 Yiping Xiong
Yiping Xiong Duanyang Zhou1
Duanyang Zhou1 Kai Zheng
Kai Zheng Wenchuan Bi
Wenchuan Bi Yun Dong
Yun Dong- 1School of Pharmaceutical Sciences, Health Science Center, Shenzhen University, Shenzhen, China
- 2School of Pharmacy and Food Sciences, Zhuhai College of Science and Technology, Zhuhai, China
Glutamate-induced neuroexcitotoxicity could be related to the pathophysiology of some neurodegenerative diseases including Parkinson’s disease and Alzheimer’s disease. Extracellular ATP exerts a wide variety of functions, such as attenuating Aβ-mediated toxicity, inhibiting N-Methyl-D-Aspartate (NMDA) receptor subunit combinations, and aggravating ischemic brain injury. However, the effect of extracellular ATP on glutamate-induced neuroexcitotoxicity remains largely unknown. Herein, we showed that extracellular ATP prevented the glutamate-induced excitotoxicity via binding to its P2Y1 receptors. We found that excessive glutamate triggered cellular reactive oxygen species (ROS) overproduction and mitochondrial membrane potential damage, which were significantly attenuated by extracellular ATP. Besides, glutamate activated autophagy, as illustrated by the increased protein level of autophagic marker LC3II and decreased level of p62, and glutamate-induced neuroexcitotoxicity could be completely abolished by autophagy inhibitor chloroquine. In addition, we revealed that extracellular ATP activated Erk1/2 signaling to suppress autophagy and to exert its neuroprotective effects, which was further reduced by autophagy agonist rapamycin and the selective Erk1/2 inhibitor PD0325901. Taken together, our findings suggest that extracellular ATP binding to P2Y1 receptors protected against glutamate-induced excitotoxicity via Erk1/2-mediated autophagy inhibition, implying the potential of ATP for the treatment of neurodegenerative disorders.
Introduction
Neuroexcitotoxicity caused by glutamate is related to pathogenesis in a variety of neurodegenerative diseases, including Parkinson’s disease (PD) and Alzheimer’s disease (AD) (Yang et al., 2014; Sabogal-Guáqueta et al., 2019; Chen et al., 2020). Glutamate acts as an excitatory neurotransmitter and exert many functions in mammalian brain, such as learning and memory, via modulating Na+, Ca2+ and K+ influx through its several receptors (Ribeiro et al., 2017). Excessive glutamate release induces the overactivation of glutamate receptors and high levels of Ca2+ entry into neural cells, which in turn triggers excitotoxicity and ultimately neural death. Many in vitro experiments have demonstrated that glutamate at high concentrations induces neural cell death. For instance, glutamate resulted in an increase of mitophagy and high levels of zinc ion in HT-22 cells (Jin et al., 2018), or induced apoptosis in the rat primary cortical neurons (Hu et al., 2013) and SH-SY5Y cells (Xiong et al., 2021). Evidence showed that glutamate induced excitotoxicity by increasing reactive oxygen species (ROS) in cultured primary neurons (Kritis et al., 2015). Therefore, it is of importance to identify a mechanism that exerts neuroprotective effects against glutamate-induced neuroexcitotoxicity.
Adenosine triphosphate (ATP) has been reported to play important roles in the central nervous system (CNS). Massive extracellular release of ATP is involved in the etiopathology of some neurodegenerative disorders. For instance, extracellular ATP activated astrocytes to mediate motor neuron death (Gandelman et al., 2010). Extracellular ATP also contributed to photoreceptor neurodegeneration in rodents and induced intracellular alpha-synuclein to damage lysosome functions (Puthussery and Fletcher, 2009; Gan et al., 2015). However, increasing evidence has showed that extracellular ATP could be neuroprotective. An analogue of ATP, ATP-γ-S-(α, β-CH2), rescued PC12 cells from oxidative stress injury and amyloid beta-induced toxicity (Danino et al., 2015). Besides, extracellular ATP administration protected astrocytes against H2O2-induced oxidative injury (Shinozaki et al., 2005), or activated glutamate receptor to modulate synaptic plasticity in the hippocampus (Yamazaki and Fujii, 2015). Importantly, ATP has recently been demonstrated as an endogenous inhibitor of N-methyl-D-aspartate (NMDA) receptors against glutamate-induced mitochondrial membrane potential disruption in cultured rat hippocampal neurons (Fujikawa et al., 2012). Therefore, these actual results suggest that extracellular ATP acts opposite effects in the CNS: neuroprotection and neurodegeneration. Although ATP has been reported to be neuroprotective against glutamate, the contribution of extracellular ATP on excitotoxicity induced by glutamate and its underlying mechanism in SH-SY5Y neuroblastoma cells still maintain largely unknown.
In the present study, we investigated the potential of extracellular ATP on glutamate-induced excitotoxicity and explored the underlying molecular mechanisms in SH-SY5Y cells. We clearly demonstrated that extracellular ATP suppress autophagy to attenuate glutamate-induced excitotoxicity via Erk1/2 signaling. Our study therefore highlighted that ATP could be as an effective agent in the treatment of some neurodegenerative diseases.
Materials and Methods
Reagents and Antibodies
Extracellular ATP, PD0325901, rapamycin, and chloroquine (CQ) were purchased from MedChemExpress (MCE, San Rafael, CA, United States), and L-glutamate was obtained from Sigma–Aldrich (St. Louis, MO, United States). MRS2500 tetraammonium salt was purchased from Tocris (Bio-techne, Shanghai, China). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin, trypsin and phosphate-buffered saline (PBS) were purchased from Gibco (Thornton, NSW, Australia). The Cell Counting kit (CCK-8) was purchased from Dojindo Laboratory (Kumamoto, Japan). The JC-1 assay kit (5,50,6,60-tetrachloro-1,10,3,30-tetraethyl-imidacarbocyanine iodide) and RIPA lysis buffer were obtained from Beyotime Biotech (China). The ROS assay kit (DCFH-DA) was purchased from Meilun (China). The FITC-labeled Annexin V Apoptosis Detection kit was purchased from BD Biosciences (Canada). The antibodies of p-Erk1/2, Erk1/2, p-Akt, Akt, LC3, SQSTM1/p62, GAPDH, FITC and horseradish peroxidase (HRP)-conjugated goat anti-rabbit/mouse antibody were obtained from Cell Signaling Technology (Danvers, MA, United States). The second antibody conjugated anti-mouse IgG and conjugated anti-rabbit IgG were purchased from Thermo. Anti-P2Y1 receptor antibody was purchased from Alomone Labs (Jerusalem BioPark, Israel).
Cell Culture and Treatment
Human neuroblastoma cell SH-SY5Y cells were purchased from The Global Bioresource Center (ATCC, United States) and were cultured in DMEM with the addition of 10% FBS, 100 U/mL penicillin and 100 U/mL streptomycin at 37°C in a humidified atmosphere of 95% air and 5% CO2 incubator. SH-SY5Y cells were plated at a density of 2 × 104/well in 96-well plates and 2 × 105/well in 6-well plates for 24 h. Thereafter, SH-SY5Y cells were pretreated with either PBS or extracellular ATP followed by a stimulation of 10 mM glutamate for 24 h and experimental analyses, such as cell viability, mitochondrial membrane potential, ROS generation and protein expression, were performed.
Cell Viability
Cell viability was quantified by CCK-8 kit following the manufacturer’s instruction. After SH-SY5Y cells seeded in 96-well plates were treated with extracellular ATP and/or glutamate for 24 h, 10 μL CCK-8 solution was added into each well and incubated at 37°C for 1 h. Thereafter, the absorbance at 450 nm was determined using a microplate reader (BioTek).
Measurement of Intracellular Reactive Oxygen Species
The intracellular levels of reactive oxygen species (ROS) were measured with probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA, ROS assay kit). Briefly, the treated SH-SY5Y cells were washed twice with PBS and incubated in serum-free medium with 10 μM DCFH-DA at 37°C for 30 min. Fluorescent images were captured in six different areas of each well under a fluorescent microscope (AxioVert A1, Zeiss, Germany) and measured using Image-J v1.8.0 software. The percentage of ROS-positive cells was calculated.
Measurement of Mitochondrial Membrane Potential
The level of mitochondrial membrane potential was measured by a commercial cyanine JC-1 assay kit, either as red fluorescent J-aggregates or as green fluorescent J-monomers, as previous (Dong et al., 2020). J-aggregates at higher mitochondrial concentrations reflect higher membrane potential whereas J-monomers at lower mitochondrial concentrations indicate lost membrane potential. Accordingly, fluctuation of mitochondrial membrane potential is represented as the J-aggregate/J-monomer (red/green) fluorescence intensity ratio. The treated SH-SY5Y cells were washed with PBS and then incubated with 1 mL JC-1 solution (5 μM) at 37°C for 30 min. Subsequently, images of the cells were captured in six different areas of each well under the fluorescence microscope. The changes in mitochondrial membrane potential were presented as the red/green fluorescence intensity ratio using Image-J v1.8.0 software.
Immunofluorescence
After each treatment, SH-SY5Y cells were fixed with 4% paraformaldehyde for 15 min. After twice washing, the cells were permeabilized with 0.5% Triton X-100 and blocked with 5% BSA. Cells were incubated with LC3 antibody or SQSTM1/p62 antibody (1:1,000) in blocking solution for 24 h and then incubated with FITC conjugated anti-mouse IgG or with FITC conjugated anti-rabbit IgG (1:2,000) for 2 h. Thereafter, DAPI (1:1,000, Beyotime, China) was incubated with cells in PBS solution for 10 min at room temperature in dark. Samples were observed under an inverted fluorescence microscope (AxioVert A1, Zeiss, Germany). The cells in six different areas of each well were photographed under fluorescent microscope. Images were analyzed with Image-J v1.8.0 software. The percentage of each group was calculated as LC3-positive or p62-positive cell number/total cell number × 100%.
Western Blot
After each treatment, cells were rinsed twice with cold PBS and lysed by RIPA lysis buffer containing proteases inhibitors (1:50, Roche, Germany), phosphatase inhibitors 1 and 2 (1:100, Roche, Germany), phenylmethanesulfonyl fluoride (PMSF, 1:100, Beyotime, China), and nuclease (1:1,000, Biolong Biotech, China) on ice. The collected cell lysates were then vortexed and the insoluble cell debris were removed by centrifugation at 12,000 × g for 10 min at 4°C. The total protein concentrations were measured using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, United States). The equal amount of protein extracts (10 μg) was separated on a 10–15% sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel and transferred onto PVDF membranes (Bio-Rad, Hercules, CA, United States). Membranes were blocked with 5% milk in Tris-buffered saline containing 0.05% (v/v) Tween 20 (TBS-T) at room temperature for 60 min and then incubated with the relevant primary antibodies at 4°C overnight. The antibodies were diluted as follows: p-Erk1/2, Erk1/2, LC3II, p62, Akt and p-Akt and GAPDH in 1:1,000 dilution. Afterward, membranes were incubated with corresponding HRP-conjugated secondary antibodies at a 1:5,000 dilution for 60 min at room temperature. Reactive bands were visualized by the Quantity One automatic imaging analysis system (Bio-Rad, Hercules, CA, United States) using enhanced chemiluminescence ECL (Millipore, Kankakee, IL, United States). Finally, the protein band intensities were calculated using Image-J v1.8.0 software.
Statistical Analysis
Statistical comparisons were performed using one-way analysis of variance (ANOVA) following by a post hoc multiple-comparison Tukey test. All quantified biological data were expressed as means ± standard error of the mean (SEM) of at least three independent experiments. Mean values were considered to be significant at *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Extracellular Adenosine Triphosphate Reduces Glutamate-Mediated Excitotoxicity via Binding to P2Y1 Receptors in SH-5Y5Y Cells
To study the vulnerability of SH-SY5Y cells to glutamate, SH-SY5Y cells were initially exposed to several concentrations of glutamate (2, 4, 6, 8, 10, 12, and 14 mM) for 24 h, and the cell viability was detected using CCK-8 kit. The result showed that glutamate caused a dose-dependent decrease in cell viability as compared to control (Figures 1A,B). Glutamate at 4 mM led to a significant decrease in cell density (P < 0.001). Glutamate of 10 mM could induce almost half SH-SY5Y cell loss and thus was applied in all the following experiments. We then examined whether extracellular ATP exerted neuroprotective effect against glutamate-induced excitotoxicity in SH-SY5Y cells as that in rat hippocampal neurons. SH-SY5Y cells were incubated with 10 mM glutamate in the presence or absence of 5 μM or 10 μM ATP for 24 h, respectively. As shown in Figures 1C,D, extracellular ATP at both concentrations significantly prevented glutamate-induced reduction of cell viability (P < 0.001, compared to the glutamate group), while 10 μM extracellular ATP did not exhibit any cytotoxicity. In addition, extracellular ATP remarkably prevented reduction of EdU incorporation induced by glutamate (Supplementary Figure 1). Importantly, we demonstrated that purinergic P2Y1 receptors of extracellular ATP were expressed in SH-SY5Y cells, and the expression of P2Y1 was not affected by all treatments (Figure 1E). However, MRS2500, the selective antagonist of P2Y1 receptors, completely abolished the neuroprotection of extracellular ATP (Figure 1F). Finally, we tested whether glutamate triggered apoptosis in SH-SY5Y cells and no apoptotic cells were observed (Supplementary Figure 2), in accordance with the previous study (Brasil et al., 2021). Taken together, these results demonstrated that extracellular ATP protects SH-SY5Y cells from glutamate-mediated excitotoxicity via the P2Y1 receptors.

Figure 1. Extracellular adenosine triphosphate (ATP) prevents glutamate-induced excitotoxicity via binding P2Y1 receptors in SH-SY5Y cells. (A) Representative images of the cell morphology after cultured SH-SY5Y cells were treated with various concentrations of glutamate (Glu) for 24 h; the control group (Ctrl) consisted of the untreated cells. Scale bar = 100 μm under 20× magnification. (B) Cell viability was detected using Cell Counting kit (CCK-8). (C) Representative images of the cell morphology after pretreated by ATP (5 and 10 μM) or phosphate-buffered saline (PBS) following by the application of 10 mM glutamate for 24 h. Scale bar = 100 μm under 20× magnification. (D) Cell viability was calculated. (E) The protein expressions of P2Y1 receptor, and the bar chart presented the levels of P2Y1 receptor in all groups. Expression of GAPDH was served as a loading control. (F) CCK-8 kit was performed to evaluated cell viability after the administration of MRS2500, and the changes were shown in histograms as percentage with control. All data represent mean ± SEM from at least three independent experiments. **P < 0.01 and ***P < 0.001 versus the glutamate group or the ATP treated control group. n.s., no significance.
Extracellular Adenosine Triphosphate Reduces Intracellular Reactive Oxygen Species Overproduction and Mitochondrial Membrane Potential Damage
Previous studies have manifested that glutamate induced excessive ROS productions and mitochondrial dysfunction to cause cell death (Atlante et al., 2001; Xu et al., 2012). Accordingly, we analyzed intracellular ROS production induced by glutamate in the presence or absence of extracellular ATP. As shown in Figures 2A,B, ROS production was significantly induced by 10 mM glutamate, which was further prevented by either 5 or 10 μM extracellular ATP. We also monitored the mitochondrial membrane potential by JC-1 staining, and found that the red/green ratio was significantly reduced by 10 mM glutamate compared to the control group (Figures 2C,D). However, administration of extracellular ATP substantially increased the red/green ratio (P < 0.001, compared to the glutamate group), suggesting that extracellular ATP could prevent mitochondrial membrane potential disruption. Therefore, extracellular ATP exerted neuroprotective effects by reducing ROS production and restoring mitochondrial functions.
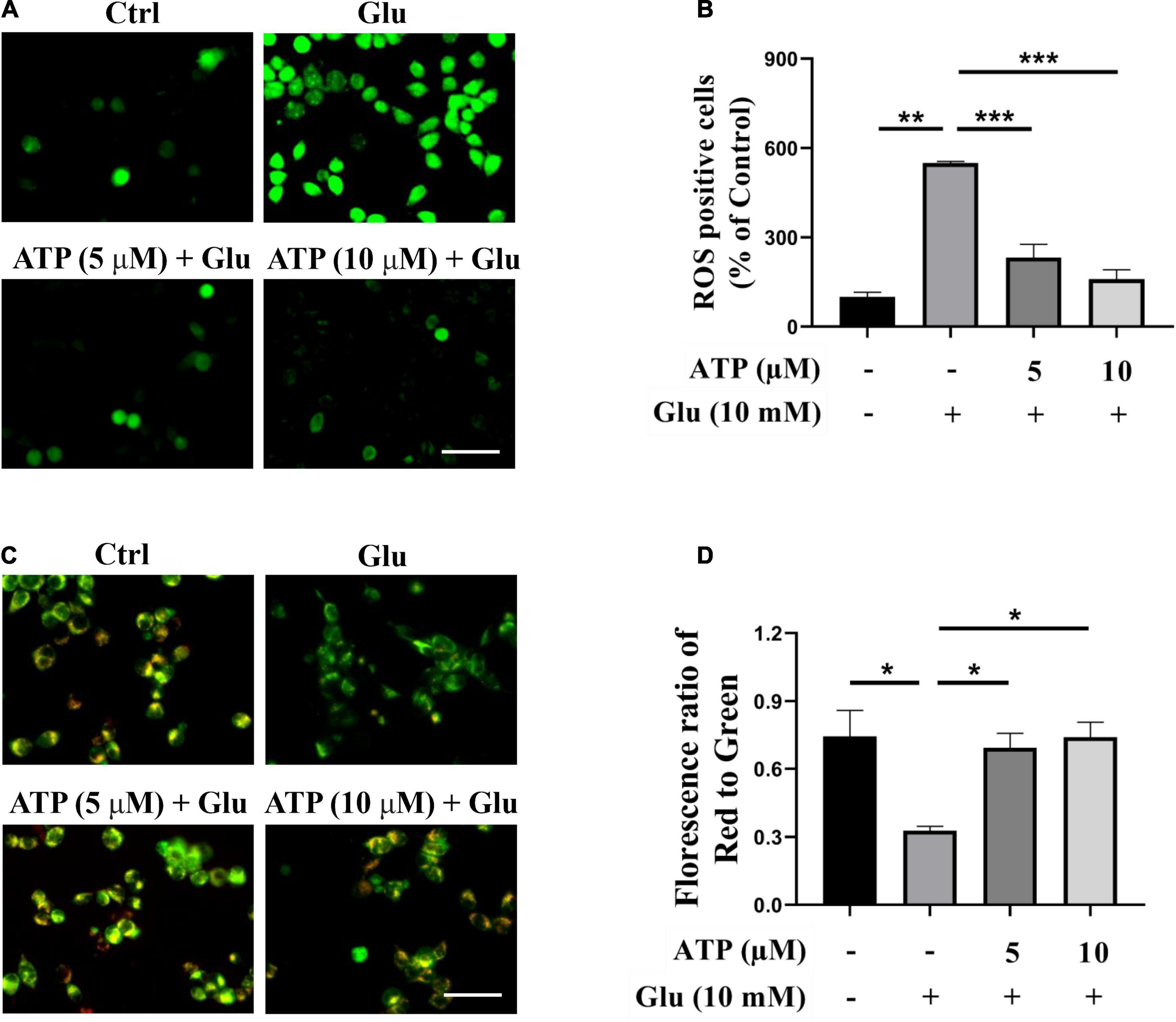
Figure 2. Extracellular ATP reduces reactive oxygen species (ROS) overgeneration and restored mitochondrial membrane potential functions induced by glutamate. Cultured SH-SY5Y cells were incubated in different concentrations (5 and 10 μM) of extracellular ATP, following by the application of 10-mM glutamate for 24 h; the control group consisted of the untreated cells. (A) SH-SY5Y cells were stained with DCFH-DA. Covered glasses with cells were observed under a fluorescent microscope. Scale bar = 100 μm under 20 × magnification. (B) Fluorescent density mean of ROS level was calibrated. (C) JC-1 staining in SH-SY5Y cells. Scale bar = 100 μm under 20× magnification. (D) The ratio of the red/green fluorescence optical density were analyzed by image J. All data in bar charts present mean ± SEM from at least three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the glutamate group or the control group.
Extracellular Adenosine Triphosphate Neuroprotects Against Glutamate-Induced Excitotoxicity via Suppressing Autophagy
There were numerous evidence showing the connections among mitochondrial damage, ROS overproduction and autophagy (Scherz-Shouval and Elazar, 2011; Li et al., 2015). To address whether autophagy was involved in glutamate-induced excitotoxicity in SH-SY5Y cells, the protein level of LC3II, a critical marker of autophagy, and the level of SQSTM1/p62 (p62), a marker for autolysosomal degradation, were analyzed. Apparently, exposure to glutamate significantly increased the level of LC3II compared to the control group (P < 0.001) whereas the expression of p62 was downregulated (Figure 3A), suggesting that glutamate activated autophagy process. Treatment with chloroquine (CQ), an autophagic inhibitor, further upregulated the amount of LC3II and p62, indicating the activation of autophagy by glutamate (Figure 3A). Furthermore, CQ treatments significantly increased cell viability and prevented ROS overproduction induced by glutamate (Figures 3B,C). Administration of CQ also significantly ameliorated the mitochondrial membrane potential disruptions (Figure 3D). Therefore, these above results clearly demonstrated that glutamate activates autophagy to cause excitotoxicity. Importantly, we found that extracellular ATP restored the activated autophagy by glutamate, as evidenced by decreased expression of LC3II and upregulated expression of p62 (Figure 3E). The immunofluorescence results showed glutamate-induced increased LC3II expression and decreased p62 expression were significantly prevented by extracellular ATP (Figures 3F,G). These suggested that ATP not only deceased autophagosome accumulation and suppressed autolysosomal degradation. Consistently, when cells were simultaneously pretreated with extracellular ATP as well as rapamycin, an agonist of autophagy, the protective effects of ATP against glutamate-induced excitotoxicity disappeared (Figures 3H–J and Supplementary Figure 3). CQ did not significantly increase ATP neuroprotection against glutamate-induced excitotoxicity (Supplementary Figure 4). Taken together, these results suggested that extracellular ATP suppresses autophagy to inhibit glutamate-induced excitotoxicity.
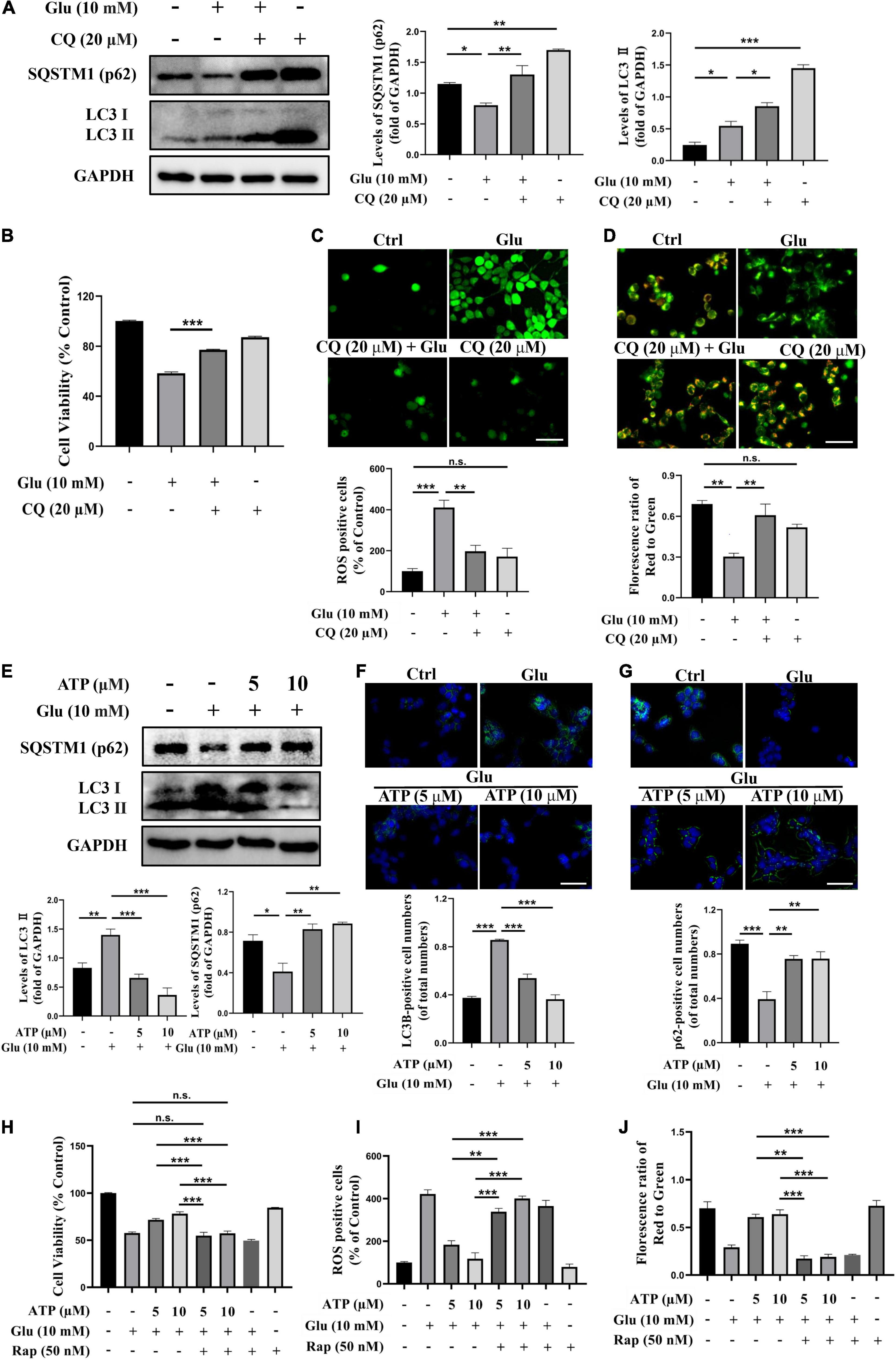
Figure 3. Extracellular ATP prevents glutamate-induced excitotoxicity via suppressing autophagy in SH-SY5Y cells. (A) The protein expression of LC3 and p62 were determined by western blot in SH-SY5Y cells treated with 10-mM Glu in the presence or absence of chloroquine (CQ) (20 μM), and expression of GAPDH was served as loading control. The quantitation of LC3 and p62 expressions were calibrated. (B) Cell viability was evaluated. (C) Treated SH-SY5Y cells were stained with DCFH-DA, and the changes were shown in histograms as percentage with control. Scale bar, 100 μm under 20× magnification. (D) Treated SH-SY5Y cells were stained using JC-1, and the changes were shown in histograms as percentage with control. Scale bar, 100 μm under 20× magnification. (E) The protein expressions of LC3 and p62 were determined by western blot in SH-SY5Y cells treated with 10-mM Glu in the presence of ATP (5 and 10 μM), and expression of GAPDH was served as loading control. The quantitation of LC3II and p62 expressions was calibrated. (F) LC3II and (G) QSTM1/p62 detected using immunocytochemistry were photographed under fluorescent microscope, and images were analyzed. (H) Cell viability was detected. (I) ROS-positive cells were calibrated. (J) Treated SH-SY5Y cells were stained using JC-1, and the changes were presented. All data in bar charts present mean ± SEM from at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus the ATP treated group or the glutamate group. n.s., no significance.
Erk1/2 Signaling Pathway Is Involved in the Neuroprotective Effects of Extracellular Adenosine Triphosphate
Next, we examined the possible roles of several signaling pathways implicated in the ATP-mediated protective response to glutamate-induced neuroexcitotoxicity, such as PI3K/Akt and Erk1/2 signaling pathways (Ali et al., 2018; Chen et al., 2020; Liu et al., 2021). We monitored the protein levels of phosphorylated Erk1/2 (p-Erk1/2) and phosphorylated Akt (p-Akt), and found that the levels of p-Erk1/2 were down-regulated by glutamate in SH-SY5Y cells; whereas the treatments of extracellular ATP significantly increased the expression of p-Erk1/2 (Figure 4A), suggesting that ATP activates Erk1/2 to antagonize glutamate. However, the expression of p-Akt was not affected by extracellular ATP and glutamate (Supplementary Figures 5E,F). To further explore the potential of Erk1/2 signaling in ATP neuroprotection against glutamate-mediate excitotoxicity, PD0325901, a selective Erk1/2 inhibitor, was employed. As shown in Figure 4B, the enhancement of cell viability treated by extracellular ATP against glutamate was totally abolished by PD0325901. In addition, PD0325901 prevented the decrease of ROS generation treated by ATP (Figure 4C), as well as inhibited the ATP-mediated recovery of mitochondrial membrane potential (Figure 4D). Furthermore, PD0325901 prevented the ATP-promoted increase in EdU incorporation (Supplementary Figures 5C,D). Together, these results suggested that the activated Erk1/2 pathway is involved in the neuroprotective effects of extracellular ATP against glutamate-induced excitotoxicity.
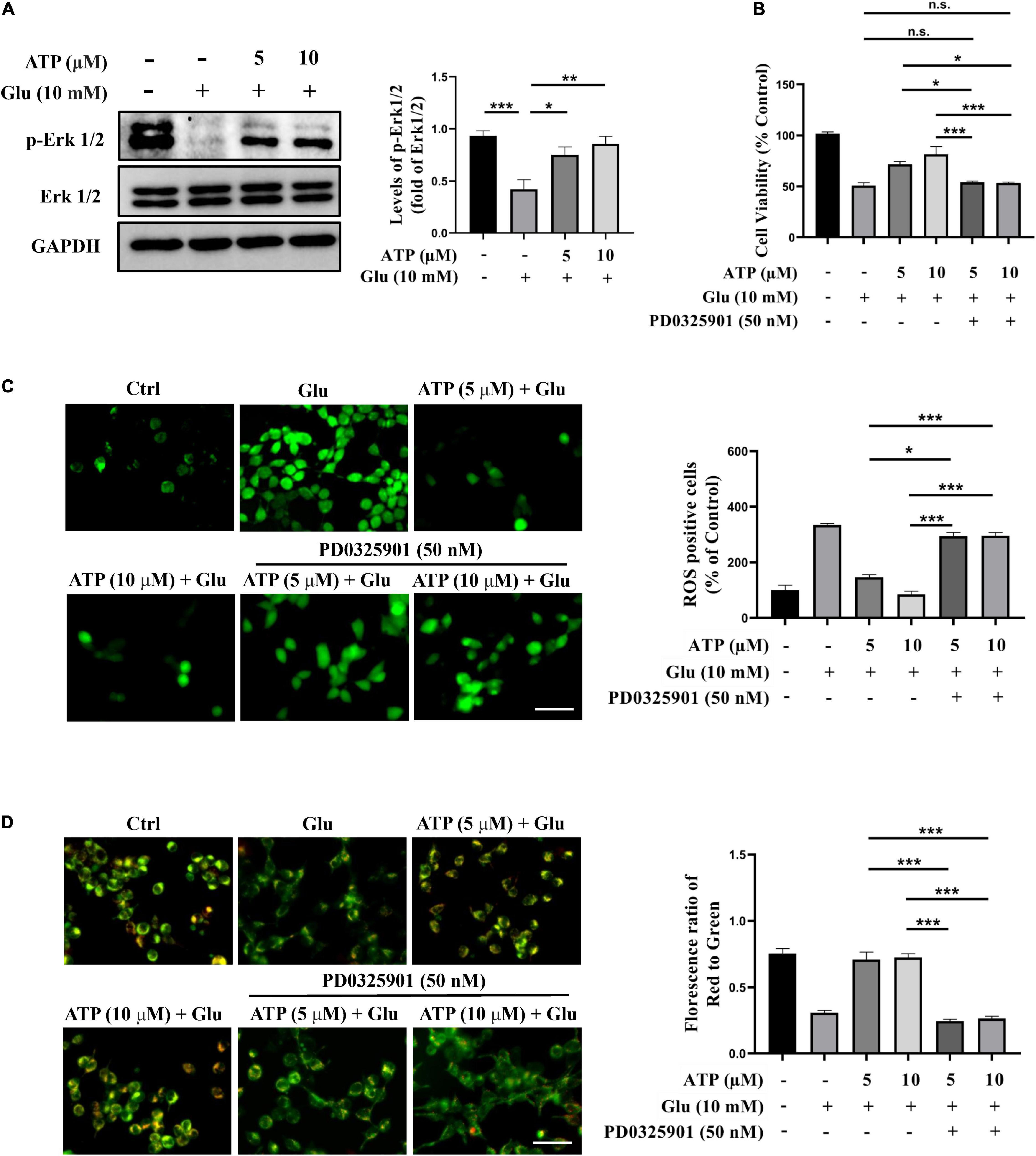
Figure 4. The neuroprotective effects of extracellular ATP against glutamate involves Erk1/2 signaling pathway. Cultured SH-SY5Y cells were treated with 10 mM Glu in the presence or absence of ATP (5 and 10 μM). The control group consists of the untreated cells. (A) The protein expression of p-Erk1/2 and Erk1/2 were determined by western blotting, and the quantitation of p-Erk1/2 and Erk1/2 expressions were calibrated. Expression of GAPDH was served as loading control. (B) CCK-8 kit was performed to evaluated cell viability. (C) All treated SH-SY5Y cells were stained with DCFH-DA, and fluorescent density mean of ROS level was calibrated. Scale bar = 100 μm under 20× magnification. (D) All treated SH-SY5Y cells were stained with JC-1 kit, and the ratio of the red/green fluorescence optical density were analyzed. Scale bar = 100 μm under 20× magnification. All data in bar charts present mean ± SEM from at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus the ATP treated group or the glutamate group. n.s., no significance.
Extracellular Adenosine Triphosphate Suppresses Autophagy via Erk1/2 Signaling Pathway
To further definite the neuroprotective mechanism of extracellular ATP against glutamate, we examined the connection between Erk1/2 and autophagy inhibition. SH-SY5Y cells were treated with glutamate and ATP in the presence or absence of PD0325901, and protein levels of LC3II levels and p62 were analyzed. Apparently, PD0325901 treatment entirely reversed the expression levels of LC3II and p62 mediated by extracellular ATP (Figures 5A–C). However, autophagy inducer rapamycin did not influence the expression of p-Erk1/2 affected by extracellular ATP (Figures 5D,E). Therefore, we concluded that extracellular ATP activates Erk1/2 signaling pathway to inhibit glutamate-mediated activation of autophagy.
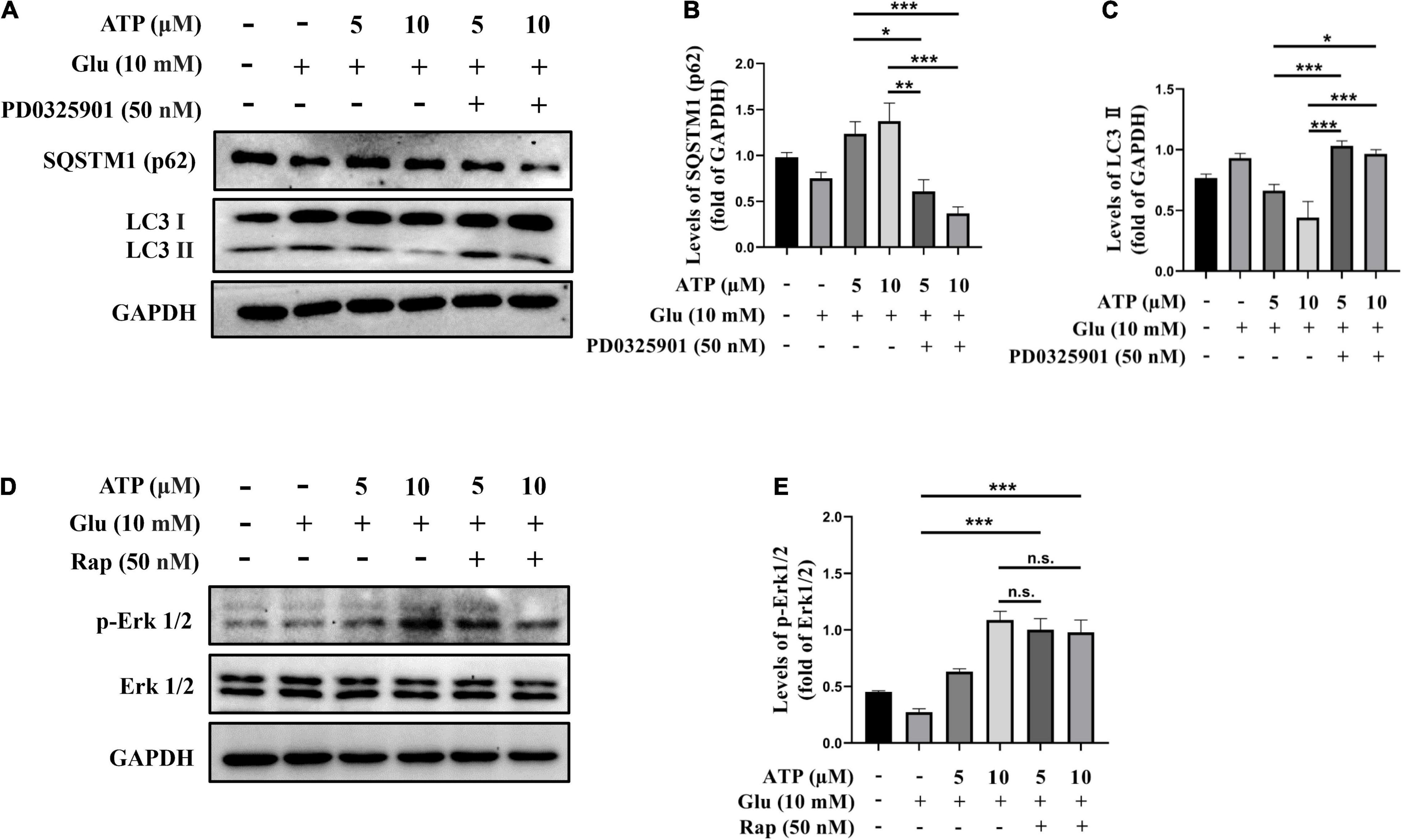
Figure 5. Upregulated p-Erk1/2 by extracellular ATP prevents autophagy induced by glutamate. Cultured SH-SY5Y cells were treated with or without ATP (5 and 10 μM), followed by the application of glutamate (10 mM) in the presence or absence of PD0325901 (50 nM) or rapamycin (50 nM) for 24 h; the untreated cells was the control group. (A) The expressions of LC3 and p62 were determined by western blot. Expression of GAPDH served as loading control. The quantitation of (B) p62 expression and (C) LC3 expression were calibrated. (D) The expressions of p-Erk1/2 and Erk1/2 were determined by western blot. Expression of GAPDH served as loading control. (E) The quantitation of p-Erk1/2 expression was calibrated. All data in bar charts present mean ± SEM from at least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 versus the glutamate group or the ATP treated group. n.s., no significance.
Discussion
Increasing studies have reported that extracellular ATP could be neuroprotective in the CNS. For instance, extracellular ATP could attenuate NMDA-mediated neurotoxicity in cultured hippocampal neurons by inhibiting the heterologous expression of NMDA receptors (Ortinau et al., 2003). Extracellular ATP regulated amyloid precursor protein processing to inhibit Aβ production (Chan et al., 2008). Enhancement of ATP production could ameliorate cognitive dysfunctions in a PD mouse model (Haga et al., 2019). In current study, we demonstrated that extracellular ATP protected SH-SY5Y cells from glutamate-induced excitotoxicity via activating Erk1/2 to suppress lethally autophagy. These studies suggest that extracellular ATP might be able to develop as a therapeutic strategy for neurodegenerative disorders.
Neuroexcitotoxicity caused by high concentration of glutamate is one of pivotal pathological features in neurodegenerative disorders (Hugon et al., 1996; Binvignat and Olloquequi, 2020). Abnormally high concentration of glutamate stimulates the excessive activation of N-methyl-D-aspartate (NMDA) subtype of ionotropic L-glutamate receptors, subsequently results in an excessive Ca2+ influx, which is taken up by mitochondria and subsequently triggers excitatory biochemical processes and death pathways (Rothstein, 1996; Jia et al., 2015). Furthermore, glutamate-mediated excitotoxicity involves ROS overproduction that leads to neuroinflammation and synaptic dysfunctions in rat brain regions (Khan et al., 2021). In the present study, we also demonstrated that glutamate remarkably caused ROS generation in SH-SY5Y cells (Figures 2A,B), which was consistent with previous studies that glutamate caused oxidative damage in SH-SY5Y cells (Sun et al., 2010; Lee et al., 2019). Massive ROS production aggressed mitochondria and led to mitochondrial dysfunctions (Sinha et al., 2013; Angelova and Abramov, 2018). Our previous study has shown that excessive ROS caused the loss of mitochondrial membrane potential and activated mitochondria-dependent cell deficits in HT-22 cells (Dong et al., 2020). Besides, ROS overproduction stimulated by glutamate could result in lipid peroxidation and mitochondrial dysfunctions to the detriment of cellular growth (Lee et al., 2019). Kushnareva et al. (2005) reported that excessive glutamate caused a reduction of mitochondrial ATP and an imbalance of homeostasis that precede commitment to neuronal death. Consistent with previous studies, we observed that exposure to 10 mM glutamate for 24 h indeed induced disruptions of mitochondrial membrane potential in SH-SY5Y cells (Figures 2C,D). Furthermore, we showed that 10 mM glutamate prevented cell proliferation by EdU staining (Supplementary Figure 1). Finally, glutamate induced HT-22 cell ferroptosis mediated by massive ROS production (Jiang et al., 2020). However, our data showed that glutamate could neither affect glutathione peroxidase 4 levels nor glutathione expression, which suggested that glutamate-induced excitotoxicity in SH-SY5Y cells was not related to ferroptosis (data not shown). Flow cytometer analysis showed that glutamate could not induce SH-SY5Y cell apoptosis (Supplementary Figure 2). Characteristics of cell injury in response to glutamate shown in these studies suggest that glutamate-induced excitotoxicity is associated to multiple molecular mechanisms. Together, we confirmed that glutamate induced excessive ROS generation, triggered mitochondrial dysfunctions, and prevented cell proliferation.
Adenosine triphosphate, a well-known intracellular energy currency, is a signaling molecule that influences cell proliferation, differentiation, survival, myelination as well as repairment in the CNS (Fields and Burnstock, 2006; Burnstock, 2007). Extracellular ATP not only counteracted H2O2-promoted cell death in astrocytes (Shinozaki et al., 2006), but also ameliorated cognitive impairments in a mouse model with PD (Haga et al., 2019). Notably, extracellular ATP has been implicated to act two opposite roles when it combines and activates different purine receptors (P2X receptors and P2Y receptors) in ischemic brain injury: pathogenesis and neuroprotection. For instance, P2 × 1, P2 × 2, P2 × 4, P2 × 7 receptors participated in the pathogenesis of cerebral ischemia whereas P2Y1 receptor could induce neuroinflammatory responses to neuroprotect against ischemic neuronal injury (Koizumi et al., 2007; Tu and Wang, 2009; Fukumoto et al., 2019). These studies demonstrated that the activation of different purine receptors could be crucial for extracellular ATP neuroprotection. Our current study elucidated that extracellular ATP protected SH-SY5Y cells from glutamate-induced neuroexcitotoxicity. The statistical results showed that several concentrations of extracellular ATP (2, 5, 10, 20, and 40 μM) could significantly rescue SH-SY5Y cells from 10 mM glutamate-mediated excitotoxicity, including increasing cell viability (Supplementary Figure 1A). The neuroprotective effects of extracellular ATP at 10, 20, and 40 μM were almost similar, and 2-μM ATP could not significantly prevent glutamate-induced cell loss. Therefore, extracellular ATP at 5 and 10 μM were applied the following experiments. The observation further showed that ATP not only inhibited ROS generation but also recovered mitochondrial functions (Figure 2). Importantly, we found that P2Y1 receptors expressed in SH-SY5Y cells, and its selective antagonist, MRS2500, absolutely eliminated neuroprotective effects of extracellular ATP against glutamate (Figures 1E,F), which was supported by a previous study that activated P2Y1 receptors were neuroprotective (Fukumoto et al., 2019). Evidence, on the contrary, demonstrated that P2Y1 receptor blockade could normalized activation of astrocytes and then attenuated cognitive dysfunctions in AD mouse models (Reichenbach et al., 2018), indicating that activation of P2Y1 receptor could mediate neurodegeneration. The opposite roles of P2Y1 receptors in neurodegeneration and neuroprotection might be due to the activation of P2Y1 receptors in different neural cells: neuroprotection in neuronal cells and neurodegeneration in glial cells. Evidence showed that P2Y2 receptor is believed to be neuroprotective under inflammatory conditions (Weisman et al., 2012). To explore whether ATP prevented glutamate-induced neuroexcitotoxicity by its binding to P2Y2 receptors, diquafosol tetrasodium, the agonist of P2Y2 receptor, was applied. Our data showed that diquafosol tetrasodium could not affect glutamate-mediated excitotoxicity in SH-SY5Y cell, indicating that the neuroprotective effects of extracellular ATP were not related to P2Y2 receptor (data not shown). In neurons, both ATP and its secondary metabolite ADP turn to AMP through CD39 or E-NTPDase and then is hydrolyzed into adenosine by CD73 or 5′ nucleotidase. Adenosine is widely valued in the treatment of the CNS diseases (Liu et al., 2019). In order to verify ATP neuroprotective effects, ADP was applied to pretreat SH-SY5Y cells incubated by glutamate, and CCK-8 assay showed that ADP did not rescue SH-SY5Y cells from glutamate-induced excitotoxicity (data not shown). Taken together, we concluded that extracellular ATP binding to its P2Y1 receptors protected from glutamate-induced excitotoxicity in SH-SY5Y cells.
Under normal physiological conditions, autophagy maintains neuronal homeostasis and neuronal survival (Mizushima et al., 2008). Autophagy has been implicated to play a dominant role in neurodegenerative disorders, such as AD and PD. Autophagy could promote the metabolism of Aβ and the assembling of tau, and thus dysfunctions of autophagy might lead to the progress of AD (Zhu et al., 2013; Li et al., 2017). Moreover, autophagy processes could alter PD-associated genes, like SNCA, GBA, ATP13A2, and FBXO7 (Lu et al., 2020). These studies suggested that selective agonists of autophagy might be developed as potential drugs for neurodegenerative disorders. However, excessive autophagy induces oxidative damage, neuroinflammatory injury, and then exacerbates neurodegeneration. Evidence has revealed that high concentrations of glutamate induced autophagy via the activation of the lysosomal Ca2+ channels TPC1 and TPC2 in neural cells (Pereira et al., 2017). In this study, we found that 10 mM glutamate significantly induced an increase of LC3II and a decrease of p62/SQATM1 (Figure 3A), indicating that autophagy could be involved in glutamate-induced excitotoxicity. An inhibitor of autophagy, CQ, significantly suppressed glutamate-induced excitotoxicity, including increasing cell viability, decreasing ROS generation, and recovering mitochondrial functions (Figures 3B–D). Our results demonstrated that glutamate-induced excitotoxicity was mediated by autophagy, which was supported by a previous study (Pereira et al., 2017). Importantly, the treatments of ATP were able to suppress autophagy induced by glutamate (Figure 3 and Supplementary Figure 3). A study demonstrated that knockdown or pharmacological inhibition of purinergic P2Y12 receptors suppressed autophagy process in advanced atherosclerosis (Pi et al., 2021), which supported that extracellular ATP could suppress autophagy processes. Similarly, high concentrations of adenosine, one of ATP metabolites, inhibited autophagy in β cells (Israeli et al., 2018). Opposite to our study, a literature showed that extracellular ATP could be though P2 × 7 receptors to trigger autophagy in primary microglia (Fabbrizio et al., 2017). In summary, these studies implied that extracellular ATP could exert different functions through activating different receptors in a certain condition. In the current study, we confirmed that the neuroprotective effects of ATP against glutamate-induced excitotoxicity was via preventing autophagy.
Multiple mechanisms were involved in the neuroprotective effects of extracellular ATP. For instance, extracellular ATP binding to P2Y1 receptors protected pancreatic duct epithelial cells via cAMP signaling pathway (Seo et al., 2016). Extracellular ATP binding to P2Y13 purinergic receptors could neuroprotect astrocytes via PI3K/Akt/GSK3 pathway (Carrasquero et al., 2009). Also, extracellular ATP attenuated stroke via inhibiting glutamate release to neuroprotection of antioxidants against (Hurtado et al., 2003). Our current study demonstrated extracellular ATP neuroprotected against glutamate-mediated excitotoxicity through Erk1/2 signaling pathway in SH-SY5Y cells (Figure 4). PD0325901, a selective antagonist of Erk1/2 remarkably abolished the neuroprotective effects of extracellular ATP. Consistently, activation of Erk1/2 promoted PC12 cell survival by blocking the expression of the pro-apoptotic factor, BH3-only protein Bim (Terasawa et al., 2009). A previous study showed that activated ATP-dependent potassium channels promoted glioma cell proliferation by upregulated Erk1/2 signaling pathway (Huang et al., 2009), which supported our findings that extracellular ATP pretreatment via Erk1/2 signaling pathway promoted SH-SY5Y cell proliferation (Supplementary Figure 5). Interestingly, extracellular ATP induced the S-phase cell cycle arrest of oral squamous cell carcinoma SAS cells via its binding to P2Y1 receptors-mediated Erk1/2 signaling (Lau et al., 2021). This research showed the antitumor effects of extracellular ATP were through interaction with P2Y1 receptors to promote Erk1/2 signaling, which was opposite to its effects in SH-SY5Y cells shown in the current study. This might be due to extracellular ATP exerted multiple functions in different microenvironments. Notably, our results showed that extracellular ATP though upregulation of p-Erk1/2 expression prevented autophagy against glutamate excitotoxicity (Figure 5). Similarly, Sun et al. (2019) demonstrated that active MEK1 protected against H2O2-induced autophagy though Erk1/2 signaling pathway in neonatal rat cardiac ventricular cardiomyocytes. Evidence, on the contrary, demonstrated that activation of Erk1/2 could contribute to glutamate-induced oxidative injury in primary cortical neurons (Stanciu et al., 2000; Sun et al., 2019). Another study also showed that PD98059, a blockade of Erk1/2 pathway, suppressed H2O2-induced cell death (Bhat and Zhang, 1999). These studies illustrate that Erk1/2 signaling pathway can promote either neuronal survival or neuronal death under different paradigms. One possible explanation is that the magnitude and duration of Erk1/2 activation determines the different downstream effectors exerting the death-promoting capacity and cellular outcomes (Sun et al., 2019).
The current research is only based on non-clinical in vitro experiments, and a combination of in vivo and in vitro experiments is still needed to further explore the role of extracellular ATP as a potential candidate against excitotoxicity.
Conclusion
We clearly demonstrated that extracellular ATP binding to P2Y1 receptors prevented glutamate-mediated neuroexcitotoxicity through activating Erk1/2 to suppress lethally autophagy in SH-SY5Y cells (Supplementary Figure 6). Our present findings provide new insights into the neuroprotective molecular mechanism of extracellular ATP and extend the therapeutic strategy of neurodegenerative diseases.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
YX performed most of the experiments. YD conceived the project and designed the experiments. DZ, KZ, and WB participated in data analysis. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Shenzhen Science and Technology Innovation Committee for Basic Research (JCYJ20210324093610027), Shenzhen Science and Technology Innovation Committee for Basic Research (JCYJ20170818144044 876), and Shenzhen Research Foundation for Advanced Talents (827/000403).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2022.901688/full#supplementary-material
References
Ali, T., Kim, T., Rehman, S. U., Khan, M. S., Amin, F. U., Khan, M., et al. (2018). Natural dietary supplementation of anthocyanins via PI3K/Akt/Nrf2/HO-1 pathways mitigate oxidative stress, neurodegeneration, and memory impairment in a mouse model of Alzheimer’s disease. Mol. Neurobiol. 55, 6076–6093. doi: 10.1007/s12035-017-0798-6
Angelova, P. R., and Abramov, A. Y. (2018). Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. 592, 692–702. doi: 10.1002/1873-3468.12964
Atlante, A., Calissano, P., Bobba, A., Giannattasio, S., Marra, E., and Passarella, S. (2001). Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett. 497, 1–5. doi: 10.1016/s0014-5793(01)02437-1
Bhat, N. R., and Zhang, P. (1999). Hydrogen peroxide activation of multiple mitogen-activated protein kinases in an oligodendrocyte cell line: role of extracellular signal-regulated kinase in hydrogen peroxide-induced cell death. J. Neurochem. 72, 112–119. doi: 10.1046/j.1471-4159.1999.0720112.x
Binvignat, O., and Olloquequi, J. (2020). Excitotoxicity as a target against neurodegenerative processes. Curr. Pharm. Design 26, 1251–1262. doi: 10.2174/1381612826666200113162641
Brasil, F. B., de Almeida, F. J. S., Luckachaki, M. D., Dall’Oglio, E. L., and de Oliveira, M. R. (2021). Astaxanthin prevents mitochondrial impairment in the dopaminergic SH-SY5Y cell line exposed to glutamate-mediated excitotoxicity: role for the Nrf2/HO-1/CO-BR axis. Eur. J. Pharmacol. 908:174336. doi: 10.1016/j.ejphar.2021.174336
Burnstock, G. (2007). Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 87, 659–797. doi: 10.1152/physrev.00043.2006
Carrasquero, L. M., Delicado, E. G., Bustillo, D., Gutiérrez-Martín, Y., Artalejo, A. R., and Miras-Portugal, M. T. (2009). P2X7 and P2Y13 purinergic receptors mediate intracellular calcium responses to BzATP in rat cerebellar astrocytes. J. Neurochem. 110, 879–889. doi: 10.1111/j.1471-4159.2009.06179.x
Chan, S. L., Kim, W. S., Kwok, J. B., Hill, A. F., Cappai, R., Rye, K. A., et al. (2008). ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J. Neurochem. 106, 793–804. doi: 10.1111/j.1471-4159.2008.05433.x
Chen, X., Bao, G., and Liu, F. (2020). Inhibition of USP15 prevent glutamate-induced oxidative damage by activating Nrf2/HO-1 signaling pathway in HT22 cells. Cell. Mol. Neurobiol. 40, 999–1010. doi: 10.1007/s10571-020-00789-3
Danino, O., Grossman, S., and Fischer, B. (2015). ATP-γ-S-(α,β-CH2) protects against oxidative stress and amyloid beta toxicity in neuronal culture. Biochem. Biophys. Res. Commun. 460, 446–450.
Dong, Y., Bi, W., Zheng, K., Zhu, E., Wang, S., Xiong, Y., et al. (2020). Nicotine prevents oxidative stress-induced hippocampal neuronal injury through α7-nAChR/Erk1/2 signaling pathway. Front. Mol. Neurosci. 13:557647. doi: 10.3389/fnmol.2020.557647
Fabbrizio, P., Amadio, S., Apolloni, S., and Volonté, C. (2017). P2X7 receptor activation modulates autophagy in SOD1-G93A mouse microglia. Front. Cell. Neurosci. 11:249. doi: 10.3389/fncel.2017.00249
Fields, R. D., and Burnstock, G. (2006). Purinergic signalling in neuron-glia interactions. Nat. Rev. Neurosci. 7, 423–436. doi: 10.1038/nrn1928
Fujikawa, K., Nakamichi, N., Kato, S., Fukumori, R., Hida, M., Takarada, T., et al. (2012). Delayed mitochondrial membrane potential disruption by ATP in cultured rat hippocampal neurons exposed to N-methyl-D-aspartate. J. Pharmacol. Sci. 119, 20–29. doi: 10.1254/jphs.12034fp
Fukumoto, Y., Tanaka, K. F., Parajuli, B., Shibata, K., Yoshioka, H., Kanemaru, K., et al. (2019). Neuroprotective effects of microglial P2Y(1) receptors against ischemic neuronal injury. J. Cereb. Blood Flow Metab. 39, 2144–2156. doi: 10.1177/0271678X18805317
Gan, M., Moussaud, S., Jiang, P., and McLean, P. J. (2015). Extracellular ATP induces intracellular alpha-synuclein accumulation via P2X1 receptor-mediated lysosomal dysfunction. Neurobiol. Aging 36, 1209–1220. doi: 10.1016/j.neurobiolaging.2014.10.037
Gandelman, M., Peluffo, H., Beckman, J. S., Cassina, P., and Barbeito, L. (2010). Extracellular ATP and the P2X7 receptor in astrocyte-mediated motor neuron death: implications for amyotrophic lateral sclerosis. J. Neuroinflammation 7:33. doi: 10.1186/1742-2094-7-33
Haga, H., Matsuo, K., Yabuki, Y., Zhang, C., Han, F., and Fukunaga, K. (2019). Enhancement of ATP production ameliorates motor and cognitive impairments in a mouse model of MPTP-induced Parkinson’s disease. Neurochem. Int. 129:104492. doi: 10.1016/j.neuint.2019.104492
Hu, S., Cui, W., Mak, S., Tang, J., Choi, C., Pang, Y., et al. (2013). Bis(propyl)-cognitin protects against glutamate-induced neuro-excitotoxicity via concurrent regulation of NO, MAPK/ERK and PI3-K/Akt/GSK3β pathways. Neurochem. Int. 62, 468–477. doi: 10.1016/j.neuint.2013.01.022
Huang, L., Li, B., Li, W., Guo, H., and Zou, F. (2009). ATP-sensitive potassium channels control glioma cells proliferation by regulating ERK activity. Carcinogenesis 30, 737–744. doi: 10.1093/carcin/bgp034
Hugon, J., Vallat, J. M., and Dumas, M. (1996). [Role of glutamate and excitotoxicity in neurologic diseases]. Rev. Neurol. 152, 239–248.
Hurtado, O., De Cristóbal, J., Sánchez, V., Lizasoain, I., Cárdenas, A., Pereira, M. P., et al. (2003). Inhibition of glutamate release by delaying ATP fall accounts for neuroprotective effects of antioxidants in experimental stroke. FASEB J. 17, 2082–2084. doi: 10.1096/fj.02-1086fje
Israeli, T., Riahi, Y., Saada, A., Yefet, D., Cerasi, E., Tirosh, B., et al. (2018). Opposing effects of intracellular versus extracellular adenine nucleotides on autophagy: implications for β-cell function. J. Cell Sci. 131:15.
Jia, M., Njapo, S. A., Rastogi, V., and Hedna, V. S. (2015). Taming glutamate excitotoxicity: strategic pathway modulation for neuroprotection. CNS Drugs 29, 153–162. doi: 10.1007/s40263-015-0225-3
Jiang, T., Cheng, H., Su, J., Wang, X., Wang, Q., Chu, J., et al. (2020). Gastrodin protects against glutamate-induced ferroptosis in HT-22 cells through Nrf2/HO-1 signaling pathway. Toxicol. Vitro 62:104715. doi: 10.1016/j.tiv.2019.104715
Jin, M. F., Ni, H., and Li, L. L. (2018). Leptin maintained zinc homeostasis against glutamate-induced excitotoxicity by preventing mitophagy-mediated mitochondrial activation in HT22 hippocampal neuronal cells. Front. Neurol. 9:322. doi: 10.3389/fneur.2018.00322
Khan, I., Saeed, K., Jo, M. G., and Kim, M. O. (2021). 17-β estradiol rescued immature rat brain against glutamate-induced oxidative stress and neurodegeneration via regulating Nrf2/HO-1 and MAP-kinase signaling pathway. Antioxidants (Basel) 10:12. doi: 10.3390/antiox10060892
Koizumi, S., Shigemoto-Mogami, Y., Nasu-Tada, K., Shinozaki, Y., Ohsawa, K., Tsuda, M., et al. (2007). UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 446, 1091–1095. doi: 10.1038/nature05704
Kritis, A. A., Stamoula, E. G., Paniskaki, K. A., and Vavilis, T. D. (2015). Researching glutamate-induced cytotoxicity in different cell lines: a comparative/collective analysis/study. Front. Cell. Neurosci. 9:91. doi: 10.3389/fncel.2015.00091
Kushnareva, Y. E., Wiley, S. E., Ward, M. W., Andreyev, A. Y., and Murphy, A. N. (2005). Excitotoxic injury to mitochondria isolated from cultured neurons. J. Biol. Chem. 280, 28894–28902. doi: 10.1074/jbc.M503090200
Lau, C. C., Aminuddin, A., Chan, K. M., Paterson, I. C., Law, L. M., and Ng, P. Y. (2021). Extracellular ATP induced S-phase cell cycle arrest via P2Y receptor-activated ERK signaling in poorly differentiated oral squamous cell carcinoma SAS cells. Life 11:16. doi: 10.3390/life11111170
Lee, H. J., Spandidos, D. A., Tsatsakis, A., Margina, D., Izotov, B. N., and Yang, S. H. (2019). Neuroprotective effects of Scrophularia buergeriana extract against glutamate-induced toxicity in SH-SY5Y cells. Int. J. Mol. Med. 43, 2144–2152.
Li, L., Tan, J., Miao, Y., Lei, P., and Zhang, Q. (2015). ROS and autophagy: interactions and molecular regulatory mechanisms. Cell. Mol. Neurobiol. 35, 615–621. doi: 10.1007/s10571-015-0166-x
Li, Q., Liu, Y., and Sun, M. (2017). Autophagy and Alzheimer’s disease. Cell. Mol. Neurobiol. 37, 377–388.
Liu, S., Liu, C., Xiong, L., Xie, J., Huang, C., Pi, R., et al. (2021). Icaritin alleviates glutamate-induced neuronal damage by inactivating GluN2B-containing NMDARs through the ERK/DAPK1 pathway. Front. Neurosci. 15:525615. doi: 10.3389/fnins.2021.525615
Liu, Y. J., Chen, J., Li, X., Zhou, X., Hu, Y. M., Chu, S. F., et al. (2019). Research progress on adenosine in central nervous system diseases. CNS Neurosci. Therap. 25, 899–910. doi: 10.1111/cns.13190
Lu, J., Wu, M., and Yue, Z. (2020). Autophagy and Parkinson’s disease. Adv. Exp. Med. Biol. 1207, 21–51.
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Ortinau, S., Laube, B., and Zimmermann, H. (2003). ATP inhibits NMDA receptors after heterologous expression and in cultured hippocampal neurons and attenuates NMDA-mediated neurotoxicity. J. Neurosci. 23, 4996–5003. doi: 10.1523/JNEUROSCI.23-12-04996.2003
Pereira, G. J., Antonioli, M., Hirata, H., Ureshino, R. P., Nascimento, A. R., Bincoletto, C., et al. (2017). Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget 8, 12730–12740. doi: 10.18632/oncotarget.14404
Pi, S., Mao, L., Chen, J., Shi, H., Liu, Y., Guo, X., et al. (2021). The P2RY12 receptor promotes VSMC-derived foam cell formation by inhibiting autophagy in advanced atherosclerosis. Autophagy 17, 980–1000. doi: 10.1080/15548627.2020.1741202
Puthussery, T., and Fletcher, E. (2009). Extracellular ATP induces retinal photoreceptor apoptosis through activation of purinoceptors in rodents. J. Comp. Neurol. 513, 430–440. doi: 10.1002/cne.21964
Reichenbach, N., Delekate, A., Breithausen, B., Keppler, K., Poll, S., Schulte, T., et al. (2018). P2Y1 receptor blockade normalizes network dysfunction and cognition in an Alzheimer’s disease model. J. Exp. Med. 215, 1649–1663. doi: 10.1084/jem.20171487
Ribeiro, F. M., Vieira, L. B., Pires, R. G., Olmo, R. P., and Ferguson, S. S. (2017). Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol. Res. 115, 179–191. doi: 10.1016/j.phrs.2016.11.013
Sabogal-Guáqueta, A. M., Hobbie, F., Keerthi, A., Oun, A., Kortholt, A., Boddeke, E., et al. (2019). Linalool attenuates oxidative stress and mitochondrial dysfunction mediated by glutamate and NMDA toxicity. Biomed. Pharmacother. 118:109295. doi: 10.1016/j.biopha.2019.109295
Scherz-Shouval, R., and Elazar, Z. (2011). Regulation of autophagy by ROS: physiology and pathology. Trends Biochem. Sci. 36, 30–38. doi: 10.1016/j.tibs.2010.07.007
Seo, J. B., Jung, S. R., Hille, B., and Koh, D. S. (2016). Extracellular ATP protects pancreatic duct epithelial cells from alcohol-induced damage through P2Y1 receptor-cAMP signal pathway. Cell Biol. Toxicol. 32, 229–247. doi: 10.1007/s10565-016-9331-3
Shinozaki, Y., Koizumi, S., Ishida, S., Sawada, J., Ohno, Y., and Inoue, K. (2005). Cytoprotection against oxidative stress-induced damage of astrocytes by extracellular ATP via P2Y1 receptors. Glia 49, 288–300. doi: 10.1002/glia.20118
Shinozaki, Y., Koizumi, S., Ohno, Y., Nagao, T., and Inoue, K. (2006). Extracellular ATP counteracts the ERK1/2-mediated death-promoting signaling cascades in astrocytes. Glia 54, 606–618. doi: 10.1002/glia.20408
Sinha, K., Das, J., Pal, P. B., and Sil, P. C. (2013). Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 87, 1157–1180. doi: 10.1007/s00204-013-1034-4
Stanciu, M., Wang, Y., Kentor, R., Burke, N., Watkins, S., Kress, G., et al. (2000). Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 275, 12200–12206. doi: 10.1074/jbc.275.16.12200
Sun, M. H., Chen, X. C., Han, M., Yang, Y. N., Gao, X. M., Ma, X., et al. (2019). Cardioprotective effects of constitutively active MEK1 against H(2)O(2)-induced apoptosis and autophagy in cardiomyocytes via the ERK1/2 signaling pathway. Biochem. Biophys. Res. Commun. 512, 125–130. doi: 10.1016/j.bbrc.2019.03.008
Sun, Z. W., Zhang, L., Zhu, S. J., Chen, W. C., and Mei, B. (2010). Excitotoxicity effects of glutamate on human neuroblastoma SH-SY5Y cells via oxidative damage. Neurosci. Bull. 26, 8–16. doi: 10.1007/s12264-010-0813-7
Terasawa, K., Ichimura, A., Sato, F., Shimizu, K., and Tsujimoto, G. (2009). Sustained activation of ERK1/2 by NGF induces microRNA-221 and 222 in PC12 cells. FEBS J. 276, 3269–3276. doi: 10.1111/j.1742-4658.2009.07041.x
Tu, J., and Wang, L. P. (2009). Therapeutic potential of extracellular ATP and P2 receptors in nervous system diseases. Neurosci. Bull. 25, 27–32. doi: 10.1007/s12264-009-1020-2
Weisman, G. A., Ajit, D., Garrad, R., Peterson, T. S., Woods, L. T., Thebeau, C., et al. (2012). Neuroprotective roles of the P2Y(2) receptor. Purinergic Signal. 8, 559–578. doi: 10.1007/s11302-012-9307-6
Xiong, S., Ma, M., Xu, Y., Wei, F., Gu, Q., He, X., et al. (2021). Protective effects of peptide FK18 against neuro-excitotoxicity in SH-SY5Y cells. Exp. Therap. Med. 21:451. doi: 10.3892/etm.2021.9880
Xu, M. F., Xiong, Y. Y., Liu, J. K., Qian, J. J., Zhu, L., and Gao, J. (2012). Asiatic acid, a pentacyclic triterpene in Centella asiatica, attenuates glutamate-induced cognitive deficits in mice and apoptosis in SH-SY5Y cells. Acta Pharmacol. Sin. 33, 578–587. doi: 10.1038/aps.2012.3
Yamazaki, Y., and Fujii, S. (2015). Extracellular ATP modulates synaptic plasticity induced by activation of metabotropic glutamate receptors in the hippocampus. Biomed. Res. 36, 1–9. doi: 10.2220/biomedres.36.1
Yang, E. J., Kim, G. S., Jun, M., and Song, K. S. (2014). Kaempferol attenuates the glutamate-induced oxidative stress in mouse-derived hippocampal neuronal HT22 cells. Food Funct. 5, 1395–1402. doi: 10.1039/c4fo00068d
Keywords: neuroexcitotoxicity, autophagy, extracellular ATP, neuroprotection, Erk1/2 signaling pathway
Citation: Xiong Y, Zhou D, Zheng K, Bi W and Dong Y (2022) Extracellular Adenosine Triphosphate Binding to P2Y1 Receptors Prevents Glutamate-Induced Excitotoxicity: Involvement of Erk1/2 Signaling Pathway to Suppress Autophagy. Front. Neurosci. 16:901688. doi: 10.3389/fnins.2022.901688
Received: 28 March 2022; Accepted: 13 May 2022;
Published: 07 June 2022.
Edited by:
Abel Santamaria, Manuel Velasco Suárez Instituto Nacional de Neurología y Neurocirugía, MexicoReviewed by:
Yuefan Yang, Fourth Military Medical University, ChinaShao Li, Dalian Medical University, China
Copyright © 2022 Xiong, Zhou, Zheng, Bi and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun Dong, eXVuZG9uZ0BzenUuZWR1LmNu; orcid.org/0000-0002-5658-3896