 Kristyna Kolarikova1
Kristyna Kolarikova1 Radek Vodicka1*Radek Vrtel1Julia Stellmachova2Martin Prochazka2
Radek Vodicka1*Radek Vrtel1Julia Stellmachova2Martin Prochazka2 Katerina Mensikova3
Katerina Mensikova3 Petr Kanovsky4
Petr Kanovsky4
- 1Department of Medical Genetics, University Hospital Olomouc, Olomouc, Czechia
- 2Department of Medical Genetics, Faculty of Medicine and Dentistry, Palacký University Olomouc, Olomouc, Czechia
- 3Department of Neurology, Faculty of Medicine and Dentistry, Palacký University Olomouc, Olomouc, Czechia
- 4Department of Neurology, University Hospital Olomouc, Olomouc, Czechia
Parkinsonism belongs to the most common neurodegenerative disease. Genetic predisposition could be one of the significant risk factor for disease development. It has been described higher prevalence of parkinsonism in large pedigree from southeastern Moravia region. The study aims were to select accessible subfamily trios from the pedigree suitable for segregation genetic analyses to perform whole exome sequencing (WES) in trio individuals and further to evaluate genetic variants in the each trio. We used IonTorrent platform for WES for five subfamily trios (1–5). Each trio included two affected and one healthy person (as control). Found variants were filtered with respect to MAF < 1% (minor allele frequency), variants effect (based on prediction tools) and disease filter (Parkinsonism responsible genes). Finally, the variants from each trio were assessed with respect to the presence in the patients. There were found no one founder mutation in the subfamilies from the pedigree. Trio 1 shares two variants with trio 2:MC1R:c.322G > A (p.A108T) and MTCL1:c.1445C > T (p.A482V), trio 3 shares two variants with trio 5: DNAJC6:c.1817A > C (p.H606P) and HIVEP3:c.3856C > A (p.R1286W). In trios 4 and 5, there were found two variants in gene CSMD1:c.3335A > G (p.E1112G) and c.4071C > G (p.I1357M) respectively. As the most potentially damaging, we evaluated the non-shared variant SLC18A2:c.583G > A (p.G195S). The variant could affect dopamine transport in dopaminergic neurons. The study of the parkinsonism genetic background in isolated Moravian population suggested that there could be significant accumulation of many risk genetic factors. For verification of the variants influence, it would be appropriate to perform a more extensive population study and suitable functional analysis.
Introduction
Parkinson’s disease (PD) is one of the most frequent neurodegenerative disorders. In addition to typical symptoms such as resting tremor, rigidity and bradykinesia, there are other symptoms of parkinsonism: hallucinations, postural instability, dementia etc. (Hughes et al., 2002). The most cases of parkinsonism are sporadic and there is affected about 1,5% population over 65 years. In this case, the disease is probably caused by the combination of genetic, environmental and epigenetic risk factors. The familiar form represents 5–10% cases with Mendelian type of inheritance (Kalinderi et al., 2016). Nowadays, there were described more than 90 genes associated with dominant or recessive inheritance of parkinsonism namely SNCA (Campêlo et al., 2017), LRRK2 (Ortega et al., 2021), VPS35 (Mir et al., 2018), Parkin (Yi et al., 2019; Bandres-Ciga et al., 2020), DJ-1 (Dolgacheva et al., 2019), PINK1 (Ando et al., 2017), DNAJC6 (Köroğlu et al., 2013), ADH1C (Buervenich et al., 2005), PLA2G6 (Lin et al., 2018), EIF4G1 (Chartier-Harlin et al., 2011), ATP13A2 (Park et al., 2015) belong to the most important.
The next generation sequencing methods can reveal further genes that could be potential risk factors for parkinsonism.
Despite the many already described causal genes and mutations, genetic predisposition is still unclear in the most of patients. Therefore, it is suitable to change the method strategy from targeted to whole exome (WES) or genome sequencing. It would prepare a field for finding of novel causal or risk genes and variants. NOTCH4 (Yemni et al., 2019), TNK2, TNR (Farlow et al., 2016), NUS1 (Guo et al., 2018), and SORL1 (Xiromerisiou et al., 2021) belong to the potential candidate genes recently identified by WES. Combination of WES data and linkage analysis can be used for identification of novel candidate genes in many diseases (Gazal et al., 2016; Toma et al., 2020).
In our previous epidemiology study, we described higher prevalence of parkinsonism in southeastern Moravia region (Hornacko) compared with general population. This region includes 10 villages, where the local people have their own specific traditions (such as dances, folk art, local dialect and religion) and migration out of the region was rare. Due to many years of territorial and social isolation, it was hypothesized that the accumulation of genetic factors may contribute to higher prevalence of PD in the region (Mensikova et al., 2013).
Thanks to our detailed study, 11 generation pedigree from the Hornacko was compiled with the help of witnesses, registry offices and local general practitioners (Figure 1; Mensikova et al., 2013). Based on that, we were looking for patients from pedigree to receive material for genetic analysis. It was possible to select 5 family trios (two affected and one healthy individual) in subfamilies from the large pedigree.

Figure 1. Pedigree of family from Hornacko. Clear circle/square sign living unaffected female/male; black circle/square sign living affected female/male; symbol with a diagonal line is for deceased individual. Highlighted individuals were accessible for WES.
The study aims were to choose accessible trios from the large pedigree suitable for segregation genetic analyses and perform WES and to call and evaluate variants using two software (Ion Reporter and Ingenuity Variant Analysis) and filtering based on genes association with the disease (parkinsonism and other neurodegenerative diseases) and variants co-occurrence in the patients within particular trio and across the whole pedigree.
Materials and Methods
The study was approved by the Ethics Committee of the Palacký University and University Hospital Olomouc, Czechia. The patients were informed in detail about the study and they all signed informed consent. In our study, 10 patients (8 females and 2 males) and 5 unaffected individuals (3 females and 2 males) were included. The average age of female patients was 67 ± 12.2 years and the average age of male patients was 71 ± 14.1 years. The youngest patient was 56 years and the oldest was 88 years. The average age of controls was 71.8 ± 14.9 years. The youngest control was 51 and the oldest was 87 years. The each trio was composed from 3 family members: two patients and one healthy individual (case assessment is described in Mensikova et al., 2014). We assume autosomal dominant inheritance with reduced penetrance and variable expressivity. The relationships in individual trios: trio 1 (patient number 1 is mother; number 2 is her daughter; control is mother’s brother), trio 2 (patient number 3 is father; number 4 is his daughter; control is father’s brother), trio 3 (patients number 5 and 6 are siblings; control is healthy mother), trio 4 (patient number 7 is mother; number 8 is her daughter; control is mother’s brother), trio 5 (patient number 9 is daughter; number 10 is her mother; control is healthy son). The detailed demographic data are in Table 1. Patients clinical data are described in Table 2. The DNA was isolated in all patients and controls from peripheral blood using salting out method (Miller et al., 1988). WES was performed by commercial company (SEQme, s.r.o., Dobris, Czechia) on Ion Torrent platform. Libraries were prepared using Ion Ampliseq Exome kit (Ion AmpliSeq 2.0 Library, according to manual). Emulsion PCR was done with template kit Ion PI Hi-Q OT2 200. Samples were barcoded to enable to load 3 samples on one Ion PI™ Chip. For sequencing, Ion PI Hi-Q Sequencing 200 kit was used. As reference genome was determinated GRCh37. Sequencing data process includes two parts. For the first part is used Torrent Suite server, where are loaded raw data from sequencer. Raw data (received on the basis of pH change) are converted to single number per well per flow. The next step is base caller, when converted data are translated into base sequence into an unaligned BAM file. For alignment step, there is used Torrent Mapping Alignment Program which performs mapping against reference sequence and it creates BAM files. The second part includes uploading BAM files to Ion Reporter, where is performed variant calling anotation and variants filtering (Vodicka et al., 2020).

Table 1. Demographic data of patients with neurodegenerative parkinsonism.
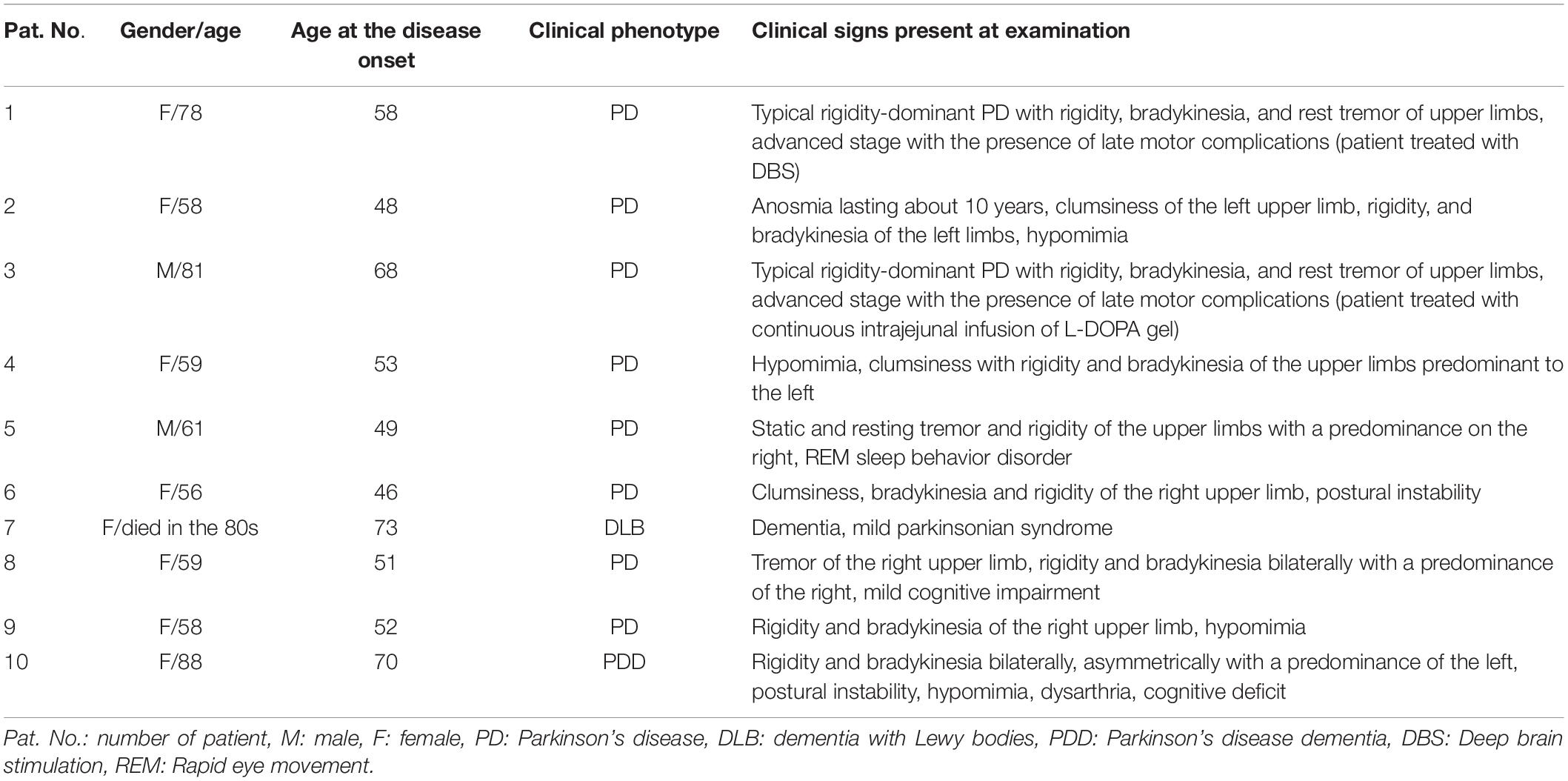
Table 2. Summary of patient clinical data.
The first step of data analysis was selection of variants common only in affected individuals in each trio. All found variants were evaluated and filtered out by two independent software:
1. Ion Reporter - minor allele frequency (MAF) < 1%, Disease research area: Parkinsonian disorders, PD, Neurodegenerative diseases and functional score: SIFT 0.00–0.05 or PolyPhen-2 0.15–1.0 and Grantham 0.0–215.
2. Ingenuity Variant Analysis - minor allele frequency < 1%, variant effects and biological context (Parkinsonism responsible genes).
Genes included in filter Disease research are (Parkinsonian disorders, PD, Neurodegenerative diseases): ADH1C, AHCY, AP2A2, APBB2, APOD, ARAP2, ATP13A2, ATP6AP2, C9ORF72, CACNA1D, CAPN2, CHCHD2, CHGA, CHMP2B, CKM, CLMN, COQ2, CSMD1, CYP2D6, DCTN1, DNAJC13, DNAJC26, DNAJC6, DNMT1, DRD1, EIF2AK3, EIF4G1, FBXO7, FUS, GALC, GBA, GIGYF2, GNAS, GPC6, GRIA1, HIVEP3, HTR1A, HTRA2, LRRK2, MAPT, MC1R, MOBP, MTCL1, NLRC4, NOS1, PARK10, PARK12, PARK16, PARK3, PARK7, PEPD, PINK1, PLA2G6, PODXL, PRGN, PRKN, PRX, PSD4, PTRHD1, RAB39B, RAB7L1, RIC3, RTN4, SHC2, SLC18A2, SLC20A2, SLC2A3, SLC2A4, SLC6A1, SNCA, SNCB, SORL1, SPTBN2, SQSTM1, STX6, SYNE1, SYNJ1, TARDBP, TBK1, TIA1, TMEM230, TNR, TPPP2, TRIM11, UBB, UCHL1, VCP, VPS13C, and VPS35.
The most important (potentially risk or pathogenic) variants were confirmed by Sanger sequencing.
Results
In all 15 samples, 99% of targets were covered 1-20× and 90% of targets were covered more than 20×. Average analyzed variants number was about 70,000 with mean depth about 75 (the number of variant in each trio is described in Table 3). The variants potentially associated with neurodegenerative disorders (rare, undescribed, evolutionary conserved and variants assessed by at least one prediction tool as damaging) are described in Table 4. Moreover, we found sharing of some variants within individual trios across the pedigree. In the trio 1 and trio 2 were found two variants in gene MC1R:NM_002386.3:c.322G > A (p.A108T) and MTCL1:NM_015210.3:c.1445C > T (p.A482V), Trio 3 shares two variants with trio 5: DNAJC6:NM_001256864.1:c.1817A > C (p.H606P) and HIVEP3:NM_024503.4:c.3856C > A (p.R1286W). In trios 4 and 5, there were found two variants in gene CSMD1:NM_033225.5:c.3335A > G (p.E1112G) and c.4071C > G (p.I1357M) rand in the gene MTCL1:NM_015210.3:c.1445C > T (p.A482V). In the trio 3 and trio 5 were found two variants in gene DNAJC6:NM_001256864.1:c.1817A > C (p.H606P) and in gene HIVEP3:NM_024503.4:c.3856C > A (p.R1286W). In the trio 4 and trio 5, there were found two different variants NM_033225.5:c.3335A > G (p.E1112G) and c.4071C > G (p.I1357M) in the same gene CSMD1.
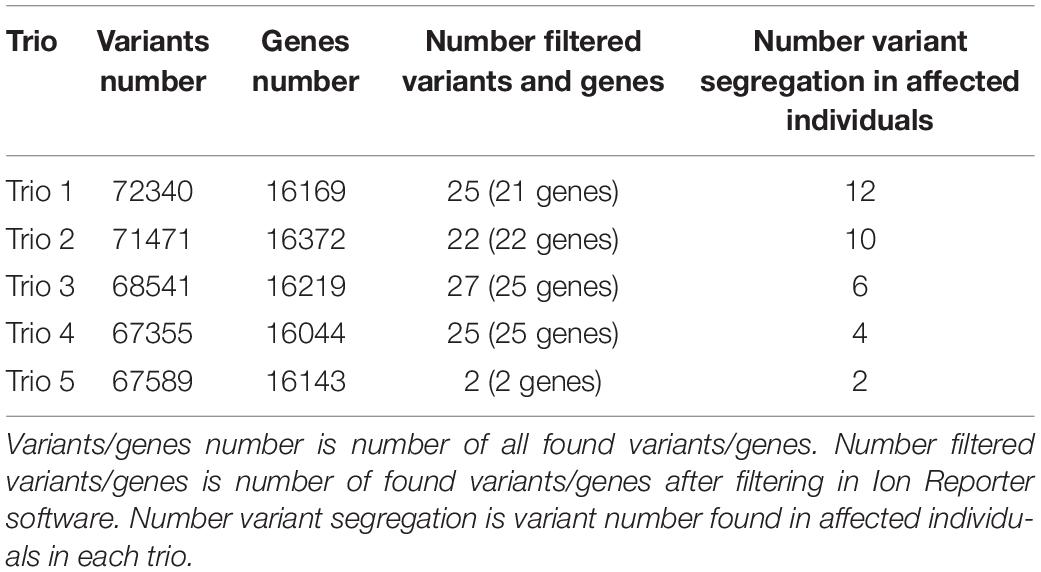
Table 3. Number of variants in individual trios.
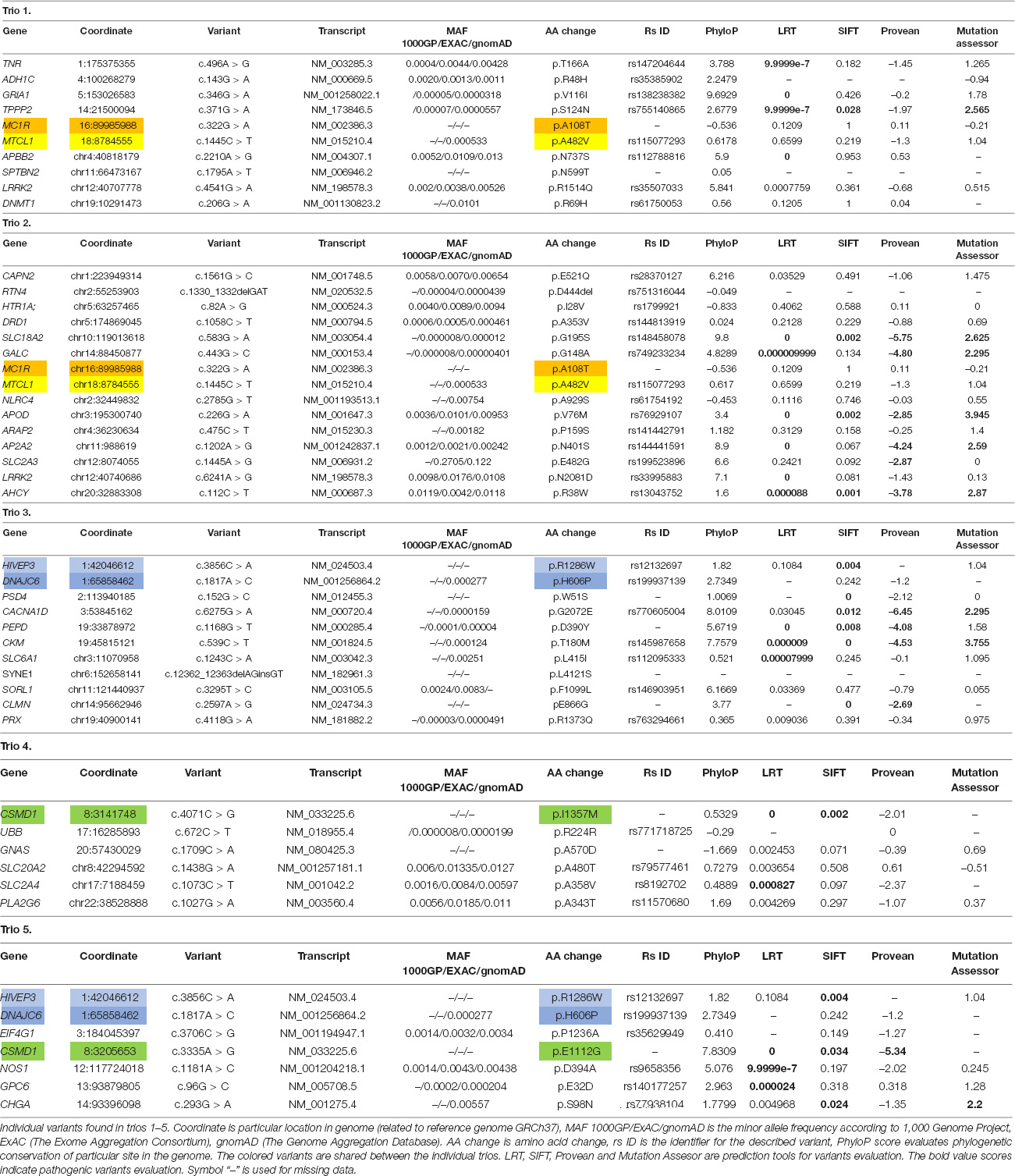
Table 4. Found variants in individual trios.
Discussion
In our whole exome study, we did not find any founder mutation across the large pedigree. There were analyzed the gene regions known to be associated with parkinsonism.
Further, we found some rare variants which were found in more than one trio and which could contribute to development of neurodegeneration disorders.
MC1R gene (OMIM 155555) encodes melanocortin 1 receptor, it is important gene for pigmentation. Study Marti et al. (2015) described MC1R gene as a risk factor for PD and association of the variant p.R160W and PD in Spain population (Marti et al., 2015). But this finding was not confirmed (Gan-Or et al., 2016). Individuals with light hair and homozygotes for the variant p.R151C have higher risk for PD compared with wild type (Gao et al., 2009).
The found variant c.322G > A was evaluated as benign according to used prediction tools and the genomic site is phylogenetic unconserved. The missense variant leads to exchange hydrophobic to polar amino acid. The variant has not yet been described.
MTCL1 gene (OMIM 615766) encodes protein which is important for microtubule bundles and it interacts with MARK2 (Sato et al., 2013). MARK2 kinase affected affinity of tau protein for microtubules by its phosphorylation (Schwalbe et al., 2013).
According to prediction tools, the found variant c.1445C > T is benign, but its frequency is very rare in population. The genomic site is weakly phylogenetic conserved. There has not been any publication about the variant association with neurodegenerative disorders.
HIVEP3 gene (OMIM 606649) encodes protein included in HIV enhancer-binding protein family. It can change transcription via the κB enhancer motif (Allen et al., 2002). In HIVs patients was described decreased levels of cerebrospinal fluid dopamine (Lopez et al., 1999).
The found variant c.3856C > A was evaluated as benign according to prediction tools LRT and Mutation Assessor, but SIFT evaluated it as damaging. The variant leads to exchange positive charged amino acid to hydrophobic. The genomic site is phylogenetic conserved. Thus, the variant c.3856C > A could affect the protein function.
DNAJC6 gene (OMIM 608375) is associated with juvenile parkinsonism (Edvardson et al., 2012, Köroğlu et al., 2013). It encodes auxilin which is important for clathrin-mediated endocytosis (Yim et al., 2010). Probably, dysfunction in neuronal endocytic processes could contribute to parkinsonism development.
The found variant c.1817A > C is located in genomic site which is phylogenetic conserved. The variant leads to exchange basic polar amino acid to non-polar. Prediction tools SIFT and Provean evaluated it as benign. The variant frequency is very rare in population.
CSMD1 gene (OMIM 608397) is associated with schizophrenia risk (Håvik et al., 2011). It is synthetized in developing CNS (central nervous system) and epithelial tissues. The protein is important for complement activation and inflammation in developing CNS (Kraus et al., 2006). Based on WES, CSMD1 gene was described in association with familial PD (Ruiz-Martínez et al., 2017).
The found undescribed variant c.3335A > G is located in high phylogenetic conserved site and it leads to exchange acid to neutral amino acid. All of used prediction tools evaluated the variant as damaging. Based on that, the variant could leads to affect protein function.
Other found variant c.4071C > G leads to exchange within hydrophobic amino acid in weakly conserved genomic site. The prediction tool Provean evaluated the variant as benign, but LRT and SIFT evaluated it as damaging.
SLC18A2 gene (OMIM 193001, also known as VMAT2) encodes ATPase antiporter transmitting monoamines- serotonin, dopamine and norepinephrine into vesicles to transport them out of cell (Liu and Edwards, 1997). Increased cytosolic dopamine and its metabolites are neurotoxic for neuronal cells (reduction leads to neuroprotection) (Mosharov et al., 2009). Brighina et al. (2013) described two SNPs in the VMAT2 promoter region in connection with a reduced risk of PD. It is assumed that increased levels of VMAT2 contribute to protection against the disease (Brighina et al., 2013).
The variant c.583G > A is located in high phylogenetic conserved genomic site and it is very rare in population. It leads to exchange hydrophobic to polar amino acid. All of used prediction tools evaluated it as damaging. In project GnomAD exomes, there were no found homozygous allele, it could indicates likely pathogenic effect (according to ACMG Classification, criteria for classifying pathogenic variants – PMS2 rule). The variant is located in disulfide bond domain, which is important for efficient monoamine transport (Thiriot et al., 2002). We assume that the variant could affect dopamine transport from dopaminergic cells and expose the cells to dopamine cytotoxicity.
Conclusion
The WES could contribute to the finding of variants responsible for development of many diseases. Generally, the evaluation of variants from the WES using different prediction tools is often not uniform and the final assessment should be taken with caution and in combination with functional assays or segregation analysis. However, even one prediction tool, strong evolution conservation or population rarity could indicate and highlight potentially risk variant.
Based on the study result, we suppose heterogenous genetic background in the development of parkinsonism in Hornacko region. According to the prediction tools, the most interesting variant seems to be SLC18A2:NM_003054.4 c.583G > A (p.G195S, rs148458078).
Our study is limited by amount of samples and it is not possible to exclude the effect of other genetic causes that were not detected by used method and filter setting. The WES cannot capture intronic variants and large genomic rearrangements. The larger population study is necessary for verification of our results.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/785725.
Ethics Statement
The studies involving human participants were reviewed and approved by the Palacký University and University Hospital Olomouc. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
KK and RVr: trio analysis and evaluation of variants. RVo: trio analysis and evaluation of variants and coordination of writing manuscript. JS: genetic consultation of patients. MP: genetic consultation of patients and coordination of writing manuscript. KM: neurological consultation of patients. PK: neurological consultation of patients and coordination of writing manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by a grant from the Ministry of Health of the Czech Republic (Grant No. 15-32715A), by grants from the Palacký University Medical School Internal Grant Agency (IGA LF 2018-009, IGA LF 2020-017, and IGA LF 2022-014), by an institutional support from the Ministry of Health of the Czech Republic: conceptual development of research organization MH CZ – DRO (FNOL, 00098892), and by the European Regional Development Fund – Project ENOCH (No. CZ.02.1.01/0.0/0.0/16_019/0000868).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Allen, C. E., Mak, C. H., and Wu, L. C. (2002). The kappa B transcriptional enhancer motif and signal sequences of V(D)J recombination are targets for the zinc finger protein HIVEP3/KRC: a site selection amplification binding study. BMC Immunol. 22:10. doi: 10.1186/1471-2172-3-10
Ando, M., Fiesel, F. C., Hudec, R., Caulfield, T. R., Ogaki, K., Górka-Skoczylas, P., et al. (2017). The PINK1 p.I368N mutation affects protein stability and ubiquitin kinase activity. Mol. Neurodegener. 12:32. doi: 10.1186/s13024-017-0174-z
Bandres-Ciga, S., Diez-Fairen, M., Kim, J. J., and Singleton, A. B. (2020). Genetics of Parkinson’s disease: an introspection of its journey towards precision medicine. Neurobiol. Dis. 137:104782. doi: 10.1016/j.nbd.2020.104782
Brighina, L., Riva, C., Bertola, F., Saracchi, E., Fermi, S., Goldwurm, S., et al. (2013). Analysis of vesicular monoamine transporter 2 polymorphisms in Parkinson’s disease. Neurobiol. Aging 34, 1712.e9–13. doi: 10.1016/j.neurobiolaging.2012.12.020
Buervenich, S., Carmine, A., Galter, D., Shahabi, H. N., Johnels, B., Holmberg, B., et al. (2005). A rare truncating mutation in ADH1C (G78Stop) shows significant association with Parkinson disease in a large international sample. Arch. Neurol. 62, 74–78. doi: 10.1001/archneur.62.1.74
Campêlo, C. L. C., Cagni, F. C., de Siqueira Figueredo, D., Oliveira, L. G. Jr., Silva-Neto, A. B., Macêdo, P. T., et al. (2017). Variants in SNCA gene are associated with parkinson’s disease risk and cognitive symptoms in a brazilian sample. Front. Aging Neurosci. 20:198. doi: 10.3389/fnagi.2017.00198
Chartier-Harlin, M. C., Dachsel, J. C., Vilariño-Güell, C., Lincoln, S. J., Leprêtre, F., Hulihan, M. M., et al. (2011). Translation initiator EIF4G1 mutations in familial Parkinson disease. Am. J. Hum. Genet. 89, 398–406. doi: 10.1016/j.ajhg.2011.08.009
Dolgacheva, L. P., Berezhnov, A. V., Fedotova, E. I., Zinchenko, V. P., and Abramov, A. Y. (2019). Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 51, 175–188. doi: 10.1007/s10863-019-09798-4
Edvardson, S., Cinnamon, Y., Ta-Shma, A., Shaag, A., Yim, Y. I., Zenvirt, S., et al. (2012). A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One 7:e36458. doi: 10.1371/journal.pone.0036458
Farlow, J. L., Robak, L. A., Hetrick, K., Bowling, K., Boerwinkle, E., Coban-Akdemir, Z. H., et al. (2016). Whole-Exome sequencing in familial parkinson disease. JAMA Neurol. 73, 68–75. doi: 10.1001/jamaneurol.2015.3266
Gan-Or, Z., Mohsin, N., Girard, S. L., Montplaisir, J. Y., Ambalavanan, A., Strong, S., et al. (2016). The role of the melanoma gene MC1R in Parkinson disease and REM sleep Behavior Disorder. Neurobiol. Aging 43, 180.e7–180.e13. doi: 10.1016/j.neurobiolaging.2016.03.029.
Gao, X., Simon, K. C., Han, J., Schwarzschild, M. A., and Ascherio, A. (2009). Genetic determinants of hair color and Parkinson’s disease risk. Ann. Neurol. 65, 76–82. doi: 10.1002/ana.21535
Gazal, S., Gosset, S., Verdura, E., Bergametti, F., Guey, S., Babron, M. C., et al. (2016). Can whole-exome sequencing data be used for linkage analysis? Eur. J. Hum. Genet. 24, 581–586. doi: 10.1038/ejhg.2015.143
Guo, J. F., Zhang, L., Li, K., Mei, J. P., Xue, J., Chen, J., et al. (2018). Coding mutations in NUS1 contribute to Parkinson’s disease. Proc. Natl. Acad. Sci. U S A. 115, 11567–11572. doi: 10.1073/pnas.1809969115
Håvik, B., Le Hellard, S., Rietschel, M., Lybæk, H., Djurovic, S., Mattheisen, M., et al. (2011). The complement control-related genes CSMD1 and CSMD2 associate to schizophrenia. Biol. Psychiatry 70, 35–42. doi: 10.1016/j.biopsych.2011.01.030
Hughes, A. J., Daniel, S. E., Ben-Shlomo, Y., and Lees, A. J. (2002). The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 125, 861–870. doi: 10.1093/brain/awf080
Kalinderi, K., Bostantjopoulou, S., and Fidani, L. (2016). The genetic background of Parkinson’s disease: current progress and future prospects. Acta Neurol. Scand. 134, 314–326. doi: 10.1111/ane.12563
Köroğlu, Ç, Baysal, L., Cetinkaya, M., Karasoy, H., and Tolun, A. (2013). DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism. Relat. Disord. 19, 320–324. doi: 10.1016/j.parkreldis.2012.11.006
Kraus, D. M., Elliott, G. S., Chute, H., Horan, T., Pfenninger, K. H., Sanford, S. D., et al. (2006). CSMD1 is a novel multiple domain complement-regulatory protein highly expressed in the central nervous system and epithelial tissues. J. Immunol. 176, 4419–4430. doi: 10.4049/jimmunol.176.7.4419
Lin, G., Lee, P. T., Chen, K., Mao, D., Tan, K. L., Zuo, Z., et al. (2018). Phospholipase PLA2G6, a parkinsonism-associated gene, affects Vps26 and Vps35, retromer function, and ceramide levels, similar to α-Synuclein gain. Cell Metab. 28, 605–618.e6. doi: 10.1016/j.cmet.2018.05.019.
Liu, Y., and Edwards, R. H. (1997). The role of vesicular transport proteins in synaptic transmission and neural degeneration. Annu. Rev. Neurosci. 20, 125–156. doi: 10.1146/annurev.neuro.20.1.125
Lopez, O. L., Smith, G., Meltzer, C. C., and Becker, J. T. (1999). Dopamine systems in human immunodeficiency virus-associated dementia. Neuropsychiatry Neuropsychol. Behav. Neurol. 12, 184–192.
Marti, T., Puig-Butille, J. A., Potrony, M., Badenas, C., Milà, M., Malvehy, J., et al. (2015). The MC1R melanoma risk variant p.R160W is associated with Parkinson disease. Ann. Neurol. 77, 889–894. doi: 10.1002/ana.24373
Mensikova, K., Kanovsky, P., Kaiserova, M., Mikulicova, L., Vastik, M., Hlustik, P., et al. (2013). Prevalence of neurodegenerative parkinsonism in an isolated population in south-eastern moravia, czech republic. Eur. J. Epidemiol. 28, 833–836. doi: 10.1007/s10654-013-9823-x
Mensikova, K., Kaňovský, P., Otruba, P., Kaiserová, M., Vastik, M., Hlustik, P., et al. (2014). Epidemiological study of neurodegenerative Parkinsonism in “Hornacko”, a specific region of the south-eastern moravia, czech republic. Czech Slovak Neurol. Neurosurg. 110, 714–720. doi: 10.14735/amcsnn2014714
Miller, S. A., Dykes, D. D., and Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16:1215. doi: 10.1093/nar/16.3.1215
Mir, R., Tonelli, F., Lis, P., Macartney, T., Polinski, N. K., Martinez, T. N., et al. (2018). The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem. J. 475, 1861–1883. doi: 10.1042/BCJ20180248
Mosharov, E. V., Larsen, K. E., Kanter, E., Phillips, K. A., Wilson, K., Schmitz, Y., et al. (2009). Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 62, 218–229. doi: 10.1016/j.neuron.2009.01.033
Ortega, R. A., Wang, C., Raymond, D., Bryant, N., Scherzer, C. R., Thaler, A., et al. (2021). Association of dual LRRK2 G2019S and GBA variations with parkinson disease progression. JAMA Netw Open 4:e215845. doi: 10.1001/jamanetworkopen.2021.5845
Park, J. S., Blair, N. F., and Sue, C. M. (2015). The role of ATP13A2 in Parkinson’s disease: clinical phenotypes and molecular mechanisms. Mov. Disord. 30, 770–779. doi: 10.1002/mds.26243
Ruiz-Martínez, J., Azcona, L. J., Bergareche, A., Martí-Massó, J. F., and Paisán-Ruiz, C. (2017). Whole-exome sequencing associates novel CSMD1 gene mutations with familial Parkinson disease. Neurol. Genet. 3:e177. doi: 10.1212/NXG.0000000000000177
Sato, Y., Akitsu, M., Amano, Y., Yamashita, K., Ide, M., Shimada, K., et al. (2013). The novel PAR-1-binding protein MTCL1 has crucial roles in organizing microtubules in polarizing epithelial cells. J. Cell Sci. 126, 4671–4683. doi: 10.1242/jcs.127845
Schwalbe, M., Biernat, J., Bibow, S., Ozenne, V., Jensen, M. R., Kadavath, H., et al. (2013). Phosphorylation of human Tau protein by microtubule affinity-regulating kinase 2. Biochemistry 52, 9068–9079. doi: 10.1021/bi401266n
Thiriot, D. S., Sievert, M. K., and Ruoho, A. E. (2002). Identification of human vesicle monoamine transporter (VMAT2) lumenal cysteines that form an intramolecular disulfide bond. Biochemistry 20, 6346–6353. doi: 10.1021/bi015779j
Toma, C., Díaz-Gay, M., Franch-Expósito, S., Arnau-Collell, C., Overs, B., Muñoz, J., et al. (2020). Using linkage studies combined with whole-exome sequencing to identify novel candidate genes for familial colorectal cancer. Int. J. Cancer 146, 1568–1577. doi: 10.1002/ijc.32683
Vodicka, R., Kolarikova, K., Vrtel, R., Mensikova, K., Kanovsky, P., and Prochazka, M. (2020). “Evaluating basic next-generation sequencing parameters in relation to true/false positivity findings of rare variants in an isolated population from the Czech Republic South-Eastern Moravia Region with a high incidence of parkinsonism,” in Bioinformatics and Biomedical Engineering. IWBBIO 2020. Lecture Notes in Computer Science, Vol. 12108, eds Rojas, I., Valenzuela, O., Rojas, F., Herrera, L., and F. Ortuño (Cham: Springer), 562–568. doi: 10.1007/978-3-030-45385-5_50
Xiromerisiou, G., Bourinaris, T., Houlden, H., Lewis, P. A., Senkevich, K., Hammer, M., et al. (2021). SORL1 mutation in a Greek family with Parkinson’s disease, and dementia. Ann. Clin. Transl. Neurol. 8, 1961–1969. doi: 10.1002/acn3.51433
Yemni, E. A., Monies, D., Alkhairallah, T., Bohlega, S., Abouelhoda, M., Magrashi, A., et al. (2019). Integrated analysis of whole exome sequencing and copy number evaluation in Parkinson’s Disease. Sci. Rep. 9:3344. doi: 10.1038/s41598-019-40102-x
Yi, W., MacDougall, E. J., Tang, M. Y., Krahn, A. I., Gan-Or, Z., Trempe, J. F., et al. (2019). The landscape of Parkin variants reveals pathogenic mechanisms and therapeutic targets in Parkinson’s disease. Hum. Mol. Genet. 28, 2811–2825. doi: 10.1093/hmg/ddz080
Keywords: whole-exome sequencing, parkinsonism, neurodegenerative disorders, trio analysis, SLC18A2 gene
Citation: Kolarikova K, Vodicka R, Vrtel R, Stellmachova J, Prochazka M, Mensikova K and Kanovsky P (2022) Whole Exome Sequencing Study in Isolated South-Eastern Moravia (Czechia) Population Indicates Heterogenous Genetic Background for Parkinsonism Development. Front. Neurosci. 16:817713. doi: 10.3389/fnins.2022.817713
Received: 18 November 2021; Accepted: 22 February 2022;
Published: 17 March 2022.
Edited by:
Partha Sarathi Sarkar, University of Texas Medical Branch at Galveston, United StatesReviewed by:
Victoria Fernandez, Washington University in St. Louis, United StatesMaria Shadrina, Institute of Molecular Genetics of National Research Centre «Kurchatov Institute», Russia
Copyright © 2022 Kolarikova, Vodicka, Vrtel, Stellmachova, Prochazka, Mensikova and Kanovsky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Radek Vodicka, UmFkZWsuVm9kaWNrYUBmbm9sLmN6