021). The altered anatomical distribution of ACE2 in the brain with Alzheimer’s disease pathology. Front. Cell Develop. Biol. 9:e684874. doi: 10.3389/fcell.2021.684874
Danielyan, L., Klein, R., Hanson, L. R., Buadze, M., Schwab, M., Gleiter, C. H., et al. (2010). Protective effects of intranasal losartan in the app/PS1 transgenic mouse model of alzheimer disease. Rejuvenation Res. 13, 195–201. doi: 10.1089/rej.2009.0944
Davies, N. M., Kehoe, P. G., Ben-Shlomo, Y., and Martin, R. M. (2011). Associations of anti-hypertensive treatments with alzheimer’s disease, vascular dementia, and other dementias. J. Alzheimers Dis. 26, 699–708. doi: 10.3233/jad-2011-110347
Deng, Z., Jiang, J., Wang, J., Pan, D., Zhu, Y., Li, H., et al. (2022). Angiotensin receptor blockers are associated with a lower risk of progression from mild cognitive impairment to dementia. Hypertension 79, 2159–2169. doi: 10.1161/hypertensionaha.122.19378
Ding, Q., Shults, N. V., Gychka, S. G., Harris, B. T., and Suzuki, Y. J. (2021). Protein expression of angiotensin-converting enzyme 2 (ACE2) is upregulated in brains with alzheimer’s disease. Int. J. Mol. Sci. 22:1687. doi: 10.3390/ijms22041687
Dong, Y. F., Kataoka, K., Tokutomi, Y., Nako, H., Nakamura, T., Toyama, K., et al. (2011). Perindopril, a centrally active angiotensin-converting enzyme inhibitor, prevents cognitive impairment in mouse models of alzheimer’s disease. FASEB J. 25, 2911–2920. doi: 10.1096/fj.11-182873
Elkins, J. S., Douglas, V. C., and Johnston, S. C. (2004). Alzheimer disease risk and genetic variation in ace: a meta-analysis. Neurology 62, 363–368. doi: 10.1212/01.wnl.0000106823.72493.ff
Ellul, J., Archer, N., Foy, C. M., Poppe, M., Boothby, H., Nicholas, H., et al. (2006). The effects of commonly prescribed drugs in patients with alzheimer’s disease on the rate of deterioration. J. Neurol. Neurosurg. Psychiatry 78, 233–239. doi: 10.1136/jnnp.2006.104034
Fakhoury, M. (2018). Microglia and astrocytes in Alzheimer’s disease: implications for therapy. Curr. Neuropharmacol. 16, 508–518. doi: 10.2174/1570159x15666170720095240
Fournier, D., Luft, F. C., Bader, M., Ganten, D., and Andrade-Navarro, M. A. (2012). Emergence and evolution of the renin–angiotensin–aldosterone system. J. Mol. Med. 90, 495–508. doi: 10.1007/s00109-012-0894-z
Gabrawy, M. M., Campbell, S., Carbone, M. A., Morozova, T. V., Arya, G. H., Turlapati, L. B., et al. (2019). Lisinopril preserves physical resilience and extends life span in a genotype-specific manner in drosophila melanogaster. J. Gerontol. Ser. A 74, 1844–1852. doi: 10.1093/gerona/glz152
Gao, Y., O’Caoimh, R., Healy, L., Kerins, D. M., Eustace, J., Guyatt, G., et al. (2013). Effects of centrally acting ACE inhibitors on the rate of cognitive decline in dementia. BMJ Open 3:e002881. doi: 10.1136/bmjopen-2013-002881
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. doi: 10.1016/s0006-291x(84)80190-4
Glover, Z., Hodges, M. D., Dravecz, N., Cameron, J., Askwith, H., Shirras, A., et al. (2019). Loss of angiotensin-converting enzyme-related (ACER) peptidase disrupts behavioural and metabolic responses to diet in drosophila melanogaster. J. Exp. Biol. 222:jeb194332. doi: 10.1242/jeb.194332
Goedert, M., and Ghetti, B. (2007). Alois Alzheimer: his life and times. Brain Pathol. 17, 57–62. doi: 10.1111/j.1750-3639.2007.00056.x
Gomes, L. A., Hipp, S. A., Rijal Upadhaya, A., Balakrishnan, K., Ospitalieri, S., Koper, M. J., et al. (2019). AΒ-induced acceleration of alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 138, 913–941. doi: 10.1007/s00401-019-02053-5
Götz, J., Chen, F., van Dorpe, J., and Nitsch, R. M. (2001). Formation of neurofibrillary tangles in P301L tau transgenic mice induced by AΒ42 fibrils. Science 293, 1491–1495. doi: 10.1126/science.1062097
Gouveia, F., Camins, A., Ettcheto, M., Bicker, J., Falcão, A., Cruz, M. T., et al. (2022). Targeting brain renin-angiotensin system for the prevention and treatment of Alzheimer’s disease: past, present and future. Ageing Res. Rev. 77:101612. doi: 10.1016/j.arr.2022.101612
Graeber, M. B., Kösel, S., Egensperger, R., Banati, R. B., Müller, U., Bise, K., et al. (1997). Rediscovery of the case described by Alois Alzheimer in 1911: historical, histological and molecular genetic analysis. Neurogenetics 1, 73–80. doi: 10.1007/s100480050011
Greeve, I., Kretzschmar, D., Tschäpe, J. A., Beyn, A., Brellinger, C., Schweizer, M., et al. (2004). Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in TransgenicDrosophila. J. Neurosci. 24, 3899–3906. doi: 10.1523/jneurosci.0283-04.2004
Haapasalo, A., and Kovacs, D. M. (2011). The many substrates of presenilin/γ-secretase. J. Alzheimers Dis. 25, 3–28. doi: 10.3233/jad-2011-101065
Hajjar, I., Catoe, H., Sixta, S., Boland, R., Johnson, D., Hirth, V., et al. (2005). Cross-sectional and longitudinal association between antihypertensive medications and cognitive impairment in an elderly population. J. Gerontol. Ser. A Biol. Med. Sci. 60, 67–73. doi: 10.1093/gerona/60.1.67
Hajjar, I., Hart, M., Chen, Y.-L., Mack, W., Milberg, W., Chui, H., et al. (2012). Effect of antihypertensive therapy on cognitive function in early executive cognitive impairment: a double-blind randomized clinical trial. Arch. Intern. Med. 172, 442–444. doi: 10.1001/archinternmed.2011.1391
Hajjar, I. M., Keown, M., Lewis, P., and Almor, A. (2008). Angiotensin converting enzyme inhibitors and cognitive and functional decline in patients with alzheimer’s disease: an observational study. Am. J. Alzheimer Dis. Other Dement. 23, 77–83. doi: 10.1177/1533317507309803
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hemming, M. L., Selkoe, D. J., and Farris, W. (2007). Effects of prolonged angiotensin-converting enzyme inhibitor treatment on amyloid β-protein metabolism in mouse models of alzheimer disease. Neurobiol. Dis. 26, 273–281. doi: 10.1016/j.nbd.2007.01.004
Holtzman, D. M., Morris, J. C., and Goate, A. M. (2011). Alzheimer’s disease: the challenge of the second century. Sci. Transl. Med. 3:77sr1. doi: 10.1126/scitranslmed.3002369
Houard, X., Williams, T. A., Michaud, A., Dani, P., Isaac, R. E., Shirras, A. D., et al. (1998). The drosophila melanogaster-related angiotensin-i-converting enzymes Acer and Ance. Distinct enzymic characteristics and alternative expression during pupal development. Eur. J. Biochem. 257, 599–606. doi: 10.1046/j.1432-1327.1998.2570599.x
Hubert, C., Houot, A. M., Corvol, P., and Soubrier, F. (1991). Structure of the angiotensin I-converting enzyme gene. Two alternate promoters correspond to evolutionary steps of a duplicated gene. J. Biol. Chem. 266, 15377–15383. doi: 10.1016/s0021-9258(18)98626-6
Hurst, D., Rylett, C. M., Isaac, R. E., and Shirras, A. D. (2003). The drosophila angiotensin-converting enzyme homologue ance is required for spermiogenesis. Dev. Biol. 254, 238–247. doi: 10.1016/s0012-1606(02)00082-9
Iwatsubo, T., Odaka, A., Suzuki, N., Mizusawa, H., Nukina, N., and Ihara, Y. (1994). Visualization of AΒ42(43) and AΒ40 in senile plaques with end-specific AΒ monoclonals: evidence that an initially deposited species is AΒ42(43). Neuron 13, 45–53. doi: 10.1016/0896-6273(94)90458-8
Jackson, L. D., Eldahshan, W., Fagan, S., and Ergul, A. (2018). Within the brain: the renin angiotensin system. Int. J. Mol. Sci. 19:876. doi: 10.3390/ijms19030876
Jeon, Y., Lee, J. H., Choi, B., Won, S.-Y., and Cho, K. S. (2020). Genetic dissection of alzheimer’s disease using drosophila models. Int. J. Mol. Sci. 21:884. doi: 10.3390/ijms21030884
Kang, J., Lemaire, H.-G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K.-H., et al. (1987). The precursor of alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. doi: 10.1038/325733a0
Kehoe, P. G., Russ, C., McIlroy, S., Williams, H., Holmans, P., Holmes, C., et al. (1999). Variation in DCP1, encoding ace, is associated with susceptibility to alzheimer disease. Nat. Genet. 21, 71–72. doi: 10.1038/5009
Kehoe, P. G., Turner, N., Howden, B., Jarutyte, L., Clegg, S. L., Malone, I. B., et al. (2021). Safety and efficacy of losartan for the reduction of brain atrophy in clinically diagnosed alzheimer’s disease (the radar trial): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 20, 895–906. doi: 10.1016/s1474-4422(21)00263-5
Kehoe, P. G., Wong, S., Palmer, L. E., and Miners, J. S. (2016). Angiotensin-converting enzyme 2 is reduced in alzheimer’s disease in association with increasing amyloid-β and tau pathology. Alzheimers Res. Ther. 8:50. doi: 10.1186/s13195-016-0217-7
Kölsch, H., Jessen, F., Freymann, N., Kreis, M., Hentschel, F., Maier, W., et al. (2005). Ace I/D polymorphism is a risk factor of alzheimer’s disease but not of vascular dementia. Neurosci. Lett. 377, 37–39. doi: 10.1016/j.neulet.2004.11.062
Krstic, D., and Knuesel, I. (2012). Deciphering the mechanism underlying late-onset alzheimer disease. Nat. Rev. Neurol. 9, 25–34. doi: 10.1038/nrneurol.2012.236
Kumar, V., Kim, S.-H., and Bishayee, K. (2022). Dysfunctional glucose metabolism in alzheimer’s disease onset and potential pharmacological interventions. Int. J. Mol. Sci. 23:9540. doi: 10.3390/ijms23179540
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic meta-analysis of diagnosed alzheimer’s disease identifies new risk loci and implicates AΒ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. doi: 10.1038/s41588-019-0358-2
Kuperstein, I., Broersen, K., Benilova, I., Rozenski, J., Jonckheere, W., Debulpaep, M., et al. (2010). Neurotoxicity of alzheimer’s disease AΒ peptides is induced by small changes in the AΒ42 to AΒ40 ratio. EMBO J. 29, 3408–3420. doi: 10.1038/emboj.2010.211
Lee, S.-H., Gomes, S. M., Ghalayini, J., Iliadi, K. G., and Boulianne, G. L. (2020). Angiotensin converting enzyme inhibitors and angiotensin receptor blockers rescue memory defects in drosophila-expressing alzheimer’s disease-related transgenes independently of the canonical renin angiotensin system. eNeuro 7:0235. doi: 10.1523/eneuro.0235-20.2020
Lehmann, D. J., Cortina-Borja, M., Warden, D. R., Smith, A. D., Sleegers, K., Prince, J. A., et al. (2005). Large meta-analysis establishes the ACE insertion-deletion polymorphism as a marker of alzheimer’s disease. Am. J. Epidemiol. 162, 305–317. doi: 10.1093/aje/kwi202
Li, N.-C., Lee, A., Whitmer, R. A., Kivipelto, M., Lawler, E., Kazis, L. E., et al. (2010). Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ 340:b5465. doi: 10.1136/bmj.b5465
Liao, F.-T., Chang, C.-Y., Su, M.-T., and Kuo, W.-C. (2013). Necessity of angiotensin-converting enzyme-related gene for cardiac functions and longevity of drosophila melanogaster assessed by optical coherence tomography. J. Biomed. Opt. 19:1. doi: 10.1117/1.jbo.19.1.011014
Liu, S., Ando, F., Fujita, Y., Liu, J., Maeda, T., Shen, X., et al. (2019). A clinical dose of angiotensin-converting enzyme (ACE) inhibitor and heterozygous ACE deletion exacerbate alzheimer’s disease pathology in mice. J. Biol. Chem. 294, 9760–9770. doi: 10.1074/jbc.ra118.006420
Luo, L., Tully, T., and White, K. (1992). Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for APPL gene. Neuron 9, 595–605. doi: 10.1016/0896-6273(92)90024-8
MacLachlan, R., Kehoe, P. G., and Miners, J. S. (2022). Dysregulation of ace-1 in normal aging and the early stages of Alzheimer’s disease. J. Gerontol. Ser. A 77, 1775–1783. doi: 10.1093/gerona/glac083
Mak, K., Yang, F., Vinters, H. V., Frautschy, S. A., and Cole, G. M. (1994). Polyclonals to β-amyloid(1–42) identify most plaque and vascular deposits in alzheimer cortex, but not striatum. Brain Res. 667, 138–142. doi: 10.1016/0006-8993(94)91725-6
Marioni, R. E., Harris, S. E., Zhang, Q., McRae, A. F., Hagenaars, S. P., Hill, W. D., et al. (2018). Gwas on family history of Alzheimer’s disease. Transl. Psychiatry 8:99. doi: 10.1038/s41398-018-0150-6
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., and Cummings, J. L. (2015). Alzheimer’s disease. Nat. Rev. Dis. Primers. 1:15056. doi: 10.1038/nrdp.2015.56
McDonald, M. J., and Rosbash, M. (2001). Microarray analysis and organization of circadian gene expression in drosophila. Cells 107, 567–578. doi: 10.1016/s0092-8674(01)00545-1
Miller, D. L., Papayannopoulos, I. A., Styles, J., Bobin, S. A., Lin, Y. Y., Biemann, K., et al. (1993). Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of alzheimer′s disease. Arch. Biochem. Biophys. 301, 41–52. doi: 10.1006/abbi.1993.1112
Miners, J. S., Ashby, E., van Helmond, Z., Chalmers, K. A., Palmer, L. E., Love, S., et al. (2008). Angiotensin-converting enzyme (ACE) levels and activity in alzheimer’s disease, and relationship of perivascular ACE-1 to cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 34, 181–193. doi: 10.1111/j.1365-2990.2007.00885.x
Narain, Y. (2000). The ace gene and alzheimer’s disease susceptibility. J. Med. Genet. 37, 695–697. doi: 10.1136/jmg.37.9.695
Newcombe, E. A., Camats-Perna, J., Silva, M. L., Valmas, N., Huat, T. J., and Medeiros, R. (2018). Inflammation: the link between comorbidities, genetics, and alzheimer’s disease. J. Neuroinflammation 15:276. doi: 10.1186/s12974-018-1313-3
Niccoli, T., Cabecinha, M., Tillmann, A., Kerr, F., Wong, C. T., Cardenes, D., et al. (2016). Increased glucose transport into neurons rescues AΒ toxicity in drosophila. Curr. Biol. 26:2550. doi: 10.1016/j.cub.2016.09.018
Nichols, E., Steinmetz, J. D., Vollset, S. E., Fukutaki, K., Chalek, J., Abd-Allah, F., et al. (2022). Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the global burden of disease study 2019. The Lancet Public Health 7, e105–e125. doi: 10.1016/s2468-2667(21)00249-8
O’Caoimh, R., Healy, L., Gao, Y., Svendrovski, A., Kerins, D. M., Eustace, J., et al. (2014). Effects of centrally acting angiotensin converting enzyme inhibitors on functional decline in patients with alzheimer’s disease. J. Alzheimers Dis. 40, 595–603. doi: 10.3233/jad-131694
Ongali, B., Nicolakakis, N., Tong, X.-K., Aboulkassim, T., Papadopoulos, P., Rosa-Neto, P., et al. (2014). Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer’s disease model. Neurobiol. Dis. 68, 126–136. doi: 10.1016/j.nbd.2014.04.018
Ouk, M., Wu, C.-Y., Rabin, J. S., Jackson, A., Edwards, J. D., Ramirez, J., et al. (2021). The use of angiotensin-converting enzyme inhibitors vs. angiotensin receptor blockers and cognitive decline in Alzheimer’s disease: the importance of blood-brain barrier penetration and Apoe Ε4 carrier status. Alzheimers Res. Ther. 13:43. doi: 10.1186/s13195-021-00778-8
Papadopoulos, P., Tong, X.-K., Imboden, H., and Hamel, E. (2016). Losartan improves cerebrovascular function in a mouse model of alzheimer’s disease with combined overproduction of amyloid-β and transforming growth factor-β1. J. Cereb. Blood Flow Metab. 37, 1959–1970. doi: 10.1177/0271678x16658489
Paul, M., Poyan Mehr, A., and Kreutz, R. (2006). Physiology of local renin-angiotensin systems. Physiol. Rev. 86, 747–803. doi: 10.1152/physrev.00036.2005
Periz, G., and Fortini, M. E. (2004). Functional reconstitution of secretase through coordinated expression of presenilin, Nicastrin, APH-1, and Pen-2. J. Neurosci. Res. 77, 309–322. doi: 10.1002/jnr.20203
Prüßing, K., Voigt, A., and Schulz, J. B. (2013). Drosophila melanogaster as a model organism for Alzheimer’s disease. Mol. Neurodegener. 8:35. doi: 10.1186/1750-1326-8-35
Reardon, S. (2023). FDA approves alzheimer’s drug Lecanemab amid safety concerns. Nature 613, 227–228. doi: 10.1038/d41586-023-00030-3
Royea, J., Zhang, L., Tong, X.-K., and Hamel, E. (2017). Angiotensin IV receptors mediate the cognitive and cerebrovascular benefits of losartan in a mouse model of alzheimer’s disease. J. Neurosci. 37, 5562–5573. doi: 10.1523/jneurosci.0329-17.2017
Rylett, C. M., Walker, M. J., Howell, G. J., Shirras, A. D., and Isaac, R. E. (2007). Male accessory glands of drosophila melanogaster make a secreted angiotensin I-converting enzyme (ance), suggesting a role for the peptide-processing enzyme in seminal fluid. J. Exp. Biol. 210, 3601–3606. doi: 10.1242/jeb.009035
Salzet, M., Deloffre, L., Breton, C., Vieau, D., and Schoofs, L. (2001). The angiotensin system elements in invertebrates. Brain Res. Rev. 36, 35–45. doi: 10.1016/s0165-0173(01)00063-7
Savaskan, E., Hock, C., Olivieri, G., Bruttel, S., Rosenberg, C., Hulette, C., et al. (2001). Cortical alterations of angiotensin converting enzyme, angiotensin II and AT1 receptor in Alzheimer’s dementia. Neurobiol. Aging 22, 541–546. doi: 10.1016/s0197-4580(00)00259-1
Shen, X. Z., Xiao, H. D., Li, P., Lin, C. X., Fuchs, S., and Bernstein, K. E. (2008). Tissue specific expression of angiotensin converting enzyme: a new way to study an old friend. Int. Immunopharmacol. 8, 171–176. doi: 10.1016/j.intimp.2007.08.010
Sink, K. M., Leng, X., Williamson, J., Kritchevsky, S. B., Yaffe, K., Kuller, L., et al. (2009). Angiotensin-converting enzyme inhibitors and cognitive decline in older adults with hypertension. Arch. Intern. Med. 169, 1195–1202. doi: 10.1001/archinternmed.2009.175
Solfrizzi, V., Scafato, E., Frisardi, V., Seripa, D., Logroscino, G., Kehoe, P. G., et al. (2013). Angiotensin-converting enzyme inhibitors and incidence of mild cognitive impairment. The Italian longitudinal study on aging. Age 35, 441–453. doi: 10.1007/s11357-011-9360-z
Soto, M. E., Abellan van Kan, G., Nourhashemi, F., Gillette-Guyonnet, S., Cesari, M., Cantet, C., et al. (2013). Angiotensin-converting enzyme inhibitors and Alzheimer’s disease progression in older adults: results from the Réseau Sur la maladie d’alzheimer français cohort. J. Am. Geriatr. Soc. 61, 1482–1488. doi: 10.1111/jgs.12415
Takami, M., Nagashima, Y., Sano, Y., Ishihara, S., Morishima-Kawashima, M., Funamoto, S., et al. (2009). Gamma-secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J. Neurosci. 29, 13042–13052. doi: 10.1523/jneurosci.2362-09.2009
Takeda, S., Sato, N., Takeuchi, D., Kurinami, H., Shinohara, M., Niisato, K., et al. (2009). Angiotensin receptor blocker prevented β-amyloid-induced cognitive impairment associated with recovery of neurovascular coupling. Hypertension 54, 1345–1352. doi: 10.1161/hypertensionaha.109.138586
Tampi, R. R., Forester, B. P., and Agronin, M. (2021). Aducanumab: evidence from clinical trial data and controversies. Drugs Cont. 10, 1–9. doi: 10.7573/dic.2021-7-3
Taylor, C. A. M., Coates, D., and Shirras, A. D. (1996). The ACER gene of drosophila codes for an angiotensin-converting enzyme homologue. Gene 181, 191–197. doi: 10.1016/s0378-1119(96)00503-3
Thomas, J., Smith, H., Smith, C. A., Coward, L., Gorman, G., de Luca, M., et al. (2021). The angiotensin-converting enzyme inhibitor lisinopril mitigates memory and motor deficits in a drosophila model of alzheimer’s disease. Pathophysiology 28, 307–319. doi: 10.3390/pathophysiology28020020
Torika, N., Asraf, K., Apte, R. N., and Fleisher-Berkovich, S. (2018). Candesartan ameliorates brain inflammation associated with alzheimer’s disease. CNS Neurosci. Ther. 24, 231–242. doi: 10.1111/cns.12802
Torika, N., Asraf, K., Cohen, H., and Fleisher-Berkovich, S. (2017). Intranasal Telmisartan ameliorates brain pathology in five familial alzheimer’s disease mice. Brain Behav. Immun. 64, 80–90. doi: 10.1016/j.bbi.2017.04.001
Torika, N., Asraf, K., Roasso, E., Danon, A., and Fleisher-Berkovich, S. (2016). Angiotensin converting enzyme inhibitors ameliorate brain inflammation associated with microglial activation: possible implications for Alzheimer’s disease. J. Neuroimmune Pharmacol. 11, 774–785. doi: 10.1007/s11481-016-9703-8
Trigiani, L. J., Royea, J., Lacalle-Aurioles, M., Tong, X.-K., and Hamel, E. (2018). Pleiotropic benefits of the angiotensin receptor blocker candesartan in a mouse model of Alzheimer disease. Hypertension 72, 1217–1226. doi: 10.1161/hypertensionaha.118.11775
van de Hoef, D. L., Hughes, J., Livne-Bar, I., Garza, D., Konsolaki, M., and Boulianne, G. L. (2009). Identifying genes that interact with drosophila presenilin and amyloid precursor protein. Genesis 47, 246–260. doi: 10.1002/dvg.20485
van der Kant, R., and Goldstein, L. S. B. (2015). Cellular functions of the amyloid precursor protein from development to dementia. Dev. Cell 32, 502–515. doi: 10.1016/j.devcel.2015.01.022
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., et al. (1999). Β-secretase cleavage of alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Wang, J., Ho, L., Chen, L., Zhao, Z., Zhao, W., Qian, X., et al. (2007). Valsartan lowers brain β-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J. Clin. Investig. 117, 3393–3402. doi: 10.1172/jci31547
Wu, C.-H., Mohammadmoradi, S., Chen, J. Z., Sawada, H., Daugherty, A., and Lu, H. S. (2018). Renin-angiotensin system and cardiovascular functions. Arterioscler. Thromb. Vasc. Biol. 38, e108–e116. doi: 10.1161/atvbaha.118.311282
Yim, H. E., and Yoo, K. H. (2008). Renin-angiotensin system - considerations for hypertension and kidney. Electrol. Blood Press. 6, 42–50. doi: 10.5049/ebp.2008.6.1.42
Yurgel, M. E., Shah, K. D., Brown, E. B., Burns, C., Bennick, R. A., DiAngelo, J. R., et al. (2018). ade2 functions in the drosophila fat body to promote sleep. G3 Genes|Genomes|Genetics 8, 3385–3395. doi: 10.1534/g3.118.200554
Zheng, W., Tian, E., Liu, Z., Zhou, C., Yang, P., Tian, K., et al. (2022). Small molecule angiotensin converting enzyme inhibitors: a medicinal chemistry perspective. Front. Pharmacol. 13:e968104. doi: 10.3389/fphar.2022.968104
Keywords: renin angiotensin system, Alzheimer’s disease, neurodegeneration, Drosophila, amyloid, angiotensin converting enzyme
Citation: Ghalayini J and Boulianne GL (2023) Deciphering mechanisms of action of ACE inhibitors in neurodegeneration using Drosophila models of Alzheimer’s disease. Front. Neurosci. 17:1166973. doi: 10.3389/fnins.2023.1166973
Edited by:
Udai Pandey, University of Pittsburgh Medical Center, United StatesReviewed by:
Wei-Ling Tsou, Wayne State University, United StatesCopyright © 2023 Ghalayini and Boulianne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabrielle L. Boulianne, Z2JvdWxAc2lja2tpZHMuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.