 Musa O. Iliyasu
Musa O. Iliyasu Sunday A. Musa2
Sunday A. Musa2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 11 April 2023
Sec. Neurodegeneration
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1081938
This article is part of the Research Topic The Biochemistry of Amyloids in Neurodegenerative Diseases, Volume II View all 15 articles
Alzheimer’s disease (AD) is a progressive neurodegenerative condition characterized by tau pathology and accumulations of neurofibrillary tangles (NFTs) along with amyloid-beta (Aβ). It has been associated with neuronal damage, synaptic dysfunction, and cognitive deficits. The current review explained the molecular mechanisms behind the implications of Aβ aggregation in AD via multiple events. Beta (β) and gamma (γ) secretases hydrolyzed amyloid precursor protein (APP) to produce Aβ, which then clumps together to form Aβ fibrils. The fibrils increase oxidative stress, inflammatory cascade, and caspase activation to cause hyperphosphorylation of tau protein into neurofibrillary tangles (NFTs), which ultimately lead to neuronal damage. Acetylcholine (Ach) degradation is accelerated by upstream regulation of the acetylcholinesterase (AChE) enzyme, which leads to a deficiency in neurotransmitters and cognitive impairment. There are presently no efficient or disease-modifying medications for AD. It is necessary to advance AD research to suggest novel compounds for treatment and prevention. Prospectively, it might be reasonable to conduct clinical trials with unclean medicines that have a range of effects, including anti-amyloid and anti-tau, neurotransmitter modulation, anti-neuroinflammatory, neuroprotective, and cognitive enhancement.
The deposition of aggregated amyloid-beta (Aβ) peptide is the hallmark of Alzheimer’s disease (AD), a neurodegenerative disorder that progresses over time (Riyaz Basha et al., 2005). Due to changes in the brain and the formation of plaques and tangles, it has been linked to neuronal damage and death (Kehoe et al., 2009). AD is the most prevalent type of dementia, accounting for 60% to 70% of dementia cases among older people (Burns and Iliffe, 2009; Zhang et al., 2018).
AD symptoms include short-term memory loss, as well as a progressive decline in the patient’s capacity for thought, judgment, problem-solving, communication, and self-care (Mount and Downton, 2006; Prasansuklab and Tencomnao, 2013). The daily life of an AD patient is also impacted by symptoms including confusion, impatience, aggression, mood swings, personality and behavior changes, issues with attention and spatial orientation, and loss of long-term memory (Prasansuklab and Tencomnao, 2013).
The fifth-leading cause of death in persons over 65 years is AD (Winston, 2020). Over 26.6 million individuals worldwide suffer from it, and its prevalence is significantly increasing yearly (Prince et al., 2014; Olajide and Sarker, 2020). More than 106 million AD patients are anticipated to exist worldwide by 2050. The disease will affect 1 in 85 people, according to estimates (Brookmeyer et al., 2007), as the population ages and environmental factors take effect (Prince et al., 2014).
AD is a leading cause of disability and life reliance among elderly adults worldwide (Nichols et al., 2019), and has a profound influence on individuals, their families, and societies at large (Winston, 2020). The estimated cost of dementia in 2015 was $818 billion, or 1.1% of the GDP (Youssef et al., 2019). In 2020, it was anticipated that treating AD would cost $305 billion in total, and as the population ages, the amount is expected to rise to more than $1 trillion (Winston, 2020). The expense of dementia globally is predicted to reach $2 trillion by 2030 (Wimo et al., 2017). There are currently no effective or disease-modifying medications for AD (Fu et al., 2019). Many of the clinical trials failed in recent years, however, quite a number of the trials are under evaluation. It is essential to advance AD research to suggest new compounds for treatment and prevention. The objective of the current review is to describe the mechanisms behind the implications of Aβ aggregation in AD using multiple pathways. The literature data published between the years 1993 and 2020 were collected using PubMed and Scopus.
The intracellular cleavage of the amyloid precursor protein (APP) by the proteolytic enzymes beta-(β-) secretase and gamma-(γ-) secretase produces the short peptide known as Aβ, which has 40–42 amino acids (Prasansuklab and Tencomnao, 2013). The APP is localized at neuronal synapses and is abundantly expressed in the brain (Thinakaran and Koo, 2008; O’brien and Wong, 2011). It has been linked to synaptic plasticity, cell–cell or cell-matrix interactions, neuroprotection, and regulation of neuronal cell development (Storey and Cappai, 1999).
However, aggregation of Aβ, produced from the cleavage of the amyloidogenic pathway causes neurotoxicity. Most of the body’s cells, including vascular endothelial cells, thyroid epithelial cells, and neuronal and nonneuronal cultured cells, produce Aβ monomers (Schmitt et al., 1995; Fukumoto et al., 1999; Hayes et al., 2002; Kitazume et al., 2010). Although compared to other cell types, neuronal cells appear to produce more Aβ (Fukumoto et al., 1999), demonstrating the possibility that the Aβ-peptide is crucial for maintaining proper CNS physiology. According to the increased long-term potentiation (LTP) mediated by Aβ40, there is a theory that Aβ may play a crucial role in synaptic structural-functional plasticity that underlies learning and memory (Koudinov and Koudinova, 2005).
According to the amyloid hypothesis, which explains why synaptic dysfunction and neurodegeneration are brought on by the aggregation of the Aβ-peptide (Van Dyck, 2018). The main contributing factor to AD is errors in the mechanisms directing Aβ formation, accumulation, or elimination. Aβ aggregation stages impair cell-to-cell communication and stimulate the immune system, which then causes inflammation and eventually kills brain cells.
The APP is processed in two distinct pathways as shown in Figure 1. Nonamyloidogenic pathway: The α-secretase enzyme first cleaves APP within the Aβ domain, and then γ-secretase cleaves at the C-terminus. Amyloidogenic pathway: Instead of α-secretase, β-secretase (BACE1) cleaves APP first at the N-terminus of the Aβ domain, and γ-secretase then cleaves it at the C-terminus. The Aβ amylogenic peptides are produced by this chain of events, which then assemble into oligomers to create extracellular neurotoxic plaques in the brain. A similar APP intracellular C-terminal domain (AICD) is released from both pathways (Thinakaran and Koo, 2008). When compared to other fragments, Aβ is chemically “stickier” than those formed by APP proteolytic processes. Small clusters (oligomers) are formed by the fragments initially, followed by chains of clusters (fibrils), and finally “mats” of fibrils (beta-sheets). The final stage is the forming of plaques which contain clusters of beta-sheets and other chemicals (Jung et al., 2010). The amyloid cascade hypothesis (ACH) explains AD pathogenesis from the outcome of two significant facts: (i) Identification of Aβ as a key component of senile plaques (SPs). (ii) Mutations of APP genes and the presenilin 1 and 2 genes (PSEN1 and PSEN2) which are typically detected at the early stage of AD. As a result, it is believed that the emergence of Aβ within SPs is caused by these mutations, which also cause dementia and neuronal cell death (Reitz, 2012).

Figure 1. Beta-amyloid formation from the proteolytic digestion of the APP. AICD: APP intracellular C-terminal domain (Thinakaran and Koo, 2008).
The aggregation of Aβ causes the formation of neurofibrillary tangles (NFTs) from hyperphosphorylation of tau and its accumulation into tangles is another pathological cause of AD (McGleenon et al., 2009). In normal conditions, tau supports neuronal structures and functions in the brain (Kolarova et al., 2012). However, under pathological circumstances, tau became excessively hyperphosphorylated and aggregated into fibrils known as neurofibrillary tangles. The accumulation of abnormal tau and tangles in neurons leads to neurotoxicity and neuronal degeneration (Gómez-Isla et al., 1997). In addition to the formation of NFTs, Tau phosphorylation impairs tau’s ability to bind microtubules, which impacts neuronal activities such as axonal transport and mitochondrial respiration (Ittner and Götz, 2011). Microtubule depolymerization, self-aggregation, and detachment caused by tau hyperphosphorylation result in neuronal cell death (Suganthy et al., 2016).
AD pathogenesis starts from the deposition of Aβ which trigger SPs formation, followed by the death of neurons due to NFTs formation (Armstrong, 2011). Neurotoxic mechanisms in the pathology of AD include aberrant protein aggregation, dysfunction of mitochondrial, decreased neurotransmitter production, inflammation, and oxidative stress (Figure 2). However, the buildup of Aβ and the aggregation of tau are the two most prevalent etiologic models of Alzheimer’s pathogenesis (Bloom, 2014).
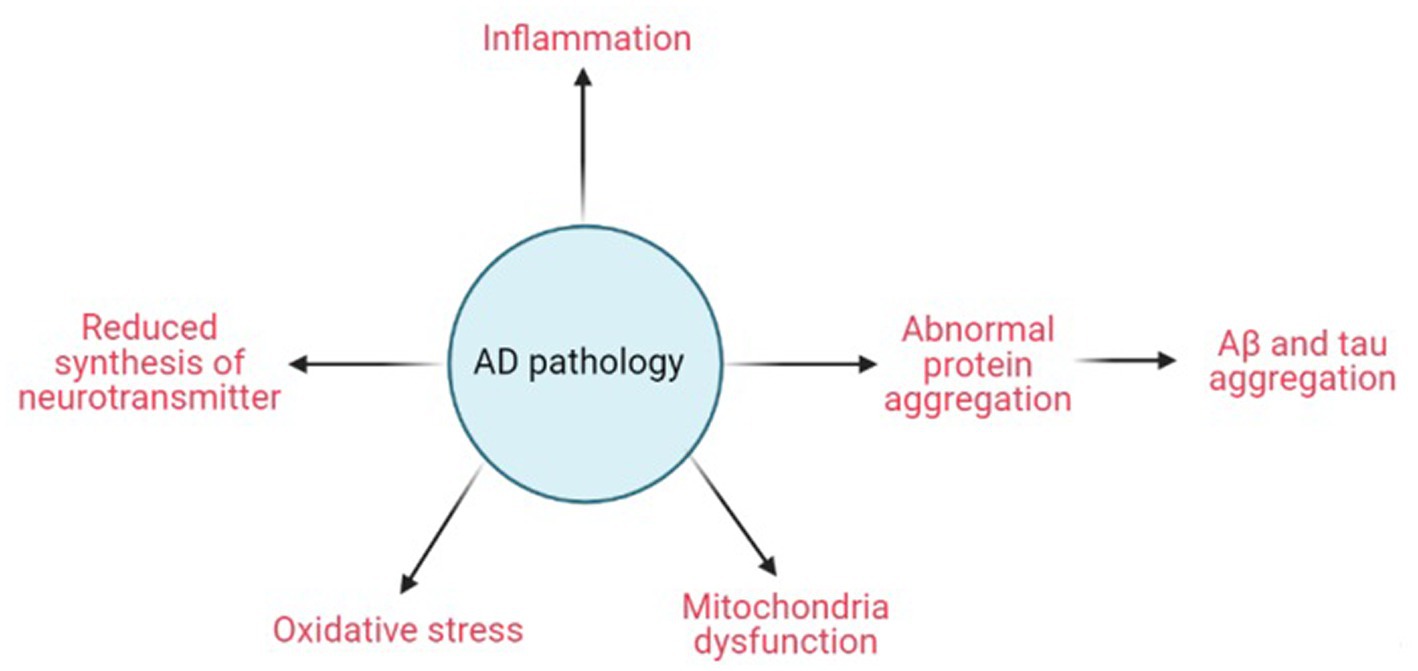
Figure 2. Schematic diagram of AD pathology. Created with BioRender.com.
The neuropathological events in AD patients are the result of the toxicity of amyloid oligomers and fibrils, which are from the aggregated forms of Aβ. The bodies regulate the amyloid level via a variety of methods as Aβ accumulates. The concentration of Aβ-peptide is controlled in healthy brain tissue by its production from APP; the influx across the blood–brain barrier (BBB) via its interaction with the receptor for advanced glycation end products (RAGE; Deane et al., 2003, 2009); and its clearance via the low-density lipoprotein receptor-related protein-1 (LRP1) from the brain and enzymatic breakdown in the brain (Selkoe, 2001; Deane et al., 2003, 2009). Additionally, the levels of Aβ affect how RAGE is expressed. RAGE is upregulated when there is excessive Aβ synthesis, and this leads to neurotoxicity (Prasansuklab and Tencomnao, 2013) as shown in Figure 3. Thus, impairments in these regulatory processes may cause excessive Aβ-peptide to build up and deposit in the brains of AD patients. By binding to Aβ12-28 residues, apolipoprotein E (ApoE) regulates Aβ’s accumulation and lessens its clearance (Prasansuklab and Tencomnao, 2013; Zhang et al., 2018) from the brain (Sagare et al., 2007). Three isoforms of ApoE such as ApoE4 (E4), ApoE3 (E3), and ApoE2 (E2; Liu et al., 2013), regulate cholesterol levels in various ways to influence γ-secretase activity and Aβ synthesis (Osenkowski et al., 2008). According to Bales et al. (2009) and Castellano et al. (2011), the brain Aβ levels and amyloid plaque loading rely on the ApoE isoforms, demonstrating the modulatory involvement of ApoE in Aβ metabolism, aggregation, and deposition (Liu et al., 2013). The differential lipidation status exhibited by ApoE isoforms affects Aβ clearance. The ApoE particles may seize Aβ and stimulate cellular uptake and degradation of ApoE-Aβ complexes (Kim et al., 2009). Aβ clearance at the blood–brain barrier is inhibited by ApoE in an isoform-dependent manner (E4 > E3 and E2). According to studies, E4 is less effective than E3 and E2 at mediating the clearance of Aβ (Deane et al., 2008; Jiang et al., 2008).
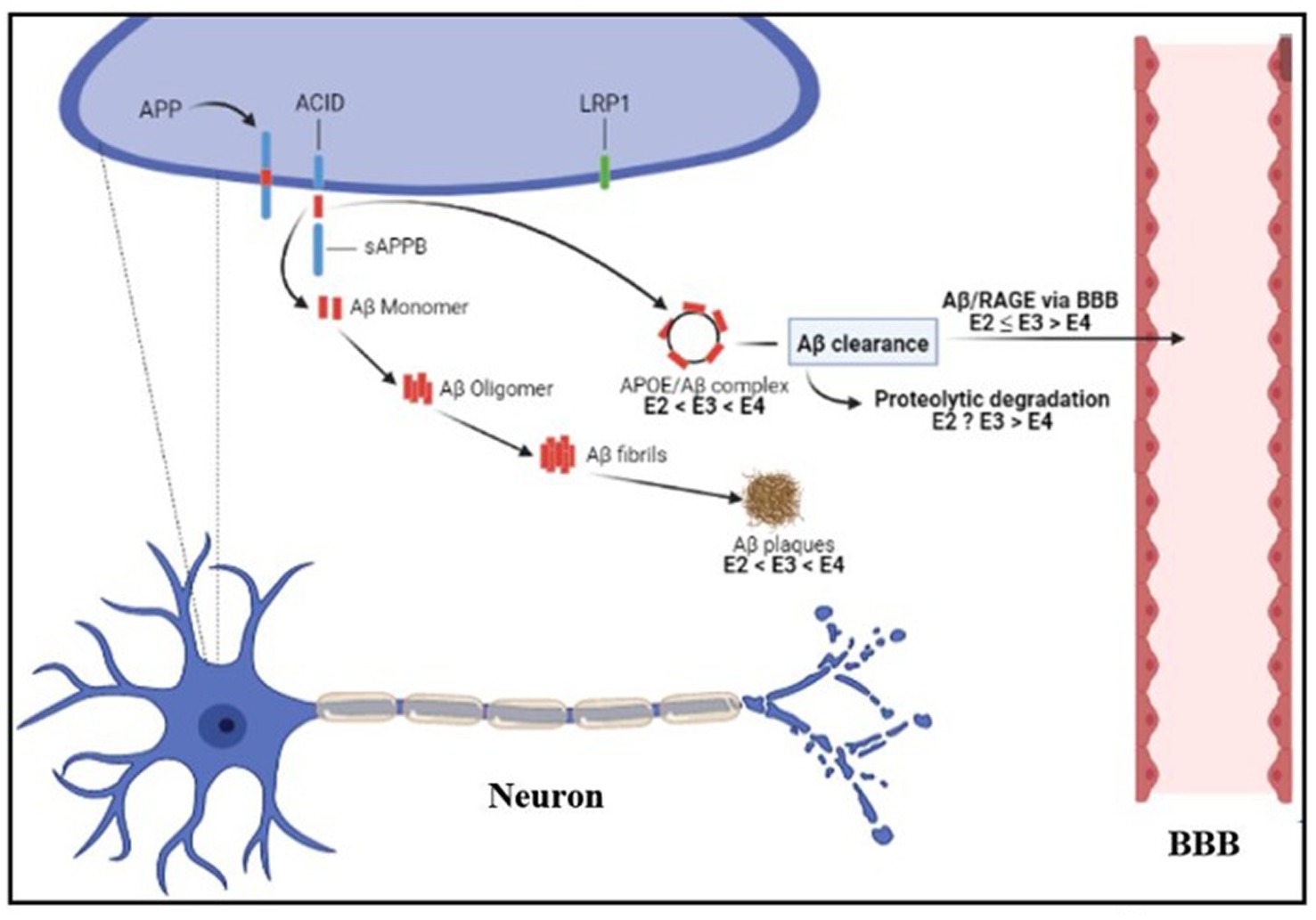
Figure 3. Diagrammatic representation of the regulatory systems for Aβ in an AD patient’s brain. Aβ, amyloid-beta; BBB, Blood–brain barrier; RAGE, Receptor for advanced glycation end products; AICD, APP intracellular C-terminal domain; APP, Amyloid precursor protein; ApoE, apolipoprotein E; E4, ApoE; E3, ApoE3; E2, ApoE2. Created with BioRender.com.
Strong neurotoxic candidates that alter Aβ and tau aggregation include metal dyshomeostasis (Figure 4). Metal ions’ effects on the aggregation of Aβ and tau have been elucidated. Metals like Zn2+, Cu2+, Fe3+, Mn2+, Pb2+, Cd2+, Hg2+, and Al3+ stimulate amyloidogenic pathways and Aβ aggregation. [red arrow] (O’brien and Wong, 2011). The neurotoxic Aβ-peptide produced by the cleavage of the APP by β- and γ-secretase is secreted into the extracellular space where it spontaneously changes into amyloid plaques. On the other hand, as seen in Figure 4A, the presence of Mg2+, Fe2+, and Li2+ inhibits the production of Aβ [blue arrow] (O’brien and Wong, 2011).
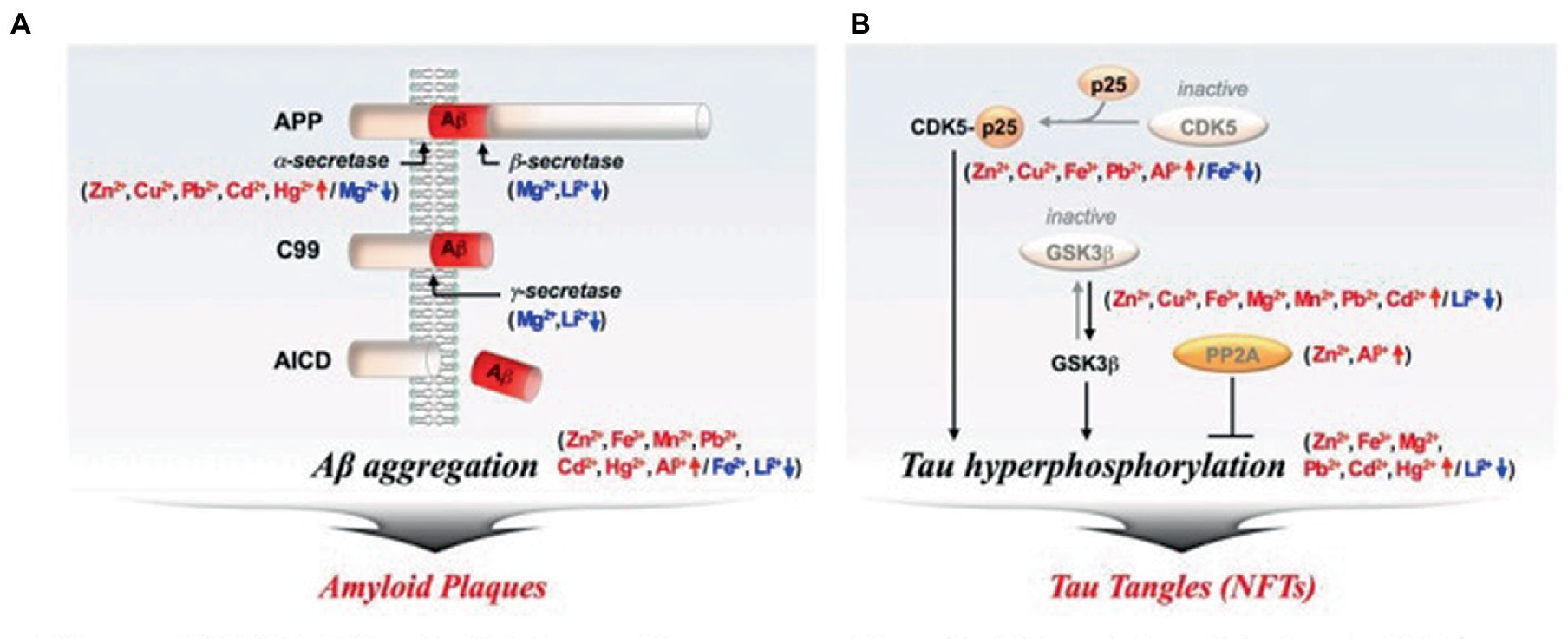
Figure 4. Metal ions’ effects on the aggregation of Aβ and tau. (A) Amyloid Plaques, (B) Tau tangle. Aβ, amyloid-beta; CDK5, Cyclin-dependent kinase; GSK-3β, Glycogen synthase kinase-3beta; NFTs, Neurofibrillary tangles; PP2A, protein phosphatase 2A (O’brien and Wong, 2011).
Tau hyperphosphorylation and aggregation are promoted by metal ions like Zn2+, Cu2+, Fe3+, Mg2+, Mn2+, Pb2+, Cd2+, Hg2+, and Al3+ [red arrow]. Numerous kinases, including glycogen synthase kinase-3 beta (GSK-3β; Rankin et al., 2007), cyclin-dependent kinase 5 (CDK-5), and others, strongly phosphorylate tau (Kimura et al., 2014). If protein phosphatase 2A (PP2A) is not activated, the hyperphosphorylation of tau may persist (Goedert, 1993). Tau that has been hyperphosphorylated forms NFTs. As depicted in Figure 4B, metal ions like Fe2+, and Li2+, however, lessen tau hyperphosphorylation [blue arrow].
The polymeric forms of Aβ cause alterations in biochemical components and brain cell activities that lead to neuropathology associated with AD symptoms. According to reports, one of the earliest clinical manifestations of AD is increased oxidative stress. Hydrogen peroxide (H2O2) created due to the reduction of metal ions by Aβ-peptides served as a mediator of the oxidative stress as shown in Figure 5 (Huang et al., 1999; Atwood et al., 2003). Aβ-peptides act as powerful oxidation catalysts and can capture transition metal ions like Cu, Fe, and Zn (Miura et al., 2000). In addition, it was shown that Aβ was toxic to neuronal cultures, and Cu2+ ions made it more toxic (Cuajungco et al., 2000). Reactive oxygen species can be produced by the Aβ/Cu(Fe) complexes as a toxin mediator (Huang et al., 1999). Furthermore, AD brains have an extracellular and intracellular accumulation of metal ions with high concentrations of Aβ plaques (Lovell et al., 1998; Religa et al., 2006), which produced free radicals. Because of lipid peroxidation and oxidative protein modification, several biomolecules in the AD brain experience conformational and structural changes that impair their ability to function, which, in turn, affects a variety of cellular processes (Qi et al., 2005). By upregulating the expression of the BACE1 gene, increasing oxidative stress enhances APP processing and ultimately increases Aβ generation (Tong et al., 2005; Coma et al., 2008; Quiroz-Baez et al., 2009). This causes oxidative stress and endoplasmic reticulum (ER) stress by increasing ROS and the accompanying rise in abnormal APP and phosphorylated tau. The ER function can be severely damaged by long-term ER stress, which also causes apoptotic signaling (Ogata et al., 2006; Kouroku et al., 2007). Aβ promotes Ca2+ release from neurons’ ER Ca2+ pools, increasing intracellular free Ca2+ (Zhou et al., 2020). Increased expression of the NR2B subunit of NMDAR causes a rise in Ca2+ ion concentration in extrasynaptic regions (Jusko et al., 2008), which, then increases the level of intracellular endoplasmic reticulum Ca2+ production (Yin et al., 1994). Studies revealed that Ca2+ overload could increase ER stress and facilitate mitochondrial Ca2+ uptake by suppressing the expression of the anti-apoptotic protein B-cell lymphoma 2 (Bcl2) and increasing the phosphorylation of extracellular regulated protein kinases (Erk) protein, which would ultimately lead to cytotoxicity and cellular apoptosis (Hajnóczky et al., 2003; Zieg et al., 2008; Zhang et al., 2018).
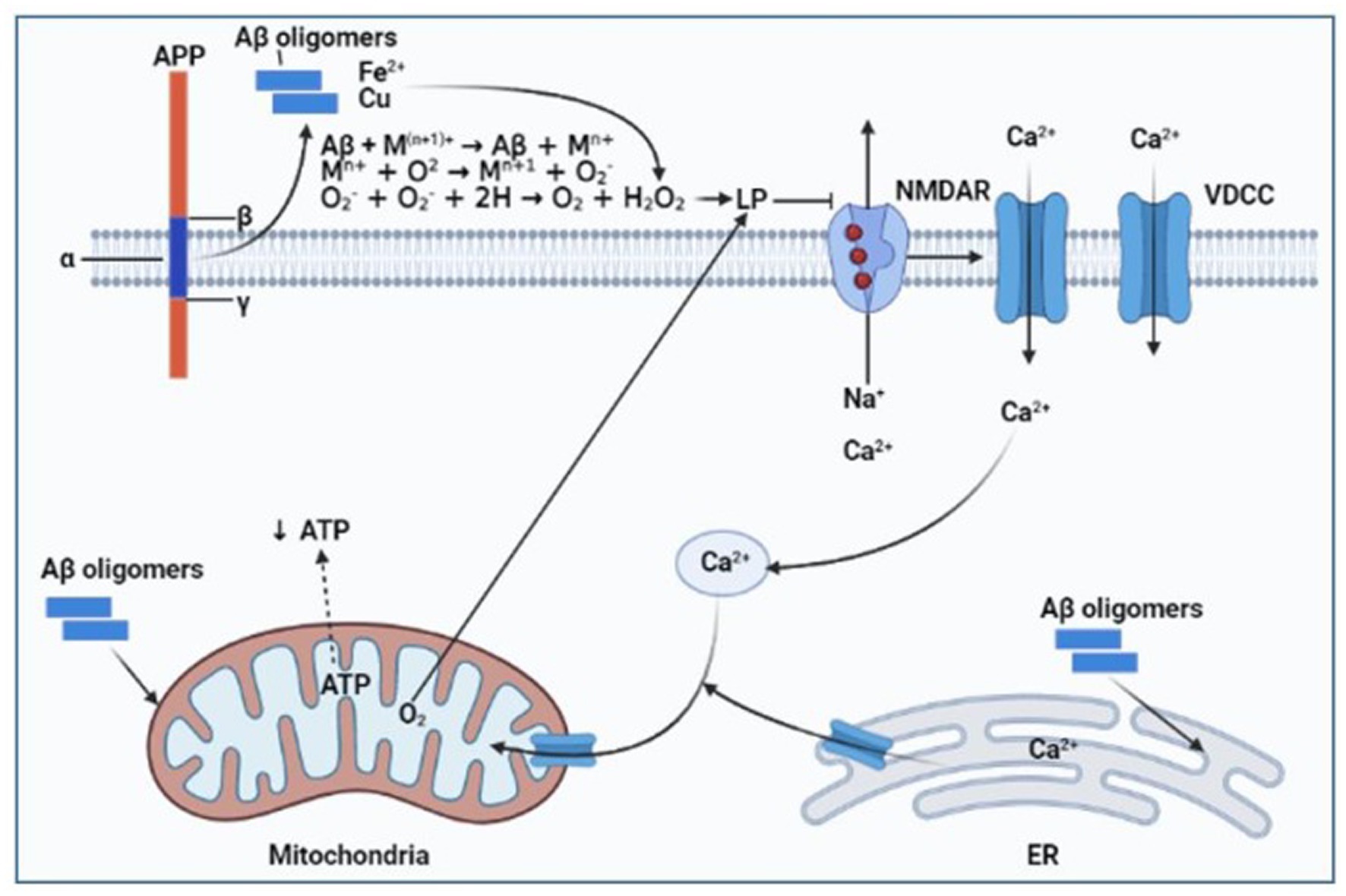
Figure 5. Diagram showing how Aβ and metal ions combine to cause oxidative stress in AD. Aβ, amyloid-beta; LP, Lipid peroxidation; NMDAR, N-methyl-D-aspartate receptor; VDCC, Voltage-dependent calcium channel; ER, endoplasmic reticulum; APP, Amyloid precursor protein; ATP, Adenosine triphosphate. Created with BioRender.com.
The expression of pro-inflammatory cytokines was increased in response to neuropathological insults induced by Aβ and its interaction with vascular RAGE (Deane et al., 2008). Microglia enhance the clearance of Aβ, but a constant generation of Aβ causes the microglia to become chronically activated, which promotes more amyloid deposition (Hickman et al., 2018). According to Kim and Choi (2015), exposure to Aβ results in microglial activation, which, in turn, causes the generation of reactive oxygen species and neurotoxic pro-inflammatory cytokines. Tau hyperphosphorylation is a result of ROS-activating p38 mitogen-activated protein kinases (p38 MAPK; Giraldo et al., 2014). p38 MAPK has been linked to neuroinflammation and AD due to its ability to activate nuclear factor-B (NF-κB; Kheiri et al., 2018), a master regulator of neuroinflammation gene transcription in the brains of AD patients (Chen et al., 2012; Liao et al., 2016; Olajide and Sarker, 2020). But data indicate that nuclear factor E2-related factor 2 (Nrf2) is negatively regulated by NF-κB (Liu et al., 2008; Kim and Vaziri, 2010; Yu et al., 2011). Substantial evidence connects the activation of the Nrf2 protection mechanism to NF-κB-mediated inflammatory actions (Nair et al., 2008; Sandberg et al., 2014). To uphold the aforementioned finding, Rojo et al. (2010) showed that cyclooxygenase-2 (COX-2), inducible nitric oxide synthases (iNOS), IL-6, and TNF-α levels are elevated when microglia are activated in Nrf2-deficient rats. Ramsey et al. (2007) first noticed this, reporting that the hippocampus of AD patients’ brains had lower amounts of Nrf2 than normal. According to Lee and Kim (2017), through the activation of p38 MAPK, Aβ plaques cause neuronal impairments such as mitochondrial dysfunction, apoptosis, tau phosphorylation, and synaptic dysfunction; the primary cause of neuroinflammation in AD is increased microglial p38 MAPK signaling brought on by Aβ, which results in the production of pro-inflammatory mediators such interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), cyclooxygenase-2 (COX-2), and inducible nitric oxide synthase (iNOS); the pathophysiology of the AD brain is worsened by the production of IL-1β from microglia, which increases p38 MAPK activation in neurons and astrocytes; Aβ plaques and IL-1β generated an increase in P38 MAPK activation in astrocytes. By releasing iNOS, COX-2, and TNF-α, this activation accelerates neuroinflammation (Figure 6).
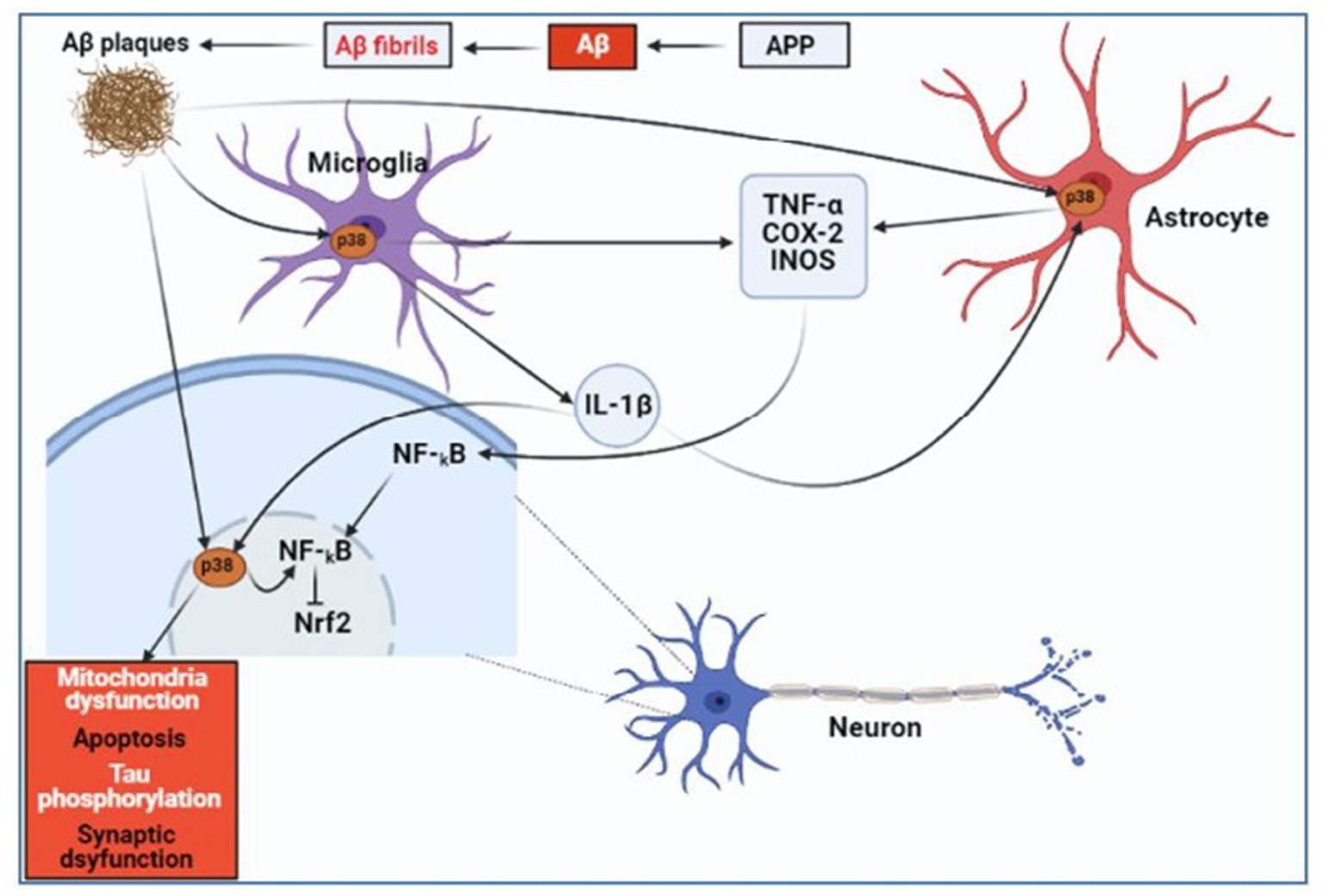
Figure 6. Diagrammatic representation of how Aβ causes neuroinflammation in AD. Aβ, amyloid-beta; NF-κB, Nuclear factor-κB; p38 MAPK, p38 Mitogen-activated protein kinases; Nrf2, Nuclear factor E2-related factor 2; IL-1β, Interleukin-1β; TNF-α, Tumor necrosis factor-α; COX-2, Cyclooxygenase-2; and iNOS, Inducible nitric oxide synthase (Schnöder et al., 2016; Lee and Kim, 2017). Created with BioRender.com.
As a neurotransmitter, acetylcholine (Ach) aids in the communication between nerve cells and is essential for memory and learning processes (Kihara and Shimohama, 2004; Francis, 2005). A report revealed that Alzheimer’s patients have reduced amounts of Ach in their brains (Kihara and Shimohama, 2004). Ach is decreased because oxidative stress is induced and inflammatory cytokines are produced by Aβ (Esposito et al., 2006). Free radicals produced due to amyloid peptides have been shown to lower the concentration of Ach by causing cholinergic neurons in the hippocampus to degenerate (Vinod et al., 2009). Additionally, acetylcholinesterase (AChE) activity increases and deactivates acetylcholine in synaptic clefts in the vicinity of amyloid plaques (Mordn et al., 1993; Sberna et al., 1997). According to another study, the amyloid peptide inhibits the production of acetylcholine (ACh) by causing choline to seep through cell membranes (Ehrenstein et al., 1997). Ach deficiency caused cognitive impairment and ultimately AD (Parent et al., 2013; Deture and Dickson, 2019) as shown in Figure 7.
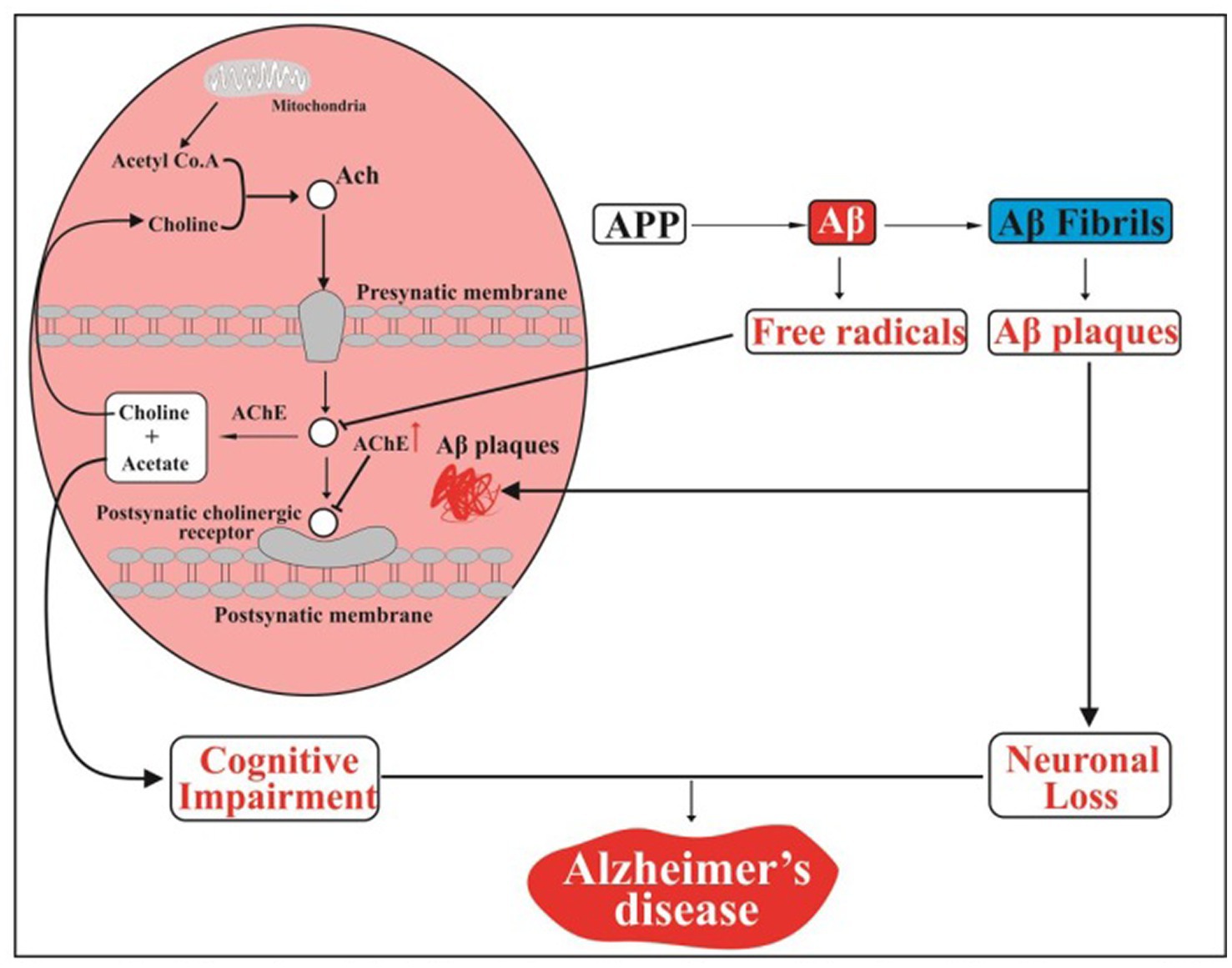
Figure 7. Aβ and acetylcholine interactions in an AD schematic diagram. APP, Amyloid precursor protein; Aβ, Amyloid-beta; Ach, acetylcholine; AChE, acetylcholinesterase (DeTure and Dickson, 2019). Created with BioRender.com.
According to Yiannopoulou and Papageorgiou (2013), the formation of amyloid oligomers, which mediates the amyloid cascade, is primarily responsible for neurotoxicity. The main pathophysiologic pillars are oxidation, inflammation, excessive glutamate, and tau hyperphosphorylation. Anti-amyloid disease-modifying treatments (DMTs) have therefore concentrated on three main mechanisms of action (MOAs), including reducing the formation of Aβ42, reducing the burden of Aβ-plaque, and promoting Aβ clearance (Yiannopoulou and Papageorgiou, 2020). Hence, inhibiting the formation of A𝛽-peptide accumulation and tau hyperphosphorylation may be part of the treatment for AD (Mendiola-Precoma et al., 2016). Physical exercise, a healthy diet, and mental stimulation are further AD prevention strategies (Nelson and Tabet, 2015).
Acetylcholinesterase inhibitors (AChEIs), such as rivastigmine, donepezil, and galantamine, are clinically effective in increasing the availability of acetylcholine at synapses and thereby inhibiting cognitive decline in AD (Andrieu et al., 2015; Hampel et al., 2018; Cummings et al., 2019). Nevertheless, diarrhea, nausea, and vomiting are some of the typical negative effects of AChEIs on the digestive system (Yiannopoulou and Papageorgiou, 2020). Memantine, which was approved in 2003, selectively binds to open calcium channels that are controlled by NMDA receptors, inhibiting NMDA-mediated ion flux and reducing pathologically excessive glutamate levels (Yiannopoulou and Papageorgiou, 2013; Matsunaga et al., 2015; Cummings et al., 2019). Memantine also reduces the activity of glycogen synthase kinase 3𝛽 (GSK-3𝛽), which, in turn, reduces tau phosphorylation (Prentice et al., 2015; Folch et al., 2016).
Despite extensive and expensive trials, the Food and Drug Administration (FDA) has not approved any DMTs or new medications for AD since 2003 (Anderson et al., 2017; Hukins et al., 2019). The β-secretase (BACE) inhibitors, lanabecestat (Burki, 2018), verubecestat (Egan et al., 2019), and atabecestat (Henley et al., 2019), as well as the anti-amyloid agents such as semagacestat (Doody et al., 2013), bapineuzumab (Vandenberghe et al., 2016), and solanezumab (Neurology, 2016), failed in recent phase 3 clinical trials. The acknowledged explanations for the numerous failures include inadequate understanding of the pathophysiology, inappropriate drug doses, late therapies in disease progression, and wrong therapeutic targets (Gauthier et al., 2016).
The current review explained the molecular mechanisms of Aβ mediating AD via multiple events, including Aβ production and accumulation, tau hyperphosphorylation, metal dyshomeostasis, oxidative stress, neuroinflammation, and inhibition of acetylcholine production. There are presently no efficient or disease-modifying medications for AD. Some of the clinical trials targeting the above events failed in recent years, however, quite a number of the trials are under evaluation. It is necessary to advance AD research to suggest novel compounds for treatment and prevention. Prospectively, it might be reasonable to conduct clinical trials with unclean medicines that have a range of effects, including anti-amyloid, anti-tau, neurotransmitter modulation, anti-neuroinflammatory, neuroprotective, and cognitive enhancement.
MI wrote the manuscript. SM, SO, and AI edited, approved, and concluded the manuscript. All authors contributed to the article and approved the submitted version.
We acknowledge Musa Mustapha’s advice. Biorender (https://biorender.com) was used to create some of the manuscript’s figures. Additionally, we give credit to the creators of some of the figures included in this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Anderson, R. M., Hadjichrysanthou, C., Evans, S., and Wong, M. M. (2017). Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 390, 2327–2329. doi: 10.1016/S0140-6736(17)32399-1
Andrieu, S., Coley, N., Lovestone, S., Aisen, P. S., and Vellas, B. (2015). Prevention of sporadic Alzheimer’s disease: lessons learned from clinical trials and future directions. Lancet Neurol. 14, 926–944. doi: 10.1016/S1474-4422(15)00153-2
Armstrong, R. A. (2011). The pathogenesis of alzheimer’s disease: A reevaluation of the “amyloid cascade hypothesis”. Int. J. Alzheimers Dis. 2011, 1–6. doi: 10.4061/2011/630865
Atwood, C. S., Obrenovich, M. E., Liu, T., Chan, H., Perry, G., Smith, M. A., et al. (2003). Amyloid-β: A chameleon walking in two worlds: A review of the trophic and toxic properties of amyloid-β. Brain Res. Rev. 43, 1–16. doi: 10.1016/S0165-0173(03)00174-7
Bales, K. R., Liu, F., Wu, S., Lin, S., Koger, D., DeLong, C., et al. (2009). Human APOE isoform-dependent effects on brain β-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29, 6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer’s disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Brookmeyer, R., Johnson, E., Ziegler-Graham, K., and Arrighi, H. M. (2007). Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 3, 186–191. doi: 10.1016/j.jalz.2007.04.381
Burki, T. (2018). Alzheimer’s disease research: the future of BACE inhibitors. Lancet 391:2486. doi: 10.1016/S0140-6736(18)31425-9
Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., DeMattos, R. B., Patterson, B. W., et al. (2011). Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3:89ra57. doi: 10.1126/scitranslmed.3002156
Chen, C. H., Zhou, W., Liu, S., Deng, Y., Cai, F., Tone, M., et al. (2012). Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 15, 77–90. doi: 10.1017/S1461145711000149
Coma, M., Guix, F. X., Ill-Raga, G., Uribesalgo, I., Alameda, F., Valverde, M. A., et al. (2008). Oxidative stress triggers the amyloidogenic pathway in human vascular smooth muscle cells. Neurobiol. Aging 29, 969–980. doi: 10.1016/j.neurobiolaging.2007.01.009
Cuajungco, M. P., Goldstein, L. E., Nunomura, A., Smith, M. A., Lim, J. T., Atwood, C. S., et al. (2000). Evidence that the β-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of Aβ by zinc. J. Biol. Chem. 275, 19439–19442. doi: 10.1074/jbc.C000165200
Cummings, J., Lee, G., Ritter, A., Sabbagh, M., and Zhong, K. (2019). Alzheimer’ s disease drug development pipeline: 2019. Alzheimers Dement. 5, 272–293. doi: 10.1016/j.trci.2019.05.008
Deane, R., Bell, R. D., Sagare, A., and Zlokovic, B. V. (2009). Clearance of amyloid-β peptide across the blood-brain barrier: implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 8, 16–30. doi: 10.2174/187152709787601867
Deane, R., du Yan, S., Submamaryan, R. K., LaRue, B., Jovanovic, S., Hogg, E., et al. (2003). RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 9, 907–913. doi: 10.1038/nm890
Deane, R., Sagare, A., Hamm, K., Parisi, M., Lane, S., Finn, M. B., et al. (2008). apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Investig. 118, 4002–4013. doi: 10.1172/JCI36663
Deture, M. A., and Dickson, D. W. (2019). The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 14, 32–18. doi: 10.1186/s13024-019-0333-5
Doody, R. S., Raman, R., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2013). A phase 3 trial of Semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 369, 341–350. doi: 10.1056/nejmoa1210951
Egan, M. F., Kost, J., Tiffini Voss, M. D., Yuki Mukai, M. D., Paul, S., Aisen, M. D., et al. (2019). Randomized trial of Verubecestat for prodromal Alzheimer’s disease. N. Engl. J. Med. 380, 1408–1420. doi: 10.1056/NEJMoa1812840.Randomized
Ehrenstein, G., Galdzicki, Z., and Lange, G. D. (1997). The choline-leakage hypothesis for the loss of acetylcholine in Alzheimer’s disease. Biophys. J. 73, 1276–1280. doi: 10.1016/S0006-3495(97)78160-8
Esposito, L., Raber, J., Kekonius, L., Yan, F., Yu, G. Q., Bien-Ly, N., et al. (2006). Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci. 26, 5167–5179. doi: 10.1523/JNEUROSCI.0482-06.2006
Folch, J., Petrov, D., Ettcheto, M., Abad, S., Sánchez-López, E., García, M. L., et al. (2016). Current research therapeutic strategies for Alzheimer’s disease treatment. Neural Plast. 63, 390–401. doi: 10.1016/0012-1606(78)90143-4
Francis, P. T. (2005). The interplay of neurotransmitters in Alzheimer’s disease. CNS Spectr. 10, 6–9. doi: 10.1017/s1092852900014164
Fu, W. Y., Wang, X., and Ip, N. Y. (2019). Targeting neuroinflammation as a therapeutic strategy for Alzheimer’s disease: mechanisms, drug candidates, and new opportunities. ACS Chem. Nerosci. 10, 872–879. doi: 10.1021/acschemneuro.8b00402
Fukumoto, H., Tomita, T., Matsunaga, H., Ishibashi, Y., Saido, T. C., and Iwatsubo, T. (1999). Primary cultures of neuronal and non-neuronal rat brain cells secrete similar proportions of amyloid β peptides ending at Aβ40 and Aβ42. Neuroreport 10, 2965–2969. doi: 10.1097/00001756-199909290-00017
Gauthier, S., Albert, M., Fox, N., Goedert, M., Kivipelto, M., Mestre-Ferrandiz, J., et al. (2016). Why has therapy development for dementia failed in the last two decades? Alzheimers Dement. 12, 60–64. doi: 10.1016/j.jalz.2015.12.003
Giraldo, E., Lloret, A., Fuchsberger, T., and Viña, J. (2014). Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol. 2, 873–877. doi: 10.1016/j.redox.2014.03.002
Goedert, M. (1993). Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 16, 460–465. doi: 10.1016/0166-2236(93)90078-Z
Gómez-Isla, T., Hollister, R., West, H., Mui, S., Growdon, J. H., Petersen, R. C., et al. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann. Neurol. 41, 17–24. doi: 10.1002/ana.410410106
Hajnóczky, G., Davies, E., and Madesh, M. (2003). Calcium signaling and apoptosis. Biochem. Biophys. Res. Commun. 304, 445–454. doi: 10.1016/S0006-291X(03)00616-8
Hampel, H., Mesulam, M., Cuello, A. C., Farlow, M. R., Giacobini, E., Grossberg, G. T., et al. (2018). The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141, 1917–1933. doi: 10.1093/brain/awy132
Hayes, G. M., Howlett, D. R., and Griffin, G. E. (2002). Production of β-amyloid by primary human Foetal mixed brain cell cultures and its modulation by exogenous soluble β-amyloid. Neuroscience 113, 641–646. doi: 10.1016/S0306-4522(02)00191-4
Henley, D., Raghavan, N., Sperling, R., Aisen, P., Raman, R., and Romano, G. (2019). Preliminary results of a trial of Atabecestat in preclinical Alzheimer’s disease. N. Engl. J. Med. 380, 1483–1485. doi: 10.1056/NEJMc1813435
Hickman, S., Izzy, S., Sen, P., Morsett, L., and El Khoury, J. (2018). Microglia in neurodegeneration. Nat. Neurosci. 21, 1359–1369. doi: 10.1038/s41593-018-0242-x
Huang, X., Cuajungco, M. P., Atwood, C. S., Hartshorn, M. A., Tyndall, J. D. A., Hanson, G. R., et al. (1999). Cu(II) potentiation of Alzheimer aβ neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 274, 37111–37116. doi: 10.1074/jbc.274.52.37111
Hukins, D., Macleod, U., and Boland, J. W. (2019). Identifying potentially inappropriate prescribing in older people with dementia: a systematic review. Eur. J. Clin. Pharmacol. 75, 467–481. doi: 10.1007/s00228-018-02612-x
Ittner, L. M., and Götz, J. (2011). Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 12, 67–72. doi: 10.1038/nrn2967
Jiang, Q., Lee, C. Y. D., Mandrekar, S., Wilkinson, B., Cramer, P., Zelcer, N., et al. (2008). ApoE promotes the proteolytic degradation of Aβ. Neuron 58, 681–693. doi: 10.1016/j.neuron.2008.04.010
Jung, S. H., Murphy, E. A., McClellan, J. L., Carmichael, M. D., and Davis, J. M. (2010). The dietary flavonoid quercetin decreases neuroinflammation in a mouse model of Alzheimer’s disease. FASEB J. 24:604.17. doi: 10.1096/FASEBJ.24.1_SUPPLEMENT.604.17
Jusko, T. A., Henderson, C. R., Lanphear, B. P., Cory-Slechta, D. A., Parsons, P. J., and Canfield, R. L. (2008). Blood lead concentration <10 μg/dL and child intelligence at 6 years of age. Environ. Health Perspect. 116, 243–248. doi: 10.1289/ehp.10424
Kehoe, P. G., Miners, S., and Love, S. (2009). Angiotensins in Alzheimer’s disease—friend or foe? Trends Neurosci. 32, 619–628. doi: 10.1016/j.tins.2009.07.006
Kheiri, G., Dolatshahi, M., Rahmani, F., and Rezaei, N. (2018). Role of p38/MAPKs in Alzheimer’s disease: implications for amyloid beta toxicity targeted therapy. Rev. Neurosci. 30, 9–30. doi: 10.1515/REVNEURO-2018-0008
Kihara, T., and Shimohama, S. (2004). Alzheimer’s disease and acetylcholine receptors. Acta Neurobiol. Exp. 64, 99–105.
Kim, J., Basak, J. M., and Holtzman, D. M. (2009). The role of Apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303. doi: 10.1016/j.neuron.2009.06.026
Kim, E. K., and Choi, E. J. (2015). Compromised MAPK signaling in human diseases: an update. Arch. Toxicol. 89, 867–882. doi: 10.1007/S00204-015-1472-2
Kim, H. J., and Vaziri, N. D. (2010). Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Renal Physiol. 298, F662–F671. doi: 10.1152/ajprenal.00421.2009
Kimura, T., Whitcomb, D. J., Jo, J., Regan, P., Piers, T., Heo, S., et al. (2014). Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philosoph Transac R Soc B Biol Sci 369:20130144. doi: 10.1098/rstb.2013.0144
Kitazume, S., Tachida, Y., Kato, M., Yamaguchi, Y., Honda, T., Hashimoto, Y., et al. (2010). Brain endothelial cells produce amyloid β from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J. Biol. Chem. 285, 40097–40103. doi: 10.1074/jbc.M110.144626
Kolarova, M., García-Sierra, F., Bartos, A., Ricny, J., and Ripova, D. (2012). Structure and pathology of tau protein in Alzheimer’s disease. Int. J. Alzheimers Dis. 2012, 1–13. doi: 10.1155/2012/731526
Koudinov, A. R., and Koudinova, N. V. (2005). Cholesterol homeostasis failure as a unifying cause of synaptic degeneration. J. Neurol. Sci. 229-230, 233–240. doi: 10.1016/j.jns.2004.11.036
Kouroku, Y., Fujita, E., Tanida, I., Ueno, T., Isoai, A., Kumagai, H., et al. (2007). ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 14, 230–239. doi: 10.1038/sj.cdd.4401984
Lee, J. K., and Kim, N. J. (2017). Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer’s disease. Molecules 22, 1–23. doi: 10.3390/molecules22081287
Liao, Y., Qi, X.-L., Cao, Y., Yu, W.-F., Ravid, R., Winblad, B., et al. (2016). Elevations in the levels of NF-κB and inflammatory chemotactic factors in the brains with Alzheimer’s disease—one mechanism may involve α3 nicotinic acetylcholine receptor. Curr. Alzheimer Res. 13, 1290–1301. doi: 10.2174/1567205013666160703174254
Liu, C. C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, F., Nguyen, J. L., Hulleman, J. D., Li, L., and Rochet, J. C. (2008). Mechanisms of DJ-1 neuroprotection in a cellular model of Parkinson’s disease. J. Neurochem. 105, 2435–2453. doi: 10.1111/j.1471-4159.2008.05333.x
Lovell, M. A., Robertson, J. D., Teesdale, W. J., Campbell, J. L., and Markesbery, W. R. (1998). Copper, iron and zinc in Alzheimer’s disease senile plaques. Int. J. Surg. Pathol. 15, 252–257. doi: 10.1177/1066896907302118
Matsunaga, S., Kishi, T., and Iwata, N. (2015). Memantine monotherapy for Alzheimer’s disease:A systematic review and meta-analysis. PLoS One 10, 1–16. doi: 10.1371/journal.pone.0123289
McGleenon, B. M., Dynan, K. B., and Passmore, A. P. (2009). Acetylcholinesterase inhibitors and alzheimer’s disease. Encycloped Neurosci 2, 9–13. doi: 10.1016/B978-008045046-9.01129-3
Mendiola-Precoma, J., Berumen, L. C., Padilla, K., and Garcia-Alcocer, G. (2016). Therapies for prevention and treatment of Alzheimer’s disease. Biomed. Res. Int. 2016, 1–17. doi: 10.1155/2016/2589276
Miura, T., Suzuki, K., Kohata, N., and Takeuchi, H. (2000). Metal binding modes of Alzheimer’s amyloid β-peptide in insoluble aggregates and soluble complexes. Biochemistry 39, 7024–7031. doi: 10.1021/bi0002479
Mordn, M. A., Mufson, E. J., G6mez-Ramos, P., and Morcillo, A. (1993). Acta Heuropathologlca regular papers Colocalization of cholinesterases with 5 amyloid protein in aged and Alzheimer’s brains*. Acta Neuropathol. 85, 362–369. doi: 10.1007/BF00334445
Mount, C., and Downton, C. (2006). Alzheimer: progress o profit. Nat. Med. 12, 780–784. doi: 10.1038/nm0706-780
Nair, S., Doh, S. T., Chan, J. Y., Kong, A. N., and Cai, L. (2008). Regulatory potential for concerted modulation of Nrf2- and Nfkb1-mediated gene expression in inflammation and carcinogenesis. Br. J. Cancer 99, 2070–2082. doi: 10.1038/SJ.BJC.6604703
Nelson, L., and Tabet, N. (2015). Slowing the progression of Alzheimer’s disease; what works? Ageing Res. Review. 23, 193–209. doi: 10.1016/j.arr.2015.07.002
Neurology, T. L. (2016). Solanezumab: too late in mild Alzheimer’ s disease? Lancet Neurol. 16:97. doi: 10.1016/S1474-4422(16)30395-7
Nichols, E., Szoeke, C. E. I., Vollset, S. E., Abbasi, N., Abd-Allah, F., Abdela, J., et al. (2019). Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 18, 88–106. doi: 10.1016/S1474-4422(18)30403-4
O’brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Sci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613
Ogata, M., Hino, S., Saito, A., Morikawa, K., Kondo, S., Kanemoto, S., et al. (2006). Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26, 9220–9231. doi: 10.1128/mcb.01453-06
Olajide, O. A., and Sarker, S. D. (2020). Alzheimer’s disease: natural products as inhibitors of neuroinflammation. Inflammopharmacology 28, 1439–1455. doi: 10.1007/s10787-020-00751-1
Osenkowski, P., Ye, W., Wang, R., Wolfe, M. S., and Selkoe, D. J. (2008). Direct and potent regulation of γ-secretase by its lipid microenvironment. J. Biol. Chem. 283, 22529–22540. doi: 10.1074/jbc.M801925200
Parent, M. J., Bedard, M.-A., Aliaga, A., Minuzzi, L., Mechawar, N., Soucy, J.-P., et al. (2013). Cholinergic depletion in Alzheimer’s disease shown by [18F]FEOBV autoradiography. Int J Mol Imaging 2013, 1–6. doi: 10.1155/2013/205045
Prasansuklab, A., and Tencomnao, T. (2013). Amyloidosis in Alzheimer’s disease: the toxicity of amyloid Beta (Aβ), mechanisms of its accumulation and implications of medicinal plants for therapy. Evid. Based Complement. Alternat. Med. 2013, 1–10. doi: 10.1155/2013/413808
Prentice, H., Modi, J. P., and Wu, J. Y. (2015). Mechanisms of neuronal protection against Excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid. Med. Cell. Longev. 2015, 1–7. doi: 10.1155/2015/964518
Prince, M., Albanese, E., Guerchet, M., and Prina, M. (2014). World Alzheimer report 2014: Dementia and risk reduction. An Analysis of Protective and Modifiable Factors. Alzheimers Disease International. London.
Qi, X. L., Xiu, J., Shan, K. R., Xiao, Y., Gu, R., Liu, R. Y., et al. (2005). Oxidative stress induced by beta-amyloid peptide1-42 is involved in the altered composition of cellular membrane lipids and the decreased expression of nicotinic receptors in human SH-SY5Y neuroblastoma cells. Neurochem. Int. 46, 613–621. doi: 10.1016/j.neuint.2005.02.007
Quiroz-Baez, R., Rojas, E., and Arias, C. (2009). Oxidative stress promotes JNK-dependent amyloidogenic processing of normally expressed human APP by differential modification of α-, β- and γ-secretase expression. Neurochem. Int. 55, 662–670. doi: 10.1016/j.neuint.2009.06.012
Ramsey, C. P., Glass, C. A., Montgomery, M. B., Lindl, K. A., Ritson, G. P., Chia, L. A., et al. (2007). Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 66, 75–85. doi: 10.1097/nen.0b013e31802d6da9
Rankin, C. A., Sun, Q., and Gamblin, T. C. (2007). Tau phosphorylation by GSK-3ß promotes tangle-like filament morphology. Mol. Neurodegener. 2, 1–14. doi: 10.1186/1750-1326-2-12
Reitz, C. (2012). Alzheimer’s disease and the amyloid cascade hypothesis: A critical review. Int. J. Alzheimers Dis. 2012:369808. doi: 10.1155/2012/369808
Religa, D., Strozyk, D., Cherny, R. A., Volitakis, I., Haroutunian, V., Winblad, B., et al. (2006). Elevated cortical zinc in Alzheimer disease. Neurology 67, 69–75. doi: 10.1212/01.wnl.0000223644.08653.b5
Riyaz Basha, M., Wei, W., Bakheet, S. A., Benitez, N., Siddiqi, H. K., Ge, Y.-W., et al. (2005). The fetal basis of Amyloidogenesis: exposure to Lead and latent overexpression of amyloid precursor protein and-amyloid in the aging brain. J. Neurosci. 25, 823–829. doi: 10.1523/JNEUROSCI.4335-04.2005
Rojo, A. I., Innamorato, N. G., Martín-Moreno, A. M., De Ceballos, M. L., Yamamoto, M., and Cuadrado, A. (2010). Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 58, 588–598. doi: 10.1002/GLIA.20947
Sagare, A., Deane, R., Bell, R. D., Johnson, B., Hamm, K., Pendu, R., et al. (2007). Clearance of amyloid-β by circulating lipoprotein receptors. Nat. Med. 13, 1029–1031. doi: 10.1038/nm1635
Sandberg, M., Patil, J., D’Angelo, B., Weber A, S. G., and Mallard, C. (2014). NRF2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology 79, 298–306. doi: 10.1016/j.neuropharm.2013.11.004.NRF2-regulation
Sberna, G., Sáez-Valero, J., Beyreuther, K., Masters, C. L., and Small, D. H. (1997). The amyloid β-protein of Alzheimer’s disease increases acetylcholinesterase expression by increasing intracellular calcium in embryonal carcinoma P19 cells. J. Neurochem. 69, 1177–1184. doi: 10.1046/j.1471-4159.1997.69031177.x
Schmitt, T. L., Steiner, E., Klingler, P., Lassmann, H., and Grubeck-Loebenstein, B. (1995). Thyroid epithelial cells produce large amounts of the Alzheimer β-amyloid precursor protein (APP) and generate potentially amyloidogenic APP fragments. J. Clin. Endocrinol. Metab. 8, 3513–3519.
Schnöder, L., Hao, W., Qin, Y., Liu, S., Tomic, I., Liu, X., et al. (2016). Deficiency of neuronal p38α MAPK attenuates amyloid pathology in Alzheimer disease mouse and cell models through facilitating lysosomal degradation of BACE1. J. Biol. Chem. 291, 2067–2079. doi: 10.1074/jbc.M115.695916
Selkoe, D. J. (2001). Clearing the brain’s amyloid cobwebs. Neuron 32, 177–180. doi: 10.1016/S0896-6273(01)00475-5
Storey, E., and Cappai, R. (1999). The amyloid precursor protein of Alzheimer’s disease and the Aβ peptide. Neuropathol. Appl. Neurobiol. 25, 81–97. doi: 10.1046/j.1365-2990.1999.00164.x
Suganthy, N., Devi, K. P., Nabavi, S. F., Braidy, N., and Nabavi, S. M. (2016). Bioactive effects of quercetin in the central nervous system: focusing on the mechanisms of actions. Biomed. Pharmacother. 84, 892–908. doi: 10.1016/j.biopha.2016.10.011
Thinakaran, G., and Koo, E. H. (2008). Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619. doi: 10.1074/JBC.R800019200
Tong, Y., Zhou, W., Fung, V., Christensen, M. A., Qing, H., Sun, X., et al. (2005). Oxidative stress potentiates BACE1 gene expression and Aβ generation. J. Neural Transm. 112, 455–469. doi: 10.1007/s00702-004-0255-3
Van Dyck, C. H. (2018). Anti-amyloid-β monoclonal antibodies for Alzheimer’s disease: pitfalls and promise. Physiol. Behav. 83, 311–319. doi: 10.1016/j.biopsych.2017.08.010.Anti-Amyloid-
Vandenberghe, R., Rinne, J. O., Boada, M., Katayama, S., Scheltens, P., Vellas, B., et al. (2016). Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res. Ther. 8:18. doi: 10.1186/s13195-016-0189-7
Vinod, T., Kuhad, A., Bishnoi, M., and Chopra, K. (2009). Chronic treatment with tocotrienol, an isoform of vitamin E, prevents intracerebroventricular streptozotocin-induced cognitive impairment and oxidative-nitrosative stress in rats. Pharmacol. Biochem. Behav. 93, 183–189. doi: 10.1016/j.pbb.2009.05.009
Wimo, A., Elenn Guerchet, M., Ali, G.-C., Wu, Y.-T., Prina, A. M., Winblad, B., et al. (2017). The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimers Dement. 13, 1–7. doi: 10.1016/j.jalz.2016.07.150
Winston, W. (2020). Economic burden of Alzheimer disease and managed care considerations. Am. J. Manag. Care 26, S177–S183. doi: 10.37765/AJMC.2020.88482
Yiannopoulou, K. G., and Papageorgiou, S. G. (2013). Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 6, 19–33. doi: 10.1177/1756285612461679
Yiannopoulou, K. G., and Papageorgiou, S. (2020). Current and future treatments in Alzheimer disease: an update. J. Central Nervous Syst Dis 12, 117957352090739–117957352090712. doi: 10.1177/1179573520907397
Yin, J. C. P., Wallach, J. S., Del Vecchio, M., Wilder, E. L., Zhou, H., Quinn, W. G., et al. (1994). Induction of a dominant negative CREB transgene specifically blocks long-term memory in drosophila. Cells 79, 49–58. doi: 10.1016/0092-8674(94)90399-9
Youssef, E.-H. H., Wiley, R. E., Khoury, C. P., Daya, R. P., Ballard, C., Evans, A. R., et al. (2019). Tip of the iceberg: assessing the global socioeconomic costs of Alzheimer’s disease and related dementias and strategic implications for stakeholders. J. Alzheimers Dis. 70, 323–341. doi: 10.3233/JAD-190426
Yu, M., Li, H., Liu, Q., Liu, F., Tang, L., Li, C., et al. (2011). Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell. Signal. 23, 883–892. doi: 10.1016/J.CELLSIG.2011.01.014
Zhang, X., Fu, Z., Meng, L., He, M., and Zhang, Z. (2018). The early events that initiate β-amyloid aggregation in Alzheimer’s disease. Front. Aging Neurosci. 10, 1–13. doi: 10.3389/fnagi.2018.00359
Zhou, F., Du, G., Xie, J., Gu, J., Jia, Q., Fan, Y., et al. (2020). RyRs mediate lead-induced neurodegenerative disorders through calcium signaling pathways. Sci. Total Environ. 701:134901. doi: 10.1016/j.scitotenv.2019.134901
Keywords: Alzheimer’s disease, amyloid-beta, tau protein, oxidative stress, neuroinflammation, acetylcholine, mechanisms
Citation: Iliyasu MO, Musa SA, Oladele SB and Iliya AI (2023) Amyloid-beta aggregation implicates multiple pathways in Alzheimer’s disease: Understanding the mechanisms. Front. Neurosci. 17:1081938. doi: 10.3389/fnins.2023.1081938
Edited by:
Jinghui Luo, Paul Scherrer Institut (PSI), SwitzerlandReviewed by:
Shivam Gupta, Novartis Institutes for BioMedical Research, United StatesCopyright © 2023 Iliyasu, Musa, Oladele and Iliya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Musa O. Iliyasu, aWxpeWFzdS5tb0Brc3UuZWR1Lm5n; bW9pbGl5YXN1QGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.