 Kaleb Dee Miles
Kaleb Dee Miles Caleb Andrew Doll*
Caleb Andrew Doll*- Department of Pediatrics, Section of Developmental Biology, University of Colorado School of Medicine, Children’s Hospital Colorado, Aurora, CO, United States
Developmental changes in ionic balance are associated with crucial hallmarks in neural circuit formation, including changes in excitation and inhibition, neurogenesis, and synaptogenesis. Neuronal excitability is largely mediated by ionic concentrations inside and outside of the cell, and chloride (Cl–) ions are highly influential in early neurodevelopmental events. For example, γ-aminobutyric acid (GABA) is the main inhibitory neurotransmitter of the mature central nervous system (CNS). However, during early development GABA can depolarize target neurons, and GABAergic depolarization is implicated in crucial neurodevelopmental processes. This developmental shift of GABAergic neurotransmission from depolarizing to hyperpolarizing output is induced by changes in Cl– gradients, which are generated by the relative expression of Cl– transporters Nkcc1 and Kcc2. Interestingly, the GABA polarity shift is delayed in Fragile X syndrome (FXS) models; FXS is one of the most common heritable neurodevelopmental disorders. The RNA binding protein FMRP, encoded by the gene Fragile X Messenger Ribonucleoprotein-1 (Fmr1) and absent in FXS, appears to regulate chloride transporter expression. This could dramatically influence FXS phenotypes, as the syndrome is hypothesized to be rooted in defects in neural circuit development and imbalanced excitatory/inhibitory (E/I) neurotransmission. In this perspective, we summarize canonical Cl– transporter expression and investigate altered gene and protein expression of Nkcc1 and Kcc2 in FXS models. We then discuss interactions between Cl– transporters and neurotransmission complexes, and how these links could cause imbalances in inhibitory neurotransmission that may alter mature circuits. Finally, we highlight current therapeutic strategies and promising new directions in targeting Cl– transporter expression in FXS patients.
Introduction
Fragile X syndrome (FXS) is one of the most common heritable neurodevelopmental disorders, which is characterized by intellectual disability, epilepsy, and behavioral symptoms such as anxiety and hyperactivity (Bhakar et al., 2012; Nelson et al., 2013). The etiology stems from a trinucleotide expansion mutation in the 5′ untranslated region of the FMR1 gene, thus leading to the loss of fragile X messenger ribonucleoprotein (FMRP) (Verkerk et al., 1991). FMRP plays essential roles in a variety of neurodevelopmental processes, including synaptogenesis, cell fate specification, and differentiation (Lu et al., 2004; Antar et al., 2006; Doll et al., 2021). Canonically, FMRP functions as an RNA binding protein (RBP), regulating the expression of an array of mRNAs associated with autism spectrum disorders and synaptic processes (Darnell et al., 2011; Ascano et al., 2012). It is estimated that 1 in 7,000 males and 1 in 11,000 females have FXS, and approximately 25–33% of individuals with FXS also meet the criteria for autism spectrum disorders (ASD) by displaying strong overlap in behavioral symptoms (Hunter et al., 2014; Kaufmann et al., 2017). With the rising prevalence and complex nature of these disorders, researchers are faced with many challenges to decipher the pathophysiology and develop therapeutics (Newschaffer et al., 2005; Contractor et al., 2021).
An established hallmark of FXS is hyperactivity, which has been associated with imbalanced excitation and inhibition (E/I ratios) in neural circuits. As the major inhibitory neurotransmitter in the brain, GABAergic influence on FXS pathogenesis represents a key component of this theory. Indeed, there is evidence that reduced GABAergic output in FXS results in hypoinhibition and imbalanced E/I output (Gibson et al., 2008; Olmos-Serrano et al., 2010; Goncalves et al., 2013). However, motor circuits begin to form in embryogenesis, and neurotransmission in embryonic stages is distinct from the mature nervous system. For example, in early neurodevelopment the main “inhibitory” neurotransmitters, GABA and glycine, can depolarize receptive neurons (Zhang et al., 2006, 2010; Reynolds et al., 2008). This excitatory influence on immature neurons is associated with crucial neurodevelopmental processes, including neural stem cell proliferation, cell migration, neurite outgrowth, synapse formation, and network oscillations, all of which represent critical hallmarks in neural circuit formation (Behar et al., 2000; Liu et al., 2005; Akerman and Cline, 2006; Ben-Ari et al., 2007; Cancedda et al., 2007; Wang and Kriegstein, 2008). As development ensues, GABA reception undergoes a polarity shift from excitatory to inhibitory output, which is dictated by the relative expression of chloride ion (Cl–) transporters and Cl– dynamics (Rivera et al., 1999; Yamada et al., 2004; Watanabe and Fukuda, 2015). However, it has been shown in models of FXS, ASD, and other neurodevelopmental disorders that this polarity shift is disrupted or delayed (Talos et al., 2012; Duarte et al., 2013; He et al., 2014; Tyzio et al., 2014; Banerjee et al., 2016; Ruffolo et al., 2016; Tang et al., 2016; Amin et al., 2017; Li et al., 2017; Hinz et al., 2019). Importantly, persistent depolarization by GABA and glycine could influence epileptic and repetitive behaviors observed in these disorders (Duarte et al., 2013; He et al., 2014; Tyzio et al., 2014; Banerjee et al., 2016; Tang et al., 2016; Huo et al., 2017; Hinz et al., 2019).
Researchers have attempted to restore excitatory/inhibitory (E/I) balance in FXS and ASD by manipulating GABAergic and glutamatergic (Glu) neurotransmission. Collectively, animal studies for both GABA and Glu modulators have produced promising results by promoting proper dendritic spine development, reducing severity of seizures, and ameliorating repetitive behaviors (He et al., 2002; Silverman et al., 2010; Henderson et al., 2012; Wei et al., 2012). Subsequent GABA modulator clinical trials assessing acamprosate, arbaclofen, riluzole/risperidone, and valproate have collectively shown improved social behavior, hyperactivity, communication skills, compulsive symptoms, and irritability in pediatric and some young adult subjects (Hellings et al., 2005; Hollander et al., 2006; Erickson et al., 2010, 2011a,b; Berry-Kravis et al., 2012). Other approaches have targeted Cl– transporters (Nkcc1 and Kcc2) by selectively inhibiting Nkcc1 function or enhancing Kcc2 expression to restore the E/I balance (Guida et al., 2015; He et al., 2019; Tang et al., 2019; Urbanska et al., 2019). Although we lack the space to review mechanisms of Cl– transporter expression, previous reviews have detailed Cl– transporter transcription, post-translational modifications, and function (Watanabe and Fukuda, 2015; Schulte et al., 2018); these studies have contributed to the development of new therapeutic strategies and represent a true success story in the interplay between basic science and the clinic. We also recognize a few of the excellent reviews on GABA and chloride transporter associations to neurodevelopmental disorders (Paluszkiewicz et al., 2011; Schulte et al., 2018; Liu et al., 2022).
This is a developmental perspective on chloride transporter links to FXS and associated neurodevelopmental disorders. We also address the development of spinal networks, which may be especially pertinent to the field as FXS, ASDs, and NDDs are all associated with motor challenges. We first summarize the canonical Cl– transporter expression and function in typical neurodevelopment and in FXS. We then discuss roles for depolarizing GABAergic signaling in critical developmental processes. Next, we present non-canonical roles for Cl– transporters, which could also influence inhibitory neurotransmission. Finally, we highlight the current developments and advancements that establish Cl– transporters as therapeutic targets. It is important to note that our primary focus is on GABAergic signaling, though glycinergic transmission—most predominant in the spinal cord—is also depolarizing in immature neurons (Wu et al., 1992; Rivera et al., 1999).
Chloride transporter expression underlies the GABAergic polarity shift
Inhibitory neurotransmission is largely dependent on electrochemical ion gradients established at the plasma membrane of neurons receptive to GABAergic and glycinergic signaling. The primary inhibitory receptors of the CNS, GABAA receptors and glycinergic receptors, are ionotropic anion permeable channels that allow the passage of chloride (Cl–) across the plasma membrane (Kaila, 1994; Kirsch, 2006). The Cl– gradient is mainly established by two Cl– transporters: Nkcc1, a Cl– importer; and Kcc2, a Cl– exporter. Therefore, the expression and activity of both transporters influences intracellular Cl– concentration, which ultimately determines the polarity of GABAergic and glycinergic transmission. In immature neurons, Nkcc1 expression is greater than Kcc2, resulting in a higher intracellular concentration of Cl– (Rivera et al., 1999). As GABA receptors are Cl– permeable, GABA signaling can depolarize immature neurons (reflecting Cl– efflux) resulting in a higher propensity toward neuronal excitation (Kaila, 1994; Dzhala et al., 2005; Moore et al., 2017). In contrast, increased Kcc2 function lowers intracellular Cl– content in more mature neurons, which now receive GABAergic signaling in a hyperpolarizing (inhibitory) manner (Figure 1). The developmental upregulation of Kcc2 expression is a conserved process, as described in humans, rats, mice, zebrafish, and C. elegans (Gulyás et al., 2001; Hübner et al., 2001; Tanis et al., 2009; Zhang et al., 2010; Huo et al., 2017). The polarity-switch is crucial for the formation of inhibitory networks that limit excitability in the nervous system.
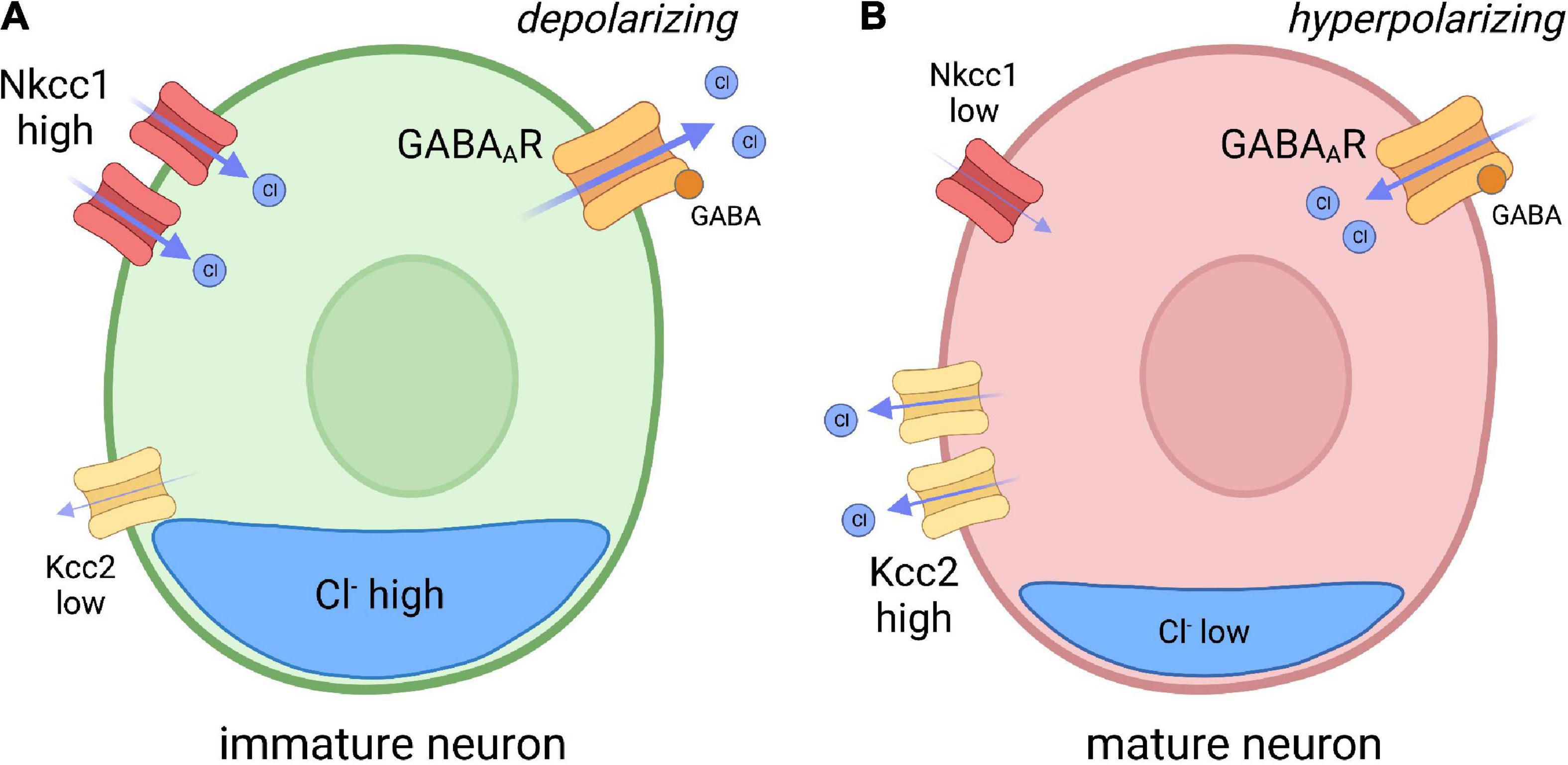
Figure 1. Model depicting chloride (CI) transporter expression in immature and mature neurons. (A) In immature neurons, the Nkcc1 Cl– importer is highly expressed, while the Kcc2 Cl– exporter expression is low, resulting in high internal chloride concentration. As GABA A receptors (GABAAR) are Cl– permeable, GABA reception spurs depolarization of the membrane through Cl– efflux. (B) In mature neurons, Kcc2 expression is increased, resulting in reduced internal Cl– concentration compared to immature neurons. GABA binding to GABAAR in mature neurons therefore drives hyperpolarization. Inspired by Ben-Ari, Trends in Neurosciences, 2017.
Chloride transporter expression is regulated by regional and temporal mechanisms that appear to coincide with the maturation of distinct neural networks. In the rodent brain, Nkcc1 is expressed in the precursors of the neuroepithelium, while Kcc2 is primarily expressed in differentiated neurons (Li et al., 2002; Wang et al., 2002; Stein et al., 2004). Kcc2 expression also correlates with the progressive maturation of different brain regions, as it is first expressed in the caudal spinal cord and medulla and then proceeds to more rostral structures in the brain (Li et al., 2002; Stein et al., 2004; Watanabe and Fukuda, 2015). Interestingly, the reversal potential of GABA type A receptor- (GABAAR-) mediated currents varies among neuronal populations and brain structures, which reflects the diverse expression and function of the transporters (Watanabe and Fukuda, 2015). For example, Kcc2 is expressed in motor neurons during early embryonic stages when GABAergic/glycinergic neurotransmission is still depolarizing (Hübner et al., 2001; Delpy et al., 2008). This may suggest that Nkcc1-mediated chloride import is more predominant at this stage, prior to the inhibitory influence of GABA in more mature stages.
Changes in chloride transporter expression and function are also associated with neurodevelopmental disorders that share common patient phenotypes, most notably epilepsy. The GABA polarity shift is delayed in FXS and other neurodevelopmental disorders, and there is evidence that dysregulation of the Cl– transporters may underlie altered inhibitory transmission (Talos et al., 2012; He et al., 2014; Banerjee et al., 2016; Amin et al., 2017). In Fmr1 knockout mice, Nkcc1 protein expression is upregulated in the cortex at P10, which may account for altered chloride homeostasis and delayed polarity shift in these animals (He et al., 2014). In addition, inhibition of Nkcc1 function via bumetanide during the critical period of somatosensory development restores sensory deficits and neuronal morphology in Fmr1 knockout mice (He et al., 2019). Although it is unclear if FMRP directly regulates the translation of Nkcc1 and Kcc2, FMRP binds the transcripts encoding these proteins, Slc12a2 and Slc12a5, respectively (Darnell et al., 2011). Intriguingly, mutations in SLC12A2 (encoding NKCC1) are linked with neurodevelopmental disorders, and genetic variation in SLC12A5 (encoding KCC2) is associated with epilepsy and autism (Kahle et al., 2014; Puskarjov et al., 2014; Merner et al., 2015; McNeill et al., 2020; Stödberg et al., 2020).
Chloride transporter misexpression is also noted in related neurodevelopmental disorders. Some patients with tuberous sclerosis (a condition often co-morbid with seizures) show upregulated Nkcc1 and downregulated Kcc2 in cortical tubers (Talos et al., 2012; Ruffolo et al., 2016). Hippocampal neurons from DiGeorge syndrome model mice also show increased expression of Nkcc1 and reduced expression of Kcc2 (Amin et al., 2017). Kcc2 downregulation is also observed in VPA-induced rodent models of autism and neurons derived from patients with Rett syndrome (Duarte et al., 2013; Banerjee et al., 2016; Tang et al., 2016; Hinz et al., 2019). Finally, both rodent and human temporal lobe epilepsy studies show upregulated Nkcc1 and downregulated Kcc2 (Karlócai et al., 2016; Auer et al., 2020; Hampel et al., 2021). These findings are especially important as these neurodevelopment disorders share a common comorbidity of increased seizure susceptibility, which is speculated to be a cortical manifestation of runaway hyperexcitability produced by E/I imbalance (Canitano, 2007; González and Bautista, 2009; Glaze et al., 2010; He et al., 2014; Ruffolo et al., 2016). Taken together, many neurodevelopmental disorders that share common hyperexcitable phenotypes are associated with misregulated chloride transporter expression.
The influence of depolarizing GABA and glycine on neurodevelopment
Cell proliferation and synaptogenesis
The GABAergic influence on neurodevelopment begins with roles in regulating neurogenesis (Figure 2). GABA can act as a cell cycle inhibitor in neural precursors, organotypic brain slices, and in the subventricular zone (SVZ) of adult mice (Nguyen et al., 2003; Liu et al., 2005). Immature neuroblasts in the SVZ express non-synaptic GABA that drives depolarizing GABAAR reception in GFAP-expressing progenitor cells, which ultimately leads to a reduction in the number of proliferative stem cells (Liu et al., 2005). This shows that newly generated GABAergic neurons restrict neurogenesis by inhibiting stem cell proliferation (Liu et al., 2005; Platel et al., 2008). Moreover, premature reversal of the chloride gradient through global overexpression of Kcc2 in zebrafish hinders the production of spinal neurons and disrupts axonal development, in a mechanism that likely involves glycinergic depolarization (Reynolds et al., 2008). These studies show critical roles for depolarizing GABAergic/glycinergic signaling in the generation of neuronal cell types.
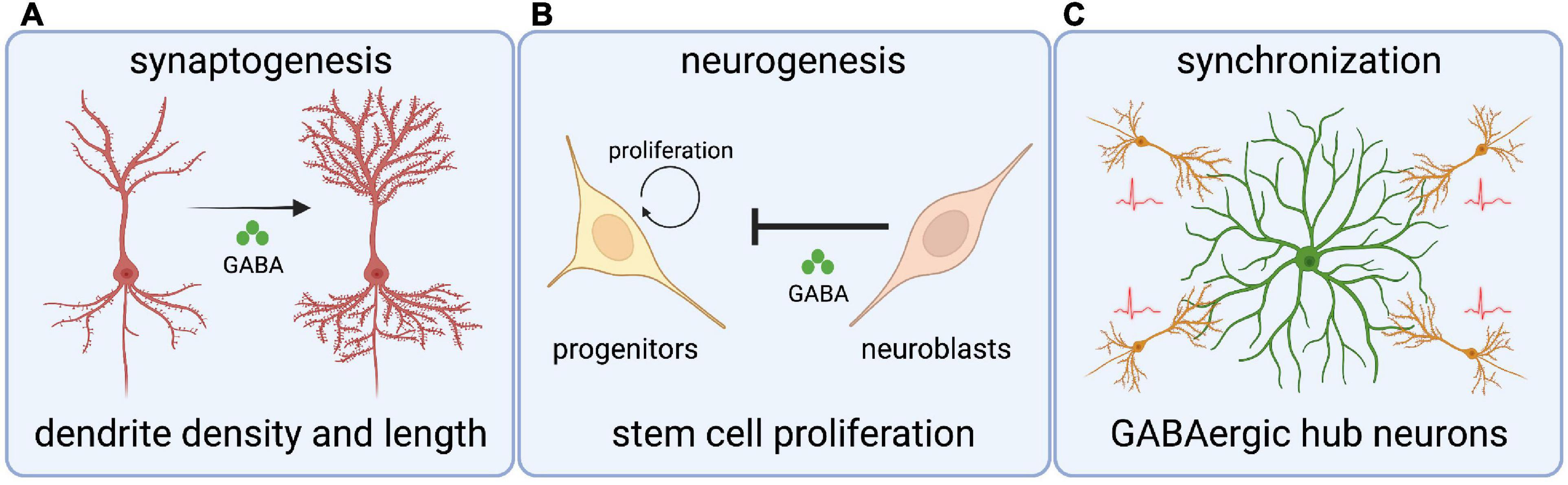
Figure 2. Roles for chloride transporters and depolarizing GABA in crucial neurodevelopmental processes. (A) Depolarizing GABA (through Cl– transporters) regulates dendritic spine length and density in cortical neurons. (B) GABA signaling from neuroblasts inhibits stem cell proliferation in the subventricular zone. (C) GABAergic hub interneurons synchronize nascent hippocampal networks through giant depolarizing potentials.
Synaptogenesis is an intricate, multistep process that requires many cell intrinsic and extrinsic factors. Several studies have investigated the role of depolarizing GABA on synaptogenesis by manipulating the Cl– gradient. For example, premature expression of Kcc2 in a subpopulation of ventricular progenitors and progenitor-derived cortical neurons leads to neurons with fewer and shorter dendrites, indicating that depolarizing GABAergic signaling mediates morphological maturation (Cancedda et al., 2007). In contrast, inhibition of Nkcc1 leads to decreased dendrite density and increased dendrite length in newly born cortical neurons (Wang and Kriegstein, 2008). In line with this, disruption of depolarizing GABA in the dentate gyrus of adult mouse hippocampus alters the morphology of newly generated granule neurons (Ge et al., 2006). Finally, GABAAR activation through depolarizing GABA is essential for synaptic inputs on newborn cortical neurons and in adult mouse granule neurons (Ge et al., 2006; Wang and Kriegstein, 2008).
Pioneering GABA signaling can also influence E/I balance in developing neural circuits, as a premature GABA polarity shift in Xenopus tectal neurons and rat cortical neurons leads to an increase of inhibitory inputs compared to excitatory inputs (Chudotvorova et al., 2005; Akerman and Cline, 2006); a similar mechanism occurs in the mammalian cortex (Wang and Kriegstein, 2008). Lastly, depolarizing GABAergic neurotransmission also drives calcium influx by relieving the voltage dependent Mg2+ block, thereby initiating intracellular signaling cascades needed for glutamatergic synapse development (Leinekugel et al., 1995; Akerman and Cline, 2006; Ben-Ari et al., 2007). In summary, depolarizing GABA influences the critical neurodevelopmental mechanisms of neurogenesis and synaptogenesis.
Neural circuit formation
It is important to note that GABAergic currents often mature before glutamatergic counterparts, and GABA is the first and sole source of neurotransmission in various brain structures (Chen et al., 1996; Van Den Pol et al., 1998; Tyzio et al., 1999; Gao and Van Den Pol, 2001; Hennou et al., 2002; Gozlan and Ben-Ari, 2003; Johnson et al., 2003; Wang and Kriegstein, 2008). Therefore, the timing and degree of depolarizing GABAergic signaling in immature neurons is vital for the formation of neural networks. For example, GABAergic hub interneurons play essential roles in synchronizing hippocampal networks by orchestrating the generation of giant depolarizing potentials (GDP); GDP represent the first synapse-driven patterns of activity that help drive the synchronous network activity required for synaptic plasticity (Bonifazi et al., 2009; Picardo et al., 2011). Interestingly, stimulation of hippocampal hub neurons reduces network oscillations and in extreme cases stimulation completely desynchronizes activity (Bonifazi et al., 2009). While this data suggests considerable complexity in hub neuron physiology, it ultimately shows the influence on these pioneer GABAergic cells on the emergence of patterned activity in the brain.
The development of motor circuits in the spinal cord also appear to require synchronizing influence by GABAergic interneurons. GABA and glycine depolarize immature motor neurons in the rodent spinal cord (Wu et al., 1992); these neurotransmitters also drive spontaneous motor neuron activity (Nishimaru et al., 1996). In zebrafish, premature overexpression of Kcc2 reduces early locomotion, which suggests that early motor networks must form prior to the reversal of the chloride gradient (Reynolds et al., 2008). Moreover, GABAergic ventrolateral descending (VeLD) interneurons are highly active in early zebrafish embryogenesis and are physiologically coupled with other spinal neurons, including motor neurons; VeLD appear to act as central pattern generators in generating the earliest spontaneous movements, as inhibition of VeLD activity disrupts the integration of neurons into the motor network (Saint-Amant and Drapeau, 2000; Warp et al., 2012). Taken together, depolarizing activity from GABAergic neurons provides essential synchronizing influence on the emergence of motor circuits.
Foundational alterations in the formation of motor circuits in embryogenesis could have lasting consequences. Hyperexcitable motor behavior is a principal phenotype in various FXS models at many stages of development and maturity (Kim et al., 2014; Shamay-Ramot et al., 2015; Sørensen et al., 2015; Hu et al., 2020; Liu et al., 2022). Depolarizing GABAergic interneurons are crucial for the formation of neural circuits, and the GABAergic polarity shift is hindered in FXS. Importantly, GABAAR expression and GABAAR-mediated inhibition is actually reduced in FXS patients and models and is theorized to underlie hyperexcitable behavior (El Idrissi et al., 2005; D’Hulst et al., 2006; Morin-Parent et al., 2019). It is important to note that depolarizing glycinergic contributions to neurodevelopment are far less explored in FXS models and it is unclear if glycinergic signaling provides an influence that is distinct from GABA. However, altered glycinergic reception is linked to ASD in a mechanism that appears rooted in embryogenesis (Pilorge et al., 2016). Although it is difficult to link developmental roles for GABAergic/glycinergic signaling with later inhibitory requirements, new FXS models must integrate cellular and molecular approaches in embryonic stages with behavioral and physiological readouts in progressive developmental timepoints.
Non-canonical roles for the chloride transporter Kcc2
In addition to ion transport function, Kcc2 also plays structural roles in the generation and function of dendritic spines, as Kcc2 mediates actin dynamics by interacting with β-PIX (β isoform of Rac/Cdc42 guanine nucleotide exchange factor), and with the aid of Rac1 GTPase controls the phosphorylation of Cofilin-1, an actin regulating protein (Chevy et al., 2015; Llano et al., 2015). Cofilin function includes the severing of actin filaments which leads to increased filament turnover, and cofilin-mediated action allows the insertion of AMPA receptors during chemically induced long-term potentiation (LTP) (Bamburg and Bernstein, 2010; Gu et al., 2010). Kcc2 also binds 4.1N, a cytoskeleton-associated protein that regulates lateral diffusion of both Kcc2 and AMPA receptors at excitatory synapses (Li et al., 2007; Gauvain et al., 2011; Chamma et al., 2013). Interestingly, Kcc2 overexpression or downregulation leads to changes in synaptogenesis, including dendritic spines that resemble those in patients with FXS and Fmr1 KO mice (Irwin et al., 2001; Li et al., 2007; Fiumelli et al., 2013; Awad et al., 2018). This phenotype is speculated to contribute to FXS symptoms and severity, as modulation of actin cytoskeleton dynamics ameliorates seizures, rescues behavioral abnormalities (hyperactivity and repetitive movements), and reverses spine abnormalities displayed in Fmr1 KO mice (Dolan et al., 2013). Kcc2 also physically interacts with a variety of membrane and scaffolding proteins, such as metabotropic GABAB receptors; the inhibitory post-synaptic scaffold Gephyrin, required for clustering of GABAergic and glycinergic receptors at the postsynaptic membrane; the GluK2 and Neto2 subunits of Kainate glutamatergic receptors (KAR), implicated in both the kinetics and pre and postsynaptic modulation; and Task-3 (KCNK9) leak potassium channels, linked to network oscillations (Ivakine et al., 2013; Mahadevan et al., 2014; Wright et al., 2017; Goutierre et al., 2019; Kesaf et al., 2020; Al Awabdh et al., 2022). These non-canonical roles for Kcc2 establish the Cl– exporter as a persistent and crucial regulator of neuronal morphology and synaptic function.
Pharmacological approaches and future therapeutics
Persistent changes in excitatory/inhibitory balance in patients with FXS has led to pharmaceutical strategies to restore E/I balance through modulation of GABAergic or glutamatergic neurotransmission. Limited GABA modulator clinical trials (acamprosate, arbaclofen, riluzole/risperidone, and valproate), have shown mixed and inconclusive results, but include improved social behavior, hyperactivity, communication skills, compulsive symptoms, and irritability, with no serious adverse effects (Wu et al., 1992; Kaila, 1994; Dzhala et al., 2005; Hellings et al., 2005; Hollander et al., 2006; Kirsch, 2006; Paluszkiewicz et al., 2011; Guida et al., 2015; Moore et al., 2017; Schulte et al., 2018; He et al., 2019; Tang et al., 2019; Urbanska et al., 2019; Liu et al., 2022). In addition, some glutamatergic antagonists (riluzole, aripiprazole, memantine, phenobam, mavoglurant, and basimglurant) have shown promising results in clinical trials, especially regarding hyperactivity, irritability, and social deficits, though outcomes were highly variable in the treatment of core symptoms (Findling et al., 2014; Ichikawa et al., 2017; Wink et al., 2018; Hardan et al., 2019). Optimal therapeutics must address the complexity of Glu/GABA signaling, the heterogeneity of ASDs, and how ASD pathophysiology varies across age (Masi et al., 2017; Ure et al., 2018).
Alternative pharmaceutical approaches have directly targeted chloride homeostasis to restore E/I balance via drugs targeted to Nkcc1 or Kcc2. In multiple FXS and ASD animal models the Nkcc1 inhibitor bumetanide rescues GABAergic neurotransmission as well as social and repetitive behaviors (Tyzio et al., 2014; Amin et al., 2017; Zeidler et al., 2017; Savardi et al., 2020). As mentioned previously, bumetanide also rescues somatosensory deficits and morphological changes in the Fmr1 knockout mouse (He et al., 2019). A recent clinical trial on bumetanide in children and adolescents with ASD showed promising results by improving symptoms of ASD; however, several limitations such as short observation periods, lack of older ASD participants, and exclusion of participants with comorbidities (epilepsy) should be considered (Lemonnier et al., 2017). Other clinical trials with bumetanide have produced mixed results, which may be due to the blood-brain barrier, diuretic effects, and/or ototoxicity in infants (likely reflecting the ubiquitous expression of Nkcc1) (Löscher et al., 2013; Ben-Ari et al., 2016; Römermann et al., 2017; Nussbaum and Perazella, 2019; Crutel et al., 2021). Therefore, further testing is needed to determine efficient and safe dosages for chronic treatment. Accordingly, additional bumetanide prodrugs and analogs (BUM1, BUM5, bumepamine, azosemide, STS66, BUM9, ARN23746) have been developed to optimize selectivity and brain penetration (Töllner et al., 2014; Erker et al., 2016; Brandt et al., 2018; Hampel et al., 2018; Huang et al., 2019; Savardi et al., 2020).
In related NDDs, additional approaches to minimize hyperexcitable circuit function have focused on enhancement of the chloride exporter Kcc2. These pharmacological approaches include FDA approved Kcc2 expression-enhancing compounds such as BIO (inhibitor of GSK3b), KW-2449 (inhibitor of Flit3), piperine (agonist of TRPV1), and resveratrol (activator of SIRT1) (Guida et al., 2015; Tang et al., 2019; Urbanska et al., 2019). For example, KW-2449 and BIO can hyperpolarize neurons cultured from human Rett Syndrome patients to values comparable to wild type, and restore morphology (Tang et al., 2019). In addition, treatment of Mecp2 knockout mice (a model of Rett syndrome) with KW-2449 or piperine rescues respiratory and locomotion phenotypes (Tang et al., 2019). These results in Rett Syndrome models may represent promise for FXS and other NDDs that also display altered Cl– transporter expression. However, more studies are needed to elucidate the mechanisms of action and potential side effects, and approaches to promote Kcc2 expression and function must also include membrane targeting to achieve transporter function. Although these approaches are in infancy, this neuron-specific transporter has perhaps the most promise as a pharmaceutical target, as Kcc2 function can directly minimize chloride content and reduce hyperexcitability.
Discussion
What is the link between FMRP and chloride balance? Changes in chloride homeostasis in developing neurons underlies the fascinating polarity shift that occurs in the maturation of inhibitory circuits; this maturation is delayed in FXS, which may have foundational consequences on the formation and function of the nervous system. We speculate that in the absence of FMRP, deficient neuronal differentiation leaves immature neurons locked in an elevated chloride state, and thus more prone to excitation. There are several clues pointing to this mechanism: 1) Nkcc1 inclusion and Kcc2 exclusion from precursor cells (Li et al., 2002; Wang et al., 2002; Stein et al., 2004); 2) elevated Nkcc1 expression in FXS knockout mice (He et al., 2014); 3) deficient differentiation in the absence of FMRP, as seen in neural precursor cells and spinal cell subtypes (Luo et al., 2010; Edens et al., 2019; Doll et al., 2021; Raj et al., 2021). As FMRP appears to bind both Slc12a2 and Slc12a5 (the genes encoding Nkcc1 and Kcc2) (Darnell et al., 2011), there could be a direct regulatory role for the RNA binding protein in the transport/localization, stability, or translation of these transcripts. The mechanism could also be indirect through microRNA-based repression of Nkcc1 (Liu et al., 2015; Lippi et al., 2016). Given regional and temporal differences in transporter expression during development, rescuing chloride imbalance on a global level will be especially challenging.
Is there a link or point of convergence between Cl– transporter function and disparate NDDs? Although a diverse family of NDDs (FXS, Rett Syndrome, DiGeorge syndrome, Dravet syndrome, epilepsy, etc.) are linked to unique genetic loci, they have common symptoms and comorbidities and are associated with altered Cl– transporter expression. In other words, changes in the crucial neurodevelopmental polarity shift are common to disease states with vast genetic heterogeneity. Given these commonalities across NDDs, we predict that the progression of transporter expression is linked with neuronal maturation, such that alterations in neurogenesis and differentiation in unique disease states lie upstream of chloride homeostasis. Although this suggests independent genetic mechanisms underlie chloride balance, it also grants broader therapeutic opportunities through refined drugs targeting these transporters.
Is there a link between depolarizing GABAergic/glycinergic function in embryogenesis to deficient inhibition at maturity? Following the neuro-archeology concept formulated by Ben-Ari, if synchronizing GABAergic neurons maintain a persistent depolarizing influence they could drive extended immature currents and oscillations in the developing brain that would ultimately perturb the developmental sequence and operation of functional networks (Ben-Ari, 2008, 2014). We speculate that this could present runaway hyperexcitability in the form of cognitive impairment and seizure susceptibility shown in neurodevelopmental disabilities and various forms of epilepsy (Giachello and Baines, 2015). Neural circuits develop hierarchically, within narrow critical periods that shape connectivity and long-term function (Hensch, 2004; LeBlanc and Fagiolini, 2011). Although many of these critical periods have been dissected through elegant developmental studies, it is still very difficult to link early neurodevelopmental events (neurogenesis, fate specification, cell migration, etc.) with long term functional output (behavior). The restoration of E/I balance represents a main tenet of FXS and ASD modeling and entails either a restoration of inhibition or a dampening of excitation. While many studies have shown deficient GABAergic signaling in established FXS circuits, roles for FMRP in early development, prior to the polarity shift, are less explored.
Data availability statement
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
KM: research and writing. CD: editing and visualization. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by the United States National Institute of Health (NIH) grant R21 NS117886 to CD.
Acknowledgments
We would like to thank Chase Barker and members of the Bruce Appel laboratory for insightful discussion. Figures were created with Biorender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akerman, C. J., and Cline, H. T. (2006). Depolarizing GABAergic conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J. Neurosci. 26, 5117–5130. doi: 10.1523/JNEUROSCI.0319-06.2006
Al Awabdh, S., Donneger, F., Goutierre, M., Séveno, M., Vigy, O., Weinzettl, P., et al. (2022). Gephyrin interacts with the K-Cl cotransporter KCC2 to regulate its surface expression and function in cortical neurons. J. Neurosci. 42, 166–182. doi: 10.1523/JNEUROSCI.2926-20.2021
Amin, H., Marinaro, F., Tonelli, D. D. P., and Berdondini, L. (2017). Developmental excitatory-to-inhibitory GABA-polarity switch is disrupted in 22q11.2 deletion syndrome: A potential target for clinical therapeutics. Sci. Rep. 7:15752. doi: 10.1038/s41598-017-15793-9
Antar, L. N., Li, C., Zhang, H., Carroll, R. C., and Bassell, G. J. (2006). Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol. Cell. Neurosci. 32, 37–48. doi: 10.1016/j.mcn.2006.02.001
Ascano, M., Mukherjee, N., Bandaru, P., Miller, J. B., Nusbaum, J. D., Corcoran, D. L., et al. (2012). FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386. doi: 10.1038/nature11737
Auer, T., Schreppel, P., Erker, T., and Schwarzer, C. (2020). Functional characterization of novel bumetanide derivatives for epilepsy treatment. Neuropharmacology 162:107754. doi: 10.1016/j.neuropharm.2019.107754
Awad, P. N., Amegandjin, C. A., Szczurkowska, J., Carriço, J. N., Fernandes Do Nascimento, A. S., Baho, E., et al. (2018). KCC2 regulates dendritic spine formation in a brain-region specific and BDNF dependent manner. Cereb. Cortex 28, 4049–4062. doi: 10.1093/cercor/bhy198
Bamburg, J. R., and Bernstein, B. W. (2010). Roles of ADF/cofilin in actin polymerization and beyond. F1000 Biol. Rep. 2:62. doi: 10.3410/B2-62
Banerjee, A., Rikhye, R. V., Breton-Provencher, V., Tangb, X., Li, C., Li, K., et al. (2016). Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 113, E7287–E7296. doi: 10.1073/pnas.1615330113
Behar, T. N., Schaffner, A. E., Scott, C. A., Greene, C. L., and Barker, J. L. (2000). GABA receptor antagonists modulate postmitotic cell migration in slice cultures of embryonic rat cortex. Cereb. Cortex 10, 899–909. doi: 10.1093/cercor/10.9.899
Ben-Ari, Y. (2008). Neuro-archaeology: Pre-symptomatic architecture and signature of neurological disorders. Trends Neurosci. 31, 626–636. doi: 10.1016/j.tins.2008.09.002
Ben-Ari, Y. (2014). The GABA excitatory/inhibitory developmental sequence: A personal journey. Neuroscience 279, 187–219. doi: 10.1016/j.neuroscience.2014.08.001
Ben-Ari, Y., Damier, P., and Lemonnier, E. (2016). Failure of the nemo trial: Bumetanide is a promising agent to treat many brain disorders but not newborn seizures. Front. Cell. Neurosci. 10:90. doi: 10.3389/fncel.2016.00090
Ben-Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. G. A. B. A. (2007). A pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284. doi: 10.1152/physrev.00017.2006
Berry-Kravis, E. M., Hessl, D., Rathmell, B., Zarevics, P., Cherubini, M., Walton-Bowen, K., et al. (2012). Effects of STX209 (Arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: A randomized, controlled, phase 2 trial. Sci. Transl. Med. 4:152ra127. doi: 10.1126/scitranslmed.3004214
Bhakar, A. L., D̈olen, G., and Bear, M. F. (2012). The pathophysiology of fragile X (and what it teaches us about synapses). Annu. Rev. Neurosci. 35, 417–443. doi: 10.1146/annurev-neuro-060909-153138
Bonifazi, P., Goldin, M., Picardo, M. A., Jorquera, I., Cattani, A., Bianconi, G., et al. (2009). GABAergic hub neurons orchestrate synchrony in developing hippocampal networks. Science 326, 1419–1424. doi: 10.1126/science.1175509
Brandt, C., Seja, P., Töllner, K., Römermann, K., Hampel, P., Kalesse, M., et al. (2018). Bumepamine, a brain-permeant benzylamine derivative of bumetanide, does not inhibit NKCC1 but is more potent to enhance phenobarbital’s anti-seizure efficacy. Neuropharmacology 143, 186–204. doi: 10.1016/j.neuropharm.2018.09.025
Cancedda, L., Fiumelli, H., Chen, K., and Poo, M. M. (2007). Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J. Neurosci. 27, 5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007
Canitano, R. (2007). Epilepsy in autism spectrum disorders. Eur. Child Adolesc. Psychiatry 16, 61–66. doi: 10.1007/s00787-006-0563-2
Chamma, I., Heubl, M., Chevy, Q., Renner, M., Moutkine, I., Eugène, E., et al. (2013). Activity-dependent regulation of the K/Cl transporter KCC2 membrane diffusion, clustering, and function in hippocampal neurons. J. Neurosci. 33, 15488–15503. doi: 10.1523/JNEUROSCI.5889-12.2013
Chen, G., Trombley, P. Q., and Van Den Pol, A. N. (1996). Excitatory actions of GABA in developing rat hypothalamic neurones. J. Physiol. 494, 451–464. doi: 10.1113/jphysiol.1996.sp021505
Chevy, Q., Heubl, M., Goutierre, M., Backer, S., Moutkine, I., Eugène, E., et al. (2015). KCC2 gates activity-driven AMPA receptor traffic through Cofilin phosphorylation. J. Neurosci. 35, 15772–15786. doi: 10.1523/JNEUROSCI.1735-15.2015
Chudotvorova, I., Ivanov, A., Rama, S., Hübner, C. A., Pellegrino, C., Ben-Ari, Y., et al. (2005). Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J. Physiol. 566, 671–679. doi: 10.1113/jphysiol.2005.089821
Contractor, A., Ethell, I. M., and Portera-Cailliau, C. (2021). Cortical interneurons in autism. Nat. Neurosci. 24, 1648–1659. doi: 10.1038/s41593-021-00967-6
Crutel, V., Lambert, E., Penelaud, P. F., Albarrán Severo, C., Fuentes, J., Rosier, A., et al. (2021). Bumetanide oral liquid formulation for the treatment of children and adolescents with autism spectrum disorder: Design of two phase III studies (SIGN trials). J. Autism Dev. Disord. 51, 2959–2972. doi: 10.1007/s10803-020-04709-8
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y. S., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
Delpy, A., Allain, A. E., Meyrand, P., and Branchereau, P. (2008). NKCC1 cotransporter inactivation underlies embryonic development of chloride-mediated inhibition in mouse spinal motoneuron. J. Physiol. 586, 1059–1075. doi: 10.1113/jphysiol.2007.146993
D’Hulst, C., De Geest, N., Reeve, S. P., Van Dam, D., De Deyn, P. P., Hassan, B. A., et al. (2006). Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 1121, 238–245. doi: 10.1016/j.brainres.2006.08.115
Dolan, B. M., Duron, S. G., Campbell, D. A., Vollrath, B., Shankaranarayana Rao, B. S., Ko, H.-Y., et al. (2013). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. U.S.A. 110, 5671–5676. doi: 10.1073/pnas.1219383110
Doll, C. A., Scott, K., and Appel, B. (2021). Fmrp regulates oligodendrocyte lineage cell specification and differentiation. Glia 69, 2349–2361. doi: 10.1002/glia.24041
Duarte, S. T., Armstrong, J., Roche, A., Ortez, C., Pérez, A., del Mar O’Callaghan, M., et al. (2013). Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with rett syndrome. PLoS One 8:e68851. doi: 10.1371/journal.pone.0068851
Dzhala, V. I., Talos, D. M., Sdrulla, D. A., Brumback, A. C., Mathews, G. C., Benke, T. A., et al. (2005). NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 11, 1205–1213. doi: 10.1038/nm1301
Edens, B. M., Vissers, C., Su, J., Arumugam, S., Xu, Z., Shi, H., et al. (2019). FMRP modulates neural differentiation through m6A-dependent mRNA nuclear export. Cell Rep. 28, 845–854.e5. doi: 10.1016/j.celrep.2019.06.072
El Idrissi, A., Ding, X. H., Scalia, J., Trenkner, E., Brown, W. T., and Dobkin, C. (2005). Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci. Lett. 377, 141–146. doi: 10.1016/j.neulet.2004.11.087
Erickson, C. A., Early, M., Stigler, K. A., Wink, L. K., Mullett, J. E., and McDougle, C. J. (2011a). An open-label naturalistic pilot study of acamprosate in youth with autistic disorder. J. Child Adolesc. Psychopharmacol. 21, 565–569. doi: 10.1089/cap.2011.0034
Erickson, C. A., Mullett, J. E., and McDougle, C. J. (2010). Brief report: Acamprosate in fragile X syndrome. J. Autism Dev. Disord. 40, 1412–1416. doi: 10.1007/s10803-010-0988-9
Erickson, C. A., Weng, N., Weiler, I. J., Greenough, W. T., Stigler, K. A., Wink, L. K., et al. (2011b). Open-label riluzole in fragile X syndrome. Brain Res. 1380, 264–270. doi: 10.1016/j.brainres.2010.10.108
Erker, T., Brandt, C., Töllner, K., Schreppel, P., Twele, F., Schidlitzki, A., et al. (2016). The bumetanide prodrug BUM5, but not bumetanide, potentiates the antiseizure effect of phenobarbital in adult epileptic mice. Epilepsia 57, 698–705. doi: 10.1111/epi.13346
Findling, R. L., Mankoski, R., Timko, K., Lears, K., McCartney, T., McQuade, R. D., et al. (2014). A randomized controlled trial investigating the safety and efficacy of aripiprazole in the long-term maintenance treatment of pediatric patients with irritability associated with autistic disorder. J. Clin. Psychiatry 75, 22–30. doi: 10.4088/JCP.13m08500
Fiumelli, H., Briner, A., Puskarjov, M., Blaesse, P., Belem, B. J., Dayer, A. G., et al. (2013). An ion transport-independent role for the cation-chloride cotransporter kcc2 in dendritic spinogenesis in vivo. Cereb. Cortex 23, 378–388. doi: 10.1093/cercor/bhs027
Gao, X. B., and Van Den Pol, A. N. (2001). GABA, not glutamate, a primary transmitter driving action potentials in developing hypothalamic neurons. J. Neurophysiol. 85, 425–434. doi: 10.1152/jn.2001.85.1.425
Gauvain, G., Chamma, I., Chevy, Q., Cabezas, C., Irinopoulou, T., Bodrug, N., et al. (2011). The neuronal K-Cl cotransporter KCC2 influences postsynaptic AMPA receptor content and lateral diffusion in dendritic spines. Proc. Natl. Acad. Sci. U.S.A. 108, 15474–15479. doi: 10.1073/pnas.1107893108
Ge, S., Goh, E. L. K., Sailor, K. A., Kitabatake, Y., Ming, G. L., and Song, H. (2006). GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439, 589–593. doi: 10.1038/nature04404
Giachello, C. N. G., and Baines, R. A. (2015). Inappropriate neural activity during a sensitive period in embryogenesis results in persistent seizure-like behavior. Curr. Biol. 25, 2964–2968. doi: 10.1016/j.cub.2015.09.040
Gibson, J. R., Bartley, A. F., Hays, S. A., and Huber, K. M. (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 100, 2615–2626. doi: 10.1152/jn.90752.2008
Glaze, D. G., Percy, A. K., Skinner, S., Motil, K. J., Neul, J. L., Barrish, J. O., et al. (2010). Epilepsy and the natural history of Rett syndrome. Neurology 74, 909–912. doi: 10.1212/WNL.0b013e3181d6b852
Goncalves, J. T., Anstey, J. E., Golshani, P., and Portera-Cailliau, C. (2013). Circuit level defects in the developing neocortex of Fragile X mice. Nat. Neurosci. 16, 903–909. doi: 10.1038/nn.3415
González, W., and Bautista, R. E. D. (2009). Seizures and EEG findings in an adult patient with DiGeorge syndrome: A case report and review of the literature. Seizure 18, 648–651. doi: 10.1016/j.seizure.2009.07.003
Goutierre, M., Al Awabdh, S., Donneger, F., François, E., Gomez-Dominguez, D., Irinopoulou, T., et al. (2019). KCC2 regulates neuronal excitability and hippocampal activity via interaction with task-3 channels. Cell Rep. 28, 91–103.e7. doi: 10.1016/j.celrep.2019.06.001
Gozlan, H., and Ben-Ari, Y. (2003). Interneurons are the source and the targets of the first synapses formed in the rat developing hippocampal circuit. Cereb. Cortex 13, 684–692. doi: 10.1093/cercor/13.6.684
Gu, J., Lee, C. W., Fan, Y., Komlos, D., Tang, X., Sun, C., et al. (2010). /cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat. Neurosci. 13, 1208–1215. doi: 10.1038/nn.2634
Guida, N., Laudati, G., Anzilotti, S., Secondo, A., Montuori, P., Di Renzo, G., et al. (2015). Resveratrol via sirtuin-1 downregulates RE1-silencing transcription factor (REST) expression preventing PCB-95-induced neuronal cell death. Toxicol. Appl. Pharmacol. 288, 387–398. doi: 10.1016/j.taap.2015.08.010
Gulyás, A. I., Sík, A., Payne, J. A., Kaila, K., and Freund, T. F. (2001). The KCl cotransporter, KCC2, is highly expressed in the vicinity of excitatory synapses in the rat hippocampus. Eur. J. Neurosci. 13, 2205–2217. doi: 10.1046/j.0953-816X.2001.01600.x
Hampel, P., Johne, M., Gailus, B., Vogel, A., Schidlitzki, A., Gericke, B., et al. (2021). Deletion of the Na-K-2Cl cotransporter NKCC1 results in a more severe epileptic phenotype in the intrahippocampal kainate mouse model of temporal lobe epilepsy. Neurobiol. Dis. 152:105297. doi: 10.1016/j.nbd.2021.105297
Hampel, P., Römermann, K., MacAulay, N., and Löscher, W. (2018). Azosemide is more potent than bumetanide and various other loop diuretics to inhibit the sodium-potassium-chloride-cotransporter human variants hNKCC1A and hNKCC1B. Sci. Rep. 8:9877. doi: 10.1038/s41598-018-27995-w
Hardan, A. Y., Hendren, R. L., Aman, M. G., Robb, A., Melmed, R. D., Andersen, K. A., et al. (2019). Efficacy and safety of memantine in children with autism spectrum disorder: Results from three phase 2 multicenter studies. Autism 23, 2096–2111. doi: 10.1177/1362361318824103
He, Q., Arroyo, E. D., Smukowski, S. N., Xu, J., Piochon, C., Savas, J. N., et al. (2019). Critical period inhibition of NKCC1 rectifies synapse plasticity in the somatosensory cortex and restores adult tactile response maps in fragile X mice. Mol. Psychiatry 24, 1732–1747. doi: 10.1038/s41380-018-0048-y
He, Q., Nomura, T., Xu, J., and Contractor, A. (2014). The developmental switch in GABA polarity is delayed in fragile X mice. J. Neurosci. 34, 446–450. doi: 10.1523/JNEUROSCI.4447-13.2014
He, Y., Benz, A., Fu, T., Wang, M., Covey, D. F., Zorumski, C. F., et al. (2002). Neuroprotective agent riluzole potentiates postsynaptic GABAA receptor function. Neuropharmacology 42, 199–209. doi: 10.1016/S0028-3908(01)00175-7
Hellings, J. A., Weckbaugh, M., Nickel, E. J., Cain, S. E., Zarcone, J. R., Reese, R. M., et al. (2005). A double-blind, placebo-controlled study of valproate for aggression in youth with pervasive developmental disorders. J. Child Adolesc. Psychopharmacol. 15, 682–692. doi: 10.1089/cap.2005.15.682
Henderson, C., Wijetunge, L., Kinoshita, M. N., Shumway, M., Hammond, R. S., Postma, F. R., et al. (2012). Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 4:152ra128. doi: 10.1126/scitranslmed.3004218
Hennou, S., Khalilov, I., Diabira, D., Ben-Ari, Y., and Gozlan, H. (2002). Early sequential formation of functional GABAA and glutamatergic synapses on CA1 interneurons of the rat foetal hippocampus. Eur. J. Neurosci. 16, 197–208. doi: 10.1046/j.1460-9568.2002.02073.x
Hensch, T. K. (2004). Critical period regulation. Annu. Rev. Neurosci. 27, 549–579. doi: 10.1146/annurev.neuro.27.070203.144327
Hinz, L., Torrella Barrufet, J., and Heine, V. M. (2019). KCC2 expression levels are reduced in post mortem brain tissue of Rett syndrome patients. Acta Neuropathol. Commun. 7:196. doi: 10.1186/s40478-019-0852-x
Hollander, E., Soorya, L., Wasserman, S., Esposito, K., Chaplin, W., and Anagnostou, E. (2006). Divalproex sodium vs. placebo in the treatment of repetitive behaviours in autism spectrum disorder. Int. J. Neuropsychopharmacol. 9, 209–213. doi: 10.1017/S1461145705005791
Hu, J., Chen, L., Yin, J., Yin, H., Huang, Y., and Tian, J. (2020). Hyperactivity, memory defects, and craniofacial abnormalities in zebrafish fmr1 mutant larvae. Behav. Genet. 50, 152–160. doi: 10.1007/s10519-020-09995-7
Huang, H., Bhuiyan, M. I. H., Jiang, T., Song, S., Shankar, S., Taheri, T., et al. (2019). A novel Na+-K+-Cl- cotransporter 1 inhibitor STS66* reduces brain damage in mice after ischemic stroke. Stroke 50, 1021–1025. doi: 10.1161/STROKEAHA.118.024287
Hübner, C. A., Stein, V., Hermans-Borgmeyer, I., Meyer, T., Ballanyi, K., and Jentsch, T. J. (2001). Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30, 515–524. doi: 10.1016/s0896-6273(01)00297-5
Hunter, J., Rivero-Arias, O., Angelov, A., Kim, E., Fotheringham, I., and Leal, J. (2014). Epidemiology of fragile X syndrome: A systematic review and meta-analysis. Am. J. Med. Genet. Part A 164, 1648–1658. doi: 10.1002/ajmg.a.36511
Huo, Q., Chen, M., He, Q., Zhang, J., Li, B., Jin, K., et al. (2017). Prefrontal cortical GABAergic dysfunction contributes to aberrant UP-state duration in APP knockout mice. Cereb. Cortex 27, 4060–4072. doi: 10.1093/cercor/bhw218
Ichikawa, H., Mikami, K., Okada, T., Yamashita, Y., Ishizaki, Y., Tomoda, A., et al. (2017). Aripiprazole in the treatment of irritability in children and adolescents with autism spectrum disorder in Japan: A randomized, double-blind, placebo-controlled study. Child Psychiatry Hum. Dev. 48, 796–806. doi: 10.1007/s10578-016-0704-x
Irwin, S. A., Patel, B., Idupulapati, M., Harris, J. B., Crisostomo, R. A., Larsen, B. P., et al. (2001). Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: A quantitative examination. Am. J. Med. Genet. 98, 161–167. doi: 10.1002/1096-8628(20010115)98:2<161::aid-ajmg1025>3.0.co;2-b
Ivakine, E. A., Acton, B. A., Mahadevan, V., Ormond, J., Tang, M., Pressey, J. C., et al. (2013). Neto2 is a KCC2 interacting protein required for neuronal Cl- regulation in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 110, 3561–3566. doi: 10.1073/pnas.1212907110
Johnson, J., Tian, N., Caywood, M. S., Reimer, R. J., Edwards, R. H., and Copenhagen, D. R. (2003). Vesicular neurotransmitter transporter expression in developing postnatal rodent retina: GABA and glycine precede glutamate. J. Neurosci. 23, 518–529. doi: 10.1523/jneurosci.23-02-00518.2003
Kahle, K. T., Merner, N. D., Friedel, P., Silayeva, L., Liang, B., Khanna, A., et al. (2014). Genetically encoded impairment of neuronal KCC 2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 15, 766–774. doi: 10.15252/embr.201438840
Kaila, K. (1994). Ionic basis of GABAA receptor channel function in the nervous system. Prog. Neurobiol. 42, 489–537. doi: 10.1016/0301-0082(94)90049-3
Karlócai, M. R., Wittner, L., Tóth, K., Maglóczky, Z., Katarova, Z., Rásonyi, G., et al. (2016). Enhanced expression of potassium-chloride cotransporter KCC2 in human temporal lobe epilepsy. Brain Struct. Funct. 221, 3601–3615. doi: 10.1007/s00429-015-1122-8
Kaufmann, W. E., Kidd, S. A., Andrews, H. F., Budimirovic, D. B., Esler, A., Haas-Givler, B., et al. (2017). Autism spectrum disorder in fragile X syndrome: Cooccurring conditions and current treatment. Pediatrics 139, S194–S206. doi: 10.1542/peds.2016-1159F
Kesaf, S., Khirug, S., Dinh, E., Saez Garcia, M., Soni, S., Orav, E., et al. (2020). The kainate receptor subunit GluK2 interacts with KCC2 to promote maturation of dendritic spines. Front. Cell. Neurosci. 14:252. doi: 10.3389/fncel.2020.00252
Kim, L., He, L., Maaswinkel, H., Zhu, L., Sirotkin, H., and Weng, W. (2014). Anxiety, hyperactivity and stereotypy in a zebrafish model of fragile X syndrome and autism spectrum disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 55, 40–49. doi: 10.1016/j.pnpbp.2014.03.007
Kirsch, J. (2006). Glycinergic transmission. Cell Tissue Res. 326, 535–540. doi: 10.1007/s00441-006-0261-x
LeBlanc, J. J., and Fagiolini, M. (2011). Autism: A “critical period” disorder? Neural Plast. 2011:921680. doi: 10.1155/2011/921680
Leinekugel, X., Tseeb, V., Ben-Ari, Y., and Bregestovski, P. (1995). Synaptic GABAA activation induces Ca2+ rise in pyramidal cells and interneurons from rat neonatal hippocampal slices. J. Physiol. 487, 319–329. doi: 10.1113/jphysiol.1995.sp020882
Lemonnier, E., Villeneuve, N., Sonie, S., Serret, S., Rosier, A., Roue, M., et al. (2017). Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 7, e1056–e1059. doi: 10.1038/tp.2017.10
Li, H., Khirug, S., Cai, C., Ludwig, A., Blaesse, P., Kolikova, J., et al. (2007). KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron 56, 1019–1033. doi: 10.1016/j.neuron.2007.10.039
Li, H., Tornberg, J., Kaila, K., Airaksinen, M. S., and Rivera, C. (2002). Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur. J. Neurosci. 16, 2358–2370. doi: 10.1046/j.1460-9568.2002.02419.x
Li, Y., Zhou, Y., Peng, L., and Zhao, Y. (2017). Reduced protein expressions of cytomembrane GABAARβ3 at different postnatal developmental stages of rats exposed prenatally to valproic acid. Brain Res. 1671, 33–42. doi: 10.1016/j.brainres.2017.06.018
Lippi, G., Fernandes, C. C., Ewell, L. A., John, D., Romoli, B., Curia, G., et al. (2016). MicroRNA-101 regulates multiple developmental programs to constrain excitation in adult neural networks. Neuron 92, 1337–1351. doi: 10.1016/j.neuron.2016.11.017
Liu, T., Wan, R. P., Tang, L. J., Liu, S. J., Li, H. J., Zhao, Q. H., et al. (2015). MicroRNA profile in fmr1 knockout mice reveals microrna expression alterations with possible roles in fragile X syndrome. Mol. Neurobiol. 51, 1053–1063. doi: 10.1007/s12035-014-8770-1
Liu, X., Kumar, V., Tsai, N. P., and Auerbach, B. D. (2022). Hyperexcitability and homeostasis in fragile X syndrome. Front. Mol. Neurosci. 14:805929. doi: 10.3389/fnmol.2021.805929
Liu, X., Wang, Q., Haydar, T. F., and Bordey, A. (2005). Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat. Neurosci. 8, 1179–1187. doi: 10.1038/nn1522
Llano, O., Smirnov, S., Soni, S., Golubtsov, A., Guillemin, I., Hotulainen, P., et al. (2015). KCC2 regulates actin dynamics in dendritic spines via interaction with β-PIX. J. Cell Biol. 209, 671–686. doi: 10.1083/jcb.201411008
Löscher, W., Puskarjov, M., and Kaila, K. (2013). Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 69, 62–74.
Lu, R., Wang, H., Liang, Z., Ku, L., O’Donnell, W. T., Li, W., et al. (2004). The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc. Natl. Acad. Sci. U.S.A. 101, 15201–15206. doi: 10.1073/pnas.0404995101
Luo, Y., Shan, G., Guo, W., Smrt, R. D., Johnson, E. B., Li, X., et al. (2010). Fragile X mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 6:e1000898. doi: 10.1371/journal.pgen.1000898
Mahadevan, V., Pressey, J. C., Acton, B. A., Uvarov, P., Huang, M. Y., Chevrier, J., et al. (2014). Kainate receptors coexist in a functional complex with KCC2 and regulate chloride homeostasis in hippocampal neurons. Cell Rep. 7, 1762–1770. doi: 10.1016/j.celrep.2014.05.022
Masi, A., DeMayo, M. M., Glozier, N., and Guastella, A. J. (2017). An overview of autism spectrum disorder, heterogeneity and treatment options. Neurosci. Bull. 33, 183–193. doi: 10.1007/s12264-017-0100-y
McNeill, A., Iovino, E., Mansard, L., Vache, C., Baux, D., Bedoukian, E., et al. (2020). SLC12A2 variants cause a neurodevelopmental disorder or cochleovestibular defect. Brain 143, 2380–2387. doi: 10.1093/brain/awaa176
Merner, N. D., Chandler, M. R., Bourassa, C., Liang, B., Khanna, A. R., Dion, P., et al. (2015). Regulatory domain or CPG site variation in SLC12A5, encoding the chloride transporter KCC2, in human autism and schizophrenia. Front. Cell. Neurosci. 9:386. doi: 10.3389/fncel.2015.00386
Moore, Y. E., Kelley, M. R., Brandon, N. J., Deeb, T. Z., and Moss, S. J. (2017). Seizing control of KCC2: A new therapeutic target for epilepsy. Trends Neurosci. 40, 555–571. doi: 10.1016/j.tins.2017.06.008
Morin-Parent, F., Champigny, C., Lacroix, A., Corbin, F., and Lepage, J. F. (2019). Hyperexcitability and impaired intracortical inhibition in patients with fragile-X syndrome. Transl. Psychiatry 9:312. doi: 10.1038/s41398-019-0650-z
Nelson, D. L., Orr, H. T., and Warren, S. T. (2013). The unstable repeats-Three evolving faces of neurological disease. Neuron 77, 825–843. doi: 10.1016/j.neuron.2013.02.022
Newschaffer, C. J., Falb, M. D., and Gurney, J. G. (2005). National autism prevalence trends from United States special education data. Pediatrics 115, 277–282. doi: 10.1542/peds.2004-1958
Nguyen, L., Malgrange, B., Breuskin, I., Bettendorff, L., Moonen, G., Belachew, S., et al. (2003). Autocrine/paracrine activation of the GABAA receptor inhibits the proliferation of neurogenic polysialylated neural cell adhesion molecule-positive (PSA-NCAM+) precursor cells from postnatal striatum. J. Neurosci. 23, 3278–3294. doi: 10.1523/jneurosci.23-08-03278.2003
Nishimaru, H., Iizuka, M., Ozaki, S., and Kudo, N. (1996). Spontaneous motoneuronal activity mediated by glycine and GABA in the spinal cord of rat fetuses in vitro. J. Physiol. 497, 131–143. doi: 10.1113/jphysiol.1996.sp021755
Nussbaum, E. Z., and Perazella, M. A. (2019). Diagnosing acute interstitial nephritis: Considerations for clinicians. Clin. Kidney J. 12, 808–813. doi: 10.1093/ckj/sfz080
Olmos-Serrano, J. L., Paluszkiewicz, S. M., Martin, B. S., Kaufmann, W. E., Corbin, J. G., and Huntsman, M. M. (2010). Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J. Neurosci. 30, 9929–9938. doi: 10.1523/JNEUROSCI.1714-10.2010
Paluszkiewicz, S. M., Martin, S., and Huntsman, M. (2011). Fragile X syndrome: The GABAergic system and circuit dysfunction. Dev. Neurosci. 33, 349–364. doi: 10.1159/000329420
Picardo, M. A., Guigue, P., Bonifazi, P., Batista-Brito, R., Allene, C., Ribas, A., et al. (2011). Pioneer GABA cells comprise a subpopulation of hub neurons in the developing hippocampus. Neuron 71, 695–709. doi: 10.1016/j.neuron.2011.06.018
Pilorge, M., Fassier, C., Le Corronc, H., Potey, A., Bai, J., De Gois, S., et al. (2016). Genetic and functional analyses demonstrate a role for abnormal glycinergic signaling in autism. Mol. Psychiatry 21, 936–945. doi: 10.1038/mp.2015.139
Platel, J. C., Dave, K. A., and Bordey, A. (2008). Control of neuroblast production and migration by converging GABA and glutamate signals in the postnatal forebrain. J. Physiol. 586, 3739–3743. doi: 10.1113/jphysiol.2008.155325
Puskarjov, M., Seja, P., Heron, S. E., Williams, T. C., Ahmad, F., Iona, X., et al. (2014). A variant of KCC2 from patients with febrile seizures impairs neuronal Cl- extrusion and dendritic spine formation. EMBO Rep. 15, 723–729. doi: 10.1002/embr.201438749
Raj, N., McEachin, Z. T., Harousseau, W., Zhou, Y., Zhang, F., Merritt-Garza, M. E., et al. (2021). Cell-type-specific profiling of human cellular models of fragile X syndrome reveal PI3K-dependent defects in translation and neurogenesis. Cell Rep. 35:108991. doi: 10.1016/j.celrep.2021.108991
Reynolds, A., Brustein, E., Liao, M., Mercado, A., Babilonia, E., Mount, D. B., et al. (2008). Neurogenic role of the depolarizing chloride gradient revealed by global overexpression of KCC2 from the onset of development. J. Neurosci. 28, 1588–1597. doi: 10.1523/JNEUROSCI.3791-07.2008
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Römermann, K., Fedrowitz, M., Hampel, P., Kaczmarek, E., Töllner, K., Erker, T., et al. (2017). Multiple blood-brain barrier transport mechanisms limit bumetanide accumulation, and therapeutic potential, in the mammalian brain. Neuropharmacology 117, 182–194. doi: 10.1016/j.neuropharm.2017.02.006
Ruffolo, G., Iyer, A., Cifelli, P., Roseti, C., Mühlebner, A., van Scheppingen, J., et al. (2016). Functional aspects of early brain development are preserved in tuberous sclerosis complex (TSC) epileptogenic lesions. Neurobiol. Dis. 95, 93–101. doi: 10.1016/j.nbd.2016.07.014
Saint-Amant, L., and Drapeau, P. (2000). Motoneuron activity patterns related to the earliest behavior of the zebrafish embryo. J. Neurosci. 20, 3964–3972. doi: 10.1523/jneurosci.20-11-03964.2000
Savardi, A., Borgogno, M., Narducci, R., La Sala, G., Ortega, J. A., Summa, M., et al. (2020). Discovery of a small molecule drug candidate for selective NKCC1 inhibition in brain disorders. Chem 6, 2073–2096.
Schulte, J. T., Wierenga, C. J., and Bruining, H. (2018). Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev. 90, 260–271. doi: 10.1016/j.neubiorev.2018.05.001
Shamay-Ramot, A., Khermesh, K., Porath, H. T., Barak, M., Pinto, Y., Wachtel, C., et al. (2015). Fmrp interacts with adar and regulates RNA editing, synaptic density and locomotor activity in zebrafish. PLoS Genet. 11:e1005702. doi: 10.1371/journal.pgen.1005702
Silverman, J. L., Tolu, S. S., Barkan, C. L., and Crawley, J. N. (2010). Repetitive self-grooming behavior in the BTBR mouse model of autism is blocked by the mGluR5 antagonist MPEP. Neuropsychopharmacology 35, 976–989. doi: 10.1038/npp.2009.201
Sørensen, E. M., Bertelsen, F., Weikop, P., Skovborg, M. M., Banke, T., Drasbek, K. R., et al. (2015). Hyperactivity and lack of social discrimination in the adolescent Fmr1 knockout mouse. Behav. Pharmacol. 26, 733–740. doi: 10.1097/FBP.0000000000000152
Stein, V., Hermans-Borgmeyer, I., Jentsch, T. J., and Hübner, C. A. (2004). Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J. Comp. Neurol. 468, 57–64. doi: 10.1002/cne.10983
Stödberg, T., Magnusson, M., Lesko, N., Wredenberg, A., Munoz, D. M., Stranneheim, H., et al. (2020). SLC12A2 mutations cause NKCC1 deficiency with encephalopathy and impaired secretory epithelia. Neurol. Genet. 6:e478. doi: 10.1212/NXG.0000000000000478
Talos, D. M., Sun, H., Kosaras, B., Joseph, A., Folkerth, R. D., Poduri, A., et al. (2012). Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann. Neurol. 71, 539–551. doi: 10.1002/ana.22696
Tang, X., Drotar, J., Li, K., Clairmont, C. D., Brumm, A. S., Sullins, A. J., et al. (2019). Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med. 11:eaau0164. doi: 10.1126/scitranslmed.aau0164
Tang, X., Kim, J., Zhou, L., Wengert, E., Zhang, L., Wu, Z., et al. (2016). KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 113, 751–756. doi: 10.1073/pnas.1524013113
Tanis, J. E., Bellemer, A., Moresco, J. J., Forbush, B., and Koelle, M. R. (2009). The potassium chloride cotransporter KCC-2 coordinates development of inhibitory neurotransmission and synapse structure in Caenorhabditis elegans. J. Neurosci. 29, 9943–9954. doi: 10.1523/JNEUROSCI.1989-09.2009
Töllner, K., Brandt, C., Töpfer, M., Brunhofer, G., Erker, T., Gabriel, M., et al. (2014). A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann. Neurol. 75, 550–562. doi: 10.1002/ana.24124
Tyzio, R., Nardou, R., Ferrari, D. C., Tsintsadze, T., Shahrokhi, A., Eftekhari, S., et al. (2014). Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343, 675–679. doi: 10.1126/science.1247190
Tyzio, R., Represa, A., Jorquera, I., Ben-Ari, Y., Gozlan, H., and Aniksztejn, L. (1999). The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of the apical dendrite. J. Neurosci. 19, 10372–10382. doi: 10.1523/jneurosci.19-23-10372.1999
Urbanska, M., Kazmierska-Grebowska, P., Kowalczyk, T., Caban, B., Nader, K., Pijet, B., et al. (2019). GSK3β activity alleviates epileptogenesis and limits GluA1 phosphorylation. EBioMedicine 39, 377–387. doi: 10.1016/j.ebiom.2018.11.040
Ure, A., Rose, V., Bernie, C., and Williams, K. (2018). Autism: One or many spectrums? J. Paediatr. Child Health 54, 1068–1072. doi: 10.1111/jpc.14176
Van Den Pol, A. N., Gao, X. B., Patrylo, P. R., Ghosh, P. K., and Obrietan, K. (1998). Glutamate inhibits GABA excitatory activity in developing neurons. J. Neurosci. 18, 10749–10761. doi: 10.1523/jneurosci.18-24-10749.1998
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi: 10.1016/0092-8674(91)90397-h
Wang, C., Shimizu-Okabe, C., Watanabe, K., Okabe, A., Matsuzaki, H., Ogawa, T., et al. (2002). Developmental changes in KCC1, KCC2, and NKCC1 mRNA expressions in the rat brain. Dev. Brain Res. 139, 59–66. doi: 10.1016/S0165-3806(02)00536-9
Wang, D. D., and Kriegstein, A. R. (2008). GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J. Neurosci. 28, 5547–5558. doi: 10.1523/JNEUROSCI.5599-07.2008
Warp, E., Agarwal, G., Wyart, C., Friedmann, D., Oldfield, C. S., Conner, A., et al. (2012). Emergence of patterned activity in the developing zebrafish spinal cord. Curr. Biol. 22, 93–102. doi: 10.1016/j.cub.2011.12.002
Watanabe, M., and Fukuda, A. (2015). Development and regulation of chloride homeostasis in the central nervous system. Front. Cell. Neurosci. 9:371. doi: 10.3389/fncel.2015.00371
Wei, H., Dobkin, C., Sheikh, A. M., Malik, M., Brown, W. T., and Li, X. (2012). The therapeutic effect of memantine through the stimulation of synapse formation and dendritic spine maturation in autism and fragile X syndrome. PLoS One 7:e36981. doi: 10.1371/journal.pone.0036981
Wink, L. K., Adams, R., Horn, P. S., Tessier, C. R., Bantel, A. P., Hong, M., et al. (2018). Randomized placebo-controlled cross-over pilot study of riluzole for drug-refractory irritability in autism spectrum disorder. J. Autism Dev. Disord. 48, 3051–3060. doi: 10.1007/s10803-018-3562-5
Wright, R., Newey, S. E., Ilie, A., Wefelmeyer, W., Raimondo, J. V., Ginham, R., et al. (2017). Neuronal chloride regulation via KCC2 is modulated through a GABAB receptor protein complex. J. Neurosci. 37, 5447–5462. doi: 10.1523/JNEUROSCI.2164-16.2017
Wu, W. L., Ziskind-Conhaim, L., and Sweet, M. A. (1992). Early development of glycine- and GABA-mediated synapses in rat spinal cord. J. Neurosci. 12, 3935–3945. doi: 10.1523/jneurosci.12-10-03935.1992
Yamada, J., Okabe, A., Toyoda, H., Kilb, W., Luhmann, H. J., and Fukuda, A. (2004). Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J. Physiol. 557, 829–841. doi: 10.1113/jphysiol.2004.062471
Zeidler, S., de Boer, H., Hukema, R. K., and Willemsen, R. (2017). Combination therapy in fragile x syndrome; possibilities and pitfalls illustrated by targeting the mGluR5 and GABA pathway simultaneously. Front. Mol. Neurosci. 10:368. doi: 10.3389/fnmol.2017.00368
Zhang, L.-L., Fina, M. E., and Vardi, N. (2006). Regulation of KCC2 and NKCC during development: Membrane insertion and differences between cell types. J. Comp. Neurol. 499, 132–143. doi: 10.1002/cne.21100
Keywords: fragile X syndrome, GABA, chloride transporters, inhibition, excitation
Citation: Miles KD and Doll CA (2022) Chloride imbalance in Fragile X syndrome. Front. Neurosci. 16:1008393. doi: 10.3389/fnins.2022.1008393
Received: 31 July 2022; Accepted: 20 September 2022;
Published: 12 October 2022.
Edited by:
Elizabeth Anne McCullagh, Oklahoma State University, United StatesReviewed by:
Anis Contractor, Northwestern University, United StatesZhanyan Fu, Broad Institute, United States
Copyright © 2022 Miles and Doll. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caleb Andrew Doll, Y2FsZWIuZG9sbEBjdWFuc2NodXR6LmVkdQ==