 Masashi Mizuguchi
Masashi Mizuguchi Akiko Shibata
Akiko Shibata Mariko Kasai
Mariko Kasai Ai Hoshino
Ai Hoshino- 1Department of Developmental Medical Sciences, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
- 2Department of Pediatrics, National Rehabilitation Center for Children With Disabilities, Tokyo, Japan
- 3Laboratory for Brain Development and Disorders, RIKEN Center for Brain Science, Tokyo, Japan
- 4Department of Pediatrics, Saitama Citizens Medical Center, Saitama, Japan
- 5Department of Neuropediatrics, Tokyo Metropolitan Neurological Hospital, Fuchu, Japan
Acute encephalopathy is a constellation of syndromes in which immune response, metabolism and neuronal excitation are affected in a variable fashion. Most of the syndromes are complex disorders, caused or aggravated by multiple, genetic and environmental risk factors. Environmental factors include pathogenic microorganisms of the antecedent infection such as influenza virus, human herpesvirus-6 and enterohemorrhagic Escherichia coli, and drugs such as non-steroidal anti-inflammatory drugs, valproate and theophylline. Genetic factors include mutations such as rare variants of the SCN1A and RANBP2 genes, and polymorphisms such as thermolabile CPT2 variants and HLA genotypes. By altering immune response, metabolism or neuronal excitation, these factors complicate the pathologic process. On the other hand, some of them could provide promising targets to prevent or treat acute encephalopathy.
Introduction
Acute encephalopathy is a severe brain complication of infection, characterized clinically by acute onset of severe and long-lasting disturbance of consciousness, usually accompanied by seizures. The pathologic substrate of acute encephalopathy is diffuse or widespread, non-inflammatory brain edema, which can be visualized by neuroimaging techniques such as cranial magnetic resonance imaging (MRI) and computed tomography (CT). Acute encephalopathy consists of multiple syndromes, between which there are both similarities and differences (Mizuguchi et al., 2007). Acute encephalopathy may occur at any age but is most common in infancy and childhood. The onset is usually preceded by common infectious diseases, mostly febrile, such as influenza, exanthem subitum and rotavirus gastroenteritis. The incidence of each syndrome is variable to a great extent among countries and ethnicities.
Previous studies have provided numerous pieces of information on the involvement of many risk factors in the etiology and pathogenesis of acute encephalopathy. Environmental factors include pathogens of the antecedent infection, drugs and toxins, whereas genetic factors include gene mutations and polymorphisms. The aim of this review is to show a comprehensive list of the factors, and to describe how they cause or aggravate brain edema to cause acute encephalopathy.
Acute encephalopathy syndromes
Acute encephalopathy is classified in two ways: microbiologic classification based on the pathogen of antecedent infection, such as influenza-associated encephalopathy, human herpesvirus-6 (HHV-6)-associated encephalopathy, rotavirus-associated encephalopathy and severe acute respiratory syndrome coronavirus-2 (SARS CoV-2)-associated encephalopathy, and syndromic classification based on the clinical and neuroimaging features (Table 1), such as acute necrotizing encephalopathy (ANE), acute encephalopathy with biphasic seizures and late reduced diffusion (AESD), clinically mild encephalitis/encephalopathy with a reversible splenial lesion (MERS) and febrile infection-related epilepsy syndrome (FIRES, also known as acute encephalitis with refractory, repetitive partial seizures) (Mizuguchi et al., 2007; Mizuguchi et al., 2021; Figure 1). Among these syndromes, prognosis varies to a large extent. For example, ANE is characterized by a high fatality (26–28%) and a high rate of neurologic sequelae (45–56%), AESD by a low fatality (1–2%) and a high rate of neurologic sequelae (61–66%), and MERS by a low fatality (0%) and a low rate of neurologic sequelae (5–7%) (Hoshino et al., 2012; Kasai et al., 2020).
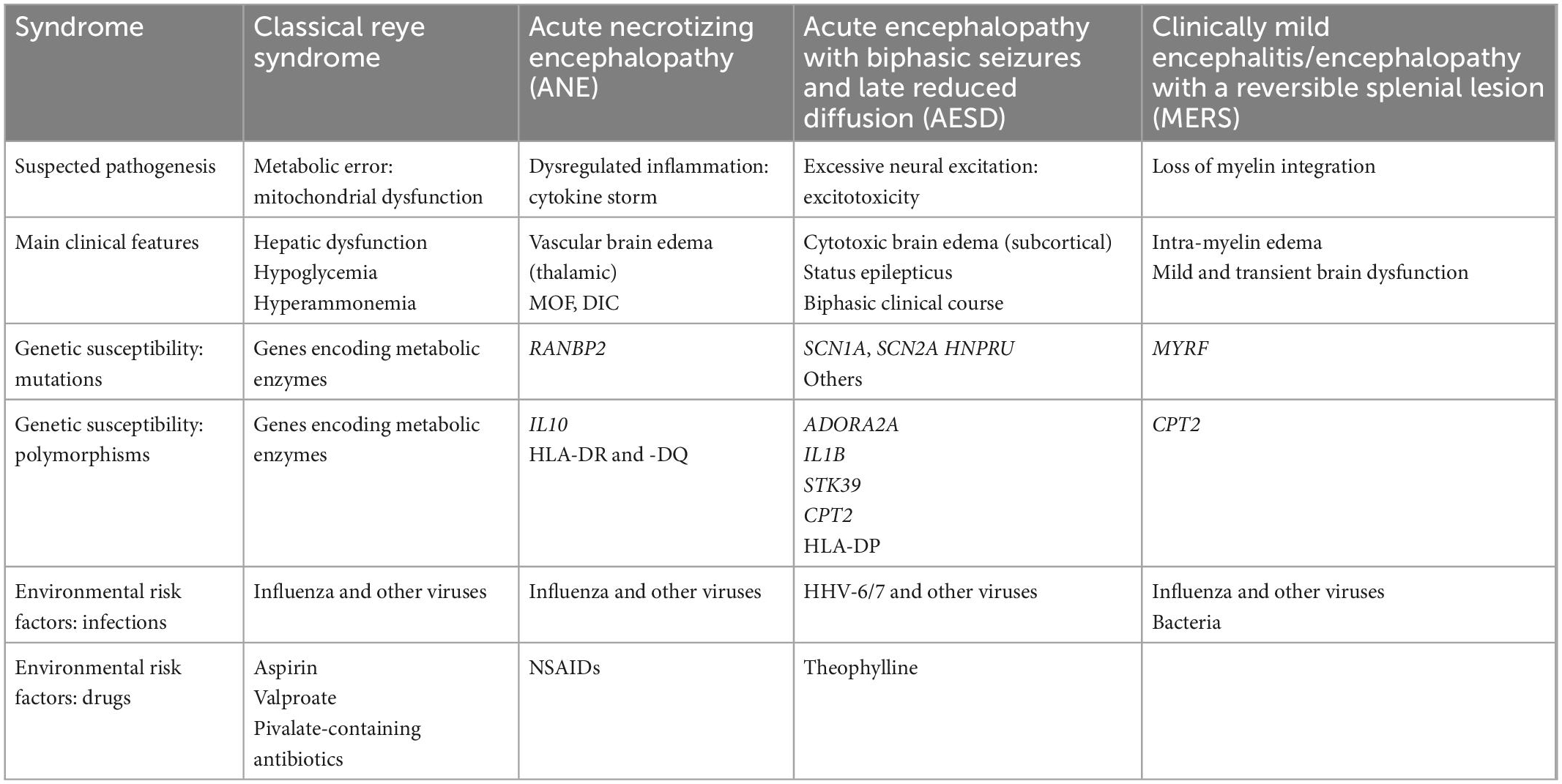
Table 1. Major acute encephalopathy syndromes: Pathogenesis, clinical features and risk factors.
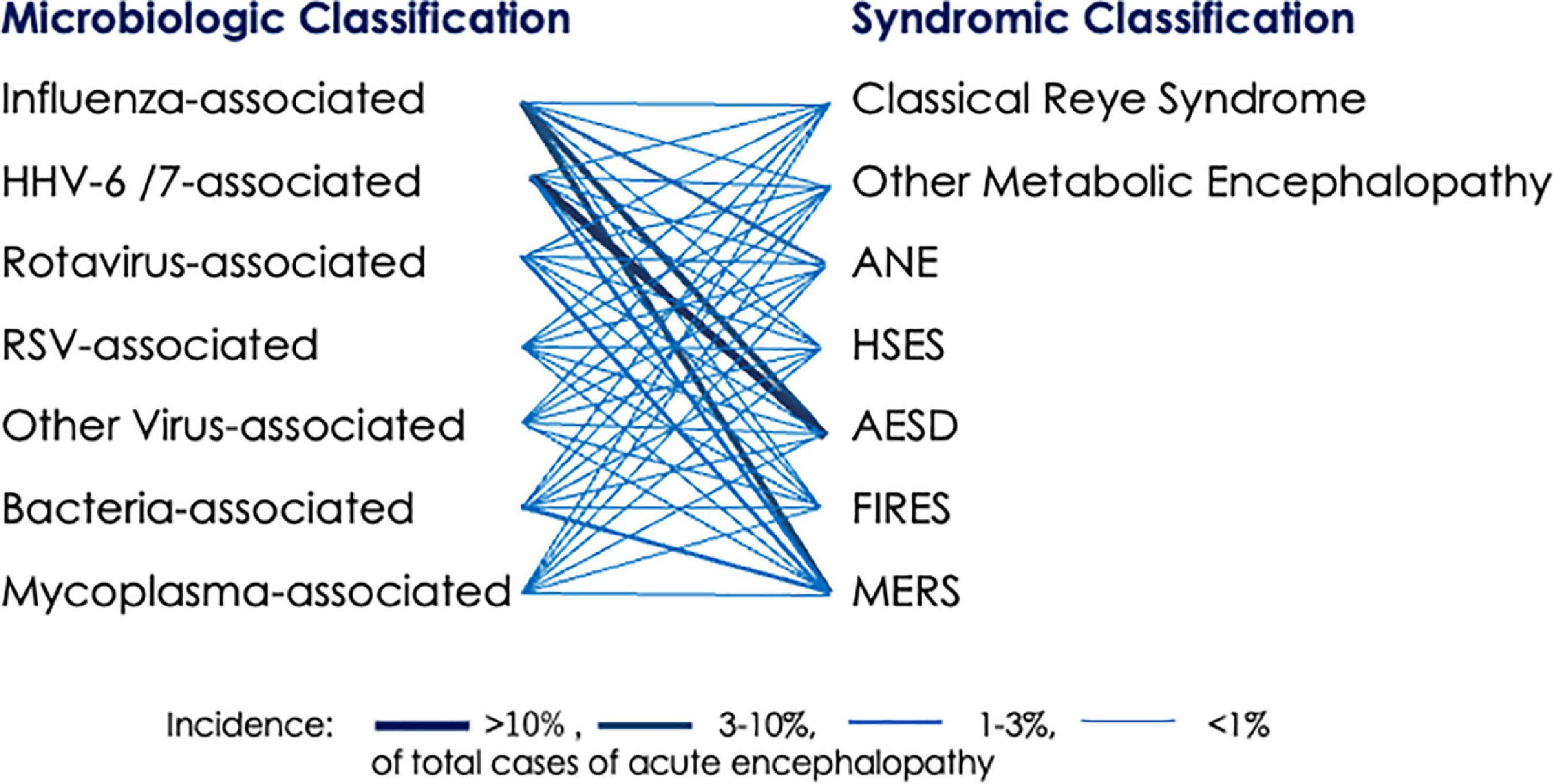
Figure 1. Association between pathogens of antecedent infection and encephalopathy syndromes. A bold line indicates high incidence, based on epidemiologic data in Japan (Hoshino et al., 2012; Kasai et al., 2020). ANE, acute necrotizing encephalopathy; AESD, acute encephalopathy with biphasic seizures and late reduced diffusion; FIRES, febrile infection-related epilepsy syndrome; HHV, human herpesvirus; HSES, hemorrhagic shock and encephalopathy syndrome; MERS, clinically mild encephalitis/encephalopathy with reversible splenial lesion; RSV, respiratory syncytial virus.
Pathogenesis of acute encephalopathy
In severe syndromes, there are three major pathogenetic events: dysregulated immune responses, defective energy metabolism and excessive neuronal excitation (Figure 2). Clinical and laboratory findings of ANE include signs of systemic inflammatory response syndrome such as multiple organ failure (MOF) and disseminated intravascular coagulation (DIC) (Mizuguchi et al., 2007), as well as very high levels of inflammatory cytokines in the serum (Ichiyama et al., 2003a,b), whereas neuroimaging findings characteristic of ANE are symmetric brain lesions in the bilateral thalamus, showing hemorrhage in the center and vasogenic edema in the periphery (Mizuguchi et al., 2002). Based on these findings, the main pathomechanism of ANE is considered to be dysregulated immune responses, or cytokine storm in which the brain is damaged as a primary target organ.
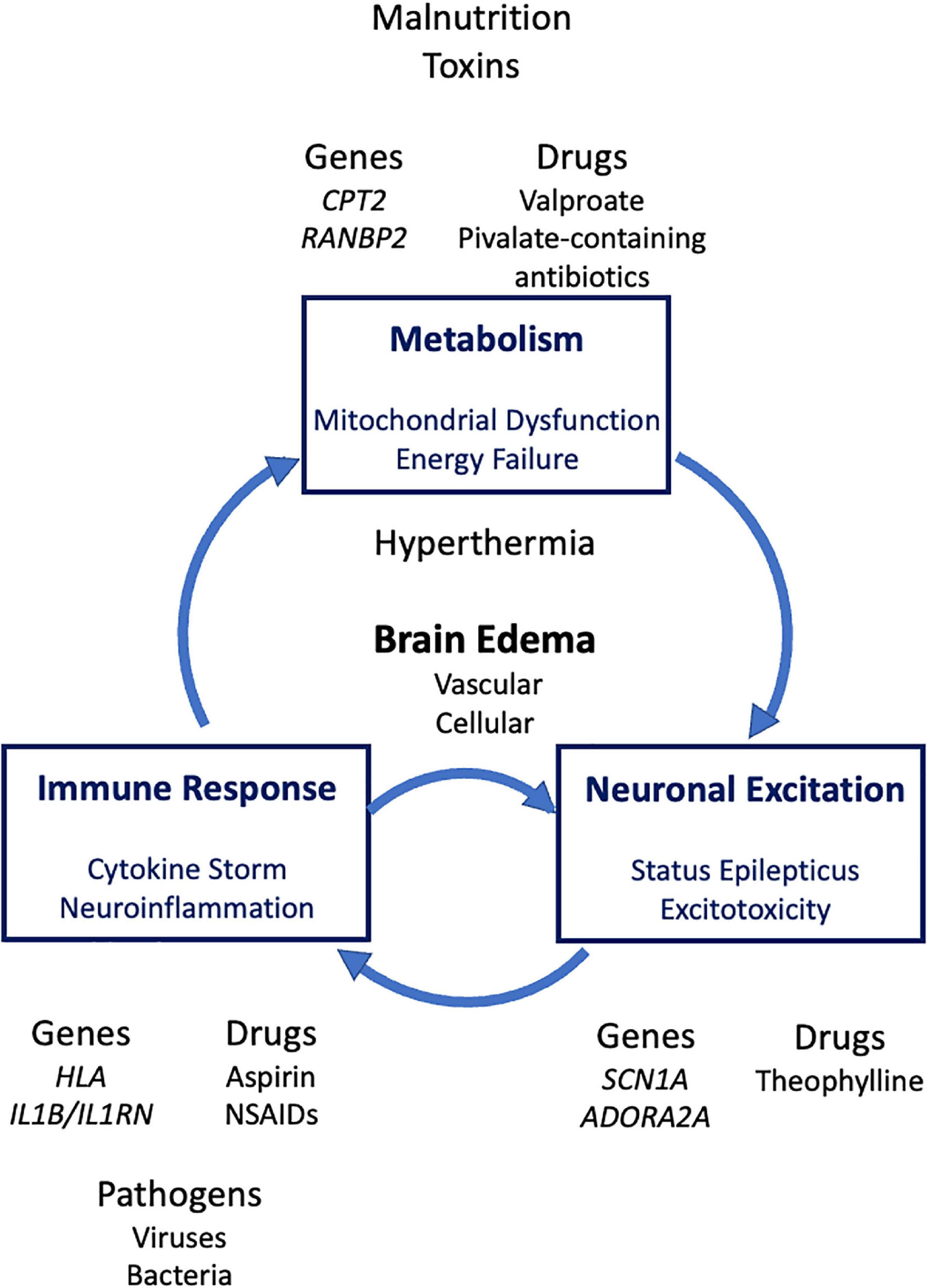
Figure 2. Genetic and environmental risk factors and major pathologic processes of severe acute encephalopathy. HLA, human leukocyte antigen; NSAIDs, non-steroidal anti-inflammatory drugs.
Biochemical findings of classical Reye syndrome include hyperammonemia, hypoglycemia and free fatty acidemia. Previous studies, mostly in the 20th century, have proven this syndrome as a transient disorder of mitochondria that regulate urea cycle, gluconeogenesis and fatty acid oxidation (Visentin et al., 1995).
Clinical picture of AESD is characterized by biphasic course consisting of acute stage (within 1 week after onset) and subacute stage (1 week–1 month after onset), and by delayed appearance of cerebral lesions representing cytotoxic edema in the subcortical white matter (Takanashi et al., 2006). Neuronal apoptosis is suggested by the serial change of cytochrome c in the cerebrospinal fluid (Hosoya et al., 2005), and excitotoxicity by an increase in glutamate in the subcortical lesion demonstrated by magnetic resonance spectroscopy (Takanashi et al., 2009; Takanashi et al., 2015). Thus, AESD is considered to be caused by excessive neuronal excitation.
The three pathogenetic processes are mutually related. First, in classical Reye syndrome, proinflammatory cytokines, such as tumor necrosis factor (TNF), mediate the metabolic effects of toxins and drugs causative of this syndrome (Larrick and Kunkel, 1986), indicating that cytokine storm impairs mitochondrial function. Second, mitochondrial encephalopathy syndromes, such as Leigh syndrome and mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS), are usually manifested with severe epileptic seizures, since neurons are the most energy-consuming cell type vulnerable to energy failure due to mitochondrial dysfunction. Third, severe epileptic seizures or status epilepticus induces proinflammatory responses in the brain (Vezzani and Granata, 2005; Vezzani et al., 2008), which in turn affect blood-brain barrier (Oby and Janigro, 2006). Conversely, proinflammatory molecules such as interleukin 1β (IL-1β) and TNF increase neuronal excitability (Nabbout et al., 2011). IL-1β plays a major role in the precipitation of seizures associated with fever (Heida et al., 2009). Taken together, inflammation is both a cause and consequence of seizures. Dysregulated immune response and excessive neuronal excitation form a “small” vicious cycle that further fosters neuroinflammation and status epilepticus (Nabbout et al., 2011; Figure 2).
Based on these relationships, the three pathogenetic changes, inflammatory, metabolic and neuronal (epileptic), may form a “large” vicious cycle (Figure 2). Two or three of them may co-exist in very severe syndromes of acute encephalopathy. For example, hemorrhagic shock and encephalopathy syndrome shows both findings of cytokine storm such as fever, shock, DIC, and MOF (Rinka et al., 2008), and those of neuronal over-excitation such as “electrical storms” on electroencephalogram (EEG), a severe form of subclinical status epilepticus (Harden et al., 1991), and “bright tree appearance” on magnetic resonance imaging, cerebral subcortical lesions of reduced diffusion mimicking those of AESD (Kuki et al., 2021).
Acute encephalopathy as a complex disorder
The onset and evolution of pathologic process in acute encephalopathy involve multiple initiating and aggravating factors, some of which are environmental. Pathogens of antecedent infections, typically with high fever, are an essential trigger. Fasting (malnutrition) and/or excessive protein intake may also trigger metabolic encephalopathies including classical Reye syndrome. Drugs and toxins may worsen acute encephalopathy. For example, aspirin and aflatoxin play major roles in the pathogenesis of classical Reye syndrome (De Vivo, 1985).
The involvement of genetic factors is suggested by the uneven geographic distribution of cases. As described below, the commonest pathogen of acute encephalopathy are common viruses such as influenza virus, HHV-6/7 and rotavirus, all of which are distributed worldwide. On the other hand, the incidence of various encephalopathy syndromes is highly variable among countries and ethnicities. For example, AESD is the most common syndrome in Japan (Hoshino et al., 2012; Kasai et al., 2020), but is rarely reported from other countries. The number of sporadic cases of ANE is much larger in east Asian countries than in west Asia, Europe, and America, whereas that of familial/recurrent ANE (ANE1) is much smaller. These geographic and ethnic differences, together with the presence of rare familial cases, suggest genetic susceptibility to acute encephalopathy.
When the total sum of effects of these genetic and environmental factors exceeds a certain threshold, acute encephalopathy may occur. This notion applies even to an encephalopathy syndrome showing Mendelian inheritance, given the low penetrance (40–50%) of ANE1 (an autosomal dominantly inherited disorder caused by a gene mutation) in which the involvement of other genetic (thermolabile variants of a mitochondrial enzyme) and environmental factors (influenza virus) are described (Neilson et al., 2003; Neilson et al., 2004; Ohashi et al., 2021).
Infectious agents
Viral infections
In the majority of acute encephalopathy cases, the antecedent infection is caused by common viruses that often affect young children to produce high fever. According to a Japanese survey on the epidemiology of acute encephalopathy conducted twice (the first in 2010 and second in 2017), the two most common viruses were influenza virus and HHV-6/7, followed by rotavirus and respiratory syncytial virus (RSV) (Hoshino et al., 2012; Kasai et al., 2020).
Association of viruses with syndromes is non-specific, and any virus can cause any syndrome. For example, clinical manifestation of ANE shows no difference between influenza virus and other viruses (Okumura et al., 2009). However, the Japanese survey has noted strong epidemiologic associations between exanthem subitum (HHV-6/7) and AESD, between influenza and ANE, between influenza and MERS, and between rotavirus gastroenteritis and MERS (Figure 1; Hoshino et al., 2012). The reason for these associations remains obscure, but there are two possible explanations. One is that different viruses elicit host immune responses with distinct cytokine profiles, thereby triggering certain syndromes of acute encephalopathy. Another is the relationship between the age predilection of a viral infection and age-dependent vulnerability of developing brain to specific types of insults, based on the degree of brain maturation.
Influenza virus is the commonest causative pathogen of acute encephalopathy. In Japan, age distribution of influenza encephalopathy shows its peak at 1 year, and median at 6 years, suggesting that the risk of severe brain complication (acute encephalopathy) is higher in primary infection than in re-infections. Males and females are equally affected. The seasonality of influenza-associated encephalopathy is the same as that of influenza. The incidence of encephalopathy tends to be higher in A(H3N2) epidemic than in A(H1N1) epidemic (Mizuguchi et al., 2007). In 2009, however, pandemic A(H1N1) was associated with a remarkable increase in cases of influenza-associated encephalopathy (Kasai et al., 2020). With regard to syndromes, influenza is the commonest pathogen in MERS and ANE (Hoshino et al., 2012).
Human herpesvirus-6/7 is the second commonest pathogen of acute encephalopathy. Age distribution shows a sharp peak at 1 year of age. Males and females are equally affected. Seasonal and yearly fluctuation of incidence is small. AESD is by far the commonest syndrome of HHV-6/7 encephalopathy, whereas HHV-6/7 is the commonest pathogen in AESD (Hoshino et al., 2012).
Severe acute respiratory syndrome coronavirus-2 has recently been added to the list of pathogenic viruses of antecedent infections. COVID-19 is occasionally complicated by severe acute encephalopathy, including ANE, hemorrhagic shock, and encephalopathy syndrome, and encephalopathy with fulminant cerebral edema (LaRovere et al., 2021; Ray et al., 2021). COVID-19-associated multiple inflammatory syndrome in children (MIS-C) or pediatric inflammatory syndrome (PIMS) is often complicated by acute encephalopathy with lesions in the splenium of corpus callosum characteristic of MERS (Abdel-Mannan et al., 2020; Lindan et al., 2021).
Bacterial infections
Acute encephalopathy is a severe brain complication of bacterial gastroenteritis caused by enterohemorrhagic Escherichia coli, Shigella, Salmonella, Yersinia, Campylobacter, and Bacillus cereus. Clinical manifestations of the bacterial encephalopathy are largely different from those of viral encephalopathy, since toxins characteristic of each bacteria species, such as verotoxin (Shiga toxin), Salmonella toxin and endotoxins, play major roles in the pathogenesis of encephalopathy (Mizuguchi et al., 2001; Lingwood, 2020).
On the other hand, acute focal glomerular nephritis (AFGN) is often complicated by MERS, a syndrome usually associated with influenza virus, rotavirus and other viruses. In AFGN-associated MERS, increased cytokines and chemokines in the blood and cerebrospinal fluid are implicated (Kometani et al., 2014; Okada et al., 2022). According to the Japanese epidemiological survey, bacterial infections including AFGN account for 3–4% of MERS cases (Hoshino et al., 2012). By contrast, antecedent bacterial infections are very rare in cases of AESD and ANE (Yamaguchi et al., 2018; Huber et al., 2020).
Drugs
Drugs affecting immune responses and/or metabolism
Diclofenac sodium and mefenamic acid are non-steroidal anti-inflammatory drugs (NSAIDs) with a potent antipyretic effect. A Japanese study on the epidemiology of influenza-associated encephalopathy revealed that the use of these NSAIDs was associated with an increase in mortality (Nagao et al., 2008). Many of the fatal cases had cytokine storm, typically ANE (Ichiyama et al., 2003a). To explore the reason for this seemingly paradoxical aggravation, an in vitro study found an increase in nitric oxide production in astrocytes activated by proinflammatory cytokines such as IL-1β and TNF (Kakita et al., 2009). NSAIDs inhibit cyclooxygenase and induce proinflammatory cytokines, which in turn damages cerebrovascular endothelial cells, causing brain edema.
Aspirin (salicylate), an antipyretic which had widely been used for febrile infections of children until the 1970’s, may trigger or aggravate acute encephalopathy including classical Reye syndrome (Hurwitz et al., 1985). Since the 1980’s when warnings were issued about the use of aspirin, the incidence of this syndrome has sharply declined (Belay et al., 1999). In the pathogenesis of aspirin-induced encephalopathy, multiple mechanisms are involved. First, aspirin enhance the release of IL-1 and TNF from macrophage, which in turn impair metabolism in the liver (Larrick and Kunkel, 1986). Second, aspirin can directly inhibit hepatic mitochondria (Trost and Lemasters, 1997; Pessayre et al., 1999).
Drugs affecting mitochondrial metabolism
Sodium valproate (VPA), a widely used antiepileptic drug, may elicit adverse effects to cause hepatotoxicity and encephalopathy, or classical Reye syndrome (Baganz and Dross, 1994; Verrotti et al., 2002; Segura-Bruna et al., 2006). Other risk factors of this encephalopathy include infants under 2 years of age, other antiepileptic drugs (phenytoin, phenobarbital, and topiramate), metabolic diseases (urea cycle disorders), excessive protein intake and malnutrition (Honeycutt et al., 1992; Kay et al., 1986). Hyperammonemia, a common finding in valproate-induced encephalopathy, is caused either by inhibition by valproyl CoA of carbamyl phosphate synthase-1, a key enzyme of urea cycle, or by secondary carnitine deficiency disturbing mitochondrial metabolism such as beta-oxidation (Warter et al., 1983; Kay et al., 1986; Duarte et al., 1993; Fromenty and Pessayre, 1995).
Glycerol and fructose, an osmotic agent injected intravenously to treat cerebral edema, may paradoxically cause acute encephalopathy in neonates, undernourished infants, and congenital metabolic disorders of glycerol and fructose such as fructose-1,6-bisphosphatse deficiency (Hasegawa et al., 2003).
Pivalate-containing antibiotics may cause secondary hypocarnitinemia, occasionally leading to acute encephalopathy with or without hypoglycemia (Stanley, 2004; Kobayashi et al., 2016). Other risk factors include congenital metabolic disorders, valproate treatment, blood and peritoneal dialysis, total parenteral nutrition, dietary restriction, anti-cancer agents, liver failure, neuromuscular disorders, severe motor and intellectual disabilities, anorexia and malnutrition (Lokrantz et al., 2004; Makino et al., 2007).
Drugs affecting neuronal excitation
Theophylline, a xanthine derivative, is a non-selective, competitive antagonist of adenosine. There are in the human brain anti-excitatory adenosine A1 receptor (Fukuda et al., 2010) and pro-excitatory A2A receptor (Fukuda et al., 2011). Since the action of A1 receptor predominate over that of A2A receptor, a net effect of adenosine is anti-excitatory (an endogenous anticonvulsant) and that of theophylline pro-excitatory. In addition, theophylline inhibits pyridoxal kinase, causing vitamin B6 deficiency and a decrease in gamma-aminobutyric acid (GABA) (Ubbink et al., 1989). In Japan, theophylline had widely been used as a bronchodilator to treat an acute attack of bronchial asthma and “asthmatic bronchitis,” an acute respiratory infection of children with wheezing, until the 2000’s, when the Japanese Guidelines for the Treatment and Management of Bronchial Asthma of Children were revised. Triggered by fever, some Japanese children taking theophylline had convulsive seizures, which were often prolonged and refractory to intravenous benzodiazepines, the first-line anti-convulsant used in pediatric emergency rooms (Nakada et al., 1983; Yoshikawa, 2007; Korematsu et al., 2008). Super-refractory seizures often required treatment with a high-dose, intravenous barbiturate under mechanical ventilation. After several weeks of intensive care, most of the patients were left with severe neurologic handicaps including severe motor and intellectual disabilities and intractable epilepsy. Many of them were diagnosed with AESD (Saitoh et al., 2015b) and others with unclassified encephalopathy (Shiihara et al., 2006).
Susceptibility genes
In a small number of familial cases, acute encephalopathy shows Mendelian inheritance. To date, two syndromes, ANE1 and familial/recurrent MERS (encephalopathy with reversible myelin vacuolation), have been documented to be inherited in an autosomal dominant fashion. Previous studies to explore their genetic background adopted hypothesis-free approaches: genome-wide linkage analysis followed by high-throughput sequencing for ANE1, and whole exome sequencing analysis for familial/recurrent MERS, both of which successfully discovered their causative genes (Neilson et al., 2004; Neilson et al., 2009; Kurahashi et al., 2018).
In a large number of sporadic cases, acute encephalopathy is a complex disorder in which multiple genetic factors are likely involved. Most genetic studies have adopted a case-control association study. Because of a small sample size resulting from the low incidence of acute encephalopathy, few studies have adopted hypothesis-free approaches, such as genome-wide association study (GWAS) (Kasai et al., 2022a). The vast majority of studies have used a candidate gene approach to search for common and/or rare variants (mutations and/or polymorphisms), based either on common disease-common variants hypothesis and/or common disease-multiple rare variants hypothesis (Shibata et al., 2020). In consideration of the suspected pathomechanism of acute encephalopathy described above, genes related to immune responses, metabolism and neuronal excitation have been selected as candidate genes in many of the genetic studies.
Genes regulating immune response
Human leukocyte antigen (HLA) genotypes include HLA class I (HLA-A, -C, and -B) and HLA class II (HLA-DR, -DQ, and -DP). The encoded molecules play an important role in the modulation of immune responses and self versus non-self recognition. Japanese studies identified DRB1*09:01 and DQB1*03:03 as risk alleles for sporadic ANE (Hoshino et al., 2016), and DPB1*04:01:01 as a protective allele for AESD (Kasai et al., 2022b). The association of DQB1*03:03 and ANE was also reported in a Korean case (Oh et al., 2004). DRB1*09:01 is common in east Asia but rare in European populations, which may partially account for the high incidence of ANE in east Asia.
Interleukin-1β and its receptors constitute a critical pathway both in neuroinflammation and neuroprotection. In the promotor of the IL1B gene, there is an upstream variant, rs16944 (IL1B-511T > C), which is associated with high expression of IL-1β (Aguet et al., 2020). Previous studies in Asian countries demonstrated a positive (risk) association of rs16944 with febrile seizures (FS) (Tsai et al., 2002; Kira et al., 2005; Yu et al., 2018). By contrast, a recent study showed a negative (protective) association of rs16944 with AESD (Shibata et al., 2022). AESD has a genetic background distinct from FS, although the clinical presentation of AESD at its onset is indistinguishable from that of prolonged FS.
Interleukin 1 receptor antagonist (IL-1Ra), an endogenous antagonist of IL-1β, is encoded by the IL1RN gene. A variable number of tandem repeats (VNTR) polymorphism in IL1RN intron 2 is associated with many chronic inflammatory diseases and with FS (Tsai et al., 2002). A Japanese study found an association of a VNTR allele, RN2 (two repeats), and FIRES (Saitoh et al., 2016). Since an RN2 allele is associated with reduced IL1RN mRNA expression and enhanced IL-1β production (Santtila et al., 1998; Korthagen et al., 2012), RN2 may confer susceptibility to excessive neuroinflammation.
Interleukin 10 (IL-10), an anti-inflammatory cytokine, is encoded by the IL10 gene. A genetic polymorphism in its promotor region is a combination of two SNPs, rs1800871 and rs180072, which show complete linkage and are associated with a reduced production of IL-10. Our recent study demonstrated an association of this polymorphism with ANE (Ishii and Hirose, 2018).
Toll-like receptor 3 (TLR3), a pattern recognition receptor for double stranded RNAs, activates innate immune responses upon viral infections. A loss-of-function mutation of the TLR3 gene was found in a single case of influenza-associated encephalopathy (Hidaka et al., 2006), and in multiple cases of herpes simplex virus encephalitis (Zhang et al., 2007).
Serine/threonine kinase 39 (STK39), an activator of the p38 mitogen-activated protein kinase (MAPK) pathway, mediates cellular stress-activated signals (Johnston et al., 2000), and is involved in the pathogenesis of brain edema (Kahle et al., 2008). A GWAS study in Japan individuals has recently identified a variant in intron located in the enhancer region of the STK39 gene as a candidate susceptibility factor of AESD (Kasai et al., 2022a).
Genes regulating metabolism
Inherited metabolic errors of fatty acids, organic acids, carbohydrates and the urea cycle may be clinically manifested with acute encephalopathy (Mizuguchi et al., 2007). In particular, disorders of energy metabolism often show coma and/or convulsion because the brain is one of the most energy- or ATP-consuming organs in the human body. Common biochemical findings of metabolic encephalopathy include hypoglycemia, lactic acidosis, ketonuria, metabolic acidosis, hyperammonemia, hypocarnitinemia, and hepatic dysfunction (Mizuguchi et al., 2021).
Many causative genes of these metabolic disorders are listed as genetic risk factors of acute encephalopathy. Most of them are located on auto-chromosome, some on X chromosome, and only a few on mitochondrial DNA.
Carnitine palmitoyltransferase II (CPT2), a key enzyme of lipid metabolism located on the mitochondrial inner membrane, catabolizes acylcarnitine and produces acyl-CoA, which in turn is catabolized through β-oxidation to produce ATP. Homozygous mutations of the CPT2 gene may cause a Reye-like syndrome (Vianey-Saban et al., 1993). Several polymorphisms in exon 4 and exon 5 of the CPT2 gene causes thermolability, a severe loss of enzymatic activity at high body temperature despite a minimal or mild reduction at normal body temperature. Several studies in Japan have shown an association of the CPT2 thermolabile polymorphism, in particular rs2229291 (F352C) in exon 4, with acute encephalopathy. Earlier studies reported its associations with severe syndromes, such as influenza-associated and other encephalopathy with fatal or poor outcome (Chen et al., 2005; Yao et al., 2008; Kubota et al., 2012), AESD and ANE (Shinohara et al., 2011), whereas later studies demonstrated those with a mild syndrome, MERS, and with entire acute encephalopathy (Shibata et al., 2019). The rs2229291 variant is common in east Asians but not reported in Caucasians (NCBI),1 which may account for the high incidence of acute encephalopathy in east Asia.
Ran-binding protein 2 (RANBP2), or nucleoporin 358, is a component of the nuclear pore complex involved in nucleocytoplasmic transport, pro-inflammatory signaling and mitochondrial trafficking. Missense mutations in the RANBP2 gene have been found in ANE1 (Neilson et al., 2009; Bergamino et al., 2012; Sell et al., 2016; Hu et al., 2022). Studies on the pathogenesis of ANE1 have hypothesized the impact of RANBP2 mutation on immune response and energy metabolism (Cho et al., 2012; Levine et al., 2020). In support of the metabolic hypothesis, a recent study demonstrated an attenuated ability of mutated RANBP2 to bind to cytochrome c oxidase copper chaperone, COX11 (Shibata et al., 2021), which may affect glucose metabolism because RANBP2, when bound to COX11, suppresses its inhibitory activity over hexokinase 1 (Aslanukov et al., 2006). Pathologically, bilateral symmetric brain lesions in ANE1 resembles those in Leigh syndrome, a mitochondrial encephalopathy, which may be explained by mitochondrial dysfunction common to both conditions.
Genes regulating neuronal excitation
The α1 (Nav1.1) and α2 (Nav1.2) subunits of neuronal voltage-gated sodium channels are encoded by the SCN1A and SCN2A genes, respectively. Mutations of these genes cause various epileptic syndromes of variable clinical presentation. Dravet syndrome is a severe epilepsy characterized clinically by fever sensitivity, whose main genetic cause is missense, truncation, indel and microdeletion mutations of the SCN1A gene. In patients with Dravet syndrome, the incidence of acute encephalopathy is high, and acute encephalopathy is an important cause of death (Takayanagi et al., 2010; Tang et al., 2011; Okumura et al., 2012). Vaccination, particularly for pertussis, may also trigger acute encephalopathy in children having a SCN1A mutation (Berkovic et al., 2006). Notably, acute encephalopathy may occur in patients with a SCN1A mutation but without Dravet syndrome, many of whom being apparently healthy prior to the onset of encephalopathy. In a Japanese cohort of 87 patients with severe encephalopathy including ANE and AESD, but without underlying neurological disorders, three reportedly had SCN1A missense mutations (Saitoh et al., 2012). Another study demonstrated an association of SCN1A non-synonymous variants with AESD. Many of these variants were located in the transmembrane segments or pore-forming regions of the SCN1A molecule (Shibata et al., 2020).
Mutations of the SCN2A gene has been found in a small number of Japanese cases of acute encephalopathy including FIRES, AESD, ANE and AESD (Kobayashi et al., 2012; Fukasawa et al., 2015; Saitoh et al., 2015a). A polymorphism in SCN2A intron 22, rs 1864885, has shown a possible association with FIRES (Saitoh et al., 2016).
Adenosine A2A receptor (ADORA2A) enhances excitatory neurotransmitter release, and its experimental activation in the brain lowers the threshold of hyperthermia-induced seizures in rat pups (Fukuda et al., 2011). There are in the ADORA2A gene four SNPs, rs2298383, rs5751876, rs35320474, and rs4822492, which show complete linkage and comprise only two haplotypes (A and B) in Japanese population. A case-control association study showed that the risk of AESD is higher in AA than in AB and BB diplotypes. Since ADORA2A mRNA expression and cAMP production is higher in AA, this diplotype likely alters the intracellular adenosine/cAMP cascade, thereby promoting seizures and excitotoxic damage in AESD (Shinohara et al., 2013).
Voltage-dependent calcium channel a-1 (CACNA1A) is a subunit of P/Q type calcium channel distributed throughout the brain. Mutations in the CACNA1A gene are clinically manifested with diverse phenotypes including familial hemiplegic migraine and episodic ataxia. Several cases of migraine reportedly had hemiconvulsion-hemiplegia syndrome, an encephalopathy syndrome showing a significant overlap with AESD (Yamazaki et al., 2011; Ohmura et al., 2012).
The α3-subunit of the Na(+)/K(+) -ATPase (ATP1A3) is an electrogenic cation pump in the brain regulating concentration gradients of sodium and potassium ions across the plasma membrane. Mutations in the ATPA1A3 gene clinically present with multiple phenotypes including alternating hemiplegia of childhood, rapid-onset dystonia parkinsonism, cerebellar ataxia, areflexia, pes cavus, optic atrophy, sensorineural hearing loss (CAPOS) syndrome and early-infantile epileptic encephalopathy. Specific ATPA1A3 variants may cause acute encephalopathy: relapsing encephalopathy with cerebellar ataxia (Dard et al., 2015; Biela et al., 2021).
Rho-related BTB domain-containing protein 2 (RHOBTB2) is an atypical Rho GTPase and a substrate for the cullin-3-based ubiquitin ligase complex that recruits target proteins for degradation. Mutations in the RHOBTB2 gene cause epileptic encephalopathy, which is often complicated by febrile status epilepticus followed by acute encephalopathy (Belal et al., 2018).
Protocadherin 19 (PCDH19) is a transmembrane regulating cell-cell contact and involved in neuronal proliferation, migration and synaptic function. Mutations in the PCDH19 gene on X chromosome cause neurodevelopmental PCDH-clustering epilepsy syndrome in female hemizygotes. A female patient with a missense mutation in PCDH19 reportedly had acute encephalopathy mimicking FIRES (Specchio et al., 2011).
Heterogeneous nuclear ribonucleoprotein U (HNPRU) is a component of spliceosome that plays a role in brain development. Mutations in the HNPRU gene cause early-onset epileptic encephalopathy, which is occasionally complicated by AESD (Shimada et al., 2018).
Hamartin and tuberin, encoded by TSC1 and TSC2, respectively, form a complex to play a pivotal role in the mammalian target of rapamycin (mTOR) pathway. Mutations in these genes cause tuberous sclerosis complex (TSC), showing focal cortical dysplasia (cortical tubers) and epilepsy often resistant to antiepileptic drugs. Triggered by febrile infections, TSC is often complicated by febrile status epilepticus followed by acute encephalopathy, including AESD (Numoto et al., 2021). Other congenital syndromes with disorders of cerebral cortical development are occasionally complicated by AESD and other syndromes of acute encephalopathy (Hirayama et al., 2017).
Genes involved in thermoregulation
The neurotrophic tyrosine kinase receptor 1 (NTRK1) is a neurotrophin receptor involved in neuronal survival and differentiation. Mutations in the NTRK1 gene cause hereditary sensory and autonomic neuropathy type IV (HSAN-IV), or congenital insensitivity to pain with anhidrosis (Indo et al., 1996). Common symptoms of HSAN-IV include recurrent episodes of hyperthermia and unexplained fever, leading occasionally to acute encephalopathy resembling heatstroke (Iwanaga et al., 1996).
The α9 (Nav1.7) subunit of neuronal voltage-gated sodium channels (SCN9A) is expressed in somatic and visceral neurons and in sympathetic ganglion neurons, and plays a role in pain signaling. Mutations in the SCN9A gene may cause erythermalgia, a rare neuropathy characterized by attacks of burning pain in the extremities in response to warm stimuli. Hypothermia, an occasional complication of erythermalgia, reportedly lead to fungal pneumonia which in turn triggered the onset of MERS (Takahashi et al., 2007).
The α10 (Nav1.8) subunit of neuronal voltage-gated sodium channels (SCN10A) is expressed in nociceptive neurons, and is involved in pain sensation. Biallelic mutations in the SCN10A genes may cause epileptic encephalopathy, neuromuscular disease, bradycardia and/or anhidrosis. A patient with compound missense mutations reportedly had FIRES (Kambouris et al., 2016).
Genes maintaining myelin sheath
Myelin regulatory factor (MYRF) is a transcription factor necessary for oligodendrocyte differentiation and the maintenance of mature oligodendrocytes and myelin structure (Emery et al., 2009; Koenning et al., 2012). A heterozygous missense mutation of the MYRF gene causes familial/recurrent MERS, or encephalopathy with reversible myelin vaculotion (Kurahashi et al., 2018). Except for the familial occurrence showing Mendelian, autosomal dominant inheritance, this condition is clinically indistinguishable from MERS type 2, in which intra-myelinic edema is suggested as the pathologic substrate (Takanashi, 2009). Interestingly, an X-linked demyelinating disorder, X-linked Charcot-Marie-Tooth disease type 1 (CMTX1), is occasionally complicated by familial/recurrent, transient leukoencephalopathy showing reversible, cerebral white matter and callosal lesions mimicking those of MERS type 2. CMTX1 is caused by mutations in the GJB1 (gap junction beta 1) gene encoding another factor for myelin maintenance, connexin 32. Often triggered by a febrile infection, many male hemizygotes and several female heterozygotes reportedly had transient leukoencephalopathy, whose main clinical findings are not disturbance of consciousness, but motor and sensory signs (Hanemann et al., 2003; Sato et al., 2012; Wang and Yin, 2016).
Conclusion
This review tried to provide a comprehensive list of risk factors of acute encephalopathy, and described many viruses, drugs and gene variants which are involved, either as initiating or aggravating factors, at the onset or during the evolution of pathogenetic process leading to brain edema. The risk factors were classified according to the main pathogenetic event in which each factor is involved. Due to the limitation inherent to this approach, however, this review failed to describe some risk factors of acute encephalopathy complicating certain neurologic disorders whose pathomechanism is complex and/or poorly understood, such as neonatal hypoxic-ischemic encephalopathy, periventricular leukomalacia, congenital cytomegalovirus infection, Prader-Willi syndrome, congenital adrenal hyperplasia and incontinentia pigmenti (Abe et al., 2011, 2016; Hirayama et al., 2017).
The complex associations of these factors with pathogenetic mechanisms have multiple aspects. First, a single risk factor may be associated with multiple syndromes of different mechanisms. For example, a thermolabile CPT2 variant, a risk factor in energy metabolism, is associated not only with metabolic encephalopathy, but also with other syndromes such as AESD and MERS. Second, multiple risk factors of different categories may converge to develop a single syndrome. For example, common variants of three genes, IL1B (immune response), CPT2 (metabolism) and ADORA2A (neuronal excitation), are all associated with AESD.
The complex pathomechanism of acute encephalopathy may hamper its prevention and treatment. On the other hand, every risk factor could be a candidate target for preventive measures and/or therapeutic intervention. For example, vaccines against influenza virus and rotavirus may ameliorate the incidence, mortality and morbidity of influenza- and rotavirus-associated encephalopathy, respectively. Restriction in the use of aspirin caused a dramatic decrease in classical Reye syndrome (Belay et al., 1999). Molecular target therapies against proinflammatory cytokines may improve the outcome of severe syndromes: anakinra and tocilizumab for FIRES (Aledo-Serrano et al., 2022) and tocilizumab for ANE (Koh et al., 2019). The attenuation of CPT2 in acute encephalopathy may be rescued by a lipid-lowering agent, bezafibrate (Yao et al., 2011). In conclusion, studies to explore various risk factors should suggest many candidate targets for improved prevention and treatment of acute encephalopathy in the future.
Author contributions
MM contributed to the writing of the manuscript. AS, MK, and AH contributed to the refinement of the manuscript. All authors read and approved the submitted version.
Funding
This research was supported by a Grant-in-Aid for Scientific Research, No. 15H04872, from the Japan Society for the Promotion of Sciences and a Grant-in-Aid for Policy Research on Intractable Diseases, No. 21FC1005, from the National Institute of Public Health, Japan.
Acknowledgments
The authors are grateful to the Collaborative Research Supporting Committee of the Japanese Society of Child Neurology for promoting clinical and genetic studies of acute encephalopathy in Japan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Abdel-Mannan, O., Eyre, M., Löbel, U., Bamford, A., Eltze, C., Hameed, B., et al. (2020). Neurologic and radiographic findings associated with COVID-19 infection in children. JAMA Neurol. 77, 1440–1445. doi: 10.1001/jamaneurol.2020.2687
Abe, S., Okumura, A., Hamano, S., Tanaka, M., Shiihara, T., Aizaki, K., et al. (2011). Early infantile manifestations of incontinentia pigmenti mimicking acute encephalopathy. Brain Dev. 33, 28–34. doi: 10.1016/j.braindev.2010.04.002
Abe, Y., Sakai, T., Okumura, A., Akaboshi, S., Fukuda, M., Haginoya, K., et al. (2016). Manifestations and characteristics of congenital adrenal hyperplasia-associated encephalopathy. Brain Dev. 38, 638–647. doi: 10.1016/j.braindev.2016.01.007
Aguet, F., Barbeira, A., Bonazzola, R., Brown, A., Castel, S., Jo, B., et al. (2020). The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330. doi: 10.1126/science.aaz1776
Aledo-Serrano, A., Hariramani, R., Gonzalez-Martinez, A., Álvarez-Troncoso, J., Toledano, R., Bayat, A., et al. (2022). Anakinra and tocilizumab in the chronic phase of febrile infection-related epilepsy syndrome (FIRES): Effectiveness and safety from a case-series. Seizure 100, 51–55. doi: 10.1016/j.seizure.2022.06.012
Aslanukov, A., Bhowmick, R., Guruju, M., Oswald, J., Raz, D., Bush, R. A., et al. (2006). RanBP2 modulates Cox11 and hexokinase I activities and haploinsufficiency of RanBP2 causes deficits in glucose metabolism. PLoS Genet. 2:e177. doi: 10.1371/journal.pgen.0020177
Baganz, M. D., and Dross, P. E. (1994). Valproic acid-induced hyperammonemic encephalopathy: MR appearance. AJNR Am. J. Neuroradiol. 15, 1779–1781.
Belal, H., Nakashima, M., Matsumoto, H., Yokochi, K., Taniguchi-Ikeda, M., Aoto, K., et al. (2018). De novo variants in RHOBTB2, an atypical Rho GTPase gene, cause epileptic encephalopathy. Hum. Mutat. 39, 1070–1075. doi: 10.1002/humu.23550
Belay, E. D., Bresee, J. S., Holman, R. C., Khan, A. S., Shahriari, A., and Schonberger, L. B. (1999). Reye’s syndrome in the United States from 1981 through 1997. N. Engl. J. Med. 340, 1377–1382. doi: 10.1056/NEJM199905063401801
Bergamino, L., Capra, V., Biancheri, R., Rossi, A., Tacchella, A., Ambrosini, L., et al. (2012). Immunomodulatory therapy in recurrent acute necrotizing encephalopathy ANE1: Is it useful? Brain Dev. 34, 384–391. doi: 10.1016/j.braindev.2011.08.001
Berkovic, S. F., Harkin, L., McMahon, J. M., Pelekanos, J. T., Zuberi, S. M., Wirrell, E. C., et al. (2006). De-novo mutations of the sodium channel gene SCN1A in alleged vaccine encephalopathy: A retrospective study. Lancet Neurol. 5, 488–492. doi: 10.1016/S1474-4422(06)70446-X
Biela, M., Rydzanicz, M., Szymanska, K., Pieniawska-Smiech, K., Lewandowicz-Uszynska, A., Chruszcz, J., et al. (2021). Variants of ATP1A3 in residue 756 cause a separate phenotype of relapsing encephalopathy with cerebellar ataxia (RECA)-Report of two cases and literature review. Mol. Genet. Genomic Med. 9:e1772. doi: 10.1002/mgg3.1772
Chen, Y., Mizuguchi, H., Yao, D., Ide, M., Kuroda, Y., Shigematsu, Y., et al. (2005). Thermolabile phenotype of carnitine palmitoyltransferase II variations as a predisposing factor for influenza-associated encephalopathy. FEBS Lett. 579, 2040–2044. doi: 10.1016/j.febslet.2005.02.050
Cho, K. I., Searle, K., Webb, M., Yi, H., and Ferreira, P. A. (2012). Ranbp2 haploinsufficiency mediates distinct cellular and biochemical phenotypes in brain and retinal dopaminergic and glia cells elicited by the Parkinsonian neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Cell. Mol. Life Sci. 69, 3511–3527. doi: 10.1007/s00018-012-1071-9
Dard, R., Mignot, C., Durr, A., Lesca, G., Sanlaville, D., Roze, E., et al. (2015). Relapsing encephalopathy with cerebellar ataxia related to an ATP1A3 mutation. Dev. Med. Child Neurol. 57, 1183–1186. doi: 10.1111/dmcn.12927
Duarte, J., Macias, S., Coria, F., Fernandez, E., and Claveria, L. E. (1993). Valproate-induced coma: Case report and literature review. Ann. Pharmacother. 27, 582–583. doi: 10.1177/106002809302700510
Emery, B., Agalliu, D., Cahoy, J. D., Watkins, T. A., Dugas, J. C., Mulinyawe, S. B., et al. (2009). Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 138, 172–185. doi: 10.1016/j.cell.2009.04.031
Fromenty, B., and Pessayre, D. (1995). Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol. Ther. 67, 101–154. doi: 10.1016/0163-7258(95)00012-6
Fukasawa, T., Kubota, T., Negoro, T., Saitoh, M., Mizuguchi, M., Ihara, Y., et al. (2015). A case of recurrent encephalopathy with SCN2A missense mutation. Brain Dev. 37, 631–634. doi: 10.1016/j.braindev.2014.10.001
Fukuda, M., Suzuki, Y., Hino, H., Kuzume, K., Morimoto, T., and Ishii, E. (2010). Adenosine A1 receptor blockage mediates theophylline-associated seizures. Epilepsia 51, 483–487. doi: 10.1111/j.1528-1167.2009.02382.x
Fukuda, M., Suzuki, Y., Hino, H., Morimoto, T., and Ishii, E. (2011). Activation of central adenosine A(2A) receptors lowers the seizure threshold of hyperthermia-induced seizure in childhood rats. Seizure 20, 156–159. doi: 10.1016/j.seizure.2010.11.012
Hanemann, C. O., Bergmann, C., Senderek, J., Zerres, K., and Sperfeld, A.-D. (2003). Transient, recurrent, white matter lesions in X-linked Charcot-Marie-Tooth disease with novel connexin 32 mutation. Arch. Neurol. 60, 605–609. doi: 10.1001/archneur.60.4.605
Harden, A., Boyd, S. G., Cole, G., and Levin, M. (1991). EEG features and their evolution in the acute phase of haemorrhagic shock and encephalopathy syndrome. Neuropediatrics 22, 194–197. doi: 10.1055/s-2008-1071440
Hasegawa, Y., Kikawa, Y., Miyamoto, J., Sugimoto, S., Adachi, M., Ohura, T., et al. (2003). Intravenous glycerol should not be used in patients with unrecognized fructose-1,6-giphosphate deficiency. Pediatr. Int. 45, 5–9.
Heida, J. G., Moshé, S. L., and Pittman, Q. J. (2009). The role of interleukin-1β in febrile seizures. Brain Dev 31, 388–393. doi: 10.1016/j.braindev.2008.11.013
Hidaka, F., Matsuo, S., Muta, T., Takeshige, K., Mizukami, T., and Nunoi, H. (2006). A missense mutation of the Toll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin. Immunol. 119, 188–194. doi: 10.1016/j.clim.2006.01.005
Hirayama, Y., Saito, Y., and Maegaki, Y. Status Epilepticus Study Group. (2017). “Symptomatic” infection-associated acute encephalopathy in children with underlying neurological disorders. Brain Dev. 39, 243–247. doi: 10.1016/j.braindev.2016.09.014
Honeycutt, D., Callahan, K., Rutledge, L., and Evans, B. (1992). Heterozygote ornithine transcarbamylase deficiency presenting as symptomatic hyperammonemia during initiation of valproate therapy. Neurology 42, 666–668. doi: 10.1212/wnl.42.3.666
Hoshino, A., Saitoh, M., Miyagawa, T., Kubota, M., Takanashi, J. I., Miyamoto, A., et al. (2016). Specific HLA genotypes confer susceptibility to acute necrotizing encephalopathy. Genes Immun. 17, 367–369. doi: 10.1038/gene.2016.32
Hoshino, A., Saitoh, M., Oka, A., Okumura, A., Kubota, M., Saito, Y., et al. (2012). Epidemiology of acute encephalopathy in Japan, with emphasis on the association of viruses and syndromes. Brain Dev. 34, 337–343. doi: 10.1016/j.braindev.2011.07.012
Hosoya, M., Nunoi, H., Aoyama, M., Kawasaki, Y., and Suzuki, H. (2005). Cytochrome c and tumor necrosis factor-alpha values in serum and cerebrospinal fluid of patients with influenza-associated encephalopathy. Pediatr. Infect. Dis. J. 24, 467–470. doi: 10.1097/01.inf.0000160995.07461.b8
Hu, Y., Tian, Z., Zhao, B., Dong, C., and Cao, L. (2022). A novel variation in RANBP2 associated with infection-triggered familial acute necrotizing encephalopathy. Neurol. Sci. 43, 3973–3977. doi: 10.1007/s10072-022-06033-8
Huber, J. N., Berg, A. D., and Bula-Rudas, F. (2020). Acute necrotizing encephalopathy due to Streptococcus pneumoniae: An uncommon pathogen in a devastating disease. Pediatr. Neurol. 108, 126–127. doi: 10.1016/j.pediatrneurol.2020.03.002
Hurwitz, E. S., Barrett, M. J., Bregman, D., Gunn, W. J., Schonberger, L. B., Fairweather, W. R., et al. (1985). Public Health Service study on Reye’s syndrome and medications. Report of the pilot phase. N. Engl. J. Med. 313, 849–857. doi: 10.1056/NEJM198510033131403
Ichiyama, T., Endo, S., Kaneko, M., Isumi, H., Matsubara, T., and Furukawa, S. (2003a). Serum cytokine concentrations of influenza-associated acute necrotizing encephalopathy. Pediatr. Int. 45, 734–736. doi: 10.1111/j.1442-200x.2003.01822.x
Ichiyama, T., Isumi, H., Ozawa, H., Matsubara, T., Morishima, T., and Furukawa, S. (2003b). Cerebrospinal fluid and serum levels of cytokines and soluble tumor necrosis factor receptor in influenza virus-associated encephalopathy. Scand. J. Infect. Dis. 35, 59–61. doi: 10.1080/0036554021000026986
Indo, Y., Tsuruta, M., Hayashida, Y., Karim, M. A., Ohta, K., Kawano, T., et al. (1996). Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat. Genet. 13, 485–488. doi: 10.1038/ng0896-485
Ishii, A., and Hirose, S. (2018). “Genetic background of encephalopathy,” in Acute encephalopathy and encephalitis in infancy and its related disorders, eds H. Yamanouchi, S. L. Moshe, and A. Okumura (St. Louis, MO: Elsevier), 45–52.
Iwanaga, R., Matsuishi, T., Ohnishi, A., Nakashima, M., Abe, T., Ohtaki, E., et al. (1996). Serial magnetic resonance images in a patient with congenital sensory neuropathy with anhidrosis and complications resembling heat stroke. J. Neurol. Sci. 142, 79–84. doi: 10.1016/0022-510x(96)00152-9
Johnston, A. M., Naselli, G., Gonez, L. J., Martin, R. M., Harrison, L. C., and DeAizpurua, H. J. (2000). SPAK, a STE20/SPS1-related kinase that activates the p38 pathway. Oncogene 19, 4290–4297. doi: 10.1038/sj.onc.1203784
Kahle, K. T., Staley, K. J., Nahed, B. V., Gamba, G., Hebert, S. C., Lifton, R. P., et al. (2008). Roles of the cation-chloride cotransporters in neurological disease. Nat. Clin. Pract. Neurol. 4, 490–503. doi: 10.1038/ncpneuro0883
Kakita, H., Aoyama, M., Hussein, M. H., Kato, S., Suzuki, S., Ito, T., et al. (2009). Diclofenac enhances proinflammatory cytokine-induced nitric oxide production through NF-kappaB signaling in cultured astrocytes. Toxicol. Appl. Pharmacol. 238, 56–63. doi: 10.1016/j.taap.2009.04.014
Kambouris, M., Thevenon, J., Soldatos, A., Cox, A., Stephen, J., Ben-Omran, T., et al. (2016). Biallelic SCN10A mutations in neuromuscular disease and epileptic encephalopathy. Ann. Clin. Transl. Neurol. 4, 26–35. doi: 10.1002/acn3.372
Kasai, M., Omae, Y., Kawai, Y., Shibata, A., Hoshino, A., Mizuguchi, M., et al. (2022a). GWAS identifies candidate susceptibility loci and microRNA biomarkers for acute encephalopathy with biphasic seizures and late reduced diffusion. Sci. Rep. 12:1332. doi: 10.1038/s41598-021-04576-y
Kasai, M., Omae, Y., Khor, S. S., Shibata, A., Hoshino, A., Mizuguchi, M., et al. (2022b). Protective association of HLA-DPB1*04:01:01 with acute encephalopathy with biphasic seizures and late reduced diffusion identified by HLA imputation. Genes Immun. 23, 123–128. doi: 10.1038/s41435-022-00170-y
Kasai, M., Shibata, A., Hoshino, A., Maegaki, Y., Yamanouchi, H., Takanashi, J., et al. (2020). Epidemiological changes of acute encephalopathy in Japan based on national surveillance for 2014-2017. Brain Dev. 42, 508–514. doi: 10.1016/j.braindev.2020.04.006
Kay, J. D., Hilton-Jones, D., and Hyman, N. (1986). Valproate toxicity and ornithine carbamoyltransferase deficiency. Lancet 2, 1283–1284. doi: 10.1016/s0140-6736(86)92714-5
Kira, R., Torisu, H., Takemoto, M., Nomura, A., Sakai, Y., Sanefuji, M., et al. (2005). Genetic susceptibility to simple febrile seizures: Interleukin-1b promoter polymorphisms are associated with sporadic cases. Neurosci. Lett. 384, 239–244. doi: 10.1016/j.neulet.2005.04.097
Kobayashi, H., Fukuda, S., Yamada, S., Hasegawa, Y., Takahashi, T., Puvesuren, J., et al. (2016). Clinical features of carnitine deficiency secondary to pivalate-conjugated antibiotic therapy. J. Pediatr. 173, 183–187. doi: 10.1016/j.jpeds.2016.02.080
Kobayashi, K., Ohzono, H., Shinohara, M., Saitoh, M., Ohmori, I., Ohtsuka, Y., et al. (2012). Acute encephalopathy with a novel point mutation in the SCN2A gene. Epilepsy Res. 102, 109–112. doi: 10.1016/j.eplepsyres.2012.04.016
Koenning, M., Jackson, S., Hay, C. M., Faux, C., Kilpatrick, T. J., Willingham, M., et al. (2012). Myelin gene regulatory factor is required for maintenance of myelin and mature oligodendrocyte identity in the adult CNS. J. Neurosci. 32, 12528–12542. doi: 10.1523/JNEUROSCI.1069-12.2012
Koh, J. C., Murugasu, A., Krishnappa, J., and Thomas, T. (2019). Favorable outcomes with early interleukin 6 receptor blockade in severe acute necrotizing encephalopathy of childhood. Pediatr. Neurol. 98, 80–84. doi: 10.1016/j.pediatrneurol.2019.04.009
Kometani, H., Kawatani, M., Ohta, G., Okazaki, S., Ogura, K., Yasutomi, M., et al. (2014). Marked elevation of interleukin-6 in mild encephalopathy with a reversible splenial lesion (MERS) associated with acute focal bacterial nephritis caused by Enterococcus faecalis. Brain Dev. 36, 551–553. doi: 10.1016/j.braindev.2013.07.012
Korematsu, S., Miyahara, H., Nagakura, T., Suenobu, S., and Izumi, T. (2008). Theophylline-associated seizures and their clinical characterizations. Pediatr. Int. 50, 95–98. doi: 10.1111/j.1442-200X.2007.02524.x
Korthagen, N. M., van Moorsel, C. H., Kazemier, K. M., Ruven, H. J., and Grutters, J. C. (2012). IL1RN genetic variations and risk of IPF: A meta-analysis and mRNA expression study. Immunogenetics 64, 371–377. doi: 10.1007/s00251-012-0604-6
Kubota, M., Chida, J., Hoshino, H., Ozawa, H., Koide, A., Kashii, H., et al. (2012). Thermolabile CPT II variants and low blood ATP levels are closely related to severity of acute encephalopathy in Japanese children. Brain Dev. 34, 20–27. doi: 10.1016/j.braindev.2010.12.012
Kuki, I., Inoue, T., Nukui, M., Okazaki, S., Kawawaki, H., Ishikawa, J., et al. (2021). MRI findings at neurological onset predict neurological prognosis in hemorrhagic shock and encephalopathy syndrome. J. Neurol. Sci. 430:120010. doi: 10.1016/j.jns.2021.120010
Kurahashi, H., Azuma, Y., Masuda, A., Okuno, T., Nakahara, E., Imamura, T., et al. (2018). MYRF is associated with encephalopathy with reversible myelin vacuolization. Ann. Neurol. 83, 98–106. doi: 10.1002/ana.25125
LaRovere, K. L., Riggs, B. J., Poussaint, T. Y., Young, C. C., Newhams, M. M., Maamari, M., et al. (2021). Neurologic involvement in children and adolescents hospitalized in the United States for COVID-19 or multisystem inflammatory syndrome. JAMA Neurol. 78, 536–547. doi: 10.1001/jamaneurol.2021.0504
Larrick, J. W., and Kunkel, S. L. (1986). Is Reye’s syndrome caused by augmented release of tumor necrosis factor? Lancet 2, 132–133. doi: 10.1016/s0140-6736(86)91947-1
Levine, J. M., Ahsan, N., Ho, E., and Santoro, J. D. (2020). Genetic acute necrotizing encephalopathy associated with RANBP2: Clinical and therapeutic implications in pediatrics. Mult. Scler. Relat. Disord. 43, 102194. doi: 10.1016/j.msard.2020.102194
Lindan, C. E., Mankad, K., Ram, D., Kociolek, L. K., Slivera, V. M., Boddaert, N., et al. (2021). Neuroimaging manifestations in children with SARS-CoV-2 infection: A multinational, multicentre collaborative study. Lancet Child Adolesc. Health 5, 167–177. doi: 10.1016/S2352-4642(20)30362-X
Lingwood, C. (2020). Verotoxin receptor-based pathology and therapies. Front. Cell. Infect. Microbiol. 10:123. doi: 10.3389/fcimb.2020.00123
Lokrantz, C.-M., Eriksson, B., Rosén, I., and Asztely, F. (2004). Hyperammonemic encephalopathy induced by a combination of valproate and pivmecillinam. Acta Neurol. Scand. 109, 297–301. doi: 10.1046/j.1600-0404.2003.00227.x
Makino, Y., Sugiura, T., Sugiyama, N., and Koyama, N. (2007). Carnitine-associated encephalopathy caused by long-term treatment with an antibiotic containing pivalic acid. Pediatrics 120, e739–e741. doi: 10.1542/peds.2007-0339
Mizuguchi, M., Hayashi, M., Nakano, I., Kuwashima, M., Yoshida, K., Nakai, Y., et al. (2002). Concentric structure of thalamic lesions of acute necrotizing encephalopathy. Neuroradiology 44, 489–493. doi: 10.1007/s00234-002-0773-3
Mizuguchi, M., Ichiyama, T., Imataka, G., Okumura, A., Goto, T., Sakuma, H., et al. (2021). Guidelines for the diagnosis and treatment of acute encephalopathy in childhood. Brain Dev. 43, 2–31. doi: 10.1016/j.braindev.2020.08.001
Mizuguchi, M., Sugatani, J., Maeda, T., Momoi, T., Arima, K., Takashima, S., et al. (2001). Cerebrovascular damage in young rabbits after intravenous administration of Shiga toxin 2. Acta Neuropathol. 102, 306–312. doi: 10.1007/s004010100384
Mizuguchi, M., Yamanouchi, H., Ichiyama, T., and Shiomi, M. (2007). Acute encephalopathy associated with influenza and other viral infections. Acta Neurol. Scand. 115, 45–56. doi: 10.1111/j.1600-0404.2007.00809.x
Nabbout, R., Vezzani, A., Dulac, O., and Chiron, C. (2011). Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol. 10, 99–108. doi: 10.1016/S1474-4422(10)70214-3
Nagao, T., Morishima, T., Kimura, H., Yokota, S., Yamashita, N., Ichiyama, T., et al. (2008). Prognostic factors in influenza-associated encephalopathy. Infect. Dis. J. 27, 384–389. doi: 10.1097/INF.0b013e318162a13b
Nakada, T., Kwee, I. L., Lerner, A. M., and Remler, M. P. (1983). Theophylline-induced seizures: Clinical and pathophysiologic aspects. West. J. Med. 138, 371–374.
Neilson, D. E., Adams, M. D., Orr, C. M., Schelling, D. K., Eiben, R. M., Kerr, D. S., et al. (2009). Infection-triggered familial or recurrent cases of acute necrotizing encephalopathy caused by mutations in a component of the nuclear pore, RANBP2. Am. J. Hum. Genet. 84, 44–51. doi: 10.1016/j.ajhg.2008.12.009
Neilson, D. E., Eiben, R. M., Waniewski, S., Hoppel, C. L., Varnes, M. E., Bangert, B. A., et al. (2003). Autosomal dominant acute necrotizing encephalopathy. Neurology 61, 226–230. doi: 10.1212/01.wnl.0000073544.28775.1a
Neilson, D. E., Feiler, H. S., Wilhelmsen, K. C., Lynn, A., Eiben, R. M., Kerr, D. S., et al. (2004). Autosomal dominant acute necrotizing encephalopathy maps to 2q12.1-2q13. Ann. Neurol. 55, 291–294. doi: 10.1002/ana.10849
Numoto, S., Kurahashi, H., Sato, A., Kubota, M., Shiihara, T., Okanishi, T., et al. (2021). Acute encephalopathy in children with tuberous sclerosis complex. Orphanet J. Rare Dis. 16, 5. doi: 10.1186/s13023-020-01646-8
Oby, E., and Janigro, D. (2006). The blood-brain barrier and epilepsy. Epilepsia 47, 1761–1774. doi: 10.1111/j.1528-1167.2006.00817.x
Oh, H. H., Kwon, S. H., Kim, C. W., Choe, B. H., Ko, C. W., Jung, H. D., et al. (2004). Molecular analysis of HLA class II-associated susceptibility to neuroinflammatory diseases in Korean children. J. Korean Med. Sci. 19, 426–430. doi: 10.3346/jkms.2004.19.3.426
Ohashi, E., Hayakawa, I., Murofushi, Y., Kawai, M., Suzuki-Muromoto, S., Abe, Y., et al. (2021). Recurrent acute necrotizing encephalopathy in a boy with RANBP2 mutation and thermolabile CPT2 variant: The first case of ANE1 in Japan. Brain Dev. 43, 873–878. doi: 10.1016/j.braindev.2021.04.009
Ohmura, K., Suzuki, Y., Saito, Y., Wada, T., Goto, M., and Seto, S. (2012). Sporadic hemiplegic migraine presenting as acute encephalopathy. Brain Dev. 34, 691–695. doi: 10.1016/j.braindev.2011.11.002
Okada, T., Fujita, Y., Imataka, G., Takase, N., Tada, H., Sakuma, H., et al. (2022). Increased cytokines/chemokines and hyponatremia as a possible cause of clinically mild encephalitis/encephalopathy with a reversible splenial lesion associated with acute focal bacterial nephritis. Brain Dev. 44, 30–35. doi: 10.1016/j.braindev.2021.07.008
Okumura, A., Abe, S., Kidokoro, H., and Mizuguchi, M. (2009). Acute necrotizing encephalopathy: A comparison between influenza and non-influenza cases. Microbiol. Immunol. 53, 277–280. doi: 10.1111/j.1348-0421.2009.00124.x
Okumura, A., Uematsu, M., Imataka, G., Tanaka, M., Okanishi, T., Kubota, T., et al. (2012). Acute encephalopathy in children with Dravet syndrome. Epilepsia 53, 79–86. doi: 10.1111/j.1528-1167.2011.03311.x
Pessayre, D., Mansouri, A., Hauozi, D., and Fromenty, B. (1999). Hepatotoxicity due to mitochondrial dysfunction. Cell. Biol. Toxicol. 15, 367–373. doi: 10.1023/a:1007649815992
Ray, S. T. J., Abdel-Mannan, O., Sa, M., Fuller, C., Wood, G. K., Pysden, K., et al. (2021). Neurological manifestations of SARS-CoV-2 infection in hospitalised children and adolescents in the UK: A prospective national cohort study. Lancet Child Adolesc. Health 5, 631–641. doi: 10.1016/S2352-4642(21)00193-0
Rinka, H., Yoshida, T., Kubota, T., Tsuruwa, M., Fuke, A., Yoshimoto, A., et al. (2008). Hemorrhagic shock and encephalopathy syndrome–the markers for an early HSES diagnosis. BMC Pediatr. 8:43. doi: 10.1186/1471-2431-8-43
Saitoh, M., Shinohara, M., Ishii, A., Ihara, Y., Hirose, S., Shiomi, M., et al. (2015b). Clinical and genetic features of acute encephalopathy in children taking theophylline. Brain Dev. 37, 463–470. doi: 10.1016/j.braindev.2014.07.010
Saitoh, M., Ishii, A., Ihara, Y., Hoshino, A., Terashima, H., Kubota, M., et al. (2015a). Missense mutations in sodium channel SCN1A and SCN2A predispose children to encephalopathy with severe febrile seizures. Epilepsy Res. 117, 1–6. doi: 10.1016/j.eplepsyres.2015.08.001
Saitoh, M., Kobayashi, K., Ohmori, I., Tanaka, Y., Tanaka, K., Inoue, T., et al. (2016). Cytokine-related and sodium channel polymorphism as candidate predisposing factors for childhood encephalopathy FIRES/AERRPS. J. Neurol. Sci. 368, 272–276. doi: 10.1016/j.jns.2016.07.040
Saitoh, M., Shinohara, M., Hoshino, H., Kubota, M., Amemiya, K., Takanashi, J.-I., et al. (2012). Mutations of the SCN1A gene in acute encephalopathy. Epilepsia 53, 558–564. doi: 10.1111/j.1528-1167.2011.03402.x
Santtila, S., Savinainen, K., and Hurme, M. (1998). Presence of the IL-1RA allele 2 (IL1RN*2) is associated with enhanced IL-1β production in vitro. Scand. J. Immunol. 47, 195–198. doi: 10.1046/j.1365-3083.1998.00300.x
Sato, K., Kubo, S., Fujii, H., Okamoto, M., Takahashi, K., Takamatsu, K., et al. (2012). Diffusion tensor imaging and magnetic resonance spectroscopy of transient cerebral white matter lesions in X-linked Charcot-Marie-Tooth disease. J. Neurol. Sci. 316, 178–180. doi: 10.1016/j.jns.2012.01.017
Segura-Bruna, N., Rodoriguez-Campello, A., Puente, V., and Roquer, J. (2006). Valproate-induced hyperammonemic encephalopathy. Acta Neurol. Scand. 114, 1–7. doi: 10.1111/j.1600-0404.2006.00655.x
Sell, K., Storch, K., Hahn, G., Lee-Kirsch, M. A., Ramantani, G., Jackson, S., et al. (2016). Variable clinical course in acute necrotizing encephalopathy and identification of a novel RANBP2 mutation. Brain Dev. 38, 777–780. doi: 10.1016/j.braindev.2016.02.007
Shibata, A., Kasai, M., Hoshino, A., Miyagawa, T., Matsumoto, H., Yamanaka, G., et al. (2019). Thermolabile polymorphism of carnitine palmitoyltransferase 2: A genetic risk factor of overall acute encephalopathy. Brain Dev. 41, 862–869. doi: 10.1016/j.braindev.2019.07.008
Shibata, A., Kasai, M., Hoshino, A., and Mizuguchi, M. (2022). Association of IL-1B rs16944 polymorphism with acute encephalopathy with biphasic seizures and late reduced diffusion is opposite to that of febrile seizures. Front. Neurol. 13:891721. doi: 10.3389/fneur.2022.891721
Shibata, A., Kasai, M., Hoshino, A., Tanaka, T., and Mizuguchi, M. (2021). RANBP2 mutation causing autosomal dominant acute necrotizing encephalopathy attenuates its interaction with COX11. Neurosci. Lett. 763:136173. doi: 10.1016/j.neulet.2021.136173
Shibata, A., Kasai, M., Terashima, H., Hoshino, A., Miyagawa, T., Kikuchi, K., et al. (2020). Case-control association study of rare nonsynonymous variants of SCN1A and KCNQ2 in acute encephalopathy with biphasic seizures and late reduced diffusion. J. Neurol. Sci. 414:116808. doi: 10.1016/j.jns.2020.116808
Shiihara, T., Kato, M., Ichiyama, T., Takahashi, Y., Tanuma, N., Miyata, R., et al. (2006). Acute encephalopathy with refractory status epilepticus: Bilateral mesial temporal and claustral lesions, associated with a peripheral marker of oxidative DNA damage. J. Neurol. Sci. 250, 159–161. doi: 10.1016/j.jns.2006.07.002
Shimada, S., Oguni, H., Otani, Y., Nishikawa, A., Ito, S., Eto, K., et al. (2018). An episode of acute encephalopathy with biphasic seizures and late reduced diffusion followed by hemiplegia and intractable epilepsy observed in a patient with a novel frameshift mutation in HNRNPU. Brain Dev. 40, 813–818. doi: 10.1016/j.braindev.2018.05.010
Shinohara, M., Saitoh, M., Nishizawa, D., Ikeda, K., Hirose, S., Takanashi, J.-I., et al. (2013). ADORA2A polymorphism predisposes children to encephalopathy with febrile status epilepticus. Neurology 80, 1571–1576. doi: 10.1212/WNL.0b013e31828f18d8
Shinohara, M., Saitoh, M., Takanashi, J., Yamanouchi, H., Kubota, M., Goto, T., et al. (2011). Carnitine palmitoyl transferase II polymorphism is associated with multiple syndromes of acute encephalopathy with various infectious diseases. Brain Dev. 33, 512–517. doi: 10.1016/j.braindev.2010.09.002
Specchio, N., Fusco, L., and Vigevano, F. (2011). Acute-onset epilepsy triggered by fever mimicking FIRES (febrile infection-related epilepsy syndrome): The role of protocadherin 19 (PCDH19) gene mutation. Epilepsia 52, e172–e175. doi: 10.1111/j.1528-1167.2011.03193.x
Stanley, C. A. (2004). Carnitine deficiency disorders in children. Ann. N. Y. Acad. Sci. 1033, 42–51. doi: 10.1196/annals.1320.004
Takahashi, K., Saitoh, M., Hoshino, H., Mimaki, M., Yokoyama, Y., Takamizawa, M., et al. (2007). A case of primary erythermalgia, wintry hypothermia and encephalopathy. Neuropediatrics 38, 157–159. doi: 10.1055/s-2007-990265
Takanashi, J. (2009). Two newly proposed infectious encephalitis/encephalopathy syndromes. Brain Dev. 31, 521–528. doi: 10.1016/j.braindev.2009.02.012
Takanashi, J., Mizuguchi, M., Terai, M., and Barkovich, A. J. (2015). Disrupted glutamate-glutamine cycle in acute encephalopathy with biphasic seizures and late reduced diffusion. Neuroradiology 57, 1163–1168. doi: 10.1007/s00234-015-1573-x
Takanashi, J., Oba, H., Barkovich, A. J., Tada, H., Tanabe, Y., Yamanouchi, H., et al. (2006). Diffusion MRI abnormalities after prolonged febrile seizures with encephalopathy. Neurology 66, 1304–1309. doi: 10.1212/01.wnl.0000210487.36667.a5
Takanashi, J., Tada, H., Terada, H., and Barkovich, A. J. (2009). Excitotoxicity in acute encephalopathy with biphasic seizures and late reduced diffusion. AJNR Am. J. Neuroradiol. 30, 132–135. doi: 10.3174/ajnr.A1247
Takayanagi, M., Haginoya, K., Umehara, N., Kitamura, T., Numata, Y., Wakusawa, K., et al. (2010). Acute encephalopathy with a truncation mutation in the SCN1A gene: A case report. Epilepsia 51, 1886–1888. doi: 10.1111/j.1528-1167.2010.02600.x
Tang, S., Lin, J. P., Hughes, E., Siddiqui, A., Lim, M., and Lascelles, K. (2011). Encephalopathy and SCN1A mutations. Epilepsia 52, e26–e30. doi: 10.1111/j.1528-1167.2011.03019.x
Trost, L. C., and Lemasters, J. J. (1997). Role of the mitochondrial permeability transition in salicylate toxicity to cultured rat hepatocytes: Implications for the pathogenesis of Reye’s syndrome. Toxicol. Appl. Pharmacol. 147, 431–441. doi: 10.1006/taap.1997.8313
Tsai, F. J., Hsieh, Y. Y., Chang, C. C., Lin, C. C., and Tsai, C. H. (2002). Polymorphisms for interleukin 1β exon 5 and interleukin 1 receptor antagonist in Taiwanese children with febrile convulsions. Arch. Pediatr. Adolesc. Med. 156, 545–548. doi: 10.1001/archpedi.156.6.545
Ubbink, J. B., Delport, R., Becker, P. J., and Bissbort, S. (1989). Evidence of a theophylline-induced vitamin B6 deficiency caused by noncompetitive inhibition of pyridoxal kinase. Lab. Clin. Med. 113, 15–22.
Verrotti, A., Trotta, D., Morgese, G., and Chiarelli, F. (2002). Valproate-induced hyperammonemic encephalopathy. Metab. Brain Dis. 17, 367–373. doi: 10.1023/a:1021918104127
Vezzani, A., Balosso, S., and Ravizza, T. (2008). The role of cytokines in the pathophysiology of epilepsy. Brain Behav. Immun. 22, 797–803. doi: 10.1016/j.bbi.2008.03.009
Vezzani, A., and Granata, T. (2005). Brain inflammation in epilepsy: Experimental and clinical evidence. Epilepsia 46, 1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x
Vianey-Saban, C., Mousson, B., Bertrand, C., Stamm, D., Dumoulin, R., Zabot, M. T., et al. (1993). Carnitine palmitoyl transferase I deficiency presenting as a Reye-like syndrome without hypoglycaemia. Eur. J. Pediatr. 152, 334–338. doi: 10.1007/BF01956748
Visentin, M., Salmona, M., and Tacconi, M. T. (1995). Reye’s and Reye-like syndromes, drug-related diseases? (Causative agents, etiology, pathogenesis, and therapeutic approaches). Drug Metab. Rev. 27, 517–539. doi: 10.3109/03602539508998334
Wang, Y., and Yin, F. (2016). A review of X-linked Charcot-Marie-Tooth Disease. J. Child Neurol. 31, 761–772. doi: 10.1177/0883073815604227
Warter, J. M., Marescaux, C., Brandt, C., Rumbach, L., Micheletti, G., Chabrier, G., et al. (1983). Sodium valproate associated with phenobarbital: Effects on ammonia metabolism in humans. Epilepsia 24, 628–633. doi: 10.1111/j.1528-1157.1983.tb03428.x
Yamaguchi, H., Nagase, H., Ito, Y., Matsunoshita, N., Mizutani, M., Matsushige, T., et al. (2018). Acute focal bacterial nephritis characterized by acute encephalopathy with biphasic seizures and late reduced diffusion. J. Infect. Chemother. 24, 932–935. doi: 10.1016/j.jiac.2018.04.007
Yamazaki, S., Ikeno, K., Abe, T., Tohyama, J., and Adachi, Y. (2011). Hemiconvulsion-hemiplegia-epilepsy syndrome associated with CACNA1A S218L mutation. Pediatr. Neurol. 45, 193–196. doi: 10.1016/j.pediatrneurol.2011.04.010
Yao, D., Mizuguchi, H., Yamaguchi, M., Yamada, H., Chida, J., Shikata, K., et al. (2008). Thermal instability of compound variants of carnitine palmitoyltransferase II and impaired mitochondrial fuel utilization in influenza-associated encephalopathy. Hum. Mutat. 29, 718–727. doi: 10.1002/humu.20717
Yao, M., Yao, D., Yamaguchi, M., Chida, J., Yao, D., and Kido, H. (2011). Bezafibrate upregulates carnitine palmitoyltransferase II expression and promotes mitochondrial energy crisis dissipation in fibroblasts of patients with influenza-associated encephalopathy. Mol. Genet. Metab. 104, 265–272. doi: 10.1016/j.ymgme.2011.07.009
Yoshikawa, H. (2007). First-line therapy for theophylline-associated seizures. Acta Neurol. Scand. Suppl. 186, 57–61. doi: 10.1111/j.1600-0404.2007.00810.x
Yu, X., Zhang, N., Liu, S., Xi, Z., and Zhang, Y. (2018). Polymorphisms in the interleukin- 1b (IL-1B) and interleukin-1a (IL-1A) genes on risk of febrile seizures: A meta-analysis. Neurol. Sci. 39, 1529–1536. doi: 10.1007/s10072-018-3449-4
Keywords: acute encephalopathy, infection, drug, immune response, metabolism, neuronal excitation, susceptibility gene
Citation: Mizuguchi M, Shibata A, Kasai M and Hoshino A (2023) Genetic and environmental risk factors of acute infection-triggered encephalopathy. Front. Neurosci. 17:1119708. doi: 10.3389/fnins.2023.1119708
Received: 09 December 2022; Accepted: 09 January 2023;
Published: 24 January 2023.
Edited by:
Go Kawano, St. Mary’s Hospital, JapanReviewed by:
Kazuhiro Muramatsu, Jichi Medical University, JapanHideyuki Iwayama, Aichi Medical University, Japan
Copyright © 2023 Mizuguchi, Shibata, Kasai and Hoshino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masashi Mizuguchi,  YnJhZGV2QG0udS10b2t5by5hYy5qcA==
YnJhZGV2QG0udS10b2t5by5hYy5qcA==