
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 07 July 2022
Sec. Neuropharmacology
Volume 16 - 2022 | https://doi.org/10.3389/fnins.2022.938089
This article is part of the Research Topic Neuroprotective Mechanisms and Translational Approaches in the Retina View all 9 articles
 Yu Zhu1,2
Yu Zhu1,2 Bowen Cao2,3
Bowen Cao2,3 Arianna Tolone1
Arianna Tolone1 Jie Yan1,2
Jie Yan1,2 Gustav Christensen1,2
Gustav Christensen1,2 Blanca Arango-Gonzalez3
Blanca Arango-Gonzalez3 Marius Ueffing3*
Marius Ueffing3* François Paquet-Durand1*
François Paquet-Durand1*Therapy development for neurodegenerative diseases of the retina constitutes a major unmet medical need, and this may be particularly relevant for inherited diseases of the retina, which are largely untreatable to this day. Therapy development necessitates appropriate models to improve the understanding of the underlying degenerative mechanisms, as well as for the testing and evaluation of novel treatment approaches. This review provides an overview of various in vitro model systems used to study retinal neuroprotection. The in vitro methods and technologies discussed range from primary retinal cell cultures and cell lines, to retinal organoids and organotypic retinal explants, to the cultivation of whole eyeballs. The advantages and disadvantages of these methods are compared and evaluated, also in view of the 3R principles (i.e., the refinement, reduction, and replacement of live animal testing), to identify suitable in vitro alternatives for in vivo experimentation. The article further expands on the use of in vitro models to test and evaluate neuroprotective treatments and to aid the development of retinal drug delivery systems. Among the pharmacological agents tested and characterized in vitro are such that interfere with aberrant cyclic guanosine monophosphate (cGMP) -signaling or such that inhibit the activities of poly (ADP-ribose) polymerase (PARP), histone deacetylases (HDAC), calpain-type proteases, as well as unfolded protein response-related stress. We then introduce nanoparticle-based drug delivery systems and discuss how different in vitro systems may be used to assess their efficacy in the treatment of retinal diseases. The summary provides a brief comparison of available in vitro models and relates their advantages and limitations to the various experimental requirements, for instance, for studies into disease mechanisms, novel treatments, or retinal toxicity. In many cases, combinations of different in vitro models may be required to obtain a comprehensive view of the efficacy of a given retinal neuroprotection approach.
The retina is a light-sensitive neuronal tissue located at the posterior part of the eyeball. It is arranged in three layers of cells, namely the outer nuclear layer (ONL), the inner nuclear layer (INL), and the retinal ganglion cell (RGC) layer. The ONL harbors photoreceptors, which are unique neurons dedicated to converting light into electrochemical signals and as such essential for vision. Two different types of photoreceptors are distinguished: Rod photoreceptors respond to dim light and enable vision at night, whereas cone photoreceptors respond to bright daylight and mediate high-resolution and color vision (Kolb et al., 2001).
The retina may suffer from a variety of neurodegenerative diseases, including common diseases such as glaucoma or age-related macular degeneration (AMD) and rare, inherited retinal degeneration (IRD). While glaucoma affects RGCs that form the optic nerve and relay visual information to the brain, AMD and IRD affect photoreceptors. Accordingly, IRD is typically caused by mutations in genes expressed in rod photoreceptors (Berger et al., 2010). However, rod degeneration is usually followed by a secondary loss of cones, leading to a characteristic two-stage disease progression, in which first night vision and then high-acuity daylight vision is lost (Kennan et al., 2005; Guadagni et al., 2015). In AMD, on the other hand, the disease phenotype appears to be linked primarily to a loss of cone photoreceptors, even though rods are also affected by the disease (Curcio et al., 2000).
To this day, neurodegenerative diseases of the retina remain poorly treatable, especially when it comes to diseases affecting photoreceptors, which are essentially untreatable (Power et al., 2020a). This situation creates a strong need for models that can accurately reproduce disease pathogenesis and allow the testing of new treatments for retinal neuroprotection. For the purposes of this review, we will discuss in vitro models for retinal diseases, focusing especially on models for photoreceptor diseases.
Cell cultures are in many ways ideal for scientific studies: They are relatively easy to handle, inexpensive, and relatively homogenous in their cellular composition. Most cell lines can be propagated almost indefinitely, although, with increasing passage number, changes in culture properties and composition can occur and should be considered. In the context of IRD research, cell cultures can be used, for instance, for initial drug screenings before translating into in vivo experiments (Vighi et al., 2018). Often such studies involve the overexpression of mutant IRD genes critical to retinal function as a way to understand the pathological mechanisms induced by these mutations (McKeone et al., 2014). Among these, mutations in the rhodopsin (RHO) gene may be the most studied and are classified into more than seven classes based on their subcellular properties and biochemical characteristics (The human gene mutations database1; information retrieved in June 2022). This section describes the application of different cellular models for research into retinal disease pathologies, focusing on IRD and photoreceptor-related diseases.
661W cells are an immortalized mouse cell line derived from transgenic mice expressing the SV40 T antigen under the control of the human inter-photoreceptor retinol-binding protein promoter (Tan et al., 2004). They express a number of cone photoreceptor markers, such as blue and green cone opsin, transducin, and cone-arrestin, yet at much lower levels than mature photoreceptors. 661W cells also display a neuronal shape and protrusion formation, showing some morphological resemblance with photoreceptors (Tan et al., 2004). They have been widely used to study cone degeneration (Mencl et al., 2018). One recent study also showed that 661W cells might be used for studying retinal ciliopathies as these cells grow long primary cilia similar in structure to those in cone photoreceptor outer segments and localize many cilium proteins to the axoneme, membrane, and transition zone (Wheway et al., 2019). On the other hand, 661W cells also express rod photoreceptor and bipolar cell markers (Mencl et al., 2018). This cell line was successfully used for studies of retinitis pigmentosa (RP) caused by rhodopsin mutations (Surgucheva et al., 2005). However, 661W cells have specific limitations, for example, relatively a low transfection efficiency of around 10% (Griciuc, 2010) and a low protein expression level for photoreceptor-specific proteins (Wheway et al., 2019).
Remarkably, a recent study reported the generation of a new clone derived from 661W cells, named 661W-A11, which stably overexpresses the neural retina leucine zipper (NRL) transcription factor, driving cell differentiation toward the rod photoreceptor fate (Huang et al., 2021). Furthermore, compared with 661W cells, 661W-A11 cell lines showed a significant increase in the expression of rod-specific genes but not of cone-specific genes, which makes this new cell line potentially suitable as a cell culture model to study IRD and the development of high-throughput in vitro drug screening systems. Altogether, the 661W cell line may mimic certain characteristics of photoreceptors, allowing for exploration of specific aspects of retinal disease mechanisms, yet it is still far from representing primary photoreceptors.
The Y-79 cell line was originally established from a human retinoblastoma tumor and is thought to be derived from primitive multipotential retinoblasts (Reid et al., 1974; Kyritsis et al., 1984). Like 661W cells, the Y-79 line has also been used as a model for photoreceptors. Y-79 cells are usually maintained in suspension culture in an undifferentiated state, while some cells can be stimulated to express characteristics of differentiated, mature retinal cell types (Kyritsis et al., 1985). Y-79 cells express a comparable mRNA level of rod PDE6A and cone PDE6C genes while the transcript encoding the rod PDE6B subunit was 10 times more abundant. This made the Y-79 line be a good model for studying transcriptional regulation of rod-specific genes (Di Polo and Farber, 1995). However, later studies reported that Y-79 cells were not able to express functionally active cone PDE6A protein and suggested that the expression of a fully active stable PDE6 enzyme required other post-transcriptional events that did not occur or were inhibited in Y-79 cells (White et al., 2004). Moreover, this cell line appears to be difficult to transfect and lacks reproducibility of gene transfer, reducing the utility of these cells for scientific research (White et al., 2001).
Since the generation of sensory neurons such as photoreceptors remains a challenge, reprogramming other cell types into photoreceptor-like cells has been developed as an alternative method. Two major approaches have been considered, gene-induced and chemical (or pharmacological) reprogramming. An example of gene-induced reprogramming is the introduction of human NEUROD (NRD) or NGN1 genes to hTERT RPE-1 (human telomerase-immortalized retinal pigmented epithelial) and ARPE-19 cell cultures, respectively, or the transfection of NGN1 or NGN3 genes into primary retinal pigment epithelial (RPE) cell cultures (from mouse or porcine origin), changing gene expression and cellular morphologies, suggesting that they are responsive to the reprogramming (Yan et al., 2013).
Furthermore, chemical reprogramming of fibroblasts by administering a set of small molecules [valproic acid (V), CHIR99021 (a GSK3 inhibitor; C), RepSox (R), and forskolin (F), together denoted VCRF] has been shown to induce the transformation of fibroblasts into rod photoreceptor-like cells based on gene expression profiling (Mahato et al., 2020).
Besides that, single colonies derived from the pigmented ciliary margin cells of adult mouse eyes have been generated. They can clonally proliferate in vitro to form spheroid colonies of cells that can differentiate into retinal-specific cell types, including rod photoreceptors, bipolar neurons, and Müller glial cells (MGCs) (Tropepe et al., 2000). This method has been applied in IRD research, using retinoic acid to drive the differentiation into photoreceptor cells (Sanges et al., 2006). While reprogramming somatic cells with stem cell-like behavior remains a possibility for retinal research, the more recent discovery of induced pluripotent stem cells has opened further and potentially more interesting possibilities (see organoid cultures below).
SK-N-SH is a human neuroblastoma cell line developed in the early 1970s (Biedler et al., 1973) that expresses multiple neurochemical markers and exhibits a neuronal phenotype (Biedler et al., 1978). Furthermore, SK-N-SH cells respond to numerous insults, including β-amyloid overexpression, mitochondrial permeability transition, and serum deprivation, indicating that this cell line may be useful in the assessment of neurotoxicity and neuroprotection (Ba et al., 2003). Accordingly, SK-N-SH cells have been used as a model for identifying and characterizing modulators of mutant rhodopsin processing and aggregation (Chapple and Cheetham, 2003; Mendes and Cheetham, 2008; Griciuc et al., 2010; Athanasiou et al., 2017). Besides, this cell line was suggested as an in vitro model for studying neuroprotection mechanisms in retinal degenerative diseases (Watters and Dorsa, 1998; Green et al., 2001; Wen et al., 2004; Wang et al., 2006). Limitations of the SK-N-SH cell line include low transfection efficiencies of around 15% (Griciuc, 2010) and low expression of retina-specific genes and proteins.
HEK293 cells and COS cells are common in vitro models that have been widely used in cell biology and biotechnology research for many years. The HEK293 immortalized cell line was derived from human embryonic kidney cells from a female fetus (Graham et al., 1977; Graham, 1992; Kavsan et al., 2011). They exhibit high transfection efficiency and can produce proteins most similar to those naturally synthesized in humans (Dumont et al., 2016). Several further cell lines have been derived from HEK293 cells, including HEK293T (Lin et al., 2014), HEK293E (Kim et al., 2009b), HEK293H (Bloom et al., 2001), and HEK293S (Lin et al., 2014). Among these, HEK293T and HEK293S appear to be the most commonly used hosts.
COS cells are fibroblast-like cell lines derived from the kidney tissue of the African green monkey (Chlorocebus aethiops), obtained by immortalizing CV-1 cells (Jensen et al., 1964) by transformation with an origin defective mutant of SV40 virus that can produce large T antigen (Gluzman, 1981). Three COS lines were created (COS-1, COS-3, and COS-7), of which two are commonly used (COS-1 and COS-7).
While HEK293 and COS cells are non-neuronal in origin, they have been used extensively in neurodegeneration research. For instance, in neurodegenerative diseases, such as Parkinson’s, Alzheimer’s, and Huntington’s disease, HEK293 and COS cells are often used together to characterize the pathology and define protein accumulation (Hering et al., 2004; Lammich et al., 2004; So et al., 2013; Magno et al., 2019). Likewise, in IRD-research causative mutations often lead to protein conformational defects, for instance, in the RHO or PDE6 genes. Such mutants can be easily overexpressed in HEK293 and COS7 cells for experimental investigation. Consequently, numerous studies based on these two cell lines examined protein behavior in IRD (Mendes and Cheetham, 2008; Griciuc et al., 2010; Gopalakrishna et al., 2016).
The MIO-M1 cell line (Moorfields/Institute of Ophthalmology-Müller 1) was the first immortalized human MGCs line derived from a female corneal donor (Limb et al., 2002b). Cell cultures derived from the MIO-M1 line are often characterized as MGCs, based on the expression of specific makers and morphological analysis. However, the effect of immortalization, high passage number, and expression on non-MGC markers have received little attention. Indeed MIO-M1 cells have been shown to express neural stem cell markers such as tubulin, SOX2, PAX6, CHX10, and NOTCH1, when exposed to various extracellular matrix and growth factors in vitro (Lawrence et al., 2007). Other investigations found MIO-M1 to express features of mature cells, producing hepatocyte growth factor and vascular endothelial growth factor (Hollborn et al., 2005) and matrix metalloproteinases 1, 2, and 9 (Limb et al., 2002a; Limb et al., 2005). Altogether, these findings suggest that MIO-M1 cells are not truly representative of MGCs, limiting the usefulness of this cell line for investigations on MGC physiology.
The role of microglial cells in retinal degenerative diseases appears ambiguous, but the general consensus seems to be that these cells are mostly involved in secondary pathologies rather than the primary changes causing IRD (Sancho-Pelluz et al., 2008; Ferrer-Martin et al., 2015; Funatsu et al., 2022). Nevertheless, the relatively recently introduced BV2 and MG5 microglial cell lines may provide new attractive opportunities for studying microglial properties and the possible roles of these cells in photoreceptor degeneration (Chumsakul et al., 2020; Ozaki et al., 2022).
For decades, researchers have attempted to isolate different retinal cell types from a variety of different species, such as fish, amphibians, rodents, primates, and humans (Han et al., 2000). The physical separation of retinal cells from each other typically requires enzymatic and mechanical intervention. After dissociation, cell types may be identified by their morphology, specific markers, or characteristic measurements, such as the transepithelial resistance, to assess RPE cell confluence and cell density (Skaper, 2012). The primary cell cultures mentioned in this review include rod and cone photoreceptors, MGCs, and RPE cells.
The culture of primary photoreceptor cells has been challenging for many years. While various protocols and methods have been tried (Han et al., 2000), including protocols used for MGC culture (see below), the viability of isolated photoreceptors is typically limited to 1–2 days. Early in 1972, protocols for isolating photoreceptors from turtles were already available (Lam, 1972). This was followed by the addition of neurotrophic factors (Skaper, 2012; Forouzanfar et al., 2020) to the cultures, such as the ciliary neurotrophic factor (CNTF) and brain-derived neurotrophic factor (BDNF), to promote the structural integrity and survival of photoreceptor cells. Isolated cone photoreceptor cultures obtained from rat retina based on a peanut agglutinin (PNA) lectin–panning procedure (Skaper, 2012) show viability typically limited to 1–2 days. Hence, there are significant challenges when trying to obtain adequate photoreceptor cultures, including the short-time survival after isolation and the loss of photoreceptor-specific structures and compartments (i.e., inner and outer segments and ribbon synapse) (Yang et al., 2001).
Müller glial cells form a network that connects other retinal cells, extending vertically through the entire retina, from the RGCs to photoreceptors. MGCs play a crucial role in metabolism, support, and modulation of neuronal excitability by releasing and transporting neurotransmitters (Eastlake et al., 2021). They may also show progenitor cell characteristics in the adult retina (Lawrence et al., 2007). Moreover, MGCs can respond to retinal injury by secreting neuroprotective factors reducing rod photoreceptor cell death in IRD mouse models (Del Rio et al., 2011; Roche et al., 2016).
Primary MGCs are used relatively often cultivated after enzymatic and mechanical isolation being relatively similar across different species (Hicks and Courtois, 1990; Han et al., 2000; Liu et al., 2017). MGC isolation after dissociation of primary retinal cells is facilitated greatly by the fact that MGCs are essentially the only cell type that survives for more than 2 days in culture, while all other neuronal cell types (e.g., photoreceptors, see above) rapidly die off. However, after 1–2 days in culture, primary MGCs rapidly dedifferentiate and assume a fibroblast-like phenotype (Hauck et al., 2003).
The RPE consists of pigmented cells essential for photoreceptor cell survival. They are highly polarized cells that mediate the recycling of the photopigment retinal, phagocytose the rod- and cone- outer segments (Schnichels et al., 2021), and protect photoreceptors against photooxidation (Strauss, 2005). RPE cells are linked by tight junctions and form a monolayer that constitutes a part of the outer blood-retinal barrier (BRB), which regulates the movement of solutes and nutrients from the choroid to the sub-retinal space (Campbell and Humphries, 2012). Moreover, they are immunocompetent (at least in part) and provide the molecular and cellular interphase between the neuroretina and the choriocapillaris (Armento et al., 2021b). Immortalized RPE cells, such as the RPE-1 cell line, have been widely used for experimental purposes, especially in AMD research, as they degenerate in this disease (Armento et al., 2021a).
The RPE is located at the back of the eyeball, making in vivo studies difficult. However, RPE cells lend themselves to in vitro studies since they typically preserve their complete functional phenotype and cell contacts (Oswald and Baranov, 2018). The first human RPE culture goes back to 1973 (Mannagh et al., 1973), and since then, many strategies for RPE culturing have been developed (Skaper, 2012). Common problems of RPE cultures include pigmentation loss, low cell-substrate adhesion, alterations in cell morphology, and low long-term viability of cells (Blenkinsop et al., 2013). In part to overcome these issues, different cell lines have been developed that can mimic most of the RPEs properties.
The ARPE-19 cell line was developed from the RPE cell layer of a human eye post-mortem (Kozlowski, 2015). This cell line displays many of the properties of native RPE cells; for example, the cobblestone morphology, which is characteristic of RPE cells (Dunn et al., 1996), the formation of polarized structures on porous filter supports (Dunn et al., 1996), the expression of cellular retinaldehyde-binding protein (CRALBP), one of the RPE-selective markers (Ablonczy and Crosson, 2007). Because of these RPE-like properties, the ARPE-19 cell line has frequently been used in the research of retinal disorders, including AMD (Kozlowski, 2015), RP (Hulleman et al., 2016), retinal ciliopathies, and Leber’s congenital amaurosis (van Wijk et al., 2009). Similarly, another cell strain, hTERT RPE-1, an hTERT immortalized female RPE cell line, is also routinely used in molecular biology studies of retinal ciliopathies (Adams et al., 2007; Spalluto et al., 2013). The findings suggest that hTERT RPE-1 cells could serve as a model system for studying the molecular pathways, including reciliation in the late G1 of the cell cycle and those that stimulate cilium disassembly.
The ground-breaking work of Yamanaka and colleagues (Takahashi and Yamanaka, 2006) made it possible to generate induced pluripotent stem cells (iPSC) from somatic cells. iPSCs can generate most cell types and tissues of an organism. This has given rise to numerous applications, including the generation of human stem cell-derived retinal RPE cells and retinal organoids. Concerning the retina, iPSCs can differentiate into RPE cells and cells of the neural retina, including rods and cones. The process of harvesting somatic cells and reprogramming them into virus-free human iPSCs (hiPSC) and from there to retinal organoids has been standardized in various laboratories (Liebau et al., 2019; Cobb et al., 2021).
A variety of signaling pathway modulators promote differentiation to retinal cells (e.g., the TGFß, Wnt, Nodal, and BMP signaling) in a defined time frame that takes up to 52 weeks to complete (Achberger et al., 2019b). Furthermore, animal experiments and even clinical trials have been initiated with the transplantation of stem cell-derived retinal cells into the diseased or degenerated retina (Mandai et al., 2017). As such, hiPSC-derived organoids can serve as a human model system to study cell development, function, and disease mechanisms but also represent a new source of individual cell material for future cell-based therapies. Moreover, the hiPSC-derived retinal organoids obtained from individual donors suffering, for instance, from IRD, can provide retina-like structures mimicking the disease. Such organoids feature not only the genetic set-up of the donor but also the retinal context, including retinal anatomy and physiology, as well as light reception.
Three-dimensional organoids derived from hiPSC resemble rudimentary optic vesicle-like structures with a retinal layering similar to in vivo conditions (Nakano et al., 2012; Zhong et al., 2014). The resulting organoids contain the most relevant retinal cell types in a physiological layering (Achberger et al., 2019a). As such, three-dimensional retinal organoids from hiPSC cells comprise all main cell types of the retina in a layered manner, such as photoreceptors, amacrine, horizontal, bipolar cells, as well as RGCs and MGCs (Canto-Soler et al., 2016). An exception is the RPE, which forms amorphous structures apart from the neuroretina. While attempts have been made to co-culture retinal organoids with RPE, these have so far had only limited success (Achberger et al., 2019b). Still, immunofluorescence shows the outer and inner limiting membrane and synaptic connections between the neuronal layers (Achberger et al., 2019a).
Nevertheless, retinal organoids are still facing a variety of limitations: They lack functional maturation of differentiated cells, especially with respect to mature photoreceptors, which do not grow proper outer segments and do not connect well with the RPE. Microglial cells are missing in retinal organoids, and RGCs do not reach full maturation and are unable to bundle to an optic nerve-like structure or connect to the brain. The inner limiting membrane produced in an interplay between RGCs and MGCs end-feet lacks the Bruch’s membrane, separating the retina from the choroid and choriocapillaris. Also, the inner retinal vasculature is lacking. As such, and due to the lack of physiological perfusion from the bloodstream, the delivery of nutrients, metabolites, and oxygen remains entirely artificial.
Cell lines and primary cell cultures are suitable for studying intrinsic cell characteristics but do not allow for the investigation of complex interactions between different cell types arranged in specific structures and organizational units. Yet, the retina is a complex network governed by a multitude of cell-to-cell interactions, both under physiological and pathological conditions. Here, organotypic retinal explant cultures allow long-term studies on a retina that has conserved its normal histotypic context and that can be easily intervened for experimental purposes (Caffe et al., 1989; Caffé et al., 1993). Over the last decades, several different methods have been used. For instance, the culture of complete mouse eyecups allowed to maintain retinal structures and photoreceptor viability for around 6 days in culture (Müller et al., 2017). The explanted retina cultured without RPE, displayed characteristic morphological alterations and extensive photoreceptor cell death starting between 2 to 4 days in vitro (Ferrer-Martín et al., 2014; Müller, 2019). Nevertheless, retinal explants cultured without RPE but with the vitreous remaining attached to the inner limiting membrane have been used for short-term (48 h) studies into drug delivery through the vitreoretinal interface (Tavakoli et al., 2020). In addition, the whole retinal vasculature degenerates rapidly in the absence of blood supply.
The approaches mentioned above were suitable only for investigations limited to in vitro duration of a few days. For longer-term studies, organotypic retinal explants with intact RPE were developed in the late 1980s (Caffe et al., 1989). The preparation of retinal explants (Belhadj et al., 2020) includes the separation of the retina from choroid and sclera, the removal of lenses, iris, ciliary body, vitreous, and optic nerve, and the cutting of the retinal cup into a four-leaf clover shape to flatten the retina on the culturing membrane. The cultivation of the retina in defined R16 medium, free of serum and antibiotics, allows culture periods of at least 4 weeks. An important aspect of such retinal explants is that they must be cultured on a suitable porous membrane in an air-liquid interface, such that the retina is covered by only a thin film of liquid created by the surface tension of water. This is critical to ensure sufficient oxygenation of the retina. Retinal explants prepared in this way may be used for different experimental applications and technologies, such as histology, patch-clamp recording (Moritoh et al., 2010), or multi-electrode-array (MEA) recording (Reinhard et al., 2014).
While organotypic retinal explants with RPE cannot replace in vivo experimentation completely, there are many merits compared to in vivo experiments, including but not limited to: lower experimental complexity, cost-effectiveness, the ability of experimental manipulations under entirely controlled conditions, the avoidance of animal pain and suffering, and a reduction of animals needed since retinas from both eyes can be used (Belhadj et al., 2020). These advantages make retinal explants an almost ideal and indispensable system for identifying and studying new potential treatments for retinal diseases. A limitation, however, is the relatively complex explantation and dissection procedure that requires appropriate training and experience. In addition, some of the limitations mentioned for organoid cultures (see above; lack of perfusion, microglial cells, Bruch’s membrane) likewise apply to organotypic retinal explants.
Before the event of modern cell and organ culture systems, in vitro studies on the retina typically employed whole eye cultures, starting in the second half of the 19th century, often using frog eyes (Kuhne and Steiner, 1880; Kühne, 1881; Brücke and Garten, 1907). A limitation in these studies was that the electroretinographic response (ERG) to a light stimulus was typically lost within 1 h from enucleation, a phenomenon attributed to tissue degeneration. However, the ERG response could be preserved for up to 24 h when isolated frog eyes were kept under a 100% oxygen atmosphere (Bauereisen et al., 1956), indicating that sufficient oxygenation was critical to retinal function. Similar studies in whole-eye cultures obtained from rabbits (Böck et al., 1964) and rats (Suga, 1972) demonstrated the importance of temperature and sufficient glucose supply for retinal function.
In an attempt to control all these parameters, in 1960, vascular perfusion of the entire mammalian eye after enucleation was first carried out (Lele and Grimes, 1960). Afterward, vascular perfusion was applied to different mammalian species (Niemeyer, 2001; Koeberle et al., 2006), including rats, rabbits, dogs, monkeys, cats, and bovines. This allowed the recording of retinal function using an ERG, light-evoked optic nerve response, and standing potential. Perfused cat eyes could be cultured for 8–10 h while still displaying light-responsiveness as assessed via ERG (Niemeyer, 1975). With perfusion beyond 9 h, photoreceptors swelled slightly, while RPE cells, RGCs, and other cell types in the INL showed mild and moderate loss (Koeberle et al., 2006). Perfusion times over 10 h led to retinal detachments and overall function loss (Niemeyer, 2001).
Among the advantages of using vascular perfusion of whole eyes are the possibilities to combine the monitoring of electrophysiological activity with the arterial application of an intervention without influence by extraocular regulation, such as systemic vascular changes. In addition, the structural integrity and the pathophysiological performance of the retina allowed studies on the pharmacology of vascular dynamics and studies into acute retinal toxicity (Niemeyer, 2001). Nevertheless, an inevitable disadvantage of whole-eye culture is the relatively limited time frame for experimental interventions (from minutes to several hours), which does not allow investigating longer-term effects.
The mechanisms underlying retinal neurodegenerative disorders are complex and far from being completely understood. In vitro models, such as cell cultures and organotypic retinal explants, are widely used for IRD research as these models are suitable for studying components of the photoreceptor cell death pathway, such as cyclic guanosine monophosphate (cGMP), calpain-type proteases, histone deacetylases (HDAC) and poly (ADP-ribose) polymerase (PARP).
Increased cGMP levels have been connected to retinal photoreceptor degeneration already in the early 1970s (Farber and Lolley, 1974). Moreover, high levels of cGMP have been observed in different animal models for IRD, suggesting that excessive cGMP signaling may be a common event in several mutations causing photoreceptor degeneration (Power et al., 2020a). One way to study the mechanism of cell death triggered by abnormal cGMP accumulation is the inhibition of its two prototypic targets: Cyclic nucleotide-gated (CNG) channels and protein kinase G (PKG), both of which have been connected to photoreceptor cell death (Farber and Lolley, 1974; Paquet-Durand et al., 2009). Both photoreceptor-like cell cultures and organotypic retinal explant cultures have been employed to target CNG channels and PKG and have contributed valuable insights into the photoreceptor death pathway (Vighi et al., 2018; Tolone et al., 2021; Das et al., 2022).
Recently, PKG inhibition mediated by cGMP analogs has attracted the interest of IRD researchers as a potential neuroprotective strategy to slow down photoreceptor death (Paquet-Durand et al., 2019). cGMP analogs mimic the overall structure of cGMP while carrying substitutions in certain residues that enable them to inhibit specific cGMP-signaling targets (Schwede et al., 2000). PKG-inhibiting cGMP analogs carry a phosphorothioate configured as Rp, which allows them to counteract PKG activation (Zhao et al., 1997). Two novel cGMP analogs with strong protective abilities on degenerating photoreceptors have recently been identified using in vitro systems: Photoreceptor-like cell cultures allowed an initial screening of several compounds, while organotypic retinal explants cultures derived from different IRD mouse models confirmed the neuroprotective capacities of these cGMP analogs (Vighi et al., 2018; Tolone et al., 2021). Furthermore, the use of retinal explant cultures treated with inhibitory cGMP analogs also led to the identification of novel PKG targets that may be involved in the cell death mechanism of IRD (Roy et al., 2021; Roy et al., 2022).
Poly (ADP-ribose) Polymerases are DNA repair enzymes (Ko and Ren, 2012), comprising a family of 17 isoforms that share a conserved catalytic domain (Krishnakumar and Kraus, 2010). The best-characterized isoform within this family is PARP-1, which catalyzes the PARylation of target proteins by consuming nicotinamide adenine dinucleotide (NAD+) (Houtkooper et al., 2010; Morales et al., 2014; Bai, 2015; Lord and Ashworth, 2017; Murata et al., 2019). Over-activation of PARP forces the cell to synthesize NAD+ using salvage pathways, which may cause subsequent ATP depletion (Houtkooper et al., 2010; Xie et al., 2020). Hence, persistent PARP activation results in a specific form of cell death termed PARthanatos (David et al., 2009).
In in vitro retinal cultures derived from rd1 mice, PARP inhibition reduced the number of cells exhibiting death markers (Paquet-Durand et al., 2007; Sahaboglu et al., 2016; Sahaboglu et al., 2020; Yan et al., 2022), suggesting the neuroprotective effect of PARP inhibitors, and highlighting the importance of a novel cell death pathway that is triggered by high cGMP-levels and likely related to PARthanatos (Yan et al., 2021). The success of PARP inhibitors in in vitro experiments makes PARP inhibition appear as an attractive strategy for therapy development, notably because a number of already clinically approved PARP inhibitors offer accelerated and low-cost progress in disease combating. This may be of particular interest in the case of rare IRD-type diseases such as RP (Sahaboglu et al., 2020).
Histone function is modulated by multiple posttranslational modifications, including reversible acetylation of the amino-terminal ε-group of lysine residues. Histone acetylation is tightly controlled by a balance between the opposing activities of histone acetyltransferases (HATs) and HDACs. There are 18 potential human HDACs grouped into four classes (Seto and Yoshida, 2014).
With the help of in vitro retinal cultures, IRD was reported to be associated with excessive HDAC activation, and in vitro HDAC inhibition strongly reduced photoreceptor cell death (Sancho-Pelluz et al., 2010; El Bahhaj et al., 2014). Since then, HDACs have been considered part of the cGMP-dependent cell death mechanism (Arango-Gonzalez et al., 2014). On the other hand, sirtuin-type HDACs (class III HDACs) are considered to be neuroprotective (Jiang et al., 2011; Gomes et al., 2018; Xu et al., 2018). Given the pleiotropic effects of HDAC activity, HDAC inhibitors, such as trichostatin A (TSA), suberoylanilide hydroxamic acid (SAHA), or valproic acid (VPA), often target multiple HDACs, and the biological consequences are often unpredictable and underappreciated (Witt et al., 2009; Falkenberg and Johnstone, 2014). Here, in vitro experimentation may offer a rapid and economical way of solving essential open questions for the therapeutic development of HDAC inhibitors for IRD.
Calpain belongs to a 15-member family of the calcium-dependent thiol proteases (Perrin and Huttenlocher, 2002; Curcio et al., 2016). The best-characterized calpains in the central nervous system are two distinct, heterodimeric subtypes: μ-calpain and m-calpain, also known as calpain-1 and calpain-2 (Cheng et al., 2018). Like HDACs, calpains may show pleiotropic effects, in which calpain-1 could be neuroprotective, while calpain-2 could be neurodegenerative (Baudry and Bi, 2016; Baudry, 2019; Wang et al., 2020). In addition, over-activated calpain has been reported in rd1 photoreceptors in vivo, and calpain inhibition with the endogenous calpain inhibitor calpastatin on in vitro rd1 retinal explant cultures attenuated photoreceptor degeneration (Paquet-Durand et al., 2006; Power et al., 2020b; Yan et al., 2022), which suggests that calpains form part of IRD cell death mechanisms. The only specific inhibitor for classical calpains currently available, calpastatin, blocks calpain-1 and calpain-2, calpain-8, calpain-9, and calpain-8/9 (Wendt et al., 2004; Hata et al., 2007; Hata et al., 2010; Ono et al., 2016). Thus, further in vitro studies are needed to further elucidate the diverse roles of different calpain isoforms during IRD.
In addition to the mechanisms and drug categories mentioned above, other pharmacological approaches targeting common events observed in IRD have been validated using in vitro studies. Physiological and pathophysiological processes such as proteostasis, neuroinflammatory response, and mitochondrial homeostasis are affected in IRD, generating the chance to identify multiple molecular targets for pharmacological intervention studies. In this context, a particularly interesting therapeutic strategy is to attenuate the stress induced by perturbance of proteostasis, that can arise, once proper protein folding, transport, and degradation of proteins are compromised. For instance, mutations in the RHO gene encoding for rhodopsin, that cause protein misfolding and aggregation, are the most common cause of autosomal dominant retinitis pigmentosa (adRP) (Sen et al., 2021c). Pharmacological targeting of proteostasis regulators has been proven neuroprotective for rhodopsin mutations. Inhibition of valosin-containing protein (VCP) through a small molecule inhibitor significantly reduced cell death in photoreceptors, restored physiological rhodopsin localization in the outer segment, and improved retinal function in organotypic retinal explants derived from RhoP23H transgenic rats and RhoP23H knock-in mice (Arango-Gonzalez et al., 2020; Sen et al., 2021a,c). As proof of concept, the delivery of VCP siRNA using reverse magnetofection in organotypic cultures of RhoP23H transgenic retinas effectively prevented photoreceptor cell death and attenuated retinal degeneration in vitro (Sen et al., 2021b). Other small molecules newly identified that rescued the transport of rhodopsin include the chaperon YC-001 which protected Abca4–/– * Rdh8–/– double-mutant mice from bright light-induced photoreceptor death (Chen et al., 2018).
Recombinant neurotrophic factors have also been tested in organotypic cultures for photoreceptor neuroprotection. This included CNTF, BDNF, and glial cell-line derived neurotrophic factor (GDNF). CNTF is one of the most studied neuroprotective agents with acknowledged potential in treating diseases of the posterior eye segment (Itkonen et al., 2020) and has been found to promote cone survival (Dutt et al., 2010; Lipinski et al., 2011). BDNF delayed photoreceptor cell loss and increased the number of photoreceptor rows (Pinzon-Duarte et al., 2004); however, it was much more effective when combined with CNTF in rd1 mouse retinal explants (Caffé et al., 2001; Azadi et al., 2007). GDNF is a member of the transforming growth factor β (TGFβ) family that promotes the survival, proliferation, and differentiation of neurons, RGCs, and photoreceptors (Kolomeyer and Zarbin, 2014). When combined with CNTF, GDNF effectively attenuated the secondary loss of cone photoreceptors on retinal explants derived from RHO knockout mice (Lipinski et al., 2011). Possibly, GDNF executes its neuroprotective effects on photoreceptors indirectly via MGCs (Hauck et al., 2006). Upon GDNF stimulation isolated RMGs secrete a mixture of factors that prolong the viability of photoreceptors in mouse retinas in vitro (Del Rio et al., 2011). Upon analyzing the MGCs conditioned medium using proteomics- and transcriptomic approaches, several macromolecules with photoreceptor protective properties have been identified (Hauck et al., 2014; von Toerne et al., 2014). Among these compounds, Osteopontin (OPN) and Cysteine-rich angiogenic inducer 61 (Cyr61) have shown protective effects on rd1 photoreceptors in vitro (Del Rio et al., 2011; Kucharska et al., 2014).
To treat retinal diseases, intravitreal injections are commonly used, for instance, in the treatment of AMD (Garcia-Quintanilla et al., 2019) to get the administered drugs in close proximity to the target tissue. However, for most treatments, multiple injections are necessary, which increases the risk of complications (Shima et al., 2008). The administration of drugs combined with a drug delivery system (DDS) can be beneficial in improving the pharmacokinetic profile with the aim of reducing the injection frequency and limiting systemic side effects (Zeng et al., 1993; Zhang et al., 2010; Bochot and Fattal, 2012; Kamaleddin, 2017). In this regard, nanoparticles have gained interest since they can be engineered small enough to diffuse freely within the vitreous (Lee et al., 2017) and permeate relevant ocular barriers (Kim et al., 2009a). For systemic applications, targeting moieties on the nanoparticle surface can help the drug accumulate in a specific tissue or cell type in the body (Hu et al., 2019).
Since most DDS typically exhibit an improvement over a free drug solution in vivo, the challenge of evaluating the potential of DDS without using living animals is that suitable in vitro systems should closely emulate the in vivo conditions. A common strategy seems to be taking relevant excised ocular tissue from dead animals and investigating the DDS properties in this system (Eriksen et al., 2017; Tavakoli et al., 2021). An obvious source of the tissue samples is leftover eyes during meat production at slaughterhouses. For instance, bovine eyes have been used to prepare retinal explant cultures with the vitreous intact (vitreoretinal explant cultures) to investigate the diffusion of nanoparticles through the vitreoretinal interface (Peynshaert et al., 2017; Tavakoli et al., 2020). Here, it was found that liposomes surface-grafted with poly (ethylene glycol) polymers need to be less than 100 nm in diameter to diffuse into the retina. When the vitreous is conserved during the explantation procedure, the explant cultures are more restrictive to the nanoparticle’s ability to diffuse into the retina (Peynshaert et al., 2017). Still, it is not certain how active the cells are in such explant cultures. If active cell uptake is important for the application, retinal explant cultures derived right after animal sacrifice might be more suitable. For mouse-derived retinas, the culture can be kept viable for up to 4 weeks (see above). The long-term viability of the retina also allows for the implementation of appropriate nanoparticle cytotoxicity studies (Prajapati et al., 2021).
To analyze topical applications, the permeation of DDS through the cornea and mucus barrier has been investigated using Franz-diffusion cells loaded with appropriate ex vivo tissues (Bao et al., 2018; Gomez-Segura et al., 2020).
For studies into the pharmacokinetic profile of drugs in DDS, a 2-chamber flow-based system called PK-Eye™ has been developed to estimate the anterior elimination half-life of intravitreally administered drugs entirely free from biological tissue (Awwad et al., 2017). Evidence suggests that predictions on the elimination of small lipophilic molecules are less accurate, e.g., the half-life of triamcinolone acetonide was found to be 26–28 days in the model, while this was shown to be considerably faster in humans (15.4–18.6 days) due to the elimination route through the retinal-choroid-sclera pathway, which the model does not take into account (Awwad et al., 2015). The half-life of proteins like ranibizumab, on the other hand, was more accurately represented and found to be about 8 days, which is in the same range as in humans (7–9 days) (Awwad et al., 2015). Nanoparticles are primarily cleared through the anterior route (Del Amo et al., 2017), suggesting that the PK-Eye™ model might be useful for estimating their intraocular elimination half-life.
In summary, multiple in vitro settings have been employed in the literatures, depending on which specific property of the DDS is being investigated (e.g., cell uptake, pharmacokinetics, permeation through ocular tissue barriers). The information gained from such in vitro models can give useful insight into the DDS behavior and predict whether they might yield successful in vivo results.
A large variety of very diverse in vitro systems is available to model human retinal diseases. Each of these systems comes with its own set of benefits and limitations, and in practice, it will be important to precisely define the experimental questions at hand to choose the appropriate in vitro model then. In Table 1 we have summarized the main in vitro systems currently available for research into retinal degeneration and neuroprotection.
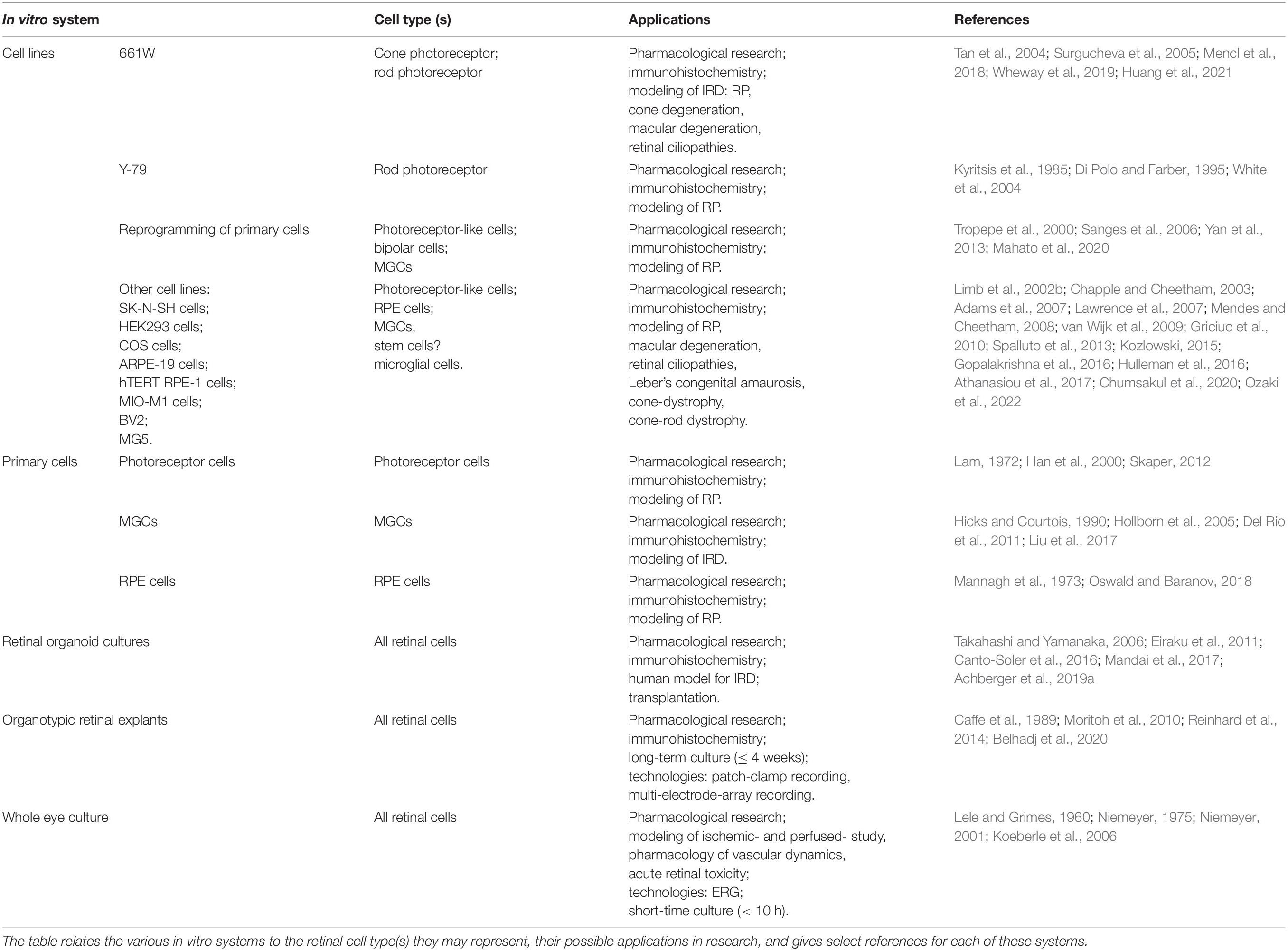
Table 1. Overview of in vitro test systems available for retinal neuroprotection research.
Notably, cell culture-based systems may be useful to study the intrinsic properties of proteins, as well as their interaction networks, and to compare these with those of corresponding mutant proteins, provided that the cell culture chosen displays a sufficiently high expression of the proteins in question. More complex systems such as co-cultures, retinal organoids, or organotypic retinal explants should be employed if the research questions demand a retinal systems-oriented approach, concerning interdependencies between cell types. Complex in vitro culture systems may also be required when highly differentiated primary retinal tissue is to be studied since immortalized cell culture-based systems may lack retina-specific proteins with respect to quantity and quality. The study of even more complex interactions with other organ systems may eventually require the use of in vivo animal models. This is especially true for cases where developmental processes or defects are the focus of a study or where the intact eye has to be considered once translational therapy-oriented studies are being conducted.
Nevertheless, the use of in vitro test systems features a variety of advantages, including easier and faster molecular and genetic manipulation, reduced experimentation time, and reduced costs compared to animal experimentation. Moreover, hiPSC and their derivatives enable the study of molecular disease mechanisms and the development of therapies in a human and patient-specific context. Last but not least, in vitro test systems limit the number of animals used in experiments and replace animal experiments with in vitro alternatives, such that live animal research may only be needed in the final stages of pre-clinical development. In this way, clever combinations of in vitro systems with limited in vivo testing may significantly contribute to advance the implementation of the 3R principles, i.e., the replacement, the reduction, and the refinement of live animal testing for retinal research (Russell and Burch, 1959).
AT, BA-G, BC, FP-D, GC, JY, MU, and YZ: writing—review and editing. All authors contributed to one or more chapters of the manuscript, read, and agreed to the published version of the manuscript.
This research was funded by Charlotte and Tistou Kerstan Foundation, the Zinke Heritage Foundation, the German Research Council (DFG: PA1751/10-1), the German Ministry for research and Education (BMBF: TargetRD, 16GW0267K), the European Union (EU: transMed, H2020-2017-765441), and the University of Tübingen Open Access publishing fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Norman Rieger for excellent technical assistance and Thomas Wheeler-Schilling for helpful comments on the manuscript.
IRD, inherited retinal degeneration; cGMP, cyclic guanosine monophosphate; PARP, poly (ADP-ribose) polymerase; HDAC, histone deacetylases; RP, retinitis pigmentosa; ONL, outer nuclear layer; INL, inner nuclear layer; RPE, retinal pigment epithelial; RGC, retinal ganglion cell; MGC, Müller glial cell; CNTF, ciliary neurotrophic factor; iPSC, induced pluripotent stem cells; PKG, protein kinase G; DDS, drug delivery system.
Ablonczy, Z., and Crosson, C. E. (2007). VEGF modulation of retinal pigment epithelium resistance. Exp. Eye Res. 85, 762–771.
Achberger, K., Haderspeck, J. C., Kleger, A., and Liebau, S. (2019a). Stem cell-based retina models. Adv. Drug Deliv. Rev. 140, 33–50.
Achberger, K., Probst, C., Haderspeck, J., Bolz, S., Rogal, J., Chuchuy, J., et al. (2019b). Merging organoid and organ-on-a-chip technology to generate complex multi-layer tissue models in a human retina-on-a-chip platform. Elife 8:e46188. doi: 10.7554/eLife.46188
Adams, N. A., Awadein, A., and Toma, H. S. (2007). The retinal ciliopathies. Ophthalmic. Genet. 28, 113–125.
Arango-Gonzalez, B., Sen, M., Guarascio, R., Ziaka, K., del Amo, E. M., Hau, K., et al. (2020). Inhibition of VCP preserves retinal structure and function in autosomal dominant retinal degeneration. BioRxiv [Preprint]. doi: 10.1101/2020.11.17.384669
Arango-Gonzalez, B., Trifunovic, D., Sahaboglu, A., Kranz, K., Michalakis, S., Farinelli, P., et al. (2014). Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PLoS One 9:e112142. doi: 10.1371/journal.pone.0112142
Armento, A., Murali, A., Marzi, J., Almansa-Garcia, A. C., Arango-Gonzalez, B., Kilger, E., et al. (2021a). Complement Factor H Loss in RPE Cells Causes Retinal Degeneration in a Human RPE-Porcine Retinal Explant Co-Culture Model. Biomolecules 11:1621. doi: 10.3390/biom11111621
Armento, A., Schmidt, T. L., Sonntag, I., Merle, D. A., Jarboui, M. A., Kilger, E., et al. (2021b). CFH Loss in Human RPE Cells Leads to Inflammation and Complement System Dysregulation via the NF-kappaB Pathway. Int. J. Mol. Sci. 22:8727. doi: 10.3390/ijms22168727
Athanasiou, D., Aguila, M., Opefi, C. A., South, K., Bellingham, J., Bevilacqua, D., et al. (2017). Rescue of mutant rhodopsin traffic by metformin-induced AMPK activation accelerates photoreceptor degeneration. Hum. Mol. Genet. 26, 305–319. doi: 10.1093/hmg/ddw387
Awwad, S., Day, R. M., Khaw, P. T., Brocchini, S., and Fadda, H. M. (2017). Sustained release ophthalmic dexamethasone: in vitro in vivo correlations derived from the PK-Eye. Int. J. Pharm. 522, 119–127. doi: 10.1016/j.ijpharm.2017.02.047
Awwad, S., Lockwood, A., Brocchini, S., and Khaw, P. T. (2015). The PK-Eye: A Novel In Vitro Ocular Flow Model for Use in Preclinical Drug Development. J. Pharm. Sci. 104, 3330–3342. doi: 10.1002/jps.24480
Azadi, S., Johnson, L. E., Paquet-Durand, F., Perez, M. T., Zhang, Y., Ekström, P. A., et al. (2007). CNTF+BDNF treatment and neuroprotective pathways in the rd1 mouse retina. Brain Res. 1129, 116–129. doi: 10.1016/j.brainres.2006.10.031
Ba, F., Pang, P. K., and Benishin, C. G. (2003). The establishment of a reliable cytotoxic system with SK-N-SH neuroblastoma cell culture. J. Neurosci. Methods 123, 11–22. doi: 10.1016/s0165-0270(02)00324-2
Bai, P. (2015). Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 58, 947–958. doi: 10.1016/j.molcel.2015.01.034
Bao, Q., Newman, B., Wang, Y., Choi, S., and Burgess, D. J. (2018). In vitro and ex vivo correlation of drug release from ophthalmic ointments. J. Control Release 276, 93–101. doi: 10.1016/j.jconrel.2018.03.003
Baudry, M. (2019). Calpain-1 and Calpain-2 in the Brain: Dr. Jekill and Mr Hyde? Curr. Neuropharmacol. 17, 823–829. doi: 10.2174/1570159X17666190228112451
Baudry, M., and Bi, X. (2016). Calpain-1 and Calpain-2: The Yin and Yang of Synaptic Plasticity and Neurodegeneration. Trends Neurosci. 39, 235–245. doi: 10.1016/j.tins.2016.01.007
Bauereisen, E., Kuchler, G., Pilz, A., and Sickel, W. (1956). Oxygen pressure and b-wave of electroretinogram of isolated frog eyes. Pflugers Arch. Gesamte. Physiol. Menschen Tiere 263, 566–576. doi: 10.1007/BF00362174
Belhadj, S., Tolone, A., Christensen, G., Das, S., Chen, Y., and Paquet-Durand, F. (2020). Long-Term, Serum-Free Cultivation of Organotypic Mouse Retina Explants with Intact Retinal Pigment Epithelium. J. Vis. Exp. 165:p.e61868. doi: 10.3791/61868
Berger, W., Kloeckener-Gruissem, B., and Neidhardt, J. (2010). The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 29, 335–375.
Biedler, J. L., Helson, L., and Spengler, B. A. (1973). Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 33, 2643–2652.
Biedler, J. L., Roffler-Tarlov, S., Schachner, M., and Freedman, L. S. (1978). Multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones. Cancer Res. 38, 3751–3757.
Blenkinsop, T. A., Salero, E., Stern, J. H., and Temple, S. (2013). The culture and maintenance of functional retinal pigment epithelial monolayers from adult human eye. Methods Mol. Biol. 945, 45–65.
Bloom, F. R., Price, P., Lao, G., Xia, J. L., Crowe, J. H., Battista, J. R., et al. (2001). Engineering mammalian cells for solid-state sensor applications. Biosens. Bioelectron. 16, 603–608.
Bochot, A., and Fattal, E. (2012). Liposomes for intravitreal drug delivery: a state of the art. J. Control. Release 161, 628–634.
Böck, J., Bornschein, H., and Hommer, K. (1964). The influence of the environmental temperature on the retinal survival time of enucleated eyeballs. Vision Res. 4, 609–625. doi: 10.1016/0042-6989(64)90047-1
Brücke, E. T., and Garten, S. (1907). Zur vergleichenden Physiologie der Netzhautströme. Arch. Für Die Gesamte Physiol. Des Menschen und Der Tiere 120, 290–348.
Caffé, A. R., Söderpalm, A. K., Holmqvist, I., and van Veen, T. (2001). A combination of CNTF and BDNF rescues rd photoreceptors but changes rod differentiation in the presence of RPE in retinal explants. Invest Ophthalmol. Vis. Sci. 42, 275–282.
Caffé, A. R., Söderpalm, A., and van Veen, T. (1993). Photoreceptor-specific protein expression of mouse retina in organ culture and retardation of rd degeneration in vitro by a combination of basic fibroblast and nerve growth factors. Curr. Eye Res. 12, 719–726. doi: 10.3109/02713689308995767
Caffe, A. R., Visser, H., Jansen, H. G., and Sanyal, S. (1989). Histotypic differentiation of neonatal mouse retina in organ culture. Curr. Eye Res. 8, 1083–1092. doi: 10.3109/02713688908997401
Campbell, M., and Humphries, P. (2012). The blood-retina barrier: tight junctions and barrier modulation. Adv. Exp. Med. Biol. 763, 70–84.
Campbell, P., and Chader, J. (1989). Y-79 retinoblastoma cells Isolation and characterization of clonal lineages. Exp. Eye Res. 48, 77–85. doi: 10.1016/0014-4835(89)90021-3
Canto-Soler, V., Flores-Bellver, M., and Vergara, M. N. (2016). Stem Cell Sources and Their Potential for the Treatment of Retinal Degenerations. Invest Ophthalmol. Vis. Sci. 57, ORSFd1–ORSFd9.
Chapple, J. P., and Cheetham, M. E. (2003). The chaperone environment at the cytoplasmic face of the endoplasmic reticulum can modulate rhodopsin processing and inclusion formation. J. Biol. Chem. 278, 19087–19094. doi: 10.1074/jbc.M212349200
Chen, Y., Jastrzebska, B., Golczak, M., Gulati, S., Tang, H., Seibel, W., et al. (2018). A novel small molecule chaperone of rod opsin and its potential therapy for retinal degeneration. Nat. Commun. 9:1976. doi: 10.1038/s41467-018-04261-1
Cheng, S. Y., Wang, S. C., Lei, M., Wang, Z., and Xiong, K. (2018). Regulatory role of calpain in neuronal death. Neural. Regen. Res. 13, 556–562.
Chumsakul, O., Wakayama, K., Tsuhako, A., Baba, Y., Takai, Y., Kurose, T., et al. (2020). Apigenin Regulates Activation of Microglia and Counteracts Retinal Degeneration. J. Ocul. Pharmacol. Ther. 36, 311–319. doi: 10.1089/jop.2019.0163
Cobb, H., Aparicio-Domingo, S., and Canto-Soler, M. V. (2021). Transitioning into GMP-Compliance: Alternative Methods for Producing Retinal Organoids for Transplantation. Transl. Vision Sci. Technol. 10:9. doi: 10.1167/tvst.10.10.9
Curcio, C. A., Owsley, C., and Jackson, G. R. (2000). Spare the rods, save the cones in aging and age-related maculopathy. Invest Ophthalmol. Vis. Sci. 41, 2015–2018.
Curcio, M., Salazar, I. L., Mele, M., Canzoniero, L. M., and Duarte, C. B. (2016). Calpains and neuronal damage in the ischemic brain: The swiss knife in synaptic injury. Prog. Neurobiol. 143, 1–35.
Das, S., Popp, V., Power, M., Groeneveld, K., Melle, C., Rogerson, L., et al. (2022). Redefining the role of Ca2+ -permeable channels in photoreceptor degenerationusing diltiazem. Cell Death Dis. 13, 1–13. doi: 10.1038/s41419-021-04482-1
David, K. K., Andrabi, S. A., Dawson, T. M., and Dawson, V. L. (2009). Parthanatos, a messenger of death. Front. Biosci. 14:1116–1128. doi: 10.2741/3297
Del Amo, E. M., Rimpela, A. K., Heikkinen, E., Kari, O. K., Ramsay, E., Lajunen, T., et al. (2017). Pharmacokinetic aspects of retinal drug delivery. Prog. Retin Eye Res. 57, 134–185.
Del Rio, P., Irmler, M., Arango-Gonzalez, B., Favor, J., Bobe, C., Bartsch, U., et al. (2011). GDNF-induced osteopontin from Muller glial cells promotes photoreceptor survival in the Pde6brd1 mouse model of retinal degeneration. Glia 59, 821–832. doi: 10.1002/glia.21155
Di Polo, A., and Farber, D. B. (1995). Rod photoreceptor-specific gene expression in human retinoblastoma cells. Proc. Natl. Acad. Sci. U. S. A. 92, 4016–4020.
Dumont, J., Euwart, D., Mei, B., Estes, S., and Kshirsagar, R. (2016). Human cell lines for biopharmaceutical manufacturing: history, status, and future perspectives. Crit. Rev. Biotechnol. 36, 1110–1122. doi: 10.3109/07388551.2015.1084266
Dunn, K. C., Aotaki-Keen, A. E., Putkey, F. R., and Hjelmeland, L. M. (1996). ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp. Eye Res. 62, 155–169.
Dutt, K., Cao, Y., and Ezeonu, I. (2010). Ciliary neurotrophic factor: a survival and differentiation inducer in human retinal progenitors. Vitro Cell. Develop. Biol. Anim. 46, 635–646.
Eastlake, K., Lamb, W., Luis, J., Khaw, P., Jayaram, H., and Limb, G. (2021). Prospects for the application of Müller glia and their derivatives in retinal regenerative therapies. Prog. Retin. Eye Res. 85:100970. doi: 10.1016/j.preteyeres.2021.100970
Eiraku, M., Takata, N., Ishibashi, H., Kawada, M., Sakakura, E., Okuda, S., et al. (2011). Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472, 51–56.
El Bahhaj, F., Dekker, F. J., Martinet, N., and Bertrand, P. (2014). Delivery of epidrugs. Drug Disc. Today 19, 1337–1352.
Eriksen, A. Z., Brewer, J., Andresen, T. L., and Urquhart, A. J. (2017). The diffusion dynamics of PEGylated liposomes in the intact vitreous of the ex vivo porcine eye: A fluorescence correlation spectroscopy and biodistribution study. Int. J. Pharm. 522, 90–97. doi: 10.1016/j.ijpharm.2017.03.003
Falkenberg, K. J., and Johnstone, R. W. (2014). Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 13, 673–691.
Farber, D. B., and Lolley, R. N. (1974). Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science 186, 449–451.
Ferrer-Martín, R. M., Martín-Oliva, D., Sierra, A., Carrasco, M. C., Martín-Estebané, M., Calvente, R., et al. (2014). Microglial cells in organotypic cultures of developing and adult mouse retina and their relationship with cell death. Exp. Eye Res. 121, 42–57. doi: 10.1016/j.exer.2014.02.015
Ferrer-Martin, R. M., Martin-Oliva, D., Sierra-Martin, A., Carrasco, M. C., Martin-Estebane, M., Calvente, R., et al. (2015). Microglial Activation Promotes Cell Survival in Organotypic Cultures of Postnatal Mouse Retinal Explants. PLoS One 10:e0135238. doi: 10.1371/journal.pone.0135238
Forouzanfar, F., Shojapour, M., Aghili, Z. S., and Asgharzade, S. (2020). Growth Factors as Tools in Photoreceptor Cell Regeneration and Vision Recovery. Curr. Drug Targets 21, 573–581. doi: 10.2174/1389450120666191121103831
Funatsu, J., Murakami, Y., Shimokawa, S., Nakatake, S., Fujiwara, K., Okita, A., et al. (2022). Circulating inflammatory monocytes oppose microglia and contribute to cone cell death in retinitis pigmentosa. PNAS Nexus 1:gac003. doi: 10.1093/pnasnexus/pgac003
Garcia-Quintanilla, L., Luaces-Rodriguez, A., Gil-Martinez, M., Mondelo-Garcia, C., Maronas, O., Mangas-Sanjuan, V., et al. (2019). Pharmacokinetics of Intravitreal Anti-VEGF Drugs in Age-Related Macular Degeneration. Pharmaceutics 11:365.
Gluzman, Y. (1981). SV40-transformed simian cells support the replication of early SV40 mutants. Cell 23, 175–182.
Gomes, B. A. Q., Silva, J. P. B., Romeiro, C. F. R., Dos Santos, S. M., Rodrigues, C. A., Goncalves, P. R., et al. (2018). Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxid. Med. Cell Longev. 2018:8152373.
Gomez-Segura, L., Parra, A., Calpena-Campmany, A. C., Gimeno, A., Gomez, de Aranda, I., et al. (2020). Ex Vivo Permeation of Carprofen Vehiculated by PLGA Nanoparticles through Porcine Mucous Membranes and Ophthalmic Tissues. Nanomaterials 10:355. doi: 10.3390/nano10020355
Gopalakrishna, K. N., Boyd, K., Yadav, R. P., and Artemyev, N. O. (2016). Aryl Hydrocarbon Receptor-interacting Protein-like 1 Is an Obligate Chaperone of Phosphodiesterase 6 and Is Assisted by the gamma-Subunit of Its Client. J. Biol. Chem. 291, 16282–16291. doi: 10.1074/jbc.M116.737593
Graham, F. L., Smiley, J., Russell, W. C., and Nairn, R. (1977). Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36, 59–74.
Green, P. S., Yang, S. H., Nilsson, K. R., Kumar, A. S., Covey, D. F., and Simpkins, J. W. (2001). The nonfeminizing enantiomer of 17beta-estradiol exerts protective effects in neuronal cultures and a rat model of cerebral ischemia. Endocrinology 142, 400–406. doi: 10.1210/endo.142.1.7888
Griciuc, A. (2010). Molecular and Functional Analysis of the ERAD Effector VCP in Cellular and Drosophila Models for Retinitis Pigmentosa. Germany: Technische Universität München.
Griciuc, A., Aron, L., Piccoli, G., and Ueffing, M. (2010). Clearance of Rhodopsin(P23H) aggregates requires the ERAD effector VCP. Biochim. Biophys. Acta 1803, 424–434.
Guadagni, V., Novelli, E., Piano, I., Gargini, C., and Strettoi, E. (2015). Pharmacological approaches to retinitis pigmentosa: A laboratory perspective. Prog. Retin Eye Res. 48, 62–81.
Han, Y., Jacoby, R. A., and Wu, S. M. (2000). Morphological and electrophysiological properties of dissociated primate retinal cells. Brain Res. 875, 175–186. doi: 10.1016/s0006-8993(00)02614-7
Hata, S., Abe, M., Suzuki, H., Kitamura, F., Toyama-Sorimachi, N., Abe, K., et al. (2010). Calpain 8/nCL-2 and calpain 9/nCL-4 constitute an active protease complex, G-calpain, involved in gastric mucosal defense. PLoS Genet. 6:e1001040. doi: 10.1371/journal.pgen.1001040
Hata, S., Doi, N., Kitamura, F., and Sorimachi, H. (2007). Stomach-specific calpain, nCL-2/calpain 8, is active without calpain regulatory subunit and oligomerizes through C2-like domains. J. Biol. Chem. 282, 27847–27856. doi: 10.1074/jbc.M703168200
Hauck, S. M., Kinkl, N., Deeg, C. A., Swiatek-de Lange, M., Schoffmann, S., and Ueffing, M. (2006). GDNF family ligands trigger indirect neuroprotective signaling in retinal glial cells. Mol. Cell. Biol. 26, 2746–2757.
Hauck, S. M., Suppmann, S., and Ueffing, M. (2003). Proteomic profiling of primary retinal Müller glia cells reveals a shift in expression patterns upon adaptation to in vitro conditions. Glia 44, 251–263. doi: 10.1002/glia.10292
Hauck, S. M., von Toerne, C., and Ueffing, M. (2014). The neuroprotective potential of retinal Muller glial cells. Adv. Exp. Med. Biol. 801, 381–387.
Hering, R., Strauss, K. M., Tao, X., Bauer, A., Woitalla, D., Mietz, E. M., et al. (2004). Novel homozygous p.E64D mutation in DJ1 in early onset Parkinson disease (PARK7). Hum. Mutat. 24, 321–329. doi: 10.1002/humu.20089
Hicks, D., and Courtois, Y. (1990). The growth and behaviour of rat retinal Müller cells in vitro 1. An improved method for isolation and culture. Exp. Eye Res. 51, 119–129.
Hollborn, M., Tenckhoff, S., Jahn, K., Iandiev, I., Biedermann, B., Schnurrbusch, U. E., et al. (2005). Changes in retinal gene expression in proliferative vitreoretinopathy: glial cell expression of HB-EGF. Mol. Vis. 11, 397–413.
Houtkooper, R. H., Cantó, C., Wanders, R. J., and Auwerx, J. (2010). The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 31, 194–223. doi: 10.1210/er.2009-0026
Hu, Y., Gaillard, P. J., de Lange, E. C. M., and Hammarlund-Udenaes, M. (2019). Targeted brain delivery of methotrexate by glutathione PEGylated liposomes: How can the formulation make a difference? Eur. J. Pharm. Biopharm. 139, 197–204. doi: 10.1016/j.ejpb.2019.04.004
Huang, L., Kutluer, M., Adani, E., Comitato, A., and Marigo, V. (2021). New In Vitro Cellular Model for Molecular Studies of Retinitis Pigmentosa. Int. J. Mol. Sci. 22:6440.
Hulleman, J. D., Nguyen, A., Ramprasad, V., Murugan, S., Gupta, R., Mahindrakar, A., et al. (2016). A novel H395R mutation in MKKS/BBS6 causes retinitis pigmentosa and polydactyly without other findings of Bardet-Biedl or McKusick-Kaufman syndrome. Mol. Vision 22, 73–81.
Itkonen, J., Annala, A., Tavakoli, S., Arango-Gonzalez, B., Ueffing, M., Toropainen, E., et al. (2020). Characterization, Stability, and in Vivo Efficacy Studies of Recombinant Human CNTF and Its Permeation into the Neural Retina in ex Vivo Organotypic Retinal Explant Culture Models. Pharmaceutics 12:611. doi: 10.3390/pharmaceutics12070611
Jensen, F. C., Girardi, A. J., Gilden, R. V., and Koprpwski, H. (1964). Infection of human and simian tissue cultures with rous sarcoma virus. Proc. Natl. Acad. Sci. U. S. A. 52, 53–59.
Jiang, M., Wang, J., Fu, J., Du, L., Jeong, H., West, T., et al. (2011). Neuroprotective role of Sirt1 in mammalian models of Huntington’s disease through activation of multiple Sirt1 targets. Nat. Med. 18, 153–158. doi: 10.1038/nm.2558
Kamaleddin, M. A. (2017). Nano-ophthalmology: Applications and considerations. Nanomedicine 13, 1459–1472. doi: 10.1016/j.nano.2017.02.007
Kavsan, V. M., Iershov, A. V., and Balynska, O. V. (2011). Immortalized cells and one oncogene in malignant transformation: old insights on new explanation. BMC Cell Biol. 12:23. doi: 10.1186/1471-2121-12-23
Kennan, A., Aherne, A., and Humphries, P. (2005). Light in retinitis pigmentosa. Trends Genet. 21, 103–110.
Kim, H., Robinson, S. B., and Csaky, K. G. (2009a). Investigating the Movement of Intravitreal Human Serum Albumin Nanoparticles in the Vitreous and Retina. Pharm. Res. 26, 329–337. doi: 10.1007/s11095-008-9745-6
Kim, K.-S., Kim, M. S., Moon, J. H., Jeong, M. S., Kim, J., Lee, G. M., et al. (2009b). Enhancement of recombinant antibody production in HEK 293E cells by WPRE. Biotech. Bioproc. Eng. 14:633.
Ko, H. L., and Ren, E. C. (2012). Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules 2, 524–548.
Koeberle, M. J., Hughes, P. M., Skellern, G. G., and Wilson, C. G. (2006). Pharmacokinetics and disposition of memantine in the arterially perfused bovine eye. Pharm. Res. 23, 2781–2798. doi: 10.1007/s11095-006-9106-2
Kolb, H., Nelson, R., Ahnelt, P., and Cuenca, N. (2001). Cellular organization of the vertebrate retina. Prog. Brain Res. 131, 3–26.
Kolomeyer, A. M., and Zarbin, M. A. (2014). Trophic factors in the pathogenesis and therapy for retinal degenerative diseases. Surv. Ophthalmol. 59, 134–165.
Kozlowski, M. R. (2015). The ARPE-19 cell line: mortality status and utility in macular degeneration research. Curr. Eye Res. 40, 501–509. doi: 10.3109/02713683.2014.935440
Krishnakumar, R., and Kraus, W. L. (2010). The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol. Cell 39, 8–24. doi: 10.1016/j.molcel.2010.06.017
Kucharska, J., Del Rio, P., Arango-Gonzalez, B., Gorza, M., Feuchtinger, A., Hauck, S. M., et al. (2014). Cyr61 activates retinal cells and prolongs photoreceptor survival in rd1 mouse model of retinitis pigmentosa. J. Neurochem. 130, 227–240. doi: 10.1111/jnc.12704
Kühne, W. (1881). Über electrische Vorgänge im Sehorgane. Heidelberg: Carl Winter’s Universitätsbuchhandlung.
Kyritsis, A. P., Tsokos, M., Triche, T. J., and Chader, G. J. (1984). Retinoblastoma–origin from a primitive neuroectodermal cell? Nature 307, 471–473. doi: 10.1038/307471a0
Kyritsis, A. P., Wiggert, B., Lee, L., and Chader, G. J. (1985). Butyrate enhances the synthesis of interphotoreceptor retinoid-binding protein (IRBP) by Y-79 human retinoblastoma cells. J. Cell. Physiol. 124, 233–239. doi: 10.1002/jcp.1041240210
Lam, D. M. (1972). Biosynthesis of acetylcholine in turtle photoreceptors. Proc. Natl. Acad. Sci. U. S. A. 69, 1987–1991.
Lammich, S., Schöbel, S., Zimmer, A. K., Lichtenthaler, S. F., and Haass, C. (2004). Expression of the Alzheimer protease BACE1 is suppressed via its 5’-untranslated region. EMBO Rep. 5, 620–625. doi: 10.1038/sj.embor.7400166
Lawrence, J. M., Singhal, S., Bhatia, B., Keegan, D. J., Reh, T. A., Luthert, P. J., et al. (2007). MIO-M1 cells and similar muller glial cell lines derived from adult human retina exhibit neural stem cell characteristics. Stem Cells 25, 2033–2043. doi: 10.1634/stemcells.2006-0724
Lee, J., Goh, U., Lee, H.-J., Kim, J., Jeong, M., and Park, J.-H. (2017). Effective Retinal Penetration of Lipophilic and Lipid-Conjugated Hydrophilic Agents Delivered by Engineered Liposomes. Mol. Pharm. 14, 423–430. doi: 10.1021/acs.molpharmaceut.6b00864
Lele, P. P., and Grimes, P. (1960). The role of neural mechanisms in the regulation of intraocular pressure in the cat. Exp. Neurol. 2, 199–220. doi: 10.1016/0014-4886(60)90009-1
Liebau, S., Russ, H. A., and Kleger, A. (2019). Stem cell derived organoids in human disease and development. Stem Cells Int. 2019:7919427.
Limb, G. A., Daniels, J. T., Pleass, R., Charteris, D. G., Luthert, P. J., and Khaw, P. T. (2002a). Differential expression of matrix metalloproteinases 2 and 9 by glial Müller cells: response to soluble and extracellular matrix-bound tumor necrosis factor-alpha. Am. J. Pathol. 160, 1847–1855. doi: 10.1016/s0002-9440(10)61131-5
Limb, G. A., Matter, K., Murphy, G., Cambrey, A. D., Bishop, P. N., Morris, G. E., et al. (2005). Matrix metalloproteinase-1 associates with intracellular organelles and confers resistance to lamin A/C degradation during apoptosis. Am. J. Pathol. 166, 1555–1563. doi: 10.1016/S0002-9440(10)62371-1
Limb, G. A., Salt, T. E., Munro, P. M., Moss, S. E., and Khaw, P. T. (2002b). In vitro characterization of a spontaneously immortalized human Müller cell line (MIO-M1). Invest Ophthalmol. Vis. Sci. 43, 864–869.
Lin, Y. C., Boone, M., Meuris, L., Lemmens, I., Van Roy, N., Soete, A., et al. (2014). Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 5:4767. doi: 10.1038/ncomms5767
Lipinski, D. M., Singh, M. S., and MacLaren, R. E. (2011). Assessment of cone survival in response to CNTF, GDNF, and VEGF165b in a novel ex vivo model of end-stage retinitis pigmentosa. Invest. Ophthalmol. Visual Sci. 52, 7340–7346. doi: 10.1167/iovs.11-7996
Liu, X., Tang, L., and Liu, Y. (2017). Mouse müller cell isolation and culture. Bio-protocol 7:e2429.
Lord, C. J., and Ashworth, A. (2017). PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158.
Magno, L., Lessard, C. B., Martins, M., Lang, V., Cruz, P., Asi, Y., et al. (2019). Alzheimer’s disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res. Ther. 11:16. doi: 10.1186/s13195-019-0469-0
Mahato, B., Kaya, K. D., Fan, Y., Sumien, N., Shetty, R. A., Zhang, W., et al. (2020). Pharmacologic fibroblast reprogramming into photoreceptors restores vision. Nature 581, 83–88. doi: 10.1038/s41586-020-2201-4
Mandai, M., Watanabe, A., Kurimoto, Y., Hirami, Y., Morinaga, C., Daimon, T., et al. (2017). Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 376, 1038–1046.
Mannagh, J., Arya, D. V., and Irvine, A. R. (1973). Tissue culture of human retinal pigment epithelium. Invest. Ophthalmol. Visual Sci. 12, 52–64.
Mencl, S., Trifunoviæ, D., Zrenner, E., and Paquet-Durand, F. (2018). PKG-Dependent Cell Death in 661W Cone Photoreceptor-like Cell Cultures (Experimental Study). Adv. Exp. Med. Biol. 1074, 511–517. doi: 10.1007/978-3-319-75402-4_63
Mendes, H. F., and Cheetham, M. E. (2008). Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 17, 3043–3054. doi: 10.1093/hmg/ddn202
Morales, J., Li, L., Fattah, F. J., Dong, Y., Bey, E. A., Patel, M., et al. (2014). Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot Gene. Expr. 24, 15–28.
Moritoh, S., Tanaka, K. F., Jouhou, H., Ikenaka, K., and Koizumi, A. (2010). Organotypic tissue culture of adult rodent retina followed by particle-mediated acute gene transfer in vitro. PLoS One 5:e12917. doi: 10.1371/journal.pone.0012917
Müller, B., Wagner, F., Lorenz, B., and Stieger, K. (2017). Organotypic Cultures of Adult Mouse Retina: Morphologic Changes and Gene Expression. Invest. Ophthalmol. Vis. Sci. 58, 1930–1940. doi: 10.1167/iovs.16-20718
Murata, M. M., Kong, X., Moncada, E., Chen, Y., Imamura, H., Wang, P., et al. (2019). NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell 30, 2584–2597. doi: 10.1091/mbc.E18-10-0650
Nakano, T., Ando, S., Takata, N., Kawada, M., Muguruma, K., Sekiguchi, K., et al. (2012). Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10, 771–785. doi: 10.1016/j.stem.2012.05.009
Niemeyer, G. (2001). Retinal research using the perfused mammalian eye. Prog. Retin Eye Res. 20, 289–318.
Ono, Y., Saido, T. C., and Sorimachi, H. (2016). Calpain research for drug discovery: challenges and potential. Nat. Rev. Drug Discov. 15, 854–876.
Oswald, J., and Baranov, P. (2018). Regenerative medicine in the retina: from stem cells to cell replacement therapy. Ther. Adv. Ophthalmol. 10:2515841418774433.
Ozaki, E., Delaney, C., Campbell, M., and Doyle, S. L. (2022). Minocycline suppresses disease-associated microglia (DAM) in a model of photoreceptor cell degeneration. Exp. Eye Res. 217:108953. doi: 10.1016/j.exer.2022.108953
Paquet-Durand, F., Azadi, S., Hauck, S. M., Ueffing, M., van Veen, T., and Ekström, P. (2006). Calpain is activated in degenerating photoreceptors in the rd1 mouse. J. Neurochem. 96, 802–814.
Paquet-Durand, F., Hauck, S. M., van Veen, T., Ueffing, M., and Ekström, P. (2009). PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J. Neurochem. 108, 796–810. doi: 10.1111/j.1471-4159.2008.05822.x
Paquet-Durand, F., Marigo, V., and Ekström, P. (2019). RD Genes Associated with High Photoreceptor cGMP-Levels (Mini-Review). Adv. Exp. Med. Biol. 1185, 245–249. doi: 10.1007/978-3-030-27378-1_40
Paquet-Durand, F., Silva, J., Talukdar, T., Johnson, L. E., Azadi, S., van Veen, T., et al. (2007). Excessive activation of poly(ADP-ribose) polymerase contributes to inherited photoreceptor degeneration in the retinal degeneration 1 mouse. J. Neurosci. 27, 10311–10319. doi: 10.1523/JNEUROSCI.1514-07.2007
Peynshaert, K., Devoldere, J., Forster, V., Picaud, S., Vanhove, C., De Smedt, S. C., et al. (2017). Toward smart design of retinal drug carriers: a novel bovine retinal explant model to study the barrier role of the vitreoretinal interface. Drug Deliv. 24, 1384–1394. doi: 10.1080/10717544.2017.1375578
Pinzon-Duarte, G., Arango-Gonzalez, B., Guenther, E., and Kohler, K. (2004). Effects of brain-derived neurotrophic factor on cell survival, differentiation and patterning of neuronal connections and Muller glia cells in the developing retina. Eur. J. Neurosci. 19, 1475–1484. doi: 10.1111/j.1460-9568.2004.03252.x
Power, M. J., Rogerson, L. E., Schubert, T., Berens, P., Euler, T., and Paquet-Durand, F. (2020b). Systematic spatiotemporal mapping reveals divergent cell death pathways in three mouse models of hereditary retinal degeneration. J. Comp. Neurol. 528, 1113–1139. doi: 10.1002/cne.24807
Power, M., Das, S., Schütze, K., Marigo, V., Ekström, P., and Paquet-Durand, F. (2020a). Cellular mechanisms of hereditary photoreceptor degeneration - Focus on cGMP. Prog. Retin Eye Res. 74:100772.
Prajapati, M., Christensen, G., Paquet-Durand, F., and Loftsson, T. (2021). Cytotoxicity of β-Cyclodextrins in Retinal Explants for Intravitreal Drug Formulations. Molecules 26:1492. doi: 10.3390/molecules26051492
Reid, T. W., Albert, D. M., Rabson, A. S., Russell, P., Craft, J., Chu, E. W., et al. (1974). Characteristics of an established cell line of retinoblastoma. J. Natl. Cancer Inst. 53, 347–360.
Reinhard, K., Tikidji-Hamburyan, A., Seitter, H., Idrees, S., Mutter, M., Benkner, B., et al. (2014). Step-by-step instructions for retina recordings with perforated multi electrode arrays. PLoS One 9:e106148. doi: 10.1371/journal.pone.0106148
McKeone, R., Wikstrom, M., Kiel, M. C., and Elizabeth Rakoczy, P. E. (2014). Assessing the correlation between mutant rhodopsin stability and the severity of retinitis pigmentosa. Mol. Vision 20, 183–199.
Roche, S. L., Wyse-Jackson, A. C., Byrne, A. M., Ruiz-Lopez, A. M., and Cotter, T. G. (2016). Alterations to retinal architecture prior to photoreceptor loss in a mouse model of retinitis pigmentosa. Int. J. Dev. Biol. 60, 127–139. doi: 10.1387/ijdb.150400tc
Roy, A., Groten, J., Marigo, V., Tomar, T., and Hilhorst, R. (2021). Identification of novel substrates for cGMP dependent protein kinase (PKG) through kinase activity profiling to understand its putative role in inherited retinal degeneration. Int. J. Mol. Sci. 22:1180. doi: 10.3390/ijms22031180
Roy, A., Tolone, A., Hilhorst, R., Groten, J., Tomar, T., and Paquet-Durand, F. (2022). Kinase activity profiling identifies putative downstream targets of cGMP/PKG signaling in inherited retinal neurodegeneration. Cell Death Discov. 8:93. doi: 10.1038/s41420-022-00897-7
Yan, R. T., Li, X., Jian, H., Clyde, G., and Shu, Z. W. (2013). Photoreceptor-like cells from reprogramming cultured mammalian RPE cells. Mol. Vision 19, 1178–1187.
Russell, W. M. S., and Burch, R. L. (1959). The Principles of Humane Experimental Technique. London: Methuen & Co. Limited.
Sahaboglu, A., Barth, M., Secer, E., Amo, E. M., Urtti, A., Arsenijevic, Y., et al. (2016). Olaparib significantly delays photoreceptor loss in a model for hereditary retinal degeneration. Sci. Rep. 6:39537. doi: 10.1038/srep39537
Sahaboglu, A., Miranda, M., Canjuga, D., Avci-Adali, M., Savytska, N., Secer, E., et al. (2020). Drug repurposing studies of PARP inhibitors as a new therapy for inherited retinal degeneration. Cell Mol. Life Sci. 77, 2199–2216. doi: 10.1007/s00018-019-03283-2
Sancho-Pelluz, J., Alavi, M. V., Sahaboglu, A., Kustermann, S., Farinelli, P., Azadi, S., et al. (2010). Excessive HDAC activation is critical for neurodegeneration in the rd1 mouse. Cell Death Dis. 1:e24. doi: 10.1038/cddis.2010.4
Sancho-Pelluz, J., Wunderlich, K. A., Rauch, U., Romero, F. J., van Veen, T., Limb, G. A., et al. (2008). Sialoadhesin expression in intact degenerating retinas and following transplantation. Invest. Ophthalmol. Vis. Sci. 49, 5602–5610. doi: 10.1167/iovs.08-2117
Sanges, D., Comitato, A., Tammaro, R., and Marigo, V. (2006). Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc. Natl. Acad. Sci. U. S. A. 103, 17366–17371. doi: 10.1073/pnas.0606276103
Schnichels, S., Paquet-Durand, F., Löscher, M., Tsai, T., Hurst, J., Joachim, S. C., et al. (2021). Retina in a dish: Cell cultures, retinal explants and animal models for common diseases of the retina. Prog. Retin Eye Res. 81:100880. doi: 10.1016/j.preteyeres.2020.100880
Schwede, F., Maronde, E., Genieser, H., and Jastorff, B. (2000). Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol. Ther. 87, 199–226. doi: 10.1016/s0163-7258(00)00051-6
Sen, M., Al-Amin, M., Kicková, E., Sadeghi, A., Puranen, J., Urtti, A., et al. (2021a). Retinal neuroprotection by controlled release of a VCP inhibitor from self-assembled nanoparticles. J. Control Release 339, 307–320. doi: 10.1016/j.jconrel.2021.09.039
Sen, M., Bassetto, M., Poulhes, F., Zelphati, O., Ueffing, M., and Arango-Gonzalez, B. (2021b). Efficient Ocular Delivery of VCP siRNA via Reverse Magnetofection in RHO P23H Rodent Retina Explants. Pharmaceutics 13:225. doi: 10.3390/pharmaceutics13020225
Sen, M., Kutsyr, O., Cao, B., Bolz, S., Arango-Gonzalez, B., and Ueffing, M. (2021c). Pharmacological Inhibition of the VCP/Proteasome Axis Rescues Photoreceptor Degeneration in RHO(P23H) Rat Retinal Explants. Biomolecules 11:1528. doi: 10.3390/biom11101528
Seto, E., and Yoshida, M. (2014). Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 6:a018713.
Shima, C., Sakaguchi, H., Gomi, F., Kamei, M., Ikuno, Y., Oshima, Y., et al. (2008). Complications in patients after intravitreal injection of bevacizumab. Acta Ophthalmol. 86, 372–376.
Skaper, S. D. (2012). Isolation and culture of rat cone photoreceptor cells. Methods Mol. Biol. 846, 147–158.
So, P. P., Khodr, C. E., Chen, C.-D., and Abraham, C. R. (2013). Comparable dimerization found in wildtype and familial Alzheimer’s disease amyloid precursor protein mutants. Am. J. Neurodegen. Dis. 2, 15–28.
Spalluto, C., Wilson, D. I., and Hearn, T. (2013). Evidence for reciliation of RPE1 cells in late G1 phase, and ciliary localisation of cyclin B1. FEBS Open Bio. 3, 334–340. doi: 10.1016/j.fob.2013.08.002
Suga, S. (1972). Studies on the electroretinogram of the isolated rat eyeball. Nippon Ganka Gakkai Zasshi 76, 884–892.
Surgucheva, I., Ninkina, N., Buchman, V. L., Grasing, K., and Surguchov, A. (2005). Protein aggregation in retinal cells and approaches to cell protection. Cell Mol. Neurobiol. 25, 1051–1066.
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676.
Tan, E., Ding, X. Q., Saadi, A., Agarwal, N., Naash, M. I., and Al-Ubaidi, M. R. (2004). Expression of cone-photoreceptor-specific antigens in a cell line derived from retinal tumors in transgenic mice. Invest. Ophthalmol. Vis. Sci. 45, 764–768. doi: 10.1167/iovs.03-1114
Tavakoli, S., Kari, O. K., Turunen, T., Lajunen, T., Schmitt, M., Lehtinen, J., et al. (2021). Diffusion and Protein Corona Formation of Lipid-Based Nanoparticles in the Vitreous Humor: Profiling and Pharmacokinetic Considerations. Mol. Pharm. 18, 699–713. doi: 10.1021/acs.molpharmaceut.0c00411
Tavakoli, S., Peynshaert, K., Lajunen, T., Devoldere, J., Amo, E. M. D., Ruponen, M., et al. (2020). Ocular barriers to retinal delivery of intravitreal liposomes: Impact of vitreoretinal interface. J. Control. Release 328, 952–961. doi: 10.1016/j.jconrel.2020.10.028
Tolone, A., Haq, W., Fachinger, A., Rentsch, A., Herberg, F. W., Schwede, F., et al. (2021). Retinal degeneration: Multilevel protection of photoreceptor and ganglion cell viability and function with the novel PKG inhibitor CN238. BioRxiv [Preprint]. doi: 10.1101/2021.08.05.455191
Tropepe, V., Coles, B. L., Chiasson, B. J., Horsford, D. J., Elia, A. J., McInnes, R. R., et al. (2000). Retinal stem cells in the adult mammalian eye. Science 287, 2032–2036.
van Wijk, E., Kersten, F. F., Kartono, A., Mans, D. A., Brandwijk, K., Letteboer, S. J., et al. (2009). Usher syndrome and Leber congenital amaurosis are molecularly linked via a novel isoform of the centrosomal ninein-like protein. Hum. Mol. Genet. 18, 51–64. doi: 10.1093/hmg/ddn312
Vighi, E., Trifunovic, D., Veiga-Crespo, P., Rentsch, A., Hoffmann, D., Sahaboglu, A., et al. (2018). Combination of cGMP analogue and drug delivery system provides functional protection in hereditary retinal degeneration. Proc. Natl. Acad. Sci. U. S. A. 115, E2997–E3006. doi: 10.1073/pnas.1718792115
von Toerne, C., Menzler, J., Ly, A., Senninger, N., Ueffing, M., and Hauck, S. M. (2014). Identification of a novel neurotrophic factor from primary retinal Muller cells using stable isotope labeling by amino acids in cell culture (SILAC). Mol. Cell Proteomics 13, 2371–2381. doi: 10.1074/mcp.M113.033613
Wang, X., Dykens, J. A., Perez, E., Liu, R., Yang, S., Covey, D. F., et al. (2006). Neuroprotective effects of 17β-estradiol and nonfeminizing estrogens against H2O2 toxicity in human neuroblastoma SK-N-SH cells. Mol. Pharm. 70, 395–404. doi: 10.1124/mol.106.022384
Wang, Y., Liu, Y., Bi, X., and Baudry, M. (2020). Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death. Cells 9:2698. doi: 10.3390/cells9122698
Watters, J. J., and Dorsa, D. M. (1998). Transcriptional effects of estrogen on neuronal neurotensin gene expression involve cAMP/protein kinase A-dependent signaling mechanisms. J. Neurosci. 18, 6672–6680. doi: 10.1523/JNEUROSCI.18-17-06672.1998
Wen, Y., Perez, E. J., Green, P. S., Sarkar, S. N., and Simpkins, J. W. (2004). nNOS is involved in estrogen mediated neuroprotection in neuroblastoma cells. Neuroreport 15, 1515–1518. doi: 10.1097/01.wnr.0000131674.92694.96
Wendt, A., Thompson, V. F., and Goll, D. E. (2004). Interaction of calpastatin with calpain: a review. Biol. Chem. 385, 465–472.
Wheway, G., Nazlamova, L., Turner, D., and Cross, S. (2019). 661W Photoreceptor Cell Line as a Cell Model for Studying Retinal Ciliopathies. Front. Genet. 10:308. doi: 10.3389/fgene.2019.00308
White, J. B., Taylor, R. E., and Pittler, S. J. (2001). Reproducible high efficiency gene transfer into Y79 retinoblastoma cells using adenofection. J. Neurosci. Methods 106, 1–7. doi: 10.1016/s0165-0270(00)00368-x
White, J. B., Thompson, W. J., and Pittler, S. J. (2004). Characterization of 3’,5’ cyclic nucleotide phosphodiesterase activity in Y79 retinoblastoma cells: absence of functional PDE6. Mol. Vis. 10, 738–749.
Witt, O., Deubzer, H. E., Milde, T., and Oehme, I. (2009). HDAC family: What are the cancer relevant targets? Cancer Lett. 277, 8–21.
Xie, N., Zhang, L., Gao, W., Huang, C., Huber, P. E., Zhou, X., et al. (2020). NAD(+) metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther. 5:227.
Xu, J., Jackson, C. W., Khoury, N., Escobar, I., and Perez-Pinzon, M. A. (2018). Brain SIRT1 Mediates Metabolic Homeostasis and Neuroprotection. Front. Endocrinol. 9:702. doi: 10.3389/fendo.2018.00702
Yan, J., Chen, Y., Zhu, Y., and Paquet-Durand, F. (2021). Programmed Non-Apoptotic Cell Death in Hereditary Retinal Degeneration: Crosstalk between cGMP-Dependent Pathways and PARthanatos? Int. J. Mol. Sci. 22:10567. doi: 10.3390/ijms221910567
Yan, J., Günter, A., Das, S., Mühlfriedel, R., Michalakis, S., Jiao, K., et al. (2022). Inherited Retinal Degeneration: PARP-Dependent Activation of Calpain Requires CNG Channel Activity. Biomolecules 12:455. doi: 10.3390/biom12030455
Yang, J. H., Gross, R. L., Basinger, S. F., and Wu, S. M. (2001). Apoptotic cell death of cultured salamander photoreceptors induced by cccp: CsA-insensitive mitochondrial permeability transition. J. Cell Sci. 114, 1655–1664. doi: 10.1242/jcs.114.9.1655
Zeng, S., Hu, C., Wei, H., Lu, Y., Zhang, Y., Yang, J., et al. (1993). Intravitreal Pharmacokinetics of Liposome-encapsulated Amikacin in a Rabbit Model. Ophthalmology 100, 1640–1644. doi: 10.1016/s0161-6420(93)31423-5
Zhang, R., He, R., Qian, J., Guo, J., Xue, K., and Yuan, Y.-F. (2010). Treatment of Experimental Autoimmune Uveoretinitis with Intravitreal Injection of Tacrolimus (FK506) Encapsulated in Liposomes. Invest. Ophthalmol. Vis. Sci. 51, 3575–3582. doi: 10.1167/iovs.09-4373
Zhao, J., Trewhella, J., Corbin, J., Francis, S., Mitchell, R., Brushia, R., et al. (1997). Progressive cyclic nucleotide-induced conformational changes in the cGMP-dependent protein kinase studied by small angle X-ray scattering in solution. J. Biol. Chem. 272, 31929–31936. doi: 10.1074/jbc.272.50.31929
Keywords: retinitis pigmentosa (RP), protein kinase G (PKG), neurodegeneration, toxicity testing, drug development
Citation: Zhu Y, Cao B, Tolone A, Yan J, Christensen G, Arango-Gonzalez B, Ueffing M and Paquet-Durand F (2022) In vitro Model Systems for Studies Into Retinal Neuroprotection. Front. Neurosci. 16:938089. doi: 10.3389/fnins.2022.938089
Received: 06 May 2022; Accepted: 20 June 2022;
Published: 07 July 2022.
Edited by:
Markus Tschopp, University of Bern, SwitzerlandCopyright © 2022 Zhu, Cao, Tolone, Yan, Christensen, Arango-Gonzalez, Ueffing and Paquet-Durand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marius Ueffing, bWFyaXVzLnVlZmZpbmdAdW5pLXR1ZWJpbmdlbi5kZQ==; François Paquet-Durand, ZnJhbmNvaXMucGFxdWV0LWR1cmFuZEB1bmktdHVlYmluZ2VuLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.