 Yan Tao
Yan Tao Yusuke Murakami
Yusuke Murakami Demetrios G. Vavvas2
Demetrios G. Vavvas2 Koh-Hei Sonoda
Koh-Hei Sonoda- 1Department of Ophthalmology, Graduate School of Medical Sciences, Kyushu University, Fukuoka, Japan
- 2Ines and Frederick Yeatts Retinal Research Laboratory, Retina Service, Department of Ophthalmology, Massachusetts Eye and Ear, Harvard Medical School, Boston, MA, United States
Necroptosis mediates the chronic inflammatory phenotype in neurodegeneration. Receptor-interacting protein kinase (RIPK) plays a pivotal role in the induction of necroptosis in various cell types, including microglia, and it is implicated in diverse neurodegenerative diseases in the central nervous system and the retina. Targeting RIPK has been proven beneficial for alleviating both neuroinflammation and degeneration in basic/preclinical studies. In this review, we discuss the role of necroptosis in retinal degeneration, including (1) the molecular pathways involving RIPK, (2) RIPK-dependent microglial activation and necroptosis, and (3) the interactions between necroptosis and retinal neuroinflammation/degeneration. This review will contribute to a renewed focus on neuroinflammation induced by necroptosis and to the development of anti-RIPK drugs against retinal degeneration.
Introduction
Neurodegenerative diseases are a complex group of disorders involving the processes of cell death and inflammation cell death inflammation, and include Alzheimer's and Parkinson's disease, amyotrophic lateral sclerosis (ALS) and retinal degeneration (Tansey and Goldberg, 2010; Heneka et al., 2015; Ito et al., 2016; Kauppinen et al., 2016). Inflammation is a multicellular process by which immune cells defend against pathogens and repair injury through a series of molecular and cellular changes, including the release of pro-inflammatory cytokines, apoptosis, and necroptosis (Wallach et al., 2014). Inflammation is strongly associated with cell death, especially that in a necrotic form (Wallach et al., 2014). Damage associated molecular patterns (DAMPs) generated by necrotic cells, such as interleukin-1α (IL-1α), tumor necrosis factor α (TNF-α), and high-mobility group box 1 protein (HMGB1) (Rock and Kono, 2008; Yanai et al., 2009; Wallach et al., 2014), strongly exacerbate inflammation.
Microglia are the first line of defense in the central nervous system (CNS), and exert both cytotoxic and cytoprotective effects at the crossroads between homeostasis and disease (Cherry et al., 2014; Lloyd et al., 2019; Rodríguez-Gómez et al., 2020). Persistent activation of microglia can trigger necroptosis, a type of necrosis induced by the activation of receptor-interacting protein kinase 1 (RIPK1)/RIPK3 (Weinlich et al., 2017; Galluzzi et al., 2018), and necroptosis in turn induces secretion of proinflammatory DAMPs and mediates chronic inflammation (Rodríguez-Gómez et al., 2020). It has been shown that microglial necroptosis executed by RIPK1/3 contributes to neuronal damage as well as brain regeneration (Welser et al., 2010; Ofengeim et al., 2017; He et al., 2021).
This review focuses on RIPK-dependent necroptosis which orchestrates neuroinflammation and degeneration in the CNS and retina. We also discuss the potency of anti-necroptosis therapy, which may inhibit excessive neuroinflammation and promote neuronal survival and regeneration.
Necroptosis and Ripk Signaling
Necroptosis—A Programmed Necrotic Cell Death
Necrosis is a type of cell death that has morphologic features of cellular swelling, cell membrane rupture and the release of intracellular molecules including DAMPs (Kaczmarek et al., 2013; Galluzzi et al., 2018; Yuan et al., 2019). Although necrosis was traditionally deemed as a passive process of cell death, it is now known that some necrosis is molecularly regulated, and differential mechanisms that underlie programmed necrosis have been identified (e.g., necroptosis, pyroptosis, and ferroptosis). Necroptosis was originally described in 2005 as necrotic cell death mediated by the activation of RIPK1 (Degterev et al., 2005). The stimuli that induce necroptosis include TNF-α (Degterev et al., 2008), Fas ligand (FasL) (Holler et al., 2000), TNF-related apoptosis-inducing ligand (TRAIL) (Holler et al., 2000), the interferons (IFNs) (Thapa et al., 2013; Dillon et al., 2014), and ligands for pathogen recognition receptors (PRRs), such as toll-like receptor 3 (TLR3) (Bermejo and Rey-Bellet, 1975), TLR4 and DNA-dependent activator of interferon regulatory factors (DAI) (Upton et al., 2012; Wang et al., 2014; Huang et al., 2015).
Structure and Function of RIPK
RIPK1 is a multi-functional protein that regulates cell survival, inflammation, and cell death. It is composed of three domains, an N-terminal kinase domain for phosphorylation, an intermediate domain containing an RIP homotypic interaction motif (RHIM) for RIPK1/RIPK3 interaction and pro-inflammatory action, and a C-terminal death domain (DD) for recruiting TNFα receptor 1 (TNFR1) and the TNFR1-associated death domain (TRADD) protein (Figure 1) (Yuan et al., 2019). RIPK3, a key molecule for RIPK1/RIPK3 phosphorylation, also has an N-terminal kinase domain and RHIM domain, but lacks the DD in its C-terminus (Figure 1) (Wu et al., 2014). Mixed lineage kinase domain-like protein (MLKL) consists of an N-terminal 4-helix bundle (4HB) for interacting with the membrane and the first brace helix, an intermediate brace region with two helices involved in MLKL oligomerization, and a C-terminal pseudokinase domain (PsKD) for interaction with phosphorylated RIPK3 kinase domains (Figure 1) (Murphy et al., 2013; Quarato et al., 2016; Petrie et al., 2018, 2019). MLKL forms oligomers and the 4HB insert into the plasma membrane through phosphatidylinositol phosphate (PIP)-binding sites, resulting in membrane disruption and necroptosis induction (Dondelinger et al., 2014; Su et al., 2014; Quarato et al., 2016; Petrie et al., 2019). Therefore, the phosphorylation of MLKL and the membrane translocation of MLKL oligomers are the optimal markers for necroptosis (Fricker et al., 2018).
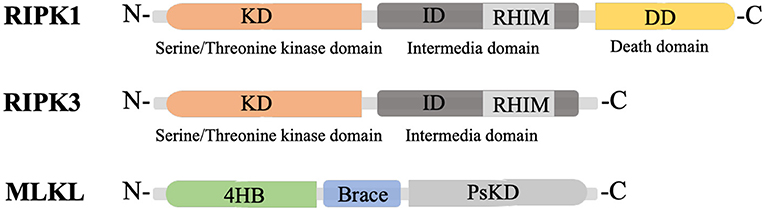
Figure 1. Structure of RIPK1, RIPK3 and MLKL. KD, N-terminal kinase domain; ID, intermediate domain; RHIM, RIP homotypic interaction motif; DD, death domain; 4HB, 4-helix bundle; PsKD, pseudokinase domain.
Among several stimuli for necroptosis, TNFR1 signaling is the most thoroughly investigated and proceeds as follows (Figure 2) (Seo et al., 2019). First, TNF binding to its receptor initiates the formation of complex I at the cell membrane. TNFR1 recruits RIPK1, TRADD, TNF receptor associated factor 2 (TRAF2) and cellular inhibitor of apoptosis 1/2 (cIAP1/2). cIAP1/2 initiates the ubiquitination process on the complex I (Bertrand et al., 2008; Varfolomeev et al., 2008; Annibaldi et al., 2018; Seo et al., 2019), and K63 poly-ubiquitination (Ub) of RIPK1 links many molecules, thereby forming a larger complex I. The TGF-β activated kinase 1 (TAK1) complex phosphorylates inhibitor of NF-κB kinase β (IKKβ) to activate the nuclear factor-κB (NF-κB) pathway (Jha et al., 2019; Seo et al., 2019). The NF-κB-dependent production of pro-survival genes, such as cIAP1, A20 and caspase-8-like inhibitory protein (c-FLIPL), protects cells from RIPK1-independent death. A20 is a deubiquitinating enzyme that prevents NF-κB activation and restricts TNF-induced apoptosis (Onizawa et al., 2015). cIAP1 and c-FLIPL facilitate NF-κB activation and inhibit necroptosis. NF-κB activation also modulates immune response via the production of proinflammatory molecules (Jha et al., 2019; Jensen et al., 2020).
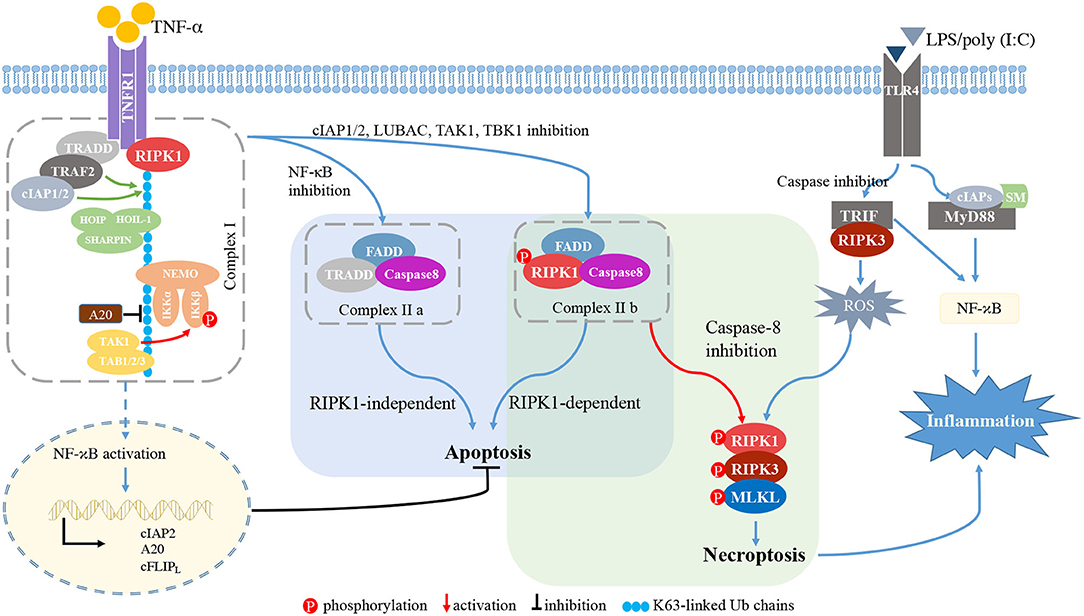
Figure 2. RIPK signaling. TNFR1 recruits RIPK1, TRADD, TRAF2, and cIAP1/2. cIAP1/2 initiates the ubiquitination process on complex I. The K63 Ub chain of RIPK1 links molecules in the large TNF complex I. The TAK1 complex phosphorylates IKKβ to induce NF-κB activation. When NF-κB, the complex of cIAP1/2, or LUBAC, TAK1, or TBK1 is inhibited, membrane-bound complex I is dissociated and complex II is formed. If NF-κB is inhibited, TRADD, FADD and caspase-8 form complex IIa, inducing RIPK1-independent apoptosis. When inhibition of cIAP1/2, TAK1, TBK1, or LUBAC occurs, RIPK1, FADD, caspase-8, and c-FLIPL form complex IIb, inducing RIPK1-dependent apoptosis. Procaspase-8/cFLIPL heterodimers prevent necroptosis. Upon caspase-8 inhibition, complex IIb initiates RIPK1- and RIPK3-dependent necroptosis. Activated RIPK1 heterodimerizes with RIPK3 and RIPK3 phosphorylates MLKL, thereby driving the polymerization of RIPK1, RIPK3 and MLKL. This RIPK1-RIPK3-MLKL complex is called a “necrosome.” In macrophages/microglia, LPS or poly(I:C) is recognized by TLR4 and mediates the interaction between TRIF and RIPK3. In the presence of caspase inhibitor, the TRIF/RIPK3 complex induces ROS accumulation, and subsequently triggers necroptosis independent of NF-κB activation. TLR4 also recruits MyD88 and cIAPs to activate the NF-κB pathway, thereby inducing pro-inflammatory cytokines. TNF-α, tumor necrosis factor-α; TNFR1, TNFα receptor 1; RIPK1/3, receptor-interacting protein kinases 1/3; TRADD, TNFR1-associated death domain protein; TRAF2, TNF receptor associated factor 2; cIAP1/2, cellular inhibitor of apoptosis 1/2; HOIL-1, haem-oxidized iron-regulatory protein 2 ubiquitin ligase-1; HOIP, HOIL-1 interacting protein; SHARPIN, SHANK-associated RH domain-interacting protein; NF-κB, nuclear factor-κB; NEMO, cell death-protective nuclear factor-κB essential modulator; IKKα/β, inhibitor of NF-κB kinase α/β; TAB2/3, TAK1-binding protein 2 and 3; FADD, Fas associated via death domain; cFLIPL, the long isoform of cellular FLICE-like inhibitory protein; MLKL, mixed lineage kinase domain-like protein; TIRF, TIR-domain containing adapter-inducing interferon-b; MYD88, myeloid differentiation primary response gene 88; SMs, SMAC mimetics; ROS, reactive oxygen species.
The TNFR1-mediated signaling pathway can induce apoptosis. Once the NF-κB pathway or complex I is inhibited by cIAP1/2 or by the linear ubiquitination assembly complex (LUBAC), TAK1, or TANK binding kinase 1 (TBK1), the membrane-bound complex I is dissociated and cytosolic complex IIa and IIb are formed. TRADD, FADD and caspase-8 form complex IIa and mediate RIPK1-independent apoptosis (Dondelinger et al., 2016; Annibaldi and Meier, 2018). Complex IIb is composed of RIPK1, FADD, caspase-8 and c-FLIPL, and initiates RIPK1-dependent apoptosis (Cho et al., 2009; Oberst et al., 2011; Sun et al., 2012).
Upon caspase-8 inhibition, complex IIa contributes to necroptotic induction by recruiting RIPK1, and complex IIb initiates RIPK1- and RIPK3-dependent necroptosis. Activated RIPK1 heterodimerizes with RIPK3 and RIPK3 phosphorylates MLKL, thereby polymerizing RIPK1, RIPK3 and MLKL (Sun et al., 2012; Zhao et al., 2012). This RIPK1-RIPK3-MLKL complex, called a “necrosome,” triggers the subsequent cell lysis. A20 inhibits formation of the RIPK1-RIPK3 complexes by deubiquitinating RIPK3 (Onizawa et al., 2015; Galluzzi et al., 2018).
The PRR family can also initiate necroptosis. TLR4 recruits the adaptor molecules myeloid differentiation primary response gene 88 (MyD88) and TIR-domain containing adapter-inducing interferon-b (TRIF), while TRIF is the sole adaptor molecule for TLR3 (Kawai and Akira, 2010). Stimulation of TLR4/TLR3 via LPS or poly(I:C) induces the formation of the TRIF-RIPK3 complex (Kaiser and Offermann, 2005; He et al., 2011). In the presence of caspase-8 inhibitor, reactive oxygen species (ROS) accumulation occurs downstream of the TRIF-RIPK3 complex, and the subsequent c-jun N-terminal kinase (JNK) activation forms the RIPK1-RIPK3-MLKL complex and induces necroptosis independent of NF-κB activation (He et al., 2011; Kim and Li, 2013). TLR4 also recruits the adaptor molecules MyD88 and cIAPs to induce the NF-κB pathway activation and release of pro-inflammatory cytokines (Figure 2).
Interactions Between Necroptosis and Neuroinflammation
Necroptosis can trigger inflammation. Activated microglia/macrophages also induce necroptosis at the chronic inflammatory site, thereby forming a positive feedback loop. In neurodegenerative diseases, dying or dead neuronal cells release DAMPs and activate PRRs (Wallach et al., 2014). PRR signaling activates microglia and transforms them to a pro-inflammatory phenotype that produces abundant pro-inflammatory cytokines (Fernández-Velasco et al., 2014). TNF-α and PRPs-mediated signals activate intracellular pathways, such as the NF-κB activation pathway, which leads to the transcription of various pro-inflammatory molecules, and the RIPK pathway, which mediates microglial activation and necroptosis (Granger and Kolb, 1968; Liu et al., 2017).
Characteristics of Microglial Activation in Neurodegeneration
Microglia are resident macrophages in the CNS and retina that surveil the surrounding environment and maintain tissue homeostasis (Nimmerjahn et al., 2005). Microglia can dynamically change their morphology and function in response to microenvironmental alterations (Choi et al., 2021). Although, in healthy individuals, blood circulating monocytes cannot enter the CNS and retina due to the healthy blood-brain and retina-barrier, under disease conditions they can infiltrate and differentiate into macrophages.
Microglia exhibit diverse functional phenotypes depending on the disease context (Ransohoff, 2016). In the same way that peripheral macrophages are traditionally classified into two major phenotypes (Martinez and Gordon, 2014) (i.e., M1 and M2), microglia can also polarize into both pro- and anti-inflammatory states at the diseased loci. Once microglia and/or macrophages detect DAMPs, they convert to the pro-inflammatory M1-like phenotype and form a high motility amoeboid shape (Chen and Xu, 2015). Activated microglia translocate to the damage site and secrete pro-inflammatory cytokines (IL-1β, IL-6, IL-8, TNF-α, iNOS) (Kalkman and Feuerbach, 2016; Aguzzi and Zhu, 2017). After the damaging molecules are diminished, the microglia change their polarity to an anti-inflammatory M2-like phenotype, improving their phagocytotic function, secreting IL-4, IL-10, IL-13, TGF-β, and arginase 1 and clearing the cellular debris to promote recovery (Cherry et al., 2014; Park et al., 2016). Triggering-receptors-expressed-on-myeloid-cells 2 (TREM2) is one kind of microglial receptor that is highly expressed in anti-inflammatory or pro-regenerative microglia and involved in phagocytosis and immune regulation (Paloneva et al., 2002; Hickman and El Khoury, 2014; Hickman et al., 2018). Overexpression of TREM2 enhances phagocytic activity and down-regulates pro-inflammatory responses (Han et al., 2017). In contrast, deletion of TREM2 or the adaptor tyrosine kinase-binding protein (TyroBP or DAP12), which binds to TREM2, leads to an excess of the pro-inflammatory phenotype, which decreases microglial survival and causes amyloid plaque deposition in experimental models of Alzheimer's disease (AD) (Ito and Hamerman, 2012). Therefore, these findings suggest that TREM2 is a key immune regulator that mediates the homeostatic function of microglia. However, it should be noted that there are more diverse and complex phenotypes in microglia and macrophages, and their plasticity (reaction states) and diversity (subtypes) should both be considered rather than merely the two commonly phenotypes (Stratoulias et al., 2019). Single-cell level analyses will help further clarify the microglia/macrophage subsets that regulate CNS health and diseases (Ransohoff, 2016; Hammond et al., 2019; Masuda et al., 2019; Stratoulias et al., 2019).
RIPK Modulates Microglial Activation and Necroptosis in Neuroinflammation
Microglia recognize the DAMPs through pattern recognition proteins (PRPs) such as TLRs. As shown in Section 2.2, TLR signaling mediates microglial activation (i.e., M1-like polarization) as well as apoptosis/necroptosis through RIPK recruitment into the cell death complex. TNF-α released from microglia may also activate RIPK pathways in the surrounding neuronal cells as well as microglia themselves.
Soluble oligomeric amyloid β (Aβ) has been shown to stimulate TNF-α release from activated microglia, leading to the induction of neuronal necroptosis in an in vitro model of AD (Salvadores et al., 2021). Ripk3−/− and Mlkl−/− mice favor pro- to anti-inflammatory phenotype transformation of microglia in response to ischemic cortex injury (Yang et al., 2018). Therefore, RIPK may regulate necroptosis and microglia/macrophage polarization in neurodegeneration (Table 1).

Table 1. Pharmacological and genetic interventions that manage cell death and inflammation in RIPK-related pathways in CNS diseases.
RIPK may contribute to microglia/macrophage polarization via a mechanism that is at least partly independent of necroptosis (Ofengeim and Yuan, 2013; Ofengeim et al., 2015, 2017; Kondylis et al., 2017; Ueta et al., 2019). Our previous study showed that RIPK inhibition suppresses M2-like polarization through caspase activation and attenuates the formation of laser-induced choroidal neovascularization (CNV) (Ueta et al., 2019). Both in vivo and in vitro experiments have shown that infiltrating macrophages, rather than vascular endothelial cells, are the main target for catalytic inhibition of RIPK. These findings suggest that RIPK has a non-necrotic function in angiogenesis via regulation of the macrophage phenotype. Ofengeim et al. reported a non-cell death function of RIPK1—namely, mediation of the disease-associated microglia (DAM) phenotype—in AD pathology (Ofengeim et al., 2017). They showed that RIPK1 is highly expressed in the cerebral microglia of patients with AD. In a mouse model of AD and in vitro cultured microglia, RIPK1 inhibition was shown to attenuate the DAM response and to enhance phagocyte activity to clear Aβ (Ofengeim et al., 2017). Transcriptional analysis found that RIPK1 induces Cst7 expression, which regulates the lysosomal function of microglia, suggesting that RIPK1-dependent transcription disturbs the homeostatic function of microglia and leads to Aβ accumulation in AD (Ofengeim et al., 2017). Alternatively, Vince et al. demonstrated that RIPK3 directly regulates NLRP3 inflammasome activation and IL-1β secretion in macrophages (Vince et al., 2012). They showed that RIPK3 mediates IAP-antagonist-induced IL-1β secretion prior to inducing necrotic changes (Vince et al., 2012).
Ubiquitination in Microglia-Related Inflammation
Ubiquitin participates in various biological processes, including protein degradation, transcription, DNA and immune regulation (Husnjak and Dikic, 2012; Zinngrebe et al., 2014). Several E3 ubiquitin ligases are involved in the regulation of MyD88- and TRIF-dependent signaling from TLRs (Wertz and Dixit, 2010). TRAF6, one of the ubiquitin E3 ligases, mediates K63-linked ubiquitination of the IKK complex subunit, IKKγ, and activates MyD88- and TRIF-dependent NF-κB signaling (Walsh et al., 2008). A20 can modulate ubiquitination and remove K63 polyubiquitin chains from target proteins to terminate signaling, as shown in Figure 2 (Komander et al., 2009; Mohebiany et al., 2020).
Kinsella et al. (2016) identified that the Bcl-2 family protein BH3-interacting domain death agonist (Bid) strengthens the TLR4-NF-κB pro-inflammatory response by promoting K63-linked polyubiquitination of TRAF6 in microglia. In a subsequent study, they demonstrated that Bid modulates MyD88- and TRIF-dependent signaling by attenuating the cleavage of polyubiquitin chains, thereby enhancing the inflammatory response (Kinsella et al., 2018).
A study on AD demonstrated that the E3 ubiquitin ligase COP1 inhibits the activation of microglia and the release of pro-inflammatory factors by degrading the transcription factor CCAAT/enhancer binding protein beta (c/EBPβ) (Ndoja et al., 2020). These transcription factors promote gene expressions related to microglial activation and inflammation. Increased secretion of pro-inflammatory factors and neurotoxicity in COP1-deficient microglia were observed in a mouse model of tau-mediated neurodegeneration and microglia-neuronal co-cultures. Thus, COP1 is important for suppression of the pathogenic c/EBPβ-dependent gene expression process in microglia.
RIPK is also involved in K63-linked Ub. IAPs are the key E3 ubiquitin ligases that ubiquitinate RIPK1/2 and cause NF-κB activation (Figure 2) (Jensen et al., 2020). Several studies have shown that RIPK1-mediated inflammatory signaling can be inhibited by second mitochondria-derived activator of caspases (SMAC), a small-molecule antagonist of IAPs (Busca et al., 2018; Goncharov et al., 2018). SMAC mimetics can also suppress the pro-inflammatory response in TLR signaling (Figure 2) (Tseng et al., 2010). Deubiquitinating enzyme is an alternative target to modulate RIPK activation. Lu et al. recently demonstrated that microglial necroptosis is suppressed by melatonin via the regulation of A20 in a model of intracerebral hemorrhage (Lu et al., 2019). Therefore, targeting ubiquitination may be a promising anti-inflammatory strategy.
The Role of Microglial Necroptosis in Neuroinflammation
Necroptosis can occur in neurons as well as microglial cells in CNS diseases. Thadathil et al. investigated the expression of p-MLKL, a marker of necroptosis, in the brains of young and aged mice and showed that nearly 70–80% of p-MLKL immunoreactivity is observed in neurons and <10% in microglia (Thadathil et al., 2021). Blocking or inhibiting necroptosis resulted in a significant reduction in neuroinflammation in aged mice (Thadathil et al., 2021). Therefore, necroptosis may be critically important in age-associated neuroinflammation and neurodegeneration.
Ito et al. investigated the functional roles of RIPK in an amyotrophic lateral sclerosis (ALS) model. Mutations in the optineurin OPTN gene have been implicated in patients with ALS (Ito et al., 2016). Using an ALS model of optineurin (Optn)-deficient mice, they reported that RIPK1 inhibition reduces the levels of proinflammatory cytokines, including IL-1α, IL-1β, interferon-γ (IFNγ), and TNF-α, in the spinal cord (Ito et al., 2016). RNA-sequencing analysis of Optn−/−, and Optn−/−; Ripk1D138N/D138N microglia revealed that the M1-like phenotype in Optn−/− microglia is suppressed by RIPK1 inhibition (Kigerl et al., 2009; Ito et al., 2016). In addition, necrotic cell death in the spinal cord of Optn−/− mice was rescued by RIPK inhibition, which led to improved motor function (Ito et al., 2016). In pathological spinal cord sections from patients with ALS, increased co-immunostainings of RIPK1, RIPK3, MLKL, RIPK1 p-Ser14/15, p-MLKL, and microglia were observed (Ito et al., 2016). Therefore, RIPK1 may be a critical mediator of microglial activation as well as necroptosis in the axonal pathology of ALS.
The Role of Ripk in Retinal Degeneration and Neuroinflammation
Given the significant roles of the RIPK pathway in necroptosis induction and microglial activation in CNS disorders, it would be natural to expect that RIPK is involved in retinal necroptosis and inflammation. Indeed, although it was traditionally considered that apoptosis is the main form of cell death in retinal degeneration, our group revealed that RIPK-dependent necroptosis is redundantly activated when caspases are inhibited in experimental retinal detachment (RD) (Trichonas et al., 2010). Rosenbaum et al. also demonstrated that treatment with Nec-1 prevents retinal cell death in a rat model of retinal ischemia (Rosenbaum et al., 2010). It is now known that RIPK plays pivotal roles in various retinal and optic nerve disorders, including inherited retinal diseases (IRDs), age-related macular degeneration (AMD), and glaucoma, even in the absence of caspase inhibition (Trichonas et al., 2010; Murakami et al., 2012, 2014; Kataoka et al., 2015; Do et al., 2017; Kayama et al., 2018). In the discussion below, we introduce studies addressing the RIPK function in necroptosis, microglial activation, and neuroinflammation in retinal degeneration (Figure 3; Table 2).
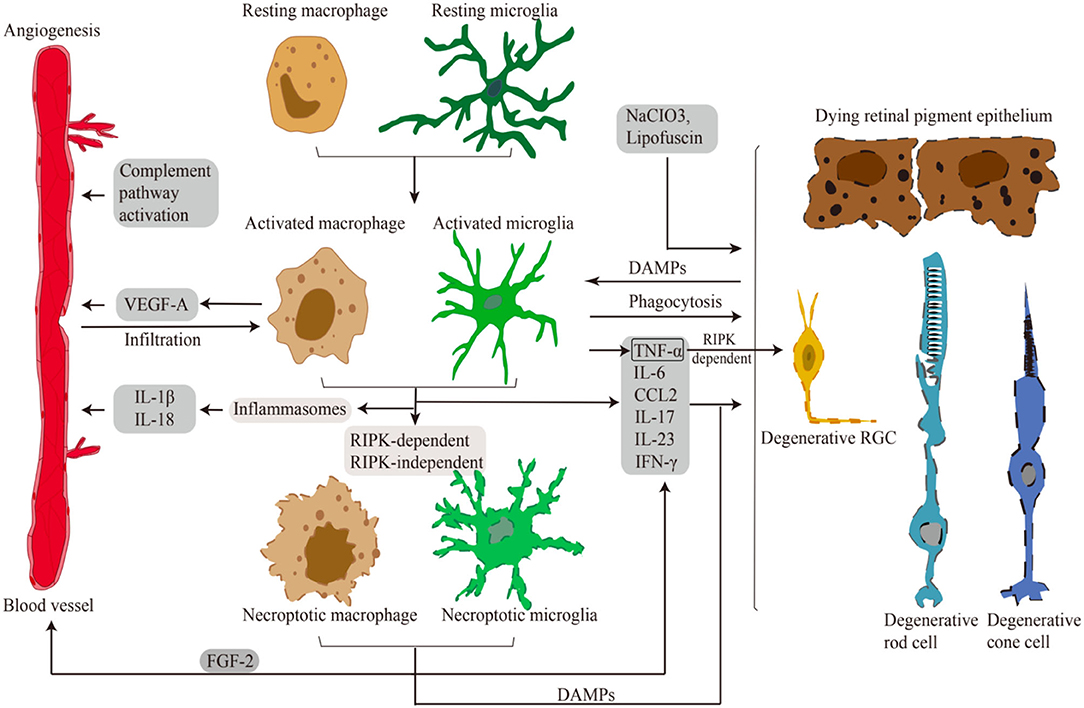
Figure 3. Microglia/Macrophages in retinal disorders. Schematic representation of necroptotic and activated microglia/macrophages in retinal necroptosis and inflammation. Various damages and DAMPs activate innate immune cells. Activated and necroptotic microglia/macrophages via inflammasome-dependent/-independent mechanisms secrete inflammatory and angiogenic mediators. These pro-inflammatory and angiogenic factors induce choroidal neovascularization. Pro-inflammatory factors and DAMPs also lead to necrotic cell death of RGCs, RPE cells and photoreceptors. Moreover, activated microglia/macrophages infiltrate to the outer retina to phagocytose dead cells and modulate inflammation.
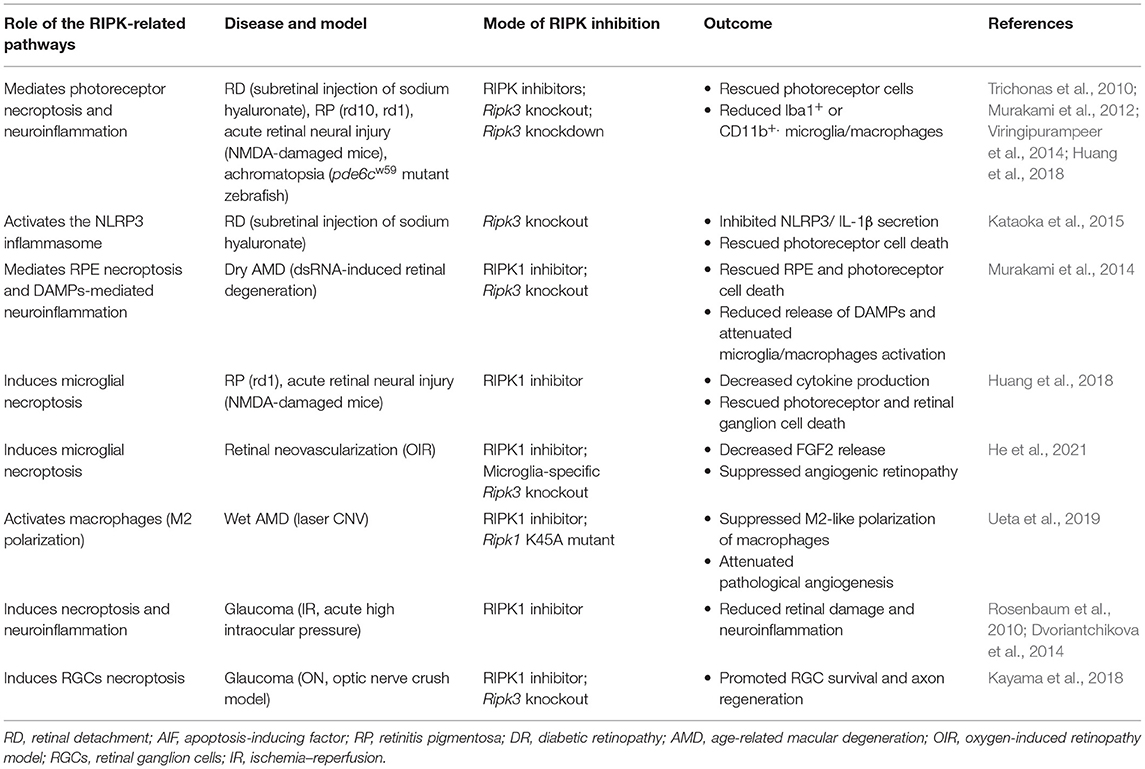
Table 2. Pharmacological and genetic interventions that manage cell death and inflammation in RIPK-related pathways in retinal diseases.
Role of RIPK in Retinal Detachment
Photoreceptor cells receive metabolic support from the underlying retinal pigment epithelium (RPE) and choroidal vessels, and when the neuroretina is physically detached from the RPE, photoreceptor cells start to die. Experimental models of RD induced by the subretinal injection of sodium hyaluronate and human samples with RD have shown that photoreceptor cell death is induced as early as 12 h and peaks at around 2–3 days after RD (Cook et al., 1995; Hisatomi et al., 2001; Arroyo et al., 2005). Hypoxia plays an important role in this process, because oxygen therapy substantially prevents photoreceptor cell loss in a cat model of RD (Lewis et al., 2004). Neuroinflammation also contributes to RD-induced retinal degeneration. Multiple cytokines/chemokines, including TNF-α, IL-1β, and MCP-1, are substantially elevated in the eyes in both experimental and human RD (Nakazawa et al., 2006a; Yoshimura et al., 2009). Moreover, genetic knockout of Mcp-1 attenuates macrophage infiltration and prevents photoreceptor cell death in a rodent model of RD (Nakazawa et al., 2007). Sweigard et al. suggested that hypoxia mediates the activation of the alternative complement pathway in the detached mouse retina, indicating a molecular link between hypoxia and neuroinflammation (Sweigard et al., 2015).
Apoptosis is the main form of photoreceptor cell death after RD (Cook et al., 1995; Hisatomi et al., 2001; Arroyo et al., 2005). Consistent with the apoptotic morphological changes, such as pyknosis, both caspase activation in intrinsic and extrinsic pathways and upregulated expression of death receptor ligands (e.g., TNF-α and Fas-L) are observed in the rat retina after RD (Zacks et al., 2003, 2004). Paradoxically, however, caspase inhibition via intravitreal injection of pan-caspase inhibitor did not lead to protection against RD-induced retinal degeneration, suggesting the presence of other or redundant effectors for the photoreceptor cell death after RD.
As described above, caspases and RIPK redundantly function as cell death effectors in TNF-α signaling. Given the evidence that RIPK-dependent necrosis is strongly induced when the caspase pathway is blocked under various physiological and pathological conditions, we hypothesized that RIPK may mediate photoreceptor cell death after RD in concert with caspases. Indeed, we found that caspase inhibition in rodent models of RD substantially reduced apoptosis of photoreceptor cells while simultaneously increasing necrotic cell death in the detached retina. This necrotic change was rescued by additional treatment with Nec-1 or Ripk3 deficiency, indicating that photoreceptor cell death after RD is redundantly regulated by at least two cell death pathways, i.e., caspase-dependent apoptosis and RIPK-dependent necroptosis (Trichonas et al., 2010). Consistent with our findings, Dong et al. demonstrated that Nec-1 inhibits RIPK phosphorylation and prevents necrotic photoreceptor cells after experimental RD (Dong et al., 2012).
RIPK inhibition in experimental RD prevents photoreceptor cell death, in addition to attenuating the infiltration of CD11b-positive microglia/macrophages in the detached retina (Trichonas et al., 2010). Kataoka et al. further investigated the roles of RIPK in inducing neuroinflammation after mouse experimental and human RD and showed that RIPK regulates NLRP3/caspase-1 inflammasome activation and prevents IL-1β secretion from subretinal microglia/macrophages (Kataoka et al., 2015). Because Vince et al. (2012) demonstrated that RIPK3 directly promotes IL-1β secretion in macrophages via inflammasome-dependent and -independent mechanisms, it may be possible that RIPK directly modulates microglia/macrophage activation after RD, in addition to indirectly activating neuroinflammation through the release of necrotic DAMPs.
Role of RIPK in Retinitis Pigmentosa
Retinitis pigmentosa (RP) is a set of hereditary retinal diseases characterized by the degeneration of rod and cone photoreceptors with genetic defects (Hartong et al., 2006). The typical progression in patients with RP begins with night blindness and loss of mid-peripheral visual field due to rod cell dysfunction and death, followed by loss of peripheral and central vision due to cone cell death (Hartong et al., 2006; Murakami et al., 2015). Many studies have shown that multiple signaling pathways lead to photoreceptor death, including apoptosis, autophagy and necroptosis (Murakami et al., 2012, 2015; Sizova et al., 2014; Athanasiou et al., 2018; Viringipurampeer et al., 2019). However, the specific mechanisms predominating in each of rod and cone photoreceptor cell death in RP remain elusive.
Rod cell death in RP is induced by mutations in the causal genes, most of which are related to rod function, structure or homeostasis, and the dead cells exhibit apoptotic morphology. In contrast, cone cells, which do not express deleterious genes, remain healthy in the early stage of disease, but also degenerate after rod cell death. The mechanisms of this secondary cone cell death have not been fully understood; however, accumulating evidence suggests that microenvironmental alterations such as neuroinflammation, oxidation and metabolic imbalance contribute to the cone cell death in RP (Yoshida et al., 2013; Olivares-González et al., 2018, 2021; Newton and Megaw, 2020). Interestingly, morphological analyses of the postmortem eyes of RP patients demonstrated some necrotic features, such as swollen cytoplasm and membrane rupture in remaining cones (To et al., 2004). Consistent with these anatomical features, our group demonstrated that RIPK is essential for induction of cone necroptosis and microglia activation in rd10 mice, a mouse model of RP induced by a missense mutation in the Pde6β gene, and both effects could be suppressed by Ripk3 deletion (Murakami et al., 2012). Subsequently, we found significantly enlarged cone cells in the macula of human RP patients using adaptive optics technology, and observed higher levels of HMGB1, a DAMP released from necrotic cells, in the vitreous of RP patients (Scaffidi et al., 2002; Murakami et al., 2015).
Consistent with our findings, Viringipurampeer et al. (2014) identified that cones expressed high levels of RIPK1 and RIPK3, and that the dying cones could be rescued by knockdown of Ripk3 in pde6cw59 mutant zebrafish. Yang et al. (2017) also revealed that RIPK1 and RIPK3 were markedly upregulated in the retinas of sigma-1 receptor (S1R)-deficient rd10 mice, accompanied with a deteriorated loss of cones. A recent study reported that the necroptosis pathway is highly activated during photoreceptor death in Pro23His (P23H) transgenic albino rats, and P23H is also the most common mutation in autosomal dominant RP (Kakavand et al., 2020). Increased phospho-MLKL has been observed in the inner and outer segments of the P23H-3 mouse retina (Kakavand et al., 2020). Furthermore, Sato et al. (2013) observed TNF-induced rod and cone necroptosis in Irbp-deficient mice. Interphotoreceptor retinoid-binding protein (IRBP) secreted by photoreceptors is important for photoreceptor survival, and IRBP mutation is associated with human RP.
Therefore, RIPK-mediated necroptosis may be a common mechanism for cone cell death in RP, and RIPK-mediated necroptosis may be a novel target to prevent the death of photoreceptors in RP patients.
Cell death and inflammation are interconnected. Indeed, we previously demonstrated that microglial activation is suppressed by Ripk3 deficiency in rd10 mice. This anti-inflammatory effect of RIPK inhibition may be explained by the reduced release of DAMPs from necrotic cone cells as well as by the attenuation of microglial activation/necroptosis.
To our knowledge, only a single report has described the implications of microglial necroptosis in both in vitro and in vivo (rd1 mice, pde6βrd1 and NMDA-damaged mice) models of retinal degeneration (Huang et al., 2018). TLR4 is involved in the activation of microglia in these models, in association with infiltration of Iba-1+/RIPK3+ necroptotic microglia at the site of degeneration. These necroptotic microglia secrete inflammation mediators, e.g., TNF-α, chemokine (C-C motif) ligand 2 (CCL2), IL-17, IL-23 and IFN-γ. Pharmacological blockade of RIPK1 has been shown to ameliorate microglial activation and retinal degeneration in both an NMDA-induced retinal neural injury model and murine BV2 microglial cells (Huang et al., 2018). These findings suggest that microglial necroptosis may regulate neuroinflammation in RP, at least in part.
Role of RIPK in Dry Age-Related Macular Degeneration
AMD is a primary cause of central visual loss in developed countries and affects primarily the elderly (Smith et al., 2001). Early AMD is characterized by pathological deposits (drusen) between the RPE and the Bruch's membrane (Al-Zamil and Yassin, 2017; Handa et al., 2019). RPE are monolayer cells maintaining the functionality of overlying photoreceptors (Boulton and Dayhaw-Barker, 2001). Widespread drusen, various pathologies such as inflammation, and various genetic and environmental factors promote RPE dysfunction and photoreceptor death (Handa et al., 2019; Miller et al., 2021). Advanced late-stage dry AMD, also known as geographic atrophy (GA), is associated with the degradation of RPE cells in a non-neovascular form (Handa et al., 2019; Miller et al., 2021). Oxidative stress is implicated in the pathogenesis of both wet and dry AMD, and it can cause blood-retinal barrier breakdown that triggers chronic inflammation (Barnett and Handa, 2013).
Alterations of many inflammatory cytokines have been detected in plasma, serum or intraocular fluid in patients with AMD, but regrettably there is no stable trend or consistency in the reported findings (Tan et al., 2020).
Infiltration of macrophages has been detected in late advanced dry AMD and neovascular AMD (Penfold et al., 1985; Lad et al., 2015). Activated immune cells such as microglia/macrophages can migrate or infiltrate to the outer nuclear layer and remove photoreceptor debris, which is believed to be related to atrophic AMD (Killingsworth et al., 1990; Gupta et al., 2003). Analysis of a single-cell transcriptomic atlas of human retinas indicated that microglia are one of the factors most predictive of AMD, which highlights the importance of microglia in AMD pathogenesis (Menon et al., 2019). A histological study of the human eyes revealed that higher numbers of macrophages are concentrated in the choroid in late dry AMD than in normal AMD (Wang et al., 2022). Taken together, these data indicate a pathogenetic role of inflammation and immune response in AMD. Yet there is still controversy and debate regarding the origin of the cells reported in histopathology as macrophages, with some studies supporting the notion that some of these cells may not be bona-fide immune cells but rather transdifferentiated RPE cells (Lad et al., 2015; Curcio and Ach, 2016).
The RIPK-dependent necrotic pathway is crucial in RPE and photoreceptor cell death in AMD. Our previous study identified that programmed necroptosis is the main mechanism for cell death of RPE in dsRNA-induced retinal degeneration, a model of dry AMD (Murakami et al., 2014). Ripk3 deficiency in this model reduced the release of DAMPs and suppressed the inflammatory response in the retina (Murakami et al., 2014). These findings suggest that RIPK-dependent necroptosis amplifies neuroinflammation by regulating the release of intracellular DAMPs (Murakami et al., 2014). Oxidative stress is one important component of AMD pathogenesis. Hanus et al. (2015) confirmed that RPE necroptosis is the predominant mechanism of NaIO3-induced RPE cell death, a model of dry AMD induced by oxidative injury. Because caspase-8 is severely downregulated in the mature RPE (Yang et al., 2007), RPE cells may be predisposed to undergo necroptosis under a stressed condition.
A study by Pan et al. (2021) reported that retinal lipofuscin activates an atypical necroptosis in RPE cells as well as macrophages/microglia. That study provided new insights into necroptosis activation in aged human retinas with AMD. Interestingly, its authors demonstrated an atypical RPE necroptotic mechanism that involves neither RIPK1 nor RIPK3, but rather is mediated by lysosomal membrane permeabilization (LMP) and subsequent MLKL phosphorylation (Pan et al., 2021). In addition to RPE cells, phospho-MLKL staining was also observed in microglia/macrophages, which phagocytosed the sloughed RPE fragments (Pan et al., 2021). These findings suggest the intimate interactions between necroptosis and neuroinflammation in the pathology of dry AMD. They also show that anti-necroptosis agents that target MLKL phosphorylation (Nec-7) and oligomerization (NSA) prevent light-independent lipofuscin-elicited necroptosis. Thus, targeting lipofuscin-induced necroptosis may be a new therapeutic strategy for dry AMD.
Role of RIPK in Wet Age-Related Macular Degeneration
CNV is the end stage of the wet form of AMD, with growth of new blood vessels between Bruch's membrane and the RPE (Hobbs and Pierce, 2021). After CNV, extravasations, hemorrhage and fibrovascular scar formation occur in the subretinal space, leading to the death of the neurosensory retina and vision loss (Al-Zamil and Yassin, 2017; Hobbs and Pierce, 2021). How does the accumulated drusen induce neovascularization? Although vascular endothelial growth factor (VEGF) has been established as the key factor that initiates and promotes CNV, neuroinflammation also modulates CNV formation (Balser et al., 2019; Uemura et al., 2021). Activated microglia/macrophages release angiogenic and anti-angiogenic factors and work with VEGF to regulate retinal and choroidal angiogenesis (Pollard, 2009; Welser et al., 2010).
Doyle et al. (2012) showed that isolated drusen from AMD donor eyes can activate the NLRP3 inflammasome and secretion of IL-1b and IL-18. In a model of wet AMD with laser-induced CNV, IL-18 activation inhibited CNV development in Nlrp3−/− mice (Doyle et al., 2012). This study indicates that NLRP3 and IL-18 play a protective role in angiogenesis. Some in vitro studies have suggested that IL-18 may inhibit vascular endothelial cell proliferation (Park et al., 2001; Kim et al., 2005, 2006). The results of numerous studies are consistent with those of Doyle et al. (2012) that IL-18 has an antiangiogenic effect (Kim et al., 2005, 2006). The findings of Qiao et al. support the notion that IL-18 plays a role in suppressing angiogenesis by promoting the regression of pathologic neovascularization (NV), and the work of Cao et al. revealed an inhibitory effect of IL-18 on FGF-induced NV (Cao et al., 1999; Qiao et al., 2007). Therefore, IL-18 neuroinflammation may function in part to repress CNV formation in AMD, though other studies have failed to observe a significant contribution of IL-18 deficiency to spontaneous CNVM formation in Vegfahyper mice (Malsy et al., 2020; Marneros, 2021).
RIPK-dependent microglial necroptosis is also involved in retinal angiogenesis. He et al. argued that microglial necroptosis may be an important biological process in the etiology of retinal angiogenesis. They identified a subpopulation of microglia that highly express RIPK3 and MLKL through single-cell RNA sequencing in an oxygen-induced retinopathy (OIR) model (He et al., 2021). This necroptotic subpopulation of microglia promoted angiogenesis by releasing intracellular FGF2 (an angiogenic factor). The extracellular release of FGF2 was reduced by Nec1 treatment or conditional deletion of Ripk3. Moreover, combined treatment with anti-RIPK1 and anti-VEGF drugs has been shown to remarkably suppress angiogenesis in an OIR model (He et al., 2021). Collectively, this evidence reveals a new mechanism by which microglial necroptosis contributes to angiogenic retinopathy.
RIPK also regulates angiogenesis via non-necroptotic modulation of macrophages. Ueta et al. demonstrated that RIPK1 inhibition suppressed angiogenesis by modulating macrophage polarization in a mouse model of laser-induced CNV and alkali injury-induced corneal neovascularization (Ueta et al., 2019). Mechanistically, we revealed that RIPK1 inhibition mediates caspase activation and suppresses the pro-angiogenic M2-like phenotype in infiltrating macrophages. Therefore, RIPK may be a potential therapeutic target of pathological angiogenesis that could both inhibit necroptosis and modulate microglia/macrophage polarization.
Role of RIPK in Glaucoma
Glaucoma is characterized by the progressive loss of retinal ganglion cells (RGCs) and is a leading cause of irreversible blindness (Quigley and Broman, 2006). Glaucomatous neurodegeneration is attributed to various pathologies, such as mechanical pressure, vascular deficiency, inflammation, oxidative stress and metabolic dysregulation (Tezel, 2021). Elevated intraocular pressure (IOP) is the major risk factor for the progression of glaucoma, and pressure-induced retinal ischemia aggravates the death of RGCs (Sellés-Navarro et al., 1996).
While apoptosis has traditionally been considered the major type of RGC death in glaucoma, attempts to rescue RGCs by regulating apoptosis in glaucoma have been unsuccessful. Accumulating evidence indicates that both apoptosis and necroptosis participate in glaucomatous RGC death, and RIPK also plays a crucial role (Lee et al., 2013; Dvoriantchikova et al., 2014; Do et al., 2017; Wang et al., 2020). Dvoriantchikova et al. demonstrated that RIPK inhibition prevented RGC necroptosis in both an in vitro and an in vivo model of ischemic injury (Dvoriantchikova et al., 2014). Do et al. reported that RIPK1 was activated in a rat model of high IOP-induced ischemic injury, and a novel RIPK1-inhibitory compound significantly attenuated RGC death in a dose-dependent manner (Do et al., 2017). Consistent with these in vivo findings, Xiong et al. revealed that RIPK-mediated necroptosis is induced by oxygen-glucose deprivation in retinal ganglion cell line 5 (RGC-5) (Liao et al., 2017). Subsequently, they found that p90 ribosomal protein S6 kinase 3 (RSK3) is an upstream regulator of RIPK3 phosphorylation, and that an RSK3 inhibitor conferred RGC protection and functional recovery in rat with high IOP-induced ischemic injury (Wang et al., 2020). Our previous study also revealed that the expression of RIPK was increased in an optic nerve (ON) crush model, and necrosis inhibitors promoted a moderate level of axonal regeneration (Kayama et al., 2018). Collectively, these findings suggest that RIP kinase-dependent necroptosis is both a novel mechanism of RGC death and a suitable therapeutic target.
As described above, neuroinflammation is one of the pathogenic mechanisms of RGC degeneration in glaucoma (Weinreb et al., 2014). TNF-α is elevated in the aqueous humor, retina and optic nerve of glaucoma patients (Yan et al., 2000; Tezel, 2008; Sawada et al., 2010) as well as in a glaucoma mouse model induced by chronic ocular hypertension (Nakazawa et al., 2006b). In the latter study in mice (Nakazawa et al., 2006b) further showed that increased IOP induces TNF-α production in the retina and optic nerve, and ultimately causes RGC loss via microglia activation (Figure 3). Moreover, they found that intravitreal TNF-α injection induced RGC death that mimics that the glaucoma, and blockade of microglial activation rescued this RGC death induced by TNF-α (Nakazawa et al., 2006b). Collectively, these findings of Nakazawa et al. (2006b) highlight the importance of TNF-α and microglial neuroinflammation in the pathology of glaucoma. Other authors similarly showed that microglia are highly activated in the optic nerve head in human eyes with glaucoma, in association with the abundant expression of TNF-α, TGF-β and matrix metalloproteinases (Neufeld, 1999; Yuan and Neufeld, 2001).
The roles of RIPK and necroptosis in the neuroinflammation of glaucoma are still elusive. Dvoriantchikova et al. demonstrated that RIPK inhibition attenuates the expression of pro-inflammatory genes such as Il-1b, Ccl5, Cxcl10, Nos2, and Cybb in a high-IOP induced ischemic injury model, suggesting that RIPK may also mediate neuroinflammation in glaucoma (Dvoriantchikova et al., 2014).
Using a neuroinflammatory model of glaucoma (TNF-α induced RGC degeneration), Ko et al. revealed that a non-canonical form of necroptosis is critical for axonal degeneration of RGCs. They showed that sterile alpha and TIR motif 1 (SARM1), an inducible NAD+ cleavage enzyme, act downstream of TNF-α and activate MLKL. In this model, MLKL does not directly induce necroptosis but rather mediates the loss of axon survival factors NMNAT2 and SCG10/STMN2, which leads to axon degeneration. Therefore, inhibition of TNF-α and/or SARM1 may suppress axon necroptosis, and these pathways may serve as alternative targets for glaucomatous neurodegeneration.
Prospects for Anti-Ripk/Necroptosis Treatment in Retinal Disorders
RIPK-dependent necroptosis has been proposed as a promising target for a variety of neurodegenerative diseases that involve both neuronal cell death and neuroinflammation. Inhibition of the RIPK pathway has been proven effective in blocking necroptosis and suppressing neuroinflammation in many of the basic experiments mentioned above, and therefore, strategies to translate these studies into clinical trials should be explored.
In preclinical studies, RIPK1 inhibitors have been the agents most studied for the treatment of CNS diseases. Such research has shown that Nec-1 administration controls the neuronal cell death induced by acute neuronal injury and improves motor and spatial memory (You et al., 2008). Nec-1 also promotes the ability of microglia to degrade Aβ in APP/PS1 mice (Ofengeim et al., 2017). The RIPK1 inhibitor GSK′547 ameliorated the aggregation of lipofuscin-like lysosomal inclusions in microglia and improved survival in a model of lysosomal storage disorder (Safaiyan et al., 2016; Cougnoux et al., 2018). RIPK is activated under various necrotic or non-necrotic pathological inflammatory conditions, and thus well-designed drugs that target certain RIPK regulation points are critical for the treatment of human CNS and peripheral pathologies.
Indeed, Nec-1s and other RIPK1 inhibitors (e.g., GSK2982772, DNL104, DNL747, and DNL788) are presently being assessed in clinical trials for neurodegenerative and autoimmune diseases (Degterev et al., 2019; Mifflin et al., 2020). A phase II study of GSK2982772 for the treatment of psoriasis and ulcerative colitis (UC) was completed. The authors reported that GSK2982772 was safe and well-tolerated when dosed at 60 mg t.i.d., and that it may have ameliorated inflammation in 65 psoriasis patients (Weisel et al., 2020). They then assessed the potential effects at higher dosages and in patients with more active disease (Weisel et al., 2020). However, GSK2982772 showed no significant difference in clinical efficacy between the treatment and placebo groups in UC patients (Weisel et al., 2021). A phase I clinical trial of DNL747 for the treatment of AD has been completed, but the results have not been disclosed. RIPK3 inhibitors have not yet entered clinical trials, but studies on these agents are developing rapidly (Jensen et al., 2020).
Although the existing results of these clinical trials show the safety of anti-RIPK drugs with currently unknown efficacy, it will be critical to develop more secure, sustained, and effective inhibitors of RIPK to combat chronic neurodegeneration. Therefore, medicines with better pharmacological designs and sustained local delivery may be required for the further development of anti-RIPK therapy to prevent blindness in patients with currently incurable retinal degeneration.
Author Contributions
YM proposed and guided the direction of the manuscript. YT and YM wrote the main body of this manuscript. K-HS and DV provided supervision of the review. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Grants-in-Aid for Scientific Research, #JP19K09952 and #JP22H03242 (to YM), a Uehara Memorial Foundation grant (to YM), a Charitable Trust Fund for Ophthalmic Research in Commemoration of Santen Pharmaceutical's Founder grant (to YM), and a Bayer Retina Award (to YM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aguzzi, A., and Zhu, C. (2017). Microglia in prion diseases. J. Clin. Invest. 127, 3230–3239. doi: 10.1172/jci90605
Al-Zamil, W. M., and Yassin, S. A. (2017). Recent developments in age-related macular degeneration: a review. Clin. Interv. Aging 12, 1313–1330. doi: 10.2147/cia.S143508
Annibaldi, A., and Meier, P. (2018). Checkpoints in TNF-induced cell death: implications in inflammation and cancer. Trends Mol. Med. 24, 49–65. doi: 10.1016/j.molmed.2017.11.002
Annibaldi, A., Wicky John, S., Vanden Berghe, T., Swatek, K. N., Ruan, J., Liccardi, G., et al. (2018). Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Mol. Cell 69, 566–580.e565. doi: 10.1016/j.molcel.2018.01.027
Arroyo, J. G., Yang, L., Bula, D., and Chen, D. F. (2005). Photoreceptor apoptosis in human retinal detachment. Am. J. Ophthalmol. 139, 605–610. doi: 10.1016/j.ajo.2004.11.046
Athanasiou, D., Aguila, M., Bellingham, J., Li, W., McCulley, C., Reeves, P. J., et al. (2018). The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 62, 1–23. doi: 10.1016/j.preteyeres.2017.10.002
Balser, C., Wolf, A., Herb, M., and Langmann, T. (2019). Co-inhibition of PGF and VEGF blocks their expression in mononuclear phagocytes and limits neovascularization and leakage in the murine retina. J. Neuroinflamm. 16, 26. doi: 10.1186/s12974-019-1419-2
Barnett, B. P., and Handa, J. T. (2013). Retinal microenvironment imbalance in dry age-related macular degeneration: a mini-review. Gerontology 59, 297–306. doi: 10.1159/000346169
Bermejo, E., and Rey-Bellet, J. (1975). Comparative study on the hypnotic effect of flurazepam and heptabarbital. Rev. Med. Suisse Romande 95, 635–641.
Bertrand, M. J., Milutinovic, S., Dickson, K. M., Ho, W. C., Boudreault, A., Durkin, J., et al. (2008). cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700. doi: 10.1016/j.molcel.2008.05.014
Boulton, M., and Dayhaw-Barker, P. (2001). The role of the retinal pigment epithelium: topographical variation and ageing changes. Eye 15(Pt 3), 384–389. doi: 10.1038/eye.2001.141
Busca, A., Konarski, Y., Gajanayaka, N., O'Hara, S., Angel, J., Kozlowski, M., et al. (2018). cIAP1/2-TRAF2-SHP-1-Src-MyD88 complex regulates lipopolysaccharide-induced IL-27 production through NF-κB activation in human macrophages. J. Immunol. 200, 1593–1606. doi: 10.4049/jimmunol.1700199
Cao, R., Farnebo, J., Kurimoto, M., and Cao, Y. (1999). Interleukin-18 acts as an angiogenesis and tumor suppressor. FASEB J. 13, 2195–2202. doi: 10.1096/fasebj.13.15.2195
Chen, M., and Xu, H. (2015). Parainflammation, chronic inflammation, and age-related macular degeneration. J. Leukoc. Biol. 98, 713–725. doi: 10.1189/jlb.3RI0615-239R
Cherry, J. D., Olschowka, J. A., and O'Banion, M. K. (2014). Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflamm. 11, 98. doi: 10.1186/1742-2094-11-98
Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. doi: 10.1016/j.cell.2009.05.037
Choi, S., Guo, L., and Cordeiro, M. F. (2021). Retinal and brain microglia in multiple sclerosis and neurodegeneration. Cells 10, 1507. doi: 10.3390/cells10061507
Cook, B., Lewis, G. P., Fisher, S. K., and Adler, R. (1995). Apoptotic photoreceptor degeneration in experimental retinal detachment. Invest. Ophthalmol. Vis. Sci. 36, 990–996.
Cougnoux, A., Clifford, S., Salman, A., Ng, S. L., Bertin, J., and Porter, F. D. (2018). Necroptosis inhibition as a therapy for Niemann-Pick disease, type C1: Inhibition of RIP kinases and combination therapy with 2-hydroxypropyl-β-cyclodextrin. Mol. Genet. Metab. 125, 345–350. doi: 10.1016/j.ymgme.2018.10.009
Curcio, C. A., and Ach, T. (2016). Macrophages or retinal pigment epithelium expressing macrophage markers in age-related macular degeneration? Comment on Lad et al. 2015. Graefes Arch. Clin. Exp. Ophthalmol. 254, 1237–1238. doi: 10.1007/s00417-016-3300-7
Degterev, A., Hitomi, J., Germscheid, M., Ch'en, I. L., Korkina, O., Teng, X., et al. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321. doi: 10.1038/nchembio.83
Degterev, A., Huang, Z., Boyce, M., Li, Y., Jagtap, P., Mizushima, N., et al. (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119. doi: 10.1038/nchembio711
Degterev, A., Ofengeim, D., and Yuan, J. (2019). Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. U.S.A. 116, 9714–9722. doi: 10.1073/pnas.1901179116
Dillon, C. P., Weinlich, R., Rodriguez, D. A., Cripps, J. G., Quarato, G., Gurung, P., et al. (2014). RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202. doi: 10.1016/j.cell.2014.04.018
Do, Y. J., Sul, J. W., Jang, K. H., Kang, N. S., Kim, Y. H., Kim, Y. G., et al. (2017). A novel RIPK1 inhibitor that prevents retinal degeneration in a rat glaucoma model. Exp. Cell Res. 359, 30–38. doi: 10.1016/j.yexcr.2017.08.012
Dondelinger, Y., Darding, M., Bertrand, M. J., and Walczak, H. (2016). Poly-ubiquitination in TNFR1-mediated necroptosis. Cell. Mol. Life Sci. 73, 2165–2176. doi: 10.1007/s00018-016-2191-4
Dondelinger, Y., Declercq, W., Montessuit, S., Roelandt, R., Goncalves, A., Bruggeman, I., et al. (2014). MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 7, 971–981. doi: 10.1016/j.celrep.2014.04.026
Dong, K., Zhu, H., Song, Z., Gong, Y., Wang, F., Wang, W., et al. (2012). Necrostatin-1 protects photoreceptors from cell death and improves functional outcome after experimental retinal detachment. Am. J. Pathol. 181, 1634–1641. doi: 10.1016/j.ajpath.2012.07.029
Doyle, S. L., Campbell, M., Ozaki, E., Salomon, R. G., Mori, A., Kenna, P. F., et al. (2012). NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 18, 791–798. doi: 10.1038/nm.2717
Dvoriantchikova, G., Degterev, A., and Ivanov, D. (2014). Retinal ganglion cell (RGC) programmed necrosis contributes to ischemia-reperfusion-induced retinal damage. Exp. Eye Res. 123, 1–7. doi: 10.1016/j.exer.2014.04.009
Fernández-Velasco, M., González-Ramos, S., and Boscá, L. (2014). Involvement of monocytes/macrophages as key factors in the development and progression of cardiovascular diseases. Biochem. J. 458, 187–193. doi: 10.1042/bj20131501
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541. doi: 10.1038/s41418-017-0012-4
Goncharov, T., Hedayati, S., Mulvihill, M. M., Izrael-Tomasevic, A., Zobel, K., Jeet, S., et al. (2018). Disruption of XIAP-RIP2 association blocks NOD2-mediated inflammatory signaling. Mol. Cell 69, 551–565.e557. doi: 10.1016/j.molcel.2018.01.016
Granger, G. A., and Kolb, W. P. (1968). Lymphocyte in vitro cytotoxicity: mechanisms of immune and non-immune small lymphocyte mediated target L cell destruction. J. Immunol. 101, 111–120.
Gupta, N., Brown, K. E., and Milam, A. H. (2003). Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp. Eye Res. 76, 463–471. doi: 10.1016/s0014-4835(02)00332-9
Hammond, T. R., Dufort, C., Dissing-Olesen, L., Giera, S., Young, A., Wysoker, A., et al. (2019). Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50, 253–271.e256. doi: 10.1016/j.immuni.2018.11.004
Han, J., Wang, M., Ren, M., and Lou, H. (2017). Contributions of triggering-receptor-expressed-on-myeloid-cells-2 to neurological diseases. Int. J. Neurosci. 127, 368–375. doi: 10.1080/00207454.2016.1264072
Handa, J. T., Bowes Rickman, C., Dick, A. D., Gorin, M. B., Miller, J. W., Toth, C. A., et al. (2019). A systems biology approach towards understanding and treating non-neovascular age-related macular degeneration. Nat. Commun. 10, 3347. doi: 10.1038/s41467-019-11262-1
Hanus, J., Anderson, C., and Wang, S. (2015). RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 24(Pt B), 286–298. doi: 10.1016/j.arr.2015.09.002
Hartong, D. T., Berson, E. L., and Dryja, T. P. (2006). Retinitis pigmentosa. Lancet 368, 1795–1809. doi: 10.1016/s0140-6736(06)69740-7
He, C., Liu, Y., Huang, Z., Yang, Z., Zhou, T., Liu, S., et al. (2021). A specific RIP3(+) subpopulation of microglia promotes retinopathy through a hypoxia-triggered necroptotic mechanism. Proc. Natl. Acad. Sci. U. S. A. 118, 1–10. doi: 10.1073/pnas.2023290118
He, S., Liang, Y., Shao, F., and Wang, X. (2011). Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 20054–20059. doi: 10.1073/pnas.1116302108
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer's disease. Lancet Neurol. 14, 388–405. doi: 10.1016/s1474-4422(15)70016-5
Hickman, S., Izzy, S., Sen, P., Morsett, L., and El Khoury, J. (2018). Microglia in neurodegeneration. Nat. Neurosci. 21, 1359–1369. doi: 10.1038/s41593-018-0242-x
Hickman, S. E., and El Khoury, J. (2014). TREM2 and the neuroimmunology of Alzheimer's disease. Biochem. Pharmacol. 88, 495–498. doi: 10.1016/j.bcp.2013.11.021
Hisatomi, T., Sakamoto, T., Murata, T., Yamanaka, I., Oshima, Y., Hata, Y., et al. (2001). Relocalization of apoptosis-inducing factor in photoreceptor apoptosis induced by retinal detachment in vivo. Am. J. Pathol. 158, 1271–1278. doi: 10.1016/s0002-9440(10)64078-3
Hobbs, S. D., and Pierce, K. (2021). “Wet age-related macular degeneration (Wet AMD),” in StatPearls (Treasure Island, FL: StatPearls Publishing).
Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., et al. (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495. doi: 10.1038/82732
Huang, Z., Wu, S. Q., Liang, Y., Zhou, X., Chen, W., Li, L., et al. (2015). RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 17, 229–242. doi: 10.1016/j.chom.2015.01.002
Huang, Z., Zhou, T., Sun, X., Zheng, Y., Cheng, B., Li, M., et al. (2018). Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 25, 180–189. doi: 10.1038/cdd.2017.141
Husnjak, K., and Dikic, I. (2012). Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 81, 291–322. doi: 10.1146/annurev-biochem-051810-094654
Ito, H., and Hamerman, J. A. (2012). TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. Eur. J. Immunol. 42, 176–185. doi: 10.1002/eji.201141679
Ito, Y., Ofengeim, D., Najafov, A., Das, S., Saberi, S., Li, Y., et al. (2016). RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608. doi: 10.1126/science.aaf6803
Jensen, S., Seidelin, J. B., LaCasse, E. C., and Nielsen, O. H. (2020). SMAC mimetics and RIPK inhibitors as therapeutics for chronic inflammatory diseases. Sci. Signal. 13. doi: 10.1126/scisignal.aax8295
Jha, N. K., Jha, S. K., Kar, R., Nand, P., Swati, K., and Goswami, V. K. (2019). Nuclear factor-kappa β as a therapeutic target for Alzheimer's disease. J. Neurochem. 150, 113–137. doi: 10.1111/jnc.14687
Kaczmarek, A., Vandenabeele, P., and Krysko, D. V. (2013). Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223. doi: 10.1016/j.immuni.2013.02.003
Kaiser, W. J., and Offermann, M. K. (2005). Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 174, 4942–4952. doi: 10.4049/jimmunol.174.8.4942
Kakavand, K., Jobling, A. I., Greferath, U., Vessey, K. A., de Iongh, R. U., and Fletcher, E. L. (2020). Photoreceptor degeneration in Pro23His transgenic rats (line 3) involves autophagic and necroptotic mechanisms. Front. Neurosci. 14, 581579. doi: 10.3389/fnins.2020.581579
Kalkman, H. O., and Feuerbach, D. (2016). Antidepressant therapies inhibit inflammation and microglial M1-polarization. Pharmacol. Ther. 163, 82–93. doi: 10.1016/j.pharmthera.2016.04.001
Kataoka, K., Matsumoto, H., Kaneko, H., Notomi, S., Takeuchi, K., Sweigard, J. H., et al. (2015). Macrophage- and RIP3-dependent inflammasome activation exacerbates retinal detachment-induced photoreceptor cell death. Cell Death Dis. 6, e1731. doi: 10.1038/cddis.2015.73
Kauppinen, A., Paterno, J. J., Blasiak, J., Salminen, A., and Kaarniranta, K. (2016). Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 73, 1765–1786. doi: 10.1007/s00018-016-2147-8
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384. doi: 10.1038/ni.1863
Kayama, M., Omura, K., Murakami, Y., Reshef, E., Thanos, A., Morizane, Y., et al. (2018). Combined inhibition of apoptosis and necrosis promotes transientneuroprotection of retinal ganglion cells and partial-axon regeneration after optic nerve damage. bioRxiv. [Preprint]. doi: 10.1101/357566
Kigerl, K. A., Gensel, J. C., Ankeny, D. P., Alexander, J. K., Donnelly, D. J., and Popovich, P. G. (2009). Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 29, 13435–13444. doi: 10.1523/jneurosci.3257-09.2009
Killingsworth, M. C., Sarks, J. P., and Sarks, S. H. (1990). Macrophages related to Bruch's membrane in age-related macular degeneration. Eye 4 (Pt 4), 613–621. doi: 10.1038/eye.1990.86
Kim, B., Lee, S., Suvas, S., and Rouse, B. T. (2005). Application of plasmid DNA encoding IL-18 diminishes development of herpetic stromal keratitis by antiangiogenic effects. J. Immunol. 175, 509–516. doi: 10.4049/jimmunol.175.1.509
Kim, J., Kim, C., Kim, T. S., Bang, S. I., Yang, Y., Park, H., et al. (2006). IL-18 enhances thrombospondin-1 production in human gastric cancer via JNK pathway. Biochem. Biophys. Res. Commun. 344, 1284–1289. doi: 10.1016/j.bbrc.2006.04.016
Kim, S. J., and Li, J. (2013). Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 4, e716. doi: 10.1038/cddis.2013.238
Kinsella, S., Fichtner, M., Watters, O., König, H. G., and Prehn, J. H. M. (2018). Increased A20-E3 ubiquitin ligase interactions in bid-deficient glia attenuate TLR3- and TLR4-induced inflammation. J. Neuroinflamm. 15, 130. doi: 10.1186/s12974-018-1143-3
Kinsella, S., König, H. G., and Prehn, J. H. (2016). Bid promotes K63-linked polyubiquitination of tumor necrosis factor receptor associated factor 6 (TRAF6) and sensitizes to mutant SOD1-induced proinflammatory signaling in microglia. eNeuro 3. doi: 10.1523/eneuro.0099-15.2016
Komander, D., Clague, M. J., and Urbé, S. (2009). Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563. doi: 10.1038/nrm2731
Kondylis, V., Kumari, S., Vlantis, K., and Pasparakis, M. (2017). The interplay of IKK, NF-κB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol. Rev. 277, 113–127. doi: 10.1111/imr.12550
Lad, E. M., Cousins, S. W., Van Arnam, J. S., and Proia, A. D. (2015). Abundance of infiltrating CD163+ cells in the retina of postmortem eyes with dry and neovascular age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 253, 1941–1945. doi: 10.1007/s00417-015-3094-z
Lee, Y. S., Dayma, Y., Park, M. Y., Kim, K. I., Yoo, S. E., and Kim, E. (2013). Daxx is a key downstream component of receptor interacting protein kinase 3 mediating retinal ischemic cell death. FEBS Lett. 587, 266–271. doi: 10.1016/j.febslet.2012.12.004
Lewis, G. P., Talaga, K. C., Linberg, K. A., Avery, R. L., and Fisher, S. K. (2004). The efficacy of delayed oxygen therapy in the treatment of experimental retinal detachment. Am. J. Ophthalmol. 137, 1085–1095. doi: 10.1016/j.ajo.2004.01.045
Liao, L., Shang, L., Li, N., Wang, S., Wang, M., Huang, Y., et al. (2017). Mixed lineage kinase domain-like protein induces RGC-5 necroptosis following elevated hydrostatic pressure. Acta Biochim. Biophys. Sin. 49, 879–889. doi: 10.1093/abbs/gmx088
Liu, T., Zhang, L., Joo, D., and Sun, S. C. (2017). NF-κB signaling in inflammation. Signal. Transduct. Target Ther. 2, 17023. doi: 10.1038/sigtrans.2017.23
Lloyd, A. F., Davies, C. L., Holloway, R. K., Labrak, Y., Ireland, G., Carradori, D., et al. (2019). Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat. Neurosci. 22, 1046–1052. doi: 10.1038/s41593-019-0418-z
Lu, J., Sun, Z., Fang, Y., Zheng, J., Xu, S., Xu, W., et al. (2019). Melatonin suppresses microglial necroptosis by regulating deubiquitinating enzyme A20 after intracerebral hemorrhage. Front. Immunol. 10, 1360. doi: 10.3389/fimmu.2019.01360
Malsy, J., Alvarado, A. C., Lamontagne, J. O., Strittmatter, K., and Marneros, A. G. (2020). Distinct effects of complement and of NLRP3- and non-NLRP3 inflammasomes for choroidal neovascularization. Elife 9. doi: 10.7554/eLife.60194
Marneros, A. G. (2021). Role of inflammasome activation in neovascular age-related macular degeneration. FEBS J. doi: 10.1111/febs.16278
Martinez, F., and Gordon, S. (2014). Martinez, FO & Gordon, S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 13. doi: 10.12703/P6-13
Masuda, T., Sankowski, R., Staszewski, O., Böttcher, C., Amann, L., Sagar, et al. (2019). Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392. doi: 10.1038/s41586-019-0924-x
Menon, M., Mohammadi, S., Davila-Velderrain, J., Goods, B. A., Cadwell, T. D., Xing, Y., et al. (2019). Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration. Nat. Commun. 10, 4902. doi: 10.1038/s41467-019-12780-8
Mifflin, L., Ofengeim, D., and Yuan, J. (2020). Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 19, 553–571. doi: 10.1038/s41573-020-0071-y
Miller, J. W., D'Anieri, L. L., Husain, D., Miller, J. B., and Vavvas, D. G. (2021). Age-related macular degeneration (AMD): a view to the future. J. Clin. Med. 10, 1124–1127. doi: 10.3390/jcm10051124
Mohebiany, A. N., Ramphal, N. S., Karram, K., Di Liberto, G., Novkovic, T., Klein, M., et al. (2020). Microglial A20 protects the brain from CD8 T-cell-mediated immunopathology. Cell Rep. 30, 1585–1597.e1586. doi: 10.1016/j.celrep.2019.12.097
Murakami, Y., Ikeda, Y., Nakatake, S., Tachibana, T., Fujiwara, K., Yoshida, N., et al. (2015). Necrotic enlargement of cone photoreceptor cells and the release of high-mobility group box-1 in retinitis pigmentosa. Cell Death Discov. 1, 15058. doi: 10.1038/cddiscovery.2015.58
Murakami, Y., Matsumoto, H., Roh, M., Giani, A., Kataoka, K., Morizane, Y., et al. (2014). Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 21, 270–277. doi: 10.1038/cdd.2013.109
Murakami, Y., Matsumoto, H., Roh, M., Suzuki, J., Hisatomi, T., Ikeda, Y., et al. (2012). Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc. Natl. Acad. Sci. U.S.A. 109, 14598–14603. doi: 10.1073/pnas.1206937109
Murphy, J. M., Czabotar, P. E., Hildebrand, J. M., Lucet, I. S., Zhang, J. G., Alvarez-Diaz, S., et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. doi: 10.1016/j.immuni.2013.06.018
Nakazawa, T., Hisatomi, T., Nakazawa, C., Noda, K., Maruyama, K., She, H., et al. (2007). Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc. Natl. Acad. Sci. U.S.A. 104, 2425–2430. doi: 10.1073/pnas.0608167104
Nakazawa, T., Matsubara, A., Noda, K., Hisatomi, T., She, H., Skondra, D., et al. (2006a). Characterization of cytokine responses to retinal detachment in rats. Mol. Vis. 12, 867–878. doi: 10.1017/S0952523806230219
Nakazawa, T., Nakazawa, C., Matsubara, A., Noda, K., Hisatomi, T., She, H., et al. (2006b). Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J. Neurosci. 26, 12633–12641. doi: 10.1523/jneurosci.2801-06.2006
Ndoja, A., Reja, R., Lee, S. H., Webster, J. D., Ngu, H., Rose, C. M., et al. (2020). Ubiquitin ligase COP1 suppresses neuroinflammation by degrading c/EBPβ in microglia. Cell 182, 1156–1169.e1112. doi: 10.1016/j.cell.2020.07.011
Neufeld, A. H. (1999). Microglia in the optic nerve head and the region of parapapillary chorioretinal atrophy in glaucoma. Arch. Ophthalmol. 117, 1050–1056. doi: 10.1001/archopht.117.8.1050
Newton, F., and Megaw, R. (2020). Mechanisms of photoreceptor death in retinitis pigmentosa. Genes. 11, 1120–1148. doi: 10.3390/genes11101120
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Oberst, A., Dillon, C. P., Weinlich, R., McCormick, L. L., Fitzgerald, P., Pop, C., et al. (2011). Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367. doi: 10.1038/nature09852
Ofengeim, D., Ito, Y., Najafov, A., Zhang, Y., Shan, B., DeWitt, J. P., et al. (2015). Activation of necroptosis in multiple sclerosis. Cell Rep. 10, 1836–1849. doi: 10.1016/j.celrep.2015.02.051
Ofengeim, D., Mazzitelli, S., Ito, Y., DeWitt, J. P., Mifflin, L., Zou, C., et al. (2017). RIPK1 mediates a disease-associated microglial response in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 114, E8788–e8797. doi: 10.1073/pnas.1714175114
Ofengeim, D., and Yuan, J. (2013). Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 14, 727–736. doi: 10.1038/nrm3683
Olivares-González, L., Martínez-Fernández de la Cámara, C., Hervás, D., Millán, J. M., and Rodrigo, R. (2018). HIF-1α stabilization reduces retinal degeneration in a mouse model of retinitis pigmentosa. FASEB J. 32, 2438–2451. doi: 10.1096/fj.201700985R
Olivares-González, L., Velasco, S., Campillo, I., and Rodrigo, R. (2021). Retinal inflammation, cell death and inherited retinal dystrophies. Int. J. Mol. Sci. 22, 2096–2113. doi: 10.3390/ijms22042096
Onizawa, M., Oshima, S., Schulze-Topphoff, U., Oses-Prieto, J. A., Lu, T., Tavares, R., et al. (2015). The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 16, 618–627. doi: 10.1038/ni.3172
Paloneva, J., Manninen, T., Christman, G., Hovanes, K., Mandelin, J., Adolfsson, R., et al. (2002). Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662. doi: 10.1086/342259
Pan, C., Banerjee, K., Lehmann, G. L., Almeida, D., Hajjar, K. A., Benedicto, I., et al. (2021). Lipofuscin causes atypical necroptosis through lysosomal membrane permeabilization. Proc. Natl. Acad. Sci. U.S.A. 118. doi: 10.1073/pnas.2100122118
Park, H., Byun, D., Kim, T. S., Kim, Y. I., Kang, J. S., Hahm, E. S., et al. (2001). Enhanced IL-18 expression in common skin tumors. Immunol. Lett. 79, 215–219. doi: 10.1016/s0165-2478(01)00278-4
Park, H. J., Oh, S. H., Kim, H. N., Jung, Y. J., and Lee, P. H. (2016). Mesenchymal stem cells enhance α-synuclein clearance via M2 microglia polarization in experimental and human parkinsonian disorder. Acta Neuropathol. 132, 685–701. doi: 10.1007/s00401-016-1605-6
Penfold, P. L., Killingsworth, M. C., and Sarks, S. H. (1985). Senile macular degeneration: the involvement of immunocompetent cells. Graefes Arch. Clin. Exp. Ophthalmol. 223, 69–76. doi: 10.1007/bf02150948
Petrie, E. J., Czabotar, P. E., and Murphy, J. M. (2019). The structural basis of necroptotic cell death signaling. Trends Biochem. Sci. 44, 53–63. doi: 10.1016/j.tibs.2018.11.002
Petrie, E. J., Sandow, J. J., Jacobsen, A. V., Smith, B. J., Griffin, M. D. W., Lucet, I. S., et al. (2018). Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat. Commun. 9, 2422. doi: 10.1038/s41467-018-04714-7
Pollard, J. W. (2009). Trophic macrophages in development and disease. Nat. Rev. Immunol. 9, 259–270. doi: 10.1038/nri2528
Qiao, H., Sonoda, K. H., Ikeda, Y., Yoshimura, T., Hijioka, K., Jo, Y. J., et al. (2007). Interleukin-18 regulates pathological intraocular neovascularization. J. Leukoc. Biol. 81, 1012–1021. doi: 10.1189/jlb.0506342
Quarato, G., Guy, C. S., Grace, C. R., Llambi, F., Nourse, A., Rodriguez, D. A., et al. (2016). Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol. Cell 61, 589–601. doi: 10.1016/j.molcel.2016.01.011
Quigley, H. A., and Broman, A. T. (2006). The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 90, 262–267. doi: 10.1136/bjo.2005.081224
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Rock, K. L., and Kono, H. (2008). The inflammatory response to cell death. Annu. Rev. Pathol. 3, 99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456
Rodríguez-Gómez, J. A., Kavanagh, E., Engskog-Vlachos, P., Engskog, M. K. R., Herrera, A. J., Espinosa-Oliva, A. M., et al. (2020). Microglia: agents of the CNS pro-inflammatory response. Cells. 9, 1717–1762. doi: 10.3390/cells9071717
Rosenbaum, D. M., Degterev, A., David, J., Rosenbaum, P. S., Roth, S., Grotta, J. C., et al. (2010). Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J. Neurosci. Res. 88, 1569–1576. doi: 10.1002/jnr.22314
Safaiyan, S., Kannaiyan, N., Snaidero, N., Brioschi, S., Biber, K., Yona, S., et al. (2016). Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 19, 995–998. doi: 10.1038/nn.4325
Salvadores, N., Moreno-Gonzalez, I., Ruiz, N. G., Quiroz, G., Vegas, L., Escandón, M., et al. (2021). Aβ oligomers trigger necroptosis-mediated neurodegeneration via microglia activation in Alzheimer's disease. bioRxiv. 10, 31–48. doi: 10.1101/2021.08.27.457960
Sato, K., Li, S., Gordon, W. C., He, J., Liou, G. I., Hill, J. M., et al. (2013). Receptor interacting protein kinase-mediated necrosis contributes to cone and rod photoreceptor degeneration in the retina lacking interphotoreceptor retinoid-binding protein. J. Neurosci. 33, 17458–17468. doi: 10.1523/jneurosci.1380-13.2013
Sawada, H., Fukuchi, T., Tanaka, T., and Abe, H. (2010). Tumor necrosis factor-alpha concentrations in the aqueous humor of patients with glaucoma. Invest. Ophthalmol. Vis. Sci. 51, 903–906. doi: 10.1167/iovs.09-4247
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195. doi: 10.1038/nature00858
Sellés-Navarro, I., Villegas-Pérez, M. P., Salvador-Silva, M., Ruiz-Gómez, J. M., and Vidal-Sanz, M. (1996). Retinal ganglion cell death after different transient periods of pressure-induced ischemia and survival intervals. A quantitative in vivo study. Invest. Ophthalmol. Vis. Sci. 37, 2002–2014.
Seo, J., Kim, M. W., Bae, K. H., Lee, S. C., Song, J., and Lee, E. W. (2019). The roles of ubiquitination in extrinsic cell death pathways and its implications for therapeutics. Biochem. Pharmacol. 162, 21–40. doi: 10.1016/j.bcp.2018.11.012
Sizova, O. S., Shinde, V. M., Lenox, A. R., and Gorbatyuk, M. S. (2014). Modulation of cellular signaling pathways in P23H rhodopsin photoreceptors. Cell. Signal. 26, 665–672. doi: 10.1016/j.cellsig.2013.12.008
Smith, W., Assink, J., Klein, R., Mitchell, P., Klaver, C. C., Klein, B. E., et al. (2001). Risk factors for age-related macular degeneration: pooled findings from three continents. Ophthalmology 108, 697–704. doi: 10.1016/s0161-6420(00)00580-7
Stratoulias, V., Venero, J. L., Tremblay, M., and Joseph, B. (2019). Microglial subtypes: diversity within the microglial community. EMBO J. 38, e101997. doi: 10.15252/embj.2019101997
Su, L., Quade, B., Wang, H., Sun, L., Wang, X., and Rizo, J. (2014). A plug release mechanism for membrane permeation by MLKL. Structure 22, 1489–1500. doi: 10.1016/j.str.2014.07.014
Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. doi: 10.1016/j.cell.2011.11.031
Sweigard, J. H., Matsumoto, H., Smith, K. E., Kim, L. A., Paschalis, E. I., Okonuki, Y., et al. (2015). Inhibition of the alternative complement pathway preserves photoreceptors after retinal injury. Sci. Transl. Med. 7, 297ra116. doi: 10.1126/scitranslmed.aab1482
Tan, W., Zou, J., Yoshida, S., Jiang, B., and Zhou, Y. (2020). The role of inflammation in age-related macular degeneration. Int. J. Biol. Sci. 16, 2989–3001. doi: 10.7150/ijbs.49890
Tansey, M. G., and Goldberg, M. S. (2010). Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 37, 510–518. doi: 10.1016/j.nbd.2009.11.004
Tezel, G. (2008). TNF-alpha signaling in glaucomatous neurodegeneration. Prog. Brain Res. 173, 409–421. doi: 10.1016/s0079-6123(08)01128-x
Tezel, G. (2021). Multifactorial pathogenic processes of retinal ganglion cell degeneration in glaucoma towards multi-target strategies for broader treatment effects. Cells. 10, 1372–1398. doi: 10.3390/cells10061372
Thadathil, N., Nicklas, E. H., Mohammed, S., Lewis, T. L. Jr., Richardson, A., and Deepa, S. S. (2021). Necroptosis increases with age in the brain and contributes to age-related neuroinflammation. Geroscience. 43, 2345–2361. doi: 10.1007/s11357-021-00448-5
Thapa, R. J., Nogusa, S., Chen, P., Maki, J. L., Lerro, A., Andrake, M., et al. (2013). Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc. Natl. Acad. Sci. U.S.A. 110, E3109–3118. doi: 10.1073/pnas.1301218110
To, K., Adamian, M., and Berson, E. L. (2004). Histologic study of retinitis pigmentosa due to a mutation in the RP13 gene (PRPC8): comparison with rhodopsin Pro23His, Cys110Arg, and Glu181Lys. Am. J. Ophthalmol. 137, 946–948. doi: 10.1016/j.ajo.2003.10.047
Trichonas, G., Murakami, Y., Thanos, A., Morizane, Y., Kayama, M., Debouck, C. M., et al. (2010). Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc. Natl. Acad. Sci. U.S.A. 107, 21695–21700. doi: 10.1073/pnas.1009179107
Tseng, P. H., Matsuzawa, A., Zhang, W., Mino, T., Vignali, D. A., and Karin, M. (2010). Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat. Immunol. 11, 70–75. doi: 10.1038/ni.1819
Uemura, A., Fruttiger, M., D'Amore, P. A., De Falco, S., Joussen, A. M., Sennlaub, F., et al. (2021). VEGFR1 signaling in retinal angiogenesis and microinflammation. Prog. Retin. Eye Res. 84, 100954. doi: 10.1016/j.preteyeres.2021.100954
Ueta, T., Ishihara, K., Notomi, S., Lee, J. J., Maidana, D. E., Efstathiou, N. E., et al. (2019). RIP1 kinase mediates angiogenesis by modulating macrophages in experimental neovascularization. Proc. Natl. Acad. Sci. U.S.A. 116, 23705–23713. doi: 10.1073/pnas.1908355116
Upton, J. W., Kaiser, W. J., and Mocarski, E. S. (2012). DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297. doi: 10.1016/j.chom.2012.01.016
Varfolomeev, E., Goncharov, T., Fedorova, A. V., Dynek, J. N., Zobel, K., Deshayes, K., et al. (2008). c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J. Biol. Chem. 283, 24295–24299. doi: 10.1074/jbc.C800128200
Vince, J. E., Wong, W. W., Gentle, I., Lawlor, K. E., Allam, R., O'Reilly, L., et al. (2012). Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36, 215–227. doi: 10.1016/j.immuni.2012.01.012
Viringipurampeer, I. A., Gregory-Evans, C. Y., Metcalfe, A. L., Bashar, E., Moritz, O. L., and Gregory-Evans, K. (2019). Cell death pathways in mutant rhodopsin rat models identifies genotype-specific targets controlling retinal degeneration. Mol. Neurobiol. 56, 1637–1652. doi: 10.1007/s12035-018-1192-8
Viringipurampeer, I. A., Shan, X., Gregory-Evans, K., Zhang, J. P., Mohammadi, Z., and Gregory-Evans, C. Y. (2014). Rip3 knockdown rescues photoreceptor cell death in blind pde6c zebrafish. Cell Death Differ. 21, 665–675. doi: 10.1038/cdd.2013.191
Wallach, D., Kang, T. B., and Kovalenko, A. (2014). Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat. Rev. Immunol. 14, 51–59. doi: 10.1038/nri3561
Walsh, M. C., Kim, G. K., Maurizio, P. L., Molnar, E. E., and Choi, Y. (2008). TRAF6 autoubiquitination-independent activation of the NFkappaB and MAPK pathways in response to IL-1 and RANKL. PLoS ONE 3, e4064. doi: 10.1371/journal.pone.0004064
Wang, J., Zhang, H., Ji, J., Wang, L., Lv, W., He, Y., et al. (2022). A histological study of atherosclerotic characteristics in age-related macular degeneration. Heliyon 8, e08973. doi: 10.1016/j.heliyon.2022.e08973
Wang, M., Wan, H., Wang, S., Liao, L., Huang, Y., Guo, L., et al. (2020). RSK3 mediates necroptosis by regulating phosphorylation of RIP3 in rat retinal ganglion cells. J. Anat. 237, 29–47. doi: 10.1111/joa.13185
Wang, X., Li, Y., Liu, S., Yu, X., Li, L., Shi, C., et al. (2014). Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. U.S.A. 111, 15438–15443. doi: 10.1073/pnas.1412767111
Weinlich, R., Oberst, A., Beere, H. M., and Green, D. R. (2017). Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18, 127–136. doi: 10.1038/nrm.2016.149
Weinreb, R. N., Aung, T., and Medeiros, F. A. (2014). The pathophysiology and treatment of glaucoma: a review. JAMA 311, 1901–1911. doi: 10.1001/jama.2014.3192
Weisel, K., Berger, S., Papp, K., Maari, C., Krueger, J. G., Scott, N., et al. (2020). Response to inhibition of receptor-interacting protein kinase 1 (RIPK1) in active plaque psoriasis: a randomized placebo-controlled study. Clin. Pharmacol. Ther. 108, 808–816. doi: 10.1002/cpt.1852
Weisel, K., Scott, N., Berger, S., Wang, S., Brown, K., Powell, M., et al. (2021). A randomised, placebo-controlled study of RIPK1 inhibitor GSK2982772 in patients with active ulcerative colitis. BMJ Open Gastroenterol. 8. doi: 10.1136/bmjgast-2021-000680
Welser, J. V., Li, L., and Milner, R. (2010). Microglial activation state exerts a biphasic influence on brain endothelial cell proliferation by regulating the balance of TNF and TGF-β1. J. Neuroinflamm. 7, 89. doi: 10.1186/1742-2094-7-89
Wertz, I. E., and Dixit, V. M. (2010). Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harb. Perspect. Biol. 2, a003350. doi: 10.1101/cshperspect.a003350
Wu, X. N., Yang, Z. H., Wang, X. K., Zhang, Y., Wan, H., Song, Y., et al. (2014). Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 21, 1709–1720. doi: 10.1038/cdd.2014.77
Yan, X., Tezel, G., Wax, M. B., and Edward, D. P. (2000). Matrix metalloproteinases and tumor necrosis factor alpha in glaucomatous optic nerve head. Arch. Ophthalmol. 118, 666–673. doi: 10.1001/archopht.118.5.666
Yanai, H., Ban, T., Wang, Z., Choi, M. K., Kawamura, T., Negishi, H., et al. (2009). HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 462, 99–103. doi: 10.1038/nature08512
Yang, H., Fu, Y., Liu, X., Shahi, P. K., Mavlyutov, T. A., Li, J., et al. (2017). Role of the sigma-1 receptor chaperone in rod and cone photoreceptor degenerations in a mouse model of retinitis pigmentosa. Mol. Neurodegener. 12, 68. doi: 10.1186/s13024-017-0202-z
Yang, J., Zhao, Y., Zhang, L., Fan, H., Qi, C., Zhang, K., et al. (2018). RIPK3/MLKL-mediated neuronal necroptosis modulates the M1/M2 polarization of microglia/macrophages in the ischemic cortex. Cereb. Cortex 28, 2622–2635. doi: 10.1093/cercor/bhy089
Yang, P., Peairs, J. J., Tano, R., Zhang, N., Tyrell, J., and Jaffe, G. J. (2007). Caspase-8-mediated apoptosis in human RPE cells. Invest. Ophthalmol. Vis. Sci. 48, 3341–3349. doi: 10.1167/iovs.06-1340
Yoshida, N., Ikeda, Y., Notomi, S., Ishikawa, K., Murakami, Y., Hisatomi, T., et al. (2013). Clinical evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology 120, 100–105. doi: 10.1016/j.ophtha.2012.07.006
Yoshimura, T., Sonoda, K. H., Sugahara, M., Mochizuki, Y., Enaida, H., Oshima, Y., et al. (2009). Comprehensive analysis of inflammatory immune mediators in vitreoretinal diseases. PLoS ONE 4, e8158. doi: 10.1371/journal.pone.0008158
You, Z., Savitz, S. I., Yang, J., Degterev, A., Yuan, J., Cuny, G. D., et al. (2008). Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 28, 1564–1573. doi: 10.1038/jcbfm.2008.44
Yuan, J., Amin, P., and Ofengeim, D. (2019). Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33. doi: 10.1038/s41583-018-0093-1
Yuan, L., and Neufeld, A. H. (2001). Activated microglia in the human glaucomatous optic nerve head. J. Neurosci. Res. 64, 523–532. doi: 10.1002/jnr.1104
Zacks, D. N., Hänninen, V., Pantcheva, M., Ezra, E., Grosskreutz, C., and Miller, J. W. (2003). Caspase activation in an experimental model of retinal detachment. Invest. Ophthalmol. Vis. Sci. 44, 1262–1267. doi: 10.1167/iovs.02-0492
Zacks, D. N., Zheng, Q. D., Han, Y., Bakhru, R., and Miller, J. W. (2004). FAS-mediated apoptosis and its relation to intrinsic pathway activation in an experimental model of retinal detachment. Invest. Ophthalmol. Vis. Sci. 45, 4563–4569. doi: 10.1167/iovs.04-0598
Zhao, J., Jitkaew, S., Cai, Z., Choksi, S., Li, Q., Luo, J., et al. (2012). Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. U.S.A. 109, 5322–5327. doi: 10.1073/pnas.1200012109
Keywords: necroptosis, neuroinflammation, microglia, retinal degeneration, RIPK
Citation: Tao Y, Murakami Y, Vavvas DG and Sonoda K-H (2022) Necroptosis and Neuroinflammation in Retinal Degeneration. Front. Neurosci. 16:911430. doi: 10.3389/fnins.2022.911430
Received: 02 April 2022; Accepted: 23 May 2022;
Published: 29 June 2022.
Edited by:
Henri Leinonen, University of Eastern Finland, FinlandCopyright © 2022 Tao, Murakami, Vavvas and Sonoda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yusuke Murakami, bXVyYWthbWkueXVzdWtlLjQwN0BtLmt5dXNodS11LmFjLmpw