 Beata Monfort
Beata Monfort Kristian Want
Kristian Want Sylvain Gervason
Sylvain Gervason Benoit D’Autréaux
Benoit D’Autréaux
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 02 March 2022
Sec. Neurodegeneration
Volume 16 - 2022 | https://doi.org/10.3389/fnins.2022.838335
This article is part of the Research TopicBasic and Clinical Mechanisms in Friedreich AtaxiaView all 9 articles
Friedreich’s ataxia (FRDA) is the most prevalent autosomic recessive ataxia and is associated with a severe cardiac hypertrophy and less frequently diabetes. It is caused by mutations in the gene encoding frataxin (FXN), a small mitochondrial protein. The primary consequence is a defective expression of FXN, with basal protein levels decreased by 70–98%, which foremost affects the cerebellum, dorsal root ganglia, heart and liver. FXN is a mitochondrial protein involved in iron metabolism but its exact function has remained elusive and highly debated since its discovery. At the cellular level, FRDA is characterized by a general deficit in the biosynthesis of iron-sulfur (Fe-S) clusters and heme, iron accumulation and deposition in mitochondria, and sensitivity to oxidative stress. Based on these phenotypes and the proposed ability of FXN to bind iron, a role as an iron storage protein providing iron for Fe-S cluster and heme biosynthesis was initially proposed. However, this model was challenged by several other studies and it is now widely accepted that FXN functions primarily in Fe-S cluster biosynthesis, with iron accumulation, heme deficiency and oxidative stress sensitivity appearing later on as secondary defects. Nonetheless, the biochemical function of FXN in Fe-S cluster biosynthesis is still debated. Several roles have been proposed for FXN: iron chaperone, gate-keeper of detrimental Fe-S cluster biosynthesis, sulfide production stimulator and sulfur transfer accelerator. A picture is now emerging which points toward a unique function of FXN as an accelerator of a key step of sulfur transfer between two components of the Fe-S cluster biosynthetic complex. These findings should foster the development of new strategies for the treatment of FRDA. We will review here the latest discoveries on the biochemical function of frataxin and the implication for a potential therapeutic treatment of FRDA.
The severe neurological disease Friedreich’s Ataxia (FRDA) is the most frequent hereditary autosomal recessive ataxia with an incidence of about 30,000 in the Caucasian population (people with European ancestry) and a carrier rate near 1:100 (Delatycki et al., 2000; Synofzik and Németh, 2018). The disease was first described by Dr. Nikolaus Friedreich in the mid-19th century, who documented the pathology of several patients suffering from “degenerative atrophy of spinal posterior column” and heart problems related to hypertrophic cardiomyopathy, which was confirmed by later studies to be the dominant cause of death characterized by left ventricular hypertrophy, smaller left ventricular diastolic diameters and volumes, increased wall thicknesses and arrhythmia (Friedreich, 1863; Tsou et al., 2011). Patients display signs of cerebellar, limb and sensory ataxia with symptoms onset generally during the first two decades of life (Cook and Giunti, 2017; Smith and Kosman, 2020). Diabetes, blindness and deafness are additional phenotypes that appear in the course of the disease but with a different incidence frequency, onset and progression (Parkinson et al., 2013; Lynch et al., 2021).
Friedreich’s ataxia is caused by mutations on chromosome 9, in the gene coding for the frataxin protein (FXN) (Chamberlain et al., 1988; Campuzano et al., 1996). The highest amount of frataxin mRNA is present in the heart, spinal cord, liver, skeletal muscle, and pancreas, which are also the most affected sites in patients with FRDA (Campuzano et al., 1996; Jiralerspong et al., 1997). The vast majority of patients (∼96%) have abnormal GAA-triplet expansions of different length on both alleles of the first intron of the frataxin gene, leading to a drastic decline in the transcription of frataxin mRNA and consequently reduced levels of frataxin (Yandim et al., 2013; Gottesfeld, 2019). About 4% of patients are heterozygotes for a GAA-triplet expansion on one allele and a point mutation or an insertion/deletion in the FXN gene on the other one (Santos et al., 2010; Delatycki and Corben, 2012; Galea et al., 2016). Frataxin levels are also decreased in compound heterozygous patients together with expression of dysfunctional forms of frataxin. The amount of remaining frataxin in FRDA patients ranges from 30 to 2%, depending on the extent of the silencing that is linked to the number of GAA-triplet repeats (Campuzano et al., 1997; Lazaropoulos et al., 2015). The onset and the severity of the neurological features also correlate with the number of GAA-triplet repeats; this is less clear for the non-neurological symptoms (diabetes, cardiomyopathy, scoliosis, and pes cavus) and atypical features (Campuzano et al., 1996; Dürr et al., 1996; Filla et al., 1996; Montermini et al., 1997; Lazaropoulos et al., 2015; Reetz et al., 2016, 2021; Indelicato et al., 2020). Intron 1 of the FXN gene typically contains up to about 30 GAA expansions. The pathological late onset starts at about 40–60 GAA on the shorter allele with a mean value of about 300 GAA. As the number of GAA increases, the onset of the disease occurs earlier, with an average value of approximately 600 GAA and up to 1,700 characterizing early onset. This suggests a direct link between the level of remaining frataxin and the severity of the neurological features (onset and state of health).
Major breakthroughs in exploring frataxin’s function came from studies of the cellular phenotypes of FRDA patient tissues (Rotig et al., 1997) and animal and cellular FRDA models from human (Liu et al., 2011), mouse (Cossee et al., 2000; Puccio et al., 2001; Miranda et al., 2002), drosophila (Calap-Quintana et al., 2018; Russi et al., 2020) and yeast (Babcock et al., 1997; Foury and Cazzalini, 1997; Duby et al., 2002; Muhlenhoff et al., 2002). The first yeast model containing a knockout mutation of Yfh1, the yeast frataxin homolog, resulted in mitochondrial iron accumulation, which led to the conclusion that Yfh1 is involved in mitochondrial iron homeostasis (Babcock et al., 1997; Foury and Cazzalini, 1997). Further observations uncovered that abnormal frataxin production led to a marked decrease in enzymatic activities of mitochondrial and extra-mitochondrial iron-sulfur (Fe-S) cluster enzymes (Foury and Cazzalini, 1997; Rotig et al., 1997; Wilson and Roof, 1997; Puccio et al., 2001; Duby et al., 2002; Muhlenhoff et al., 2002; Lu and Cortopassi, 2007; Martelli et al., 2007). Fe-S clusters are protein cofactors providing catalytic activities to numerous enzymes with essential functions in ATP production (Stiban et al., 2016), Krebs cycle (Switzer, 1989), redox catalysis (Bauerle et al., 2015; Kimura and Suzuki, 2015), protein (Kispal et al., 2005) and DNA (Torrents et al., 2002; Zhang et al., 2011, 2014) synthesis, signaling (Crack et al., 2014) as well as DNA maintenance (Fuss et al., 2015; Shi et al., 2021). Any defect in their biosynthesis thus lead to metabolic defects affecting energy production and many other cellular functions (Beilschmidt and Puccio, 2014; Stehling et al., 2014; Alsina et al., 2018b; Cardenas-Rodriguez et al., 2018). In addition to the Fe-S cluster biosynthesis defect, sensitivity to oxidative stress and decline in enzymatic activities of heme enzymes were also observed (Foury and Cazzalini, 1997; Wilson and Roof, 1997; Lesuisse et al., 2003; Lange et al., 2004; Schoenfeld et al., 2005; Al-Mahdawi et al., 2006; Lu and Cortopassi, 2007). Oxidative stress in FRDA patients is thought to result from the production of reactive oxygen species catalyzed by free iron accumulating in mitochondria (Fenton reaction) and from impaired oxidative stress signaling by the master regulator NRF2 (NF-E2-related factor) allowing reactive oxygen species to accumulate (Alsina et al., 2018a). This array of intricate phenotypes made it difficult to assign the primary function of frataxin, until it became apparent that frataxin is directly involved in the biosynthesis of Fe-S clusters and that the other phenotypes appear consequently to the Fe-S cluster biosynthesis defect (Pastore and Puccio, 2013).
Several therapeutic strategies targeting either the primary or secondary defects of FRDA are under development following two main directions: (1) restoration of FXN levels using gene expression modulators, protein stabilizers and gene therapy and (2) alleviation of secondary cellular defects such as mitochondrial functions, iron accumulation, oxidative stress and ferroptosis (Clay et al., 2019; Zesiewicz et al., 2020; Lynch and Farmer, 2021; Ocana-Santero et al., 2021; Pallardó et al., 2021; Yang et al., 2021). Hereby, pharmacologic approaches including iron-chelators, antioxidants, NRF2 activators, ferroptosis inhibitors and molecules improving mitochondrial functions have reached clinical trials. Promising results were also reported with frataxin expression activators and gene therapy. A third strategy not yet explored would be to replace FXN using FXN-mimics to target the primary consequence of FRDA. However, this strategy is still unexploited as only recently the enzymatic function of frataxin in Fe-S cluster biosynthesis has become clear. This review summarizes and critically analyses the different proposed functions of frataxin in iron metabolism, heme and Fe-S cluster biosynthesis: iron storage, iron chaperone, gate-keeper of detrimental Fe-S cluster biosynthesis over activity, sulfide production stimulator or sulfur transfer accelerator (Srour et al., 2020). The latter is the most established function and could serve as a new direction to test potential therapeutics restoring Fe-S cluster biosynthesis in patients with FRDA.
When the mutations responsible for FRDA were identified in the locus encoding the frataxin protein (FXN), its molecular function was not known (Campuzano et al., 1996). Soon after, orthologs of human FXN were identified in other organisms, such as Yfh1 in yeast and CyaY in bacteria, which has greatly contributed to the elucidation of FXN’s function. The first clue on the molecular function of FXN came from the analysis of the cellular phenotypes in FRDA patient tissues along with animal and cellular models. Among the variety of phenotypes, the most prominent and recurrent ones are the Fe-S cluster and heme deficits, iron accumulation within mitochondria, loss of mitochondrial DNA and a higher sensitivity to oxidative stress (Sanchez-Casis et al., 1976; Babcock et al., 1997; Foury and Cazzalini, 1997; Rotig et al., 1997; Wilson and Roof, 1997; Puccio et al., 2001; Muhlenhoff et al., 2002; Lesuisse et al., 2003; Schoenfeld et al., 2005). However, the severity of these phenotypes differs substantially from one organism to another and depends on the cell-type. For instance, iron accumulation, oxidative stress and heme deficiency are not systematically observed and are tissue-specific (Cossee et al., 2000; Becker et al., 2002; Muhlenhoff et al., 2002; Seznec et al., 2005; Steinkellner et al., 2017). Thereby, no definitive conclusions were drawn on the exact function of frataxin based on these observations. Moreover, it was unclear at the time which cellular phenotypes correspond to either primary or secondary effects due to the lack of FXN.
Nonetheless, these phenotypes were pointing to a key function of FXN in iron metabolism. Fe-S clusters and heme are iron-containing protein cofactors providing enzymatic activities to a multitude of proteins involved in essential biological functions. Among the multifaceted functions of Fe-S proteins are electron transfer for ATP production in mitochondria (Stiban et al., 2016) and DNA synthesis (Zhang et al., 2014), redox catalysis in numerous metabolic pathways by the radical SAM superfamily (Bauerle et al., 2015; Kimura and Suzuki, 2015), non-redox catalysis in the Krebs cycle (Switzer, 1989; Stiban et al., 2016) and protein synthesis by ribosomes (Kispal et al., 2005), iron donation (Zhang et al., 2011), sulfur donation (Lotierzo et al., 2005; Fugate and Jarrett, 2012; Mulliez et al., 2017), signaling (Crack et al., 2014) and maintenance of genome integrity (Fuss et al., 2015; Shi et al., 2021). Heme containing enzymes are also involved in electron transfer for ATP production (Santucci et al., 2019), signaling (Shimizu et al., 2019) and redox catalysis (Flohé, 2020), but also oxygen transport (Sen Gupta, 2019), xenobiotic detoxification (Esteves et al., 2021) and oxidative stress defense (Fugate and Jarrett, 2012; Aratani, 2018). It is interesting to note that in yeast strains deleted for the fxn gene, FXN is totally absent but Fe-S clusters and hemes are still produced (Babcock et al., 1997; Duby et al., 2002; Muhlenhoff et al., 2002; Lange et al., 2004). This suggests that FXN is a regulator rather than an obligatory enzyme.
The presence of granular iron deposits in the mitochondria of cardiomyocytes was one of the first findings of iron metabolism dysregulation in FRDA patients (Sanchez-Casis et al., 1976; Lamarche et al., 1980; Martelli and Puccio, 2014), which very quickly led to the suspicion that frataxin could function as an iron storage protein (Adamec et al., 2000; Cossee et al., 2000; Gakh et al., 2002). Although iron deposits were not observed in the cerebellum, hallmarks of iron dysregulation such as the modification of the expression of the iron-regulated proteins transferrin receptor 1 (TFR1), ferritins (FRTs) and ferroportin (FPN) are manifest in neurons (Martelli and Puccio, 2014). A first hypothesis on the molecular function of frataxin emerged when purified Yfh1, the yeast frataxin, was found to directly bind iron and to oligomerize in a ferritin-like shape hosting thousands of iron atoms (Adamec et al., 2000). An iron storage function was thus proposed for frataxin.
The full-length human frataxin (FXN210, 23 kDa) is translated as a precursor protein in the cytosol and targeted to mitochondria using a targeting sequence in its N-terminal domain that is cleaved upon import into mitochondria by the mitochondrial processing peptidase (MPP) in an unusual two-step process (Koutnikova et al., 1998; Condò et al., 2007; Schmucker et al., 2008). During the first step, the precursor is cleaved into an intermediate form (FXN42–210, 18 kDa) that is subsequently shortened to the mature form (FXN81–210, 14 kDa) in the second step. A minor fraction of frataxin also exists as isoforms with extra-mitochondrial localization in the cytoplasm and the nucleus that originate from alternative codon initiation and splicing events (Pianese et al., 2002; Condo et al., 2006; Xia et al., 2012; Abruzzo et al., 2013; Pérez-Luz et al., 2015; Guo et al., 2018; Agrò and Díaz-Nido, 2020; Weng et al., 2020). Two isoforms have been characterized in human that both contain an elongated N-terminus, FXN II (FXN76–210, 15 kDa) and FXN III (FXN1–5,53–210, 18 kDa), with an N-acetyl modification in the case of FXN II (Xia et al., 2012; Guo et al., 2018). They have distinct tissue distribution with FXN II mRNA more abundant in the cerebellum and FXN III mRNA in the heart (Xia et al., 2012) and N-acetylated FXN76–210 the predominant form in erythrocytes (Guo et al., 2018). Extra-mitochondrial frataxin isoforms apparently contribute to cellular fitness as was shown in cell survival (Condo et al., 2006) and respiration assays (Agrò and Díaz-Nido, 2020), but their exact biochemical functions are still unclear. In mouse, a truncated cytosolic isoform (FXN79–207) is the predominant form, suggesting that it might contribute to the FRDA phenotypes in this model (Weng et al., 2020). However, this isoform is not present in human subjects. In human, the proportions of FXN II and III vs. the canonical mature mitochondrial form (FXN81–210) have not been determined, except in the heart of FRDA patients, where the canonical FXN is the predominant form (Weng et al., 2020), which questions the role of the isoforms.
The structures of several frataxin proteins from various organisms have been solved, showing a remarkably conserved structure (Cho et al., 2000; Dhe-Paganon et al., 2000; Musco et al., 2000; Adinolfi et al., 2002; He et al., 2004). The mature form folds into an α-β sandwich structural motif composed of a six to seven stranded β-sheet forming a flat surface on one side and two α-helix on the other side (Figure 1). A high number of conserved acidic amino acids (Supplementary Figure 1) are present in the first α-helix (α1) and the edge of the following β-sheet, which defines an acidic ridge with a negatively charged surface (Figure 1). A patch of highly conserved amino acids are also gathered in the middle of the β-sheet including a strictly conserved tryptophan at position 155 (Figure 1). Eukaryotic frataxins also contain a longer N-terminal domain of about 10 amino acids (81–92) that is unstructured and poorly conserved (Adinolfi et al., 2002; Prischi et al., 2009).
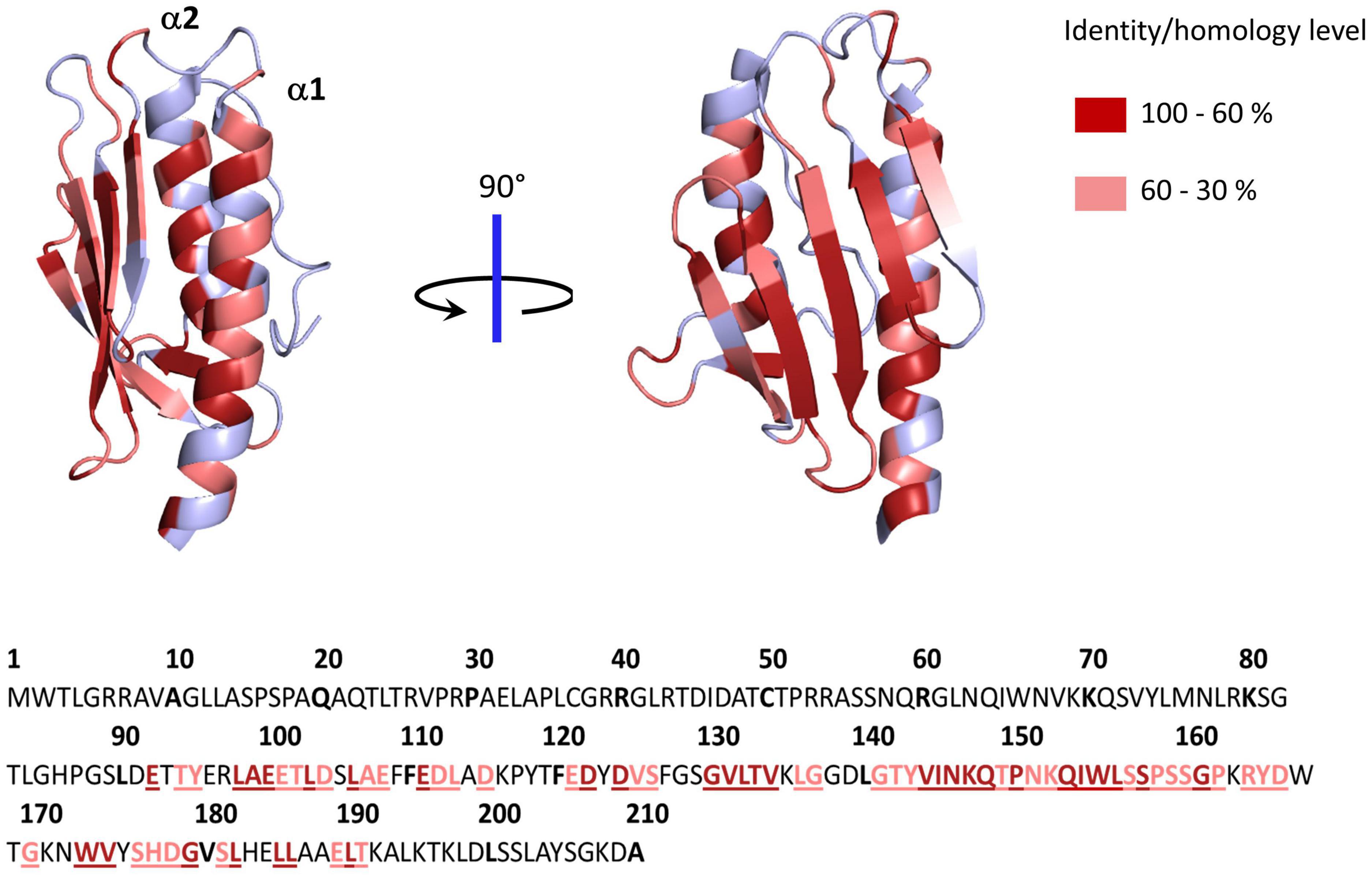
Figure 1. Structure of human frataxin (FXN) mapping conserved amino acids. Structure of human FXN (PDB code 1EKG) highlighting conserved amino acids from alignment of 50 eukaryotic and 50 prokaryotic frataxins (Supplementary Figure 1). Below is the sequence of full length human frataxin. Amino acids colored in dark red have identity/homology percentage between 100 and 60% and those in light red between 60 and 30%.
Recombinant frataxin from S. cerevisiae, E. coli and human have been shown to self-assemble into higher-order oligomers that bind iron (Adamec et al., 2000; Adinolfi et al., 2002; Gakh et al., 2002; O’Neill et al., 2005). Titration of Yfh1 with increasing iron concentrations under aerobic conditions leads to the stepwise assembly of trimers into multimers of 48–60 subunits sequestering over 2,000–3,000 atoms of iron within iron cores of 2–4 nm (Adamec et al., 2000; Gakh et al., 2002). The assembly of Yfh1 was further shown to be driven by iron oxidation and mineralization of Fe3+ ions. Conversely, iron core reduction resulted in protein disassembly. A similar iron-dependent oligomerization process was later on reported for the bacterial frataxin CyaY (Adinolfi et al., 2002). Human FXN was also found to bind multiple iron ions and to oligomerize, but in an iron independent manner via stable interactions mediated by its extended N-terminal region (Cavadini et al., 2002). Based on these data, frataxin was proposed to function as an iron storage protein maintaining iron bio-availability for Fe-S cluster and heme biosynthesis. Nevertheless, the physiological relevance of this iron-dependent oligomerization process has been questioned. First, only immature precursors of human FXN (FXN42–210, FXN55–210) were shown to oligomerize; furthermore, in iron-independent manners (Musco et al., 2000; Adinolfi et al., 2002; Cavadini et al., 2002; Schmucker et al., 2008; Gakh et al., 2010). Iron-dependent assembly of mature human FXN was only observed under strongly destabilizing conditions at 57°C (Adinolfi et al., 2002). This behavior might be linked to the higher thermal stability of human FXN compared to yeast and bacterial ones (Adinolfi et al., 2004), hence suggesting that oligomerization may be related to protein denaturation. However, a recent study estimated the internal temperature of mitochondria close to 50°C, which could promote oligomerization of mature frataxin (Chrétien et al., 2018). Moreover, recent studies revived the potential of human frataxin to oligomerize in an iron-dependent manner. This was observed by dynamic light scattering (DLS) but oligomerization occurred solely at a large iron excess (iron:FXN molar ratio of 10:1) with significantly slow kinetics (60 min for complete oligomerization) (Ahlgren et al., 2017). Unfortunately, a high-resolution structure of the human mature FXN81–210 oligomer is still lacking. Many studies reported iron-binding properties of monomeric frataxin proteins (Campbell et al., 2021). A total of twelve aspartate and glutamate residues along the conserved acidic ridge were found to bind iron but no structurally defined iron-binding site could be identified. A crystallographic structure of a Yfh1 trimer containing iron was generated by soaking the crystals of the apo-FXN trimer into an iron solution (Soderberg et al., 2013). However, neither of the residues of the acidic ridge were binding iron. Instead, the iron ion was found in an unusual configuration, apparently held by only two oxygen atoms from the amide moieties of Thr118 and Ala133 at distances of about 4 Å, a binding mode inconsistent with metal coordination. This strengthens the idea that iron binding to frataxin is non-specific and mediated by electrostatic interactions.
Further evidence against a role of frataxin as an iron storage protein under physiological conditions came from in vivo studies. In yeast cells, a Yhf1 mutant unable to perform self-assembly fully complemented a Δyfh1 strain, clearly indicating that frataxin oligomerization is not needed for its cellular function (Aloria et al., 2004). In a yeast strain accumulating iron in mitochondria, Yfh1 overexpression was neither able to prevent iron accumulation nor modify the nature of the iron minerals deposit (Seguin et al., 2010). Finally, in different mouse models of FRDA, a chronological analysis of phenotypic appearance showed that the Fe–S cluster deficit appears very early in the course of the disease, followed by the cardiac symptoms, oxidative stress and only afterward iron deposition, thus pointing to iron accumulation as a late, secondary effect of the Fe-S cluster defect (Puccio et al., 2001; Martelli and Puccio, 2014; Poburski et al., 2016).
In conclusion, these data have refuted the iron storage function of FXN. The accumulation of iron is in fact not a specific feature of frataxin deficiency but of defects in the early stages of the Fe-S cluster biosynthetic pathway due to iron misuse (Lill et al., 2012; Muhlenhoff et al., 2015). Thereby, it is now generally accepted that iron accumulation is a consequence of the Fe-S cluster defect, thus indicating that FXN is not a general ferritin-like iron storage protein but operates more directly in Fe-S cluster and/or heme biosynthesis.
While the iron storage function was disproved, frataxin was proposed to operate as an iron donor/chaperone for heme and Fe-S cluster biosynthesis. Direct interactions between FXN and ferrochelatase, the enzyme inserting iron into protoporphyrin IX (PPIX) at the last step of heme synthesis, were detected in vitro (Lesuisse et al., 2003; He et al., 2004; Yoon and Cowan, 2004; Soderberg et al., 2016). However, the interaction with ferrochelatase was not confirmed in vivo, in neither mammalian (Schmucker et al., 2011) nor yeast cells (Lange et al., 2004). Moreover, the insertion of iron into PPIX was not impaired in frataxin depleted cells from yeast (Muhlenhoff et al., 2002) and mammal (Schoenfeld et al., 2005). Rather, the defect in heme synthesis was explained by an inhibition of ferrochelatase activity by a still ill-defined mechanism as well as an increased level of zinc leading to the accumulation of Zn-PPIX (Lange et al., 2004; Schoenfeld et al., 2005). In erythroid progenitors from FRDA patients, heme biosynthesis was not altered (Morgan et al., 1979; Steinkellner et al., 2017). Altogether, these data suggest that FXN is not involved in heme biosynthesis. A defect in the insertion of iron into PPIX was reported in yeast, but in a strain where the yfh1 gene was deleted, thus totally lacking Yfh1, which may correspond to more stringent conditions allowing the accumulation of secondary defects (Lesuisse et al., 2003). This suggests that the ferrochelatase defect is a late indirect effect of frataxin deficiency. This was confirmed in a human embryonic kidney cell line through chronological investigations of the phenotypic defects caused by progressive FXN depletion revealing that the heme defect occurs much later than the Fe-S cluster defect and oxidative stress (Lu and Cortopassi, 2007). Moreover, with the notable exception of erythroid cells, the defects in heme biosynthesis do not lead to conspicuous iron accumulation in mitochondria, in contrast to both frataxin and Fe-S cluster deficiencies (Crisp et al., 2003; Chung et al., 2012; Lill et al., 2012; Muhlenhoff et al., 2015). Finally, a causal relationship between the Fe-S cluster defect and heme deficiency might exist in human and the other organisms where the ferrochelatase holds a [2Fe2S] cluster (Weerth et al., 2021). Although the function of this Fe-S cluster is still elusive, heme biosynthesis might be directly affected by the Fe-S cluster defect in these organisms.
In contrast, several independent studies reported reliable interactions both in vitro and in vivo between frataxin and two components of the Fe-S cluster assembly machinery, the scaffold protein ISCU and the cysteine desulfurase NFS1 (Gerber et al., 2003; Yoon and Cowan, 2003; Ramazzotti et al., 2004; Foury et al., 2007; Wang and Craig, 2008; Schmucker et al., 2011). Moreover, several in vitro reconstitutions of Fe-S cluster assembly machineries from a diverse range of organisms have further unveiled a functional role of frataxins in Fe-S cluster biosynthesis (Srour et al., 2020). It is now commonly accepted that the primary function of frataxin is the regulation of Fe-S cluster biogenesis whereas the heme defect, iron accumulation and oxidative stress are consequences of the Fe-S cluster defect. However, the exact function of FXN in Fe-S cluster biosynthesis remained controversial until very recently.
Strikingly, although eukaryotic frataxins are mitochondrial proteins, they are involved in both mitochondrial and extra-mitochondrial Fe-S cluster biogenesis, thus pointing to a pivotal role in this process (Figure 2A; Muhlenhoff et al., 2002; Lu and Cortopassi, 2007; Martelli et al., 2007). Indeed, in eukaryotes, the mitochondrion plays a central role in cellular Fe-S cluster biosynthesis (Lill and Freibert, 2020; Srour et al., 2020). This process is initiated in mitochondria by the Fe-S cluster assembly machinery (ISC) on a scaffold protein called ISCU. The ISC machinery synthesizes [2Fe2S] clusters on ISCU that are transferred to recipient apo-proteins (Gervason et al., 2019). A set of specialized proteins also use [2Fe2S] clusters as building blocks for the assembly of the cubane [4Fe4S] cluster on ISCA scaffold proteins (Muhlenhoff et al., 2011; Beilschmidt et al., 2017; Weiler et al., 2020). Additionally, the ISC machinery synthesizes a sulfur-containing compound of yet-undefined nature, named compound X, that is exported to the cytoplasm to sustain the production of cytoplasmic and nuclear Fe-S clusters by the cytosolic iron-sulfur cluster assembly machinery (CIA). Compound X has been proposed to be either a poly sulfur molecule or a [2Fe2S] cluster ligated by small molecules, possibly glutathione (GSH) (Schaedler et al., 2014; Li and Cowan, 2015; Pandey et al., 2019; Lill and Freibert, 2020). Frataxin is specifically involved in the synthesis of the [2Fe2S] clusters by the ISC core machinery and most likely compound X synthesis as well, since the lack of frataxin leads to a Fe-S cluster defect in the cytoplasm (Lu and Cortopassi, 2007; Martelli et al., 2007).
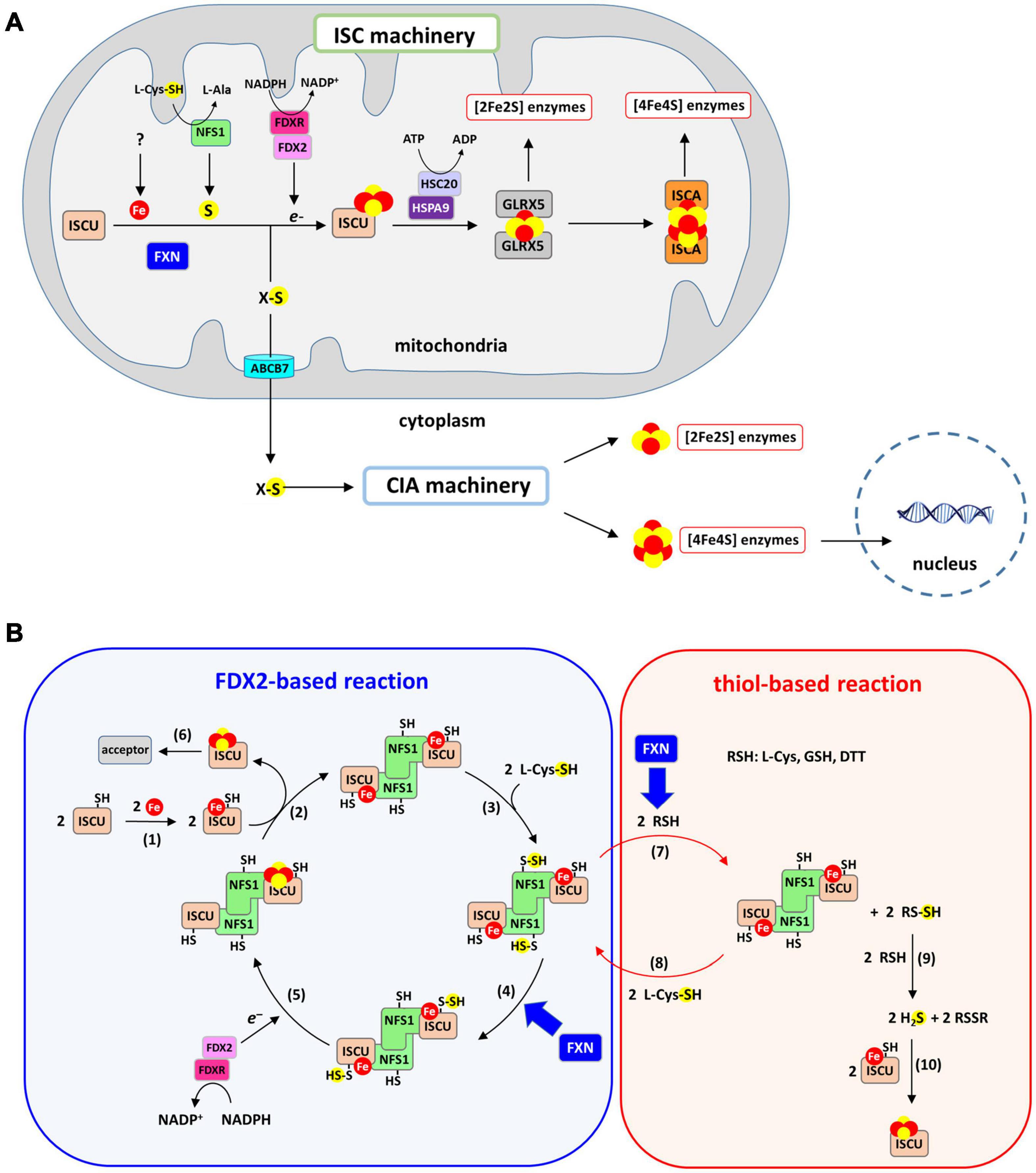
Figure 2. Iron-sulfur (Fe-S) cluster biogenesis pathway. (A) Fe-S cluster biosynthesis is initiated in mitochondria by the ISC machinery that encompasses the scaffold protein ISCU on which [2Fe2S] clusters are assembled, the cysteine-desulfurase NFS1 that provides sulfur in the form of a cysteine-bound persulfide and FDX2/FDXR that reduces the persulfide to sulfide. Frataxin accelerates the formation of the [2Fe2S] cluster on ISCU. The [2Fe2S] cluster is transferred onto GLRX5 with assistance from the ATP-dependent chaperones HSC20 and HSPA9, and then to recipient apo-proteins. Conversion to a [4Fe4S] cluster from the [2Fe2S] cluster is achieved by the ISCA proteins. The ISC machinery generates a sulfur-containing compound (compound X) that is exported by the ABCB7 transporter to the CIA machinery. [4Fe4S] clusters are assembled by the CIA machinery and delivered to their cytoplasmic and nuclear protein acceptors. (B) Mechanism of Fe-S cluster biosynthesis by the ISC machinery: (1) a ferrous iron is inserted into the assembly site of ISCU, (2) two Fe-ISCU form a heterodimer with a dimer of NFS1 (ACP and ISD11 that bind NFS1 are omitted for clarity), (3) NFS1 catalyzes the formation of persulfide on its catalytic cysteine, (4) the persulfide of NFS1 is transferred to ISCU and FXN accelerates this reaction, (5) FDX2/FDXR reduces the persulfide into sulfide, leading to formation of a [2Fe2S] cluster most likely by dimerization of ISCU, (6) the [2Fe2S] cluster carried by ISCU is transferred to acceptor proteins. Step 1–6 describe the FDX2-based reaction. A non-physiological process called the thiol-based reaction allows formation of Fe-S cluster in vitro in the absence of FDX2/FDXR via the following steps: (7) thiols (RSH) such as DTT, L-cysteine or GSH reduces the persulfide of NFS1, which leads to formation of persulfidated thiols (RSSH). FXN accelerates this persulfide cleavage by thiols. (8) the persulfide of NFS1 is regenerated by reaction with L-cysteine, (9) RSSH reacts with a second thiol to form free sulfide in the form of H2S alongside oxidized RSSR, (10) free sulfide slowly incorporates into Fe-ISCU to form [2Fe2S] and [4Fe4S] clusters by an unknown mechanism, with DTT favoring formation [4Fe4S] clusters.
A recent in vitro reconstitution of the complete ISC machinery performed in our laboratory using recombinant mouse proteins has shed new light on the mechanism of Fe-S cluster biosynthesis and the functional role of FXN (Figure 2B, blue chart) (Parent et al., 2015; Gervason et al., 2019; Srour et al., 2020). Using a biochemical assay to track sulfur processing during the assembly process within the ISC machinery, we unraveled the Fe-S cluster assembly process step-by-step (Parent et al., 2015; Gervason et al., 2019, 2021). The synthesis of Fe-S clusters is initiated by the insertion of a Fe2+ ion into the assembly site of the ISCU scaffold protein (Figure 2B, reaction 1). Sulfur is then provided by NFS1, a pyridoxal phosphate (PLP) dependent enzyme, in the form of a persulfide on its catalytic cysteine carried by a mobile loop. Two additional small proteins bind to NFS1, the LYR motif containing protein ISD11 and the acyl carrier protein ACP, which together regulate Fe-S cluster biosynthesis in response to acetyl-coA availability by stabilizing NFS1 (Van Vranken et al., 2018). NFS1 and ISCU form a heterodimeric complex (Figure 2B, reaction 2) in which the persulfide generated by NFS1 (Figure 2B, reaction 3) is transferred to ISCU when iron is present in the assembly site of ISCU (Figure 2B, reaction 4). Then, the ferredoxin FDX2, in complex with the NADPH dependent ferredoxin reductase FDXR, cleaves the persulfide into sulfide (S2–) and a [2Fe2S] cluster is formed, most likely by dimerization of ISCU (Figure 2B, reaction 5). At the final stage, the [2Fe2S] cluster is transferred to recipient apo-proteins by the ATP dependent chaperone/co-chaperone system HSPA9/HSC20 (Figure 2B, reaction 6). Several studies using reconstituted ISC machineries of mouse, human, yeast, and bacteria have reported a key function of frataxin in this process, but most of these reconstructions used incomplete ISC machineries, lacking FDX2 and FDXR (Figure 2B, red chart), which has led to a misinterpretation of the functional role of frataxin. We will first provide an overview of the results from these studies before introducing the data collected with the physiologically relevant reconstructions from complete ISC machineries.
Reconstitutions of the human and bacterial ISC machineries suggested that iron-loaded frataxin promotes the formation of Fe-S clusters by donating iron to ISCU (Yoon and Cowan, 2003; Layer et al., 2006; Colin et al., 2013). This iron chaperone function was attributed to the monomeric form of frataxin (Cook et al., 2006). However, the iron-binding site in ISCU was not characterized in these studies to ascertain that frataxin was actually donating iron to ISCU. The recent characterization of the iron binding site of mouse ISCU by spectroscopic techniques (electronic absorption, site directed mutagenesis, NMR, and Mössbauer) has enabled direct monitoring of iron insertion into ISCU (Gervason et al., 2019). It was found that ISCU directly binds iron in its assembly site without requirement of FXN or any other accessory proteins (Figure 2B, reaction 1). Another important feature of these studies that has previously been overlooked is the presence of a zinc ion in the assembly site of recombinant ISCU expressed in bacteria (Gervason et al., 2019). It is still unclear whether the zinc ion is present under physiological conditions or if it is an artifact of purification, but due to its high affinity for ISCU, it precludes iron binding. The zinc must be removed to allow iron binding. The ability of FXN to insert iron ions into zinc-containing ISCU was thus tested, but it was not able to exchange zinc with iron (Gervason et al., 2019).
Moreover, as previously mentioned, the iron binding properties of frataxin point to a non-specific fixation mode (Pastore and Puccio, 2013). First, the number of iron bound to frataxin varied between studies (from one to seven), with Fe2+ and Fe3+ binding with similar affinities. The proposed iron-binding site involves several glutamate and aspartate residues from the conserved acidic ridge (Campbell et al., 2021). However, the structure of the complex between FXN and the NFS1-ISD11-ACP-ISCU complex (Fox et al., 2019) revealed that most of these amino acids are either directly involved in the interaction with NFS1 or in the internal structure of the protein (Figure 3). Only two of them, D104 and D112, are not interacting with other amino acids, but are also not strictly conserved across species and spatially too distant to form a structurally defined metal-binding site. Altogether, these data point to non-specific iron binding via electrostatic interactions, which is not consistent with the proposed function of frataxin as an iron chaperone.
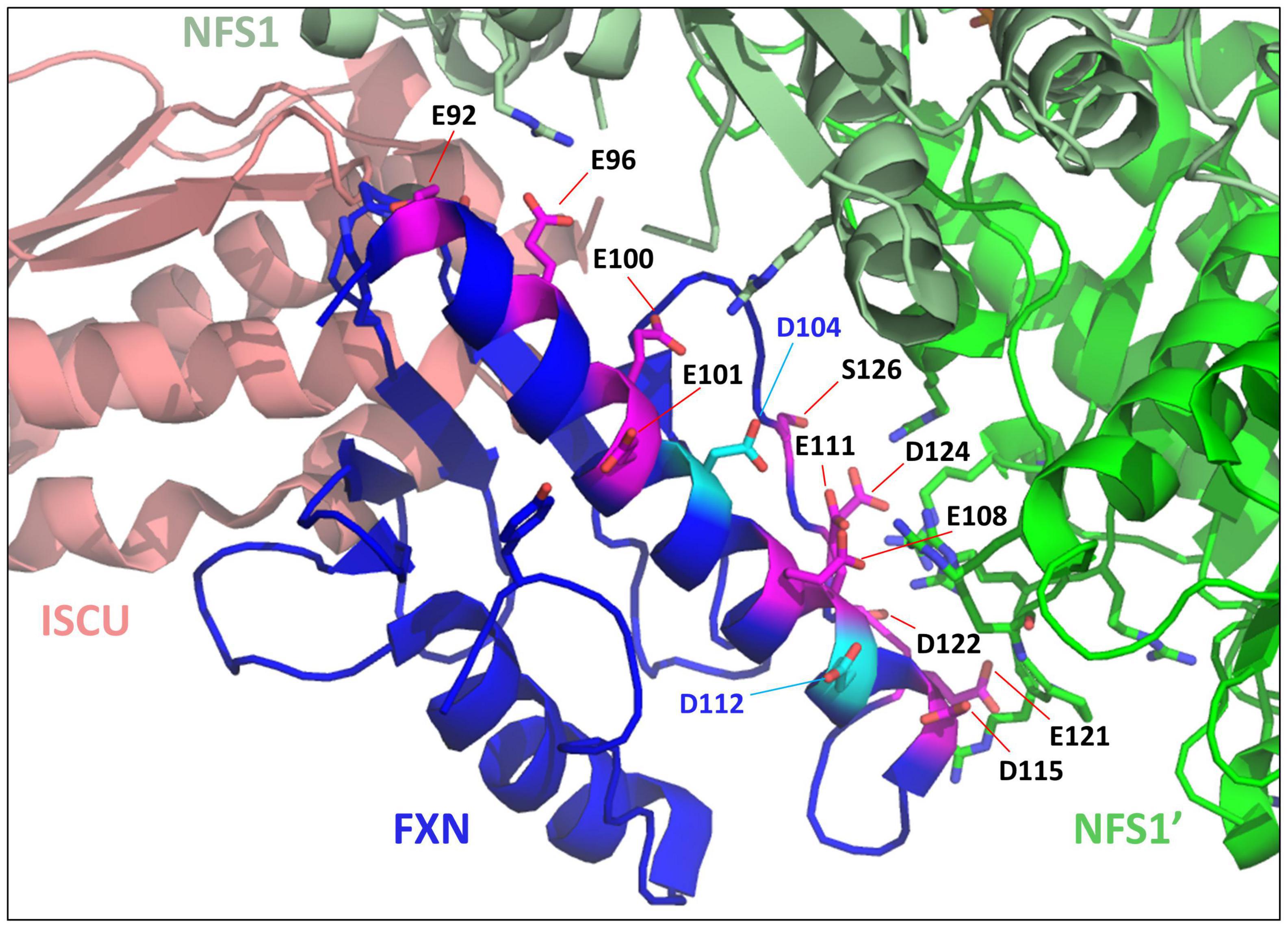
Figure 3. Mapping iron-binders amino acids of frataxin (FXN) within the NFS1-ISD11-ACP-Zn-ISCU-FXN complex. Structure of the human NFS1-ISD11-ACP-Zn-ISCU-FXN complex (PDB code 6NZU) highlighting the amino acids of FXN identified as iron-binders. The amino acids interacting with NFS1 (E96, E100, E108, E111, D115, E121, and D124) or important for the internal structure of FXN (E92, E101) are colored in pink and those not interacting with other amino acids (D104 and D112) are in cyan. ISCU is colored in salmon, the two subunits of the NFS1 dimer are in light green (NFS1) and dark green (NFS1’), FXN is in blue.
In conclusion, these data invalidate the hypothesis that frataxin is an iron donor or iron chaperone for ISCU. The main hypothesis for the iron insertion process in ISCU is that it originates from labile iron pools, which are present in the cytoplasm as well as mitochondria where they maintain iron in its reduced form (Fe2+) bound to small molecules such as GSH (Hider and Kong, 2013; Lindahl and Moore, 2016; Philpott and Jadhav, 2019; Wofford et al., 2019).
The first evidence that FXN might be involved in sulfur donation rather than iron insertion came from studies of the bacterial ISC machinery (Adinolfi et al., 2009). In vitro reconstitutions of the bacterial ISC machinery were performed with recombinant IscU and IscS, the ortholog of NFS1, with DTT as a reductant instead of Fdx, and the homolog of FDX2 to cleave the persulfide into sulfide (Adinolfi et al., 2009). Hereafter, this type of reaction will be referred to as a thiol-based reaction (Figure 2B, red chart). Surprisingly, the enzymatic assays revealed that CyaY slows sulfide production by IscS and consequently Fe-S cluster production. This inhibitory effect was enhanced in the presence of free iron thus suggesting that the regulation by CyaY is iron-dependent. A function as a “gate-keeper” preventing uncontrolled production of sulfide ions in the presence of iron was then proposed for CyaY. However, the thiol-based assays performed with the human and mouse ISC systems revealed a completely reversed effect, with FXN stimulating the rate of sulfide production by NFS1, which was correlated with an acceleration of Fe-S cluster production (Tsai and Barondeau, 2010; Colin et al., 2013). The stimulatory effect of FXN was further shown to promote formation of [4Fe4S] clusters at the expense of [2Fe2S] clusters and FXN was proposed to facilitate iron entry concomitantly to its stimulatory effect on sulfide production (Colin et al., 2013). These data thus led to a controversy on the functional role of frataxins, with a protective effect of the bacterial frataxin on one hand, and a stimulatory function for the mammalian frataxin on the other hand.
Our laboratory later showed that the mechanism of stimulation by mammalian FXN relies on the acceleration of persulfide cleavage by DTT (Figure 2B, reaction 7), which increases sulfide release and Fe-S assembly by the thiol-based pathway (Parent et al., 2015). However, the thiol-based reaction cannot be considered as physiologically relevant and thus the significance of the stimulatory effect of FXN in this process is unclear. DTT and thiols do not function as surrogates of FDX2 and Fdx because they function differently (Parent et al., 2015; Gervason et al., 2019). Using a biochemical assay to detect and quantify protein-bound persulfide, the production of the sulfide ions in thiol-based reactions was shown to result from the cleavage of the persulfide carried by NFS1 (Figure 2B, red chart), whereas it is the persulfide of ISCU that is cleaved in FDX2-based reactions (Figure 2B, blue chart) (Parent et al., 2015; Gervason et al., 2019, 2021). Other thiols, including biological ones such as cysteine and GSH, were also shown to cleave persulfide and to sustain Fe-S cluster synthesis, thus raising the possibility that these thiol-based reactions could be physiologically relevant (Parent et al., 2015; Lin et al., 2020). However, the intrinsic properties of the thiol-based reaction compared to the FDX2-based one indicated that this is not the case. The thiol-based reaction is much slower and is far less efficient than the FDX2-based one, with only about 5% of the sulfide ions actually incorporated as Fe-S clusters compared to 95% in the case of the FDX2-based reaction. This is due to the lack of confinement and coordination of sulfide production with the presence of iron in ISCU. In the thiol-based process, the sulfide ions are released in solution in a two-step process involving formation of a persulfidated thiol (Figure 2B, reaction 7) that is subsequently cleaved into sulfide by reacting with a second thiol molecule (Figure 2B, reaction 9) (Parent et al., 2015). Then, the sulfide ions react with the iron center in ISCU to generate a Fe-S cluster via a still undefined mechanism (Figure 2B, reaction 10). These successive reactions are much slower and less efficient than the FDX2-based process which relies on concerted processes. Moreover, under physiological conditions, it is likely that the sulfide ions will diffuse outside of the NFS1-ISCU complex and thus the chance to specifically form a Fe-S cluster in ISCU will be very low. In contrast, in the FDX2-based reaction, persulfide supply to ISCU and sulfide production are coupled to the presence of iron in ISCU (Figure 2B, blue chart). Under these conditions, no “free” sulfide is produced. Moreover, in contrast to the FDX2-based reaction, the thiol-based one leads to the formation of [4Fe4S] clusters in addition to [2Fe2S] clusters (Colin et al., 2013; Parent et al., 2015), with DTT enhancing formation of [4Fe4S] clusters (Fox et al., 2015), a function that is attributed to the ISCA scaffold proteins (Muhlenhoff et al., 2011; Beilschmidt et al., 2017; Gervason et al., 2019; Weiler et al., 2020), which further questioned its physiological relevance. In conclusion, although biological thiols could play the role of DTT in vivo, the thiol-based reaction is not physiologically relevant for Fe-S cluster biosynthesis, which rules out the idea that FXN stimulates Fe-S cluster biosynthesis by enhancing the cleavage of NFS1’s persulfide by thiols.
To establish the physiological function of FXN in Fe-S cluster biogenesis, the FDX2-based reaction was studied in a stepwise manner (Gervason et al., 2019). First, the in vitro reconstitution of the complete mouse ISC machinery including FDX2 (FDX2-based reaction) showed that FXN accelerates the formation of [2Fe2S] clusters. By analysing the effect of FXN at each step, we found that FXN accelerates the transfer of persulfide from NFS1 to ISCU (Figure 2B, reaction 4) (Parent et al., 2015; Gervason et al., 2019). An independent study also reported that human FXN facilitates sulfur transfer to ISCU, which points to a conserved mechanism across species (Bridwell-Rabb et al., 2014). However, Fe-S cluster assembly is a multi-step process, therefore to stimulate the whole process FXN must operate on the rate-limiting step. Persulfide transfer to ISCU was identified as the rate-limiting step in the mouse system, thereby, the stimulatory effect of FXN was directly correlated to its global stimulatory effect on the Fe-S cluster assembly process (Gervason et al., 2019). In other studies, FXN and its yeast homolog were found to stimulate the formation of the persulfide by NFS1 (Figure 2B, reaction 8), which suggest that FXN could operate at a different step of the reaction (Pandey et al., 2013; Patra and Barondeau, 2019). However, persulfide formation is not rate-limiting since kinetic studies showed that it is much faster than persulfide transfer (Parent et al., 2015; Gervason et al., 2019), which dismisses the possibility that FXN could stimulate Fe-S cluster assembly by accelerating persulfide formation on NFS1. Altogether, these mechanistic data have provided a long-awaited clarification on the functional role of frataxin by establishing that FXN stimulates Fe-S cluster biosynthesis by accelerating persulfide transfer.
Another important feature of the stimulation provided by FXN is that it accelerates the reaction and does not modify the nature of the Fe-S cluster formed, i.e., a [2Fe2S] cluster, nor the final yield of Fe-S clusters (Gervason et al., 2019), which fits the definition of an enzyme. However, since the reaction is already efficient in its absence, its functional role is best defined as an accelerator. This function is also consistent with the cellular phenotypes of yeast and mammalian cells lacking FXN, in which Fe-S clusters are still produced but at a lower level (Puccio et al., 2001; Duby et al., 2002; Muhlenhoff et al., 2002). Moreover, a number of transcription factors regulates FXN expression (Oktay et al., 2007; Guccini et al., 2011; Yandim et al., 2013; Fernández-Frías et al., 2020), which suggests that FXN could operate as a regulator to fine-tune Fe-S cluster biogenesis in response to variations in physiological conditions. Among them are Nrf2, the main regulator of the antioxidant defense (Sahdeo et al., 2014). Activation of Fe-S cluster production in response to reactive oxygen species (ROS) via the Nrf2-FXN axis could help restore Fe-S proteins damaged by ROS (D’Autreaux and Toledano, 2007). FXN is also part of the hypoxic response with both HIF-1α and HIF-2α activating FXN expression under hypoxic conditions (Oktay et al., 2007; Guccini et al., 2011). The activation of FXN expression under hypoxia might help improve mitochondrial bioenergetics under low oxygen concentration. The HIF-FXN axis also protects cells against ischemia-reperfusion episodes and is possibly critical for tumor progression (Amaral and Okonko, 2015; Nanayakkara et al., 2015; Schultz et al., 2016). Interestingly, the expression of FXN was found to be upregulated under iron-rich conditions and conversely downregulated under iron-starved conditions (Li et al., 2008). Thus, Fe-S cluster production seems to increase concomitantly with the level of available iron via modulation of FXN expression, which will amplify iron utilization by this pathway and thus prevent iron accumulation. In erythroid cells, the expression of FXN is not significantly modulated by the iron level (Becker et al., 2002), possibly to favor redirection of iron for heme biosynthesis. Therefore, the iron-dependent regulation of FXN expression might be an important regulatory circuit for iron utilization in different cellular contexts. Further studies in this direction will help better define the physiological role of FXN.
To move toward the design of drugs that could replace FXN, the next step is the elucidation of the mechanism by which FXN stimulates persulfide transfer. A key feature of this reaction is its iron-dependency, which is unique to this system (Gervason et al., 2019). Persulfide transfer reactions have been described in other sulfur insertion processes, such as the thiolation of transfer RNA, the synthesis of the molybdenum cofactor of nitrogenase and thiamine synthesis, but none of them are known to be metal-dependent (Black and Dos Santos, 2015; Leimkühler et al., 2017). In contrast, in the absence of iron in ISCU, the transfer of persulfide between NFS1 and ISCU and its acceleration by FXN are abolished (Gervason et al., 2019). This iron dependency thus appears as a central feature of Fe-S cluster biosynthesis, probably to couple sulfur acquisition by ISCU with iron availability. Thereby, understanding the role of the iron center in persulfide transfer is critical to unravel the mechanism by which FXN stimulates this reaction. Iron binds in the form of a ferrous iron (Fe2+) to the assembly site of ISCU and site directed mutagenesis suggests that the cysteine Cys35 and Cys61, the aspartate Asp37 and the histidine His103 are ligating the metal ion while Cys104, the persulfide receptor, remains unbound (Gervason et al., 2019). Unfortunately, no structure of iron-bound ISCU alone or within the NFS1-ISD11-ACP-ISCU-FXN complex has yet been reported to start assessing the structural details of persulfide transfer. A number of structures were solved but with zinc instead of iron, hereafter referred to as Zn-ISCU (Boniecki et al., 2017; Fox et al., 2019). However, since Zn-ISCU also enables persulfide transfer and its acceleration by FXN (Gervason et al., 2019), the structures solved with zinc should also be informative on the mechanism.
The NMR structure of mouse Zn-ISCU (PDB code 1WFZ) revealed a structural arrangement very similar to the one suspected for Fe-ISCU (Figure 4A). In this structure, the zinc ion is coordinated by the cysteine Cys35 and Cys61, the aspartate Asp37 and the histidine His103, while Cys104, the persulfide receptor, remains unbound but at a close distance. The crystal structure of the human NFS1-ISD11-ACP-Zn-ISCU complex shows that the catalytic cysteine of NFS1 is binding to the zinc ion through exchange with Cys35 (Figure 4B; Boniecki et al., 2017). Although the structure was not solved with a persulfidated cysteine, it is likely that the persulfide carried by NFS1 would bind in a similar fashion. This might facilitate persulfide transfer by providing an electrophilic nature to the terminal sulfur of the persulfide (also called sulfane sulfur) to allow a nucleophilic attack by Cys104. However, this structure also highlights the steric hindrance between the catalytic cysteine of NFS1 and Cys104 due to the presence of the zinc ion and its ligands. It is likely that a structural rearrangement is needed to allow persulfide transfer. Interestingly, the structure of the pentameric NFS1-ISD11-ACP-Zn-ISCU-FXN complex solved by cryoelectron microscopy showed that FXN induces a rearrangement of the coordination sphere of the zinc ion (Figure 4C; Fox et al., 2019). FXN interacts with Cys35 of ISCU via its asparagine Asn151 and its glutamine Gln148, which stabilizes Cys35 in an unbound position away from the metal center. A second interaction involves tryptophan Trp155 of FXN interacting with His103 of ISCU via a π-π stacking interface that is anchoring His103 in an unbound position away from the zinc center. A third interaction between the serine Ser157 of FXN and the main chain proline Pro99 of ISCU is twisting the α-helix carrying Cys104 and His103, which may further stabilize the (FXN) Trp155-His103(ISCU) interaction and concomitantly promote the motion of Cys104 toward the metal ion. The structural rearrangement induced by FXN thus seems to clear access between the catalytic cysteine of NFS1 and Cys104 of ISCU and to force Cys104 into closer proximity to the zinc center. Altogether, this might facilitate direct persulfide transfer to Cys104. Importantly, Trp155 is strictly conserved among frataxin proteins (Supplementary Figure 1), which strengthens the idea that the His103-Trp155 interaction is a central feature of the activation mechanism by FXN.
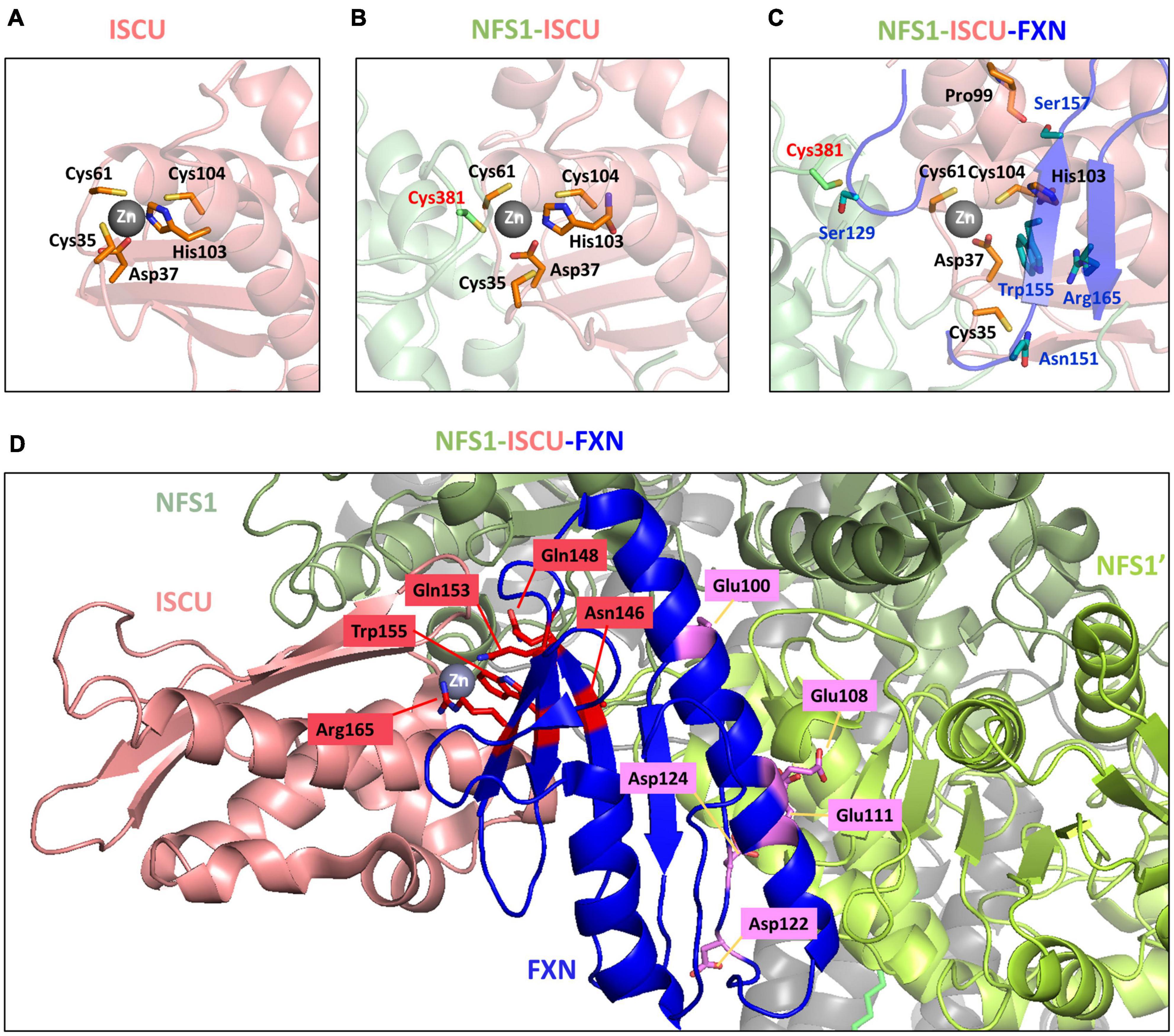
Figure 4. Structural rearrangement at the zinc site of ISCU upon binding of NFS1 and frataxin (FXN). Zinc site of panel (A) mouse ISCU (PDB code 1WFZ), (B) human NFS1-ISD11-ACP-ISCU complex (PDB code 5WLW) and (C) human NFS1-ISD11-ACP-ISCU-FXN complex (PDB code 6NZU). (D) FXN variants with impaired activity mapped on the structure of the human NFS1-ISD11-ACP-ISCU-FXN complex (PDB code 6NZU). The amino acids in red are involved in the interaction with ISCU, amino acids in purple are involved in interaction with NFS1. ISCU is colored in salmon, the two subunits of the NFS1 dimer are in light green (NFS1) and dark green (NFS1’), FXN is in blue.
Frataxin also interacts with the flexible loop of NFS1 and stabilizes the catalytic Cys381 halfway between the PLP of NFS1 and ISCU, which is difficult to rationalize with the stimulation of persulfide transfer (Fox et al., 2019). However, one has to consider that the catalytic cysteine is not persulfidated in this structure and thus this position may correspond to an intermediate state of the flexible loop returning to the PLP pocket for persulfide loading.
The analysis of point mutations in FXN provide further insight on the hypothetical mechanism proposed above. A small percentage of FRDA patients are heterozygous and contain, besides the GAA-triplet expansion, a point mutation in the Fxn gene (Santos et al., 2010; Galea et al., 2016). Two mouse models harboring the point mutations G127V and I151F, equivalent to G130V and I154F in human FXN, respectively, and recapitulating FRDA phenotypes were recently generated (Fil et al., 2020; Medina-Carbonero et al., 2022), which also helps better understand the impact of these mutations. These clinical variants affect residues that are conserved and important for structure and/or function of FXN (Table 1, clinical variants). In addition to clinical variants, other mutations impairing FXN and Yfh1 activities in vivo have been identified (Table 1, other mutants; Leidgens et al., 2010; Schmucker et al., 2011). Hereby, it is important to distinguish buried residues from those exposed to the solvent on the surface. Buried residues participate in the stability of the whole protein fold or may alter the maturation process (Table 1, core amino acids), whereas surface exposed residues might be involved in interactions with NFS1 and ISCU (Table 1, last two columns). Interestingly, the variants of surface exposed residues belong to two different domains of FXN within the NFS1-ISCU-FXN complex: the region spanning Glu96 to Asp124, corresponding to the first α-helix of FXN that is involved in the interaction with NFS1, and the region spanning Asn146 to Arg165 composing the β-sheet that is involved in the interaction with ISCU (Figure 4D). All the amino acids involved in the interaction with ISCU (N146, Q148, Q153, W155, and R165) are oriented toward the zinc center and are likely involved in the stimulation of persulfide transfer. The tryptophan Trp155 was already identified in the interaction with His103 of ISCU (Figure 4C). The asparagine Asn146, glutamine Gln153 and arginine Arg165 are packing close to the Trp155-His103 interaction and further stabilize it (Figure 4D). The glutamine Gln148 seems to cooperate with Asn151 to stabilize Cys35 of ISCU in the unbound position (Figure 4D). The analysis of the variants thus strengthen the idea that maintaining Cys35 and His103 in free positions, away from the metal center, are two critical structural features of the interaction between FXN and ISCU that promotes persulfide transfer.
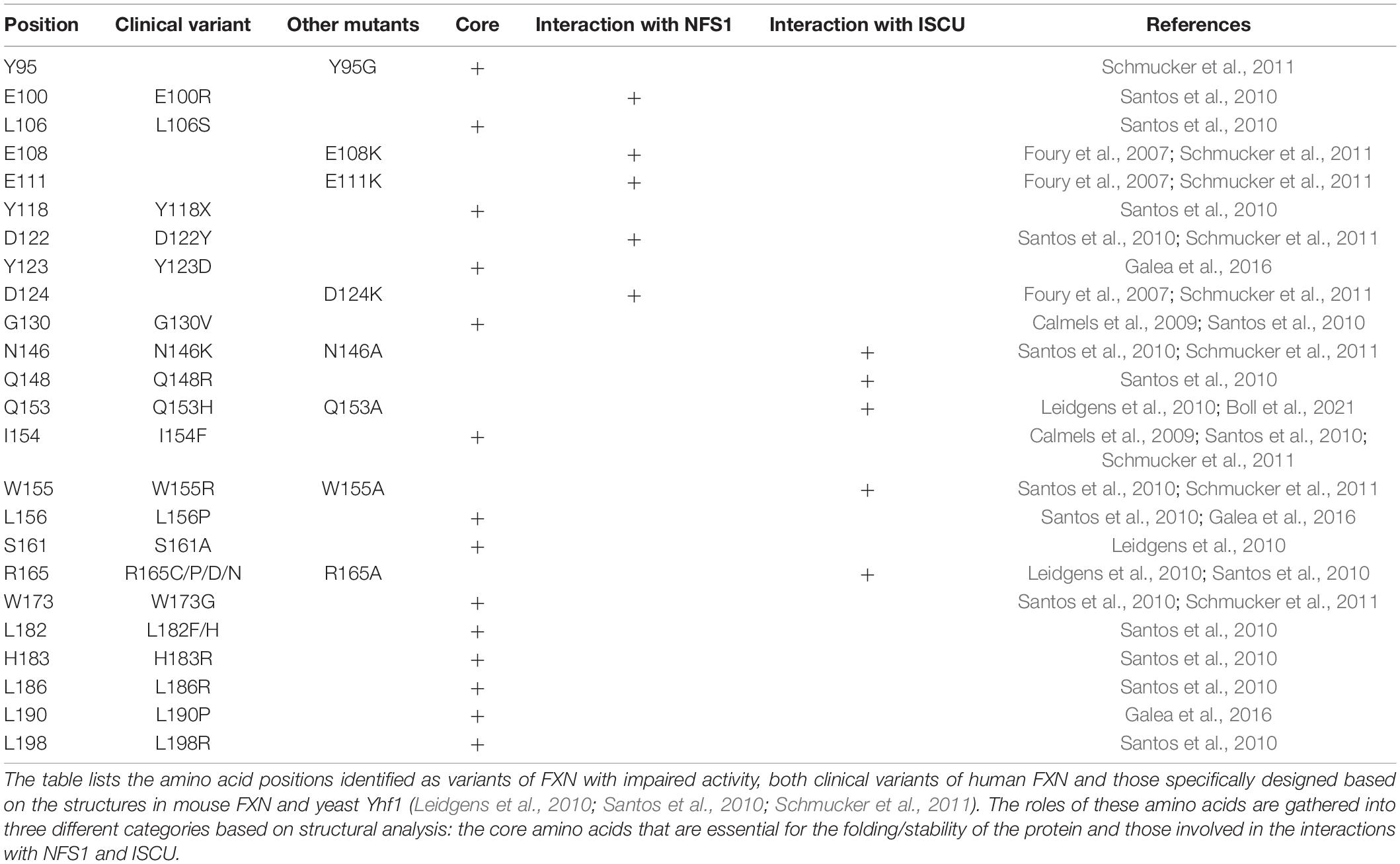
Table 1. Key amino acids of FXN identified as impaired variants.
Despite great efforts in elucidating the underlying mechanisms leading to the cellular phenotypes in FRDA patients and the search for therapeutic treatments, no cure is currently available. One of the main obstacles has remained the lack of understanding of the biochemical function of frataxin, but recent advances are opening new perspectives. The first models proposed that frataxin operates as an iron storage or iron chaperone protein, but these hypotheses were weakened by several studies disproving the ability of frataxin to store and deliver iron. Instead, frataxin was recently shown to stimulate Fe-S cluster biosynthesis by accelerating persulfide transfer from NFS1 to the scaffolding protein ISCU (Bridwell-Rabb et al., 2014; Parent et al., 2015; Gervason et al., 2019). Early exploration of phenotypes in patients as well as in cellular and animal models provided a starting point to target the symptoms appearing in the course of the disease, mainly mitochondrial dysfunction, iron accumulation, oxidative stress and lipid oxidation. Most of the drug candidates targeting these metabolic defects did not lead to significant improvement in clinical trials and were withdrawn, but some of them are still under studies (a list of current and withdrawn candidates is available at the FARA website1) (Lynch and Farmer, 2021). However, by acting exclusively on one of the multiple metabolic defects, these strategies are expected to provide only limited benefit, and compensatory effects might mitigate their therapeutic potential. These approaches are likely to be more potent in combination with each other. Among these strategies, iron chelation was shown to alleviate some of the cardiac phenotypes but was withdrawn due to worsening of the disease. Based on the assumption that frataxin may behave as a Ferritin-like iron storage or general iron donor, iron chelators such as deferiprone were developed to remove iron deposits in cardiomyocytes. Although deferiprone improved cardiac functions in clinical trials, adverse effects and worsening of the disease appeared at high doses (>20 mg/kg/day) (Pandolfo et al., 2014; Lynch and Farmer, 2021). The underlying reasons for the deleterious effects of direct iron chelation are complex. The recent findings on the functional role of frataxin as a sulfur transfer accelerator shed some new light to seize the links between iron accumulation and frataxin deficiency. Under conditions of frataxin deficiency, as sulfur donation is slowed down, iron builds up because it can no longer be used. Iron does not accumulate due to overabundance as one might predict based on the iron storage functional model. Therefore, by sequestering available iron, iron chelators further reduce Fe-S cluster biogenesis in FRDA patients, which could worsen the disease. In conclusion, although iron chelation therapy has a beneficial impact on the cardiac phenotype by clearing iron deposits, it becomes toxic probably by worsening the Fe-S clusters synthesis shortage.
The most recent strategies are now targeting the lack of FXN, which is the primary defect of FRDA, as this is expected to have the highest therapeutic benefit (Yang et al., 2021). Three different approaches are currently under development: increase of frataxin expression, delivery of recombinant frataxin protein and restoration of the frataxin locus by gene therapy. These strategies are highly promising. However, concerns regarding gene dosage will have to be considered with gene therapy and modulation of frataxin expression since frataxin overexpression was reported to be toxic in several models (Navarro et al., 2011; Wang et al., 2014; Vannocci et al., 2018; Belbellaa et al., 2020).
In addition to the strategies restoring frataxin levels, the current knowledge on the functional role of frataxin in the stimulation of sulfur donation to ISCU opens new perspectives for the development of alternative approaches. For instance, sulfur donors and drugs substituting frataxin in sulfur transfer acceleration might be of interest in the treatment of FRDA. In this context, unraveling the mechanism by which frataxin overexpression is toxic will become a key question to solve in the near future in order to adjust therapies based on frataxin substitution and the modulation of its expression.
BD’A, BM, and KW wrote the review. SG performed the protein alignments and provided the Figure 1. All authors contributed to the article and approved the submitted version.
This work was supported by grants from FARA (Friedreich’s Ataxia Research Alliance), Ataxia UK, AFAF (Association Française de l’Ataxie de Friedreich), FRM (Fondation pour la Recherche Médicale, FDT202012010541) and ANR (Agence Nationale de la Recherche, ANR-17-CE11-21).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors are grateful to David Lynch for the invitation to contribute this review to the research topic “Basic and Clinical Mechanisms in Friedreich Ataxia.”
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2022.838335/full#supplementary-material
Abruzzo, P. M., Marini, M., Bolotta, A., Malisardi, G., Manfredini, S., Ghezzo, A., et al. (2013). Frataxin mRNA Isoforms in FRDA patients and normal subjects: effect of tocotrienol supplementation. Biomed Res. Int. 2013:276808. doi: 10.1155/2013/276808
Adamec, J., Rusnak, F., Owen, W. G., Naylor, S., Benson, L. M., Gacy, A. M., et al. (2000). Iron-dependent self-assembly of recombinant yeast frataxin: implications for Friedreich ataxia. Am. J. Hum. Genet. 67, 549–562. doi: 10.1086/303056
Adinolfi, S., Iannuzzi, C., Prischi, F., Pastore, C., Iametti, S., Martin, S. R., et al. (2009). Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 16, 390–396. doi: 10.1038/nsmb.1579
Adinolfi, S., Nair, M., Politou, A., Bayer, E., Martin, S., Temussi, P., et al. (2004). The factors governing the thermal stability of frataxin orthologues: how to increase a protein’s stability. Biochemistry 43, 6511–6518. doi: 10.1021/bi036049+
Adinolfi, S., Trifuoggi, M., Politou, A. S., Martin, S., and Pastore, A. (2002). A structural approach to understanding the iron-binding properties of phylogenetically different frataxins. Hum. Mol. Genet. 11, 1865–1877. doi: 10.1093/hmg/11.16.1865
Agrò, M., and Díaz-Nido, J. (2020). Effect of mitochondrial and cytosolic FXN isoform expression on mitochondrial dynamics and metabolism. Int. J. Mol. Sci. 21:8251. doi: 10.3390/ijms21218251
Ahlgren, E. C., Fekry, M., Wiemann, M., Soderberg, C. A., Bernfur, K., Gakh, O., et al. (2017). Iron-induced oligomerization of human FXN81-210 and bacterial CyaY frataxin and the effect of iron chelators. PLoS One 12:e0188937. doi: 10.1371/journal.pone.0188937
Al-Mahdawi, S., Pinto, R. M., Varshney, D., Lawrence, L., Lowrie, M. B., Hughes, S., et al. (2006). GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 88, 580–590. doi: 10.1016/j.ygeno.2006.06.015
Aloria, K., Schilke, B., Andrew, A., and Craig, E. A. (2004). Iron-induced oligomerization of yeast frataxin homologue Yfh1 is dispensable in vivo. EMBO Rep. 5, 1096–1101. doi: 10.1038/sj.embor.7400272
Alsina, D., Ros, J., and Tamarit, J. (2018b). Nitric oxide prevents Aft1 activation and metabolic remodeling in frataxin-deficient yeast. Redox Biol. 14, 131–141. doi: 10.1016/j.redox.2017.09.001
Alsina, D., Purroy, R., Ros, J., and Tamarit, J. (2018a). Iron in Friedreich Ataxia: A central role in the pathophysiology or an epiphenomenon? Pharmaceuticals 11:89. doi: 10.3390/ph11030089
Amaral, N., and Okonko, D. O. (2015). Mitigation of myocardial ischemia-reperfusion injury via HIF-1α-frataxin signaling. Am. J. Physiol. Heart Circ. Physiol. 309, H728–H730. doi: 10.1152/ajpheart.00553.2015
Aratani, Y. (2018). Myeloperoxidase: its role for host defense, inflammation, and neutrophil function. Arch. Biochem. Biophys. 640, 47–52. doi: 10.1016/j.abb.2018.01.004
Babcock, M., Silva, D. D., Oaks, R., Davis-Kaplan, S., Jiralerspong, S., Montermini, L., et al. (1997). Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of Frataxin. Science 276, 1709–1712. doi: 10.1126/science.276.5319.1709
Bauerle, M. R., Schwalm, E. L., and Booker, S. J. (2015). Mechanistic diversity of radical S-adenosylmethionine (SAM)-dependent methylation. J. Biol. Chem. 290, 3995–4002. doi: 10.1074/jbc.R114.607044
Becker, E. M., Greer, J. M., Ponka, P., and Richardson, D. R. (2002). Erythroid differentiation and protoporphyrin IX down-regulate frataxin expression in Friend cells: characterization of frataxin expression compared to molecules involved in iron metabolism and hemoglobinization. Blood 99, 3813–3822. doi: 10.1182/blood.v99.10.3813
Beilschmidt, L. K., Ollagnier de Choudens, S., Fournier, M., Sanakis, I., Hograindleur, M. A., Clemancey, M., et al. (2017). ISCA1 is essential for mitochondrial Fe4S4 biogenesis in vivo. Nat. Commun. 8:15124. doi: 10.1038/ncomms15124
Beilschmidt, L. K., and Puccio, H. M. (2014). Mammalian Fe-S cluster biogenesis and its implication in disease. Biochimie 100, 48–60. doi: 10.1016/j.biochi.2014.01.009
Belbellaa, B., Reutenauer, L., Messaddeq, N., Monassier, L., and Puccio, H. (2020). High levels of Frataxin overexpression lead to mitochondrial and cardiac toxicity in mouse models. Mol. Ther. Methods Clin. Dev. 19, 120–138. doi: 10.1016/j.omtm.2020.08.018
Black, K. A., and Dos Santos, P. C. (2015). Shared-intermediates in the biosynthesis of thio-cofactors: mechanism and functions of cysteine desulfurases and sulfur acceptors. Biochim. Biophys. Acta 1853, 1470–1480. doi: 10.1016/j.bbamcr.2014.10.018
Boll, M., Gasca-Salda, A. D., May, S., Dávila-Ortiz de Montellano, D. J., and Monroy-Jaramillo, N. (2021). Clinical and molecular features of first Mexican Friedreich’s ataxia patients with compound heterozygous FXN mutations. Neurol. India 69, 1363–1367. doi: 10.4103/0028-3886.329555
Boniecki, M. T., Freibert, S. A., Muhlenhoff, U., Lill, R., and Cygler, M. (2017). Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex. Nat. Commun. 8:1287. doi: 10.1038/s41467-017-01497-1
Bridwell-Rabb, J., Fox, N. G., Tsai, C. L., Winn, A. M., and Barondeau, D. P. (2014). Human frataxin activates Fe-S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry 53, 4904–4913. doi: 10.1021/bi500532e
Calap-Quintana, P., Navarro, J. A., González-Fernández, J., Martínez-Sebastián, M. J., Moltó, M. D., and Llorens, J. V. (2018). Drosophila melanogaster Models of Friedreich’s Ataxia. Biomed Res. Int. 2018:5065190. doi: 10.1155/2018/5065190
Calmels, N., Schmucker, S., Wattenhofer-Donze, M., Martelli, A., Vaucamps, N., Reutenauer, L., et al. (2009). The first cellular models based on frataxin missense mutations that reproduce spontaneously the defects associated with Friedreich ataxia. PLoS One 4:e6379. doi: 10.1371/journal.pone.0006379
Campbell, C. J., Pall, A. E., Naik, A. R., Thompson, L. N., and Stemmler, T. L. (2021). Molecular Details of the Frataxin–Scaffold interaction during mitochondrial Fe–S Cluster Assembly. Int. J. Mol. Sci. 22:6006. doi: 10.3390/ijms22116006
Campuzano, V., Montermini, L., Lutz, Y., Cova, L., Hindelang, C., Jiralerspong, S., et al. (1997). Frataxin is Reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 6, 1771–1780. doi: 10.1093/hmg/6.11.1771
Campuzano, V., Montermini, L., Molto, M. D., Pianese, L., Cossee, M., Cavalcanti, F., et al. (1996). Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427. doi: 10.1126/science.271.5254.1423
Cardenas-Rodriguez, M., Chatzi, A., and Tokatlidis, K. (2018). Iron-sulfur clusters: from metals through mitochondria biogenesis to disease. J. Biol. Inorg. Chem. 23, 509–520. doi: 10.1007/s00775-018-1548-6
Cavadini, P., O’Neill, H. A., Benada, O., and Isaya, G. (2002). Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum. Mol. Genet. 11, 217–227. doi: 10.1093/hmg/11.3.217
Chamberlain, S., Shaw, J., Rowland, A., Wallis, J., South, S., Nakamura, Y., et al. (1988). Mapping of mutation causing Friedreich’s ataxia to human chromosome 9. Nature 334, 248–250. doi: 10.1038/334248a0
Cho, S.-J., Lee, M. G., Yang, J. K., Lee, J. Y., Song, H. K., and Suh, S. W. (2000). Crystal structure of Escherichia coli CyaY protein reveals a previously unidentified fold for the evolutionarily conserved frataxin family. Proc. Natl. Acad. Sci. U.S.A. 97, 8932–8937. doi: 10.1073/pnas.160270897
Chrétien, D., Bénit, P., Ha, H.-H., Keipert, S., El-Khoury, R., Chang, Y.-T., et al. (2018). Mitochondria are physiologically maintained at close to 50°C. PLoS Biol. 16:e2003992. doi: 10.1371/journal.pbio.2003992
Chung, J., Chen, C., and Paw, B. H. (2012). Heme metabolism and erythropoiesis. Curr. Opin. Hematol. 19, 156–162. doi: 10.1097/MOH.0b013e328351c48b
Clay, A., Hearle, P., Schadt, K., and Lynch, D. R. (2019). New developments in pharmacotherapy for Friedreich ataxia. Exp. Opin. Pharmacother. 20, 1855–1867. doi: 10.1080/14656566.2019.1639671
Colin, F., Martelli, A., Clemancey, M., Latour, J. M., Gambarelli, S., Zeppieri, L., et al. (2013). Mammalian frataxin controls sulfur production and iron entry during de novo Fe4S4 cluster assembly. J. Am. Chem. Soc. 135, 733–740. doi: 10.1021/ja308736e
Condò, I., Ventura, N., Malisan, F., Rufini, A., Tomassini, B., and Testi, R. (2007). In vivo maturation of human frataxin. Hum. Mol. Genet. 16, 1534–1540. doi: 10.1093/hmg/ddm102
Condo, I., Ventura, N., Malisan, F., Tomassini, B., and Testi, R. (2006). A pool of extramitochondrial frataxin that promotes cell survival. J. Biol. Chem. 281, 16750–16756. doi: 10.1074/jbc.M511960200
Cook, A., and Giunti, P. (2017). Friedreich’s ataxia: clinical features, pathogenesis and management. Bri. Med. Bull. 124, 19–30. doi: 10.1093/bmb/ldx034
Cook, J. D., Bencze, K. Z., Jankovic, A. D., Crater, A. K., Busch, C. N., Bradley, P. B., et al. (2006). Monomeric yeast frataxin is an iron-binding protein. Biochemistry 45, 7767–7777. doi: 10.1021/bi060424r
Cossee, M., Puccio, H., Gansmuller, A., Koutnikova, H., Dierich, A., LeMeur, M., et al. (2000). Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum. Mol. Genet. 9, 1219–1226. doi: 10.1093/hmg/9.8.1219
Crack, J. C., Green, J., Thomson, A. J., and Brun, N. E. L. (2014). Iron–Sulfur clusters as biological sensors: the chemistry of reactions with molecular oxygen and nitric oxide. Acc. Chem. Res. 47, 3196–3205. doi: 10.1021/ar5002507
Crisp, R. J., Pollington, A., Galea, C., Jaron, S., Yamaguchi-Iwai, Y., and Kaplan, J. (2003). Inhibition of Heme biosynthesis prevents transcription of iron uptake genes in yeast. J. Biol. Chem. 278, 45499–45506. doi: 10.1074/jbc.M307229200
D’Autreaux, B., and Toledano, M. B. (2007). ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 8, 813–824. doi: 10.1038/nrm2256
Delatycki, M. B., and Corben, L. A. (2012). Clinical features of friedreich ataxia. J. Child Neurol. 27, 1133–1137. doi: 10.1177/0883073812448230
Delatycki, M. B., Williamson, R., and Forrest, S. M. (2000). Friedreich ataxia: an overview. J. Med. Genet. 37, 1–8. doi: 10.1136/jmg.37.1.1
Dhe-Paganon, S., Shigeta, R., Chi, Y.-I., Ristow, M., and Shoelson, S. E. (2000). Crystal structure of human Frataxin. J. Biol. Chem. 275, 30753–30756. doi: 10.1074/jbc.C000407200
Duby, G., Foury, F., Ramazzotti, A., Herrmann, J., and Lutz, T. (2002). A non-essential function for yeast frataxin in iron-sulfur cluster assembly. Hum. Mol. Genet. 11, 2635–2643. doi: 10.1093/hmg/11.21.2635
Dürr, A., Cossee, M., Agid, Y., Campuzano, V., Mignard, C., Penet, C., et al. (1996). Clinical and genetic abnormalities in patients with Friedreich’s Ataxia. New Eng. J. Med. 335, 1169–1175. doi: 10.1056/nejm199610173351601
Esteves, F., Rueff, J., and Kranendonk, M. (2021). The Central Role of Cytochrome P450 in Xenobiotic Metabolism—A Brief Review on a Fascinating Enzyme Family. J. Xenobiot. 11, 94–114. doi: 10.3390/jox11030007
Fernández-Frías, I., Pérez-Luz, S., and Díaz-Nido, J. (2020). Analysis of putative epigenetic regulatory elements in the FXN Genomic Locus. Int. J. Mol. Sci. 21:3410. doi: 10.3390/ijms21103410
Fil, D., Chacko, B. K., Conley, R., Ouyang, X., Zhang, J., Darley-Usmar, V. M., et al. (2020). Mitochondrial damage and senescence phenotype of cells derived from a novel frataxin G127V point mutation mouse model of Friedreich’s ataxia. Dis. Model. Mech. 13, doi: 10.1242/dmm.045229
Filla, A., De Michele, G., Cavalcanti, F., Pianese, L., Monticelli, A., Campanella, G., et al. (1996). The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet 59, 554–560.
Flohé, L. (2020). Looking back at the early stages of redox biology. Antioxidants 9:1254. doi: 10.3390/antiox9121254
Foury, F., and Cazzalini, O. (1997). Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett. 411, 373–377. doi: 10.1016/S0014-5793(97)00734-5
Foury, F., Pastore, A., and Trincal, M. (2007). Acidic residues of yeast frataxin have an essential role in Fe–S cluster assembly. EMBO Rep. 8, 194–199. doi: 10.1038/sj.embor.7400881
Fox, N. G., Chakrabarti, M., McCormick, S. P., Lindahl, P. A., and Barondeau, D. P. (2015). The Human Iron–Sulfur Assembly Complex Catalyzes the Synthesis of [2Fe-2S] Clusters on ISCU2 That Can Be Transferred to Acceptor Molecules. Biochemistry 54, 3871–3879. doi: 10.1021/bi5014485
Fox, N. G., Yu, X., Feng, X., Bailey, H. J., Martelli, A., Nabhan, J. F., et al. (2019). Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 10:2210. doi: 10.1038/s41467-019-09989-y
Friedreich, N. (1863). “XVII. Leber degenerative Atrophie der spinalen Hinterstränge,” in Band 26, ed. R. Virchow (Berlin: De Gruyter), 391–419. doi: 10.1515/9783112391280-017
Fugate, C. J., and Jarrett, J. T. (2012). Biotin synthase: insights into radical-mediated carbon–sulfur bond formation. Biochim. Biophys. Acta 1824, 1213–1222. doi: 10.1016/j.bbapap.2012.01.010
Fuss, J. O., Tsai, C.-L., Ishida, J. P., and Tainer, J. A. (2015). Emerging critical roles of Fe–S clusters in DNA replication and repair. Biochim. Biophys. Acta 1853, 1253–1271. doi: 10.1016/j.bbamcr.2015.01.018
Gakh, O., Adamec, J., Gacy, A. M., Twesten, R. D., Owen, W. G., and Isaya, G. (2002). Physical evidence that yeast frataxin is an iron storage protein. Biochemistry 41, 6798–6804. doi: 10.1021/bi025566+
Gakh, O., Bedekovics, T., Duncan, S. F., Smith, D. Y. T., Berkholz, D. S., and Isaya, G. (2010). Normal and Friedreich ataxia cells express different isoforms of frataxin with complementary roles in iron-sulfur cluster assembly. J. Biol. Chem. 285, 38486–38501. doi: 10.1074/jbc.M110.145144
Galea, C. A., Huq, A., Lockhart, P. J., Tai, G., Corben, L. A., Yiu, E. M., et al. (2016). Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann. Neurol. 79, 485–495. doi: 10.1002/ana.24595
Gerber, J., Mühlenhoff, U., and Lill, R. (2003). An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 4, 906–911. doi: 10.1038/sj.embor.embor918
Gervason, S., Larkem, D., Mansour, A. B., Botzanowski, T., Muller, C. S., Pecqueur, L., et al. (2019). Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 10:3566. doi: 10.1038/s41467-019-11470-9
Gervason, S., Srour, B., and D’Autreaux, B. A. (2021). Fast and ratiometric method for quantification of cysteine-bound persulfides based on alkylation and gel-shift assays. Methods Mol. Biol. 2353, 191–205. doi: 10.1007/978-1-0716-1605-5_11
Gottesfeld, J. M. (2019). Molecular mechanisms and therapeutics for the GAA⋅TTC Expansion Disease Friedreich Ataxia. Neurotherapeutics 16, 1032–1049. doi: 10.1007/s13311-019-00764-x
Guccini, I., Serio, D., Condò, I., Rufini, A., Tomassini, B., Mangiola, A., et al. (2011). Frataxin participates to the hypoxia-induced response in tumors. Cell Death Dis. 2:e123. doi: 10.1038/cddis.2011.5
Guo, L., Wang, Q., Weng, L., Hauser, L. A., Strawser, C. J., Mesaros, C., et al. (2018). Characterization of a new N-terminally acetylated extra-mitochondrial isoform of frataxin in human erythrocytes. Sci. Rep. 8:17043. doi: 10.1038/s41598-018-35346-y
He, Y., Alam, S. L., Proteasa, S. V., Zhang, Y., Lesuisse, E., Dancis, A., et al. (2004). Yeast frataxin solution structure, iron binding, and ferrochelatase interaction. Biochemistry 43, 16254–16262. doi: 10.1021/bi0488193
Hider, R. C., and Kong, X. (2013). Iron speciation in the cytosol: an overview. Dalton Trans. 42, 3220–3229. doi: 10.1039/c2dt32149a
Indelicato, E., Nachbauer, W., Eigentler, A., Amprosi, M., Matteucci Gothe, R., Giunti, P., et al. (2020). Onset features and time to diagnosis in Friedreich’s Ataxia. Orphan. J. Rare Dis. 15:198. doi: 10.1186/s13023-020-01475-9
Jiralerspong, S., Liu, Y., Montermini, L., Stifani, S., and Pandolfo, M. (1997). Frataxin shows developmentally regulated tissue-specific expression in the mouse embryo. Neurobiol. Dis. 4, 103–113. doi: 10.1006/nbdi.1997.0139
Kimura, S., and Suzuki, T. (2015). Iron–sulfur proteins responsible for RNA modifications. Biochim. Biophys. Acta 1853, 1272–1283. doi: 10.1016/j.bbamcr.2014.12.010
Kispal, G., Sipos, K., Lange, H., Fekete, Z., Bedekovics, T., Janaky, T., et al. (2005). Biogenesis of cytosolic ribosomes requires the essential iron-sulphur protein Rli1p and mitochondria. EMBO J. 24, 589–598. doi: 10.1038/sj.emboj.7600541
Koutnikova, H., Campuzano, V., and Koenig, M. (1998). Maturation of Wild-Type and Mutated Frataxin by the mitochondrial processing peptidase. Hum. Mol. Genet. 7, 1485–1489. doi: 10.1093/hmg/7.9.1485
Lamarche, J. B., Côté, M., and Lemieux, B. (1980). The Cardiomyopathy of Friedreich’s ataxia morphological observations in 3 Cases. Can. J. Neurol. Sci. 7, 389–396. doi: 10.1017/s0317167100022927
Lange, H., Muhlenhoff, U., Denzel, M., Kispal, G., and Lill, R. (2004). The heme synthesis defect of mutants impaired in mitochondrial iron-sulfur protein biogenesis is caused by reversible inhibition of ferrochelatase. J. Biol. Chem. 279, 29101–29108. doi: 10.1074/jbc.M403721200
Layer, G., Ollagnier-de Choudens, S., Sanakis, Y., and Fontecave, M. (2006). Iron-sulfur cluster biosynthesis: characterization of Escherichia coli CYaY as an iron donor for the assembly of [2Fe-2S] clusters in the scaffold IscU. J. Biol. Chem. 281, 16256–16263. doi: 10.1074/jbc.M513569200
Lazaropoulos, M., Dong, Y., Clark, E., Greeley, N. R., Seyer, L. A., Brigatti, K. W., et al. (2015). Frataxin levels in peripheral tissue in Friedreich ataxia. Annals Clin. Trans. Neurol. 2, 831–842. doi: 10.1002/acn3.225
Leidgens, S., De Smet, S., and Foury, F. (2010). Frataxin interacts with Isu1 through a conserved tryptophan in its beta-sheet. Hum. Mol. Genet. 19, 276–286. doi: 10.1093/hmg/ddp495
Leimkühler, S., Bühning, M., and Beilschmidt, L. (2017). Shared sulfur mobilization routes for tRNA Thiolation and Molybdenum cofactor biosynthesis in prokaryotes and eukaryotes. Biomolecules 7:5. doi: 10.3390/biom7010005
Lesuisse, E., Santos, R., Matzanke, B. F., Knight, S. A., Camadro, J. M., and Dancis, A. (2003). Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1). Hum. Mol. Genet. 12, 879–889. doi: 10.1093/hmg/ddg096
Li, J., and Cowan, J. A. (2015). Glutathione-coordinated [2Fe-2S] cluster: a viable physiological substrate for mitochondrial ABCB7 transport. Chem. Commun. 51, 2253–2255. doi: 10.1039/c4cc09175b
Li, K., Besse, E. K., Ha, D., Kovtunovych, G., and Rouault, T. A. (2008). Iron-dependent regulation of frataxin expression: implications for treatment of Friedreich ataxia. Hum. Mol. Genet. 17, 2265–2273. doi: 10.1093/hmg/ddn127
Lill, R., and Freibert, S. A. (2020). Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu. Rev. Biochem. 89, 471–499. doi: 10.1146/annurev-biochem-013118-111540
Lill, R., Hoffmann, B., Molik, S., Pierik, A. J., Rietzschel, N., Stehling, O., et al. (2012). The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta 1823, 1491–1508. doi: 10.1016/j.bbamcr.2012.05.009
Lin, C. W., McCabe, J. W., Russell, D. H., and Barondeau, D. P. (2020). Molecular Mechanism of ISC iron-sulfur cluster biogenesis revealed by high-resolution native mass spectrometry. J. Am. Chem. Soc. 142, 6018–6029. doi: 10.1021/jacs.9b11454
Lindahl, P. A., and Moore, M. J. (2016). Labile low-molecular-mass metal complexes in mitochondria: trials and tribulations of a burgeoning field. Biochemistry 55, 4140–4153. doi: 10.1021/acs.biochem.6b00216
Liu, J., Verma, P. J., Evans-Galea, M. V., Delatycki, M. B., Michalska, A., Leung, J., et al. (2011). Generation of induced pluripotent stem cell lines from friedreich ataxia patients. Stem Cell Rev. Rep. 7, 703–713. doi: 10.1007/s12015-010-9210-x
Lotierzo, M., Tse Sum Bui, B., Florentin, D., Escalettes, F., and Marquet, A. (2005). Biotin synthase mechanism: an overview. Biochem. Soc. Trans. 33, 820–823. doi: 10.1042/bst0330820
Lu, C., and Cortopassi, G. (2007). Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts. Arch. Biochem. Biophys. 457, 111–122. doi: 10.1016/j.abb.2006.09.010
Lynch, D. R., and Farmer, G. (2021). Mitochondrial and metabolic dysfunction in Friedreich ataxia: update on pathophysiological relevance and clinical interventions. Neuronal Signal. 5:NS20200093. doi: 10.1042/ns20200093
Lynch, D. R., Schadt, K., Kichula, E., McCormack, S., and Lin, K. Y. (2021). Friedreich ataxia: multidisciplinary clinical care. J. Multidiscip. Healthc. 14, 1645–1658. doi: 10.2147/jmdh.s292945
Martelli, A., and Puccio, H. (2014). Dysregulation of cellular iron metabolism in Friedreich ataxia: from primary iron-sulfur cluster deficit to mitochondrial iron accumulation. Front. Pharmacol. 5:130. doi: 10.3389/fphar.2014.00130
Martelli, A., Wattenhofer-Donze, M., Schmucker, S., Bouvet, S., Reutenauer, L., and Puccio, H. (2007). Frataxin is essential for extramitochondrial Fe-S cluster proteins in mammalian tissues. Hum. Mol. Genet. 16, 2651–2658. doi: 10.1093/hmg/ddm163
Medina-Carbonero, M., Sanz-Alcázar, A., Britti, E., Delaspre, F., Cabiscol, E., Ros, J., et al. (2022). Mice harboring the FXN I151F pathological point mutation present decreased frataxin levels, a Friedreich ataxia-like phenotype, and mitochondrial alterations. Cell. Mol. Life Sci. 79:74. doi: 10.1007/s00018-021-04100-5
Miranda, C. J., Santos, M. M., Ohshima, K., Smith, J., Li, L., Bunting, M., et al. (2002). Frataxin knockin mouse. FEBS Lett. 512, 291–297. doi: 10.1016/s0014-5793(02)02251-2
Montermini, L., Richeter, A., Morgan, K., Justice, C. M., Julien, D., Castellotti, B., et al. (1997). Phenotypic variability in friedreich ataxia: Role of the associated GAA triplet repeat expansion. Ann. Neurol. 41, 675–682. doi: 10.1002/ana.410410518
Morgan, R. O., Naglie, G., Horrobin, D. F., and Barbeau, A. (1979). Erythrocyte protoporphyrin levels in patients with Friedreich’s and other ataxias. Can. J. Neurol. Sci. 6, 227–232. doi: 10.1017/s0317167100119699
Muhlenhoff, U., Hoffmann, B., Richter, N., Rietzschel, N., Spantgar, F., Stehling, O., et al. (2015). Compartmentalization of iron between mitochondria and the cytosol and its regulation. Eur. J. Cell. Biol. 94, 292–308. doi: 10.1016/j.ejcb.2015.05.003
Muhlenhoff, U., Richhardt, N., Ristow, M., Kispal, G., and Lill, R. (2002). The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum. Mol. Genet. 11, 2025–2036. doi: 10.1093/hmg/11.17.2025
Muhlenhoff, U., Richter, N., Pines, O., Pierik, A. J., and Lill, R. (2011). Specialized function of yeast Isa1 and Isa2 proteins in the maturation of mitochondrial [4Fe-4S] proteins. J. Biol. Chem. 286, 41205–41216. doi: 10.1074/jbc.M111.296152
Mulliez, E., Duarte, V., Arragain, S., Fontecave, M., and Atta, M. (2017). On the Role of Additional [4Fe-4S] Clusters with a Free Coordination Site in Radical-SAM Enzymes. Front. Chem. 5:17. doi: 10.3389/fchem.2017.00017
Musco, G., Stier, G., Kolmerer, B., Adinolfi, S., Martin, S., Frenkiel, T., et al. (2000). Towards a structural understanding of Friedreich’s ataxia: the solution structure of frataxin. Structure 8, 695–707. doi: 10.1016/s0969-2126(00)00158-1
Nanayakkara, G., Alasmari, A., Mouli, S., Eldoumani, H., Quindry, J., McGinnis, G., et al. (2015). Cardioprotective HIF-1α-frataxin signaling against ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 309, H867–H879. doi: 10.1152/ajpheart.00875.2014
Navarro, J. A., Llorens, J. V., Soriano, S., Botella, J. A., Schneuwly, S., Martínez-Sebastián, M. J., et al. (2011). Overexpression of Human and Fly Frataxins in Drosophila provokes deleterious effects at biochemical, physiological and developmental levels. PLoS One 6:e21017. doi: 10.1371/journal.pone.0021017
Ocana-Santero, G., Díaz-Nido, J., and Herranz-Martín, S. (2021). Future Prospects of Gene Therapy for Friedreich’s Ataxia. Int. J. Mol. Sci. 22:1815. doi: 10.3390/ijms22041815
Oktay, Y., Dioum, E., Matsuzaki, S., Ding, K., Yan, L.-J., Haller, R. G., et al. (2007). Hypoxia-inducible Factor 2 regulates expression of the mitochondrial aconitase chaperone protein Frataxin. J. Biol. Chem. 282, 11750–11756. doi: 10.1074/jbc.M611133200
O’Neill, H. A., Gakh, O., and Isaya, G. (2005). Supramolecular assemblies of human frataxin are formed via subunit-subunit interactions mediated by a non-conserved amino-terminal region. J. Mol. Biol. 345, 433–439. doi: 10.1016/j.jmb.2004.10.074
Pallardó, F. V., Pagano, G., Rodríguez, L. R., Gonzalez-Cabo, P., Lyakhovich, A., and Trifuoggi, M. (2021). Friedreich Ataxia: current state-of-the-art, and future prospects for mitochondrial-focused therapies. Trans. Res. 229, 135–141. doi: 10.1016/j.trsl.2020.08.009
Pandey, A., Gordon, D. M., Pain, J., Stemmler, T. L., Dancis, A., and Pain, D. (2013). Frataxin directly stimulates mitochondrial cysteine desulfurase by exposing substrate-binding sites, and a mutant Fe-S cluster scaffold protein with frataxin-bypassing ability acts similarly. J. Biol. Chem. 288, 36773–36786. doi: 10.1074/jbc.M113.525857
Pandey, A. K., Pain, J., Dancis, A., and Pain, D. (2019). Mitochondria export iron-sulfur and sulfur intermediates to the cytoplasm for iron-sulfur cluster assembly and tRNA thiolation in yeast. J. Biol. Chem. 294, 9489–9502. doi: 10.1074/jbc.RA119.008600
Pandolfo, M., Arpa, J., Delatycki, M. B., Le Quan Sang, K. H., Mariotti, C., Munnich, A., et al. (2014). Deferiprone in Friedreich ataxia: a 6-Month randomized controlled trial. Ann. Neurol. 76, 509–521. doi: 10.1002/ana.24248
Parent, A., Elduque, X., Cornu, D., Belot, L., Le Caer, J. P., Grandas, A., et al. (2015). Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun. 6:5686. doi: 10.1038/ncomms6686
Parkinson, M. H., Boesch, S., Nachbauer, W., Mariotti, C., and Giunti, P. (2013). Clinical features of Friedreich’s ataxia: classical and atypical phenotypes. J. Neurochem. 126, 103–117. doi: 10.1111/jnc.12317
Pastore, A., and Puccio, H. (2013). Frataxin: a protein in search for a function. J. Neurochem. 126, (Suppl. 1), 43–52. doi: 10.1111/jnc.12220
Patra, S., and Barondeau, D. P. (2019). Mechanism of activation of the human cysteine desulfurase complex by frataxin. Proc. Natl. Acad. Sci. U.S.A. 116, 19421–19430. doi: 10.1073/pnas.1909535116
Pérez-Luz, S., Gimenez-Cassina, A., Fernández-Frías, I., Wade-Martins, R., and Díaz-Nido, J. (2015). Delivery of the 135kb human frataxin genomic DNA locus gives rise to different frataxin isoforms. Genomics 106, 76–82. doi: 10.1016/j.ygeno.2015.05.006
Philpott, C. C., and Jadhav, S. (2019). The ins and outs of iron: escorting iron through the mammalian cytosol. Free Radic. Biol. Med. 133, 112–117. doi: 10.1016/j.freeradbiomed.2018.10.411
Pianese, L., Tammaro, A., Turano, M., De Biase, I., Monticelli, A., and Cocozza, S. (2002). Identification of a novel transcript of X25, the human gene involved in Friedreich ataxia. Neuro Lett. 320, 137–140. doi: 10.1016/S0304-3940(02)00048-4
Poburski, D., Boerner, J. B., Koenig, M., Ristow, M., and Thierbach, R. (2016). Time-resolved functional analysis of acute impairment of frataxin expression in an inducible cell model of Friedreich ataxia. Biol. Open 5, 654–661. doi: 10.1242/bio.017004
Prischi, F., Giannini, C., Adinolfi, S., and Pastore, A. (2009). The N-terminus of mature human frataxin is intrinsically unfolded. FEBS J. 276, 6669–6676. doi: 10.1111/j.1742-4658.2009.07381.x
Puccio, H., Simon, D., Cossee, M., Criqui-Filipe, P., Tiziano, F., Melki, J., et al. (2001). Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 27, 181–186. doi: 10.1038/84818
Ramazzotti, A., Vanmansart, V., and Foury, F. (2004). Mitochondrial functional interactions between frataxin and Isu1p, the iron–sulfur cluster scaffold protein, in Saccharomyces cerevisiae. FEBS Lett. 557, 215–220. doi: 10.1016/S0014-5793(03)01498-4
Reetz, K., Dogan, I., Hilgers, R.-D., Giunti, P., Mariotti, C., Durr, A., et al. (2016). Progression characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS): a 2 year cohort study. Lancet Neurol. 15, 1346–1354. doi: 10.1016/s1474-4422(16)30287-3
Reetz, K., Dogan, I., Hilgers, R.-D., Giunti, P., Parkinson, M. H., Mariotti, C., et al. (2021). Progression characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS): a 4-year cohort study. Lancet Neurol. 20, 362–372. doi: 10.1016/s1474-4422(21)00027-2
Rotig, A., de Lonlay, P., Chretien, D., Foury, F., Koenig, M., Sidi, D., et al. (1997). Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 17, 215–217. doi: 10.1038/ng1097-215
Russi, M., Martin, E., D’Autréaux, B., Tixier, L., Tricoire, H., and Monnier, V. A. (2020). Drosophila model of Friedreich ataxia with CRISPR/Cas9 insertion of GAA repeats in the frataxin gene reveals in vivo protection by N-acetyl cysteine. Hum. Mol. Genet. 29, 2831–2844. doi: 10.1093/hmg/ddaa170
Sahdeo, S., Scott, B. D., McMackin, M. Z., Jasoliya, M., Brown, B., Wulff, H., et al. (2014). Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich’s ataxia. Hum. Mol. Genet. 23, 6848–6862. doi: 10.1093/hmg/ddu408
Sanchez-Casis, G., Cote, M., and Barbeau, A. (1976). Pathology of the Heart in Friedreich’s Ataxia: review of the literature and report of one case. Can. J. Neurol. Sci. 3, 349–354. doi: 10.1017/s0317167100025580
Santos, R., Lefevre, S., Sliwa, D., Seguin, A., Camadro, J.-M., and Lesuisse, E. (2010). Friedreich Ataxia: molecular mechanisms, redox considerations, and therapeutic opportunities. Antioxid. Redox Signal. 13, 651–690. doi: 10.1089/ars.2009.3015
Santucci, R., Sinibaldi, F., Cozza, P., Polticelli, F., and Fiorucci, L. (2019). Cytochrome c: an extreme multifunctional protein with a key role in cell fate. Int. J. Biol. Macromol. 136, 1237–1246. doi: 10.1016/j.ijbiomac.2019.06.180
Schaedler, T. A., Thornton, J. D., Kruse, I., Schwarzlander, M., Meyer, A. J., van Veen, H. W., et al. (2014). A conserved mitochondrial ATP-binding cassette transporter exports glutathione polysulfide for cytosolic metal cofactor assembly. J. Biol. Chem. 289, 23264–23274. doi: 10.1074/jbc.M114.553438
Schmucker, S., Argentini, M., Carelle-Calmels, N., Martelli, A., and Puccio, H. (2008). The in vivo mitochondrial two-step maturation of human frataxin. Hum. Mol. Genet. 17, 3521–3531. doi: 10.1093/hmg/ddn244
Schmucker, S., Martelli, A., Colin, F., Page, A., Wattenhofer-Donze, M., Reutenauer, L., et al. (2011). Mammalian frataxin: an essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS One 6:e16199. doi: 10.1371/journal.pone.0016199
Schoenfeld, R. A., Napoli, E., Wong, A., Zhan, S., Reutenauer, L., Morin, D., et al. (2005). Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum. Mol. Genet. 14, 3787–3799. doi: 10.1093/hmg/ddi393
Schultz, R., Witte, O. W., and Schmeer, C. (2016). Increased Frataxin levels protect retinal ganglion cells after acute ischemia/reperfusion in the mouse retina in vivo. Invest. Ophthalmol. Vis. Sci. 57, 4115–4124. doi: 10.1167/iovs.16-19260
Seguin, A., Sutak, R., Bulteau, A. L., Garcia-Serres, R., Oddou, J. L., Lefevre, S., et al. (2010). Evidence that yeast frataxin is not an iron storage protein in vivo. Biochim. Biophys. Acta 1802, 531–538. doi: 10.1016/j.bbadis.2010.03.008
Sen Gupta, A. (2019). Hemoglobin-based oxygen carriers: current state-of-the-art and novel molecules. Shock 52(1S Suppl 1), 70–83. doi: 10.1097/SHK.0000000000001009
Seznec, H., Simon, D., Bouton, C., Reutenauer, L., Hertzog, A., Golik, P., et al. (2005). Friedreich ataxia: the oxidative stress paradox. Hum. Mol. Genet. 14, 463–474. doi: 10.1093/hmg/ddi042
Shi, R., Hou, W., Wang, Z.-Q., and Xu, X. (2021). Biogenesis of Iron–Sulfur Clusters and Their Role in DNA Metabolism. Front. Cell Dev. Biol. 9:735678. doi: 10.3389/fcell.2021.735678
Shimizu, T., Lengalova, A., Martínek, V., and Martínková, M. (2019). Heme: emergent roles of heme in signal transduction, functional regulation and as catalytic centres. Chem. Soc. Rev. 48, 5624–5657. doi: 10.1039/c9cs00268e
Smith, F. M., and Kosman, D. J. (2020). Molecular defects in Friedreich’s ataxia: convergence of oxidative stress and cytoskeletal abnormalities. Front. Mol. Biosci. 7:569293. doi: 10.3389/fmolb.2020.569293
Soderberg, C., Gillam, M. E., Ahlgren, E. C., Hunter, G. A., Gakh, O., Isaya, G., et al. (2016). The Structure of the Complex between Yeast Frataxin and Ferrochelatase: characterization and pre-steady state reaction of ferrous iron delivery and heme synthesis. J. Biol. Chem. 291, 11887–11898. doi: 10.1074/jbc.M115.701128
Soderberg, C. A. G., Rajan, S., Shkumatov, A. V., Gakh, O., Schaefer, S., Ahlgren, E. C., et al. (2013). The molecular basis of iron-induced oligomerization of frataxin and the role of the ferroxidation reaction in oligomerization. J. Biol. Chem. 288, 8156–8167. doi: 10.1074/jbc.M112.442285
Srour, B., Gervason, S., Monfort, B., and D’Autréaux, B. (2020). Mechanism of iron-sulfur cluster assembly: in the intimacy of iron and sulfur encounter. Inorganics 8:55. doi: 10.3390/inorganics8100055
Stehling, O., Wilbrecht, C., and Lill, R. (2014). Mitochondrial iron-sulfur protein biogenesis and human disease. Biochimie 100, 61–77. doi: 10.1016/j.biochi.2014.01.010
Steinkellner, H., Singh, H. N., Muckenthaler, M. U., Goldenberg, H., Moganty, R. R., Scheiber-Mojdehkar, B., et al. (2017). No changes in heme synthesis in human Friedreich s ataxia erythroid progenitor cells. Gene 621, 5–11. doi: 10.1016/j.gene.2017.04.014
Stiban, J., So, M., and Kaguni, L. S. (2016). Iron-sulfur clusters in mitochondrial metabolism: multifaceted roles of a simple cofactor. Biochemistry 81, 1066–1080. doi: 10.1134/s0006297916100059
Synofzik, M., and Németh, A. H. (2018). “Chapter 5 - Recessive ataxias,” in Handbook of Clinical Neurology, eds M. Manto and T. A. G. M. Huisman (Amsterdam: Elsevier), 73–89.
Torrents, E., Aloy, P., Gibert, I., and Rodríguez-Trelles, F. (2002). Ribonucleotide reductases: divergent evolution of an ancient enzyme. J. Mol. Evol. 55, 138–152. doi: 10.1007/s00239-002-2311-7
Tsai, C. L., and Barondeau, D. P. (2010). Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry 49, 9132–9139. doi: 10.1021/bi1013062
Tsou, A. Y., Paulsen, E. K., Lagedrost, S. J., Perlman, S. L., Mathews, K. D., Wilmot, G. R., et al. (2011). Mortality in Friedreich Ataxia. J. Neurol. Sci. 307, 46–49. doi: 10.1016/j.jns.2011.05.023
Van Vranken, J. G., Nowinski, S. M., Clowers, K. J., Jeong, M. Y., Ouyang, Y., Berg, J. A., et al. (2018). ACP acylation is an acetyl-CoA-dependent modification required for electron transport chain assembly. Mol. Cell 71, 567–580.e4. doi: 10.1016/j.molcel.2018.06.039
Vannocci, T., Notario Manzano, R., Beccalli, O., Bettegazzi, B., Grohovaz, F., Cinque, G., et al. (2018). Adding a temporal dimension to the study of Friedreich’s ataxia: the effect of frataxin overexpression in a human cell model. Dis. Model. Mech. 11:dmm032706. doi: 10.1242/dmm.032706
Wang, T., and Craig, E. A. (2008). Binding of Yeast Frataxin to the Scaffold for Fe-S Cluster Biogenesis, Isu. J. Biol. Chem. 283, 12674–12679. doi: 10.1074/jbc.M800399200
Wang, Y., Wang, Y., Marcus, S., and Busenlehner, L. S. (2014). The role of frataxin in fission yeast iron metabolism: Implications for Friedreich’s ataxia. Biochim. Biophys. Acta 1840, 3022–3033. doi: 10.1016/j.bbagen.2014.06.017
Weerth, R. S., Medlock, A. E., and Dailey, H. A. (2021). Ironing out the distribution of [2Fe-2S] motifs in ferrochelatases. J. Biol. Chem. 297:101017. doi: 10.1016/j.jbc.2021.101017
Weiler, B. D., Bruck, M. C., Kothe, I., Bill, E., Lill, R., and Muhlenhoff, U. (2020). Mitochondrial [4Fe-4S] protein assembly involves reductive [2Fe-2S] cluster fusion on ISCA1-ISCA2 by electron flow from ferredoxin FDX2. Proc. Natl. Acad. Sci. U.S.A. 117, 20555–20565. doi: 10.1073/pnas.2003982117
Weng, L., Laboureur, L., Wang, Q., Guo, L., Xu, P., Gottlieb, L., et al. (2020). Extra-mitochondrial mouse frataxin and its implications for mouse models of Friedreich’s ataxia. Sci. Rep. 10:15788. doi: 10.1038/s41598-020-72884-w
Wilson, R. B., and Roof, D. M. (1997). Respiratory deficiency due to loss of mitochondrial DNA in yeast lacking the frataxin homologue. Nat. Genet. 16, 352–357. doi: 10.1038/ng0897-352
Wofford, J. D., Bolaji, N., Dziuba, N., Outten, F. W., and Lindahl, P. A. (2019). Evidence that a respiratory shield in Escherichia coli protects a low-molecular-mass Fe(II) pool from O2-dependent oxidation. J. Biol. Chem. 294, 50–62. doi: 10.1074/jbc.RA118.005233
Xia, H., Cao, Y., Dai, X., Marelja, Z., Zhou, D., Mo, R., et al. (2012). Novel Frataxin isoforms may contribute to the pathological mechanism of Friedreich Ataxia. PLoS One 7:e47847. doi: 10.1371/journal.pone.0047847
Yandim, C., Natisvili, T., and Festenstein, R. (2013). Gene regulation and epigenetics in Friedreich’s ataxia. J. Neurochem. 126, 21–42. doi: 10.1111/jnc.12254
Yang, W., Thompson, B., and Kwa, F. A. A. (2021). Molecular approaches for the treatment and prevention of Friedreich’s ataxia. Drug Discov. Today. [Epub ahead of print]. doi: 10.1016/j.drudis.2021.11.003
Yoon, T., and Cowan, J. A. (2003). Iron-Sulfur cluster biosynthesis. characterization of frataxin as an iron donor for assembly of [2Fe-2S] Clusters in ISU-Type Proteins. J. Am. Chem. Soc. 125, 6078–6084. doi: 10.1021/ja027967i
Yoon, T., and Cowan, J. A. (2004). Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 279, 25943–25946. doi: 10.1074/jbc.C400107200
Zesiewicz, T. A., Hancock, J., Ghanekar, S. D., Kuo, S.-H., Dohse, C. A., and Vega, J. (2020). Emerging therapies in Friedreich’s Ataxia. Exp. Rev. Neurother. 20, 1215–1228. doi: 10.1080/14737175.2020.1821654
Zhang, Y., Li, H., Zhang, C., An, X., Liu, L., Stubbe, J., et al. (2014). Conserved electron donor complex Dre2–Tah18 is required for ribonucleotide reductase metallocofactor assembly and DNA synthesis. Proc. Nat. Acad. Sci. U.S.A. 111, E1695. doi: 10.1073/pnas.1405204111
Keywords: frataxin, iron-sulfur cluster, persulfide, Friedreich’s ataxia, therapy
Citation: Monfort B, Want K, Gervason S and D’Autréaux B (2022) Recent Advances in the Elucidation of Frataxin Biochemical Function Open Novel Perspectives for the Treatment of Friedreich’s Ataxia. Front. Neurosci. 16:838335. doi: 10.3389/fnins.2022.838335
Received: 17 December 2021; Accepted: 28 January 2022;
Published: 02 March 2022.
Edited by:
David Lynch, University of Pennsylvania, United StatesReviewed by:
M. Grazia Cotticelli, Children’s Hospital of Philadelphia, United StatesCopyright © 2022 Monfort, Want, Gervason and D’Autréaux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benoit D’Autréaux, YmVub2l0LmRhdXRyZWF1eEBpMmJjLnBhcmlzLXNhY2xheS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.