 Benjamin Klocke
Benjamin Klocke Kylie Krone
Kylie Krone Jason Tornes
Jason Tornes Carter Moore
Carter Moore Pothitos M. Pitychoutis
Pothitos M. Pitychoutis- Department of Biology, University of Dayton, Dayton, OH, United States
Calcium (Ca2+) comprises a critical ionic second messenger in the central nervous system that is under the control of a wide array of regulatory mechanisms, including organellar Ca2+ stores, membrane channels and pumps, and intracellular Ca2+-binding proteins. Not surprisingly, disturbances in Ca2+ homeostasis have been linked to neurodegenerative disorders, such as Alzheimer’s and Parkinson’s diseases. However, aberrations in Ca2+ homeostasis have also been implicated in neuropsychiatric disorders with a strong neurodevelopmental component including autism spectrum disorder (ASD) attention-deficit hyperactivity disorder (ADHD) and schizophrenia (SCZ). While plasma membrane Ca2+ channels and synaptic Ca2+-binding proteins have been extensively studied, increasing evidence suggests a prominent role for intracellular Ca2+ stores, such as the endoplasmic reticulum (ER), in aberrant neurodevelopment. In the context of the current mini-review, we discuss recent findings implicating critical intracellular Ca2+-handling regulators such as the sarco-ER Ca2+ ATPase 2 (SERCA2), ryanodine receptors (RyRs), inositol triphosphate receptors (IP3Rs), and parvalbumin (PVALB), in the emergence of ASD, SCZ, and ADHD.
1. Introduction
Neurodevelopmental disorders (e.g., autism spectrum disorder; ASD, and attention-deficit hyperactivity disorder; ADHD) and schizophrenia (SCZ), a neuropsychiatric disorder with a strong neurodevelopmental component (Birnbaum and Weinberger, 2017; Seidman and Mirsky, 2017; Rund, 2018), comprise debilitating diseases that are highly variable in their symptomatology and etiology (McGrath et al., 2008; Christensen et al., 2016; Hansen et al., 2018; Sayal et al., 2018). These disorders arise due to the complex interplay between genetic risk factors and early life environmental stressors, including prenatal complications, malnutrition, hormone imbalance, and exposure to environmental toxins (e.g., neurotoxic metals) (Wetmore and Garner, 2010; Lord et al., 2018; Li et al., 2019; Ijomone et al., 2020). Recent research efforts have sought to identify common disrupted molecular mechanisms that may lead to abnormal neurodevelopment. One such candidate which has garnered interest is the disruption of intracellular calcium (Ca2+) homeostasis.
Intracellular Ca2+ concentration is critical for orchestrating numerous cellular processes, including signal transduction and gene expression (Bootman et al., 2001; Naranjo and Mellström, 2012; Bononi et al., 2013; Brini et al., 2014; Britzolaki et al., 2018). Consequently, Ca2+ mishandling is implicated in the pathophysiology of neurodegenerative disorders (e.g., Alzheimer’s and Parkinson’s diseases) (Pchitskaya et al., 2018), while recent evidence suggests that aberrations in intracellular Ca2+ signaling may also underlie abnormal neurodevelopment (Pourtavakoli and Ghafouri-Fard, 2022). Of the major neuronal Ca2+-handling players, plasma membrane voltage-gated Ca2+ channels (e.g., Cacna1) are well-reviewed with regards to their role in neurodevelopment (Breitenkamp et al., 2015; Cupertino et al., 2016; Pourtavakoli and Ghafouri-Fard, 2022). Readers are referred to recent excellent reviews discussing the implication of critical plasma membrane Ca2+ players (e.g., CACNA1) and Ca2+-binding proteins involved in synaptic release (e.g., Synaptotagmin) in the pathophysiology of brain disorders (Breitenkamp et al., 2015; Cupertino et al., 2016; Pourtavakoli and Ghafouri-Fard, 2022). Interestingly, dysfunction of endoplasmic reticulum (ER) Ca2+ regulators such as the sarco-ER Ca2+ ATPase 2 (SERCA2), which sequesters cytosolic Ca2+ into the ER, and the Ca2+-releasing channels inositol triphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) have recently garnered interest in the pathophysiology of brain disorders (Britzolaki et al., 2018, 2020). In the context of the current mini-review, we discuss recent findings implicating aberrant ER-dependent Ca2+ homeostasis as a convergent pathophysiological mechanism in brain disorders with a strong neurodevelopmental component.
2. Autism spectrum disorders (ASD)
2.1. Ryanodine receptors (RyRs) and the fragile X messenger ribonucleoprotein 1 (FMR1)
Autism spectrum disorders is a neurodevelopmental disorder which comprises a wide array of behavioral symptoms including impaired sociability and communication skills, repetitive behaviors, and intellectual disability (Christensen et al., 2016; Lord et al., 2018). Although no single genetic factor is responsible for ASD, RyRs have been identified as a potential contributor to ASD pathology. RyRs are homotetrameric Ca2+-releasing channels expressed on the neuronal ER membrane; upon opening, the RyRs allow for the flux of Ca2+ ions from the ER stores into the cytosol (Figure 1; Abu-Omar et al., 2018). Notably, clinical studies suggest that mutations in genes coding for the different RyRs isoforms could possibly contribute to the pathophysiology of ASD. A copy number variation study has revealed a likely pathogenic duplication at 1q43, which encompasses the RYR2 gene, thus identifying RYR2 as a potential ASD risk gene (Soueid et al., 2016; Keil et al., 2019). Despite the fact that Ryr3 has been shown to contribute to synaptic plasticity and cognitive flexibility in mice (Balschun et al., 1999), an earlier clinical study did not report an association between RYR3 and ASD in a Japanese patient cohort (Tochigi et al., 2008). However, a more recent targeted sequencing and integrative analysis study of 3,195 Chinese patients with neurodevelopmental disorders exposed RYR3 as one of the six novel candidate genes to preferentially contribute to ASD (Wang T. et al., 2021).
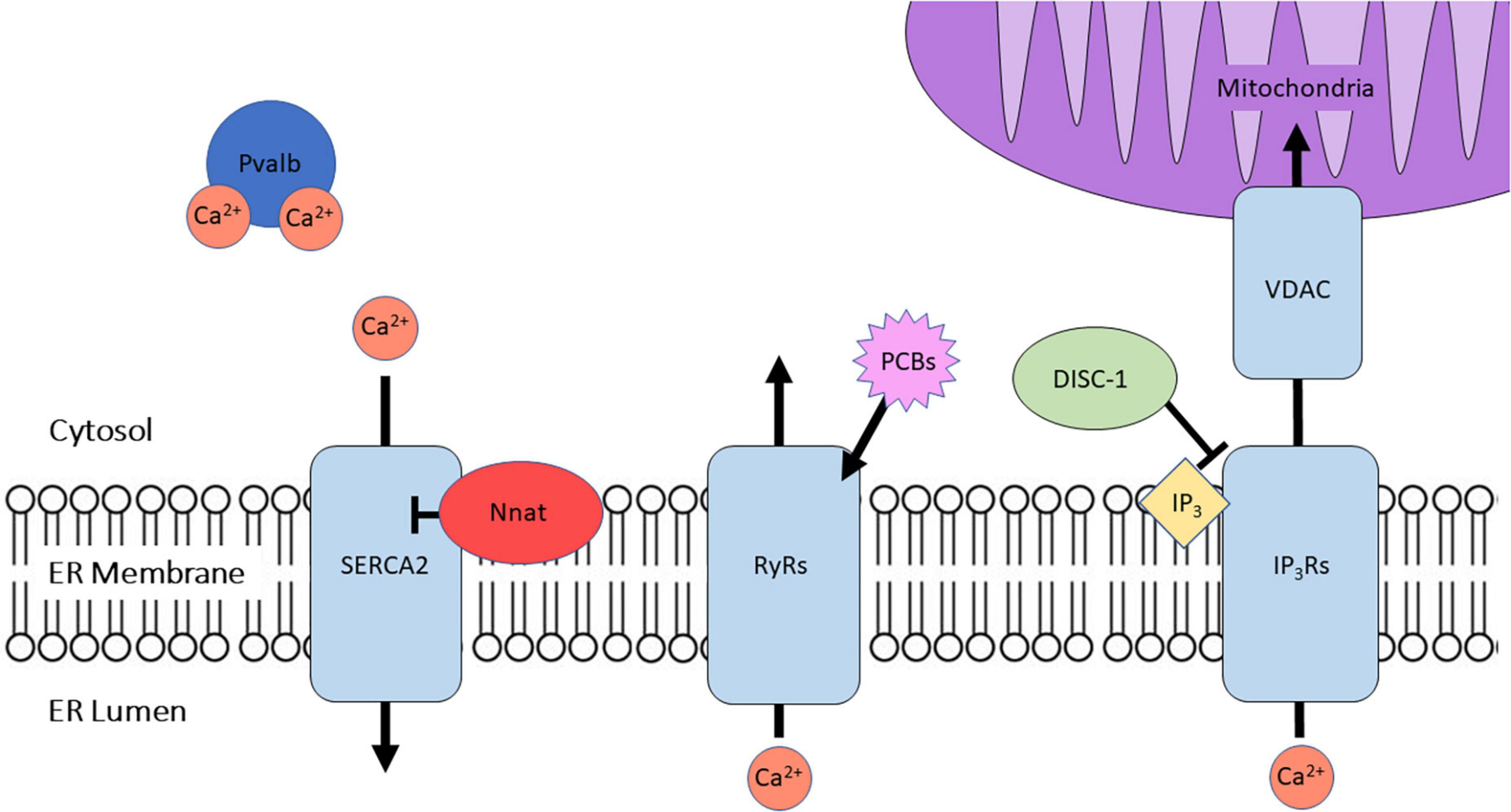
Figure 1. A summary of the proteins discussed herein and their primary role in regulating intracellular Ca2+ homeostasis. Proteins mediating ER Ca2+ efflux include ryanodine receptors (RyRs) and inositol triphosphate receptors (IP3Rs), while cytosolic Ca2+ is handled by the sarco-endoplasmic reticulum Ca2+ ATPase 2 (SERCA2), which is negatively regulated by neuronatin (Nnat). Parvalbumin (Pvalb) regulates cytosolic Ca2+ buffering via directly binding Ca2+ ions. Polychlorinated biphenyls (PCBs) target RyRs to exert their effects on Ca2+ homeostasis. IP3Rs associate with mitochondrial voltage-dependent anion channels (VDAC) in the mitochondrial-associated membranes (MAMs), and are regulated in part by disrupted in schizophrenia 1 protein (DISC-1).
Preclinical studies have provided intriguing mechanistic insights into how RyR dysfunction could affect intracellular Ca2+ homeostasis and ASD-relevant phenotypes and endophenotypes in animal models. Interestingly, mutations in the RYR1 and the fragile X messenger ribonucleoprotein 1 (FMR1) genes have both been associated with impaired Ca2+ signaling. Specifically, preclinical evidence suggests that the human T4826I-RYR1 gain-of-function mutation and the human CGG-repeat expansion in the FMR1 gene (i.e., FMR1 premutation), are both associated with elevated intracellular Ca2+ signaling; indeed, the T4826I-RYR1 gain-of-function mutation has been shown to result in increased intracellular Ca2+ concentrations in muscle cells (Barrientos et al., 2012), while murine cortical astrocytes with the FMR1 premutation displayed enhanced asynchronous Ca2+ oscillations (Chen et al., 2010; Cao et al., 2013; Robin et al., 2017). Notably, Ca2+ signaling is critical for ensuring proper dendritic morphology and synaptic connectivity. Keil et al. (2019) assessed ASD-relevant behavioral and neurobiological correlates (i.e., dendritic morphology and social behavior) in adolescent mice with the humanized T4826I-RYR1 gain-of-function mutation and with the FMR1 premutation, as well as in double mutant (DM) mice (Keil et al., 2019). Interestingly, social deficits in T4826I male and DM female mice were both accompanied by abnormal dendritic morphology (Keil et al., 2019). Based on the authors, the observed changes in dendritic morphology in these mice could be attributed to altered intracellular Ca2+ dynamics, even though additional studies are needed to yield more conclusive results (Keil et al., 2019).
Sethi et al. (2021) conducted a follow-up study to understand the interaction of Ryr1 and Fmr1 and polychlorinated-biphenyls (PCBs) exposure in ASD-like behaviors (Sethi et al., 2021). PCBs comprise environmental contaminants with established neurodevelopmental consequences that exert their neurotoxic effects by binding to the RyRs (Pessah et al., 2010). In that study, dams were orally administered a PCB mixture from 2-weeks prior to mating until pup weaning (P21). Ultrasonic vocalizations at P7 were diminished in all three mutant pup genotypes while both male and female T4826I and DM pups exhibited high spontaneous grooming behavior (Sethi et al., 2021). Further, studies from the same group found that PCBs promote synaptogenesis in cultured hippocampal neurons, as evidenced by increased dendritic spines and miniature excitatory postsynaptic currents (Lesiak et al., 2014). Importantly, these effects were found to be RyR-dependent, as treatment with either the RyR inhibitor FLA365 or RyR siRNA both rescued these effects. Taken together, these preclinical studies suggest that mutations in Ryr1 and Fmr1, two genes shown to be involved in neuronal Ca2+ handling, exert ASD-like behavioral and neuroarchitecture consequences in mice. Overall, these studies indicate that both genetic and environmental perturbation of neuronal Ca2+ homeostasis may contribute to aberrant synaptogenesis, dendritic arborization, and ultimately ASD-like behaviors.
2.2. Parvalbumin (PVALB)
Parvalbumin (PVALB) is a Ca2+-buffering protein primarily expressed in the γ-aminobutyric acid (GABA) positive interneurons of the brain that exhibit rapid burst-firing activity and are heavily dependent on intracellular Ca2+-handling (Ruden et al., 2021). While dysfunction of PVALB+ neurons is well-known to contribute to aberrant neurodevelopmental processes and ASD, the role of PVALB in maintaining the integrity of intracellular Ca2+ signaling pathways and its potential contribution to ASD has received less attention (Ruden et al., 2021). Interestingly, Pvalb–/– mice are known to exhibit an ASD-like behavioral phenotype (Wöhr et al., 2015). Recently, Janickova et al. (2020) explored the role of PVALB in regulating neuron morphology and dendritic arborization by utilizing a Pvalb–/– mouse strain in which EGFP expression was under the control of the Pvalb driver that allowed for visualization of PVALB+ neurons even in the absence of functional Pvalb expression (Janickova and Schwaller, 2020; Janickova et al., 2020). Interestingly, loss of PVALB function resulted in increased cell soma and mitochondrial size primarily in regions rich in PVALB+ neurons, such as the thalamic reticular nucleus (TRN), the molecular layer interneurons (MLI) of the cerebellum, the prefrontal cortex, and the striatum (Janickova and Schwaller, 2020; Janickova et al., 2020). Furthermore, loss of PVALB function was associated with dendritic hypertrophy in the dentate gyrus, the striatum, and the MLI, as well as by a shift of mitochondria from the central compartment of the cell to the subplasmalemmal region (Janickova and Schwaller, 2020). Taken together, these studies suggest that the impaired Ca2+ buffering brought about by the absence of PVALB may result in a compensatory proliferation and subplasmalemmal relocation of mitochondria to maintain the rapid Ca2+ dynamics these neurons rely on (Janickova and Schwaller, 2020; Ruden et al., 2021). Ultimately, this may result in enhanced dendritic arborization and oxidative stress. Although further studies are imperative, these data provide valuable insights into how PVALB-mediated Ca2+ dysfunction may induce ASD-relevant neurobiological correlates.
2.3. Inositol triphosphate receptors (IP3R)
G protein-coupled receptor (GPCR)-mediated IP3R Ca+2 signaling pathways comprise critical components of the intracellular Ca+2 handling machinery with potential implications in ASD (Berridge, 2009; Taylor and Tovey, 2010). For instance, the IP3R2 has been shown to affected by de novo copy number variants in ASD patient cohorts, while recently Ip3R2–/– mutant mice and astrocyte-specific Ip3R2 conditional knockout mice display ASD-like behaviors (Gilman et al., 2011; Wang Q. et al., 2021). Interestingly, ex vivo studies in human fibroblasts derived from patients with rare, monogenic forms of ASD (i.e., fragile X syndrome; FXS and tuberous sclerosis; TS) showed that ATP-evoked GPCR-mediated Ca2+ release from the IP3Rs was diminished in ASD fibroblasts (Schmunk et al., 2015). In a follow-up study, Schmunk et al. (2017) extended their findings by using fibroblasts from patients with sporadic ASD, as well as two additional monogenic forms of ASD (i.e., Prader–Willi syndrome and Rett syndrome), and observed a similar impaired IP3R-mediated Ca2+ signaling. Taken together, these studies suggest that depressed Ca2+ release through IP3R signaling may disrupt neurodevelopment. To our knowledge these studies have not been replicated in neural cells or in vivo models, but provide mechanistic insights into the putative implication of IP3Rs in the neurobiology of ASD.
2.4. Neuronatin (NNAT) and other genes
Neuronatin (NNAT) is a developmentally regulated ER resident protein and negative regulator of SERCA that is expressed in the brain’s PVALB + GABAergic neurons; NNAT has also been implicated in abnormal neurodevelopment, including ASD and Angelman Syndrome (AS) (Pitale et al., 2017; Vatsa et al., 2019). The miR-708, an NNAT downregulator, has been involved in the atypical Ca2+ signaling processes observed in the maternal-ubiquitin protein ligase E3A (Ube3a) deficient mouse model for AS (Vatsa et al., 2019). UBE3A plays a role in the proteasome-mediated degradation of proteins in neurons, and has thus been implicated in ASD and AS (Glessner et al., 2009; Williams et al., 2010; Yi et al., 2015; Xu et al., 2018; Lopez et al., 2019). Recently, Vatsa et al. (2019) identified miR-708 to be significantly downregulated in the cortex of maternal-Ube3a-deficient AS mice and showed that miR-708 regulates intracellular Ca2+ homeostasis by targeting NNAT (Vatsa et al., 2019). Taken together, these findings suggest that NNAT/miR-708-mediated aberrations in intracellular Ca2+ signaling may be involved in ASD/AS pathogenesis.
Interestingly, targeted sequencing and integrative analysis of 3,195 Chinese probands with several neurodevelopmental disorders exposed novel candidate genes involved in ASD, including three with relevance to Ca2+ homeostasis, namely: RYR3 [discussed in the Section “2.1. Ryanodine receptors (RyRs) and the fragile X messenger ribonucleoprotein 1 (FMR1)”], ubiquitin protein ligase E3 (UBR3), and filamin A (FLNA) (Wang T. et al., 2021). UBR3 inhibits the function of alpha 1C subunit of L-type voltage-dependent Ca2+ channel (Cav1.2) via the ubiquitin-proteasome protein degradation pathway and has been identified as a modulator of Ca2+ -induced Ca2+ release (CIRC) (Ma et al., 2020). FLNA is an actin-binding protein which regulates cytoskeletal remodeling and is regulated by Ca2+ and calmodulin, and has been shown to interact with FMR1 in long term memory processes in Drosophila (Nakamura et al., 2005; Bolduc et al., 2010; Rosa et al., 2019). Overall, these findings further support a role for intracellular Ca2+ homeostasis in ASD pathogenesis, although further research is considered imperative to confirm the contribution of these genes in neurodevelopment.
2.5. Ca2+ signaling in astrocytes
It is well established that Ca2+ signaling is also prevalent in astrocytes; while astrocytic dysfunction has been implicated in the pathophysiology of ASD, the precise mechanisms by which astrocytes contribute to disease progression and symptomatology remain elusive (Blanco-Suárez et al., 2017). The onset of ASD pathology is typically concurrent with neurodevelopmental astrocyte proliferation (Berger et al., 2013; Sigaard et al., 2016). Allen et al. (2022) sought to investigate the putative role of astrocytes in ASD pathologyClick or tap here to enter text. Upon harvesting astrocytes from organoids created by induced pluripotent stem cells (iPSCs) from ASD patients (Allen et al., 2022). Proteomic analysis revealed that “Ca2+ binding” processes were highly enriched in the altered protein networks observed in these ASD astrocytes. Follow-up two-photon live-cell imaging confirmed an exaggerated ATP-induced Ca2+ response in these ASD astrocytes. To investigate putative behavioral effects of Ca2+ disruption in ASD astrocytes, human-derived ASD astrocytes were implanted into mice at P1-3, thus generating ASD astrocyte chimeric mice. Engrafted human ASD astrocytes were found to exhibit aberrant Ca2+ fluctuations, as well as to result in ASD-relevant behaviors (i.e., enhanced repetitive behaviors in the marble burying test and impaired fear learning). Given the exaggerated Ca2+ response observed in ASD astrocytes, it was predicted that inhibition of IP3Rs would possibly restore Ca2+ signaling and function. Intriguingly, IP3R-knockdown in ASD astrocytes rescued the exaggerated Ca2+ response, hippocampal neuron network firing, and deficits in fear memory observed in chimeric mice (Allen et al., 2022). Overall, these data provide deep insights into the contribution of astrocytic Ca2+ dysregulation in the pathophysiology of ASD.
3. Schizophrenia (SCZ)
Schizophrenia is a brain disorder characterized by a constellation of symptoms including hallucinations, negative affect, and cognitive deficits (McCutcheon et al., 2020). The Disrupted in Schizophrenia–1 (DISC-1) protein is involved in numerous neuronal processes, including the regulation of dendrite morphology and neuronal migration during development (Balu and Coyle, 2011). Recent studies have suggested that DISC-1 is involved in Ca2+ regulation via the mitochondria-associated membranes (MAMs) which comprise physical connections formed between the ER IP3Rs and mitochondrial voltage-dependent anion channels (VDAC) that are involved in the transfer of Ca2+ and molecular stress signals between these two organelles (Park et al., 2017, 2015; van Vliet and Agostinis, 2018; Barazzuol et al., 2021; Means and Katz, 2021). Recent findings suggest that DISC-1 localizes to the MAM in mouse neurons, and specifically binds IP3R1 to reduce ligand-binding and subsequent Ca2+ transfer to the mitochondria in primary cortical neurons (Park et al., 2017). Upon DISC-1 dysfunction, IP3R1-mediated Ca2+ release into the MAM is disinhibited, causing a buildup of mitochondrial Ca2+ that leads to oxidative stress that ultimately impairs mitochondrial function (Park et al., 2017). Interestingly, neuronal oxidative stress has been implicated in the pathogenesis of SCZ (Emiliani et al., 2014). Taken together, this experimental evidence suggests that DISC-1 is involved in the dysregulation of Ca2+ handling in the MAMs, causing downstream mitochondrial Ca2+ hyper-accumulation and oxidative stress, shining a light on a novel mechanism by which DISC-1 may contribute to SCZ pathogenesis.
Darier’s disease is a skin condition characterized by persistent wart-like skin patches, which is due to a mutation in the SERCA2 gene that subsequently leads to Ca2+ dysfunction (Cooper and Burge, 2003). Interestingly, Darier’s disease patients have a significantly increased risk for SCZ, providing a causative link between SERCA2 and neurodevelopmental processes (Tang et al., 2010). Recently, Nakajima et al. (2021) generated a brain-specific heterozygous Serca2 loss-of-function mouse model (i.e., hetero cKO) to investigate how developmental hypofunction of Serca2 may affect SCZ-relevant behavioral and neurobiological processes (Nakajima et al., 2021). As expected, both primary hippocampal neurons and ER membranes isolated from the brain of hetero cKO mice exhibited impaired Ca2+ uptake (Nakajima et al., 2021). Hetero cKO mice exhibited impaired fear memory and enhanced exploratory behavior; moreover, microdialysis studies suggested that Serca2 hypofunction induces a hyperdopaminergic state in the nucleus accumbens (NAC) (Nakajima et al., 2021), echoing the neurochemical dopaminergic hallmarks of SCZ (McCutcheon et al., 2020). Taken together, these findings support the notion that developmental hypofunction of the Serca2 and subsequent aberrant intracellular Ca2+ handling induces SCZ-relevant behavioral and neurochemical effects.
Interestingly, recent evidence suggests an association between RyRs and SCZ. An exome sequencing study of childhood-onset SCZ patients, identified de novo variants of RYR2, which the authors highlight as a strong candidate gene given the role of RyRs in neurodevelopmental processes (Ambalavanan et al., 2016), further underscoring the putative role of RyRs in the neurobiology of neurodevelopmental disorders.
4. Attention-deficit hyperactivity disorder (ADHD)
Attention-deficit hyperactivity disorder is a neurodevelopmental disorder that is characterized by impaired attention, locomotor hyperactivity, and impulsive behaviors (Sharma and Couture, 2014). Preclinical evidence suggests that ADHD is associated with impaired intracellular Ca2+ handling; for instance, the spontaneously hypertensive rat (SHR) model of ADHD has been shown to exhibit impaired brain plasma membrane Ca2+ uptake (Horn et al., 1995; Lehohla et al., 2001). Further preclinical evidence has shown that knockout of the G-protein subunit Gβ5 (encoded by the gene Gnb5) elicits a pronounced ADHD-like hyperactive phenotype in mice (Xie et al., 2012). Moreover, a GNB5 mutation (i.e., GNB5 S81L) associated with impaired termination of DA2 receptor signaling was reported in a Saudi family presenting speech impairments and a variable ADHD diagnosis, providing initial clinical evidence for the putative role of GNB5 in the neurobiology of ADHD (Shamseldin et al., 2016). Interestingly, a recent study highlighted the role of GNB5 in store-operated Ca2+ entry (SOCE) (Kang et al., 2018). Upon depletion of ER Ca2+ stores, stromal interaction molecule 1 (STIM1), an ER Ca2+ sensor, forms a complex with the plasma membrane calcium release-activated calcium channel protein 1 (ORAI1) to initiate extracellular Ca2+ entry (Srikanth and Gwack, 2012). Kang et al. (2018) found that GNB5 expression enhances SOCE in vitro. Notably, the ability of GNB5 to enhance SOCE was found to depend on STIM1 function suggesting that GNB5 may interact with the ER Ca2+-sensing machinery to regulate Ca2+ homeostasis, although further studies are needed to determine the precise mechanisms that may underlie this process.
5. Conclusion
In the context of this mini-review, we have highlighted recent advances supporting the implication of prominent ER and cytosolic Ca2+ regulators (i.e., SERCA2, IP3Rs, RyRs, PVALB, NNAT) in the neurobiology of brain disorders with a strong neurodevelopmental component (Figure 1 and Table 1). Disease progression of monogenic brain disorders (e.g., AS, FXS) may be dependent on specific gene interactions with intracellular Ca2+ signaling mechanisms, whereas sporadic cases of SCZ, ASD, and ADHD may arise from polygenic variations that ultimately converge to the disruption of intracellular Ca2+ homeostasis and concomitant impairment of neuronal function. Further preclinical and clinical investigation is considered imperative to confirm and/or expand upon these intriguing discoveries in order to gain deep insights into the cellular and molecular Ca2+-dependent neurodevelopmental processes that are compromised in these debilitating brain diseases.
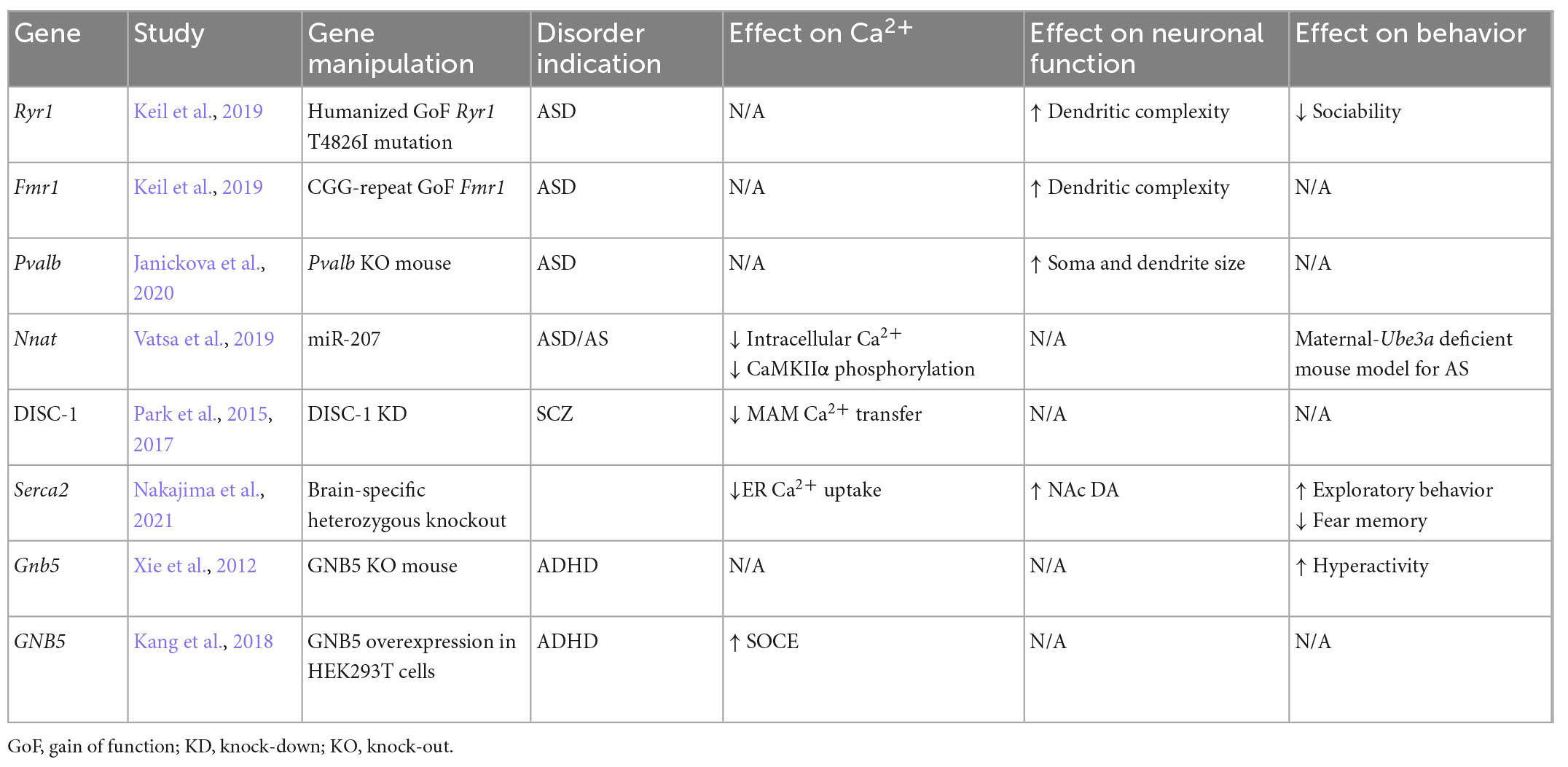
Table 1. Studies focusing on notable genes implicated in the pathophysiology of neurodevelopmental disorders and summarized findings on intracellular Ca2+ signaling, neuronal function, and behavior.
Author contributions
BK conducted the primary literature search and wrote first draft of the manuscript. KK, JT, CM, and HO wrote sections of the manuscript. PP formulated the concept and supervised the writing of the manuscript. All authors contributed to manuscript editing, revision, read, and approved the submitted version.
Funding
BK and JT were supported by the University of Dayton (UD) Graduate School and by the UD Office for Graduate Affairs through the Graduate Student Summer Fellowship (GSSF) Program. KK was supported by the UD Graduate School and the Department of Biology. CM was supported by the UD Honors Program and by the College of Arts and Sciences (CAS) Dean’s Summer Research fellowship program. HO was supported by the CAS Dean’s Summer Research fellowship program. PP was supported by funding from the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health (NIH) under award number R03NS109836. Funding sponsors had no further role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication. Publication fee was supported by the UD Office for Graduate Affairs.
Acknowledgments
This review manuscript was compiled in the context of the “Neurobiology of Disease” (BIO596) course at the University of Dayton.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abu-Omar, N., Das, J., Szeto, V., and Feng, Z. P. (2018). Neuronal ryanodine receptors in development and aging. Mol. Neurobiol. 55, 1183–1192. doi: 10.1007/s12035-016-0375-4
Allen, M., Huang, B. S., Notaras, M. J., Lodhi, A., Barrio-Alonso, E., Lituma, P. J., et al. (2022). Astrocytes derived from ASD individuals alter behavior and destabilize neuronal activity through aberrant Ca2+ signaling. Mol. Psychiatry 27, 2470–2484. doi: 10.1038/s41380-022-01486-x
Ambalavanan, A., Girard, S., Ahn, K., Zhou, S., Dionne-Laporte, A., Spiegelman, D., et al. (2016). De novo variants in sporadic cases of childhood onset schizophrenia. Eur. J. Hum. Genet. 6, 944–948. doi: 10.1038/ejhg.2015.218
Balschun, D., Wolfer, D., Bertocchini, F., Barone, V., Conti, A., Zuschratter, W., et al. (1999). Deletion of the ryanodine receptor type 3 (RyR3) impairs forms of synaptic plasticity and spatial learning. EMBO J. 19, 5264–5273. doi: 10.1093/emboj/18.19.5264
Balu, D. T., and Coyle, J. T. (2011). Neuroplasticity signaling pathways linked to the pathophysiology of schizophrenia. Neurosci. Biobehav. Rev. 35, 848–870. doi: 10.1016/j.neubiorev.2010.10.005
Barazzuol, L., Giamogante, F., and Calì, T. (2021). Mitochondria Associated Membranes (MAMs): Architecture and physiopathological role. Cell Calcium 94:102343. doi: 10.1016/j.ceca.2020.102343
Barrientos, G., Feng, W., Truong, K., Matthaei, K., Yang, T., Allen, P., et al. (2012). Gene dose influences cellular and calcium channel dysregulation in heterozygous and homozygous T4826I-RYR1 malignant hyperthermia-susceptible muscle. J. Biol. Chem. 4, 2863–2876. doi: 10.1074/jbc.M111.307926
Berger, J. M., Rohn, T. T., and Oxford, J. T. (2013). Autism as the early closure of a neuroplastic critical period normally seen in adolescence. Biol. Syst. Open Access. 1, 10.4172/2329–6577.1000118. doi: 10.4172/2329-6577.1000118
Berridge, M. J. (2009). Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 1793, 933–940. doi: 10.1016/j.bbamcr.2008.10.005
Birnbaum, R., and Weinberger, D. (2017). Genetic insights into the neurodevelopmental origins of schizophrenia. Nat. Rev. Neurosci. 18, 727–740. doi: 10.1038/nrn.2017.125
Blanco-Suárez, E., Caldwell, A. L. M., and Allen, N. J. (2017). Role of astrocyte-synapse interactions in CNS disorders. J. Physiol. 595, 1903–1916. doi: 10.1113/JP270988
Bolduc, F. V., Bell, K., Rosenfelt, C., Cox, H., and Tully, T. (2010). Fragile X mental retardation 1 and filamin A interact genetically in drosophila long-term memory. Front. Neural Circuits 3:22. doi: 10.3389/neuro.04.022.2009
Bononi, A., Bonora, M., Marchi, S., Missiroli, S., Poletti, F., Giorgi, C., et al. (2013). Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 20, 1631–1643. doi: 10.1038/cdd.2013.77
Bootman, M. D., Collins, T. J., Peppiatt, C. M., Prothero, L. S., MacKenzie, L., de Smet, P., et al. (2001). Calcium signalling – an overview. Semin. Cell Dev. Biol. 12, 3–10. doi: 10.1006/scdb.2000.0211
Breitenkamp, A., Matthes, J., and Herzig, S. (2015). Voltage-gated calcium channels and autism spectrum disorders. Curr. Mol. Pharmacol. 8, 123–132. doi: 10.2174/1874467208666150507105235
Brini, M., Calì, T., Ottolini, D., and Carafoli, E. (2014). Neuronal calcium signaling: Function and dysfunction Introduction: General principle of Ca 2+ signaling. Cell. Mol. Life Sci. 71, 2787–2814. doi: 10.1007/s00018-013-1550-7
Britzolaki, A., Saurine, J., Flaherty, E., Thelen, C., and Pitychoutis, P. M. (2018). The SERCA2: A gatekeeper of neuronal calcium homeostasis in the brain. Cell Mol. Neurobiol. 38, 981–994. doi: 10.1007/s10571-018-0583-8
Britzolaki, A., Saurine, J., Klocke, B., and Pitychoutis, P. (2020). A Role for SERCA pumps in the neurobiology of neuropsychiatric and neurodegenerative disorders. Adv. Exp. Med. Biol. 1131, 131–161. doi: 10.1007/978-3-030-12457-1_6
Cao, Z., Hulsizer, S., Cui, Y., Pretto, D. L., Kim, K. H., Hagerman, P. J., et al. (2013). Enhanced asynchronous Ca(2+) oscillations associated with impaired glutamate transport in cortical astrocytes expressing Fmr1 gene premutation expansion. J. Biol. Chem. 288, 13831–13841. doi: 10.1074/jbc.M112.441055
Chen, Y., Tassone, F., Berman, R. F., Hagerman, P. J., Hagerman, R. J., Willemsen, R., et al. (2010). Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum. Mol. Genet. 19, 196–208. doi: 10.1093/hmg/ddp479
Christensen, D. L., Baio, J., van Naarden Braun, K., Bilder, D., Charles, J., Constantino, J. N., et al. (2016). Prevalence and characteristics of autism spectrum disorder among children aged 8 years. MMWR Surveill. Summ. 65, 1–23. doi: 10.15585/mmwr.ss6503a1
Cooper, S. M., and Burge, S. M. (2003). Darier’s disease: Epidemiology, pathophysiology, and management. Am. J. Clin. Dermatol. 4, 97–105.
Cupertino, R., Kappel, D., Bandeira, C., Schuch, J., da Silva, B., Müller, D., et al. (2016). SNARE complex in developmental psychiatry: Neurotransmitter exocytosis and beyond. J. Neural Transm. 123, 867–883. doi: 10.1007/s00702-016-1514-9
Emiliani, F. E., Sedlak, T. W., and Sawa, A. (2014). Oxidative stress and schizophrenia: Recent breakthroughs from an old story. Curr. Opin. Psychiatry 27, 185–190. doi: 10.1097/YCO.0000000000000054
Gilman, S., Iossifov, I., Levy, D., Ronemus, M., Wigler, M., and Vitkup, D. (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 5, 898–907. doi: 10.1016/j.neuron.2011.05.021
Glessner, J. T., Wang, K., Cai, G., Korvatska, O., Kim, C. E., Wood, S., et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. doi: 10.1038/nature07953
Hansen, B. H., Beate, B., Skirbekk, B., Petrovski, B. E., and Kristensen, H. (2018). Neurodevelopmental disorders: Prevalence and comorbidity in children referred to mental health services. Nord. J. Psychiatry 74, 285–291. doi: 10.1080/08039488.2018.1444087
Horn, J. L., Janicki, P. K., and Franks, J. J. (1995). Diminished brain synaptic plasma membrane ca2+-atpase activity in spontaneously hypertensive rats: Association with reduced anesthetic requirements. Life Sci. 56, 427–432. doi: 10.1016/0024-3205(95)00170-B
Ijomone, O. M., Olung, N. F., Akingbade, G. T., Okoh, C. O. A., and Aschner, M. (2020). Environmental influence on neurodevelopmental disorders: Potential association of heavy metal exposure and autism. J. Trace Elem. Med. Biol. 62:126638. doi: 10.1016/j.jtemb.2020.126638
Janickova, L., and Schwaller, B. (2020). Parvalbumin-deficiency accelerates the age-dependent ROS production in Pvalb neurons in vivo: Link to neurodevelopmental disorders. Front. Cell Neurosci. 14:571216. doi: 10.3389/fncel.2020.571216
Janickova, L., Rechberger, K. F., Wey, L., and Schwaller, B. (2020). Absence of parvalbumin increases mitochondria volume and branching of dendrites in inhibitory Pvalb neurons in vivo: A point of convergence of autism spectrum disorder (ASD) risk gene phenotypes. Mol. Autism 11:7. doi: 10.1186/s13229-020-00323-8
Kang, N., Kang, J. Y., Park, S., and Shin, D. M. (2018). Increased store-operated Ca2+ entry mediated by GNB5 and STIM1. Korean J. Physiol. Pharmacol. 22, 343–348. doi: 10.4196/kjpp.2018.22.3.343
Keil, K. P., Sethi, S., Wilson, M. D., Silverman, J. L., Pessah, I. N., and Lein, P. J. (2019). Genetic mutations in Ca2+ signaling alter dendrite morphology and social approach in juvenile mice. Genes Brain Behav. 18:12526. doi: 10.1111/gbb.12526
Lehohla, M., Russell, V., and Kellaway, L. (2001). NMDA-stimulated Ca2+ uptake into barrel cortex slices of spontaneously hypertensive rats. Metab. Brain Dis. 16, 133–141. doi: 10.1023/A:1012532709306
Lesiak, A., Zhu, M., Chen, H., Appleyard, S. M., Impey, S., Lein, P. J., et al. (2014). The environmental neurotoxicant PCB 95 promotes synaptogenesis via ryanodine receptor-dependent miR132 upregulation. J. Neurosci. 34, 717–725. doi: 10.1523/JNEUROSCI.2884-13.2014
Li, M., Francis, E., Hinkle, S. N., Ajjarapu, A. S., and Zhang, C. (2019). Preconception and prenatal nutrition and neurodevelopmental disorders: A systematic review and meta-analysis. Nutrients 11:1628. doi: 10.3390/nu11071628
Lopez, S. J., Segal, D. J., and LaSalle, J. M. (2019). UBE3A: An E3 ubiquitin ligase with genome-wide impact in neurodevelopmental disease. Front. Mol. Neurosci. 11:476. doi: 10.3389/fnmol.2018.00476
Lord, C., Elsabbagh, M., Baird, G., and Veenstra-Vanderweele, J. (2018). Autism spectrum disorder. Lancet 392, 508–520. doi: 10.1016/S0140-6736(18)31129-2
Ma, X. E., Liu, B., and Zhao, C. X. (2020). Modulation of Ca2+-induced Ca2+ release by ubiquitin protein ligase E3 component n-recognin UBR3 and 6 in cardiac myocytes. Channels 14, 326–335. doi: 10.1080/19336950.2020.1824957
McCutcheon, R. A., Reis Marques, T., and Howes, O. D. (2020). Schizophrenia-an overview. JAMA Psychiatry 77, 201–210. doi: 10.1001/jamapsychiatry.2019.3360
McGrath, J., Saha, S., Chant, D., and Welham, J. (2008). Schizophrenia: A concise overview of incidence, prevalence, and mortality. Epidemiol. Rev. 30, 67–76. doi: 10.1093/epirev/mxn001
Means, R. E., and Katz, S. G. (2021). Yes, MAM! Mol. Cell Oncol. 4:1919473. doi: 10.1080/23723556.2021.1919473
Nakajima, K., Ishiwata, M., Weitemier, A. Z., Shoji, H., Monai, H., Miyamoto, H., et al. (2021). Brain-specific heterozygous loss-of-function of ATP2A2, endoplasmic reticulum Ca2+ pump responsible for Darier’s disease, causes behavioral abnormalities and a hyper-dopaminergic state. Hum. Mol. Genet. 30, 1762–1772. doi: 10.1093/hmg/ddab137
Nakamura, F., Hartwig, J. H., Stossel, T. P., and Szymanski, P. T. (2005). Ca2+ and calmodulin regulate the binding of filamin A to actin filaments. J. Biol. Chem. 280, 32426–32433. doi: 10.1074/jbc.M502203200
Naranjo, J. R., and Mellström, B. (2012). Ca2+-dependent transcriptional control of Ca2+ homeostasis. J. Biol. Chem. 287, 31674–31680. doi: 10.1074/jbc.R112.384982
Park, S. J., Jeong, J., Park, Y. U., Park, K. S., Lee, H., Lee, N., et al. (2015). Disrupted-in-schizophrenia-1 (DISC1) regulates endoplasmic reticulum calcium dynamics. Sci. Rep. 5:8694. doi: 10.1038/srep08694
Park, S. J., Lee, S. B., Suh, Y., Kim, S. J., Lee, N., Hong, J. H., et al. (2017). DISC1 modulates neuronal stress responses by gate-keeping ER-mitochondria Ca2+ transfer through the MAM. Cell Rep. 21, 2748–2759. doi: 10.1016/j.celrep.2017.11.043
Pchitskaya, E., Popugaeva, E., and Bezprozvanny, I. (2018). Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70, 87–94. doi: 10.1016/j.ceca.2017.06.008
Pessah, I. N., Cherednichenko, G., and Lein, P. J. (2010). Minding the calcium store: Ryanodine receptor activation as a convergent mechanism of PCB toxicity. Pharmacol. Ther. 125, 260–285. doi: 10.1016/j.pharmthera.2009.10.009
Pitale, P. M., Howse, W., and Gorbatyuk, M. (2017). Neuronatin protein in health and disease. J. Cell Physiol. 232, 477–481. doi: 10.1002/jcp.25498
Pourtavakoli, A., and Ghafouri-Fard, S. (2022). Calcium signaling in neurodevelopment and pathophysiology of autism spectrum disorders. Mol. Biol. Rep. 49, 10811–10823. doi: 10.1007/s11033-022-07775-6
Robin, G., López, J. R., Espinal, G. M., Hulsizer, S., Hagerman, P. J., and Pessah, I. N. (2017). Calcium dysregulation and Cdk5-ATM pathway involved in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 26, 2649–2666. doi: 10.1093/hmg/ddx148
Rosa, J. P., Raslova, H., and Bryckaert, M. (2019). Filamin A: Key actor in platelet biology. Blood 134, 1279–1288. doi: 10.1182/blood.2019000014
Ruden, J. B., Dugan, L. L., and Konradi, C. (2021). Parvalbumin interneuron vulnerability and brain disorders. Neuropsychopharmacology 46, 279–287. doi: 10.1038/s41386-020-0778-9
Rund, B. (2018). The research evidence for schizophrenia as a neurodevelopmental disorder. Scand. J. Psychol. 59, 49–58. doi: 10.1111/sjop.12414
Sayal, K., Prasad, V., Daley, D., Ford, T., and Coghill, D. (2018). ADHD in children and young people: Prevalence, care pathways, and service provision. Lancet Psychiatry 5, 175–186. doi: 10.1016/S2215-0366(17)30167-0
Schmunk, G., Boubion, B. J., Smith, I. F., Parker, I., and Gargus, J. J. (2015). Shared functional defect in IP3R-mediated calcium signaling in diverse monogenic autism syndromes. Transl. Psychiatry 5:9. doi: 10.1038/tp.2015.123
Schmunk, G., Nguyen, R., Ferguson, D., Kumar, K., Parker, I., and Gargus, J. (2017). High-throughput screen detects calcium signaling dysfunction in typical sporadic autism spectrum disorder. Sci. Rep. 7:40740. doi: 10.1038/srep40740
Seidman, L., and Mirsky, A. (2017). Evolving notions of schizophrenia as a developmental neurocognitive disorder. J. Int. Neuropsychol. Soc. 23, 881–892. doi: 10.1017/S1355617717001114
Sethi, S., Keil Stietz, K. P., Valenzuela, A. E., Klocke, C. R., Silverman, J. L., Puschner, B., et al. (2021). Developmental exposure to a human-relevant polychlorinated biphenyl mixture causes behavioral phenotypes that vary by sex and genotype in juvenile mice expressing human mutations that modulate neuronal calcium. Front. Neurosci. 15:766826. doi: 10.3389/fnins.2021.766826
Shamseldin, H. E., Masuho, I., Alenizi, A., Alyamani, S., Patil, D. N., Ibrahim, N., et al. (2016). GNB5 mutation causes a novel neuropsychiatric disorder featuring attention deficit hyperactivity disorder, severely impaired language development and normal cognition. Genome Biol. 17:195. doi: 10.1186/s13059-016-1061-6
Sharma, A., and Couture, J. (2014). A review of the pathophysiology, etiology, and treatment of attention-deficit hyperactivity disorder (ADHD). Ann. Pharmacother. 48, 209–225. doi: 10.1177/1060028013510699
Sigaard, R. K., Kjær, M., and Pakkenberg, B. (2016). Development of the cell population in the brain white matter of young children. Cereb. Cortex 26, 89–95. doi: 10.1093/cercor/bhu178
Soueid, J., Kourtian, S., Makhoul, N., Makoukji, J., Haddad, S., Ghanem, S., et al. (2016). RYR2, PTDSS1 and AREG genes are implicated in a Lebanese population-based study of copy number variation in autism. Sci. Rep. 6:19088. doi: 10.1038/srep19088
Srikanth, S., and Gwack, Y. (2012). Orai1, STIM1, and their associating partners. J. Physiol. 17, 4169–4177. doi: 10.1113/jphysiol.2012.231522
Tang, C., Chan, M., Lee, J., and Hariram, J. (2010). Darier’s disease and schizophrenia. East Asian Arch. Psychiatry 20, 190–192.
Taylor, C. W., and Tovey, S. C. (2010). IP(3) receptors: Toward understanding their activation. Cold Spring Harb. Perspect. Biol. 2:12. doi: 10.1101/cshperspect.a004010
Tochigi, M., Kato, C., Ohashi, J., Koishi, S., Kawakubo, Y., Yamamoto, K., et al. (2008). No association between the ryanodine receptor 3 gene and autism in a Japanese population. Psychiatry Clin. Neurosci. 62, 341–344. doi: 10.1111/j.1440-1819.2008.01802.x
van Vliet, A. R., and Agostinis, P. (2018). Mitochondria-associated membranes and ER stress. Curr. Top. Microbiol. Immunol. 414, 73–102. doi: 10.1007/82_2017_2
Vatsa, N., Kumar, V., Singh, B. K., Kumar, S. S., Sharma, A., and Jana, N. R. (2019). Down-regulation of miRNA-708 promotes aberrant calcium signaling by targeting neuronatin in a mouse model of angelman syndrome. Front. Mol. Neurosci. 12:35. doi: 10.3389/fnmol.2019.00035
Wang, Q., Kong, Y., and Wu, D. (2021). Impaired calcium signaling in astrocytes modulates autism spectrum disorder-like behaviors in mice. Nat. Commun. 12:3321. doi: 10.1038/s41467-021-23843-0
Wang, T., Zhang, Y., Liu, L., Wang, Y., Chen, H., Fan, T., et al. (2021). Targeted sequencing and integrative analysis of 3,195 Chinese patients with neurodevelopmental disorders prioritized 26 novel candidate genes. J. Genet. Genom. 48, 312–323. doi: 10.1016/j.jgg.2021.03.002
Wetmore, D. Z., and Garner, C. C. (2010). Emerging pharmacotherapies for neurodevelopmental disorders. J. Dev. Behav. Pediatr. 31, 564–581. doi: 10.1097/DBP.0b013e3181ee3833
Williams, C. A., Driscoll, D. J., and Dagli, A. I. (2010). Clinical and genetic aspects of Angelman syndrome. Genet. Med. 12, 385–395. doi: 10.1097/GIM.0b013e3181def138
Wöhr, M., Orduz, D., Gregory, P., Moreno, H., Khan, U., Vörckel, K. J., et al. (2015). Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Transl. Psychiatry 5:3. doi: 10.1038/tp.2015.19
Xie, K., Ge, S., Collins, V. E., Haynes, C. L., Renner, K. J., Meisel, R. L., et al. (2012). Gβ5-RGS complexes are gatekeepers of hyperactivity involved in control of multiple neurotransmitter systems. Psychopharmacology. 219, 823–834.
Xu, X., Li, C., Gao, X., Xia, K., Guo, H., Li, Y., et al. (2018). Excessive Ube3a dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res. 28, 48–68. doi: 10.1038/cr.2017.132
Keywords: autism, SERCA2, ryanodine receptors, calcium, schizophrenia, attention-deficit hyperactivity disorder (ADHD), inositol triphosphate receptor (IP3)
Citation: Klocke B, Krone K, Tornes J, Moore C, Ott H and Pitychoutis PM (2023) Insights into the role of intracellular calcium signaling in the neurobiology of neurodevelopmental disorders. Front. Neurosci. 17:1093099. doi: 10.3389/fnins.2023.1093099
Received: 08 November 2022; Accepted: 27 January 2023;
Published: 15 February 2023.
Edited by:
Apostolos Zarros, Pharmacological Research Observatory, United KingdomReviewed by:
Renata B. Cupertino, University of Vermont, United StatesYanting Chen, Shenzhen Sixth People’s Hospital, China
Copyright © 2023 Klocke, Krone, Tornes, Moore, Ott and Pitychoutis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pothitos M. Pitychoutis,  cHBpdHljaG91dGlzMUB1ZGF5dG9uLmVkdQ==
cHBpdHljaG91dGlzMUB1ZGF5dG9uLmVkdQ==