 Francesca McEwan
Francesca McEwan Jocelyn D. Glazier
Jocelyn D. Glazier Reinmar Hager
Reinmar Hager- Division of Evolution, Infection and Genomic Sciences, School of Biological Sciences, Manchester Academic Health Science Centre, Faculty of Biology, Medicine and Health, University of Manchester, Manchester, United Kingdom
The adult brain is a complex structure with distinct functional sub-regions, which are generated from an initial pool of neural epithelial cells within the embryo. This transition requires a number of highly coordinated processes, including neurogenesis, i.e., the generation of neurons, and neuronal migration. These take place during a critical period of development, during which the brain is particularly susceptible to environmental insults. Neurogenesis defects have been associated with the pathogenesis of neurodevelopmental disorders (NDDs), such as autism spectrum disorder and schizophrenia. However, these disorders have highly complex multifactorial etiologies, and hence the underlying mechanisms leading to aberrant neurogenesis continue to be the focus of a significant research effort and have yet to be established. Evidence from epidemiological studies suggests that exposure to maternal infection in utero is a critical risk factor for NDDs. To establish the biological mechanisms linking maternal immune activation (MIA) and altered neurodevelopment, animal models have been developed that allow experimental manipulation and investigation of different developmental stages of brain development following exposure to MIA. Here, we review the changes to embryonic brain development focusing on neurogenesis, neuronal migration and cortical lamination, following MIA. Across published studies, we found evidence for an acute proliferation defect in the embryonic MIA brain, which, in most cases, is linked to an acceleration in neurogenesis, demonstrated by an increased proportion of neurogenic to proliferative divisions. This is accompanied by disrupted cortical lamination, particularly in the density of deep layer neurons, which may be a consequence of the premature neurogenic shift. Although many aspects of the underlying pathways remain unclear, an altered epigenome and mitochondrial dysfunction are likely mechanisms underpinning disrupted neurogenesis in the MIA model. Further research is necessary to delineate the causative pathways responsible for the variation in neurogenesis phenotype following MIA, which are likely due to differences in timing of MIA induction as well as sex-dependent variation. This will help to better understand the underlying pathogenesis of NDDs, and establish therapeutic targets.
1. Introduction
Neuronal development is a highly orchestrated process in which the proliferation, differentiation and migration of neuronal cells allow distinct functional sub-regions to form, which eventually comprise the complex structure and function of the adult brain (Urbán and Guillemot, 2014; Mira and Morante, 2020). Compelling data suggests aberrant neurogenesis is a fundamental convergence point in the etiology of all neurodevelopmental disorders (NDDs), such as schizophrenia and autism spectrum disorder (ASD; Ernst, 2016; Fan and Pang, 2017). This is evidenced following genetic studies, which show that a high proportion of genes linked to NDDs are implicated in cellular proliferation and differentiation (Ernst, 2016).
Neurodevelopmental disorders have complex multifactorial origins; believed to be triggered by a combination of genetic and environmental factors (De Felice et al., 2015; Wilson et al., 2022). A rapidly growing and dominant hypothesis in this field is exposure to immune activation during early development in utero (Kinney et al., 2010; Feigenson et al., 2014). This was first evidenced following naturally occurring epidemics, such as the 1957 influenza epidemic in Finland, where an increased proportion of the population, who were in their second trimester of gestational development at the time of the epidemic, were later diagnosed with schizophrenia (Mednick et al., 1988; Sham et al., 1992).
Animal models have been developed to establish the mechanisms underlying the link between exposure to maternal infection and increased risk of developing NDDs (Woods et al., 2021; Bao et al., 2022). Several models involve direct administration of microorganisms, such as influenza, to the pregnant rodent (Garbett et al., 2012; Jacobsen et al., 2021). However, following the understanding that it is maternal immune activation (MIA) rather than the pathogen itself that increases the risk for NDDs (Patterson, 2002; Shi et al., 2005; Brown, 2006; Estes and McAllister, 2016), MIA is most commonly induced by bacterial or viral mimetics, lipopolysaccharide (LPS) or polyinosinic:polycytidylic acid [poly(I:C)], respectively (Meyer, 2014; Bergdolt and Dunaevsky, 2019; Woods et al., 2021; Bao et al., 2022). This stimulates the release of pro-inflammatory cytokines in the maternal plasma through activation of toll-like receptors and elicits adolescent and adult behavioral deficits reminiscent to NDD symptoms in the offspring (Meyer, 2014; Estes and McAllister, 2016; Bergdolt and Dunaevsky, 2019).
Maternal immune activation is commonly induced during mid to late gestation, which is defined as a critical period of brain development, during which neurogenesis can be influenced by adverse environmental conditions (Selemon and Zecevic, 2015; Dehorter and Del Pino, 2020). Therefore, it is reasonable to hypothesize that defects in neurogenesis are at the root of MIA-induced brain and behavioral deficits, which has been the focus of recent studies (Ben-Reuven and Reiner, 2021; Canales et al., 2021; Long et al., 2021; Tsukada et al., 2021; Loayza et al., 2022; Yu et al., 2022). However, results in outcome data often vary between MIA models, and hence the direct actions of MIA on embryonic neurogenesis and neuronal migration remain often largely unclear. This review summarizes the changes to the proliferation and differentiation of neurons, neuronal migration and cortical lamination in the embryonic rodent brain following MIA induction, and develops hypotheses about the link between these aspects of neurogenesis and aberrant brain phenotypes.
2. Modeling maternal immune activation
Animal models of human disorders should have face validity, meaning the model has similar endophenotypes to the human disease, construct validity, related to the biological deficit causing the disease, and predictive validity, defined as the similarity between model and patient in response to treatment (Crawley, 2012; Silverman et al., 2022). Strong evidence for predictive and face validity has been reported in the MIA model [reviewed extensively by Bergdolt and Dunaevsky (2019) and Haddad et al. (2020)], which is demonstrated by behavioral outputs recapitulating NDD-like symptoms in the offspring. For example, animals display altered amphetamine sensitivity when assessed through locomotor activity (Zager et al., 2012; Gray et al., 2019; Weber-Stadlbauer et al., 2021), as well as cognitive dysfunction, demonstrated by visual recognition and spatial memory deficits (Savanthrapadian et al., 2013; MacDowell et al., 2017; Richetto et al., 2017a; Lorusso et al., 2022). These are translatable to the positive and cognitive symptoms of schizophrenia, including psychotic agitation and dysfunctional working memory, respectively (Redrobe et al., 2010; van den Buuse, 2010; Batinić et al., 2016). Despite this, the biological deficits associated with MIA have been suggested to reflect specific aspects of NDDs, rather than recapitulating the biological phenotypes as a whole (Haddad et al., 2020). For example, MIA offspring brains exhibit shifts in excitatory versus inhibitory signaling systems, including defects in parvalbumin-containing γ-aminobutyric acid (GABA)ergic neurons (Zhang and van Praag, 2015; Canetta et al., 2016; Vojtechova et al., 2021), as well as alterations in the glutamatergic N-methyl-D-aspartate receptor subunit composition (Rahman et al., 2017; Hao et al., 2019). Deficits in glutamatergic and GABAergic neurotransmission are present in the brains of patients with schizophrenia and ASD (Gonzalez-Burgos et al., 2015; Balu, 2016; Rahman et al., 2020; Schoonover et al., 2020; Strube et al., 2020) and are believed to underlie the cognitive deficits associated with NDDs (Bojesen et al., 2021; Kumar et al., 2021). In comparison, few studies report changes to dopaminergic gene expression (Woods et al., 2021), which, until recently, dominated the field of schizophrenia research and remains the primary target of antipsychotic treatment (Coyle et al., 2010; Stahl, 2018; McCutcheon et al., 2020). However, current treatment lacks efficacy in ameliorating the negative and cognitive symptoms of schizophrenia (Fusar-Poli et al., 2015; McCutcheon et al., 2020), and hence, the MIA model may provide a useful tool in helping to identify therapeutic targets for these symptoms.
3. Typical neurogenesis
Neurodevelopment begins with the formation of the neural tube, where an initial pool of neuroepithelial cells (NECs) divide symmetrically until a sufficient number have formed (Figure 1; Stiles and Jernigan, 2010; Egger et al., 2011; Semple et al., 2013). At around embryonic day (E) 10–12 in the mouse telencephalon, division of NECs begins to switch from symmetric to asymmetric, forming one NEC and a radial glial cell (RGC; Dennis et al., 2016). The gradual switch from proliferation to differentiation is associated with cell cycle changes including decreased re-entry and increased cell cycle exit, as well as parameter alterations, such as lengthening of the cell cycle, or G1 phase specifically (Sommer and Rao, 2002; Ohnuma and Harris, 2003; Lancaster and Knoblich, 2012; Hardwick et al., 2015; Szűcs et al., 2020).
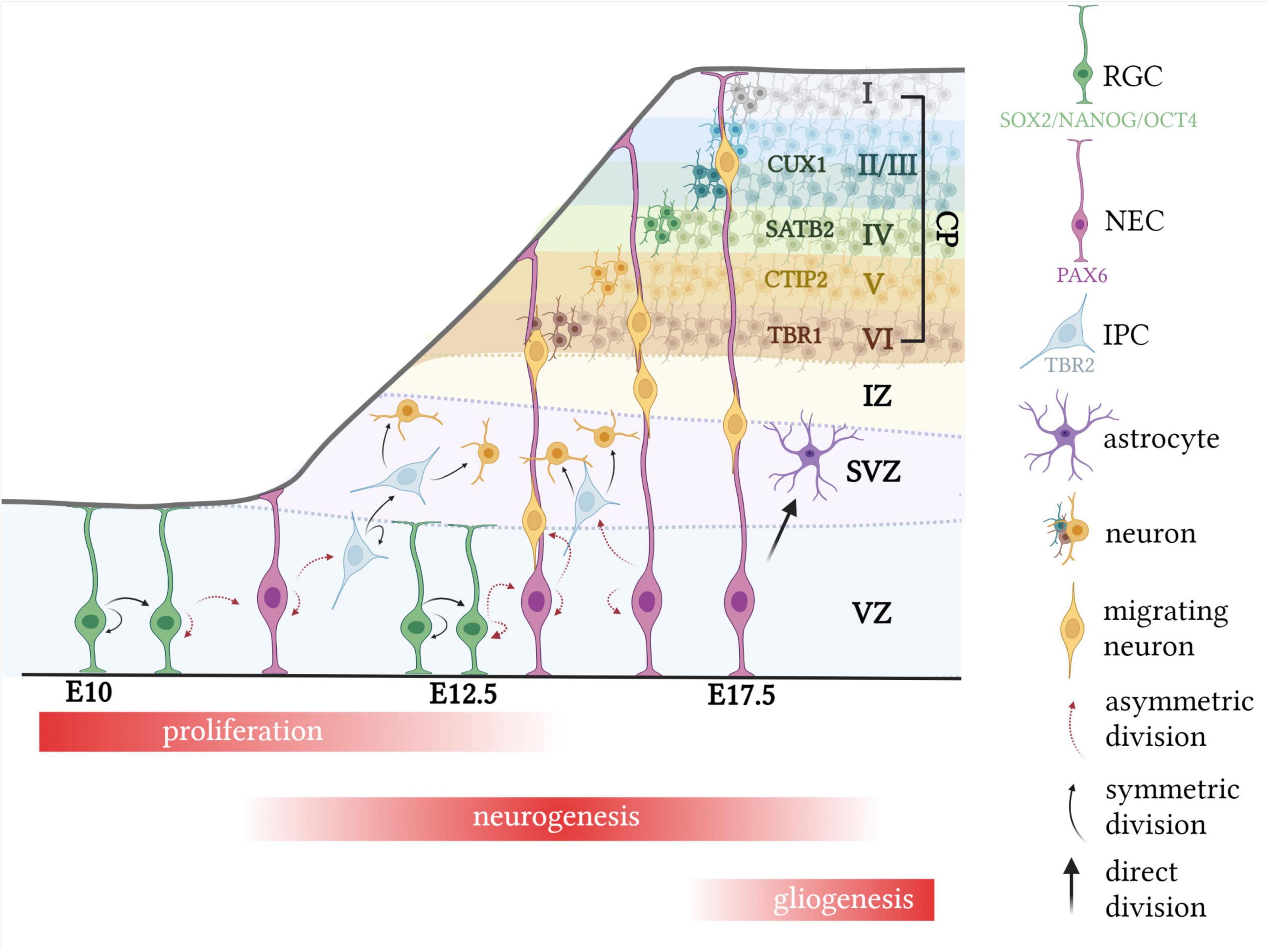
Figure 1. Typical murine corticogenesis. Following expansion of the NEC pool, division of NECs begin to switch from symmetric to asymmetric, forming one RGC and one NEC. RGCs then divide either asymmetrically to generate two RGCs, or symmetrically to form one RGC and an IPC or neuron. IPCs divide only symmetrically to form a pair of IPCs or neurons. Migrating neurons use radial glial cells as scaffolds, to migrate up to the cortical plate, which expands in an “inside-out” manner, with the deep layer IV neurons forming first, at around E12.5. During late corticogenesis, RGCs begin to directly divide into glial cells, such as astrocytes. CP, cortical plate; CTIP2, COUP TF1-interacting protein 2; CUX1, cut like homeobox 1; E, embryonic day; SATB2, special AT-rich sequence-binding protein 2; SOX2, SRY-box 2; PAX6, paired box protein Pax-6; TBR1, T-box brain transcription factor; TBR2, T-box brain protein 2; IPC, intermediate progenitor cell; NANOG, nanog homeobox; NEC, neural epithelial cell. Created with BioRender.com.
This transition is highly regulated by the expression of specific transcription factors, which may be used to trace the lineage of NEC to mature neuron (Urbán and Guillemot, 2014; Stevanovic et al., 2021). It is widely known that SRY-box 2 (SOX2), octamer-binding transcription factor 4 (OCT4) and homeobox protein NANOG (NANOG) are involved in maintaining pluripotency among stem cells (Figure 1; Ahmed et al., 2009; Desai and Pethe, 2020). The transition from NEC to RGC is associated with a decrease in expression of these pluripotent transcription factors and a concomitant increase in paired box protein pax-6 (PAX6) expression, which has received a lot of attention due to its essential role in controlling neurogenesis (Sansom et al., 2009; Suter et al., 2009; Zhang et al., 2010; Manuel et al., 2015). Despite the glial-like properties of RGCs, including certain molecular and cytological features, they can divide asymmetrically to form one RGC and either a neuron or an intermediate progenitor cell (IPC), identified as direct and indirect neurogenesis, respectively, and are responsible for the formation of all cortical neurons as well as several glial cell lineages (Figure 1; Beattie and Hippenmeyer, 2017). IPCs, also known as basal progenitors, have been uniquely associated with the subventricular zone (SVZ), which is located above the ventricular zone (VZ), and, unlike NECs and RGCs, divide only symmetrically a limited number of times (1–3) to produce neurons (Figure 1; Kowalczyk et al., 2009; Mira and Morante, 2020). For this reason, it has been suggested that IPCs function to increase the number of neurons and size of the SVZ, which becomes one of the two limited neurogenic regions in the adult (Smart et al., 2002; Götz and Huttner, 2005; Mira and Morante, 2020). RGCs, which eventually give rise to IPCs, transiently express neurogenin 2, which is the transcriptional target of T-box brain protein 2 (TBR2) and is expressed in IPCs. Hence, differentiation of RGCs to IPCs to neurons is associated with decreased PAX6 and increased TBR2 cellular density, followed by increased expression of post-mitotic neuron markers, such as neuron-specific class III beta-tubulin (TUJ1) or T-box brain transcription factor 1 (TBR1; Figure 1; Sun and Hevner, 2014; Manuel et al., 2015; Guo et al., 2021; Kim et al., 2021).
4. MIA-induced defects
Although embryonic neurogenesis and neuronal migration are highly controlled processes, they are defined as a critical period of neurodevelopment, which can be influenced by adverse environmental challenges (Fan and Pang, 2017). Accordingly, current evidence suggests MIA affects the proliferation of NECs, the differentiation of those cells into neurons and the migration of neurons to form distinct regions (De Miranda et al., 2010; Carpentier et al., 2011, 2013; Stolp et al., 2011; Gumusoglu et al., 2017; Ben-Reuven and Reiner, 2021).
4.1. Neurogenesis
Several studies show decreased proliferation in the fetal cortex acutely following MIA, demonstrated by reduced phosphohistone H3 (PHH3), a mitotically active cell marker shortly after (2–8 h) LPS administration (Table 1; Carpentier et al., 2011; Stolp et al., 2011; Kim et al., 2017; Braun et al., 2019). This is supported by nucleotide uptake studies, where the administration of a thymidine analog allows the synthesis of DNA, and hence, the proportion of cell divisions, to be tracked. For example, a reduction in single nucleotide uptake was reported when administered at 2–22 h post-LPS administration, which provides evidence for reduced proliferation in the MIA embryonic cortex (Cui et al., 2009; Stolp et al., 2011; Carpentier et al., 2013). Double-labeled thymidine studies enable a more precise determination of proliferation kinetics, where the time interval between the two nucleotides defines the cellular readout (Solius et al., 2021). Studies report decreased double-labeled cells within the first 24 h following both LPS and poly(I:C)-induced MIA, when an interval of over 10 h was used between nucleotide administration, indicating that there is a reduction in the number of cells which have re-entered the cell cycle (De Miranda et al., 2010; Carpentier et al., 2013). This is supported by findings of an increased quit fraction in these cells, which represents the proportion of cells that have left the cell cycle (Table 1; Carpentier et al., 2013). However, when an interval of 2.5 h was used, there was no significant change in the proportion of double labeled cells within the first 8 h after poly(I:C) injection and this was increased after 24 h (Ben-Reuven and Reiner, 2021). In contrast to 10 h, 2.5 h may be long enough to allow cells to exit S-phase, but not to re-enter, and hence, double-labeled cells in this instance are more likely to represent cells that have stayed in S-phase, rather than re-entered, suggesting that the length of S-phase has changed (Ben-Reuven and Reiner, 2021). Nucleotide dilution assays can also indicate proliferation state, where the nucleotide becomes diluted through cell divisions, and hence, high threshold cells represent cells that have not divided but are still in S-phase, and a low threshold signal is indicative of cells that have undergone multiple cell divisions. Studies report significantly decreased number of high threshold cells acutely following MIA in the fetal cortex (De Miranda et al., 2010; Ben-Reuven and Reiner, 2021), and De Miranda et al. (2010) showed that this was concordant with no change in the number of low threshold cells, which would be expected to increase following increased cell cycle divisions. They hence proposed that there is increased cell cycle exit, lending support to the theory of decreased proliferation acutely following MIA (De Miranda et al., 2010; Carpentier et al., 2011, 2013; Stolp et al., 2011; Ben-Reuven and Reiner, 2021).
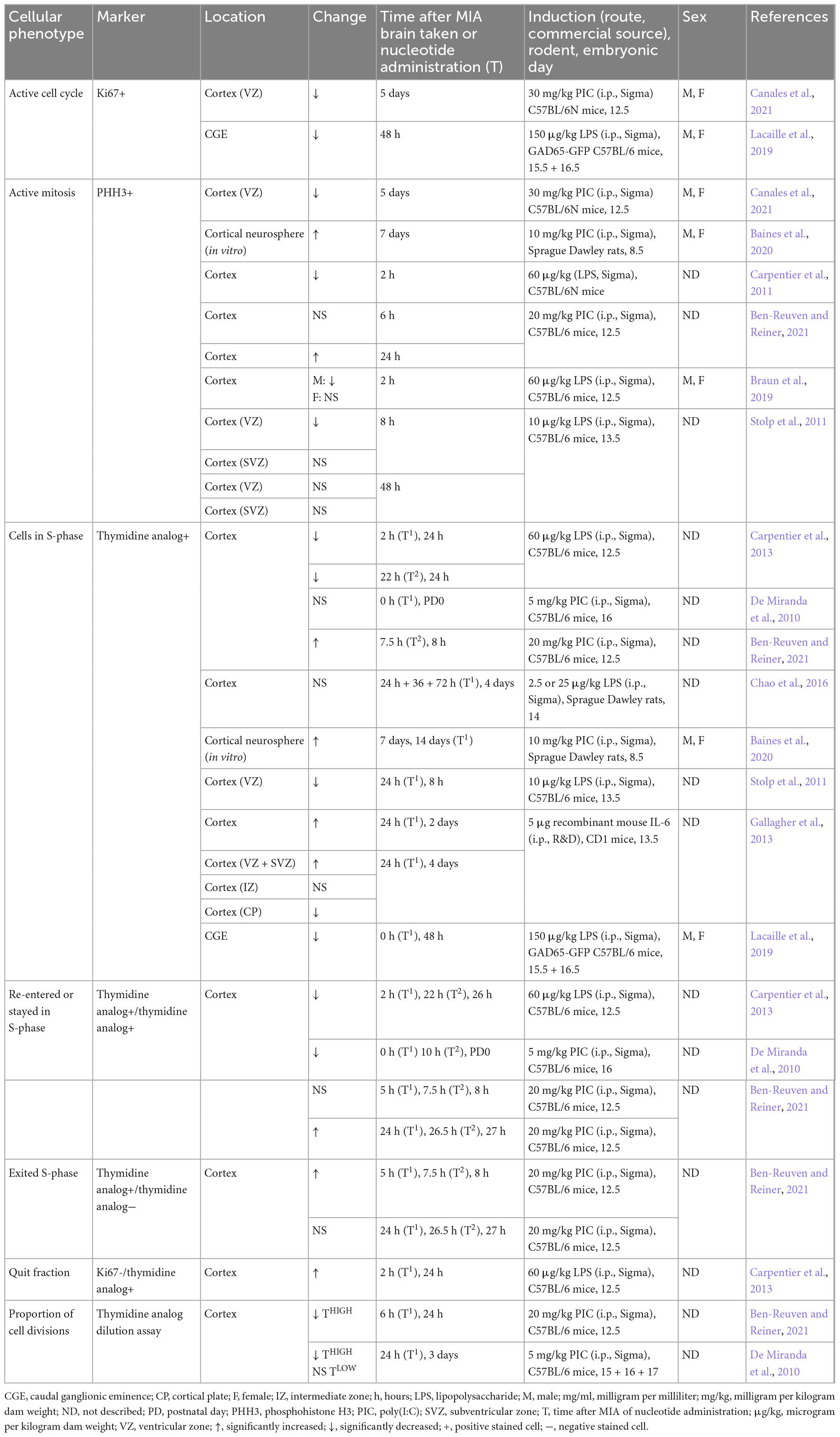
Table 1. Cell cycle phenotypes in fetal MIA offspring brains relative to vehicle controls.
The defective proliferation phenotype appears to be associated with a premature acceleration in neurogenesis, where cells are exhibiting a higher proportion of neurogenic divisions at the expense of self-renewal (Figure 2). This is demonstrated by cell cycle parameter changes recorded acutely following MIA, including a shortening of S-phase, which has been associated with a commitment to neuron production (Salomoni and Calegari, 2010; Arai et al., 2011; Mi et al., 2018; Ben-Reuven and Reiner, 2021). In accordance, it appears more NECs are dividing asymmetrically to form RGCs, IPCs or neurons, which is demonstrated by increased expansion of the RGC (PAX6) population (De Miranda et al., 2010; Tsukada et al., 2021) and elevated proportion of newly formed IPCs (Tsukada et al., 2021), contributing to indirect neurogenesis. Furthermore, studies report increased number of RGCs giving rise to a post-mitotic neuron and RGC, otherwise known as direct neurogenesis (Ben-Reuven and Reiner, 2021). Although the route of neurogenesis appears to differ between studies, where there is a discrepancy in the proportion of newly formed IPCs (Table 2; De Miranda et al., 2010; Ben-Reuven and Reiner, 2021; Tsukada et al., 2021), it is clear that MIA acutely promotes neural differentiation within the fetal brain (Figure 2).
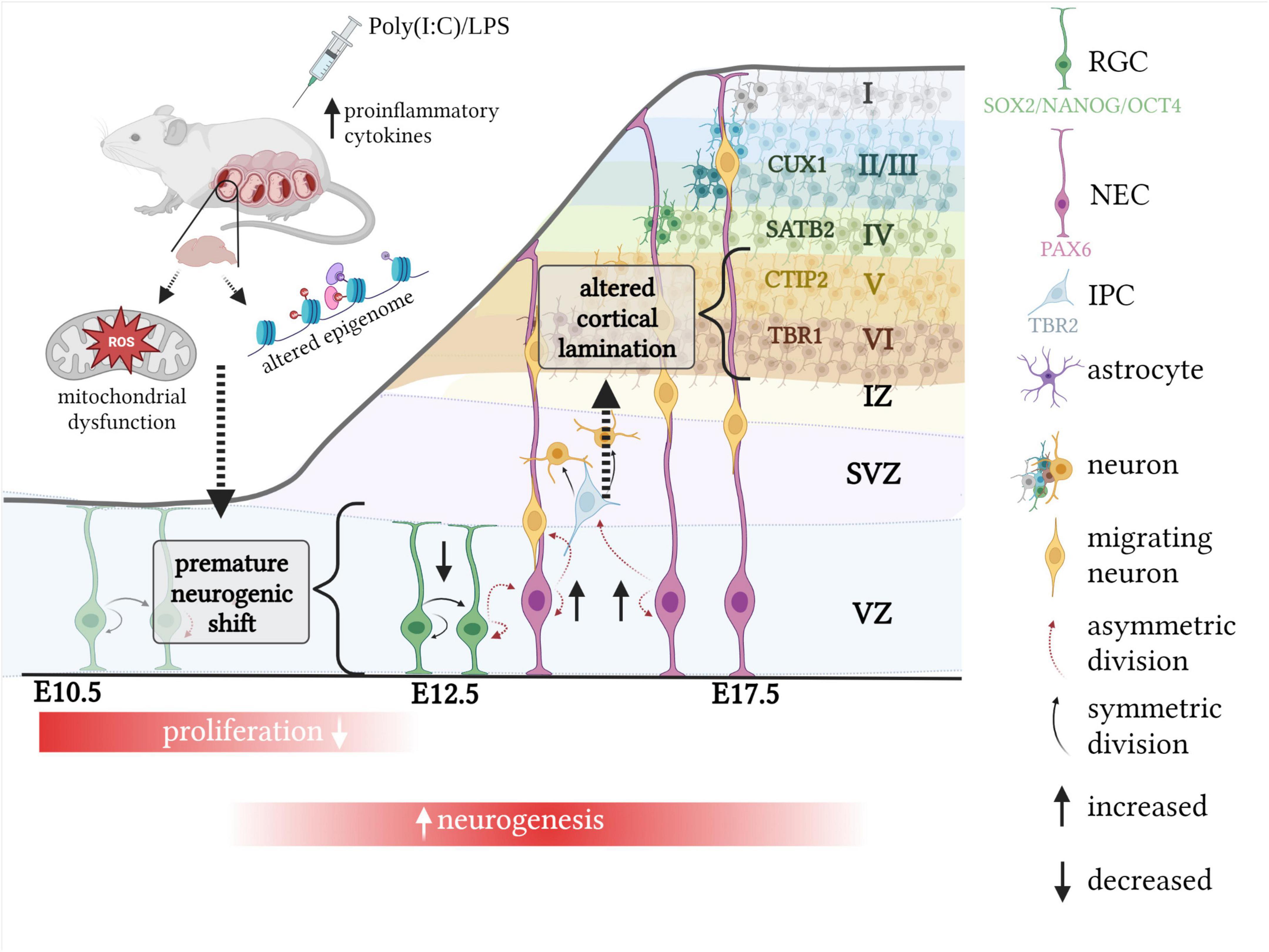
Figure 2. The impact of MIA on murine corticogenesis. Exposure to MIA whilst in utero causes an acute neurogenesis defect, associated with a decreased proportion of symmetric, proliferative divisions and increased number of asymmetric, neurogenic divisions. This results in cortical lamination defects which are dependent on the timing of MIA exposure. When induced at E12.5, increased neuron production at the expense of proliferation is associated with altered density of earlier born cortical layers, such as VI and V, which are defined by the transcription factors TBR1 and CTIP2, respectively. Mitochondrial dysfunction as well as an altered epigenome may explain the acute neurogenesis defect following MIA. Created with BioRender.com.
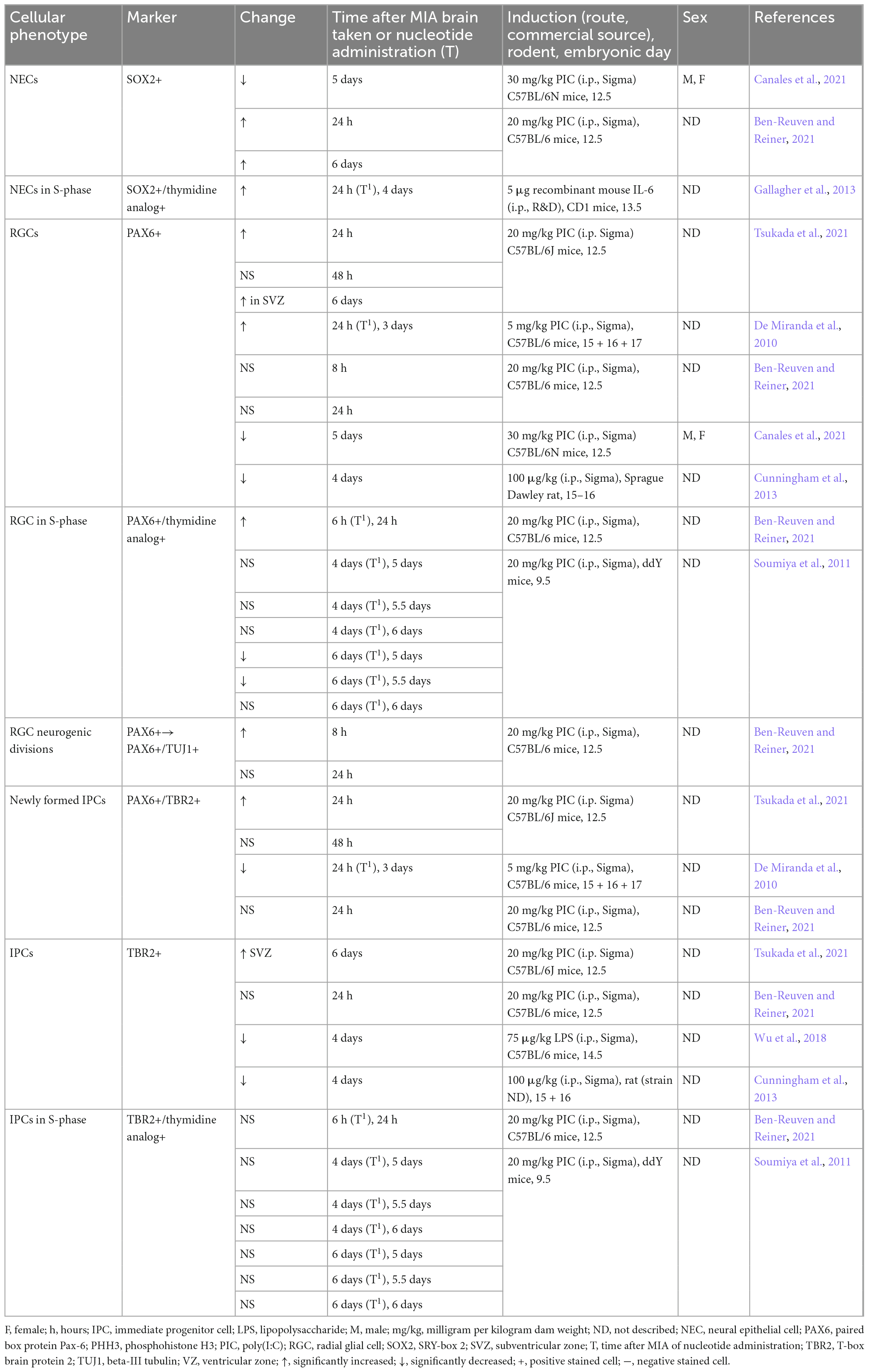
Table 2. Cell fate phenotypes in fetal MIA offspring cortex relative to controls.
Further evidence of a neurogenesis defect has been demonstrated at a delayed time point following MIA, including a reduction in proliferation after 5–6 days (Soumiya et al., 2011; Canales et al., 2021), which is in contrast to reports of increased proliferation in cortical neurospheres taken from offspring brains one week following MIA (Table 1; Baines et al., 2020). However, the use of an in vitro approach in the latter experiment may not directly reflect in vivo phenotypes, and hence, it is not known whether the differences in proliferation between these two studies is due to gestational timing differences or experimental approach. In comparison to the acute time point, dysregulated proliferation 3–5 days following MIA is predominantly associated with decreased proportion of RGCs and IPCs (Table 2; Soumiya et al., 2011; Cunningham et al., 2013; Canales et al., 2021).
It is likely that discrepancies in methodology between studies, including experimental approach, i.e., in vivo or in vitro, species and strain of model, source and dosage of immunostimulant and timing of MIA induction, contribute to differential outcomes (Kowash et al., 2019; Bao et al., 2022). Timing of MIA induction may be particularly important in this case due to the rapid and precise nature of embryonic neurogenesis (Urbán and Guillemot, 2014). The majority of studies referenced in this review expose pregnant mice to MIA at E12.5–13.5 (Maekawa et al., 2005; Osumi et al., 2008; Carpentier et al., 2011; Stolp et al., 2011; Gallagher et al., 2013; Choi et al., 2016; Braun et al., 2019; Ben-Reuven and Reiner, 2021; Canales et al., 2021; Tsukada et al., 2021), which, in typical murine neurodevelopment, coincides with significant changes in PAX6 density (Englund et al., 2005; Duan et al., 2013; Ben-Reuven and Reiner, 2021). It is thus not surprising that MIA at this time point primarily affects the PAX6 cell population. Several studies reported an acute increase in the proportion of PAX6 cells, which appears to reverse three days after MIA (De Miranda et al., 2010; Soumiya et al., 2011; Ben-Reuven and Reiner, 2021; Canales et al., 2021; Tsukada et al., 2021). In contrast, MIA at an early gestational time point, such as E8.5, exhibits a pro-proliferation phenotype (Baines et al., 2020). Differences in brain and behavioral phenotype depending on timing of gestational MIA exposure have been consistently reported and may translate to the development of distinct NDDs in humans, hence highlighting the requirement for further research (Meyer et al., 2006; Guma et al., 2022; Nakamura et al., 2022).
An increasing body of evidence suggests that there are sex-dependent effects in developing resilience or susceptibility to neurodevelopmental insults, reported in both patients and animal models (Palmer et al., 2017; Nugent et al., 2018; Hodes and Epperson, 2019; May et al., 2019; Vojtechova et al., 2021; Woodward and Coutellier, 2021). It is therefore essential for sex to be treated as an independent variable within MIA models (Coiro and Pollak, 2019). However, a number of studies referenced in this section did not indicate which sex was used for offspring experiments, nor how sex was determined (Tables 1–3), and it may therefore be assumed that in these instances, sex was disregarded, with male and female being treated as one group. Data shows a disparity in sex response to MIA in a number of developmental stages and systems (Haida et al., 2019), from the adult behavioral phenotype (Gogos et al., 2020; Zhao et al., 2021) to the acute inflammatory response (Barke et al., 2019). We may thus expect sex differences in neurogenesis. This view is further supported by evidence of decreased active mitosis in male but not female MIA cortices (Braun et al., 2019), as well as results showing that male neural progenitors have an increased tendency to differentiate in vitro (Baines et al., 2020). Hence, the pooling of both sexes into one group may often mask a sex-dependent effect, which in turn may lead to variation in proliferation phenotypes across studies.

Table 3. Cortical lamination changes in MIA fetal offspring brains in contrast to vehicle controls.
4.2. Neuronal migration
Neuronal migration is a complex process, which involves the coordination of neuronal branching and extension with cellular movement, and is guided by a number of vital signaling molecules and stimuli (Khodosevich and Monyer, 2011; Cooper, 2013; Buchsbaum and Cappello, 2019). Neurons migrate via two distinct mechanisms, radial and tangential, which are predominantly utilized by cortical projection neurons and GABAergic interneurons, respectively. Radial migration describes the process used by neurons migrating from the VZ, where radial glial “guides” are used as a scaffold for migration (Figure 1; Molyneaux et al., 2007; Stiles and Jernigan, 2010; Perez-Garcia and O’Leary, 2016). On the other hand, tangential migration involves neurons migrating from five main proliferative regions, in a manner parallel to the plial surface and perpendicular to radially migrating neurons, which thereby increases neuronal diversity in the brain (Marín, 2015).
Evidence suggests MIA offspring have neuronal migratory defects (Shi et al., 2009; Soumiya et al., 2011; Gumusoglu et al., 2017), such as a transient delay in cellular migration at E13.5–E14.5 in cortices exposed to MIA on E9.5 (Soumiya et al., 2011). GABAergic progenitors also show defective migration at E14 following administration of IL-6 to the pregnant dam (Gumusoglu et al., 2017), which may be linked to dysregulated expression of molecules required for interneuron migration, such as Nkx2.1, Nrp1, Trkb, and Arx as well as the Dlx family of genes (Oskvig et al., 2012; Nakamura et al., 2019, 2022).
Aside from these studies, there has been minimal research regarding neuronal migration in the MIA-offspring fetal brain, presumably due to the difficultly in researching cellular migration in vivo. However, neuronal migration in the murine neocortex takes place during mid-to-late gestation (Ayala et al., 2007), which often coincides with the timing of MIA insult. Further, genes involved in neuronal migration were differentially expressed in MIA prenatal non-candidate gene sets (Oskvig et al., 2012; Lombardo et al., 2018). Future research on the affected migratory processes following MIA is therefore necessary to elucidate NDD pathogenesis.
4.3. Cortical lamination
The proliferation and migration of neurons is instrumental in defining complex structures within the brain, one of which, the neocortex, is characterized by the lamination of neurons into six distinct layers (Kostović and Judaš, 2007; Guarnieri et al., 2019; Wu et al., 2021). During early murine cortical development (around E10.5), the first wave of neuronal migration from the VZ and SVZ forms the thin, preplate layer which later splits into the superficial marginal zone and the inner subplate, allowing the cortical plate to form in between (Figure 1). The cortical plate is then expanded in a tightly orchestrated, “inside-out” manner to form the characteristic six-layered structure of the cortex (Figure 1). These layers are defined by distinct cellular morphologies and densities, with each projecting to designated regions of the brain (Harris and Shepherd, 2015; Lodato and Arlotta, 2015). In brief, during early murine corticogenesis (E12.5), a wave of migrating neurons forms the corticothalamic layer, otherwise known as layer VI, which can be defined by the expression of TBR1. Layers V to II then migrate successively, in temporal waves, past earlier-born neurons, forming the subcerebral layer (V), the pyramidal layer (IV), and upper callosal layers (III-II). These layers are recognized by the expression of the transcription factors B-cell lymphoma/leukemia 11B (CTIP2), special AT-rich sequence-binding protein (SATB2), and cut like homeobox 1 (CUX1), respectively and are formed on E13.5–E14.5 (Figure 1; Molyneaux et al., 2007; Ben-Reuven and Reiner, 2021).
The densities of these layers are often altered following MIA exposure, particularly the earlier born, deep-layer neurons. However, there are differences in the direction of density change (Table 3; Gallagher et al., 2013; Chao et al., 2016; Choi et al., 2016; Wu et al., 2018; Ben-Reuven and Reiner, 2021; Canales et al., 2021), most likely due to minor discrepancies in timing of MIA induction as well as the gestational time point of lamination. The latter theory is supported by a study which administered the thymidine analog, BrdU, daily on E13.75–E16.75 after poly(I:C) on E9.5 and tracked layer density according to cell birth date. Although the deep-layer born neurons were consistently increased between E14.75 and E16.75 in MIA offspring brains, this was not evident at E13.75, and the group which had the most densely populated upper-layer fluctuated between poly(I:C) and control animals depending on embryonic day (Soumiya et al., 2011). This suggests poly(I:C) may affect the timing at which layer-specific neurons are generated (Choi et al., 2016), where there appears to be increased production of deep-layer neurons, later in corticogenesis, when this would be expected to be diminished. A number of studies support the notion of an immature phenotype in poly(I:C) animals during late gestational development, including evidence of overlapped TBR1 and CTIP2 neurons at E20, which are clearly distinguishable in vehicle controls at this time point (Chao et al., 2016), as well as localization of PAX6 and TBR2 cells in the SVZ during late corticogenesis, which would typically only be localized in the VZ at this stage (Tsukada et al., 2021).
Changes to lamination could be explained by the aforementioned acute and premature shift from symmetric to asymmetric divisions in the MIA fetal brain, where more neurons are exiting the cell cycle and are maturing to becoming CTIP2/TBR1 positive (Figure 2). These cells are typically formed at E12.5/E13.5 which coincides with timing of MIA induction of the majority of studies referenced in this review (Maekawa et al., 2005; Osumi et al., 2008; Carpentier et al., 2011; Stolp et al., 2011; Gallagher et al., 2013; Braun et al., 2019; Ben-Reuven and Reiner, 2021; Canales et al., 2021; Tsukada et al., 2021).
5. Causative mechanisms
5.1. Paired box protein pax-6
Overall, the PAX6 positive RGCs are the most affected population of cells within the embryonic brain following immune insult (Table 2), and a number of studies additionally report a change in PAX6 protein expression (De Miranda et al., 2010; Wu et al., 2018; Ben-Reuven and Reiner, 2021; Canales et al., 2021). PAX6 is a highly dose dependent transcription factor, where increased expression favors increased neurogenic divisions, yet knockout studies have also shown that it is essential for progenitor proliferation (Maekawa et al., 2005; Osumi et al., 2008; Sansom et al., 2009; Mi et al., 2018). Hence, the acute increase in PAX6 protein expression or relative intensity, which has been reported in MIA offspring (De Miranda et al., 2010; Ben-Reuven and Reiner, 2021), may be directly linked to the premature switch to neurogenic phenotype. In contrast, studies report decreased PAX6 protein expression 4–5 days following immune insult (Wu et al., 2018; Canales et al., 2021). This could be the result of a compensatory mechanism due to early depletion of the progenitor pool, therefore reversing the effect of MIA, which would support the aforementioned immature lamination at this time point.
Research indicates that PAX6, and other transcriptional regulators of neurogenesis, may be directly controlled by inflammatory mediators which are upregulated in the MIA offspring brain (Loayza et al., 2022), such as microglia (Cunningham et al., 2013) and cytokines (Walter et al., 2011; Borsini et al., 2015). However, a number of studies report no changes to inflammation status within the fetal MIA offspring brain (see Hameete et al., 2021), yet neurogenesis is defective across the prenatal timeline (Tables 1–3). Epigenetic mechanisms are believed to mediate the prolonged effects of MIA on the offspring brain and behavioral phenotypes (Bergdolt and Dunaevsky, 2019), and hence, considering epigenetics is critical for healthy neurogenesis (Albert et al., 2017; Albert and Huttner, 2018), could provide a basis for putative causative mechanisms.
5.2. Epigenetics
Epigenetic modifications, comprising DNA methylation, histone modifications and non-coding RNAs, enable a change in transcriptional response without editing the underlying DNA (Goldberg et al., 2007; Capell and Berger, 2013). As well as playing vital roles in cellular differentiation and other “typical” developmental mechanisms, epigenetic alterations contribute to a wide range of human diseases such as cancer, autoimmune disorders and NDDs (Moosavi and Motevalizadeh Ardekani, 2016). It has therefore been suggested that the epigenome serves as a “molecular bridge,” by which external triggers and stressors can modulate gene transcription and expression, and thereby contribute to an altered phenotype in the exposed individual (Nestler, 2009; Bollati and Baccarelli, 2010; Acevedo et al., 2021; Smeeth et al., 2021). The majority of studies that have researched epigenetic modifications within the MIA model focus on DNA methylation, which is consistently affected within the adult offspring brain (Labouesse et al., 2015; Richetto et al., 2017a,b; Schaafsma et al., 2017; Basil et al., 2018; Weber-Stadlbauer et al., 2021; Woods et al., 2021). However, despite the evidence that neural progenitors display dynamic changes in DNA methylation within the embryonic brain, the function of DNA methylation in neurogenesis has been questioned, where it is expected to play a lesser role in cell fate determination, and may, instead, act as a consequence of histone modifications (Stricker and Götz, 2018; Adam and Harwell, 2020).
Although there are a limited number of studies, there is some evidence that histone acetylation is dysregulated in the offspring brain following immune exposure (Tang et al., 2013; Pujol Lopez et al., 2016; Reisinger et al., 2016; Woods et al., 2021). Histone acetylation actively promotes neurogenesis through a number of histone acetyltransferases, such as the CREB binding protein and p300 co-activator family, which drive the differentiation of NECs by stimulating the transcription of pro-neural genes (Wang et al., 2010; Tsui et al., 2014; Yao and Jin, 2014; Schoof et al., 2019). Hence, a disturbance in this process, where the availability or regulation of these modifications is altered (as reported in the MIA offspring brain; Tang et al., 2013; Pujol Lopez et al., 2016; Reisinger et al., 2016), could provide a mechanism for the pro-neurogenic phenotype exhibited in MIA exposed animals. Nevertheless, as far as we know, no studies have investigated histone acetylation in the fetal MIA brain, which represents a major gap in the literature.
There are a number of hypothesized pathways in which an environmental stressor (such as MIA), may lead to altered epigenetic regulation in the exposed individual. First, regulation of epigenetic enzymes by cytokines has been reported, such as increased expression of DNA methyltransferase 1 by interleukin-6 (Braconi et al., 2010; Villagra et al., 2010), which is of interest following evidence of increased cytokine response in offspring brains acutely following insult (Woods et al., 2021). Second, availability of the substrate used by epigenetic enzymes to generate the modification may be limited. In the case of histone acetylation, acetyl-coenzyme A is utilized by histone acetyltransferases, which is produced following glycolysis in the mitochondria and is an essential intermediate of several metabolic pathways. In fact, mitochondrial biogenesis generates a number of substrates which alter epigenetic enzyme activity, including those responsible for DNA and histone modifications, as well as ATP-dependent chromatin remodelers (Wiese and Bannister, 2020). Recently, a growing body of literature has reported mitochondrial dysfunction in the MIA model (Robicsek et al., 2018; Swanepoel et al., 2018; Cieślik et al., 2020, 2021; Gyllenhammer et al., 2021; Zawadzka et al., 2021), which may provide a mechanistic pathway for altered epigenetic modifications and neurogenesis in the model.
Mitochondrial activity regulates the fate of neural progenitors, where a switch to oxidative phosphorylation from glycolysis, along with increased reactive oxygen species generation, promotes neuronal differentiation (Iwata and Vanderhaeghen, 2021). It is therefore possible that impaired mitochondrial activity, which is often concordant with elevated reactive oxygen species in the MIA model (Swanepoel et al., 2018; Cieślik et al., 2020, 2021), is controlling the acute switch from proliferative to neurogenic divisions in the embryonic brain. It is interesting to note that alternative studies of developmental insult, including mitochondrial dysfunction, intrauterine growth restriction and maternal hyperglycemia, report an almost identical phenotype in embryonic neurogenesis, where increased NECs are differentiating, at the expense of proliferation (Khacho et al., 2017; Ji et al., 2019; Brown et al., 2021). This suggests that disturbed neurogenesis may be the result of a common downstream pathway, such as oxidative stress, which has been linked to a wide range of diseases (Cenini et al., 2019; Forman and Zhang, 2021).
6. Conclusion
Neuronal development within the embryonic brain is clearly affected by MIA as demonstrated by an acute proliferation defect, which, in most cases, is concordant with increased differentiation of neurons and altered cortical lamination (Figure 2; Shi et al., 2009; De Miranda et al., 2010; Carpentier et al., 2011, 2013; Soumiya et al., 2011; Stolp et al., 2011; Chao et al., 2016; Gumusoglu et al., 2017; Wu et al., 2018; Ben-Reuven and Reiner, 2021; Canales et al., 2021).
Disturbed cortical neurogenesis has been linked to neural connectivity deficits in the postnatal offspring as well as behavioral phenotypes associated with ASD, such as reduced ultrasonic vocalizations (Wagner and MacDonald, 2021; Griffin et al., 2022), which has been reported in MIA offspring (Gzielo et al., 2021; Scott et al., 2021; Potter et al., 2023). Hence, the altered embryonic neurogenesis phenotype highlighted in this review may provide an underlying mechanism responsible for MIA-induced dysfunctional behavior. A recent review concluded that MIA causes defective hippocampal neurogenesis in the adult offspring, which is linked to defects in memory, mood and anxiety and is hypothesized to mediate susceptibility to future “hits” (Couch et al., 2021). Generation of the neurogenic niches in the adult brain, known as the SVZ and the subgranular zone, are dependent on the precise mechanisms of embryonic neurogenesis (Nicola et al., 2015; Mira and Morante, 2020). It could thus be postulated that the proliferation defect reported in this review, which remains defective at late gestational time points, is at the root of improper neurogenic niche development within the MIA model, as has been hypothesized following alternative developmental insults (Khacho et al., 2017; Brown et al., 2021). Accordingly, accelerated brain growth and dysregulated expression of genes involved in neurogenesis has been reported within ASD patients (Hazlett et al., 2011; Chow et al., 2012; Fan and Pang, 2017; Shen and Piven, 2017).
Studies of neurogenesis within the fetal MIA brain have mostly focused on the pallium (De Miranda et al., 2010; Gallagher et al., 2013; Braun et al., 2019; Canales et al., 2021), which is the main source of excitatory neurons within the cerebral cortex (Llorca and Deogracias, 2022). Yet, inhibitory interneurons, which are repeatedly reported to be affected following MIA and in NDDs (Nakamura et al., 2021; Yang et al., 2022; Yu et al., 2022), are predominantly derived from sub-pallial regions such as the caudal and medial ganglionic eminences as well as the preoptic area (Yang et al., 2022). To our knowledge, only one study has investigated neurogenesis within sub-pallial regions following MIA induction, which reported decreased expression of proliferation markers within the caudal ganglionic eminence during late gestation (Lacaille et al., 2019). Future studies will help to elucidate whether neurogenesis defects within sub-pallial regions are contributing to GABAergic deficits following MIA.
This review focused on the effect of MIA on embryonic neurogenesis and neuronal migration, which is critical for understanding the underlying mechanisms of MIA as well as establishing how MIA interacts with genetic or subsequent environmental insults to alter the neurodevelopmental trajectory. It is clear that the embryonic neurogenesis phenotype is significantly affected by the timing of MIA induction, consistent with the rapid nature of fetal brain development. Hence, further research is required to better understand how even seemingly minor alterations to the timing of immune insult affect neurogenesis and subsequent brain deficits. Sex-dependent variation should also be explored in order to understand how MIA differentially affects neurogenesis, which may be at the root of sex-specific brain and behavioral phenotypes. Future research in the field of neurogenesis will allow an improved mechanistic understanding of how MIA increases the risk of NDDs and hence will assist in therapeutic discovery.
Author contributions
FM collated the literature, prepared the manuscript, and created the figures and tables. JG and RH supervised the manuscript preparation and suggested edits for the final draft. All authors contributed to the article and approved the submitted version.
Funding
FM was supported by BBSRC grant (BB/T008725/1).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ASD, autism spectrum disorder; CTIP2, B-cell lymphoma/leukemia 11B; CUX1, cut like homeobox 1; E, embryonic day; GABA, γ -aminobutyric acid; IPC, intermediate progenitor cell; LPS, lipopolysaccharide; MIA, maternal immune activation; NANOG, homeobox protein NANOG; NDD, neurodevelopmental disorder; NEC, neuroepithelial cell; OCT4, octamer-binding transcription factor 4; PAX6, paired box protein pax-6; PHH3, phosphohistone 3; poly(I:C), polyinosinic:polycytidylic acid; RGC, radial glial cell; SATB2, special AT-rich sequence-binding protein; SOX2, SRY-box 2; SVZ, subventricular zone; TBR1, T-box brain transcription factor 1; TBR2, T-box brain transcription factor 1; TUJ1, neuron-specific class III beta-tubulin; VZ, ventricular zone.
References
Acevedo, N., Alashkar Alhamwe, B., Caraballo, L., Ding, M., Ferrante, A., Garn, H., et al. (2021). Perinatal and early-life nutrition, epigenetics, and allergy. Nutrients 13:724. doi: 10.3390/nu13030724
Adam, M. A., and Harwell, C. C. (2020). Epigenetic regulation of cortical neurogenesis; orchestrating fate switches at the right time and place. Curr. Opin. Neurobiol. 63, 146–153. doi: 10.1016/j.conb.2020.03.012
Ahmed, S., Gan, H., Lam, C. S., Poonepalli, A., Ramasamy, S., Tay, Y., et al. (2009). Transcription factors and neural stem cell self-renewal, growth and differentiation. Cell Adh. Migr. 3, 412–424. doi: 10.4161/cam.3.4.8803
Albert, M., and Huttner, W. B. (2018). Epigenetic and transcriptional pre-patterning—an emerging theme in cortical neurogenesis. Front. Neurosci. 12:359. doi: 10.3389/fnins.2018.00359
Albert, M., Kalebic, N., Florio, M., Lakshmanaperumal, N., Haffner, C., Brandl, H., et al. (2017). Epigenome profiling and editing of neocortical progenitor cells during development. EMBO J. 36, 2642–2658. doi: 10.15252/embj.201796764
Arai, Y., Pulvers, J. N., Haffner, C., Schilling, B., Nüsslein, I., Calegari, F., et al. (2011). Neural stem and progenitor cells shorten S-phase on commitment to neuron production. Nat. Commun. 2:154. doi: 10.1038/ncomms1155
Ayala, R., Shu, T., and Tsai, L.-H. (2007). Trekking across the brain: The journey of neuronal migration. Cell 128, 29–43. doi: 10.1016/j.cell.2006.12.021
Baines, K. J., Hillier, D. M., Haddad, F. L., Rajakumar, N., Schmid, S., and Renaud, S. J. (2020). Maternal immune activation alters fetal brain development and enhances proliferation of neural precursor cells in rats. Front. Immunol. 11:1145. doi: 10.3389/fimmu.2020.01145
Balu, D. T. (2016). The NMDA receptor and schizophrenia: From pathophysiology to treatment. Adv. Pharmacol. 76, 351–382. doi: 10.1016/bs.apha.2016.01.006
Bao, M., Hofsink, N., and Plösch, T. (2022). LPS versus Poly I:C model: Comparison of long-term effects of bacterial and viral maternal immune activation on the offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 322, R99–R111. doi: 10.1152/ajpregu.00087.2021
Barke, T. L., Money, K. M., Du, L., Serezani, A., Gannon, M., Mirnics, K., et al. (2019). Sex modifies placental gene expression in response to metabolic and inflammatory stress. Placenta 78, 1–9. doi: 10.1016/j.placenta.2019.02.008
Basil, P., Li, Q., Gui, H., Hui, T. C., Ling, V. H., Wong, C. C., et al. (2018). Prenatal immune activation alters the adult neural epigenome but can be partly stabilised by a n-3 polyunsaturated fatty acid diet. Transl. Psychiatry 8:125. doi: 10.1038/s41398-018-0167-x
Batinić, B., Santrač, A., Divović, B., Timić, T., Stanković, T., Obradović, A. L., et al. (2016). Lipopolysaccharide exposure during late embryogenesis results in diminished locomotor activity and amphetamine response in females and spatial cognition impairment in males in adult, but not adolescent rat offspring. Behav. Brain Res. 299, 72–80. doi: 10.1016/j.bbr.2015.11.025
Beattie, R., and Hippenmeyer, S. (2017). Mechanisms of radial glia progenitor cell lineage progression. FEBS Lett. 591, 3993–4008. doi: 10.1002/1873-3468.12906
Ben-Reuven, L., and Reiner, O. (2021). Dynamics of cortical progenitors and production of subcerebral neurons are altered in embryos of a maternal inflammation model for autism. Mol. Psychiatry 26, 1535–1550. doi: 10.1038/s41380-019-0594-y
Bergdolt, L., and Dunaevsky, A. (2019). Brain changes in a maternal immune activation model of neurodevelopmental brain disorders. Prog. Neurobiol. 175, 1–19. doi: 10.1016/j.pneurobio.2018.12.002
Bojesen, K. B., Broberg, B. V., Fagerlund, B., Jessen, K., Thomas, M. B., Sigvard, A., et al. (2021). Associations between cognitive function and levels of glutamatergic metabolites and gamma-aminobutyric acid in antipsychotic-naïve patients with schizophrenia or psychosis. Biol. Psychiatry 89, 278–287. doi: 10.1016/j.biopsych.2020.06.027
Bollati, V., and Baccarelli, A. (2010). Environmental epigenetics. Heredity 105, 105–112. doi: 10.1038/hdy.2010.2
Borsini, A., Zunszain, P. A., Thuret, S., and Pariante, C. M. (2015). The role of inflammatory cytokines as key modulators of neurogenesis. Trends Neurosci. 38, 145–157. doi: 10.1016/j.tins.2014.12.006
Braconi, C., Huang, N., and Patel, T. (2010). MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology 51, 881–890. doi: 10.1002/hep.23381
Braun, A. E., Carpentier, P. A., Babineau, B. A., Narayan, A. R., Kielhold, M. L., Moon, H. M., et al. (2019). “Females are not just ‘protected’ males”: Sex-specific vulnerabilities in placenta and brain after prenatal immune disruption. eNeuro 6:ENEURO.0358-19.2019. doi: 10.1523/ENEURO.0358-19.2019
Brown, A. S. (2006). Prenatal infection as a risk factor for schizophrenia. Schizophr. Bull. 32, 200–202. doi: 10.1093/schbul/sbj052
Brown, A. S., Wieben, M., Murdock, S., Chang, J., Dizon, M. L., Pierre, M. S., et al. (2021). Intrauterine growth restriction causes abnormal embryonic dentate gyrus neurogenesis in mouse offspring that leads to adult learning and memory deficits. eNeuro 8:ENEURO.0062-21.2021. doi: 10.1523/ENEURO.0062-21.2021
Buchsbaum, I. Y., and Cappello, S. (2019). Neuronal migration in the CNS during development and disease: Insights from in vivo and in vitro models. Development 146:dev163766. doi: 10.1242/dev.163766
Canales, C. P., Estes, M. L., Cichewicz, K., Angara, K., Aboubechara, J. P., Cameron, S., et al. (2021). Sequential perturbations to mouse corticogenesis following in utero maternal immune activation. eLife 10:e60100. doi: 10.7554/eLife.60100
Canetta, S., Bolkan, S., Padilla-Coreano, N., Song, L., Sahn, R., Harrison, N., et al. (2016). Maternal immune activation leads to selective functional deficits in offspring parvalbumin interneurons. Mol. Psychiatry 21, 956–968. doi: 10.1038/mp.2015.222
Capell, B. C., and Berger, S. L. (2013). Genome-wide epigenetics. J. Invest. Dermatol. 133:E9. doi: 10.1038/jid.2013.173
Carpentier, P. A., Dingman, A. L., and Palmer, T. D. (2011). Placental TNF-α signaling in illness-induced complications of pregnancy. Am. J. Clin. Pathol. 178, 2802–2810. doi: 10.1016/j.ajpath.2011.02.042
Carpentier, P. A., Haditsch, U., Braun, A. E., Cantu, A. V., Moon, H. M., Price, R. O., et al. (2013). Stereotypical alterations in cortical patterning are associated with maternal illness-induced placental dysfunction. J. Neurosci. 33, 16874–16888. doi: 10.1523/JNEUROSCI.4654-12.2013
Cenini, G., Lloret, A., and Cascella, R. (2019). Oxidative stress in neurodegenerative diseases: From a mitochondrial point of view. Oxid. Med. Cell. Longev. 2019:2105607. doi: 10.1155/2019/2105607
Chao, M.-W., Chen, C.-P., Yang, Y.-H., Chuang, Y.-C., Chu, T.-Y., and Tseng, C.-Y. (2016). N-acetylcysteine attenuates lipopolysaccharide-induced impairment in lamination of Ctip2-and Tbr1-expressing cortical neurons in the developing rat fetal brain. Sci. Rep. 6:32373. doi: 10.1038/srep32373
Choi, G. B., Yim, Y. S., Wong, H., Kim, S., Kim, H., Kim, S. V., et al. (2016). The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939. doi: 10.1126/science.aad0314
Chow, M. L., Pramparo, T., Winn, M. E., Barnes, C. C., Li, H.-R., Weiss, L., et al. (2012). Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet. 8:e1002592. doi: 10.1371/journal.pgen.1002592
Cieślik, M., Gąssowska-Dobrowolska, M., Jêśko, H., Czapski, G. A., Wilkaniec, A., Zawadzka, A., et al. (2020). Maternal immune activation induces neuroinflammation and cortical synaptic deficits in the adolescent rat offspring. Int. J. Mol. Sci. 21:4097. doi: 10.3390/ijms21114097
Cieślik, M., Gassowska-Dobrowolska, M., Zawadzka, A., Frontczak-Baniewicz, M., Gewartowska, M., Dominiak, A., et al. (2021). The synaptic dysregulation in adolescent rats exposed to maternal immune activation. Front. Mol. Neurosci. 13:555290. doi: 10.3389/fnmol.2020.555290
Coiro, P., and Pollak, D. D. (2019). Sex and gender bias in the experimental neurosciences: The case of the maternal immune activation model. Transl. Psychiatry 9:90. doi: 10.1038/s41398-019-0423-8
Cooper, J. A. (2013). Mechanisms of cell migration in the nervous system. J. Cell Biol. 202, 725–734. doi: 10.1083/jcb.201305021
Couch, A. C., Berger, T., Hanger, B., Matuleviciute, R., Srivastava, D. P., Thuret, S., et al. (2021). Maternal immune activation primes deficiencies in adult hippocampal neurogenesis. Brain Behav. Immun. 97, 410–422. doi: 10.1016/j.bbi.2021.07.021
Coyle, J. T., Balu, D., Benneyworth, M., Basu, A., and Roseman, A. (2010). Beyond the dopamine receptor: Novel therapeutic targets for treating schizophrenia. Dialogues Clin. Neurosci. 12, 359–382. doi: 10.31887/DCNS.2010.12.3/jcoyle
Crawley, J. N. (2012). Translational animal models of autism and neurodevelopmental disorders. Dialogues Clin. Neurosci. 14, 293–305. doi: 10.31887/DCNS.2012.14.3/jcrawley
Cui, K., Ashdown, H., Luheshi, G. N., and Boksa, P. (2009). Effects of prenatal immune activation on hippocampal neurogenesis in the rat. Schizophr. Res. 113, 288–297. doi: 10.1016/j.schres.2009.05.003
Cunningham, C. L., Martínez-Cerdeño, V., and Noctor, S. C. (2013). Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 33, 4216–4233. doi: 10.1523/JNEUROSCI.3441-12.2013
De Felice, A., Ricceri, L., Venerosi, A., Chiarotti, F., and Calamandrei, G. (2015). Multifactorial origin of neurodevelopmental disorders: Approaches to understanding complex etiologies. Toxics 3, 89–129. doi: 10.3390/toxics3010089
De Miranda, J., Yaddanapudi, K., Hornig, M., Villar, G., Serge, R., and Lipkin, W. I. (2010). Induction of Toll-like receptor 3-mediated immunity during gestation inhibits cortical neurogenesis and causes behavioral disturbances. mBio 1:e00176-10. doi: 10.1128/mBio.00176-10
Dehorter, N., and Del Pino, I. (2020). Shifting developmental trajectories during critical periods of brain formation. Front. Cell. Neurosci. 10:283. doi: 10.3389/fncel.2020.00283
Dennis, D., Picketts, D., Slack, R. S., and Schuurmans, C. (2016). Forebrain neurogenesis: From embryo to adult. Trends Dev. Biol. 9, 77–90.
Desai, D., and Pethe, P. (2020). Polycomb repressive complex 1: Regulators of neurogenesis from embryonic to adult stage. J. Cell. Physiol. 235, 4031–4045. doi: 10.1002/jcp.29299
Duan, D., Fu, Y., Paxinos, G., and Watson, C. (2013). Spatiotemporal expression patterns of Pax6 in the brain of embryonic, newborn, and adult mice. Brain Struct. Funct. 218, 353–372. doi: 10.1007/s00429-012-0397-2
Egger, B., Gold, K. S., and Brand, A. H. (2011). Regulating the balance between symmetric and asymmetric stem cell division in the developing brain. Fly 5, 237–241. doi: 10.4161/fly.5.3.15640
Englund, C., Fink, A., Lau, C., Pham, D., Daza, R. A., Bulfone, A., et al. (2005). Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J. Neurosci. 25, 247–251. doi: 10.1523/JNEUROSCI.2899-04.2005
Ernst, C. (2016). Proliferation and differentiation deficits are a major convergence point for neurodevelopmental disorders. Trends Neurosci. 39, 290–299. doi: 10.1016/j.tins.2016.03.001
Estes, M. L., and McAllister, A. K. (2016). Maternal immune activation: Implications for neuropsychiatric disorders. Science 353, 772–777. doi: 10.1126/science.aag3194
Fan, L.-W., and Pang, Y. (2017). Dysregulation of neurogenesis by neuroinflammation: Key differences in neurodevelopmental and neurological disorders. Neural Regen. Res. 12, 366–371. doi: 10.4103/1673-5374.202926
Feigenson, K. A., Kusnecov, A. W., and Silverstein, S. M. (2014). Inflammation and the two-hit hypothesis of schizophrenia. Neurosci. Biobehav. Rev. 38, 72–93. doi: 10.1016/j.neubiorev.2013.11.006
Forman, H. J., and Zhang, H. (2021). Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 20, 689–709. doi: 10.1038/s41573-021-00233-1
Fusar-Poli, P., Papanastasiou, E., Stahl, D., Rocchetti, M., Carpenter, W., Shergill, S., et al. (2015). Treatments of negative symptoms in schizophrenia: Meta-analysis of 168 randomized placebo-controlled trials. Schizophr. Bull. 41, 892–899. doi: 10.1093/schbul/sbu170
Gallagher, D., Norman, A. A., Woodard, C. L., Yang, G., Gauthier-Fisher, A., Fujitani, M., et al. (2013). Transient maternal IL-6 mediates long-lasting changes in neural stem cell pools by deregulating an endogenous self-renewal pathway. Cell Stem Cell 13, 564–576. doi: 10.1016/j.stem.2013.10.002
Garbett, K. A., Hsiao, E. Y., Kálmán, S., Patterson, P. H., and Mirnics, K. (2012). Effects of maternal immune activation on gene expression patterns in the fetal brain. Transl. Psychiatry 2:E98. doi: 10.1038/tp.2012.24
Gogos, A., Sbisa, A., Witkamp, D., and Van Den Buuse, M. (2020). Sex differences in the effect of maternal immune activation on cognitive and psychosis-like behaviour in long evans rats. Eur. J. Neurosci. 52, 2614–2626. doi: 10.1111/ejn.14671
Goldberg, A. D., Allis, C. D., and Bernstein, E. (2007). Epigenetics: A landscape takes shape. Cell 128, 635–638. doi: 10.1016/j.cell.2007.02.006
Gonzalez-Burgos, G., Cho, R. Y., and Lewis, D. A. (2015). Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol. Psychiatry 77, 1031–1040. doi: 10.1016/j.biopsych.2015.03.010
Götz, M., and Huttner, W. B. (2005). The cell biology of neurogenesis. Nat. Rev. Mol. 6, 777–788. doi: 10.1038/nrm1739
Gray, A., Tattoli, R., Dunn, A., Hodgson, D., Michie, P., and Harms, L. (2019). Maternal immune activation in mid-late gestation alters amphetamine sensitivity and object recognition, but not other schizophrenia-related behaviours in adult rats. Behav. Brain Res. 356, 358–364. doi: 10.1016/j.bbr.2018.08.016
Griffin, A., Mahesh, A., and Tiwari, V. K. (2022). Disruption of the gene regulatory programme in neurodevelopmental disorders. Biochim. Biophys. Acta Gene Regul. Mech. 1865:194860. doi: 10.1016/j.bbagrm.2022.194860
Guarnieri, F. C., De Chevigny, A., Falace, A., and Cardoso, C. (2019). Disorders of neurogenesis and cortical development. Dialogues Clin. Neurosci. 20, 255–266. doi: 10.31887/DCNS.2018.20.4/ccardoso
Guma, E., Bordeleau, M., González Ibáñez, F., Picard, K., Snook, E., Desrosiers-Grégoire, G., et al. (2022). Differential effects of early or late exposure to prenatal maternal immune activation on mouse embryonic neurodevelopment. Proc. Natl. Acad. Sci. U.S.A. 119:e2114545119. doi: 10.1073/pnas.2114545119
Gumusoglu, S. B., Fine, R. S., Murray, S. J., Bittle, J. L., and Stevens, H. E. (2017). The role of IL-6 in neurodevelopment after prenatal stress. Brain Behav. Immun. 65, 274–283. doi: 10.1016/j.bbi.2017.05.015
Guo, Z., Chen, M., Chao, Y., Cai, C., Liu, L., Zhao, L., et al. (2021). RGCC balances self-renewal and neuronal differentiation of neural stem cells in the developing mammalian neocortex. EMBO Rep. 22:e51781. doi: 10.15252/embr.202051781
Gyllenhammer, L. E., Rasmussen, J. M., Bertele, N., Halbing, A., Entringer, S., Wadhwa, P. D., et al. (2021). Maternal inflammation during pregnancy and offspring brain development: The role of mitochondria. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 7, 498–509. doi: 10.1016/j.bpsc.2021.11.003
Gzielo, K., Potasiewicz, A., Litwa, E., Piotrowska, D., Popik, P., and Nikiforuk, A. (2021). The effect of maternal immune activation on social play-induced ultrasonic vocalization in rats. Brain Sci. 11:344. doi: 10.3390/brainsci11030344
Haddad, F. L., Patel, S. V., and Schmid, S. (2020). Maternal immune activation by Poly I:C as a preclinical model for neurodevelopmental disorders: A focus on autism and schizophrenia. Neurosci. Biobehav. Rev. 113, 546–567. doi: 10.1016/j.neubiorev.2020.04.012
Haida, O., Al Sagheer, T., Balbous, A., Francheteau, M., Matas, E., Soria, F., et al. (2019). Sex-dependent behavioral deficits and neuropathology in a maternal immune activation model of autism. Transl. Psychiatry 9:124. doi: 10.1038/s41398-019-0457-y
Hameete, B. C., Fernández-Calleja, J. M., De Groot, M. W., Oppewal, T. R., Tiemessen, M. M., Hogenkamp, A., et al. (2021). The poly(I:C)-induced maternal immune activation model; a systematic review and meta-analysis of cytokine levels in the offspring. Brain Behav. Immun. Health 11:100192. doi: 10.1016/j.bbih.2020.100192
Hao, K., Su, X., Luo, B., Cai, Y., Chen, T., Yang, Y., et al. (2019). Prenatal immune activation induces age-related alterations in rat offspring: Effects upon NMDA receptors and behaviors. Behav. Brain Res. 370:111946. doi: 10.1016/j.bbr.2019.111946
Hardwick, L. J., Ali, F. R., Azzarelli, R., and Philpott, A. (2015). Cell cycle regulation of proliferation versus differentiation in the central nervous system. Cell Tissue Res. 359, 187–200. doi: 10.1007/s00441-014-1895-8
Harris, K. D., and Shepherd, G. M. (2015). The neocortical circuit: Themes and variations. Nat. Neurosci. 18, 170–181. doi: 10.1038/nn.3917
Hazlett, H. C., Poe, M. D., Gerig, G., Styner, M., Chappell, C., Smith, R. G., et al. (2011). Early brain overgrowth in autism associated with an increase in cortical surface area before age 2 years. Arch. Gen. Psychiatry 68, 467–476. doi: 10.1001/archgenpsychiatry.2011.39
Hodes, G. E., and Epperson, C. N. (2019). Sex differences in vulnerability and resilience to stress across the life span. Biol. Psychiatry 86, 421–432. doi: 10.1016/j.biopsych.2019.04.028
Iwata, R., and Vanderhaeghen, P. (2021). Regulatory roles of mitochondria and metabolism in neurogenesis. Curr. Opin. Neurobiol. 69, 231–240. doi: 10.1016/j.conb.2021.05.003
Jacobsen, H., Walendy-Gnirß, K., Tekin-Bubenheim, N., Kouassi, N. M., Ben-Batalla, I., Berenbrok, N., et al. (2021). Offspring born to influenza A virus infected pregnant mice have increased susceptibility to viral and bacterial infections in early life. Nat. Commun. 12:4957. doi: 10.1038/s41467-021-25220-3
Ji, S., Zhou, W., Li, X., Liu, S., Wang, F., Li, X., et al. (2019). Maternal hyperglycemia disturbs neocortical neurogenesis via epigenetic regulation in C57BL/6J mice. Cell Death Dis. 10:211. doi: 10.1038/s41419-019-1438-z
Khacho, M., Clark, A., Svoboda, D. S., Maclaurin, J. G., Lagace, D. C., Park, D. S., et al. (2017). Mitochondrial dysfunction underlies cognitive defects as a result of neural stem cell depletion and impaired neurogenesis. Hum. Mol. Genet. 26, 3327–3341. doi: 10.1093/hmg/ddx217
Khodosevich, K., and Monyer, H. (2011). Signaling in migrating neurons: From molecules to networks. Front. Neurosci. 5:28. doi: 10.3389/fnins.2011.00028
Kim, J.-Y., Jeong, H. S., Chung, T., Kim, M., Lee, J. H., Jung, W. H., et al. (2017). The value of phosphohistone H3 as a proliferation marker for evaluating invasive breast cancers: A comparative study with Ki67. Oncotarget 8, 65064–65076. doi: 10.18632/oncotarget.17775
Kim, M. S., Kim, D.-H., Kang, H. K., Kook, M. G., Choi, S. W., and Kang, K.-S. (2021). Modeling of hypoxic brain injury through 3D human neural organoids. Cells 10:234. doi: 10.3390/cells10020234
Kinney, D. K., Hintz, K., Shearer, E. M., Barch, D. H., Riffin, C., Whitley, K., et al. (2010). A unifying hypothesis of schizophrenia: Abnormal immune system development may help explain roles of prenatal hazards, post-pubertal onset, stress, genes, climate, infections, and brain dysfunction. Med. Hypotheses 74, 555–563. doi: 10.1016/j.mehy.2009.09.040
Kostović, I., and Judaš, M. (2007). Transient patterns of cortical lamination during prenatal life: Do they have implications for treatment? Neurosci. Biobehav. Rev. 31, 1157–1168. doi: 10.1016/j.neubiorev.2007.04.018
Kowalczyk, T., Pontious, A., Englund, C., Daza, R. A., Bedogni, F., Hodge, R., et al. (2009). Intermediate neuronal progenitors (basal progenitors) produce pyramidal–projection neurons for all layers of cerebral cortex. Cereb. Cortex 19, 2439–2450. doi: 10.1093/cercor/bhn260
Kowash, H., Potter, H., Edye, M., Prinssen, E., Bandinelli, S., Neill, J., et al. (2019). Poly(I:C) source, molecular weight and endotoxin contamination affect dam and prenatal outcomes, implications for models of maternal immune activation. Brain Behav. Immun. 82, 160–166. doi: 10.1016/j.bbi.2019.08.006
Kumar, V., Vajawat, B., and Rao, N. P. (2021). Frontal GABA in schizophrenia: A meta-analysis of 1H-MRS studies. World J. Biol. Psychiatry 22, 1–13. doi: 10.1080/15622975.2020.1731925
Labouesse, M. A., Dong, E., Grayson, D. R., Guidotti, A., and Meyer, U. (2015). Maternal immune activation induces GAD1 and GAD2 promoter remodeling in the offspring prefrontal cortex. Epigenetics 10, 1143–1155. doi: 10.1080/15592294.2015.1114202
Lacaille, H., Vacher, C.-M., Bakalar, D., O’Reilly, J. J., Salzbank, J., and Penn, A. A. (2019). Impaired interneuron development in a novel model of neonatal brain injury. eNeuro 6:ENEURO.0300-18.2019. doi: 10.1523/ENEURO.0300-18.2019
Lancaster, M. A., and Knoblich, J. A. (2012). Spindle orientation in mammalian cerebral cortical development. Curr. Opin. Neurobiol. 22, 737–746. doi: 10.1016/j.conb.2012.04.003
Llorca, A., and Deogracias, R. (2022). Origin, development, and synaptogenesis of cortical interneurons. Front. Neurosci. 16:929469. doi: 10.3389/fnins.2022.929469
Loayza, M., Lin, S., Carter, K., Ojeda, N., Fan, L.-W., Ramarao, S., et al. (2022). Maternal immune activation alters fetal and neonatal microglia phenotype and disrupts neurogenesis in mice. Pediatr. Res. doi: 10.1038/s41390-022-02239-w [Epub ahead of print].
Lodato, S., and Arlotta, P. (2015). Generating neuronal diversity in the mammalian cerebral cortex. Annu. Rev. Cell. Dev. Biol. 31, 699–720. doi: 10.1146/annurev-cellbio-100814-125353
Lombardo, M. V., Moon, H. M., Su, J., Palmer, T. D., Courchesne, E., and Pramparo, T. (2018). Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol. Psychiatry 23, 1001–1013. doi: 10.1038/mp.2017.15
Long, D., Liu, M., Li, H., Song, J., Jiang, X., Wang, G., et al. (2021). Dysbacteriosis induces abnormal neurogenesis via LPS in a pathway requiring NF-κB/IL-6. Pharmacol. Res. 167:105543. doi: 10.1016/j.phrs.2021.105543
Lorusso, J. M., Woods, R. M., Mcewan, F., Glazier, J. D., Neill, J. C., Harte, M., et al. (2022). Clustering of cognitive phenotypes identifies susceptible and resilient offspring in a rat model of maternal immune activation and early-life stress. Brain Behav. Immun. Health 25:100514. doi: 10.1016/j.bbih.2022.100514
MacDowell, K. S., Munarriz-Cuezva, E., Caso, J. R., Madrigal, J. L., Zabala, A., Meana, J. J., et al. (2017). Paliperidone reverts toll-like receptor 3 signaling pathway activation and cognitive deficits in a maternal immune activation mouse model of schizophrenia. Neuropharmacology 116, 196–207. doi: 10.1016/j.neuropharm.2016.12.025
Maekawa, M., Takashima, N., Arai, Y., Nomura, T., Inokuchi, K., Yuasa, S., et al. (2005). Pax6 is required for production and maintenance of progenitor cells in postnatal hippocampal neurogenesis. Genes Cells 10, 1001–1014. doi: 10.1111/j.1365-2443.2005.00893.x
Manuel, M. N., Mi, D., Mason, J. O., and Price, D. J. (2015). Regulation of cerebral cortical neurogenesis by the Pax6 transcription factor. Front. Cell. Neurosci. 9:70. doi: 10.3389/fncel.2015.00070
Marín, O. (2015). “Tangential migration in the telencephalon,” in The rat nervous system, ed. J. Travers (Amsterdam: Elsevier). doi: 10.1016/B978-0-12-374245-2.00003-6
May, T., Adesina, I., Mcgillivray, J., and Rinehart, N. J. (2019). Sex differences in neurodevelopmental disorders. Curr. Opin. Neurol. 32, 622–626. doi: 10.1097/WCO.0000000000000714
McCutcheon, R. A., Krystal, J. H., and Howes, O. D. (2020). Dopamine and glutamate in schizophrenia: Biology, symptoms and treatment. World Psychiatry 19, 15–33. doi: 10.1002/wps.20693
Mednick, S. A., Machon, R. A., Huttunen, M. O., and Bonett, D. (1988). Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch. Gen. Psychiatry 45, 189–192. doi: 10.1001/archpsyc.1988.01800260109013
Meyer, U. (2014). Prenatal poly(I:C) exposure and other developmental immune activation models in rodent systems. Biol. Psychiatry 75, 307–315. doi: 10.1016/j.biopsych.2013.07.011
Meyer, U., Nyffeler, M., Engler, A., Urwyler, A., Schedlowski, M., Knuesel, I., et al. (2006). The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J. Neurosci. 26, 4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006
Mi, D., Manuel, M., Huang, Y.-T., Mason, J. O., and Price, D. J. (2018). Pax6 lengthens G1 phase and decreases oscillating Cdk6 levels in murine embryonic cortical progenitors. Front. Cell. Neurosci. 12:419. doi: 10.3389/fncel.2018.00419
Mira, H., and Morante, J. (2020). Neurogenesis from embryo to adult–lessons from flies and mice. Front. Cell. Dev. Biol. 8:533. doi: 10.3389/fcell.2020.00533
Molyneaux, B. J., Arlotta, P., Menezes, J. R., and Macklis, J. D. (2007). Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 8, 427–437. doi: 10.1038/nrn2151
Moosavi, A., and Motevalizadeh Ardekani, A. (2016). Role of epigenetics in biology and human diseases. Iran. Biomed. J. 20, 246–258. doi: 10.22045/ibj.2016.01
Nakamura, J., Schroeder, A., Gibbons, A., Sundram, S., and Hill, R. (2022). Timing of maternal immune activation and sex influence schizophrenia-relevant cognitive constructs and neuregulin and GABAergic pathways. Brain Behav. Immun. 100, 70–82. doi: 10.1016/j.bbi.2021.11.006
Nakamura, J. P., Gillespie, B., Gibbons, A., Jaehne, E. J., Du, X., Chan, A., et al. (2021). Maternal immune activation targeted to a window of parvalbumin interneuron development improves spatial working memory: Implications for autism. Brain Behav. Immun. 91, 339–349. doi: 10.1016/j.bbi.2020.10.012
Nakamura, J. P., Schroeder, A., Hudson, M., Jones, N., Gillespie, B., Du, X., et al. (2019). The maternal immune activation model uncovers a role for the Arx gene in GABAergic dysfunction in schizophrenia. Brain Behav. Immun. 81, 161–171. doi: 10.1016/j.bbi.2019.06.009
Nestler, E. J. (2009). Epigenetic mechanisms in psychiatry. Biol. Psychiatry 65, 189–190. doi: 10.1016/j.biopsych.2008.10.030
Nicola, Z., Fabel, K., and Kempermann, G. (2015). Development of the adult neurogenic niche in the hippocampus of mice. Front. Neuroanat. 9:53. doi: 10.3389/fnana.2015.00053
Nugent, B. M., O’donnell, C. M., Epperson, C. N., and Bale, T. L. (2018). Placental H3K27me3 establishes female resilience to prenatal insults. Nat. Commun. 9:2555. doi: 10.1038/s41467-018-04992-1
Ohnuma, S.-I., and Harris, W. A. (2003). Neurogenesis and the cell cycle. Neuron 40, 199–208. doi: 10.1016/S0896-6273(03)00632-9
Oskvig, D. B., Elkahloun, A. G., Johnson, K. R., Phillips, T. M., and Herkenham, M. (2012). Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behav. Immun. 26, 623–634. doi: 10.1016/j.bbi.2012.01.015
Osumi, N., Shinohara, H., Numayama-Tsuruta, K., and Maekawa, M. (2008). Concise review: Pax6 transcription factor contributes to both embryonic and adult neurogenesis as a multifunctional regulator. Stem Cells 26, 1663–1672. doi: 10.1634/stemcells.2007-0884
Palmer, N., Beam, A., Agniel, D., Eran, A., Manrai, A., Spettell, C., et al. (2017). Association of sex with recurrence of autism spectrum disorder among siblings. JAMA Pediatr. 171, 1107–1112. doi: 10.1001/jamapediatrics.2017.2832
Patterson, P. H. (2002). Maternal infection: Window on neuroimmune interactions in fetal brain development and mental illness. Curr. Opin. Neurobiol. 12, 115–118. doi: 10.1016/S0959-4388(02)00299-4
Perez-Garcia, C. G., and O’Leary, D. D. (2016). Formation of the cortical subventricular zone requires MDGA1-mediated aggregation of basal progenitors. Cell Rep. 14, 560–571. doi: 10.1016/j.celrep.2015.12.066
Potter, H. G., Kowash, H. M., Woods, R. M., Revill, G., Grime, A., Deeney, B., et al. (2023). Maternal behaviours and adult offspring behavioural deficits are predicted by maternal TNFα concentration in a rat model of neurodevelopmental disorders. Brain Behav. Immun. 8, 162–175. doi: 10.1016/j.bbi.2022.12.003
Pujol Lopez, Y., Kenis, G., Stettinger, W., Neumeier, K., De Jonge, S., Steinbusch, H. W., et al. (2016). Effects of prenatal Poly I:C exposure on global histone deacetylase (HDAC) and DNA methyltransferase (DNMT) activity in the mouse brain. Mol. Biol. Rep. 43, 711–717. doi: 10.1007/s11033-016-4006-y
Rahman, M. R., Petralia, M. C., Ciurleo, R., Bramanti, A., Fagone, P., Shahjaman, M., et al. (2020). Comprehensive analysis of RNA-seq gene expression profiling of brain transcriptomes reveals novel genes, regulators, and pathways in autism spectrum disorder. Brain Sci. 10:747. doi: 10.3390/brainsci10100747
Rahman, T., Zavitsanou, K., Purves-Tyson, T., Harms, L. R., Meehan, C., Schall, U., et al. (2017). Effects of immune activation during early or late gestation on N-methyl-d-aspartate receptor measures in adult rat offspring. Front. Psychiatry 8:77. doi: 10.3389/fpsyt.2017.00077
Redrobe, J. P., Bull, S., and Plath, N. (2010). Translational aspects of the novel object recognition task in rats abstinent following sub-chronic treatment with phencyclidine (PCP): Effects of modafinil and relevance to cognitive deficits in schizophrenia. Front. Psychiatry 1:146. doi: 10.3389/fpsyt.2010.00146
Reisinger, S. N., Kong, E., Khan, D., Schulz, S., Ronovsky, M., Berger, S., et al. (2016). Maternal immune activation epigenetically regulates hippocampal serotonin transporter levels. Neurobiol. Stress 4, 34–43. doi: 10.1016/j.ynstr.2016.02.007
Richetto, J., Chesters, R., Cattaneo, A., Labouesse, M. A., Gutierrez, A. M. C., Wood, T. C., et al. (2017a). Genome-wide transcriptional profiling and structural magnetic resonance imaging in the maternal immune activation model of neurodevelopmental disorders. Cereb. Cortex 27, 3397–3413. doi: 10.1093/cercor/bhw320
Richetto, J., Massart, R., Weber-Stadlbauer, U., Szyf, M., Riva, M. A., and Meyer, U. (2017b). Genome-wide DNA methylation changes in a mouse model of infection-mediated neurodevelopmental disorders. Biol. Psychiatry 81, 265–276. doi: 10.1016/j.biopsych.2016.08.010
Robicsek, O., Ene, H. M., Karry, R., Ytzhaki, O., Asor, E., Mcphie, D., et al. (2018). Isolated mitochondria transfer improves neuronal differentiation of schizophrenia-derived induced pluripotent stem cells and rescues deficits in a rat model of the disorder. Schizophr. Bull. 44, 432–442. doi: 10.1093/schbul/sbx077
Salomoni, P., and Calegari, F. (2010). Cell cycle control of mammalian neural stem cells: Putting a speed limit on G1. Trends Cell Biol. 20, 233–243. doi: 10.1016/j.tcb.2010.01.006
Sansom, S. N., Griffiths, D. S., Faedo, A., Kleinjan, D.-J., Ruan, Y., Smith, J., et al. (2009). The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet. 5:e1000511. doi: 10.1371/journal.pgen.1000511
Savanthrapadian, S., Wolff, A. R., Logan, B. J., Eckert, M. J., Bilkey, D. K., and Abraham, W. C. (2013). Enhanced hippocampal neuronal excitability and LTP persistence associated with reduced behavioral flexibility in the maternal immune activation model of schizophrenia. Hippocampus 23, 1395–1409. doi: 10.1002/hipo.22193
Schaafsma, S. M., Gagnidze, K., Reyes, A., Norstedt, N., Månsson, K., Francis, K., et al. (2017). Sex-specific gene–environment interactions underlying ASD-like behaviors. Proc. Natl. Acad. Sci. U.S.A. 114, 1383–1388. doi: 10.1073/pnas.1619312114
Schoof, M., Launspach, M., Holdhof, D., Nguyen, L., Engel, V., Filser, S., et al. (2019). The transcriptional coactivator and histone acetyltransferase CBP regulates neural precursor cell development and migration. Acta Neuropathol. Commun. 7:199. doi: 10.1186/s40478-019-0849-5
Schoonover, K. E., Dienel, S. J., and Lewis, D. A. (2020). Prefrontal cortical alterations of glutamate and GABA neurotransmission in schizophrenia: Insights for rational biomarker development. Biomark. Neuropsychiatry 3:100015. doi: 10.1016/j.bionps.2020.100015
Scott, K. J., Tashakori-Sabzevar, F., and Bilkey, D. K. (2021). Maternal immune activation alters the sequential structure of ultrasonic communications in male rats. Brain Behav. Immun. Health 16:100304. doi: 10.1016/j.bbih.2021.100304
Selemon, L., and Zecevic, N. (2015). Schizophrenia: A tale of two critical periods for prefrontal cortical development. Transl. Psychiatry 5:e623. doi: 10.1038/tp.2015.115
Semple, B. D., Blomgren, K., Gimlin, K., Ferriero, D. M., and Noble-Haeusslein, L. J. (2013). Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 106, 1–16. doi: 10.1016/j.pneurobio.2013.04.001
Sham, P. C., O’Callaghan, E., Takei, N., Murray, G. K., Hare, E. H., and Murray, R. M. (1992). Schizophrenia following pre-natal exposure to influenza epidemics between 1939 and 1960. Br. J. Psychiatry 160, 461–466. doi: 10.1192/bjp.160.4.461
Shen, M. D., and Piven, J. (2017). Brain and behavior development in autism from birth through infancy. Dialogues Clin. Neurosci. 19, 325–333. doi: 10.31887/DCNS.2017.19.4/mshen
Shi, L., Smith, S. E., Malkova, N., Tse, D., Su, Y., and Patterson, P. H. (2009). Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav. Immun. 23, 116–123. doi: 10.1016/j.bbi.2008.07.012
Shi, L., Tu, N., and Patterson, P. H. (2005). Maternal influenza infection is likely to alter fetal brain development indirectly: The virus is not detected in the fetus. Int. J. Dev. Neurosci. 23, 299–305. doi: 10.1016/j.ijdevneu.2004.05.005
Silverman, J. L., Thurm, A., Ethridge, S. B., Soller, M. M., Petkova, S. P., Abel, T., et al. (2022). Reconsidering animal models used to study autism spectrum disorder: Current state and optimizing future. Genes Brain Behav. 21:e12803. doi: 10.1111/gbb.12803
Smart, I. H., Dehay, C., Giroud, P., Berland, M., and Kennedy, H. (2002). Unique morphological features of the proliferative zones and postmitotic compartments of the neural epithelium giving rise to striate and extrastriate cortex in the monkey. Cereb. Cortex 12, 37–53. doi: 10.1093/cercor/12.1.37
Smeeth, D., Beck, S., Karam, E. G., and Pluess, M. (2021). The role of epigenetics in psychological resilience. Lancet Psychiatry 8, 620–629. doi: 10.1016/S2215-0366(20)30515-0
Solius, G. M., Maltsev, D. I., Belousov, V. V., and Podgorny, O. V. (2021). Recent advances in nucleotide analogue-based techniques for tracking dividing stem cells: An overview. J. Biol. Chem. 297:101345. doi: 10.1016/j.jbc.2021.101345
Sommer, L., and Rao, M. (2002). Neural stem cells and regulation of cell number. Prog. Neurobiol. 66, 1–18. doi: 10.1016/S0301-0082(01)00022-3
Soumiya, H., Fukumitsu, H., and Furukawa, S. (2011). Prenatal immune challenge compromises the normal course of neurogenesis during development of the mouse cerebral cortex. J. Neurosci. Res. 89, 1575–1585. doi: 10.1002/jnr.22704
Stahl, S. M. (2018). Beyond the dopamine hypothesis of schizophrenia to three neural networks of psychosis: Dopamine, serotonin, and glutamate. CNS Spectr. 23, 187–191. doi: 10.1017/S1092852918001013
Stevanovic, M., Drakulic, D., Lazic, A., Ninkovic, D. S., Schwirtlich, M., and Mojsin, M. (2021). SOX transcription factors as important regulators of neuronal and glial differentiation during nervous system development and adult neurogenesis. Front. Mol. Neurosci. 14:654031. doi: 10.3389/fnmol.2021.654031
Stiles, J., and Jernigan, T. L. (2010). The basics of brain development. Neuropsychol. Rev. 20, 327–348. doi: 10.1007/s11065-010-9148-4
Stolp, H. B., Turnquist, C., Dziegielewska, K. M., Saunders, N. R., Anthony, D. C., and Molnár, Z. (2011). Reduced ventricular proliferation in the foetal cortex following maternal inflammation in the mouse. Brain 134, 3236–3248. doi: 10.1093/brain/awr237
Stricker, S. H., and Götz, M. (2018). DNA-methylation: Master or slave of neural fate decisions? Front. Neurosci. 12:5. doi: 10.3389/fnins.2018.00005
Strube, W., Marshall, L., Quattrocchi, G., Little, S., Cimpianu, C. L., Ulbrich, M., et al. (2020). Glutamatergic contribution to probabilistic reasoning and jumping to conclusions in schizophrenia: A double-blind, randomized experimental trial. Biol. Psychiatry 88, 687–697. doi: 10.1016/j.biopsych.2020.03.018
Sun, T., and Hevner, R. F. (2014). Growth and folding of the mammalian cerebral cortex: From molecules to malformations. Nat. Rev. Neurosci. 15, 217–232. doi: 10.1038/nrn3707
Suter, D. M., Tirefort, D., Julien, S., and Krause, K.-H. (2009). A Sox1 to Pax6 switch drives neuroectoderm to radial glia progression during differentiation of mouse embryonic stem cells. Stem Cells 27, 49–58. doi: 10.1634/stemcells.2008-0319
Swanepoel, T., Möller, M., and Harvey, B. H. (2018). N-acetyl cysteine reverses bio-behavioural changes induced by prenatal inflammation, adolescent methamphetamine exposure and combined challenges. Psychopharmacology 235, 351–368. doi: 10.1007/s00213-017-4776-5
Szűcs, E., Ducza, E., Büki, A., Kekesi, G., Benyhe, S., and Horvath, G. (2020). Characterization of dopamine D2 receptor binding, expression and signaling in different brain regions of control and schizophrenia-model Wisket rats. Brain Res. 1748:147074. doi: 10.1016/j.brainres.2020.147074
Tang, B., Jia, H., Kast, R. J., and Thomas, E. A. (2013). Epigenetic changes at gene promoters in response to immune activation in utero. Brain Behav. Immun. 30, 168–175. doi: 10.1016/j.bbi.2013.01.086
Tsui, D., Voronova, A., Gallagher, D., Kaplan, D. R., Miller, F. D., and Wang, J. (2014). CBP regulates the differentiation of interneurons from ventral forebrain neural precursors during murine development. Dev. Biol. 385, 230–241. doi: 10.1016/j.ydbio.2013.11.005
Tsukada, T., Sakata-Haga, H., Shimada, H., Shoji, H., and Hatta, T. (2021). Mid-pregnancy maternal immune activation increases Pax6-positive and Tbr2-positive neural progenitor cells and causes integrated stress response in the fetal brain in a mouse model of maternal viral infection. IBRO Neurosci. Rep. 11, 73–80. doi: 10.1016/j.ibneur.2021.07.003
Urbán, N., and Guillemot, F. (2014). Neurogenesis in the embryonic and adult brain: Same regulators, different roles. Front. Cell. Neurosci. 8:396. doi: 10.3389/fncel.2014.00396
van den Buuse, M. (2010). Modeling the positive symptoms of schizophrenia in genetically modified mice: Pharmacology and methodology aspects. Schizophr. Bull. 36, 246–270. doi: 10.1093/schbul/sbp132
Villagra, A., Sotomayor, E., and Seto, E. (2010). Histone deacetylases and the immunological network: Implications in cancer and inflammation. Oncogene 29, 157–173. doi: 10.1038/onc.2009.334
Vojtechova, I., Maleninska, K., Kutna, V., Klovrza, O., Tuckova, K., Petrasek, T., et al. (2021). Behavioral alterations and decreased number of parvalbumin-positive interneurons in Wistar rats after maternal immune activation by lipopolysaccharide: Sex matters. Int. J. Mol. Sci. 22:3274. doi: 10.3390/ijms22063274
Wagner, N. R., and MacDonald, J. L. (2021). Atypical neocortical development in the cited2 conditional knockout leads to behavioral deficits associated with neurodevelopmental disorders. Neuroscience 455, 65–78. doi: 10.1016/j.neuroscience.2020.12.009
Walter, J., Honsek, S. D., Illes, S., Wellen, J. M., Hartung, H.-P., Rose, C. R., et al. (2011). A new role for interferon gamma in neural stem/precursor cell dysregulation. Mol. Neurodegener. 6:18. doi: 10.1186/1750-1326-6-18
Wang, J., Weaver, I. C., Gauthier-Fisher, A., Wang, H., He, L., Yeomans, J., et al. (2010). CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev. Cell 18, 114–125. doi: 10.1016/j.devcel.2009.10.023
Weber-Stadlbauer, U., Richetto, J., Zwamborn, R. A., Slieker, R. C., and Meyer, U. (2021). Transgenerational modification of dopaminergic dysfunctions induced by maternal immune activation. Neuropsychopharmacology 46, 404–412. doi: 10.1038/s41386-020-00855-w
Wiese, M., and Bannister, A. J. (2020). Two genomes, one cell: Mitochondrial-nuclear coordination via epigenetic pathways. Mol. Metab. 38:100942. doi: 10.1016/j.molmet.2020.01.006
Wilson, K. D., Porter, E. G., and Garcia, B. A. (2022). Reprogramming of the epigenome in neurodevelopmental disorders. Crit. Rev. Biochem. Mol. Biol. 57, 73–112. doi: 10.1080/10409238.2021.1979457
Woods, R. M., Lorusso, J. M., Potter, H. G., Neill, J. C., Glazier, J. D., and Hager, R. (2021). Maternal immune activation in rodent models: A systematic review of neurodevelopmental changes in gene expression and epigenetic modulation in the offspring brain. Neurosci. Biobehav. Rev. 129, 389–421. doi: 10.1016/j.neubiorev.2021.07.015
Woodward, E. M., and Coutellier, L. (2021). Age-and sex-specific effects of stress on parvalbumin interneurons in preclinical models: Relevance to sex differences in clinical neuropsychiatric and neurodevelopmental disorders. Neurosci. Biobehav. Rev. 131, 1228–1242. doi: 10.1016/j.neubiorev.2021.10.031
Wu, S., Wei, T., Fan, W., Wang, Y., Li, C., and Deng, J. (2021). Cell cycle during neuronal migration and neocortical lamination. Int. J. Dev. Neurosci. 81, 209–219. doi: 10.1002/jdn.10091
Wu, Y., Qi, F., Song, D., He, Z., Zuo, Z., Yang, Y., et al. (2018). Prenatal influenza vaccination rescues impairments of social behavior and lamination in a mouse model of autism. J. Neuroinflammation 15:228. doi: 10.1186/s12974-018-1252-z
Yang, J., Yang, X., and Tang, K. (2022). Interneuron development and dysfunction. FEBS J. 289, 2318–2336. doi: 10.1111/febs.15872
Yao, B., and Jin, P. (2014). Unlocking epigenetic codes in neurogenesis. Genes Dev. 28, 1253–1271. doi: 10.1101/gad.241547.114
Yu, D., Li, T., Delpech, J.-C., Zhu, B., Kishore, P., Koshi, T., et al. (2022). Microglial GPR56 is the molecular target of maternal immune activation-induced parvalbumin-positive interneuron deficits. Sci. Adv. 8:eabm2545. doi: 10.1126/sciadv.abm2545
Zager, A., Mennecier, G., and Palermo-Neto, J. (2012). Maternal immune activation in late gestation enhances locomotor response to acute but not chronic amphetamine treatment in male mice offspring: Role of the D1 receptor. Behav. Brain Res. 232, 30–36. doi: 10.1016/j.bbr.2012.03.036
Zawadzka, A., Cieślik, M., and Adamczyk, A. (2021). The role of maternal immune activation in the pathogenesis of autism: A review of the evidence, proposed mechanisms and implications for treatment. Int. J. Mol. Sci. 22:11516. doi: 10.3390/ijms222111516
Zhang, X., Huang, C. T., Chen, J., Pankratz, M. T., Xi, J., Li, J., et al. (2010). Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell 7, 90–100. doi: 10.1016/j.stem.2010.04.017
Zhang, Z., and van Praag, H. (2015). Maternal immune activation differentially impacts mature and adult-born hippocampal neurons in male mice. Brain Behav. Immun. 45, 60–70. doi: 10.1016/j.bbi.2014.10.010
Keywords: maternal immune activation, neurogenesis, neurodevelopment, proliferation, schizophrenia, autism spectrum disorder, cortical lamination
Citation: McEwan F, Glazier JD and Hager R (2023) The impact of maternal immune activation on embryonic brain development. Front. Neurosci. 17:1146710. doi: 10.3389/fnins.2023.1146710
Received: 17 January 2023; Accepted: 15 February 2023;
Published: 06 March 2023.
Edited by:
Junhua Yang, Guangdong Pharmaceutical University, ChinaReviewed by:
Kieran Mcdermott, University of Limerick, IrelandBerislav Zlokovic, University of Southern California, United States
Copyright © 2023 McEwan, Glazier and Hager. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca McEwan, ZnJhbmNlc2NhLm1jZXdhbkBwb3N0Z3JhZC5tYW5jaGVzdGVyLmFjLnVr