
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurosci., 20 December 2022
Sec. Translational Neuroscience
Volume 16 - 2022 | https://doi.org/10.3389/fnins.2022.1060111
This article is part of the Research TopicAdvances in Theranostics of CNS Injuries and Diseases: From Basic Research to Clinical PracticeView all 10 articles
 Wang Liao1†
Wang Liao1† Haoyu Luo1†Yuting Ruan2Yingren Mai1Chongxu Liu1Jiawei Chen3Shaoqing Yang1*
Haoyu Luo1†Yuting Ruan2Yingren Mai1Chongxu Liu1Jiawei Chen3Shaoqing Yang1* Aiguo Xuan4*
Aiguo Xuan4* Jun Liu1*
Jun Liu1*Background and objective: Alzheimer’s disease (AD) is the most common type of dementia, with its pathology like beta-amyloid and phosphorylated tau beginning several years before the clinical onset. The aim is to identify genetic risk factors associated with the onset of AD.
Methods: We collected three microarray data of post-mortem brains of AD patients and the healthy from the GEO database and screened differentially expressed genes between AD and healthy control. GO/KEGG analysis was applied to identify AD-related pathways. Then we distinguished differential expressed genes between symptomatic and asymptomatic AD. Feature importance with logistic regression analysis is adopted to identify the most critical genes with symptomatic AD.
Results: Data was collected from three datasets, including 184 AD patients and 132 healthy controls. We found 66 genes to be differently expressed between AD and the control. The pathway enriched in the process of exocytosis, synapse, and metabolism and identified 19 candidate genes, four of which (VSNL1, RTN1, FGF12, and ENC1) are vital.
Conclusion: VSNL1, RTN1, FGF12, and ENC1 may be the essential genes that progress asymptomatic AD to symptomatic AD. Moreover, they may serve as genetic risk factors to identify high-risk individuals showing an earlier onset of AD.
Alzheimer’s disease (AD) is a chronic degenerative disease characterized by the extracellular deposition of beta-amyloid and intracellular accumulation of phosphorylated tau protein (Spina et al., 2021). For older adults, AD is the most common cause of dementia, with an incidence rate of approximately 1.5% among people over 65 years old and nearly 50% among people over 90 years old (Scheltens et al., 2021). Interestingly, cognitive impairment of many AD patients occurs substantial years after pathological changes, such as amyloid and neurofibrillary tangles (NFTs). In contrast, more than 20% of the aged population have an amyloid deposition. Individuals with intact cognition and neuropathology consistent with AD were called asymptomatic AD (AsymAD) (Patel et al., 2019). AsymAD is distinguishable from normal aging based on neuropathology, brain imaging, and cerebrospinal fluid biomarkers (Ayodele et al., 2021). While some of these individuals progress to symptoms related to cognition, which deviate from mild cognitive impairment (MCI), and then to AD, not all do. They are, therefore, a heterogeneous group, representing those with prodromal AD and those impervious to AD despite having pathological hallmarks. These individuals are highly likely to develop symptomatic AD. Abundant evidence has demonstrated genetic risk factors of AD, such as APP, PSEN1, PSEN2, and apolipoprotein ε4 allele (APOE4) (Lefterov et al., 2019). However, transcriptomic changes in the brain, which might reveal AD vulnerability, are currently unknown. In the present study, we identified genetic risk factors for AD onset in asymptomatic and symptomatic individuals with clear signs of AD pathology at autopsy. Understanding the fundamental changes may shed light on specific biological mechanisms involved in early pathological hallmarks of AD, providing new therapeutic targets for early intervention.
We searched the microarray sequencing datasets of AD patients’ post-mortem brain samples for 5 years on the Gene Expression Omnibus database (GEO). Then we removed the datasets with incomplete annotation of platform annotation information. Finally, we selected three datasets of the same platform [GSE139384 (Morimoto et al., 2020), GSE118553 (Patel et al., 2019), and GSE132903 (Piras et al., 2019)]. We integrated the temporal lobe data in the post-mortem brains of 184 AD patients and 132 healthy people with ComBat package (Supplementary Data Sheet 1). All the above datasets were gathered using the Illumina HumanHT-12 V4.0 platform.
We screened the differentially expressed genes (DEGs) between AD patients and control in the processed new dataset using the limma R package. We first use the lmFit function to construct the linear model and then use the contrasts fit function to calculate the contrast of the model (estimated coefficient and standard error). Next, eBayes function to compute moderated t-statistic, moderated F-statistic, and log-odds of differential expression, and use the top table function (adjust.method is ‘ fdr ’) to extract the gene table. Finally, the differential genes were screened by the standard of |logFoldChange| = 0.5, adjust P = 0.05, and displayed visually by ggplot2 and heatmap. Further, we performed weighted correlation network analysis (WGCNA) with the new dataset we got to identify the co-expression of hub genes.
All the DEGs were analyzed by GO (Gene ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis to investigate the biological mechanisms. With enrichgo and enrichKEGG functions in the clusterProfiler package, we analyzed the differential genes and logFC values to obtain significantly enriched gene functions and pathways. GO analysis was annotated by org.Hs.eg.db package, KEGG analysis set hsa as species information, p value and q value were set to 0.05, and visualize with barplot and GOChord. In order to reduce the poor enrichment results caused by the fixed threshold method for screening differential genes, we further performed GSEA analysis on the overall genes list with logFC value of the new dataset using the gseKEGG function in the clusterProfiler package, organism is hsa.
We input DEGs list into the String database1 and obtained a protein interaction network diagram and data forms. Then, we downloaded the data tables and imported them into cytoscape software, using the cytohubba plug-in to calculate the key nodes. Based on the results of MCC algorithm, the interactive network diagrams of different colors are drawn according to the calculated scores.
Differentially expressed genes were first analyzed between asymptomatic and symptomatic AD groups in the GSE118553 dataset to identify the genetic risk factors for symptomatic AD. We then overlapped these DEGs with AD-related DEGs derived from WGCNA. Feature importance with a logistic regression analysis were used to identify the predicted importance of the DEGs further. Specifically, we first sorted out the expression of immune-related differential expressed genes in the validation set. Next, the matrix was input into SPSS 26.0 software, and the binary logistic regression analysis with the forward conditional method was carried out with whether it was symptomatic AD as the dependent variable and the expression of each gene as the independent variable. Finally, the matrix of the key genes successfully incorporated into the model was input into the R language, the rms package was loaded, and the linear regression model between these genes and symptomatic AD was constructed using the lrm function. Then, the nomogram function was used to draw a nomogram representing the regression fitting to visually demonstrate the predictive power of these genes.
A total of 184 AD patients and 132 normal controls were included in this study. The demographic information is shown in Tables 1, 2. After standardized processing and removing the batch effect, the distribution of the three datasets tends to be comparable, indicating that the processed data is highly uniform and quality (Supplementary Figure 1).
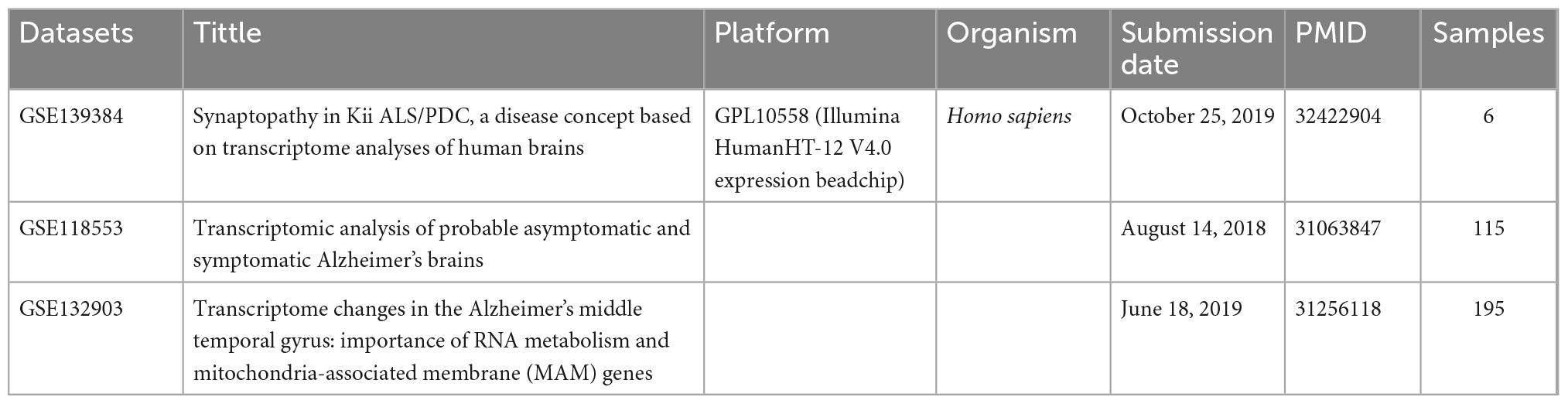
Table 1. Datasets characteristics.
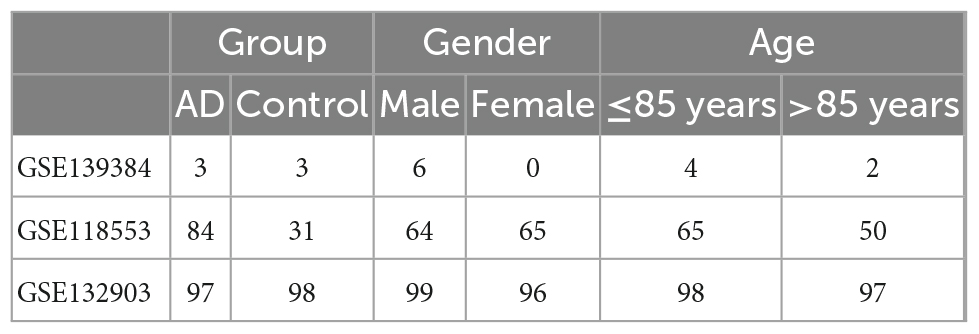
Table 2. Datasets clinical characteristics.
Nighty-two DEGs were found, including 68 down-regulated and 24 upregulated genes (Figures 1A,B). Next, we got the Median Absolute Deviation (MAD) of each gene, and the most minor top 30% genes were removed. Then we removed outlier genes and samples using the goodSampleGenes method in the R software package WGCNA (Figure 2C) and constructed a scale-free co-expression network. Then we evaluated the optimal power using the pick Soft Threshold function in the WGCNA package with a threshold of 0.9 (Figure 2A). As is shown in Figure 2B, the weighted gene network constructed with a power value of six has good connectivity. To classify genes with similar expression profiles into Gene modules, we set the gene tree’s minimum size as 30, the sensitivity as 3, and the distance less than 0.25 to synthesize modules. Finally, 18 co-expression modules were obtained, and gene clustering maps and module vector clustering maps were shown in Figures 2D,E (the gray module is considered a gene set that cannot be assigned to any module). Next, clinical information of the dataset was included to clarify its correlation with the modules (Figure 2F). Finally, based on the truncation criteria with |MM| > 0.8 and |GS| > 0.2, we selected the top five modules with the highest coefficient as the key modules: turquoise, dark turquoise, blue, brown, and black modules. Among them, 585 genes with high connectivity were extracted as hub genes, intersecting with the previously obtained 92 DEGs. A total of 66 co-expressed differential genes were left (Figure 2G).
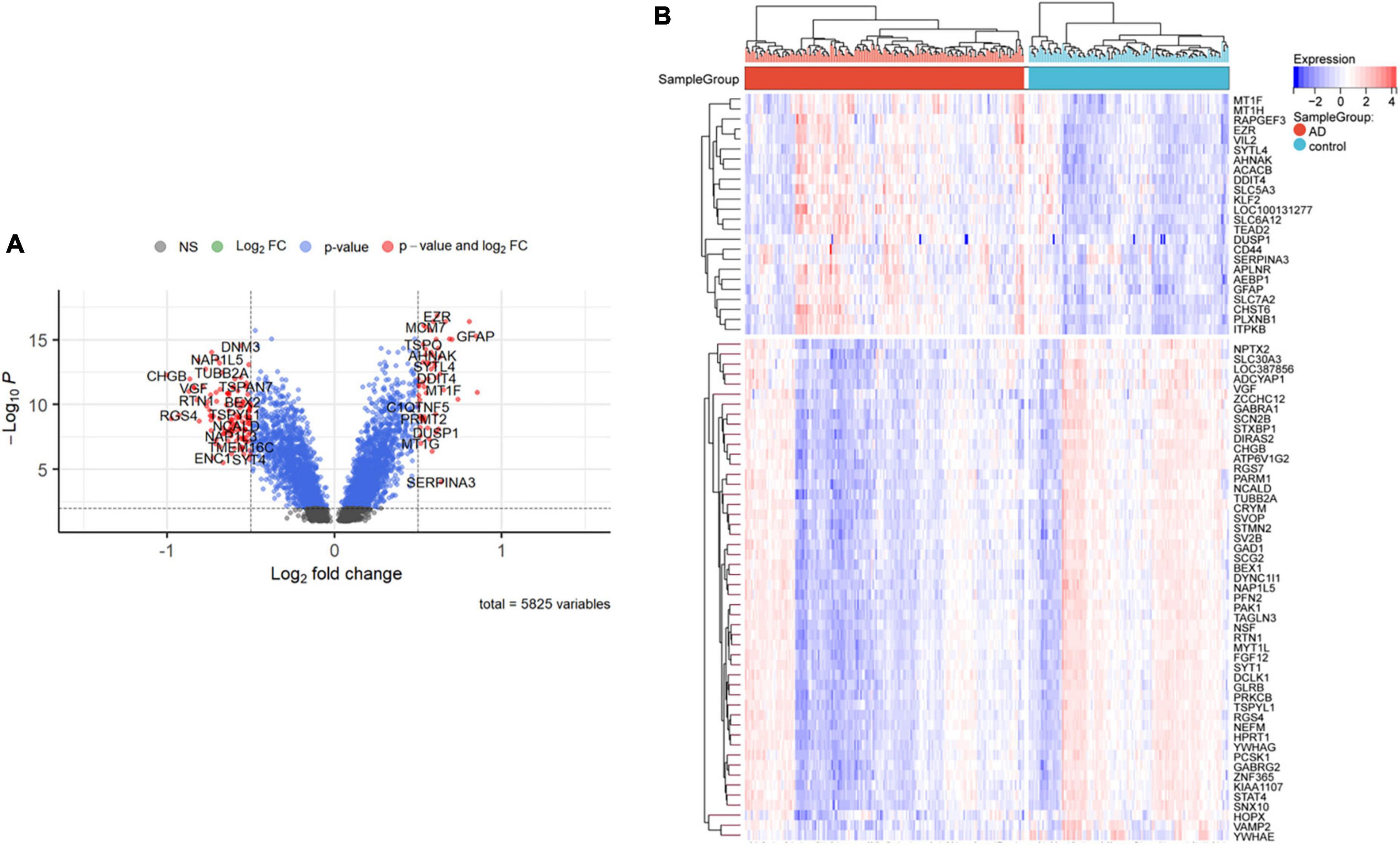
Figure 1. Differentially expressed genes between AD and control samples. (A) The right red genes represent significantly high expression in AD; the left red genes represent significantly high expression in control. (B) The heatmap shows the top 50 genes significantly expressed in AD or ND samples.
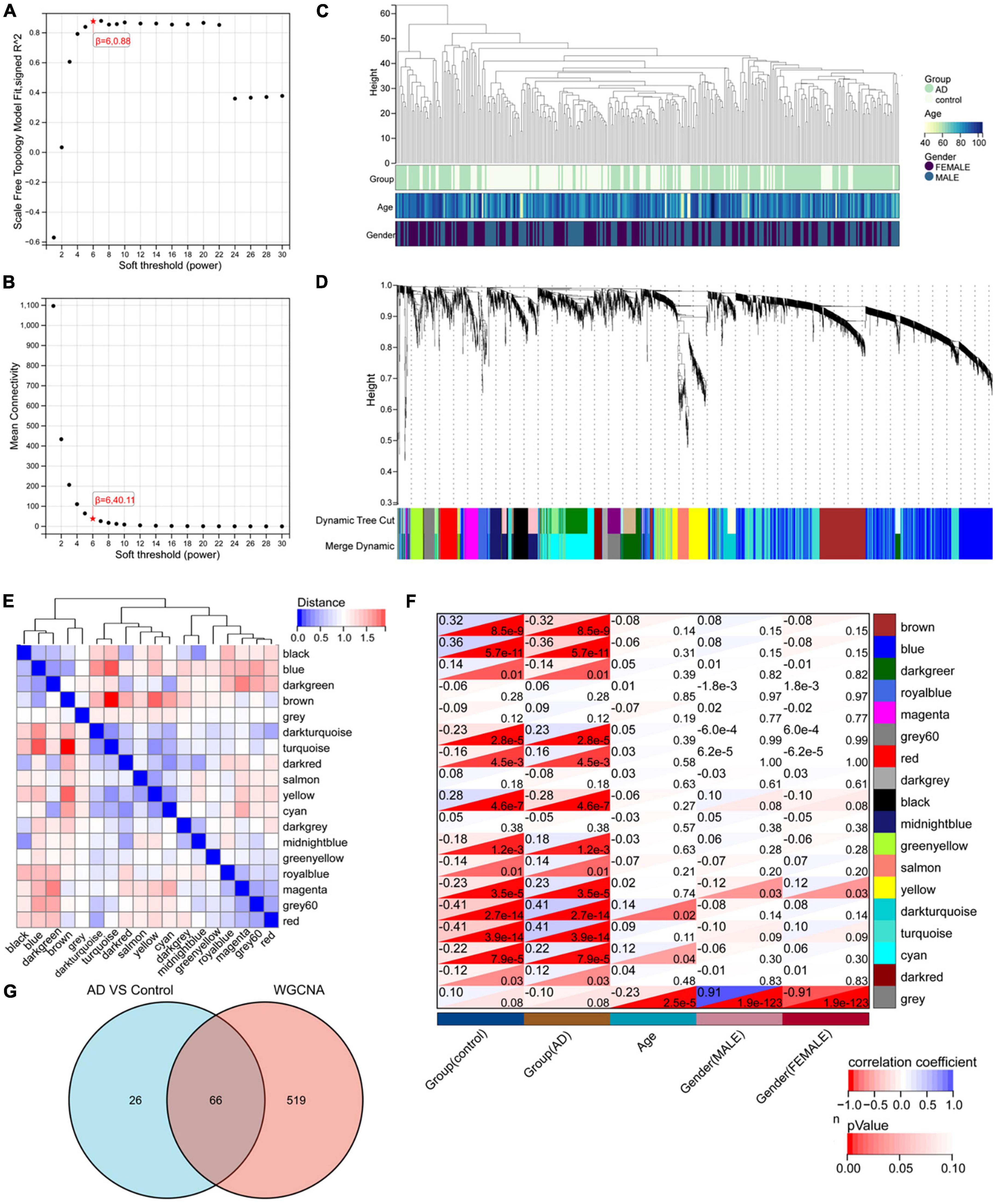
Figure 2. WGCNA analysis results. (A) The corresponding scale-free topological model fit indices at different soft threshold powers. (B) The corresponding mean connectivity values at other soft threshold powers. (C) Sample clustering. (D) Cluster dendrogram of genes. (E) Correlation map of vector clustering of 18 modules. (F) Correlation heatmap between modules and clinical features, the lower left corner of each grid is the P-value, and the upper right corner is the correlation coefficient. (G) The Wayne diagram of intersection with 585 hub genes obtained by WGCNA and 92 DEGs by limma.
We analyzed these 66 DEGs in GO and KEGG databases. We found that the GO database enriched regulating exocytosis, synapse organization, presynapse, neuron projection terminus, and transport vesicle pathways. Moreover, it is enriched in protein kinase C binding, chloride transmembrane transporter activity, and SNARE binding pathways (Figure 3A). The GO plot package chord diagram showed that SYT13, PFN2, NSF, PCLO, SYP, PRKCB, VAMP2, VSNL1, and SYT1 were significantly enriched in multiple major pathways (Figure 3B). While in the KEGG database is enriched in GABAergic synapse, synaptic vesicle cycle, insulin secretion, morphine addiction, and vasopressin-regulated water reabsorption pathways (Figure 3C). To discuss the pathway enrichment of the whole sample more comprehensively, we further performed GSEA analysis. The results showed that these genes were significantly enriched in 21 pathways, especially those associated with Alzheimer’s disease and neurodegenerative diseases (Table 3).
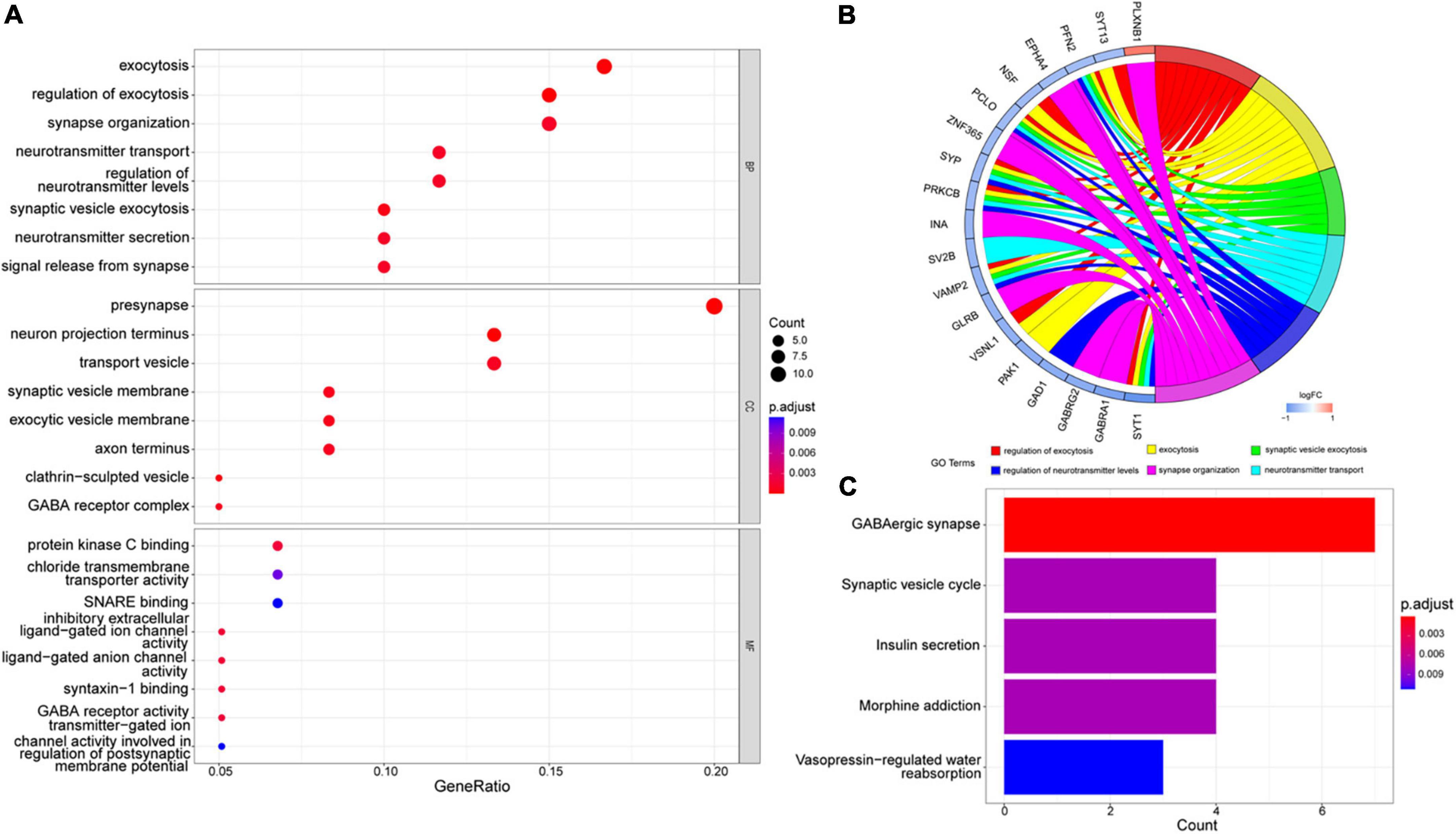
Figure 3. GO and KEGG enrichment analysis. (A) The most significant enrichment pathways in the GO database. (B) Chord diagram of GO enrichment analysis. (C) The most considerable enrichment pathways in the KEGG database.
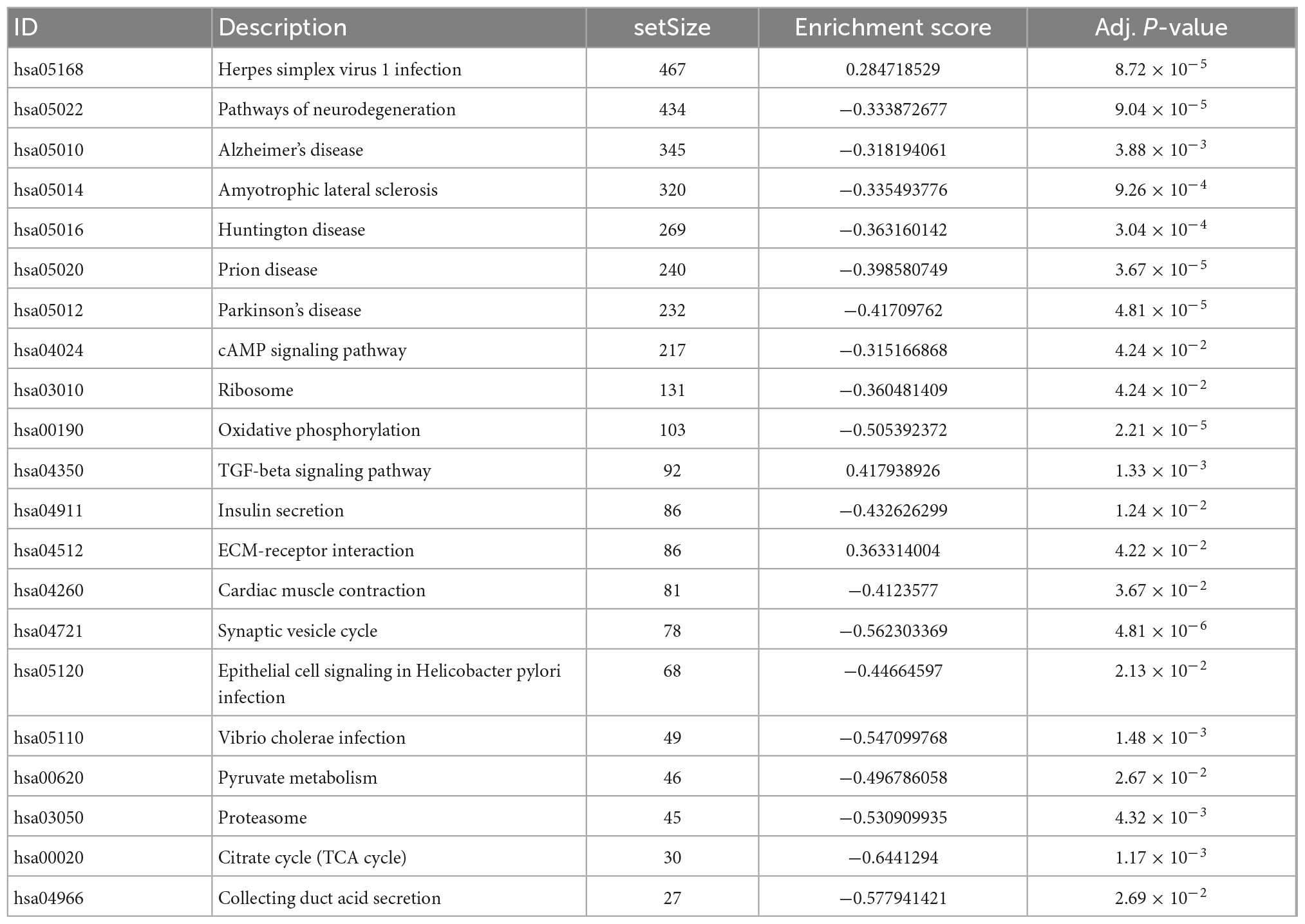
Table 3. GSEA results.
Next, 66 DEGs were further analyzed with STRING (Figure 4). After excluding 17 independent DEGs, we finally constructed a protein interaction network map according to the Maximal Clique Centrality (MCC) score, with the remaining 49 genes processed by Cytohubba. The top 10 hub genes are SYP, SYT1, GABRG2, GABRA1, SLC12A5, GAD1, SV2B, STMN2, VAMP2, and SCG2 (Table 4).
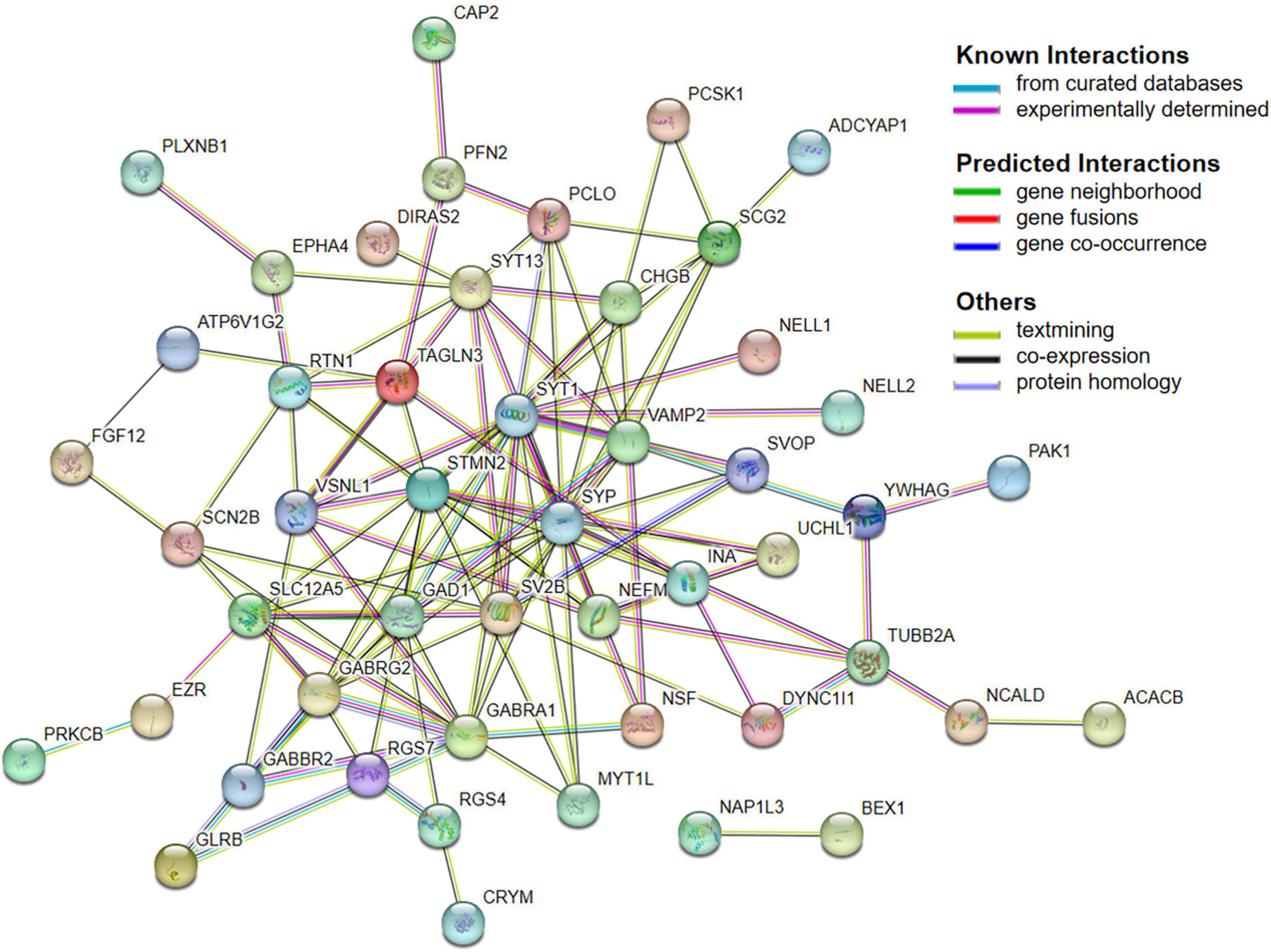
Figure 4. PPI network.
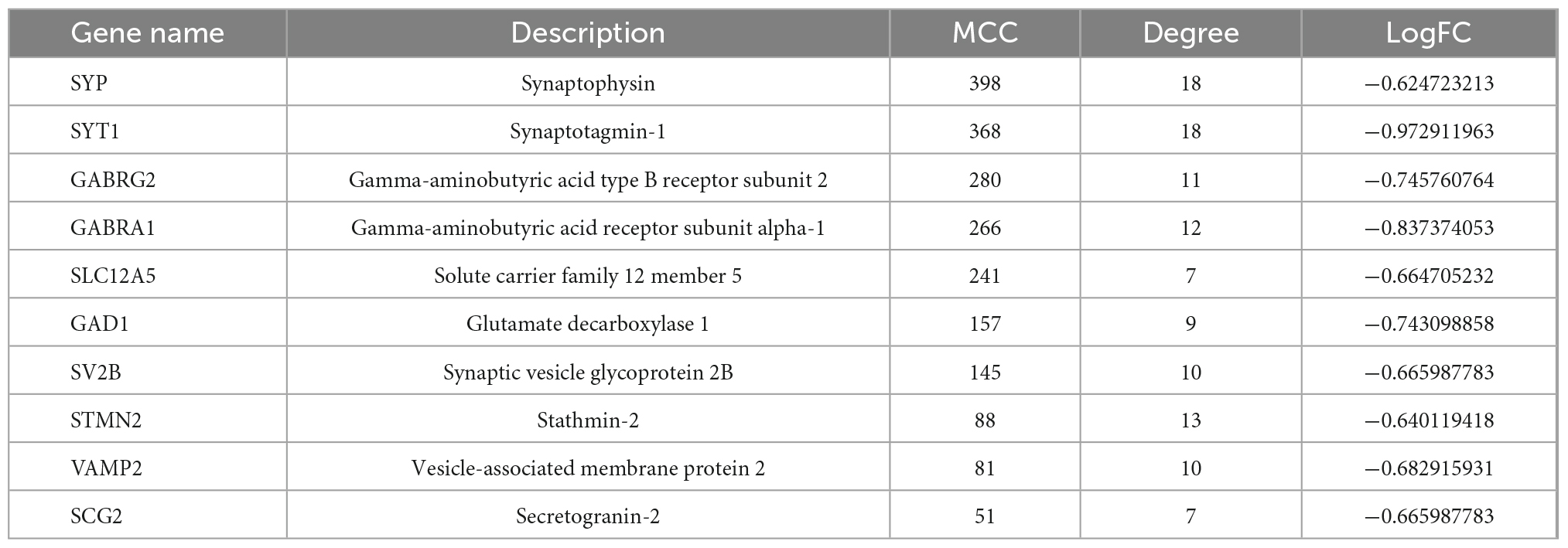
Table 4. Cytohubba results.
Firstly, we extracted the temporal lobe information from AsymAD (n = 32) and SymAD (n = 52) in GSE118553 dataset. Seven hundred and sixty-two DEGs were identified, including 30 upregulated genes and 732 down-regulated genes (Figures 5A,B). Then, we sequence overlapped these DEGs with 92 AD-specific DEGs and WGCNA hub genes and finally obtained 19 key genes, including VSNL1, TAGLN3, SYP, SVOP, SLC12A5, RTN1, PCSK1, PAK1, NSF, NEFM, NCALD, LOC387856, HPRT1, GABRG2, FGF12, ENC1, CHGB, CAP2, and ADCYAP1 (Figure 5C and Table 5).
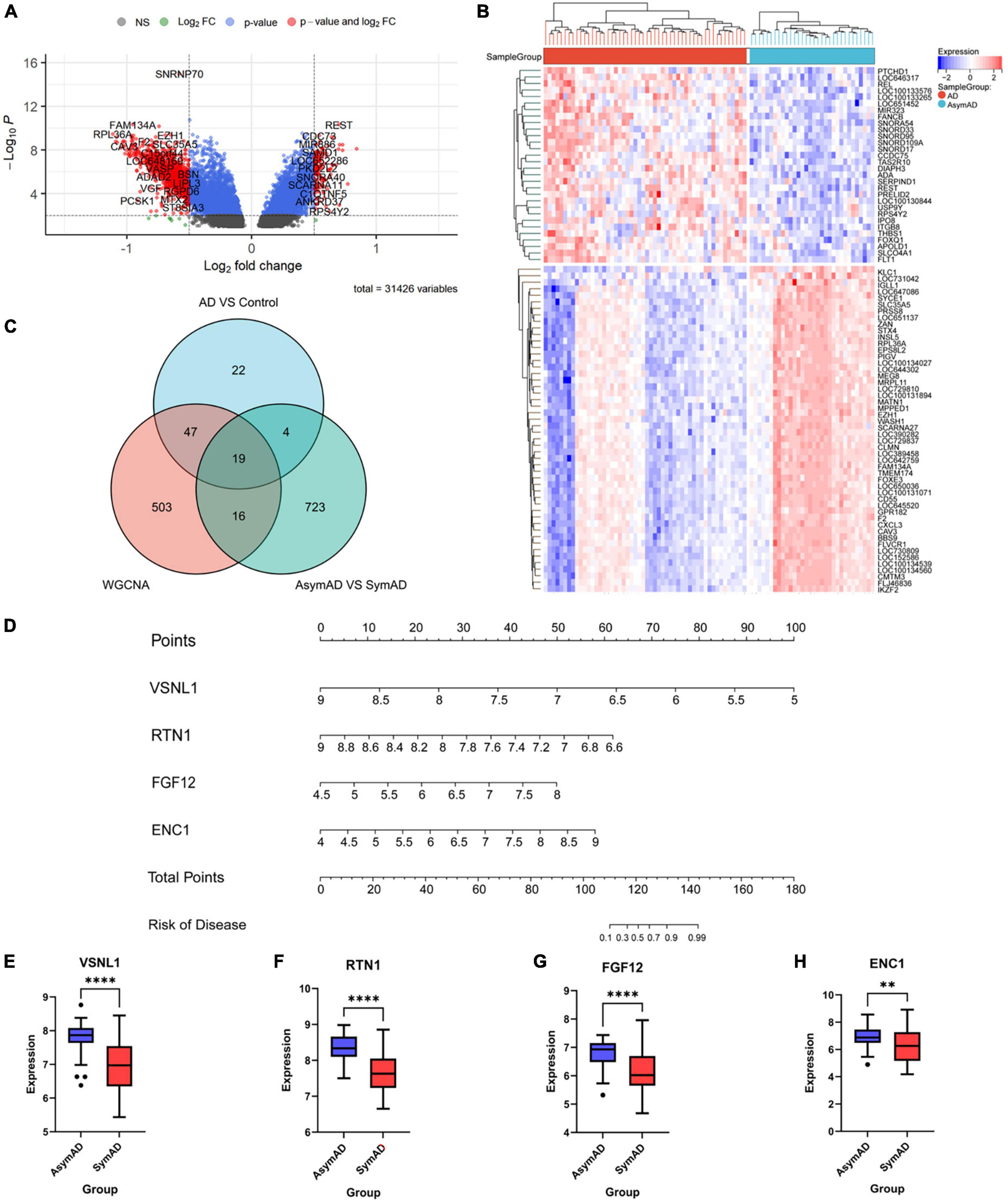
Figure 5. The gene expression difference between AsymAD and SymAD. (A) The right red genes represent significantly high expression in SymAD; the left red genes represent increased expression in AsymAD. (B) The heatmap shows the top 50 genes predominantly expressed in AsymAD or SymAD. (C) Wayne diagram to intersection DEGs of three difference analysis. (D) Nomogram of four essential genes. (E–H) Expression differences of four critical genes in AsymAD and SymAD group. ** means p-value < 0.01; **** means p-value < 0.0001.
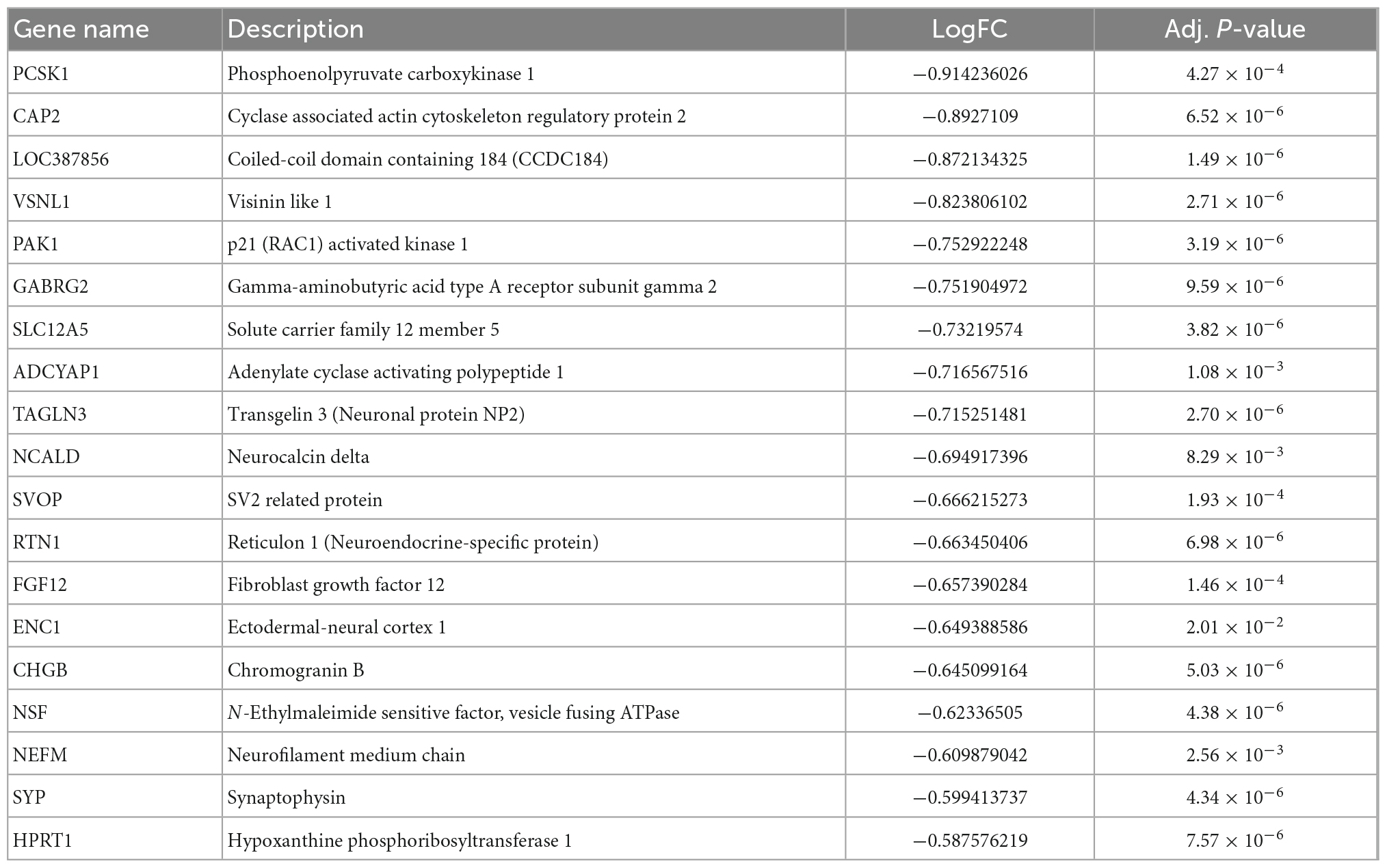
Table 5. Nineteen DEGs list.
Next, we performed a stepwise logistic regression analysis of these 19 DEGs to identify symptomatic AD risk factors and obtained four genes: VSNL1, RTN1, FGF12, and ENC1 (Table 6). Specifically, the goodness of fit and predictive ability of the model are as follows: (1) according to the Omnibus Test results, the chi-square value of the model is 54.070, and the p-value is <0.001, indicating that the model is statistically significant; (2) according to the results of Hosmer and Lemeshow Test, the p-value of the fitting model is 0.229 > 0.05, which indicates that the information in the current data has been fully extracted and the goodness of fit of the model is high; (3) according to the results of the classification table, the sensitivity of the model is 90.4% and the specificity is 80.4%. On the whole, it has a correct prediction rate of 88.1% for all samples, indicating that it has good predictive ability. Through the nomogram, we found that decreased VSNL1, RTN1, and increased FGF12 and ENC1 were positively correlated with disease risk (Figure 5D). Then, we further examined the expression levels of these four genes in disease grouping and observed that the expression of all these four genes was significantly lower in the SymAD group (Figures 5E–H).
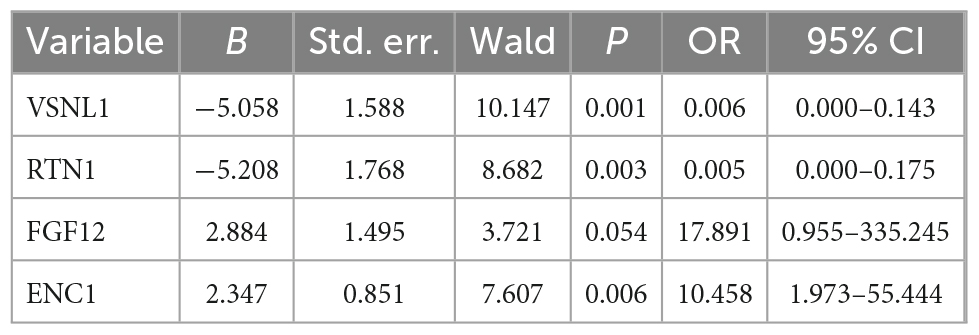
Table 6. Logistic regression analysis of four key genes.
The present study analyzed three datasets of post-mortem brain samples of AD patients and the healthy. Since the temporal lobe is demonstrated to be most related to AD, we extract the specific brain region for further analysis. A dataset with information on asymptomatic AD was applied for subsequent analysis of genetic risk factors and their onset. Differential analysis, WGCNA analysis, function enrichment pathway analysis, protein interaction network analysis, and logistic regression analysis were used to interpret the data. Finally, we found four essential genes closely related to AD, including VSNL1, RTN1, FGF12, and ENC1.
A lot of analytical methods have found many risk molecules associated with AD (Talwar et al., 2014). However, most recent studies still focus on genetic characteristics of early-onset or familial AD (Mold et al., 2020) and pay less attention to the gene expression of late-onset AD. Therefore, we firstly investigated the differential gene expression profiling between AD and the healthy. Among the 92 candidate genes, the most significantly upregulated genes were APLNR, GFAP, and AEBP1, while the most down-regulated genes were CHGB, SYT1, and RGS4. With further analysis by WGCNA, we finally obtained 66 differentially expressed genes.
Moreover, SYP and SYT1 were identified as top hub gene nodes in the protein interactions network, suggesting that these two genes play a vital role in the pathogenesis of AD. SYP has been widely demonstrated to cause cognitive disability. It mainly functions as a membrane protein of small synaptic vesicles in the central nerve system (Xiao et al., 2021). It directs the targeting of vesicle-specific membrane protein 2 (synaptobrevin) toward intracellular compartments (Chen et al., 2022). The other vital DEGs are SYT1, encoding synaptotagmin, integral membrane proteins of synaptic vesicles, and serves as a Ca2+ sensor in vesicular trafficking and exocytosis (Baker et al., 2018; Shi et al., 2020).
There are several canonical pathways in AD, including inflammatory responses, cholesterol, lipid metabolism, and endosomal vesicle recycling (Guerreiro and Hardy, 2014; Wang et al., 2021). Interestingly, metabolism and cellular biological functions were also observed in this study. It is noted that these DEGs were mainly enriched in exocytosis, synapse, and transport pathways in the GO database. Among them, the most enriched pathway in BPs is neurotransmitter transport and regulation of exocytosis. Apart from this, the most prominent pathway for enrichment in CCs is the presynapse and neuron projection terminus.
In contrast, protein kinase C binding and chloride transmembrane transporter activity are primarily significant pathways. Similarly, the most prominent enrichment in the KEGG database is the synapse, metabolism, and hormone-related pathways. All of the evidence mentioned above suggest that the DEGs between AD and controls are closely related to cell membrane function, transport, synapse, and metabolism.
Meanwhile, it is noted that AsymAD is a subtype of preclinical AD characterized as an asymptomatic at-risk state for AD, where beta-amyloids in the brain and CSF are thought to be the primary evidence (Mantzavinos and Alexiou, 2017). Basic scientists indicate that alterations in neurons, microglia, and astroglia drive the disease’s insidious progression before cognitive impairment appear (Elahi et al., 2020). In addition, data obtained through imaging studies with PiB-PET also suggests that Aβ deposits may appear up to two decades before the onset of clinical manifestations of dementia. There is growing evidence that pathological changes in AD have occurred 10 years before symptoms arise, but not all AsymADs will translate into SymADs. AsymADs are not the necessary stage of AD. Therefore, in the second part, we discussed the difference between AsymAD and SymAD to find the risk factors for converting AsymAD to SymAD and provide targets for subsequent drug treatment.
In the comparative analysis of AsymAD and SymAD, we obtained 19 genes by intersection. Among them, compared with AD genes through GeneCards2 and MalaCards3 databases, we found that CAP2, VSNL1, PAK1, SYP, ENC1, NEFM, ADCYAP1, SVOP, CHGB are related to Alzheimer’s disease; LOC387856 (CCDC184) and SYP are associated with dementia. Finally, according to logistic regression analysis, four molecules most related to clinical transformation (VSNL1, RTN1, FGF12, and ENC1) were screened out.
Visinin-like 1 (VSNL1) is a member of a subfamily of neuronal calcium sensor proteins. The encoded visinin-like protein 1 (VILIP-1) upregulates functional α4β2 nicotinic/acetylcholine receptors in hippocampal neurons (Recabarren and Alarcon, 2017). Several studies have shown that in patients with early symptomatic AD, the level of VILIP-1 in cerebrospinal fluid is closely related to whole and regional brain atrophy and is associated with amyloid load in normal individuals in cognition (Tarawneh et al., 2012). Moreover, VILIP-1 influences the intracellular neuronal signaling pathways involved in synaptic plasticity in the central nervous system. It also participates in cyclic nucleotide cascades, nicotinic signaling, and Ca2+ homeostasis, leading to neuronal loss (Groblewska et al., 2015). In this study, the expression of VSNL1 in SymAD was significantly downregulated, which was consistent with the results of Mirza and Rajeh (2017).
Reticulon 1 (RTN1) is an endoplasmic reticulum stress protein in the reticulon family involved in endocrine secretion and membrane trafficking. RTN1 is expressed predominantly in neuroendocrine tissues. Its function is mainly implicated in DNA binding or epigenetic modification, neuronal differentiation, and neurodegenerative diseases such as AD (Recabarren and Alarcon, 2017). Considerable evidence suggests that all RTN proteins and receptor NgR are engaged in the pathology change of AD by regulating the beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) function or APP processing, and thereby product amyloid β in the brain (Kulczynska-Przybik et al., 2021). Previous studies have observed a significant decrease in RTN1 expression in the frontal cortex of AD patients (Kim et al., 2000). Similarly, it was also significantly downregulated in the temporal cortex of symptomatic AD patients here.
The fibroblast growth factor 12 (FGF12) encodes the growth factor family protein involved in nervous system development and the positive regulation of voltage-gated sodium channel activity (Siekierska et al., 2016). FGF family members possess broad cell survival activities and are involved in various biological processes, including embryonic development, cell growth, morphogenesis, tissue repair, tumor growth, and invasion. The PI3K-Akt and apoptotic pathways in fibroblasts are the most relevant pathways to this gene. Gene Ontology annotations closely related FGF12 to growth factor activity and transmembrane transporter binding. This gene has been shown to play an essential role in the pathogenesis of epileptic encephalopathy (Deciphering Developmental Disorders Study, 2017).
The ectodermal-neural Cortex 1 (ENC1) gene is highly expressed in developing neurons and plays a role in the oxidative stress response as a regulator of the transcription factor Nrf2. This gene encodes a member of the kelch-related family of actin-binding proteins, which is implicated in neurite development and neuronal process formation during neuronal differentiation. In addition, ENC1 is upregulated in vitro models of neural injuries, such as oxygen-glucose deprivation or toxic intracellular protein aggregation and endoplasmic reticulum stress. Among its related pathways are the glucocorticoid receptor and wnt signaling pathway. Furthermore, based on cognitive, pathological, and genomic data, ENC1 was identified as a potential risk factor in cognitive performance and neuropathological burden in the aging population (White et al., 2017). Similar to other studies, we found a significant decrease in ENC1 expression in the temporal cortex of symptomatic AD patients (Wang et al., 2022).
In addition to the expression changes, the SNP loci of these genes are also worthy of further study. In previous researches, Hollingworth et al. (2012) found that VSNL1 (rs4038131) showed the most robust association with AD symptoms compared to controls. White et al. (2017) announced that rs76662990G locus in ENC1 was associated with slower cognitive decline in multiple domains. However, there is little evidence for the genetic locus associated with the risk of symptomatic AD, and more analysis and experimental studies are needed to gradually discover the mechanism behind it.
It is worth noting that our integrated dataset included a total of 184 patients with AD and 132 non-AD participants. The number of samples investigated ranged from 6 to 195 cases across the studies. There was no statistical difference among datasets in age. In the same way, we found that a total of 6 data sets obtained from the same platform (GPL10558) were included in the study of Zhang et al. (2019), of which the minimum sample size was 13, and the maximum was as many as 106. The three datasets we selected were also sequenced by using the same platform. Additionally, a total of 4 microarray data sets were included in the study of Yin et al. (2016). The small sample consisted of 22 cases of hepatocellular carcinoma and 21 liver tissue while the large sample covered 225 cases of hepatocellular carcinoma and 220 liver tissue. They also claimed that there was no significant difference between the datasets. We are trying to include as many datasets as possible. Sample size imbalance is inevitable as a consequence which is one of the drawbacks of such research. Researchers need to further think about how to solve this problem to improve the accuracy of analysis.
The result of the present study provides new insight into the earliest biological changes occurring in the brain before the manifestation of clinical AD symptoms. It offers new potential therapeutic targets for early disease intervention. VSNL1, RTN1, FGF12, and ENC1 may be the essential genes that progress asymptomatic AD to symptomatic AD. Moreover, they may serve as genetic risk factors to identify high-risk individuals showing an earlier onset of AD.
The original contributions presented in this study are included in this article/Supplementary material, further inquiries can be directed to the corresponding authors.
JL, SY, and AX conceived and designed the study. YR and YM analyzed the data and composition of figures. WL and HL drafted the manuscript. All authors were involved in writing of the manuscript, gave final approval of the version, and agreed to be accountable for all aspects of the work.
This study was supported by the grant from the National Natural Science Foundation of China (Nos. 82171178 and 82101271) and the Natural Science Foundation of Guangdong Province (Nos. 2021A1515010705 and 2020A1515110317).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2022.1060111/full#supplementary-material
Supplementary Figure 1 | Data standardization and quality control. (A,B) The PCA graph before and after data set processing. (C,D) The density map before and after data set processing. (E) The box plot of comparison between AD group and control group after dataset processing.
Ayodele, T., Rogaeva, E., Kurup, J. T., Beecham, G., and Reitz, C. (2021). Early-Onset Alzheimer’s disease: What is missing in research? Curr. Neurol. Neurosci. Rep. 21:4.
Baker, K., Gordon, S. L., Melland, H., Bumbak, F., Scott, D. J., Jiang, T. J., et al. (2018). SYT1-associated neurodevelopmental disorder: A case series. Brain 141, 2576–2591. doi: 10.1093/brain/awy209
Chen, G., Wang, M., Zhu, P., Wang, G., and Hu, T. (2022). Adverse effects of SYP-3343 on zebrafish development via ROS-mediated mitochondrial dysfunction. J. Hazard. Mater. 437:129382. doi: 10.1016/j.jhazmat.2022.129382
Deciphering Developmental Disorders Study (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438.
Elahi, F. M., Casaletto, K. B., La Joie, R., Walters, S. M., Harvey, D., Wolf, A., et al. (2020). Plasma biomarkers of astrocytic and neuronal dysfunction in early- and late-onset Alzheimer’s disease. Alzheimers Dement. 16, 681–695. doi: 10.1016/j.jalz.2019.09.004
Groblewska, M., Muszyński, P., Wojtulewska-Supron, A., Kulczyńska-Przybik, A., and Mroczko, B. (2015). The role of Visinin-Like Protein-1 in the pathophysiology of Alzheimer’s disease. J. Alzheimers Dis. 47, 17–32.
Guerreiro, R., and Hardy, J. (2014). Genetics of Alzheimer’s disease. Neurotherapeutics 11, 732–737.
Hollingworth, P., Sweet, R., Sims, R., Harold, D., Russo, G., Abraham, R., et al. (2012). Genome-wide association study of Alzheimer’s disease with psychotic symptoms. Mol. Psychiatry 17, 1316–1327.
Kim, S. H., Yoo, B. C., Broers, J. L., Cairns, N., and Lubec, G. (2000). Neuroendocrine-specific protein C, a marker of neuronal differentiation, is reduced in brain of patients with Down syndrome and Alzheimer’s disease. Biochem. Biophys. Res. Commun. 276, 329–334. doi: 10.1006/bbrc.2000.3464
Kulczynska-Przybik, A., Mroczko, P., Dulewicz, M., and Mroczko, B. (2021). The Implication of Reticulons (RTNs) in neurodegenerative diseases: From molecular mechanisms to potential diagnostic and therapeutic approaches. Int. J. Mol. Sci. 22:4630. doi: 10.3390/ijms22094630
Lefterov, I., Wolfe, C. M., Fitz, N. F., Nam, K. N., Letronne, F., Biedrzycki, R. J., et al. (2019). APOE2 orchestrated differences in transcriptomic and lipidomic profiles of postmortem AD brain. Alzheimers Res. Ther. 11:113. doi: 10.1186/s13195-019-0558-0
Mantzavinos, V., and Alexiou, A. (2017). Biomarkers for Alzheimer’s disease diagnosis. Curr. Alzheimer Res. 14, 1149–1154.
Mirza, Z., and Rajeh, N. (2017). Identification of electrophysiological changes in Alzheimer’s disease: A microarray based transcriptomics and molecular pathway analysis study. CNS Neurol. Disord. Drug Targets 16, 1027–1038. doi: 10.2174/1871527316666171023153837
Mold, M., Linhart, C., Gómez-Ramírez, J., Villegas-Lanau, A., and Exley, C. (2020). Aluminum and amyloid-β in familial Alzheimer’s disease. J. Alzheimers Dis. 73, 1627–1635.
Morimoto, S., Ishikawa, M., Watanabe, H., Isoda, M., Takao, M., Nakamura, S., et al. (2020). Brain transcriptome analysis links deficiencies of stress-responsive proteins to the Pathomechanism of Kii ALS/PDC. Antioxidants 9:423. doi: 10.3390/antiox9050423
Patel, H., Hodges, A. K., Curtis, C., Lee, S. H., Troakes, C., Dobson, R. J. B., et al. (2019). Transcriptomic analysis of probable asymptomatic and symptomatic Alzheimer brains. Brain Behav. Immun. 80, 644–656.
Piras, I. S., Krate, J., Delvaux, E., Nolz, J., Mastroeni, D. F., Persico, A. M., et al. (2019). Transcriptome changes in the Alzheimer’s disease middle temporal Gyrus: Importance of RNA metabolism and mitochondria-associated membrane genes. J. Alzheimers Dis. 70, 691–713. doi: 10.3233/JAD-181113
Recabarren, D., and Alarcon, M. (2017). Gene networks in neurodegenerative disorders. Life Sci. 183, 83–97.
Scheltens, P., De Strooper, B., Kivipelto, M., Holstege, H., Chételat, G., Teunissen, C. E., et al. (2021). Alzheimer’s disease. Lancet 397, 1577–1590.
Shi, Z., Zhang, K., Zhou, H., Jiang, L., Xie, B., and Wang, R. (2020). Increased miR-34c mediates synaptic deficits by targeting synaptotagmin 1 through ROS-JNK-p53 pathway in Alzheimer’s disease. Aging Cell 19:e13125.
Siekierska, A., Isrie, M., Liu, Y., Scheldeman, C., Vanthillo, N., Lagae, L., et al. (2016). Gain-of-function FHF1 mutation causes early-onset epileptic encephalopathy with cerebellar atrophy. Neurology 86, 2162–2170. doi: 10.1212/WNL.0000000000002752
Spina, S., La Joie, R., Petersen, C., Nolan, A. L., Cuevas, D., Cosme, C., et al. (2021). Comorbid neuropathological diagnoses in early versus late-onset Alzheimer’s disease. Brain 144, 2186–2198. doi: 10.1093/brain/awab099
Talwar, P., Silla, Y., Grover, S., Gupta, M., Agarwal, R., Kushwaha, S., et al. (2014). Genomic convergence and network analysis approach to identify candidate genes in Alzheimer’s disease. BMC Genomics 15:199. doi: 10.1186/1471-2164-15-199
Tarawneh, R., Lee, J. M., Ladenson, J. H., Morris, J. C., and Holtzman, D. M. (2012). CSF VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology 78, 709–719.
Wang, H., Bennett, D. A., De Jager, P. L., Zhang, Q. Y., and Zhang, H. Y. (2021). Genome-wide epistasis analysis for Alzheimer’s disease and implications for genetic risk prediction. Alzheimers Res. Ther. 13:55.
Wang, H., Zhang, Y., Zheng, C., Yang, S., Chen, X., Wang, H., et al. (2022). A 3-Gene-based diagnostic signature in Alzheimer’s disease. Eur. Neurol. 85, 6–13. doi: 10.1159/000518727
White, C. C., Yang, H. S., Yu, L., Chibnik, L. B., Dawe, R. J., Yang, J., et al. (2017). Identification of genes associated with dissociation of cognitive performance and neuropathological burden: Multistep analysis of genetic, epigenetic, and transcriptional data. PLoS Med. 14:e1002287. doi: 10.1371/journal.pmed.1002287
Xiao, Z., Yao, S., Wang, Z. M., Zhu, D. M., Bie, Y. N., Zhang, S. Z., et al. (2021). Multiparametric MRI features predict the SYP gene expression in low-grade glioma patients: A machine learning-based radiomics analysis. Front. Oncol. 11:663451. doi: 10.3389/fonc.2021.663451
Yin, F., Shu, L., Liu, X., Li, T., Peng, T., Nan, Y., et al. (2016). Microarray-based identification of genes associated with cancer progression and prognosis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 35:127.
Keywords: Alzheimer’s disease, genes, transcriptomic analysis, GEO, dementia
Citation: Liao W, Luo H, Ruan Y, Mai Y, Liu C, Chen J, Yang S, Xuan A and Liu J (2022) Identification of candidate genes associated with clinical onset of Alzheimer’s disease. Front. Neurosci. 16:1060111. doi: 10.3389/fnins.2022.1060111
Received: 02 October 2022; Accepted: 30 November 2022;
Published: 20 December 2022.
Edited by:
Guangzhi Ning, Tianjin Medical University General Hospital, ChinaReviewed by:
Li-Ching Wu, National Central University, TaiwanCopyright © 2022 Liao, Luo, Ruan, Mai, Liu, Chen, Yang, Xuan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Liu, ✉ bGl1anVuQGd6aG11LmVkdS5jbg==; Aiguo Xuan, ✉ eGFnMjAwNUAxNjMuY29t; Shaoqing Yang, ✉ MTA2OTM4MTEwNEBxcS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.