 Aditya Iyer
Aditya Iyer Arshdeep Sidhu2
Arshdeep Sidhu2 Vinod Subramaniam
Vinod Subramaniam
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 16 November 2022
Sec. Neurodegeneration
Volume 16 - 2022 | https://doi.org/10.3389/fnins.2022.1003997
This article is part of the Research Topic The Biochemistry of Amyloids in Neurodegenerative Diseases, Volume II View all 15 articles
N-α-acetylation is a frequently occurring post-translational modification in eukaryotic proteins. It has manifold physiological consequences on the regulation and function of several proteins, with emerging studies suggesting that it is a global regulator of stress responses. For decades, in vitro biochemical investigations into the precise role of the intrinsically disordered protein alpha-synuclein (αS) in the etiology of Parkinson’s disease (PD) were performed using non-acetylated αS. The N-terminus of α-synuclein is now unequivocally known to be acetylated in vivo, however, there are many aspects of this post-translational modifications that are not understood well. Is N-α-acetylation of αS a constitutive modification akin to most cellular proteins, or is it spatio-temporally regulated? Is N-α-acetylation of αS relevant to the as yet elusive function of αS? How does the N-α-acetylation of αS influence the aggregation of αS into amyloids? Here, we provide an overview of the current knowledge and discuss prevailing hypotheses on the impact of N-α-acetylation of αS on its conformational, oligomeric, and fibrillar states. The extent to which N-α-acetylation of αS is vital for its function, membrane binding, and aggregation into amyloids is also explored here. We further discuss the overall significance of N-α-acetylation of αS for its functional and pathogenic implications in Lewy body formation and synucleinopathies.
N-terminal acetylation is a post-translational modification carried out by N-terminal acetyltransferases in nascent protein chains during translation (Aksnes et al., 2019). A protein can exist in full, partial and non-acetylated form. N-terminal acetylation involves the addition of an acetyl group to the free alpha-amino group (N-α-group) of the first amino acid in the nascent protein chain by an N-terminal acetyltransferase (Nat) complex (Varland et al., 2015). To date, N-terminal acetylation is considered irreversible because an N-terminal de-acetyltransferase (Ndat) either does not exist in eukaryotic cells or remains to be discovered. We draw a clear distinction between N-terminal acetylation and N-α-acetylation in the context of this review. N-α-acetylation refers explicitly to the acetylation of the first amino acid (in most cases, methionine). In contrast, N-terminal acetylation may include the acetylation of amino acid residues in the N-terminal region in proteins comprising several amino acids. Protein acetylation also occurs on the ε-amino group of the lysine side chains (N-ε-acetylation) catalyzed by a different class of enzymes called lysine acetyltransferases (Choudhary et al., 2014) and on hydroxyl groups of tyrosine/serine/threonine referred to as O-acetylation (Yang and Grégoire, 2007). In contrast to Ndats, eukaryotic lysine deacetylases are well-known, and their functions are reviewed elsewhere (Yang and Grégoire, 2007; Choudhary et al., 2014; Xia et al., 2020). In humans, seven Nats have been identified to date — NatA, NatB, NatC, NatD, NatE, NatF, and NatH (Aksnes et al., 2019) — which are responsible for N-terminal acetylation of more than 80% of eukaryotic proteins (Arnesen et al., 2009; Johnson et al., 2010; Aksnes et al., 2016), the rest of the 20% proteome is not known to be acetylated (Ree et al., 2018). Six Nats (NatA to NatF) have broad substrate specificity, except for NatH, which is a dedicated acetylase for actin (Drazic et al., 2018). Each Nat exhibits a strong preference for specific N-terminal residues and (at least) one or two subsequent amino acids required to facilitate N-terminal acetylation (Figure 1).
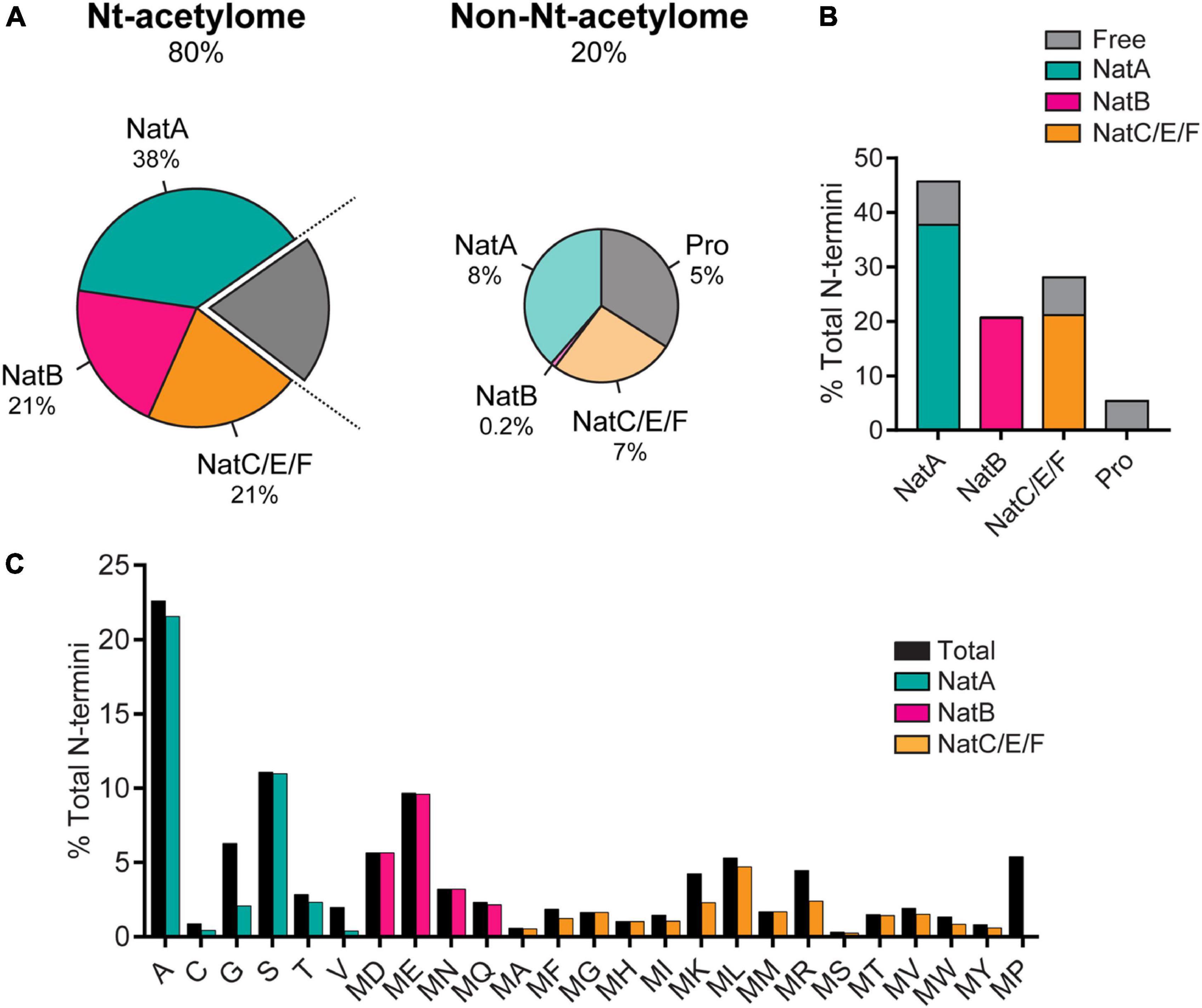
Figure 1. (A) The prevalence of N-terminal acetylation in human cells is depicted by separating the proteome into the N-terminal(Nt)-acetylome (80%) and the non-Nt-acetylome (20%). The human Nt-acetylome (the complete set of N-terminal acetylated proteins) was predicted by incorporating experimentally determined Nt-acetylation events (including NatC and NatF related data) to all SwissProt entries (version 57.8) based on the occurrence of the first two amino acids. The chance that a protein will be Nt-acetylated mainly depends on the identity of the first two amino acids. To visualize this concept, the Nt-acetylome can be grouped according to (B) NAT substrate class or (C) N-terminal amino acid frequency. NatD is not depicted due to its limited coverage. Image reproduced from Ree et al. (2018) licensed under CC BY 4.0. The figure legend is a modified excerpt of the original.
There is ample evidence in the literature that N-α-acetylation of proteins, in general, is an essential mediator of protein function, stability, and localization. N-terminal acetylation impacts protein localization and targeting (Behnia et al., 2004; Setty et al., 2004; Forte et al., 2011; Dikiy and Eliezer, 2014; Park et al., 2015), multi-protein complex formation (Scott et al., 2011; Arnaudo et al., 2013; Monda et al., 2013; Yang et al., 2013; Gao et al., 2016), protein secondary structure (Maltsev et al., 2012; Miotto et al., 2015), protein degradation (Hwang et al., 2010; Shemorry et al., 2013) and aggregation into amyloid fibrils (Kang et al., 2013; Iyer et al., 2016; Watson and Lee, 2019). Of the seven identified Nats, NatB holds particular importance in the context of diseases. NatB acetylates essential proteins at the N-terminus such as tropomyosin, actin, and alpha-synuclein (αS); is required for stability of the actin cytoskeleton; is vital for cell-cycle progression (Starheim et al., 2008), cell proliferation (Ametzazurra et al., 2008); and is implicated in diseases such as hepatocellular carcinoma and Parkinson’s disease (PD) (Polevoda and Sherman, 2003; Ametzazurra et al., 2008; Halliday et al., 2011; Neri et al., 2017).
αS is an intrinsically disordered protein found in high concentrations at the synaptic junctions of neuronal cells. Its precise role in the etiology of PD remains unknown. Several decades of research have not brought us much closer to pinning down its physiological function in eukaryotic cells. αS comprises three domains: the positively charged N-terminal region (aa 1-60) which is involved in membrane binding (Bartels et al., 2010; Lorenzen et al., 2014; Zarbiv et al., 2014; Viennet et al., 2018; Makasewicz et al., 2021), the amyloidogenic NAC domain (aa 61-95) crucial for amyloid formation (Waxman et al., 2009), and the highly charged C-terminal region (aa 96-140), that interacts with polyamines, metal ions, and cellular proteins (Antony et al., 2003; Eliezer, 2013). The observed binding of αS to phospholipid membranes is considered relevant for its function. It is also one of the proposed facilitators of the αS aggregation cascade in addition to point mutations, oxidative stress, truncations, possibly leading to neuronal cell death in PD. Like other eukaryotic proteins, αS is subjected to several post-translational modifications, including phosphorylation, ubiquitination, and acetylation; reviewed elsewhere in detail (Breydo et al., 2012; Barrett and Greenamyre, 2015; Iyer and Claessens, 2019; Zhang et al., 2019). αS is acetylated at the terminal methionine residue (N-α-acetylation) by NatB and several lysine residues in vivo (N-ε-acetylation) by other enzymes, but the physiological impact of acetylation of αS is unclear. We aim to give a critical perspective on the impact of N-α-acetylation of αS on its physiological role and pathological aggregation into amyloid fibrils.
Much before the ongoing debate over its native state, αS was widely accepted as a monomeric, intrinsically disordered protein associated with intracellular membranes and found substantially in a fibrillar state in numerous synucleinopathies. Early investigations into αS focused on the mechanism of aggregation/toxicity and possibly overlooked the role of post-translational modifications occurring in αS. The loss of a positive charge from the N-terminal methionine of αS acetylation affects its secondary structure substantially (Figure 2). N-α-acetylation is considered crucial for aggregation of αS into amyloid fibrils (Kang et al., 2012; Yang et al., 2021) and interaction with other binding partners in its native cellular environment (Zabrocki et al., 2008; Runfola et al., 2020). The N-α-acetylated form is believed to represent the functional form of the protein, and the debate over its native state being monomeric or tetrameric continues as discussed in the following sections.
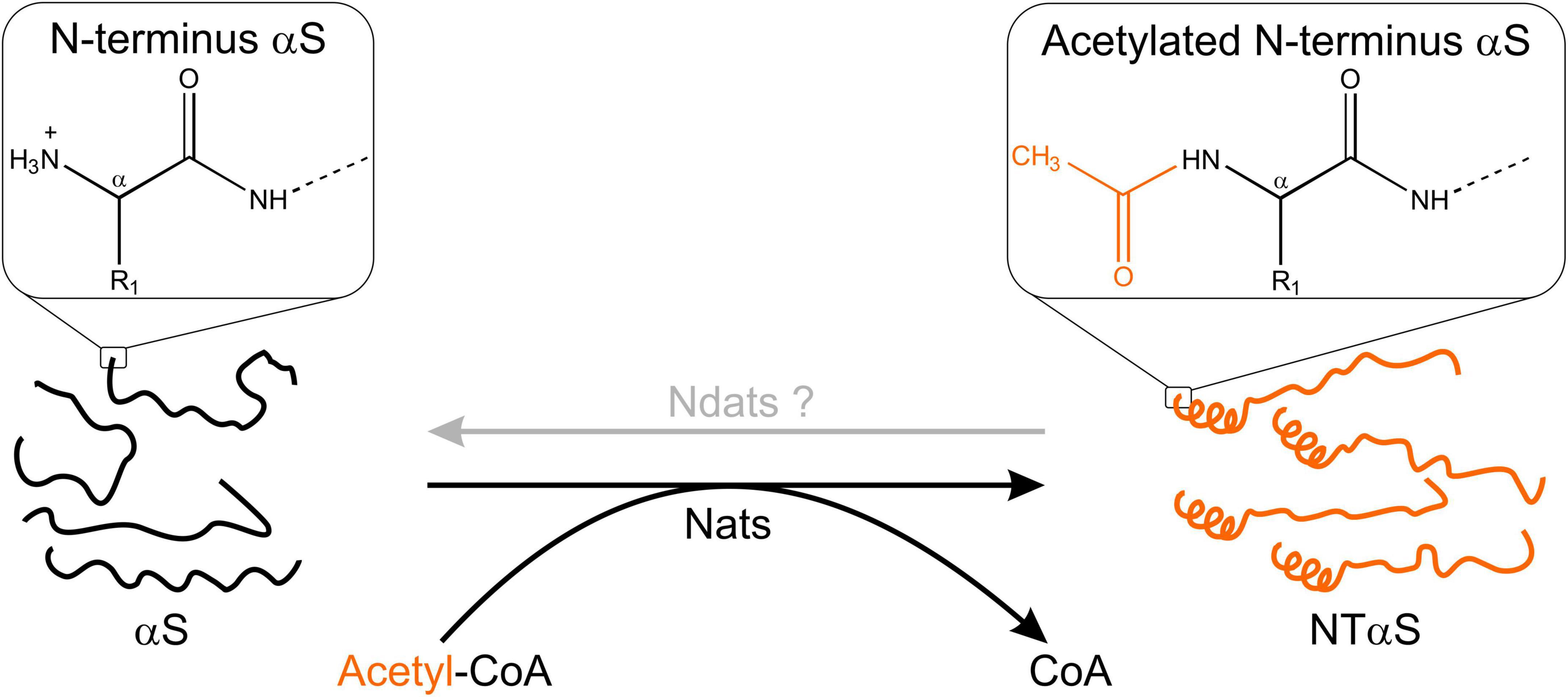
Figure 2. Schematic outline of N-terminal acetylation. N-terminal acetyltransferases (Nats) catalyze the transfer of an acetyl group (CH3CO) from acetyl-CoA (Ac-CoA) to the free α-amino group of the protein N-terminus. The transferred acetyl group eliminates a positive charge at the protein N-terminus. In the case of αS, N-α-acetylation has been shown to induce helix formation in the first 16 residues (Maltsev et al., 2012). The existence of N-terminal de-acetyltransferases (Ndats) is unknown.
The earliest report drawing attention to the presence of N-α-acetylation of αS obtained from brain cells and Lewy bodies considered it a passive post-translational modification (Figure 3; Anderson et al., 2006). Before this report, αS was mainly purified and studied from mammalian and non-mammalian sources to ascertain its genetic basis in neurogenerative diseases like PD, multiple system atrophy (MSA), Lewy body dementia, Lewy body variant of Alzheimer’s disease (LBAD), and AD. The relevance of N-α-acetylation of αS gained prominence following a report by the Selkoe group (Bartels et al., 2011), who contradicted the established view of the native state of αS as an intrinsically disordered monomer. Using numerous cell lines and an array of analytical techniques, including EM imaging, circular dichroism spectroscopy, clear-native PAGE (CN-PAGE), and sedimentation-equilibrium analytical ultracentrifugation (SE-AUC), the study reported that native/endogenous αS is an aggregation-resistant helical tetramer in dynamic equilibrium with the monomeric αS species. The study drew parallels to transthyretin amyloidosis, wherein the destabilization of a metastable tetramer in human plasma causes aberrant aggregation of monomers (Quintas et al., 1999).
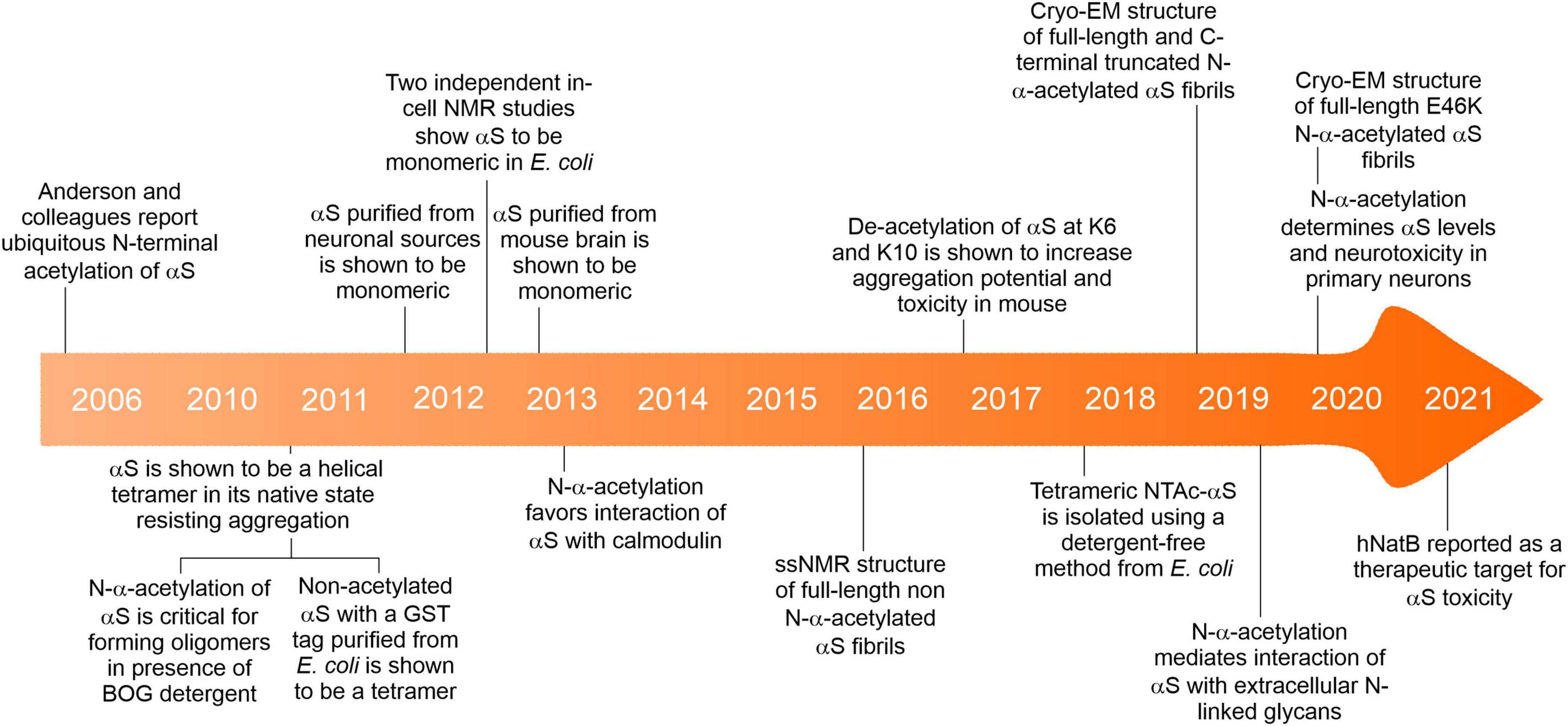
Figure 3. A brief timeline of key events about the N-α-acetylation of αS.
A widespread debate ensued challenging the tetramer hypothesis in several subsequent studies (Fauvet et al., 2012a,b; Burré et al., 2013), promptly responded to by the primary advocates of the tetramer hypothesis (Bartels and Selkoe, 2013; Dettmer et al., 2013, 2015a,2015b; Luth et al., 2015) and other groups (Ullman et al., 2011; Gurry et al., 2013; Fernández and Lucas, 2018a). The authors’ conclusion that tetrameric αS may dissociate to its monomeric form during cell lysis and widely differing protein purification protocols across research groups gained little reconciliation. The primary authors further showed that the tetrameric species was sensitive to cell-lysis protocols using in vivo cross-linking studies that showed the apparent 60-kDa tetramer does not arise from aggregation and that minor 80- and 100-kDa species accompanying varying concentrations of free monomers occurs endogenously in primary neurons as well as neuroblastoma cells that overexpress αS (Dettmer et al., 2013).
Several new questions emerged as a consequence that have been answered in part with ensuing research, while others remain contentious. Could bacterial systems employed to express and purify αS before the Selkoe report (Bartels et al., 2011) not possess the necessary physiological environment for tetramer assembly? Could N-α-acetylation of αS per se be of enough biophysical consequence to trigger the formation of aggregation-resistant tetramers? These questions were answered, in part, by a report showing that non-acetylated αS purified from Escherichia coli (E. coli) closely resembled the aforementioned tetrameric species (Wang et al., 2011). However, this construct harbored a 10-residue N-terminal fusion-protein fragment (GPLGSPEFPG) post cleavage of the Glutathione S-transferase (GST) tag that could mimic the biophysical consequences of N-α-acetylation of αS. To test if N-α-acetylation of αS in bacterial cells could lead to the formation of a tetrameric species, a bacterial co-expression system was used to generate N-α-acetylated αS (NTAc-αS). The authors determined that N-terminal acetylation and non-denaturing purification protocols, including the non-ionic detergent octyl β-D-glucopyranoside (BOG), were necessary to observe helical oligomeric αS (Trexler and Rhoades, 2012). In this co-expression system, the NatB acetylase derived from yeast is cloned into a bacterial plasmid, allowing N-terminal acetylation of NatB peptide substrates (MD, ME, MN, MQ; see Figure 1) alongside the overexpression of a target protein (Johnson et al., 2010). The CD spectrum of NTAc-αS showed a helical and presumably tetrameric form when purified in the presence of BOG, while non-acetylated or BOG-free αS was disordered and monomeric. These results implicitly contradicted the hypothesis of a folded αS tetramer in non-acetylation-competent E. coli cells used in the previous report (Wang et al., 2011). Could the detergents used during purification protocols lead to the proposed tetrameric state? Using the NatB bacterial co-expression system, Fernández and Lucas, 2018a,b demonstrated a detergent-free method to isolate recombinant tetrameric NTAc-αS. Subsequently, αS was shown to be monomeric by in-cell NMR studies in intact E. coli cells (Binolfi et al., 2012; Waudby et al., 2013) and numerous non-neuronal cells (Theillet et al., 2016). The monomer-tetramer debate is far from over but highlights the importance of how subtle environmental changes can cause significant molecular changes in αS. The physiological conditions governing the dynamic equilibrium between monomeric and tetrameric αS remain mysterious. Intuitively, an off-pathway, fibril-resistant αS tetramer can sequester aggregation-competent αS monomers. However, the irreproducibility across labs in isolating the tetrameric species, unknown factors affecting the monomer-tetramer equilibrium and tetramer stability have resulted in reluctant acceptance of its existence.
We speculate that N-α-acetylation alone or in combination with other post-translational modifications could be a regulatory step in maintaining an equilibrium between the monomeric and tetrameric states of αS. Tetrameric αS species have been purified from both endogenously expressing and overexpressing mammalian cell lines (Dettmer et al., 2013), ruling out pleiotropic effects of high concentrations. However, in gastrointestinal neuronal cells from rats, the population of tetrameric αS is absent, and these cells constitute primarily monomeric αS (Corbillé et al., 2016). Crowding within mammalian cells alone cannot explain the tetrameric state since in-cell NMR studies in bacterial cytoplasm (Binolfi et al., 2012; Waudby et al., 2013) and the periplasm (McNulty et al., 2006) that are significantly more crowded than mammalian cells (Swaminathan et al., 1997) affirm its monomeric state. Assuming there is an equilibrium between the tetrameric and monomeric species in vivo, how are the purified tetrameric αS species stably maintained, preventing their dissociation in vitro? A dynamic equilibrium between the tetrameric and monomeric state must be carefully regulated in vivo. Long-range interactions between acetyl groups and other amino acids within protein assemblies are well known (Langeberg and Scott, 2015), and transcriptional control via acetylation is one such example (Latham and Dent, 2007). It remains unclear whether N-α-acetylation, the purification methodology, the use of detergents, or the choice of a prokaryotic/eukaryotic expression system is crucial for tetramer formation. If in vivo cross-linking of tetrameric αS can be achieved, in-cell NMR studies may prove particularly useful in providing concrete evidence of such a species. In addition, how/if the distribution of the monomer-tetramer species depends on cell type and other biochemical factors needs investigation. For instance, glucocerebrosidase 1 deficiency in SH-SY5Y cells has been shown to disfavor the tetrameric αS populations over the monomeric αS population (Kim et al., 2018), while the tetrameric αS population is favored in primary neurons and erythroid cells (Dettmer et al., 2013). Addressing the N-α-acetylated state of αS is a promising avenue to probe the existence of a tetrameric species, to understand the possible mechanisms of amyloid formation, and to gain insights into the physiological function of αS.
NTAc-αS is suggested to be a physiologically relevant brain species (Bartels et al., 2011; Fauvet et al., 2012b; Burré et al., 2013; Theillet et al., 2016), and several emerging studies have benchmarked its biophysical properties with non-acetylated αS. A summary of all biophysical properties of NTAc-αS is listed in Table 1. Early solution-NMR studies with NTAc-αS revealed that N-α-acetylation of αS triggered a helical conformation in the first 16 residues (Maltsev et al., 2012; Dikiy and Eliezer, 2014) in vitro and subsequently in live neuronal and non-neuronal cells using in-cell NMR (Theillet et al., 2016). The interactions of non-acetylated αS with membranes have been studied in detail, but interactions with NTAc-αS remain relatively less explored. To the best of our knowledge, membrane binding studies have been carried out only for monomeric NTAc-αS and not for the tetrameric NTAc-αS species. It is well known that the N-terminal region (aa 1–60) of αS is involved in membrane binding. However, emerging studies show that the first 15 residues in αS largely recapitulate the binding properties of full-length αS such as partition constants, molecular mobility, and membrane insertion (Pfefferkorn et al., 2012), and removal of the first 14 residues severely compromises membrane binding (Cholak et al., 2020). How N-α-acetylation affects the membrane binding ability of αS is unclear due to conflicting results and differing solvent conditions and membrane compositions used. For example, NTAc-αS showed enhanced membrane binding in two studies (Bartels et al., 2014; Viennet et al., 2018) and no enhancement in other studies (Fauvet et al., 2012a; Maltsev et al., 2012; Iyer et al., 2016).

Table 1. Effect of N-terminal acetylation on biophysical properties of αS.
Considering that N-α-acetylation leads to loss of a positive charge from the terminal methionine residue, N-α-acetylation may affect the interaction between αS and membranes or other binding partners in the cellular milieu. Intuitively, the loss of a positive charge upon N-α-acetylation is likely to result in a decreased affinity toward anionic lipid membranes. However, N-α-acetylation of αS does not affect its binding to anionic phospholipid membranes with increasing surface charge densities but shows enhanced binding to zwitterionic phospholipid membranes in a curvature-dependent manner (Dikiy and Eliezer, 2014; Iyer et al., 2016; O’Leary et al., 2018). The observation may be reasoned as follows: N-α-acetylation of αS increases the propensity of the first 16 residues in the N-terminus to organize into helices (Figure 2). The binding of αS to lipid membranes results in a loss of conformational entropy compensated for by favorable electrostatic interactions and hydrogen bonding. Since NTAc-αS binds with a pre-existing helical conformation, the loss in conformational entropy upon binding to anionic membranes is probably lower for NTAc-αS than for the non-acetylated αS. The lower entropy cost associated with helix formation is balanced by losing the positive charge upon N-α-acetylation. The binding of non-acetylated and NTAc-αS to anionic lipid membranes is therefore comparable. In the absence of strong, attractive forces between neutral lipid membranes and αS, the effect of N-α-acetylation is likely dominated by the increased propensity of αS to fold into an amphipathic helix. Since the final helical content of both NTAc-αS and non-acetylated αS is comparable, the net free energy gain upon binding of NTAc-αS is higher with neutral lipid membranes resulting in enhanced affinity for NTAc-αS.
Although monomeric non-acetylated αS faithfully mimics NTAc-αS in specific biophysical properties like hydrodynamic radii and conformational change upon binding anionic lipid membranes, it does not reflect the importance of NTAc-αS. N-α-acetylation may have yet unknown physiological roles that may not be realized in experiments with purified proteins in vitro. For example, a recent study showed that abolishing N-α-acetylation of αS led to lower levels of αS and substantially reduced neurotoxicity in substantia nigra of rats (Vinueza-Gavilanes et al., 2020). N-α-acetylation of αS may also be possibly prevented in vivo by mutating the aspartic acid residue (D) in the second position to a proline residue (P) as recently shown for αS (Vinueza-Gavilanes et al., 2020) and numerous other proteins (Goetze et al., 2009).
Compared to its non-acetylated counterpart, NTAc-αS binds faster to model lipid membranes but forms amyloid aggregates and fibrils slower (Ruzafa et al., 2017; Cholak et al., 2020). However, in the presence of air-water interfaces, the apparent lag-time for NTAc-αS aggregation into amyloid fibrils is nearly twofold lesser than that observed with non-acetylated-αS (Viennet et al., 2018). Further, the presence of the neuronal ganglioside GM1 in model lipid membranes impaired the ability of NTAc-αS to form ThT-positive aggregates (Bartels et al., 2014). Given that the final helical content of both NTAc-αS and non-acetylated αS are comparable, the kinetic barrier for a membrane-bound helical conformation to a β-sheet conformation would also be comparable. If so, why would NTAc-αS aggregate slower on lipid membranes? Perhaps N-α-acetylation stabilizes interactions within the helical conformation and orients residues along with the interface such that αS dips further in the membrane, leading to a robust anchoring. While the above-mentioned model lipid membranes provide valuable biochemical insights, the next step must be to validate these observations in mammalian cells. Despite differences in the kinetics of membrane binding, the membrane-bound conformation and the morphology of micelle-induced aggregates of NTAc-αS are invariant with non-acetylated αS. Mimicking the biophysical consequences of N-α-acetylation of αS with or without PD familial mutations, for example, charge swap on terminal methionine, conformational restriction/stabilization of the N-terminal region, are needed to understand the monomer-tetramer equilibrium, aggregation on or in presence of lipid membranes will provide valuable mechanistic and functional insights into the role N-α-acetylation of αS.
The effect of N-α-acetylation on the structure of αS monomer and amyloid conformation has been investigated using multiple techniques in recent years. At the monomer level, N-α-acetylation does not affect the hydrodynamic radius, electrophoretic properties, and oligomerization potential of αS, suggesting minimal changes in the overall structure and biochemistry as compared to non-acetylated αS (Fauvet et al., 2012a; Kang et al., 2012; Gallea et al., 2016; Ni et al., 2019). However, NMR studies using 1H-15N HSQC show a significant difference in the chemical environment of the first nine residues and increased helical propensity of the first 12 residues (Fauvet et al., 2012a; Kang et al., 2012; Maltsev et al., 2012). The increased helicity of the N-terminus on acetylation mirrors the structural transitions observed in αS in the presence of model membranes, albeit only in a small region of the protein (Davidson et al., 1998; Eliezer et al., 2001; Georgieva et al., 2008). The acetyl carbonyl (C=O) group can participate in a hydrogen bond with the amino H (N-H) group from subsequent amino acids, which can stabilize a helix by sealing its fraying end (Fairman et al., 1989; Chakrabartty et al., 1993; Doig et al., 1994). NTAc-αS with helical N-terminus may facilitate its transition from a random coil to an α-helix in vivo, on interaction with a membrane surface, due to lower entropic cost and favorable dipole interactions associated with adding residues to an α-helix rather than initiating the helix (Zimm and Bragg, 1959; Creighton, 1993). In addition to the N-terminus, weak long-range interactions around residues 28–31, 43–46, 50, and 50–66 were also reported in acetylated αS (Kang et al., 2012). All these sites, toward the end of the N-terminal region (aa 1–60) and the beginning of the NAC region (aa 61–95) of αS, are associated with αS function and familial forms of PD (Polymeropoulos et al., 1997; Krüger et al., 1998; Zarranz et al., 2004; Lesage et al., 2013; Proukakis et al., 2013; Pasanen et al., 2014).
Histidine-50 is one of the copper (I) binding sites of αS that is mutated in the familial form of PD (H50Q mutation) (Sung et al., 2006; Proukakis et al., 2013). Non-acetylated αS binds copper via a coordination complex involving the N-terminal amine group of methionine-1, backbone and side chain of aspartate-2, and the imidazole ring of histidine-50 (Dudzik et al., 2011). A clear difference in methionine-1 and aspartate-2 environment on acetylation in NMR studies (Fauvet et al., 2012a; Kang et al., 2012) is predictive of different αS-copper interaction in acetylated and non-acetylated form. H50Q mutation in non-acetylated αS increases the aggregation of monomeric αS into amyloid structures, with minor changes in the secondary structure and negligible effect on the overall copper binding capacity (Uversky et al., 2001; Ghosh et al., 2013). Copper binding in N-α-acetylated H50Q protein (the in vivo form of H50Q mutation) is impaired, likely due to a double hit at the copper coordination complex; lack of the N-terminal amine, and the absence of the imidazole side chain at position 50 (Mason et al., 2016). Loss of copper-binding in acetylated H50Q is likely to interfere with the proposed ferrireductase activity of αS, leading to defects in metal homeostasis in vivo (Davies et al., 2011; Mezzaroba et al., 2019).
N-α-acetylated αS, like non-acetylated αS, aggregates into oligomers and fibrils under various experimental conditions (Kang et al., 2012; Maltsev et al., 2012; Gallea et al., 2016; Lima et al., 2019; Watson and Lee, 2019). There are varying reports for the effect of acetylation on both oligomers and fibrils. The extent of oligomerization of acetylated αS has been reported to be the same (Fauvet et al., 2012a; Kang et al., 2012) as well as reduced (Bu et al., 2017). Further, acetylated oligomers and fibrils show morphological and spectral features similar to unmodified αS, except for increased β-sheet and helical content in acetylated αS oligomers (Gallea et al., 2016; Iyer et al., 2016). The acetylated forms of familial PD mutants, E46K, H50Q, and A53T, show increased aggregation in 3,4-dihydroxyphenylacetaldehyde (DOPAL), dopamine, and SDS micelles, in comparison to wild-type acetylated αS (Ruzafa et al., 2017; Lima et al., 2019). Changes in fibrillization kinetics of wild-type αS upon acetylation are also ambiguous. Some studies report no significant difference (Fauvet et al., 2012a; Maltsev et al., 2012; Iyer et al., 2016), while others report slower kinetics, especially in the elongation rate (Kang et al., 2012; Ruzafa et al., 2017; Watson and Lee, 2019). This reduced elongation rate could be due to a helical secondary structure at the N-terminus that likely hinders the conversion of a monomer into the typical fibrillar β-sheet conformation (Kang et al., 2012). Acetylated αS is reported to yield distinct polymorphs (Watson and Lee, 2019) with likely increased structural homogeneity within a population (Iyer et al., 2016). The increased structural homogeneity in a fibril population may arise from a monomeric pool that is “structurally homogenous” (Figure 2). N-α-acetylation of αS results in a homogenous ensemble wherein 16 amino acids are in a helical conformation, leading to the nucleation of a homogenous population of fibrils. In a distinct polymorph, the reduced elongation rate can also be due to lower Thioflavin-T sensitivity toward acetylated αS, as Thioflavin-T fluorescence assay is sensitive to changes in topological features (Sidhu et al., 2018; Watson and Lee, 2019).
Structurally, fibrils formed by acetylated and non-acetylated αS show a mix of similar and distinct features. An overlay of four full-length αS structures, two with acetylation and two without acetylation, reveal an analogous backbone arrangement (Figure 4A; Tuttle et al., 2016; Li B. et al., 2018; Li Y. et al., 2018; Ni et al., 2019). Both types of fibril structures are formed of two protofilaments that intertwine in a twisted fibril morphology along a 21 screw axis – placing two monomers ∼180° to each other with an interaction surface in the center (Li B. et al., 2018; Li Y. et al., 2018; Ni et al., 2019). The N-α-acetylated protofilaments show a left-handed helical twist of –0.72° and a rise of ∼4.8 Å (Li Y. et al., 2018; Ni et al., 2019), while the non-acetylated protofilaments show a right-handed helical twist of 179.1° and a rise of 2.4 Å (Li B. et al., 2018). The dimer interaction surface in acetylated fibrils is formed by a hydrophobic steric zipper between residues histidine-50 to glutamate-57. Additionally, electrostatic interactions between histidine-50 and lysine-45 from one monomer and glutamate-57 from another monomer, and salt bridges between lysine-58: glutamate-61 (K58-E61) and glutamate-46: lysine-80 (E46-K80) stabilize the fibril core (Li Y. et al., 2018; Ni et al., 2019). In non-acetylated fibrils, the steric zipper is formed by residues further in the NAC region. Residues 55–62 are disordered (ssNMR studies) or do not form the steric zipper (cryoEM studies). The dimer interaction surface is formed by valine-71 to valine-82 in ssNMR studies and by glycine-68 to alanine-78 in cryoEM structures. Moreover, lysine-58 is flipped outward, resulting in the absence of the K58-E61 salt bridge (Figure 4A; Tuttle et al., 2016; Li Y. et al., 2018).
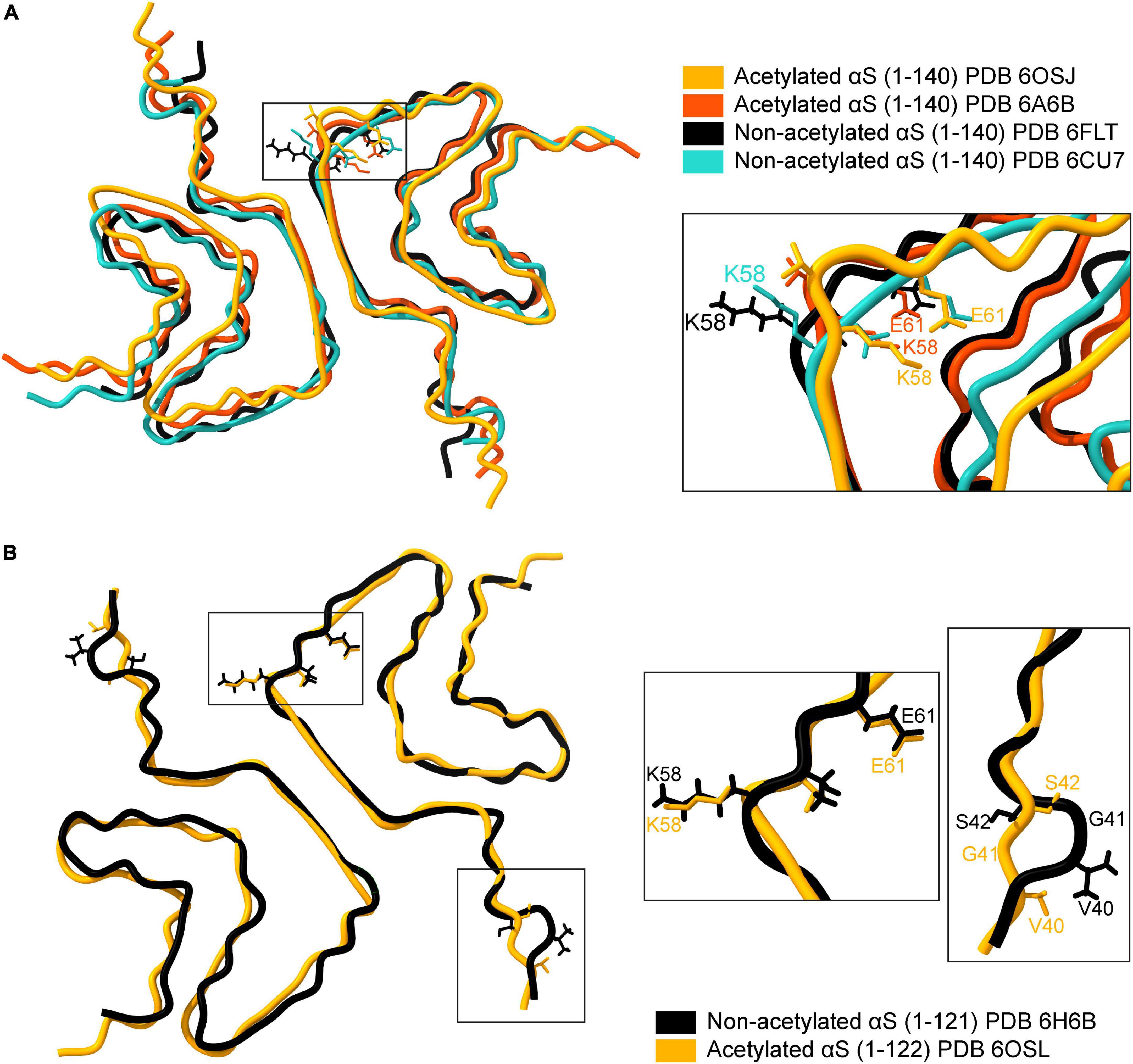
Figure 4. Comparison of available cryo-EM structures of the full-length acetylated (yellow, PDB ID: 6OSJ and orange, PDB ID:6A6B) and non-acetylated αS fibrils (black, PDB ID: 6FLT and blue, PDB ID:6CU7). (A) The backbone overlay of acetylated and non-acetylated αS fibrils is shown. The residue K58 in both acetylated αS fibrils is flipped inward, forming a salt bridge between K58-E61 (see inset). In contrast, the salt bridge is broken in acetylated αS fibrils due to the outward flip of K58. (B) The backbone overlay generated using ChimeraX of truncated (1-121/2) acetylated (yellow, PDB ID: 6OSL) and non-acetylated αS fibrils (black, PDB ID: 6H6B) depicting flipped K58 residues irrespective of acetylation state and minor loop fluctuations around the G41 residue.
The stabilizing effect of the salt-bridges on protein structure is well known, particularly in the case of αS. The compromised salt bridge between E46 and K80 side chains in an E46K variant of αS leads to a structurally homogenous yet entirely different fibril structure (consisting of one fibril species) and is more pathogenic compared to the wild-type αS (Boyer et al., 2020). A summary of all the available fibril structures of αS in PDB is listed in Table 2, with corresponding indicators for the K58-E61 salt bridge in each structure. An exciting facet of the αS fibril structure emerges concerning the K58-E61 salt bridge. Full-length N-α-acetylated αS fibrils have the K58-E61 salt bridge intact in sharp contrast to non-N-α-acetylated αS fibrils. The presence of the K58-E61 salt bridge is not influenced by N-α-acetylation alone but also by C-terminal truncation, phosphorylation of Tyr39, E46K mutation, and fibril polymorphism. The stark differences in the orientation of K58 cannot be an artifact of differing aggregation conditions since, in a single study employing identical aggregation conditions, the K58-E61 salt bridge was preserved in full-length N-α-acetylated αS fibrils and 1–103 N-α-acetylated αS fibrils but broken in 1–122 N-α-acetylated αS fibrils (Ni et al., 2019). Further, comparing structures of C-terminal truncated 1–121/2 αS fibrils suggests little or no role of N-α-acetylation on the orientation of K58 (Figure 4B). Why is the orientation of K58 sensitive to N-α-acetylation in full-length αS fibrils but not in C-terminal truncated αS fibrils? Further experiments elucidating the driving force for the K58-E61 salt bridge could be exciting and may give us a better understanding of salt-bridges in the stability of αS fibrils. It is unclear if the orientation of K58 and the salt bridge between K58-E61 is physiologically relevant to its cellular function or pathological aggregation of αS. The outward orientation of K58 may render non-acetylated, and C-terminally truncated fibrils exposed to ubiquitination or SUMOylation (signal for proteasome-induced degradation) or acetylation by lysine acetylases.

Table 2. Comparison of available αS fibril structures and an overview of K58-E61 salt bridge.
N-terminal de-acetyltransferases (Ndats) are not known in eukaryotic cells as yet, suggesting constitutional N-α-acetylation of αS by N-terminal acetyltransferases (Nats). Could it be possible that Nats decline in function or decrease in expression levels in an age-dependent manner? Such a scenario would result in a decrease in N-α-acetylated αS over time and possibly affect its function and interaction with its binding partners. It has been shown by several groups that N-α-acetylated αS fibrils are less cytotoxic compared to non-N-α-acetylated αS fibrils. Studies investigating the absolute amounts of N-α-acetylated αS and non-N-α-acetylated αS in healthy and diseased patients would be a significant step forward. The proposed hypothesis on Nats draws parallels from a study investigating the effect of the NAD-dependent deacetylase sirtuin 2 (SIRT2) on the aggregation potential and cytotoxicity of αS. The authors showed that lysine residues acetylated at the ε-amino positions in the N-terminal region of αS (K6 and K10) from mice brains could be deacetylated by SIRT2. The deacetylation event exacerbated its aggregation potential and toxicity in vitro and in the substantia nigra of rats (de Oliveira et al., 2017). Furthermore, mutating K6 and K10 residues to create αS variants that are acetylation-resistant or mimic constitutive acetylation showed that acetylation at these residues prevents αS aggregation in the substantia nigra of rats. The remarkable changes in aggregation potential and toxicity of αS in vivo resulting from acetylation of N-terminal lysine residues are intriguing. The authors proposed a model in which the age-dependent increase of SIRT2 in the brain, with the concomitant decrease of acetylated αS, leads to increased αS aggregation and the worsening of the expected defects in the autophagy-lysosome pathway (ALP) associated with aging.
The wild-type interactions of αS protofibrils are perturbed in familial PD mutations. The observation is not surprising as most of the mutations associated with the familial form of PD (H50Q, G51D, A53T, A53E) are located at the dimerization interface. The H50Q mutation disrupts the H50-K45-E57 interaction, while the E46K mutation breaks the E46-K80 salt bridge (Li B. et al., 2018). In A53T mutations, the dimerization core is formed by only two residues, Tyr-59 and Lys-60, instead of seven residues (H50-E57) in wild-type αS (Sun et al., 2020). Thus, these mutations can be expected to weaken the fibril core, resulting in morphological differences and greater fragmentation that consequently may increase seeding potential (Zhao et al., 2020a).
In structural studies, acetylated and non-acetylated αS fibrils could seed aggregation reactions and were cytotoxic (Tuttle et al., 2016; Li B. et al., 2018; Li Y. et al., 2018). In wild-type αS, acetylated αS seeds faithfully template fibril morphology across multiple seeding reactions, while non-acetylated αS seeds show poorer templating (Watson and Lee, 2019). Since the seed molecule’s conformation is critical in templating reactions, an unstable fibril core in non-acetylated αS, due to the absence of the K58-E61 salt bridge, may lead to poor templating (Sidhu et al., 2016). NMR studies show that in seeded aggregations of acetylated αS monomers with fibril seeds and off-pathway oligomers, the first 11 residues interact with the seeds in both the cases—successful templating with fibril seeds and unsuccessful templating with off-pathway oligomers. The observation suggests that the N-terminal interaction of acetylated αS is the first point of contact between a seed and a free monomer, irrespective of templating outcome (Yang et al., 2021). The differences between oligomers and fibrils from acetylated and non-acetylated αS monomers are likely due to the acetyl group. Still, some of the differences, at least, could also be due to differences in fibril preparation protocols used in each study. Differences in protein concentration; solution conditions like buffer, salt, metal ions, small molecules; agitation; incubation time have a significant effect on the kinetics and morphology of αS fibrils (Hoyer et al., 2002; Heise et al., 2005; Powers and Powers, 2006; Vilar et al., 2008; Knowles et al., 2009; Morel et al., 2010; Bousset et al., 2013; Buell et al., 2013, 2014; Sidhu et al., 2014; Buell, 2019; Panuganti and Roy, 2020). Since all the studies compared here have differences in the parameters mentioned above, a direct comparison to arrive at an empirical conclusion is challenging.
More than 300 post-translational modifications (PTMs) are known to occur in proteins (Clark et al., 2005), but a handful of these are known for αS, and their implications have been discussed in detail (Beyer, 2006; Zhang et al., 2019). These modifications include acetylation, phosphorylation, nitration, glycosylation, SUMOylation, ubiquitination, di-tyrosine crosslinking, and methionine oxidation. While the impact of PTMs in αS has been studied extensively in isolation, very few studies have considered the impact of N-α-acetylation in concert with the modifications mentioned above. Experiments in yeast show that deletion of NatB selectively increased localization of αS to cytoplasm and not plasma membrane as in wild-type yeast (Zabrocki et al., 2008). Evidence for the role of N-terminal acetylation of αS in its function are scarce and are still emerging. Since in vivo αS is universally present in the acetylated form (Bartels et al., 2011; Fauvet et al., 2012b; Burré et al., 2013; Theillet et al., 2016), all the studies with endogenous αS represent functions of acetylated αS. However, most of the studies with recombinant αS report behavior of non-acetylated αS. Only systematic comparative studies of αS behavior from endogenous and recombinant αS can delineate the effects of N-terminal acetylation. Limited studies that focus on the acetylated αS show that acetylated forms are involved in Lewy body associated pathology, metal homeostasis and synaptic function. Mass-spectrometry based studies from postmortem tissue of dementia with Lewy bodies (DLB) and PD patients, show full-length and truncated acetylated αS forms (Ac-αS1–139, Ac-αS1–119 Ac-αS1–103) and no non-acetylated forms, suggesting that in both disease and healthy conditions acetylation is present (Öhrfelt et al., 2011). This is consistent with another study that identified multiple truncated acetylated forms (Ac- αS1–6, Ac- αS13–21, Ac- αS35–43, Ac- αS46–58, Ac- αS61–80, Ac- αS81–96, Ac- αS103–119) of αS in Lewy body enriched fractions of PD patient samples (Bhattacharjee et al., 2019). In addition to brain tissues, only NTAc-αS can be detected in blood from Alzheimer’s patients and not the non-acetylated form, which is an indicator of neuronal death (Pero-Gascon et al., 2020). These studies highlight the importance to study physiologically relevant biochemistry of αS in acetylated forms to find better inhibitors for αS aggregation and to identify biomarkers.
αS is a copper binding protein with two sites for interaction with copper: Met 1-Met 5 and Ala 49-His 50 (Dudzik et al., 2011). Copper binding at Met 1–Met 5 is different for acetylated and non-acetylated αS forms. Copper binding in non-acetylated form at position Met 1–Met 5 results in a redox active state that can reduce metals while acetylated αS, though binds Cu2+, does not exhibit redox behavior (Garza-Lombó et al., 2018). The copper binding behavior of αS at the N-terminus is observed both in solution and membrane bound conformations (Dudzik et al., 2013). Since both N-terminal acetylation and copper binding increase the propensity of αS to adopt α-helical conformation, it is likely that they synergistically contribute to αS interaction with synaptic vesicles.
Post-translational modifications can be reversible or irreversible, and the regulatory dynamics of these modifications may give vital insights into protein function. Unlike reversible PTMs, like phosphorylation, glycosylation, ubiquitination, SUMOylation, methionine oxidation, nitration that may be rapidly added or removed from a protein under varied metabolic or pathologic cues, N-α-acetylation has been thought to be irreversible and occurring co-translationally. However, there is emerging evidence that acetylation of N-termini of proteins does necessarily occur co-translationally (Dormeyer et al., 2007). When 15N isotope-enriched non-acetylated αS was delivered into A2780, HeLa, RCSN-3, B65, and SK-N-SH cells using electroporation and was found to be N-α-acetylated entirely within 5 h (Theillet et al., 2016). These evidences suggest that cells prefer N-α-acetylated αS. It may be energetically more favorable for ubiquitous acetylation of αS to occur co-translationally.
There is no evidence of the existence of N-terminal de-acetyltransferases (Ndats), suggesting the irreversible nature of N-α-acetylation. This observation opens new avenues to investigate the existence of N-terminal de-acetyltransferases (Ndats) and other regulatory mechanisms that could (dys)regulate N-α-acetylation of αS. NMR studies have shown that N-α-acetylation induces stable α-helix formation in the first 16 amino acid residues in αS (Maltsev et al., 2012). N-α-acetylation of αS occurs co-translationally in eukaryotes and therefore precedes all other PTMs. Not surprisingly, the various permutations of PTMs mentioned above in αS preparations have consistently reported N-α-acetylation of αS at the least. We speculate that N-α-acetylation may “prime” αS for subsequent PTMs vital to its function and explain the cellular need to acetylate the N-terminus co-translationally. Our speculation is based on several observations: (a) N-α-acetylation led to plasma membrane localization of acetylated αS in yeast while non-acetylated αS remained in the cytoplasm. Further, the study showed decreased levels of Ser129 phosphorylation in non-acetylated αS compared to acetylated αS (Zabrocki et al., 2008). (b) Crosstalk between acetylation and other PTMs in a given protein is well known in eukaryotes (Yang and Seto, 2008) and impacts cell fate, and has implications for aging (Ree et al., 2018). For example, acetylation of histone H3 at K9/27 positions crosstalk with phosphorylation at S10/28 positions, respectively, to affect downstream gene expression (Latham and Dent, 2007). (c) The formation of a stabilized helix upon N-α-acetylation may provide lysine acetylases a helical scaffold (compared to disordered chain in non-acetylated αS) to effectively acetylate lysine residues in the 6th and 10th position in αS. Such scaffolds are well known in the context of signaling proteins and multi-protein complexes in eukaryotes (Langeberg and Scott, 2015). (d) The lack of Ndats potentially highlights the importance of N-α-acetylation of αS, with as yet unknown modes of regulation. Typically, modifications closely involved in regulatory processes are reversible processes (Martin, 2007). Examples of such reversible processes include protein (de)phosphorylation, (de)acetylation, (de)adenylylation, and (de)ADP-ribosylation. Additionally, Acetyl-CoA is a key metabolite in cellular metabolism and its consumption for the N-α-acetylation of αS indicates a necessary protein modification. (e) N-α-acetylation has been shown to inhibit protein targeting to the endoplasmic reticulum (Forte et al., 2011).
The priming role of N-α-acetylation of αS suggested here may have evaded sight as it likely does not require genomic regulation or quantitative changes in αS levels. Thus, in vivo studies investigating the impact of N-α-acetylation of αS on subsequent PTMs, especially phosphorylation, may help us understand if N-α-acetylation has a priming function. Understanding the crosstalk between N-α-acetylation and S129 phosphorylation is vital since several reports show accelerated inclusion formation and cellular toxicity in different models triggered by S129 phosphorylation (Smith et al., 2005; Sugeno et al., 2008). Additionally, more than 90% of αS deposited in Lewy bodies (LBs) in PD patients is phosphorylated at S129 while healthy individuals exhibit roughly 4% S129 phosphorylation (Arawaka et al., 2017).
The physiologically native state of αS is unquestionably N-α-acetylated. The observation has been determined exhaustively in numerous mammalian cells and organisms. It remains irrefutably an irreversible modification in αS so far. The impact of N-α-acetylation of αS in the context of pathological consequences (aggregation into toxic oligomers, fibrils, and higher-ordered aggregates) is increasingly being investigated. With the advent of cryo-EM, we are beginning to see structural details of αS fibrils at unprecedented spatial resolutions. Emerging studies are benchmarking the fibril structure of NTAc-αS housing PD familial mutations with endogenous αS fibrils isolated from diseased patients. However, despite these achievements, the impact of N-α-acetylation on the function of αS is still murky.
It is vital to understand how acetylation imbalance in αS manifests in vivo and which physiological consequences of the imbalance lead to neurotoxicity (de Oliveira et al., 2017). In this respect, a detailed proteomics study documenting the ratio of acetylated and non-acetylated αS over the progress of Lewy body formation would be remarkable. Emerging studies have shown enough evidence of N-α-acetylation affecting several downstream processes in living cells. A recent study demonstrated that N-α-acetylation of αS determines αS levels and subsequent toxicity in primary neurons (Vinueza-Gavilanes et al., 2020). Using point mutants that altered or blocked N-α-acetylation, the authors demonstrated that blocking N-α-acetylation led to a decrease in αS levels in live primary neurons and concomitantly reduced neurotoxicity. The prospect of blocking N-α-acetylation of αS by NatB is exciting, yet, maybe challenging for drug discovery strategies given that NatB acetylates ∼20% of cellular proteins. CRISPR-based strategies in the future may be able to edit the first two N-terminal amino acids and demonstrate if in vivo blocking N-α-acetylation of αS may help to decrease αS levels. Although NTAc-αS is recognized as the physiologically relevant species in healthy brain cells – in both the soluble and insoluble fractions of brain tissues of PD patients (Anderson et al., 2006) – the use of non-acetylated αS is rampant in emerging literature. The use of NTAc-αS must be encouraged, and NTAc-αS should be a gold standard for all studies investigating this multi-faceted protein (Lashuel et al., 2013) concerning conformational changes, oligomerization and aggregation propensities, lipid interactions, and other cellular binding partners.
In the future, we must focus our efforts toward elucidating (a) the effect of co-occurring N-α-acetylation and other PTMs in αS on its membrane (un)binding, oligomer/fibril structure, and corresponding aggregation kinetics, (b) the effect of co-occurring N-α-acetylation and familial PD mutations on αS function and aggregation into fibrillar structures, (c) the relation between the level of N-α-acetylation of αS and the progression rate of neurodegeneration in synucleinopathies, (d) the relation between metal-ion (dys)homeostasis and cellular models of synucleinopathies wherein levels of N-α-acetylation can be modulated, and (e) the complex relation between aggregation rates, diffusion coefficients, macromolecular crowding and N-α-acetylated αS in vivo. We may also want to investigate the interplay of regulatory factors (sirtuins) or genetic circuits triggered in PD patients and N-α-acetylation levels. While the physiological function of αS remains evasive, N-α-acetylation of αS presents an exciting path for future research.
AI, AS, and VS wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work presented here was part of a FOM-program entitled “A Single Molecule View on Protein Aggregation” (No. 127). We acknowledge generous funding from the work funded by the erstwhile Foundation for Fundamental Research on Matter (FOM), now subsumed by the Dutch Research Council (NWO). We also acknowledge support from NanoNextNL, a micro- and nanotechnology consortium of the Government of The Netherlands and 130 partners.
We thank Mireille Claessens (University of Twente, Enschede, Netherlands) for many helpful and stimulating discussions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aksnes, H., Drazic, A., Marie, M., and Arnesen, T. (2016). First things first: Vital protein marks by N-terminal acetyltransferases. Trends Biochem. Sci. 41, 746–760. doi: 10.1016/j.tibs.2016.07.005
Aksnes, H., Ree, R., and Arnesen, T. (2019). Co-translational, post-translational, and non-catalytic roles of N-terminal acetyltransferases. Mol. Cell 73, 1097–1114. doi: 10.1016/j.molcel.2019.02.007
Ametzazurra, A., Larrea, E., Civeira, M. P., Prieto, J., and Aldabe, R. (2008). Implication of human N-α-acetyltransferase 5 in cellular proliferation and carcinogenesis. Oncogene 27, 7296–7306. doi: 10.1038/onc.2008.332
Anderson, J. P., Walker, D. E., Goldstein, J. M., De Laat, R., Banducci, K., Caccavello, R. J., et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. doi: 10.1074/jbc.M600933200
Antony, T., Hoyer, W., Cherny, D., Heim, G., Jovin, T. M., and Subramaniam, V. (2003). Cellular polyamines promote the aggregation of α-synuclein. J. Biol. Chem. 278, 3235–3240. doi: 10.1074/jbc.M208249200
Arawaka, S., Sato, H., Sasaki, A., Koyama, S., and Kato, T. (2017). Mechanisms underlying extensive Ser129-phosphorylation in α-synuclein aggregates. Acta Neuropathol. Commun. 5:48. doi: 10.1186/s40478-017-0452-6
Arnaudo, N., Fernández, I. S., McLaughlin, S. H., Peak-Chew, S. Y., Rhodes, D., and Martino, F. (2013). The N-terminal acetylation of Sir3 stabilizes its binding to the nucleosome core particle. Nat. Struct. Mol. Biol. 20, 1119–1121. doi: 10.1038/nsmb.2641
Arnesen, T., Van Damme, P., Polevoda, B., Helsens, K., Evjenth, R., Colaert, N., et al. (2009). Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. U.S.A. 106, 8157–8162. doi: 10.1073/pnas.0901931106
Barrett, P. J., and Greenamyre, T. J. (2015). Post-translational modification of α-synuclein in Parkinson’s disease. Brain Res. 1628, 247–253. doi: 10.1016/j.brainres.2015.06.002
Bartels, T., Ahlstrom, L. S., Leftin, A., Kamp, F., Haass, C., Brown, M. F., et al. (2010). The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys. J. 99, 2116–2124. doi: 10.1016/j.bpj.2010.06.035
Bartels, T., Choi, J. G., and Selkoe, D. J. (2011). α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–111. doi: 10.1038/nature10324
Bartels, T., Kim, N. C., Luth, E. S., and Selkoe, D. J. (2014). N-alpha-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS One 9:e103727. doi: 10.1371/journal.pone.0103727
Bartels, T., and Selkoe, D. J. (2013). Bartels & Selkoe reply. Nature 498, E6–E7. doi: 10.1038/nature12126
Behnia, R., Panic, B., Whyte, J. R. C., and Munro, S. (2004). Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat. Cell Biol. 6, 405–413. doi: 10.1038/ncb1120
Beyer, K. (2006). α-Synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol. 112, 237–251. doi: 10.1007/s00401-006-0104-6
Bhattacharjee, P., Öhrfelt, A., Lashley, T., Blennow, K., Brinkmalm, A., and Zetterberg, H. (2019). Mass spectrometric analysis of Lewy body-enriched α-synuclein in Parkinson’s disease. J. Proteome Res. 18, 2109–2120. doi: 10.1021/acs.jproteome.8b00982
Binolfi, A., Theillet, F. X., and Selenko, P. (2012). Bacterial in-cell NMR of human α-synuclein: A disordered monomer by nature? Biochem. Soc. Trans. 40, 950–954. doi: 10.1042/BST20120096
Bousset, L., Pieri, L., Ruiz-Arlandis, G., Gath, J., Jensen, P. H., Habenstein, B., et al. (2013). Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 4:2575. doi: 10.1038/ncomms3575
Boyer, D. R., Li, B., Sun, C., Fan, W., Sawaya, M. R., Jiang, L., et al. (2019). Structures of fibrils formed by α-synuclein hereditary disease mutant H50Q reveal new polymorphs. Nat. Struct. Mol. Biol. 26, 1044–1052.
Boyer, D. R., Li, B., Sun, C., Fan, W., Zhou, K., Hughes, M. P., et al. (2020). The α-synuclein hereditary mutation E46K unlocks a more stable, pathogenic fibril structure. Proc. Natl. Acad. Sci. U.S.A. 117, 3592–3602. doi: 10.1073/pnas.1917914117
Breydo, L., Wu, J. W., and Uversky, V. N. (2012). α-Synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 1822, 261–285.
Bu, B., Tong, X., Li, D., Hu, Y., He, W., Zhao, C., et al. (2017). N-terminal acetylation preserves α-synuclein from Oligomerization by blocking intermolecular hydrogen bonds. ACS Chem. Neurosci. 8, 2145–2151. doi: 10.1021/acschemneuro.7b00250
Buell, A. K. (2019). The growth of amyloid fibrils: Rates and mechanisms. Biochem. J. 476, 2677–2703. doi: 10.1042/BCJ20160868
Buell, A. K., Galvagnion, C., Gaspar, R., Sparr, E., Vendruscolo, M., Knowles, T. P. J., et al. (2014). Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. U.S.A. 111, 7671–7676. doi: 10.1073/pnas.1315346111
Buell, A. K., Hung, P., Salvatella, X., Welland, M. E., Dobson, C. M., and Knowles, T. P. J. (2013). Electrostatic effects in filamentous protein aggregation. Biophys. J. 104, 1116–1126. doi: 10.1016/j.bpj.2013.01.031
Burré, J., Vivona, S., Diao, J., Sharma, M., Brunger, A. T., and Südhof, T. C. (2013). Properties of native brain α-synuclein. Nature 498, E4–E6. doi: 10.1038/nature12125
Chakrabartty, A., Doig, A. J., and Baldwin, R. L. (1993). Helix capping propensities in peptides parallel those in proteins. Proc. Natl. Acad. Sci. U.S.A. 90, 11332–11336. doi: 10.1073/pnas.90.23.11332
Cholak, E., Bugge, K., Khondker, A., Gauger, K., Pedraz-Cuesta, E., Pedersen, M. E., et al. (2020). Avidity within the N-terminal anchor drives α-synuclein membrane interaction and insertion. FASEB J. 34, 7462–7482. doi: 10.1096/fj.202000107R
Choudhary, C., Weinert, B. T., Nishida, Y., Verdin, E., and Mann, M. (2014). The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550. doi: 10.1038/nrm3841
Clark, R. S. B., Bayir, H., and Jenkins, L. W. (2005). Posttranslational protein modifications. Crit. Care Med. 33, S407–S409. doi: 10.1097/01.CCM.0000191712.96336.51
Corbillé, A. G., Neunlist, M., and Derkinderen, P. (2016). Cross-linking for the analysis of α-synuclein in the enteric nervous system. J. Neurochem. 139, 839–847. doi: 10.1111/jnc.13845
Creighton, T. E. (1993). Proteins: Structures and molecular properties, 2nd Edn. New York, NY: W.H.Freeman.
Davidson, W. S., Jonas, A., Clayton, D. F., and George, J. M. (1998). Stabilization of α-Synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. doi: 10.1074/jbc.273.16.9443
Davies, P., Moualla, D., and Brown, D. R. (2011). Alpha-synuclein is a cellular ferrireductase. PLoS One 6:e15814. doi: 10.1371/journal.pone.0015814
de Oliveira, R. M., Vicente Miranda, H., Francelle, L., Pinho, R., Szegö, É. M., Martinho, R., et al. (2017). The mechanism of sirtuin 2–mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol. 15:e2000374. doi: 10.1371/journal.pbio.2000374
Dettmer, U., Newman, A. J., Luth, E. S., Bartels, T., and Selkoe, D. (2013). In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and non-neural cells. J. Biol. Chem. 288, 6371–6385. doi: 10.1074/jbc.M112.403311
Dettmer, U., Newman, A. J., Von Saucken, V. E., Bartels, T., and Selkoe, D. (2015b). KTKEGV repeat motifs are key mediators of normal α-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 112, 9596–9601. doi: 10.1073/pnas.1505953112
Dettmer, U., Newman, A. J., Soldner, F., Luth, E. S., Kim, N. C., Von Saucken, V. E., et al. (2015a). Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun. 6:7314. doi: 10.1038/ncomms8314
Dikiy, I., and Eliezer, D. (2014). N-terminal Acetylation stabilizes N-terminal Helicity in Lipid- and Micelle-bound α-Synuclein and increases its affinity for Physiological Membranes. J. Biol. Chem. 289, 3652–3665. doi: 10.1074/jbc.M113.512459
Doig, A. J., Chakrabartty, A., Klingler, T. M., and Baldwin, R. L. (1994). Determination of Free Energies of N-Capping in α-Helices by Modification of the Lifson-Roig Helix-Coil Theory To Include N- and C-Capping. Biochemistry 33, 3396–3403. doi: 10.1021/bi00177a033
Dormeyer, W., Mohammed, S., Van Breukelen, B., Krijgsveld, J., and Heck, A. J. R. (2007). Targeted analysis of protein termini. J. Proteome Res. 6, 4634–4645. doi: 10.1021/pr070375k
Drazic, A., Aksnes, H., Marie, M., Boczkowska, M., Varland, S., Timmerman, E., et al. (2018). NAA80 is actin’s N-terminal acetyltransferase and regulates cytoskeleton assembly and cell motility. Proc. Natl. Acad. Sci. U.S.A. 115, 4399–4404. doi: 10.1073/pnas.1718336115
Dudzik, C. G., Walter, E. D., Abrams, B. S., Jurica, M. S., and Millhauser, G. L. (2013). Coordination of copper to the membrane-bound form of α-synuclein. Biochemistry 52, 53–60. doi: 10.1021/bi301475q
Dudzik, C. G., Walter, E. D., and Millhauser, G. L. (2011). Coordination features and affinity of the Cu2+ site in the α-synuclein protein of Parkinson’s disease. Biochemistry 50, 1771–1777. doi: 10.1021/bi101912q
Eliezer, D. (2013). The mysterious C-terminal tail of alpha-synuclein: Nanobody’s guess. J. Mol. Biol. 425, 2393–2396. doi: 10.1016/j.jmb.2013.03.031
Eliezer, D., Kutluay, E., Bussell, R., and Browne, G. (2001). Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 307, 1061–1073. doi: 10.1006/jmbi.2001.4538
Fairman, R., Shoemaker, K. R., York, E. J., Stewart, J. M., and Baldwin, R. L. (1989). Further studies of the helix dipole model: Effects of a free α−NH3+ or α−COO- group on helix stability. Proteins 5, 1–7. doi: 10.1002/prot.340050102
Fauvet, B., Fares, M. B., Samuel, F., Dikiy, I., Tandon, A., Eliezer, D., et al. (2012a). Characterization of semisynthetic and naturally N α- acetylated α-synuclein in vitro and in intact cells: Implications for aggregation and cellular properties of α-synuclein. J. Biol. Chem. 287, 28243–28262. doi: 10.1074/jbc.M112.383711
Fauvet, B., Mbefo, M. K., Fares, M. B., Desobry, C., Michael, S., Ardah, M. T., et al. (2012b). α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 287, 15345–15364. doi: 10.1074/jbc.M111.318949
Fernández, R. D., and Lucas, H. R. (2018a). Isolation of recombinant tetrameric N-acetylated α-synuclein. Protein Expr. Purif. 152, 146–154. doi: 10.1016/j.pep.2018.07.008
Fernández, R. D., and Lucas, H. R. (2018b). Mass spectrometry data confirming tetrameric α-synuclein N-terminal acetylation. Data Brief 20, 1686–1691. doi: 10.1016/j.dib.2018.09.026
Forte, G. M. A., Pool, M. R., and Stirling, C. J. (2011). N-terminal acetylation inhibits protein targeting to the endoplasmic reticulum. PLoS Biol. 9:e1001073. doi: 10.1371/journal.pbio.1001073
Gallea, J. I., Sarroukh, R., Yunes-Quartino, P., Ruysschaert, J. M., Raussens, V., and Celej, M. S. (2016). Structural remodeling during amyloidogenesis of physiological Nα-acetylated α-synuclein. Biochim. Biophys. Acta 1864, 501–510. doi: 10.1016/j.bbapap.2016.01.011
Gao, J., Barroso, C., Zhang, P., Kim, H. M., Li, S., Labrador, L., et al. (2016). N-terminal acetylation promotes synaptonemal complex assembly in C. elegans. Genes Dev. 30, 2404–2416. doi: 10.1101/gad.277350.116
Garza-Lombó, C., Posadas, Y., Quintanar, L., Gonsebatt, M. E., and Franco, R. (2018). Neurotoxicity linked to dysfunctional metal ion homeostasis and xenobiotic metal exposure: Redox signaling and oxidative stress. Antioxid. Redox Signal. 28, 1669–1703. doi: 10.1089/ars.2017.7272
Georgieva, E. R., Ramlall, T. F., Borbat, P. P., Freed, J. H., and Eliezer, D. (2008). Membrane-bound α-synuclein forms an extended helix: Long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J. Am. Chem. Soc. 130, 12856–12857. doi: 10.1021/ja804517m
Ghosh, D., Mondal, M., Mohite, G. M., Singh, P. K., Ranjan, P., Anoop, A., et al. (2013). The parkinson’s disease-associated H50Q mutation accelerates α-synuclein aggregation in vitro. Biochemistry 52, 6925–6927. doi: 10.1021/bi400999d
Goetze, S., Qeli, E., Mosimann, C., Staes, A., Gerrits, B., Roschitzki, B., et al. (2009). Identification and functional characterization of N-terminally acetylated proteins in Drosophila melanogaster. PLoS Biol. 7:e1000236. doi: 10.1371/journal.pbio.1000236
Guerrero-Ferreira, R., Taylor, N. M. I., Arteni, A. A., Kumari, P., Mona, D., Ringler, P., et al. (2019). Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. eLife 8:e48907. doi: 10.7554/eLife.48907
Guerrero-Ferreira, R., Taylor, N. M. I., Mona, D., Ringler, P., Lauer, M. E., Riek, R., et al. (2018). Cryo-EM structure of alpha-synuclein fibrils. eLife 7:e36402. doi: 10.7554/eLife.36402
Gurry, T., Ullman, O., Fisher, C. K., Perovic, I., Pochapsky, T., and Stultz, C. M. (2013). The dynamic structure of α-synuclein multimers. J. Am. Chem. Soc. 135, 3865–3872. doi: 10.1021/ja310518p
Halliday, G. M., Holton, J. L., Revesz, T., and Dickson, D. W. (2011). Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 122, 187–204. doi: 10.1007/s00401-011-0852-9
Heise, H., Hoyer, W., Becker, S., Andronesi, O. C., Riedel, D., and Baldus, M. (2005). Molecular-level secondary structure, polymorphism, and dynamics of full-length α-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. U.S.A. 102, 15871–15876. doi: 10.1073/pnas.0506109102
Hoyer, W., Antony, T., Cherny, D., Heim, G., Jovin, T. M., and Subramaniam, V. (2002). Dependence of α-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 322, 383–393. doi: 10.1016/S0022-2836(02)00775-1
Hwang, C. S., Shemorry, A., and Varshavsky, A. (2010). N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973–977. doi: 10.1126/science.1183147
Iyer, A., and Claessens, M. M. A. E. (2019). Disruptive membrane interactions of alpha-synuclein aggregates. Biochim. Biophys. Acta Proteins Proteom. 1867, 468–482. doi: 10.1016/j.bbapap.2018.10.006
Iyer, A., Roeters, S. J., Schilderink, N., Hommersom, B., Heeren, R. M. A., Woutersen, S., et al. (2016). The impact of N-terminal acetylation of α-synuclein on phospholipid membrane binding and fibril structure. J. Biol. Chem. 291, 21110–21122. doi: 10.1074/jbc.M116.726612
Johnson, M., Coulton, A. T., Geeves, M. A., and Mulvihill, D. P. (2010). Targeted amino-terminal acetylation of recombinant proteins in E. coli. PLoS One 5:e15801. doi: 10.1371/journal.pone.0015801
Kang, L., Janowska, M. K., Moriarty, G. M., and Baum, J. (2013). Mechanistic Insight into the Relationship between N-Terminal Acetylation of α-Synuclein and Fibril Formation Rates by NMR and Fluorescence. PLoS One 8:e75018. doi: 10.1371/journal.pone.0075018
Kang, L., Moriarty, G. M., Woods, L. A., Ashcroft, A. E., Radford, S. E., and Baum, J. (2012). N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 21, 911–917. doi: 10.1002/pro.2088
Kim, S., Yun, S. P., Lee, S., Umanah, G. E., Bandaru, V. V. R., Yin, X., et al. (2018). GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci. U.S.A. 115, 798–803. doi: 10.1073/pnas.1700465115
Knowles, T. P. J., Waudby, C. A., Devlin, G. L., Cohen, S. I. A., Aguzzi, A., Vendruscolo, M., et al. (2009). An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537. doi: 10.1126/science.1178250
Krüger, R., Kuhn, W., Müller, T., Woitalla, D., Graeber, M., Kösel, S., et al. (1998). Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Langeberg, L. K., and Scott, J. D. (2015). Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 16, 232–244. doi: 10.1038/nrm3966
Lashuel, H. A., Overk, C. R., Oueslati, A., and Masliah, E. (2013). The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci 14, 38–48. doi: 10.1038/nrn3406
Latham, J. A., and Dent, S. Y. R. (2007). Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 14, 1017–1024. doi: 10.1038/nsmb1307
Lesage, S., Anheim, M., Letournel, F., Bousset, L., Honoré, A., Rozas, N., et al. (2013). G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann. Neurol 73, 459–471. doi: 10.1002/ana.23894
Li, B., Ge, P., Murray, K. A., Sheth, P., Zhang, M., Nair, G., et al. (2018). Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 9:3609. doi: 10.1038/s41467-018-05971-2
Li, Y., Zhao, C., Luo, F., Liu, Z., Gui, X., Luo, Z., et al. (2018). Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res. 28, 897–903. doi: 10.1038/s41422-018-0075-x
Lima, V. D. A., Do Nascimento, L. A., Eliezer, D., and Follmer, C. (2019). Role of Parkinson’s disease-linked mutations and N-Terminal acetylation on the Oligomerization of α-Synuclein Induced by 3,4-Dihydroxyphenylacetaldehyde. ACS Chem. Neurosci. 10, 690–703. doi: 10.1021/acschemneuro.8b00498
Lorenzen, N., Lemminger, L., Pedersen, J. N., Nielsen, S. B., and Otzen, D. E. (2014). The N-terminus of α-synuclein is essential for both monomeric and oligomeric interactions with membranes. FEBS Lett. 588, 497–502. doi: 10.1016/j.febslet.2013.12.015
Luth, E. S., Bartels, T., Dettmer, U., Kim, N. C., and Selkoe, D. J. (2015). Purification of α-synuclein from human brain reveals an instability of endogenous multimers as the protein approaches purity. Biochemistry 54, 279–292. doi: 10.1021/bi501188a
Makasewicz, K., Wennmalm, S., Stenqvist, B., Fornasier, M., Andersson, A., Jönsson, P., et al. (2021). Cooperativity of α-Synuclein binding to lipid membranes. ACS Chem. Neurosci. 12, 2099–2109. doi: 10.1021/acschemneuro.1c00006
Maltsev, A. S., Ying, J., and Bax, A. (2012). Impact of N-terminal acetylation of α-synuclein on its random coil and lipid binding properties. Biochemistry 51, 5004–5013. doi: 10.1021/bi300642h
Martin, B. L. (2007). Regulation by covalent modification. eLS 1–7. doi: 10.1002/9780470015902.a0000866.pub2
Mason, R. J., Paskins, A. R., Dalton, C. F., and Smith, D. P. (2016). Copper binding and subsequent aggregation of α-Synuclein Are Modulated by N-terminal acetylation and ablated by the H50Q Missense Mutation. Biochemistry 55, 4737–4741. doi: 10.1021/acs.biochem.6b00708
McNulty, B. C., Young, G. B., and Pielak, G. J. (2006). Macromolecular crowding in the Escherichia coli periplasm maintains α-synuclein disorder. J. Mol. Biol. 355, 893–897. doi: 10.1016/j.jmb.2005.11.033
Mezzaroba, L., Alfieri, D. F., Colado Simão, A. N., and Vissoci Reiche, E. M. (2019). The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 74, 230–241. doi: 10.1016/j.neuro.2019.07.007
Miotto, M. C., Valiente-Gabioud, A. A., Rossetti, G., Zweckstetter, M., Carloni, P., Selenko, P., et al. (2015). Copper binding to the N-terminally acetylated, naturally occurring form of Alpha-Synuclein induces local helical folding. J. Am. Chem. Soc. 137, 6444–6447. doi: 10.1021/jacs.5b01911
Monda, J. K., Scott, D. C., Miller, D. J., Lydeard, J., King, D., Harper, J. W., et al. (2013). Structural conservation of distinctive N-terminal acetylation-dependent interactions across a family of mammalian NEDD8 ligation enzymes. Structure 21, 42–53. doi: 10.1016/j.str.2012.10.013
Morel, B., Varela, L., Azuaga, A. I., and Conejero-Lara, F. (2010). Environmental conditions affect the kinetics of nucleation of amyloid fibrils and determine their morphology. Biophys. J. 99, 3801–3810. doi: 10.1016/j.bpj.2010.10.039
Neri, L., Lasa, M., Elosegui-Artola, A., D’Avola, D., Carte, B., Gazquez, C., et al. (2017). NatB-mediated protein N-α-terminal acetylation is a potential therapeutic target in hepatocellular carcinoma. Oncotarget 8, 40967–40981. doi: 10.18632/oncotarget.17332
Ni, X., McGlinchey, R. P., Jiang, J., and Lee, J. C. (2019). Structural Insights into α-Synuclein Fibril Polymorphism: Effects of Parkinson’s Disease-Related C-Terminal Truncations. J. Mol. Biol. 431, 3913–3919. doi: 10.1016/j.jmb.2019.07.001
Öhrfelt, A., Zetterberg, H., Andersson, K., Persson, R., Secic, D., Brinkmalm, G., et al. (2011). Identification of novel α-synuclein isoforms in human brain tissue by using an online NanoLC-ESI-FTICR-MS method. Neurochem. Res. 36, 2029–2042. doi: 10.1007/s11064-011-0527-x
O’Leary, E. I., Jiang, Z., Strub, M. P., and Lee, J. C. (2018). Effects of phosphatidylcholine membrane fluidity on the conformation and aggregation of N-terminally acetylated α-Synuclein. J. Biol. Chem. 293, 11195–11205. doi: 10.1074/jbc.RA118.002780
Panuganti, V., and Roy, I. (2020). Oligomers, fibrils and aggregates formed by alpha-synuclein: role of solution conditions. J. Biomol. Struct. Dyn. 40, 4389–4398. doi: 10.1080/07391102.2020.1856721
Park, S. E., Kim, J. M., Seok, O. H., Cho, H., Wadas, B., Kim, S. Y., et al. (2015). Control of mammalian g protein signaling by N-terminal acetylation and the N-end rule pathway. Science 347, 1249–1252. doi: 10.1126/science.aaa3844
Pasanen, P., Myllykangas, L., Siitonen, M., Raunio, A., Kaakkola, S., Lyytinen, J., et al. (2014). A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 35:2180.e1-5. doi: 10.1016/j.neurobiolaging.2014.03.024
Pero-Gascon, R., Benavente, F., Minic, Z., Berezovski, M. V., and Sanz-Nebot, V. (2020). On-line aptamer affinity solid-phase extraction capillary electrophoresis-mass spectrometry for the analysis of blood α-Synuclein. Anal. Chem. 92, 1525–1533. doi: 10.1021/acs.analchem.9b04802
Pfefferkorn, C. M., Heinrich, F., Sodt, A. J., Maltsev, A. S., Pastor, R. W., and Lee, J. C. (2012). Depth of α-synuclein in a bilayer determined by fluorescence, neutron reflectometry, and computation. Biophys. J. 102, 613–621. doi: 10.1016/j.bpj.2011.12.051
Polevoda, B., and Sherman, F. (2003). N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol. 325, 595–622. doi: 10.1016/S0022-2836(02)01269-X
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Powers, E. T., and Powers, D. L. (2006). The kinetics of nucleated polymerizations at high concentrations: Amyloid fibril formation near and above the “supercritical concentration.”. Biophys. J. 91, 122–132. doi: 10.1529/biophysj.105.073767
Proukakis, C., Dudzik, C. G., Brier, T., MacKay, D. S., Cooper, J. M., Millhauser, G. L., et al. (2013). A novel α-synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064. doi: 10.1212/WNL.0b013e31828727ba
Quintas, A., Saraiva, M. J. M., and Britot, R. M. M. (1999). The tetrameric protein transthyretin dissociates to a non-native monomer in solution. A novel model for amyloidogenesis. J. Biol. Chem. 274, 32943–32949. doi: 10.1074/jbc.274.46.32943
Ree, R., Varland, S., and Arnesen, T. (2018). Spotlight on protein N-terminal acetylation. Exp. Mol. Med. 50, 1–13. doi: 10.1038/s12276-018-0116-z
Rossetti, G., Musiani, F., Abad, E., Dibenedetto, D., Mouhib, H., Fernandez, C. O., et al. (2016). Conformational ensemble of human α-synuclein physiological form predicted by molecular simulations. Phys. Chem. Chem. Phys. 18, 5702–5706. doi: 10.1039/c5cp04549e
Runfola, M., De Simone, A., Vendruscolo, M., Dobson, C. M., and Fusco, G. (2020). The N-terminal Acetylation of α-Synuclein changes the affinity for lipid membranes but not the structural properties of the bound state. Sci. Rep 10:204. doi: 10.1038/s41598-019-57023-4
Ruzafa, D., Hernandez-Gomez, Y. S., Bisello, G., Broersen, K., Morel, B., and Conejero-Lara, F. (2017). The influence of N-terminal acetylation on micelle-induced conformational changes and aggregation of α-Synuclein. PLoS One 12:e0178576. doi: 10.1371/journal.pone.0178576
Schweighauser, M., Shi, Y., Tarutani, A., Kametani, F., Murzin, A. G., Ghetti, B., et al. (2020). Structures of α-synuclein filaments from multiple system atrophy. Nature 585, 464–469. doi: 10.1038/s41586-020-2317-6
Scott, D. C., Monda, J. K., Bennett, E. J., Harper, J. W., and Schulman, B. A. (2011). N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science 334, 674–678. doi: 10.1126/science.1209307
Setty, S. R. G., Strochlic, T. I., Tong, A. H. Y., Boone, C., and Burd, C. G. (2004). Golgi targeting of Arf-like GTPase Arl3p requires its Nα-acetylation and the integral membrane protein Sys1p. Nat. Cell Biol. 6, 414–419. doi: 10.1038/ncb1121
Shemorry, A., Hwang, C. S., and Varshavsky, A. (2013). Control of protein quality and stoichiometries by N-Terminal acetylation and the N-End rule pathway. Mol. Cell 50, 540–551. doi: 10.1016/j.molcel.2013.03.018
Sidhu, A., Segers-Nolten, I., and Subramaniam, V. (2014). Solution conditions define morphological homogeneity of α-synuclein fibrils. Biochim. Biophys. Acta 1844, 2127–2134. doi: 10.1016/j.bbapap.2014.09.007
Sidhu, A., Segers-Nolten, I., and Subramaniam, V. (2016). Conformational compatibility is essential for heterologous aggregation of α-Synuclein. ACS Chem. Neurosci. 7, 719–727. doi: 10.1021/acschemneuro.5b00322
Sidhu, A., Vaneyck, J., Blum, C., Segers-Nolten, I., and Subramaniam, V. (2018). Polymorph-specific distribution of binding sites determines thioflavin-T fluorescence intensity in α-synuclein fibrils. Amyloid 25, 189–196. doi: 10.1080/13506129.2018.1517736
Smith, W. W., Margolis, R. L., Li, X., Troncoso, J. C., Lee, M. K., Dawson, V. L., et al. (2005). α-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J. Neurosci. 25, 5544–5552. doi: 10.1523/JNEUROSCI.0482-05.2005
Starheim, K. K., Arnesen, T., Gromyko, D., Ryningen, A., Varhaug, J. E., and Lillehaug, J. R. (2008). Identification of the human Nα-acetyltransferase complex B (hNatB): A complex important for cell-cycle progression. Biochem. J. 415, 325–331. doi: 10.1042/BJ20080658
Sugeno, N., Takeda, A., Hasegawa, T., Kobayashi, M., Kikuchi, A., Mori, F., et al. (2008). Serine 129 phosphorylation of α-synuclein induces unfolded protein response-mediated cell death. J. Biol. Chem. 283, 23179–23188. doi: 10.1074/jbc.M802223200
Sun, Y., Hou, S., Zhao, K., Long, H., Liu, Z., Gao, J., et al. (2020). Cryo-EM structure of full-length α-synuclein amyloid fibril with Parkinson’s disease familial A53T mutation. Cell Res. 30, 360–362. doi: 10.1038/s41422-020-0299-4
Sung, Y. H., Rospigliosi, C., and Eliezer, D. (2006). NMR mapping of copper binding sites in alpha-synuclein. Biochim. Biophys. Acta 1764, 5–12. doi: 10.1016/j.bbapap.2005.11.003
Swaminathan, R., Hoang, C. P., and Verkman, A. S. (1997). Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: Cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys. J. 72, 1900–1907. doi: 10.1016/S0006-3495(97)78835-0
Theillet, F. X., Binolfi, A., Bekei, B., Martorana, A., Rose, H. M., Stuiver, M., et al. (2016). Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530, 45–50. doi: 10.1038/nature16531
Trexler, A. J., and Rhoades, E. (2012). N-terminal acetylation is critical for forming α-helical oligomer of α-synuclein. Protein Sci. 21, 601–605. doi: 10.1002/pro.2056
Tuttle, M. D., Comellas, G., Nieuwkoop, A. J., Covell, D. J., Berthold, D. A., Kloepper, K. D., et al. (2016). Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 23, 409–415. doi: 10.1038/nsmb.3194
Ullman, O., Fisher, C. K., and Stultz, C. M. (2011). Explaining the structural plasticity of α-synuclein. J. Am. Chem. Soc. 133, 19536–19546. doi: 10.1021/ja208657z
Uversky, V. N., Li, J., and Fink, A. L. (2001). Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein: A possible molecular link between parkinson’s disease and heavy metal exposure. J. Biol. Chem. 276, 44284–44296. doi: 10.1074/jbc.M105343200
Varland, S., Osberg, C., and Arnesen, T. (2015). N-terminal modifications of cellular proteins: The enzymes involved, their substrate specificities and biological effects. Proteomics 15, 2385–2401. doi: 10.1002/pmic.201400619
Viennet, T., Wördehoff, M. M., Uluca, B., Poojari, C., Shaykhalishahi, H., Willbold, D., et al. (2018). Structural insights from lipid-bilayer nanodiscs link α-Synuclein membrane-binding modes to amyloid fibril formation. Commun. Biol. 1:44. doi: 10.1038/s42003-018-0049-z
Vilar, M., Chou, H. T., Lührs, T., Maji, S. K., Riek-Loher, D., Verel, R., et al. (2008). The fold of α-synuclein fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 8637–8642. doi: 10.1073/pnas.0712179105
Vinueza-Gavilanes, R., Íñigo-Marco, I., Larrea, L., Lasa, M., Carte, B., Santamaría, E., et al. (2020). N-terminal acetylation mutants affect alpha-synuclein stability, protein levels and neuronal toxicity. Neurobiol. Dis. 137:104781. doi: 10.1016/j.nbd.2020.104781
Wang, W., Perovic, I., Chittuluru, J., Kaganovich, A., Nguyen, L. T. T., Liao, J., et al. (2011). A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. U.S.A. 108, 17797–17802. doi: 10.1073/pnas.1113260108
Watson, M. D., and Lee, J. C. (2019). N-Terminal Acetylation Affects α-Synuclein Fibril Polymorphism. Biochemistry 58, 3630–3633. doi: 10.1021/acs.biochem.9b00629
Waudby, C. A., Camilloni, C., Fitzpatrick, A. W. P., Cabrita, L. D., Dobson, C. M., Vendruscolo, M., et al. (2013). In-Cell NMR characterization of the secondary structure populations of a disordered conformation of α-Synuclein within E. coli Cells. PLoS One 8:e72286. doi: 10.1371/journal.pone.0072286
Waxman, E. A., Mazzulli, J. R., and Giasson, B. I. (2009). Characterization of hydrophobic residue requirements for α-synuclein fibrillization. Biochemistry 48, 9427–9436. doi: 10.1021/bi900539p
Xia, C., Tao, Y., Li, M., Che, T., and Qu, J. (2020). Protein acetylation and deacetylation: An important regulatory modification in gene transcription (Review). Exp. Ther. Med. 20, 2923–2940. doi: 10.3892/etm.2020.9073
Yang, D., Fang, Q., Wang, M., Ren, R., Wang, H., He, M., et al. (2013). Nα-acetylated Sir3 stabilizes the conformation of a nucleosome-binding loop in the BAH domain. Nat. Struct. Mol. Biol. 20, 1116–1118. doi: 10.1038/nsmb.2637
Yang, X., Wang, B., Hoop, C. L., Williams, J. K., and Baum, J. (2021). NMR unveils an N-terminal interaction interface on acetylated-α-synuclein monomers for recruitment to fibrils. Proc. Natl. Acad. Sci. U.S.A. 118:e2017452118. doi: 10.1073/pnas.2017452118
Yang, X. J., and Grégoire, S. (2007). Metabolism, cytoskeleton and cellular signalling in the grip of protein Nε- and O-acetylation. EMBO Rep. 8, 556–562. doi: 10.1038/sj.embor.7400977
Yang, X. J., and Seto, E. (2008). Lysine Acetylation: Codified Crosstalk with Other Posttranslational Modifications. Mol. Cell 31, 449–461. doi: 10.1016/j.molcel.2008.07.002
Zabrocki, P., Bastiaens, I., Delay, C., Bammens, T., Ghillebert, R., Pellens, K., et al. (2008). Phosphorylation, lipid raft interaction and traffic of α-synuclein in a yeast model for Parkinson. Biochim. Biophys. Acta 1783, 1767–1780. doi: 10.1016/j.bbamcr.2008.06.010
Zarbiv, Y., Simhi-Haham, D., Israeli, E., Elhadi, S. A., Grigoletto, J., and Sharon, R. (2014). Lysine residues at the first and second KTKEGV repeats mediate α-Synuclein binding to membrane phospholipids. Neurobiol. Dis. 70, 90–98. doi: 10.1016/j.nbd.2014.05.031
Zarranz, J. J., Alegre, J., Gómez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The New Mutation, E46K, of α-Synuclein Causes Parkinson and Lewy Body Dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Zhang, J., Li, X., and Li, J. D. (2019). The Roles of Post-translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 13:381. doi: 10.3389/fnins.2019.00381
Zhao, K., Li, Y., Liu, Z., Long, H., Zhao, C., Luo, F., et al. (2020a). Parkinson’s disease associated mutation E46K of α-synuclein triggers the formation of a distinct fibril structure. Nat. Commun. 11:2643. doi: 10.1038/s41467-020-16386-3
Zhao, K., Lim, Y. J., Liu, Z., Long, H., Sun, Y., Hu, J. J., et al. (2020b). Parkinson’s disease-related phosphorylation at Tyr39 rearranges α-synuclein amyloid fibril structure revealed by cryo-EM. Proc. Natl. Acad. Sci. U.S.A. 117, 20305–20315. doi: 10.1073/PNAS.1922741117
Keywords: protein aggregation, fibril structure, acetylation, post-translational modifications, alpha-synuclein
Citation: Iyer A, Sidhu A and Subramaniam V (2022) How important is the N-terminal acetylation of alpha-synuclein for its function and aggregation into amyloids? Front. Neurosci. 16:1003997. doi: 10.3389/fnins.2022.1003997
Received: 26 July 2022; Accepted: 26 October 2022;
Published: 16 November 2022.
Edited by:
Wolfgang Hoyer, Heinrich Heine University Düsseldorf, GermanyReviewed by:
Thibault Viennet, Dana-Farber Cancer Institute, United StatesCopyright © 2022 Iyer, Sidhu and Subramaniam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aditya Iyer, bGlua3RvYWRpdHlhQGdtYWlsLmNvbQ==; Vinod Subramaniam, di5zdWJyYW1hbmlhbUB1dHdlbnRlLm5s
†Present Address: Aditya Iyer, Amyl Therapeutics, Liège, Belgium
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.