 Jinwen Jiang
Jinwen Jiang Qihui Wu
Qihui Wu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 18 November 2021
Sec. Neurodegeneration
Volume 15 - 2021 | https://doi.org/10.3389/fnins.2021.781722
This article is part of the Research Topic Neurodegeneration: From Disease Mechanism to Therapeutic Advancement View all 9 articles
Alzheimer’s and Parkinson’s diseases (AD and PD) are amongst top of the prevalent neurodegenerative disease. One-third of PD patients are diagnosed with dementia, a pre-symptom of AD, but the underlying mechanism is elusive. Amyloid beta (Aβ) and α-synuclein are two of the most investigated proteins, whose pathological aggregation and spreading are crucial to the pathogenesis of AD and PD, respectively. Transcriptomic studies of the mammalian central nervous system shed light on gene expression profiles at molecular levels, regarding the complexity of neuronal morphologies and electrophysiological inputs/outputs. In the last decade, the booming of the single-cell RNA sequencing technique helped to understand gene expression patterns, alternative splicing, novel transcripts, and signal pathways in the nervous system at single-cell levels, providing insight for molecular taxonomy and mechanistic targets of the degenerative nervous system. Here, we re-visited the cell-cell transmission mechanisms of Aβ and α-synuclein in mediating disease propagation, and summarized recent single-cell transcriptome sequencing from different perspectives and discussed its understanding of neurodegenerative diseases.
The deposition of β-sheet containing amyloid aggregates featured many neurodegenerative diseases, such as amyloid-β (Aβ) and tau in Alzheimer’s disease (AD), α-synuclein in Parkinson’s disease (PD), and TAR DNA-binding protein (TDP) 43 in amyotrophic lateral sclerosis (ALS), which causes inflammation, neuronal dysfunction, movement and cognition defects. These infectious amyloidogenic “seeds” are self-template dependent replication, transcellular propagation, and transmissible neuropathology between distinct brain regions. An important contribution to the neuropathology is played by the accumulation of unfolded or misfolded proteins leading to the formation of disordered (amorphous) or ordered (amyloid fibril) aggregates, so these diseases are often called “conformational diseases” (Surguchev and Surguchov, 2010). This review discussed the contribution of transcellular propagation of Aβ oligomers/fibrils in AD and α-synuclein (αSyn) aggregates in PD, in which 30–40% of PD has been diagnosed with dementia, an early symptom of AD. It also discussed the recent advance of RNA sequencing techniques in the application of neurodegenerative diseases. Unlike traditional biochemistry and molecular biology, single-cell RNA sequencing (scRNA-seq) helps to understand multiple gene expression patterns and signal pathways in the nervous system at the single-cell level, aiming to impact the development of new diagnostic and therapeutic strategies in the degenerative nervous system.
Alzheimer’s disease is the most common degenerative disease of the nervous system that occurs in the elderly. Its clinical manifestations are mainly characterized by progressive memory decline and later comprehensive cognitive dysfunction. AD patients have a long course of the disease, gradually lose the ability to live, and eventually die of complications due to the inability to take care of themselves (Tokuchi et al., 2016). The two primary cardinal lesions associated with AD are the neurofibrillary tangle and the senile plaque (Perl, 2010). The neurofibrillary tangle consists of abnormal accumulations of phosphorylated tau within the perikaryal cytoplasm of specific neurons. The senile plaque consists of a central core of beta-amyloid, a 4-kD peptide, surrounded by abnormally configured neuronal processes or neurites. Other neuropathological lesions are encountered in cases of AD, but the disease is defined and recognized by these two cardinal lesions. Other lesions include poorly understood changes such as granulovacuolar degeneration and eosinophilic rodlike bodies (Hirano bodies). The loss of synaptic components is a change that has a significant impact on cognitive function and represents another crucial morphological alteration. It is essential to recognize that distinguishing between AD, especially in its early stages, and normal aging may be complicated, particularly if one examines the brains of patients who died at an advanced old age. It is also noted that instances of pure forms of AD, in the absence of other coexistent brain disease processes, such as infarctions or PD-related lesions, are relatively uncommon.
The gross pathological manifestation of AD is brain atrophy (Rabinovici et al., 2016). The etiology and pathogenesis of AD are complex, and there is currently no effective prediction method. Previous clinical and basic research evidence around Aβ pointed out that it played a vital role in the course of AD (Selkoe and Hardy, 2016). However, none of the drugs targeting Aβ passed clinical trials, suggesting the complexity of the onset of AD. The Aβ cascade hypothesis has also been challenged. Re-recognizing the role of the Aβ cascade hypothesis in the pathogenesis of AD and exploring possible new ways of AD will be the direction of future AD research. Here, we summarized the Aβ cascade hypothesis and related AD pathology, pathophysiological process, and treatment research progress.
At present, the pathogenesis and etiology of AD are still unclear. It believes that Aβ is a polypeptide obtained by continuous hydrolysis of amyloid precursor protein (APP) under the action of β and γ secretases. The matrix precipitates and accumulates. Among them, the soluble aggregates of Aβ are considered the main form of neurotoxicity and eventually cause cognitive dysfunction (Sharma et al., 2016) and play a crucial pathological role in AD. The deposition of Aβ aggregates leads to the formation of a large number of SP in the brain.
Aβ directly acts on neurons and produces toxic effects. Moreover, it also activates microglia and astrocytes in the brain to produce cytokines, induce inflammatory response, and trigger cell death eventually. Within the chronic inflammation of the nervous system, activated microglia can engulf synapses in the early stage of AD (Goedert, 2015). In addition, the hyperphosphorylation of the microtubule-associated protein Tau in neurons can also be downstream of the excessive production and aggregation of Aβ, which in turn leads to neuronal degeneration in the brain, leading to AD-related dementia (Pooler et al., 2015).
Misfolding and aggregation of Aβ in the brain is a critical step in the course of AD (Minjarez et al., 2016). Aβ can exist in the brain in three soluble forms, including monomeric Aβ, Aβ oligomers, and fibrillar (Selkoe and Hardy, 2016). Fibrillar Aβ, also known as Aβ fibrils, can act as a pro-oxidant, induce protein oxidation, and change the activity of oxidation-sensitive enzymes (Tycko, 2016). The oligomerized Aβ can be embedded in the bilayer of the neuronal membrane to increase the membrane’s permeability, leading to an increase in reactive oxygen species in the cell, triggering mitochondrial dysfunction, and inducing lipid peroxidation. Therefore, in neurodegenerative diseases related to Aβ, the change of cell membrane permeability is a common phenomenon. In addition, more and more evidence has suggested that Aβ oligomers are the real “culprit” of AD in recent years. After Walsh et al. (2002) reported in 2002 that Aβ oligomers had a damaging effect on rats’ long-term potentiation (LTP), scientists gradually revealed that Aβ aggregation has deleterious effects on neuronal structure and cognition (Tsai et al., 2004; Cleary et al., 2005; Grutzendler et al., 2007). Shankar et al. (2008), extracted Aβ oligomers from the cerebral cortex of AD patients, added them to the hippocampal slice incubation solution, or injected them into the rat lateral ventricle, which caused the attenuation of hippocampal LTP and enhancement of long-term depression (LTD), decreased the hippocampal dendritic spine density and the decline of learning and memory ability in normal rats. This work has consolidated the critical contribution of Aβ oligomers in the pathogenesis of AD from a pre-clinical perspective.
In the normal brain, Aβ is not useless. There is experimental evidence that Aβ monomer has neurotrophic effects (Shin et al., 2015). When the brain is in a physiological state, the production and degradation of Aβ is a dynamic equilibrium process. If too much production or degradation slows down, Aβ will accumulate progressively and become an insoluble form of dense plaque, that is, SP. The removal of SP may increase the content of Aβ oligomers and accelerate neuronal degeneration (Bachhuber et al., 2015; Shi et al., 2017).
After the Aβ in the brain is produced by APP hydrolysis and metabolism, it can be degraded in the brain by glial cells and some enzymes, such as NEP and insulin degradation enzyme (IDE), and it can also be transported through the blood. Aβ is produced from the cell and secreted into the intercellular fluid, enters the cerebrospinal fluid through the brain-cerebrospinal fluid barrier, and transports from the cerebrospinal fluid to the blood and lymphatic system through the blood-brain barrier (BBB) and arachnoid villi or perivascular space (PVS), where the Aβ is cleared but also accumulated within the whole body. The lymphatic system may also pass through the lymphatic vessels of the meninges (Xiang et al., 2015). Through the BBB the transported Aβ is mediated by low-density lipoprotein receptor-related protein 1 (LRP1) (Pflanzner et al., 2011), and the transport from brain tissue fluid to cerebrospinal fluid is through the glial lymphatic system, which is a network of perivascular space and the foot processes of astrocytes surround PVS. There is clinical evidence that decreased cerebral blood flow increases the accumulation of Aβ in the brain (Mattsson et al., 2014), especially Aβ deposited in cerebral blood vessels may be one of the causes of inflammation and cytotoxic events, which can further cause BBB to be more extensive of leakage and destruction (Zenaro et al., 2017), see also Figure 1 for the transport and clearance of Aβ.
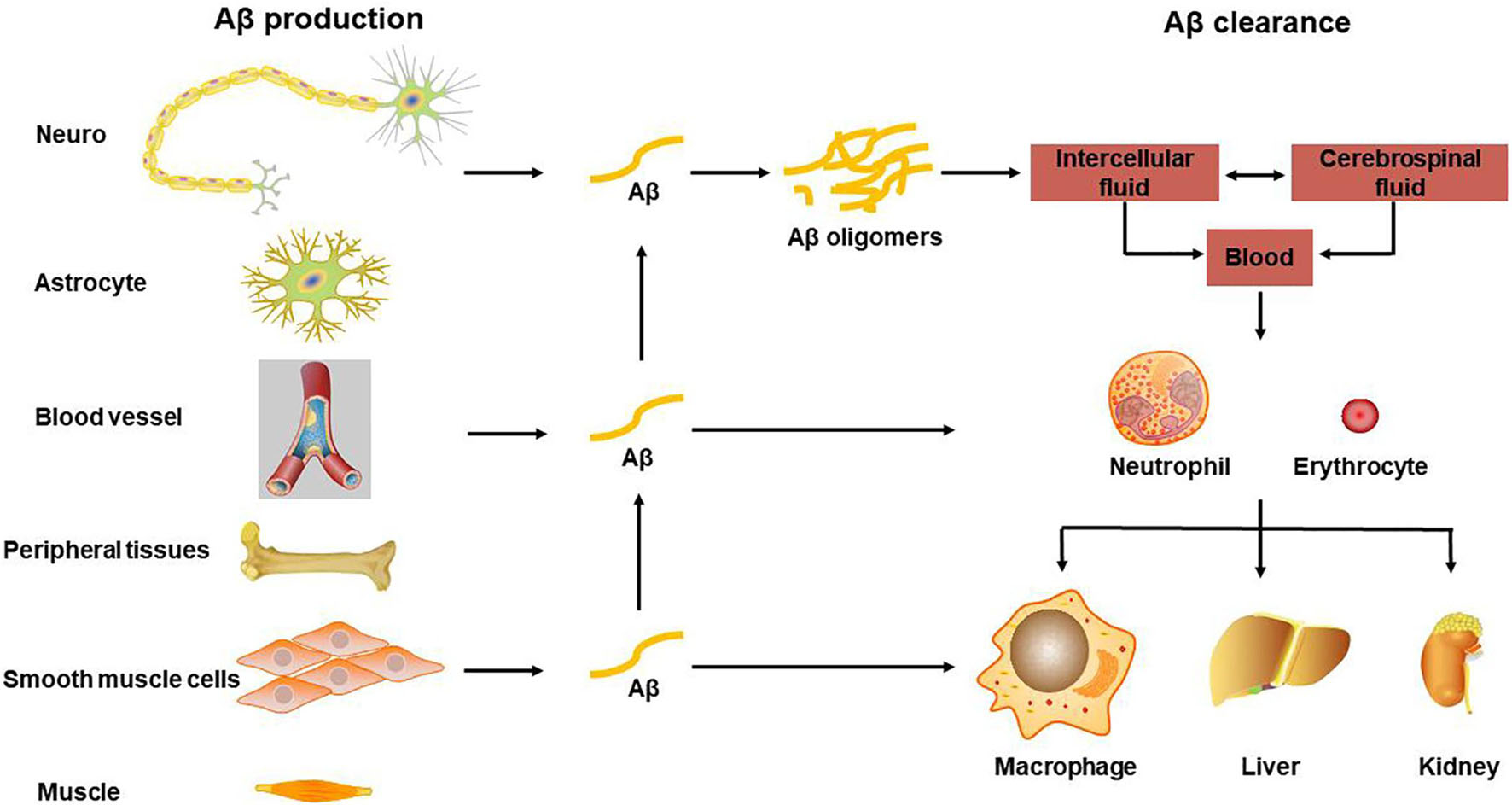
Figure 1. Transport and clearance of Aβ in brain and peripheral tissues and organs. Aβ is produced by neurons and glial cells in the brain, as well as peripheral blood vessel wall cells, skeletal muscle, and osteoblasts, etc., and secreted out of the cell; Aβ in the central nervous system is transported through brain tissue fluid, cerebrospinal fluid, and blood-brain barrier to the peripheral blood; monocytes and neutrophils swallow part of Aβ, some are degraded by Aβ degrading enzymes, and the other part is swallowed by macrophages in peripheral organs and tissues, or is excreted through bile and urine.
Parkinson’s disease is the second most prevalent neurodegenerative disease, next only to AD. Amongst the diagnostic of more than 10 million people diagnosed with PD worldwide, the major clinic lesions are degenerative dopaminergic neurons in substantia nigra. Among 55–60-year-old populations, the incidence rate of PD is exponentially increased, imposing a heavy burden on the family and society (Baiano et al., 2020). The neurodegenerative disease is common in the aged, however, the onset of PD is not clear, in which risk factors are included but not limited to genetics, environment, as well as age. Unfortunately, no effective medications for the treatment of PD are available, where most of the current strategies are preventative medicine. Since the discovery of αSyn (PARK1) in the late 1990s, currently, there are 6 major causal risk genes (SNCA, PRKN, UCHL1, PARK7, LRRK2, and PINK1) that are associated with familial PD (Blauwendraat et al., 2020). However, only 5–10% of patients with PD have a family medical history, where most of them are sporadic.
Under physiological conditions, αSyn is a soluble protein that mainly localizes on vesicles in pre-synapse. Current understanding of the functions of αSyn is still limited to vesicle trafficking, recycling, and secretion. Clinic autopsy of brains with PD revealed inclusions of Lewy bodies and Lewy neurites in the ventral tegmental area (VTA) and substantia nigra (SN) in brainstem. Later, researchers gradually recognized that the major component of Lewy bodies and Lewy neurites are phosphorylated and misfolded αSyn, together with other proteins, like ubiquitin, neurofilament, and alpha B crystallin. Tau proteins may also be present. In 1996, for the first time, the scientist observed the ultrastructure of Lewy body by electron microscopy (Forno et al., 1996). Although the discovery of αSyn in PD was known to the public for decades, the mechanisms of its function, especially under pathological condition, remains largely unclear.
For many years, prion diseases were thought to be a unique group of neurodegenerative disorders in which the normal cellular prion protein (PrPc) was recruited by infectious prion protein through “seeded” fibrillization (Stohr et al., 2012). In recent decades, mounting evidence demonstrated that a prion-like self-propagating property may apply to a wide range of diseases with protein aggregation, including αSyn, tau, Aβ, huntingtin with polyglutamine (ployQ) repeats, superoxide dismutase 1 (SOD1), and TDP43. Pathological brain-derived lysates (αSyn, tau, and TDP43) were shown to act as templates or seeds that could efficiently recruit their soluble counterparts into insoluble fibrils in wild-type primary cultures, slice cultures, as well as in vivo (Ren et al., 2009; Chen et al., 2010; Munch et al., 2011; Volpicelli-Daley et al., 2011; Stohr et al., 2012; Iba et al., 2013).
Since human brains are valuable resources and are not available in every institute, people are seeking alternative options that could recruit pathological inclusions in wild-type tissues or in vivo. About 10 years ago, recombinant αSyn monomers were reported to spontaneously aggregate into the preformed fibrils (PFFs) and induce αSyn inclusions in wild-type primary cultured hippocampal neurons and in vivo (Volpicelli-Daley et al., 2011; Luk et al., 2012), as well as in slice cultures recently (Wu et al., 2020), which mimics the pathogenesis of PD in human brains. When self-template-dependent amplification for several rounds, these amyloidogenic PFF “seeds” not only recruit endogenous αSyn but also recruit tau into aggregates both in vitro and in vivo (Guo et al., 2013). In addition, under different salt conditions, these PFFs formed functionally and structurally different “strains” with distinct neurotoxicity and biochemical properties (Patterson et al., 2019). As for deceased patients diagnosed with PD, the glial cytoplasmic inclusions (GCIs) extracted from the cerebellum are 1,000 times more potent than αSyn PFFs in inducing pathology and are 100 times more potent than Lewy body extracted from PD with dementia (Peng et al., 2018). How and why the glial milieu modifies the β-sheet folding property of αSyn and contributes to its neuropathologic function opens for further investigation.
Considering the complexity and unpredictability of the structure of spontaneously formed αSyn PFFs, the potency of neurotoxicity varies from batches to batches, as this process was affected by salt, temperature as well as pH of the reaction system (Patterson et al., 2019). The biofluid samples (CSF and blood) from patients with AD and PD contain picogram levels of aggregated αSyn and tau, which can be used as template-dependent amplification seeds. One of the amplification techniques is the real-time quaking-induced conversion (RT-QuIC) assay, which can exponentially amplify a small amount of “seeds” from various biopsies with high sensitivity and specificity, making it a useful tool to aid in the clinical diagnosis of neurodegenerative disorders (Saijo et al., 2019). RT-QuIC was first successful in the diagnosis of human prion disease and then applied to detect αSyn aggregates in the brain and CSF from dementia with Lewy body and PD patients (Fairfoul et al., 2016). In the latest study, a group of RT-QuIC reactions for 4R tauopathies has been developed, specifically PSP, CBD, and FTDP-17 MAPT (Saijo et al., 2020). This emerging template-dependent amplification technique would benefit the diagnosis of AD and PD far before the onset of clinical lesions, and investigate the underlying mechanism of the structural and functional diversities of distinct brain-derived species is another challenging topic.
When adding PFFs into cultured hippocampal neurons, they compromised synaptic miniature EPSC firing frequency, inhibited spines formation, along with decreased action potential firing rate (Wu et al., 2019), where another group observed paradoxical functional defects induced by αSyn PFFs (Froula et al., 2018). On the other hand, the neuronal hyperactivity promoted the secretion of αSyn into extracellular space in vivo (Bassil et al., 2021) as well as in vitro (Bero et al., 2011), thus facilitated the propagation of pathology in CNS. These results were confirmed by an ex vivo slice culture system recently (Guo et al., 2013). The activity-dependent regulation of protein aggregation and spreading also applies to tau and Aβ in AD animal models (Wu et al., 2016; Bassil et al., 2021). For example, in a microdialysis study, neuronal activity modulated the concentration of Aβ and tau in the extracellular interstitial fluid of mouse brains (Bero et al., 2011). By imaging the traffic of fluorescent tag labeled tau in axons within microfluidic chambers, enhancing neuronal activity increased the release of tau and facilitated tau transmission (Wang et al., 2017). Furthermore, a study that used a chemogenetic approach to modulate neuronal activity demonstrated that hyperactivity leads to increase Aβ load and was reversed by inhibiting neuronal activity in a transgenic animal in vivo (Yuan and Grutzendler, 2016). Even before the formation of extracellular plaques, neurons in the prefrontal cortex showed hyperactivity and contributed to the progression of pathology in vivo (Zott et al., 2019), and this phenomenon was confirmed by ex vivo live imaging of hippocampal slice cultures in the 5xFAD model as well (Wu et al., 2020).
When zoom-in into the donor cells, the formation and amplification of pathological seeds initialize the transmission process. Both anterograde and retrograde transport of these proteins were observed, however, the Aβ fibrils were shown to be transported ten times more efficiently than αSyn (Freundt et al., 2012; Wu et al., 2013; Brahic et al., 2016). When released outside of the neurons, both tau and αSyn fibrils can be taken up via macropinocytosis, a form of endocytosis mediated by heparan sulfate proteoglycans (HSPGs) on the cell surface (Holmes et al., 2013). In another study, a pool of 352 complementary DNAs was expressed in SH-SY5Y cells and screened for αSyn-biotin PFF–binding candidates. This large-scale screening approach identified one of the top candidates, lymphocyte-activation gene 3 (LAG3), a transmembrane protein internalized via receptor-mediated endocytosis (Mao et al., 2016). After taken up through the plasma membrane, the fibrils then traffic along the endo-lysosomal pathway, process, rupture, and release from the lysosome into the cytosol before recruiting endogenous monomeric counterparts into insoluble inclusions (Figure 2), see also (Guo and Lee, 2014; Oueslati et al., 2014; Karpowicz et al., 2017). Moreover, tunneling nanotube (TNT)-mediated cell-cell transfer of pathological tau and αSyn seeds was observed in vitro recently (Abounit et al., 2016; Tardivel et al., 2016). However, whether TNTs connecting neurons are present in vivo remains unclear.
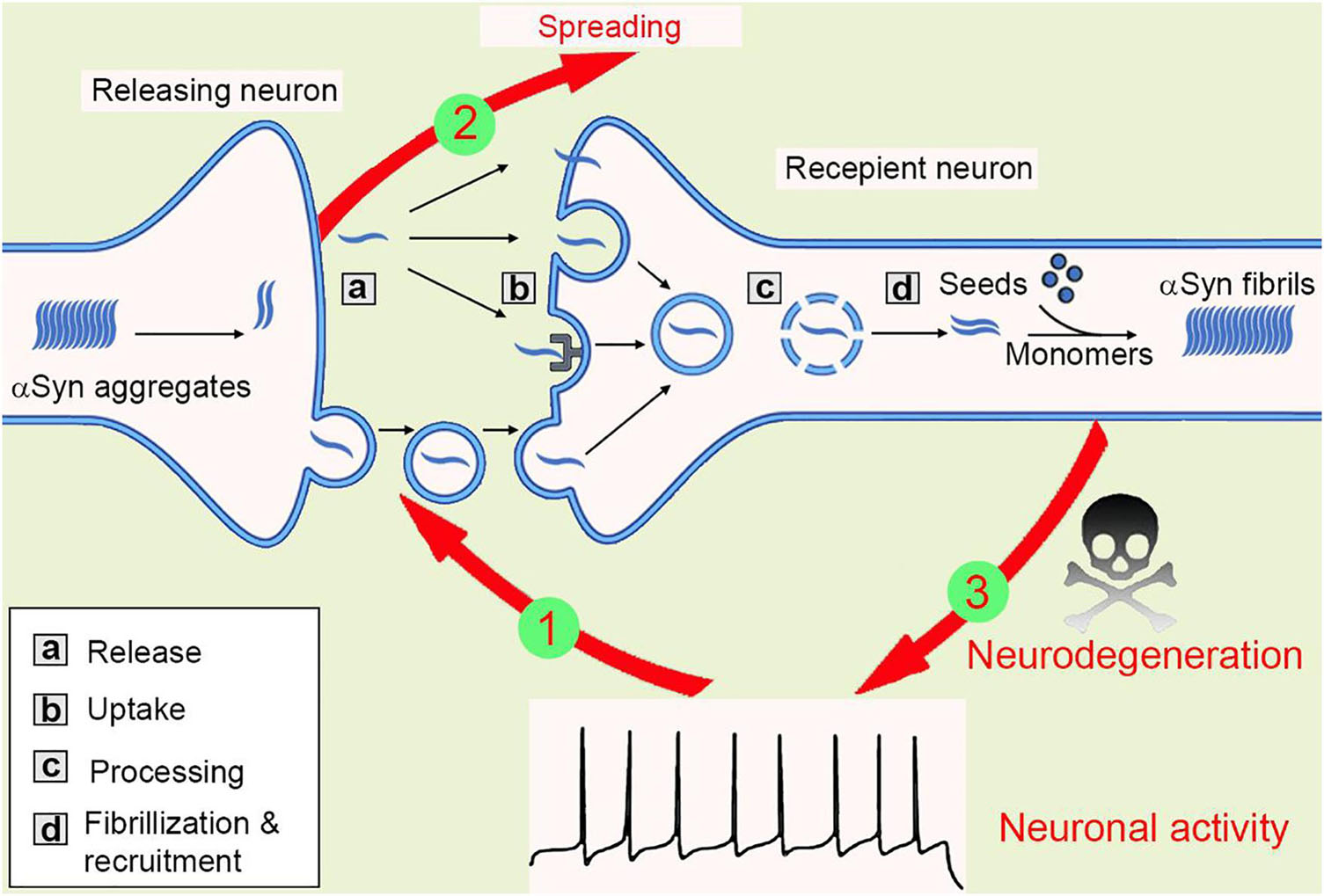
Figure 2. The spreading of αSyn aggregates between neurons. Mutations or neurotoxic environmental factors initialize the αSyn aggregation in the releasing neuron. The aggregates are released into extracellular via vesicle/exosome, and neuronal activity facilitates or inhibits this process (1). Then, the aggregates are taken up by macropinocytosis, LAG-3 receptor, or penetration into cell membrane directly. The aggregates transport inside the recipient cells, most of them will be degraded in lysosome, but some lysosomes will be ruptured and release aggregates, that are working as “seeds” to recruit endogenous αSyn monomers into new fibrils. Thus, the amyloidogenic aggregates spread between cells (2). The αSyn aggregates then compromise synaptic activity, inhibit spine formation, and finally result in neurodegeneration (3).
As for propagation and spreading of pathologies in vivo, the stereotypic progression of misfolded proteins in AD and PD follows a predictable pattern. Cross-sectional autopsy studies indicate that β-amyloid plaques first appear in the neocortex, followed by the allocortex and finally subcortical regions (Thal et al., 2002). Neurofibrillary tangles occur first in the locus coeruleus and transentorhinal area and then spread to the amygdala and interconnected neocortical brain regions (Braak and Del Tredici, 2011). Propagation of Lewy body/Lewy neurite in patients with PD is postulated to start in the brainstem and ascend toward neocortical regions with disease progression (Braak et al., 2003). The transmission of pathological tau and αSyn can occur bi-directionally within a network of interconnected populations, where the expression level of αSyn contributes to the selective vulnerability of hippocampal neuron subpopulations to fibrils-induced neurotoxicity (Luna et al., 2018).
Since the development of Tang’s single-cell RNA sequencing (scRNA-seq) method to understand the genomic transcriptome of mammalian cells in 2009 (Tang et al., 2009), this technique is booming as one of the most popular tools in the past decade. Previous scRNA-seq approaches from dissociated tissues use protease treatment at 30°C, which is known to alter the transcriptome. The brain is a complex mixture of neurons, glial and endothelial cells, making it hard to isolate the whole RNAs with traditional processes. Thus, with the development of the single nuclear RNA-seq (snRNA-seq) method, nuclei can be isolated at 4°C from tissue homogenates, which minimizes damage. Additionally, single nuclear transcriptomes can be obtained from postmortem human brain tissue stored at −80°C, making it possible to access the brain’s genetic “secrets” by RNA-seq via individual neurons. Here, we reviewed some of the recent progress of this emerging technology in the application of neurodegenerative diseases, and the latest spatial-temporal RNA-seq will also be discussed.
Poulin et al. (2016) summarized the quantitative analysis of markers in a single cell, enabling the classification of neural cells into categories (Please see Table 1 for more details and other recent literatures). With the understanding of molecular diversities of neurons via scRNA-seq, it’s getting closer to mapping the whole brain connectome at a single cell level. This will be an extremely important reference tool in the study of the nervous system and making unprecedented contributions to the field of neuroscience. Besides using it as the molecular classification tool, scRNA-seq has been largely applied in understanding mechanisms of diseases. By using microfluidic dynamic array-based scRNA-seq to simultaneously evaluate the expression of 96 genes in single neurons, Poulin et al. identified multiple novel subtypes of dopaminergic (DA) neurons which were localized in a distinct area of adult mouse brains. For example, Aldh1a1+ subtype DA neurons, located in the substantia nigra, are especially vulnerable in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD (Caudle et al., 2007).
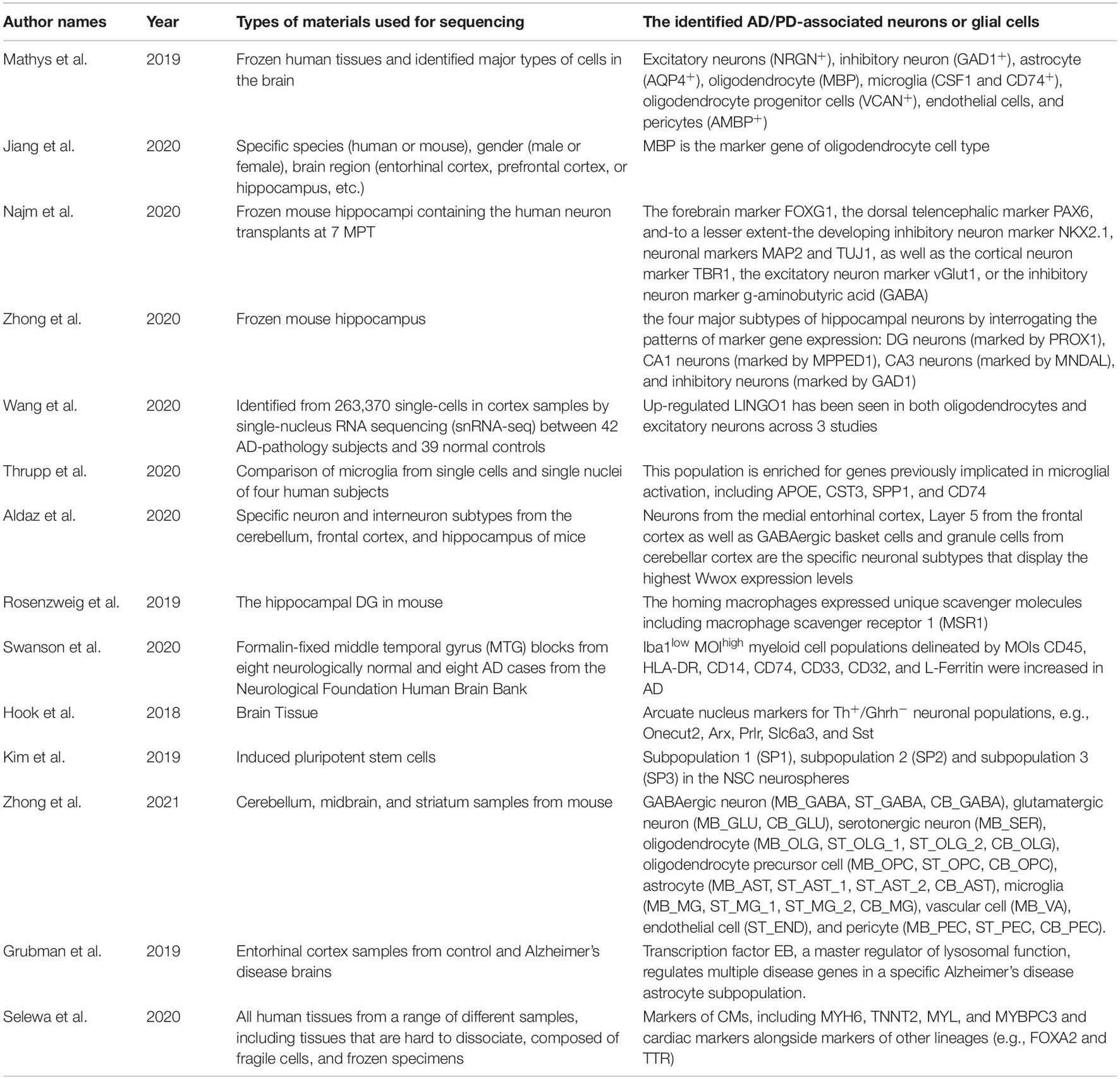
Table 1. Summary of recent studies using RNA-seq in understanding AD/PD.
The dysfunction of the striatum is associated with multiple neurodegenerative and cognitive diseases, including PD, hypersomnia, schizophrenia as well as addiction (De Deurwaerdere et al., 2017). According to the expression of D1 or D2 dopamine receptors, GABAnergic medium spiny neurons (MSNs) can be divided into D1 and D2 subtypes (Surmeier et al., 2007). By using SMART-seq2 of 1208 cells isolated from mouse striatum, the authors found that D1 and D2 MSNs can be further classified into subgroups. In addition, they also identified cell type-specific transcription and splicing factors, and these factors can change cell types by modulating transcription and gene expression profiles (Gokce et al., 2016). As a substantial neurotransmitter, dopamine plays an essential role in neurodegeneration and cognitive diseases (Caudle et al., 2007; Chen et al., 2008), thus, study the function of different types of dopaminergic neurons helps to understand the molecular basics and mechanisms of these diseases.
By analyzing single-cell transcriptomes of the GBA-N370S-PD iPSC-derived dopaminergic neurons, Lang et al. found a progression axis in response to endoplasmic reticulum (ER) stress. When analyzing differentially expressed genes (DEGs), the transcription inhibitor HDAC4 is upregulated during PD progression. In iPSC of PD patient-derived DA neurons, HDAC4 is mis-localized in the nuclear, inhibits gene expression during early progress of disease and results in defects of protein homeostasis in late-stage (Lang et al., 2019). In 2017, Ido Amit’s group discovered a new type of immune cells in the brain, disease-associated microglia (DAM), and proved that DAM is responsible to degrade dead cells and AD-related amyloid plaques. DAM is a conservative phagocytic cell in humans and mice, whose activation is dependent or independent on TREM2 (Keren-Shaul et al., 2017). The author emphasized that, only by performing scRNA-seq they can find these infrequent microglia, providing new strategies to treat AD and related disorders.
scRNA-seq has a high demand on the cell suspension viability and numbers prepared from organs or solid tissues, which means many frozen precious clinical samples (brain tissue, tumor tissue) is not applicable for this method. Thus, the snRNA-seq is another option that, to some extent, overcame this problem. As for brain tissues, scRNA-seq cannot fully analyze all types of neurons as some neurons are more easily to be affected by the dissociation process. Although the tolerance of non-neuronal cells from the neocortex of human adults is better than neurons during dissociation, more non-neuronal cells are left in single nuclear suspension. As for the neocortex of mouse brains, the proportions of layer-5 parvalbumin-positive neurons and glutaminergic neurons are lower than expected. In contrast, nuclear is more resistant to mechanical treatment and can be dissociated from frozen tissues. Single nuclear has been proofed to cover enough gene expression messages to define cell types of humans and the hippocampus of the mouse.
Previous research reported that single nuclear was isolated from frozen S1 cortex, followed with sorting with NeuN antigen, and then RNA sequenced with Fluidigm C1 system. Gene numbers and subtypes are comparable between nuclear and cells, with an average of 5,619 genes in S1 nuclear and an average of 4,797 genes in S1 cells, where nuclear has higher proportions of intron reads (Lake et al., 2018). Grubman et al. (2019) performed snRNA-seq of entorhinal cortex from deceased patients with Alzheimer’s disease and age-matched healthy controls and got 13,214 nuclear, with a median of 646 genes per cell. With AD autopsy cases, Mathys et al. (2019) performed snRNA-seq of frozen human tissues and identified major types of cells in the brain: excitatory neurons (NRGN+), inhibitory neuron (GAD1+), astrocyte (AQP4+), oligodendrocyte (MBP), microglia (CSF1+ and CD74+), oligodendrocyte progenitor cells (VCAN+), endothelial cells and pericytes (AMBP+).
The scRNA-seq technologies have enabled the use of single-cell transcriptional profiling to explore cellular heterogeneity. However, a common feature of scRNA-seq studies is that they do not resolve the spatial patterns or positional information, making it difficult to place cell-cell interactions within a broader tissue context. Harnessing in situ sequencing and quantitative analysis, the spatial transcriptomics (ST) refined these results and created a spatial subcellular map of 3D transcriptome profiles (Stahl et al., 2016; Salmen et al., 2018). In 2019, Asp et al. collected human embryonic heart tissue at different development stages (4.5–5, 6.5, and 9 post-conception weeks) and systematically analyzed subcellular resolution of regional markers and cell atlas based on ST and scRNA-seq (Asp et al., 2019). For example, in the AD mouse model, Chen et al. investigated the ST of 100 mm diameter around amyloid plaques and discovered early response genes that are enriched in myelin and oligodendrocytes, where plaque-induced genes (PIGs) involving the complement system, oxidative stress, lysosomes, and inflammation are prominent in the later phase of the disease (Chen et al., 2020). To improve spatial resolution, Slide-seq (Rodriques et al., 2019) and HDST (Vickovic et al., 2019) were developed, in which they used an alternative strategy with condensed beads-based technology, making it feasible to profile genome-wide molecular events at the single-cell level.
Overall, snRNA-seq is more universally applicable than scRNA-seq, it does not limit to fresh tissue, but also frozen ones. Additionally, a single nucleus is easier to prepare than a single cell because it requires less enzymatic digestion, thereby reducing pseudo-cell subtypes induced by mechanical stress. In terms of data analysis, snRNA-seq contains information about introns and intergenic regions, making the resolution of cell subtypes higher. SnRNA-seq cannot replace scRNA-seq. The choice of RNA-seq method depends largely on experimental conditions, such as sample, cell suspension, enzyme sensitivity and other factors. The combination uses of both scRNA-seq and snRNA-seq would be the ideal strategy to get full access to genome transcriptions.
The accumulation of αSyn aggregates in Parkinson’s and related diseases is unstoppable, and preclinical symptoms can occur about 10–20 years before the lesions of neuronal functions. Though people have developed multiple strategies to predict, investigate and slow down the process, the progression is far behind expected. Biochemistry, molecular biology, autopsy, electron microscopy, and live imaging techniques enable us to learn the underlying mechanism of protein aggregation and spreading between neurons, however, all these traditional assays are biased focused on one or two genes at a time and neglect the complexity and integrity of the biological system. In the last decade, the booming of scRNA-seq and snRNA-seq techniques lead us into a “big data” era that tens of thousands of genes and signal pathways have been uncovered within the same tissues. These big data also bring us too much “noise” and it should be more careful to choose what kind of pathways or genes that fit your hypothesis in the future. And how to use the published database to explore useful information is another recycling of resources, which, of course, will benefit and guide the preclinical research.
JJ and YL wrote and edited the manuscript. QW designed, conceived, wrote, and edited the manuscript. All authors approved of the final manuscript.
This work was supported by the Fundamental Research Funds for the Central Universities (22120210192 and 22120210368), Shanghai Pujiang Program (21PJ1412100), and National Natural Science Foundation of China (82101486).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abounit, S., Bousset, L., Loria, F., Zhu, S., de Chaumont, F., Pieri, L., et al. (2016). Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J. 35, 2120–2138. doi: 10.15252/embj.201593411
Asp, M., Giacomello, S., Larsson, L., Wu, C., Furth, D., Qian, X., et al. (2019). Wide gene expression and cell atlas of the developing human heart. Cell 179, 1647.e19–1660.e19.
Bachhuber, T., Katzmarski, N., McCarter, J. F., Loreth, D., Tahirovic, S., Kamp, F., et al. (2015). Inhibition of amyloid-beta plaque formation by alpha-synuclein. Nat. Med. 21, 802–807.
Baiano, C., Barone, P., Trojano, L., and Santangelo, G. (2020). Prevalence and clinical aspects of mild cognitive impairment in Parkinson’s disease: a meta-analysis. Mov. Disord. 35, 45–54. doi: 10.1002/mds.27902
Bassil, F., Meymand, E. S., Brown, H. J., Xu, H., Cox, T. O., Pattabhiraman, S., et al. (2021). alpha-Synuclein modulates tau spreading in mouse brains. J. Exp. Med. 218:e20192193. doi: 10.1084/jem.20192193
Bero, A. W., Yan, P., Roh, J. H., Cirrito, J. R., Stewart, F. R., Raichle, M. E., et al. (2011). Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat. Neurosci. 14, 750–756.
Blauwendraat, C., Nalls, M. A., and Singleton, A. B. (2020). The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178.
Braak, H., and Del Tredici, K. (2011). The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 121, 171–181. doi: 10.1007/s00401-010-0789-4
Braak, H., Del Tredici, K., Rub, U., de Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/s0197-4580(02)00065-9
Brahic, M., Bousset, L., Bieri, G., Melki, R., and Gitler, A. D. (2016). Axonal transport and secretion of fibrillar forms of alpha-synuclein, Abeta42 peptide and HTTExon 1. Acta Neuropathol. 131, 539–548. doi: 10.1007/s00401-016-1538-0
Caudle, W. M., Richardson, J. R., Wang, M. Z., Taylor, T. N., Guillot, T. S., McCormack, A. L., et al. (2007). Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J. Neurosci. 27, 8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007
Chen, A. K., Lin, R. Y., Hsieh, E. Z., Tu, P. H., Chen, R. P., Liao, T. Y., et al. (2010). Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J. Am. Chem. Soc. 132, 1186–1187. doi: 10.1021/ja9066207
Chen, L., Ding, Y., Cagniard, B., Van Laar, A. D., Mortimer, A., Chi, W., et al. (2008). Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J. Neurosci. 28, 425–433. doi: 10.1523/JNEUROSCI.3602-07.2008
Chen, W. T., Lu, A., Craessaerts, K., Pavie, B., Sala Frigerio, C., Corthout, N., et al. (2020). Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell 182, 976.e19–991.e19. doi: 10.1016/j.cell.2020.06.038
Cleary, J. P., Walsh, D. M., Hofmeister, J. J., Shankar, G. M., Kuskowski, M. A., Selkoe, D. J., et al. (2005). Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 8, 79–84. doi: 10.1038/nn1372
De Deurwaerdere, P., Di Giovanni, G., and Millan, M. J. (2017). Expanding the repertoire of L-DOPA’s actions: a comprehensive review of its functional neurochemistry. Prog. Neurobiol. 151, 57–100. doi: 10.1016/j.pneurobio.2016.07.002
Fairfoul, G., McGuire, L. I., Pal, S., Ironside, J. W., Neumann, J., Christie, S., et al. (2016). Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 3, 812–818. doi: 10.1002/acn3.338
Forno, L. S., DeLanney, L. E., Irwin, I., and Langston, J. W. (1996). Electron microscopy of Lewy bodies in the amygdala-parahippocampal region. Comparison with inclusion bodies in the MPTP-treated squirrel monkey. Adv. Neurol. 69, 217–228.
Freundt, E. C., Maynard, N., Clancy, E. K., Roy, S., Bousset, L., Sourigues, Y., et al. (2012). Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 72, 517–524. doi: 10.1002/ana.23747
Froula, J. M., Henderson, B. W., Gonzalez, J. C., Vaden, J. H., McLean, J. W., Wu, Y., et al. (2018). alpha-Synuclein fibril-induced paradoxical structural and functional defects in hippocampal neurons. Acta Neuropathol. Commun. 6:35. doi: 10.1186/s40478-018-0537-x
Goedert, M. (2015). NEURODEGENERATION. Alzheimer’s and Parkinson’s diseases: the prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 349:1255555. doi: 10.1126/science.1255555
Gokce, O., Stanley, G. M., Treutlein, B., Neff, N. F., Camp, J. G., Malenka, R. C., et al. (2016). Cellular taxonomy of the mouse striatum as revealed by single-cell RNA-Seq. Cell Rep. 16, 1126–1137. doi: 10.1016/j.celrep.2016.06.059
Grubman, A., Chew, G., Ouyang, J. F., Sun, G., Choo, X. Y., McLean, C., et al. (2019). A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 22, 2087–2097. doi: 10.1038/s41593-019-0539-4
Grutzendler, J., Helmin, K., Tsai, J., and Gan, W. B. (2007). Various dendritic abnormalities are associated with fibrillar amyloid deposits in Alzheimer’s disease. Ann. N.Y. Acad. Sci. 1097, 30–39. doi: 10.1196/annals.1379.003
Guo, J. L., Covell, D. J., Daniels, J. P., Iba, M., Stieber, A., Zhang, B., et al. (2013). Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154, 103–117. doi: 10.1016/j.cell.2013.05.057
Guo, J. L., and Lee, V. M. (2014). Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 20, 130–138. doi: 10.1038/nm.3457
Holmes, B. B., DeVos, S. L., Kfoury, N., Li, M., Jacks, R., Yanamandra, K., et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U.S.A. 110, E3138–E3147. doi: 10.1073/pnas.1301440110
Iba, M., Guo, J. L., McBride, J. D., Zhang, B., Trojanowski, J. Q., and Lee, V. M. (2013). Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 33, 1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013
Karpowicz, R. J. Jr., Haney, C. M., Mihaila, T. S., Sandler, R. M., Petersson, E. J., and Lee, V. M. (2017). Selective imaging of internalized proteopathic alpha-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J. Biol. Chem. 292, 13482–13497. doi: 10.1074/jbc.M117.780296
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., vir-Szternfeld, R. D., Ulland, T. K., et al. (2017). Type associated with restricting development of Alzheimer’s disease. Cell 169, 1276.e17–1290.e17.
Lake, B. B., Chen, S., Sos, B. C., Fan, J., Kaeser, G. E., Yung, Y. C., et al. (2018). Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 36, 70–80.
Lang, C., Campbell, K. R., Ryan, B. J., Carling, P., Attar, M., Vowles, J., et al. (2019). Single-cell sequencing of iPSC-dopamine neurons reconstructs disease progression and identifies hdac4 as a regulator of parkinson cell phenotypes. Cell Stem Cell 24, 93.e6–106.e6. doi: 10.1016/j.stem.2018.10.023
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O’Brien, P., Trojanowski, J. Q., et al. (2012). Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Luna, E., Decker, S. C., Riddle, D. M., Caputo, A., Zhang, B., Cole, T., et al. (2018). Differential alpha-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol. 135, 855–875. doi: 10.1007/s00401-018-1829-8
Mao, X., Ou, M. T., Karuppagounder, S. S., Kam, T. I., Yin, X., Xiong, Y., et al. (2016). Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353:aah3374.
Mathys, H., Davila-Velderrain, J., Peng, Z., Gao, F., Mohammadi, S., Young, J. Z., et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337.
Mattsson, N., Tosun, D., Insel, P. S., Simonson, A., Jack, C. R. Jr., Beckett, L. A., et al. (2014). Association of brain amyloid-beta with cerebral perfusion and structure in Alzheimer’s disease and mild cognitive impairment. Brain 137, 1550–1561. doi: 10.1093/brain/awu043
Minjarez, B., Calderon-Gonzalez, K. G., Rustarazo, M. L., Herrera-Aguirre, M. E., Labra-Barrios, M. L., Rincon-Limas, D. E., et al. (2016). Identification of proteins that are differentially expressed in brains with Alzheimer’s disease using iTRAQ labeling and tandem mass spectrometry. J. Proteomics 139, 103–121. doi: 10.1016/j.jprot.2016.03.022
Munch, C., O’Brien, J., and Bertolotti, A. (2011). Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. U.S.A. 108, 3548–3553. doi: 10.1073/pnas.1017275108
Oueslati, A., Ximerakis, M., and Vekrellis, K. (2014). Protein transmission, seeding and degradation: key steps for alpha-synuclein prion-like propagation. Exp. Neurobiol. 23, 324–336. doi: 10.5607/en.2014.23.4.324
Patterson, J. R., Polinski, N. K., Duffy, M. F., Kemp, C. J., Luk, K. C., Volpicelli-Daley, L. A., et al. (2019). Generation of alpha-synuclein preformed fibrils from monomers and use in vivo. J. Vis. Exp. 2019:148.
Peng, C., Gathagan, R. J., Covell, D. J., Medellin, C., Stieber, A., Robinson, J. L., et al. (2018). Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 557, 558–563. doi: 10.1038/s41586-018-0104-4
Pflanzner, T., Janko, M. C., Andre-Dohmen, B., Reuss, S., Weggen, S., Roebroek, A. J., et al. (2011). LRP1 mediates bidirectional transcytosis of amyloid-beta across the blood-brain barrier. Neurobiol. Aging 32, 2323.e1–2323.e11. doi: 10.1016/j.neurobiolaging.2010.05.025
Pooler, A. M., Polydoro, M., Maury, E. A., Nicholls, S. B., Reddy, S. M., Wegmann, S., et al. (2015). Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer’s disease. Acta Neuropathol. Commun. 3:14. doi: 10.1186/s40478-015-0199-x
Poulin, J. F., Tasic, B., Hjerling-Leffler, J., Trimarchi, J. M., and Awatramani, R. (2016). Disentangling neural cell diversity using single-cell transcriptomics. Nat. Neurosci. 19, 1131–1141. doi: 10.1038/nn.4366
Rabinovici, G. D., Karlawish, J., Knopman, D., Snyder, H. M., Sperling, R., and Carrillo, M. C. (2016). Testing and disclosures related to amyloid imaging and Alzheimer’s disease: common questions and fact sheet summary. Alzheimers Dement. 12, 510–515. doi: 10.1016/j.jalz.2016.03.002
Ren, P. H., Lauckner, J. E., Kachirskaia, I., Heuser, J. E., Melki, R., and Kopito, R. R. (2009). Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 11, 219–225. doi: 10.1038/ncb1830
Rodriques, S. G., Stickels, R. R., Goeva, A., Martin, C. A., Murray, E., Vanderburg, C. R., et al. (2019). Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467. doi: 10.1126/science.aaw1219
Saijo, E., Groveman, B. R., Kraus, A., Metrick, M., Orru, C. D., Hughson, A. G., et al. (2019). Ultrasensitive RT-QuIC seed amplification assays for disease-associated tau, alpha-synuclein, and prion aggregates. Methods Mol. Biol. 1873, 19–37. doi: 10.1007/978-1-4939-8820-4_2
Saijo, E., Metrick, M. A. II, Koga, S., Parchi, P., Litvan, I., Spina, S., et al. (2020). 4-Repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol. 139, 63–77. doi: 10.1007/s00401-019-02080-2
Salmen, F., Stahl, P. L., Mollbrink, A., Navarro, J. F., Vickovic, S., Frisen, J., et al. (2018). Barcoded solid-phase RNA capture for Spatial Transcriptomics profiling in mammalian tissue sections. Nat. Protoc. 13, 2501–2534. doi: 10.1038/s41596-018-0045-2
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608.
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Sharma, S., Verma, S., Kapoor, M., Saini, A., and Nehru, B. (2016). Alzheimer’s disease like pathology induced six weeks after aggregated amyloid-beta injection in rats: increased oxidative stress and impaired long-term memory with anxiety-like behavior. Neurol. Res. 38, 838–850. doi: 10.1080/01616412.2016.1209337
Shi, Q., Chowdhury, S., Ma, R., Le, K. X., Hong, S., Caldarone, B. J., et al. (2017). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 9:eaaf6295. doi: 10.1126/scitranslmed.aaf6295
Shin, M. K., Kim, H. G., and Kim, K. L. (2015). A novel brain-derived neurotrophic factor-modulating peptide attenuates Abeta1-42-induced neurotoxicity in vitro. Neurosci. Lett. 595, 63–68. doi: 10.1016/j.neulet.2015.03.070
Stahl, P. L., Salmen, F., Vickovic, S., Lundmark, A., Navarro, J. F., Magnusson, J., et al. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82. doi: 10.1126/science.aaf2403
Stohr, J., Watts, J. C., Mensinger, Z. L., Oehler, A., Grillo, S. K., DeArmond, S. J., et al. (2012). Purified and synthetic Alzheimer’s amyloid beta (Abeta) prions. Proc. Natl. Acad. Sci. U.S.A. 109, 11025–11030. doi: 10.1073/pnas.1206555109
Surguchev, A., and Surguchov, A. (2010). Conformational diseases: looking into the eyes. Brain Res. Bull. 81, 12–24. doi: 10.1016/j.brainresbull.2009.09.015
Surmeier, D. J., Ding, J., Day, M., Wang, Z., and Shen, W. (2007). D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 30, 228–235.
Tang, F., Barbacioru, C., Wang, Y., Nordman, E., Lee, C., Xu, N., et al. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382. doi: 10.1038/nmeth.1315
Tardivel, M., Begard, S., Bousset, L., Dujardin, S., Coens, A., Melki, R., et al. (2016). Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 4:117. doi: 10.1186/s40478-016-0386-4
Thal, D. R., Rub, U., Orantes, M., and Braak, H. (2002). Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. doi: 10.1212/wnl.58.12.1791
Tokuchi, R., Hishikawa, N., Sato, K., Hatanaka, N., Fukui, Y., Takemoto, M., et al. (2016). Differences between the behavioral and psychological symptoms of Alzheimer’s disease and Parkinson’s disease. J. Neurol. Sci. 369, 278–282.
Tsai, J., Grutzendler, J., Duff, K., and Gan, W. B. (2004). Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat. Neurosci. 7, 1181–1183. doi: 10.1038/nn1335
Tycko, R. (2016). Alzheimer’s disease: structure of aggregates revealed. Nature 537, 492–493. doi: 10.1038/nature19470
Vickovic, S., Eraslan, G., Salmen, F., Klughammer, J., Stenbeck, L., Schapiro, D., et al. (2019). High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987–990. doi: 10.1038/s41592-019-0548-y
Volpicelli-Daley, L. A., Luk, K. C., Patel, T. P., Tanik, S. A., Riddle, D. M., Stieber, A., et al. (2011). Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. doi: 10.1016/j.neuron.2011.08.033
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Wang, Y., Balaji, V., Kaniyappan, S., Kruger, L., Irsen, S., Tepper, K., et al. (2017). The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 12:5. doi: 10.1186/s13024-016-0143-y
Wu, J. W., Herman, M., Liu, L., Simoes, S., Acker, C. M., Figueroa, H., et al. (2013). Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870. doi: 10.1074/jbc.M112.394528
Wu, J. W., Hussaini, S. A., Bastille, I. M., Rodriguez, G. A., Mrejeru, A., Rilett, K., et al. (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19, 1085–1092. doi: 10.1038/nn.4328
Wu, Q., Shaikh, M. A., Meymand, E. S., Zhang, B., Luk, K. C., Trojanowski, J. Q., et al. (2020). Neuronal activity modulates alpha-synuclein aggregation and spreading in organotypic brain slice cultures and in vivo. Acta Neuropathol. 140, 831–849. doi: 10.1007/s00401-020-02227-6
Wu, Q., Takano, H., Riddle, D. M., Trojanowski, J. Q., Coulter, D. A., and Lee, V. M. (2019). alpha-Synuclein (alphaSyn) preformed fibrils induce endogenous alphaSyn aggregation, compromise synaptic activity and enhance synapse loss in cultured excitatory hippocampal neurons. J. Neurosci. 39, 5080–5094. doi: 10.1523/JNEUROSCI.0060-19.2019
Xiang, Y., Bu, X. L., Liu, Y. H., Zhu, C., Shen, L. L., Jiao, S. S., et al. (2015). Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 130, 487–499. doi: 10.1007/s00401-015-1477-1
Yuan, P., and Grutzendler, J. (2016). Attenuation of beta-amyloid deposition and neurotoxicity by chemogenetic modulation of neural activity. J. Neurosci. 36, 632–641. doi: 10.1523/JNEUROSCI.2531-15.2016
Zenaro, E., Piacentino, G., and Constantin, G. (2017). The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 107, 41–56.
Keywords: Alzheimer’s disease, Parkinson’s disease, Aβ cascade hypothesis, α-synuclein aggregation and spreading, transcriptomics of nervous system
Citation: Jiang J, Liu Y and Wu Q (2021) Revisit the Cellular Transmission and Emerging Techniques in Understanding the Mechanisms of Proteinopathies. Front. Neurosci. 15:781722. doi: 10.3389/fnins.2021.781722
Received: 23 September 2021; Accepted: 29 October 2021;
Published: 18 November 2021.
Edited by:
Nan Zhang, Houston Methodist Research Institute, United StatesReviewed by:
Zhentao Zhang, Renmin Hospital of Wuhan University, ChinaCopyright © 2021 Jiang, Liu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qihui Wu, cWlodWl3dUB0b25namkuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.