 Giuseppina Amadoro1,2*
Giuseppina Amadoro1,2* Valentina Latina2
Valentina Latina2 Bijorn Omar Balzamino3
Bijorn Omar Balzamino3 Rosanna Squitti4Monica Varano3
Rosanna Squitti4Monica Varano3 Pietro Calissano1
Pietro Calissano1 Alessandra Micera3*
Alessandra Micera3*- 1Institute of Translational Pharmacology (IFT)-CNR, Rome, Italy
- 2European Brain Research Institute, Rome, Italy
- 3Research Laboratories in Ophthalmology, IRCCS-Fondazione Bietti, Rome, Italy
- 4Molecular Markers Laboratory, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, Italy
Alzheimer’s disease (AD) is an age-associated neurodegenerative disease which is the most common cause of dementia among the elderly. Imbalance in nerve growth factor (NGF) signaling, metabolism, and/or defect in NGF transport to the basal forebrain cholinergic neurons occurs in patients affected with AD. According to the cholinergic hypothesis, an early and progressive synaptic and neuronal loss in a vulnerable population of basal forebrain involved in memory and learning processes leads to degeneration of cortical and hippocampal projections followed by cognitive impairment with accumulation of misfolded/aggregated Aβ and tau protein. The neuroprotective and regenerative effects of NGF on cholinergic neurons have been largely demonstrated, both in animal models of AD and in living patients. However, the development of this neurotrophin as a disease-modifying therapy in humans is challenged by both delivery limitations (inability to cross the blood–brain barrier (BBB), poor pharmacokinetic profile) and unwanted side effects (pain and weight loss). Age-related macular degeneration (AMD) is a retinal disease which represents the major cause of blindness in developed countries and shares several clinical and pathological features with AD, including alterations in NGF transduction pathways. Interestingly, nerve fiber layer thinning, degeneration of retinal ganglion cells and changes of vascular parameters, aggregation of Aβ and tau protein, and apoptosis also occur in the retina of both AD and AMD. A protective effect of ocular administration of NGF on both photoreceptor and retinal ganglion cell degeneration has been recently described. Besides, the current knowledge about the detection of essential trace metals associated with AD and AMD and their changes depending on the severity of diseases, either systemic or locally detected, further pave the way for a promising diagnostic approach. This review is aimed at describing the employment of NGF as a common therapeutic approach to AMD and AD and the diagnostic power of detection of essential trace metals associated with both diseases. The multiple approaches employed to allow a sustained release/targeting of NGF to the brain and its neurosensorial ocular extensions will be also discussed, highlighting innovative technologies and future translational prospects.
Introduction
As an integral part/extension of the central nervous system (CNS), ocular structures display several cytological, anatomical, and developmental similarities with the brain so that the retina and cerebral areas, receiving directly (visual system) and indirectly (amygdala, hippocampus, and other hypothalamic nuclei) its inputs, constitute the so-called “eye–brain connection” (Erskine and Herrera, 2014; Tirassa et al., 2018). Because of the biological connection between the brain and eyes, analogous and corresponding pathological processes can lead to dysfunction of both of them. Oxidative injury, chronic activation of inflammatory pathway(s), mitochondrial impairment, vascular changes, neurotrophin(s) imbalance, protein quality control deficits and proteostasis, metal ion dyshomeostasis, genetics causes, or a combination of them are proposed to be involved in age-related disorders featured by brain and retinal degeneration, including Alzheimer’s disease (AD) and age-related macular degeneration (AMD; Kaarniranta et al., 2011; Masuzzo et al., 2016; Xu et al., 2018; Acevedo et al., 2019; Micera et al., 2019; Mufson et al., 2019; Wang and Mao, 2021). AD is a complex and multifactorial neurodegenerative disorder representing the main cause of dementia in the elderly (Walsh and Selkoe, 2004; Querfurth and LaFerla, 2010). The extracellular neuritic plaques, composed of β-amyloid (Aβ), and the intracellular neurofibrillary tangles (NFTs), composed of hyperphosphorylated tau protein, are the two key neuropathological hallmarks associated with this disease. Both Aβ plaques and NFTs impair synaptic plasticity and neural circuit networks causing the clinical symptoms of progressive memory loss (Spires-Jones and Hyman, 2014; Masters et al., 2015). AMD is a late-onset retinal neurodegenerative disorder representing the major cause of blindness in Western countries (Fine et al., 2000; Gehrs et al., 2006; Jager et al., 2008; Çerman et al., 2015). Accumulation of insoluble extracellular aggregates called drusen in the retina, degeneration of retinal pigment epithelium (RPE) cells, and choroidal neovascularization (CNV) manifest in AMD-suffering subjects leading to irreversible loss of vision (Ambati and Fowler, 2012; van Lookeren Campagne et al., 2014).
Here, we discuss how the understanding of the common pathophysiological molecular mechanisms linking these two overlapping disorders will help in the development of novel diagnostic/prognostic biomarkers and effective therapeutic avenues. Furthermore, the power of the nerve growth factor (NGF) in maintaining the functional phenotype(s) of different neuronal populations and retinal cells along with the recent optimization of innovative strategies allowing both local and systemic delivery of this neurotrophin jointly prospects its utilization as a promising, neuroprotective option for the treatment of both AD and AMD.
Alzheimer’s Disease and AMD Pathophysiology: Similarities and Differences
Epidemiological, genetic, pathological, and clinical lines of evidence have documented a strong relationship between AD and AMD (Wang and Mao, 2021) which have prompted scientists to speculate that AMD could be a form of “Alzheimer’s disease in the eye” (Kaarniranta et al., 2011; Ohno-Matsui, 2011) (Figure 1).
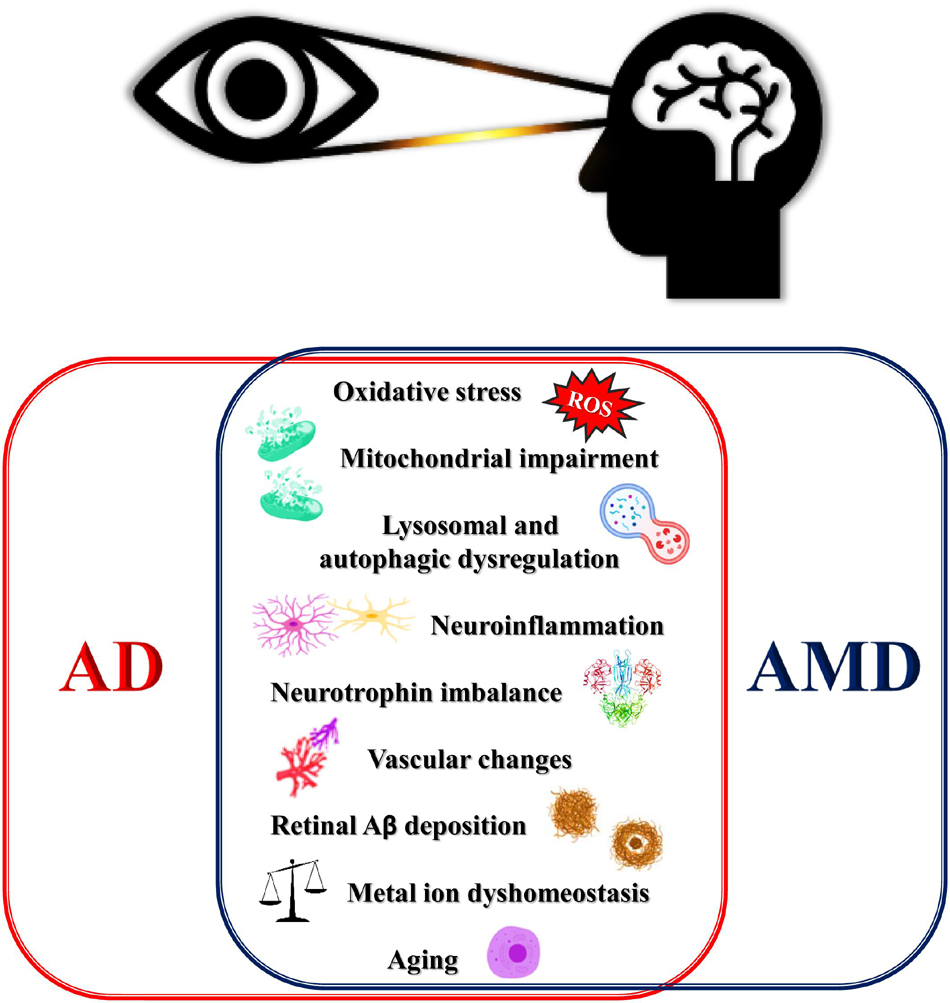
Figure 1. Schematic for overlapping degenerative mechanisms shared by AMD and AD. These pathways have been associated with brain and retina degeneration, cognitive, deficits and loss of vision. AD, Alzheimer’s disease; AMD, age-related macular degeneration; ROS, reactive oxygen species.
To better understand the burden on public health of these two age-related multifactorial neurodegenerative disorders, it is worth pointing out that the prevalence of AD is estimated to be 46 million of cases worldwide (Brookmeyer et al., 2007; Reitz and Mayeux, 2014), with the number of AD patients expected to triple in 2050 (Baumgart et al., 2015), reaching 131 million of cases (Prince et al., 2013). In a similar way, 15% (age 65–74), 25% (age 75–84), and 30% (age 85% or more) of subjects are affected with AMD (Klein et al., 2004; Wong et al., 2014). Indeed, investigations have demonstrated that, in addition to a strong genetic component, aging is the principal common risk factor for both AD and AMD (Coleman et al., 2008; Mayeux and Stern, 2012; Rudnicka et al., 2012). Genetic heritable components play a role in the development of AD, with rare autosomal mutations in the three genes APP, PSEN1, and PSEN2 and the APOE epsilon-4 allele associated with familial early-onset and sporadic late-onset AD forms, respectively (Mayeux and Stern, 2012). On the other hand, CFH (encoding for a complement inhibitor factor) and ARMS2/HTRA1 (encoding for a serine protease) are linked with AMD (Logue et al., 2014; Woo et al., 2015; Tsiloulis et al., 2016). Interestingly, genome-wide association studies (GWAS) have shown that single nucleotide polymorphisms (SNPs) in ABCA7 (encoding for ATP-binding cassette transporter subfamily A member 7), HGS (encoding for a member of the clathrin-mediated endocytosis signaling pathway), and PILRA/ZCW1P1 genes (encoding for the paired-immunoglobulin-like type 2 receptor which binds with herpes simplex virus-1 and for a zinc finger protein functioning as a histone modification, respectively) are also common in the pathogenesis of both AMD and AD (Ding et al., 2009; Kaarniranta et al., 2011). Besides, hypercholesterolemia, hypertension, atherosclerosis, obesity, diabetes, and environmental risk factors also strongly contribute to the development of both AD and AMD in mid-life (Kivipelto et al., 2001, 2002, 2005; Newman et al., 2005; Polidori et al., 2001; Seddon et al., 2005; Age-Related Eye Disease Study Research Group (AREDSRG), 2000; Kaarniranta et al., 2011; Ohno-Matsui, 2011). Relevantly, patients suffering AMD exhibit an increased risk of developing AD as compared with people without AMD (Frost et al., 2016; Wen et al., 2021), even though a direct association between these two disorders has not been confirmed (Williams et al., 2014). Besides, compelling studies have demonstrated that vision abnormalities featured by degeneration of the visual cortex and/or RGC loss or AMD-like retinal degeneration are prominent in AD patients and manifest even before clinical signs of cognitive decline (Ashok et al., 2020).
The phenotypic parallelism between AD and AMD is strongly highlighted by experimental evidence that the beta-amyloid (Aβ) peptide – the main biochemical component of extracellular senile plaques featuring the AD brain – is also found in ocular drusen deposits characterizing AMD (Anderson et al., 2004). In particular, these latter inclusions generally localized in the macula, the central region of the retina, are focal depositions of acellular debris accumulating between the basal lamina of postmitotic neuroepithelial cells called RPE – which take care of neural cells, rods, and cones – and the inner collagenous layer of the Bruch’s membrane (Usui et al., 2018; Blasiak, 2020). The majority of patients manifest a dry form of AMD which is characterized by the presence of amorphic drusens classified as “hard” and “soft” drusen. “Hard” drusen is focal and thickening of the basement membrane of RPE, whereas “soft” drusen is hardening confined to the separation of the basement membrane from Bruch’s membrane at the inner collagenous zone. On the other hand, pseudodrusen, which is associated with clinical progression to more severe stages of AMD, is a deposit located above the basement membrane of RPE (Miller, 2013). Advanced AMD manifests with progressive RPE deterioration and develops into the exudative AMD (eAMD) with CNV or non-exudative AMD (neAMD) with geographic atrophy (GA) in which drusen coalesces and causes damage to RPE and photoreceptors (Jindal, 2015; Hernandez-Zimbron et al., 2018). Interestingly, the amyloid deposits possessing a central core with non-radiating fibrils have been also detected in the inner layers of the retina of AD patients, in contrast to classical/neuritic plaques found in their brain (Koronyo-Hamaoui et al., 2011). Relevantly, just as in AD, the role of these deposits is controversial in AMD and other types of macular degeneration since it is still unclear whether they are causing the RPE dysfunction or are a mere consequence of the impairment of the RPE function (Biscetti et al., 2017). In addition to Aβ, several protein and lipid constituents of drusens – including clusterin, vitronectin, amyloid P, esterified cholesterol and phosphatidylcholine, apolipoprotein E, and inflammatory mediators, such as acute phase reactants and complement components (C5, C5b9, and C3 fragments) – are also present in insoluble cerebral aggregates of AD (Mullins et al., 2000; Crabb et al., 2002; Luibl et al., 2006; Isas et al., 2010; Wang et al., 2010). Likewise, metal elements such as Zn2+, Fe3+, Cu2+, ubiquitin, and lipofuscin account for the biochemical composition of lesions from both AD and AMD (Atwood et al., 2002; Ryhänen et al., 2009; Ohno-Matsui, 2011). Although rodents have neither functional macula nor sharp vision in the retina, an important demonstration of commonalities occurring between AMD and AD is represented by the human APOE4-expressing transgenic mice, an animal model carrying the APOE4 allelic variant which is one of the major risk factors for AD onset/progression. Interestingly, old animals of this strain, when fed a high-fat diet, exhibit an ocular phenotype which resembles several crucial features of AMD. Besides, their retinal as well as memory dysfunction is strongly attenuated by the delivery of antibody Aβ-neutralizing (Ding et al., 2008, 2011). Additional parallel pathophysiological events occurring in both AMD and AD pathogenesis encompass increased oxidative stress and mitochondrial and lysosomal dysfunctions. Owing to the relative enrichment of unsaturated lipids, the abundance of redox-active transition metals, the modest antioxidant defense, the neurotransmitter auto-oxidation and RNA oxidation, and the Ca2+ signaling along with the high energetic demand, the brain is largely susceptible to oxidative stress especially at terminal synaptic ends (Cobley et al., 2018). In a corresponding manner, the retina is particularly prone to undergo injury linked to excessive generation of reactive oxygen species (ROS) due to its high oxygen consumption, exposure to continuous light, high intracellular levels of polyunsaturated fatty acids (PUFAs) in photoreceptor outer segments, presence of photosensitizers in the RPE and neurosensory retina, and daily phagocytosis of the retinal outer segment originating from rods and cones (Beatty et al., 2000; Fine et al., 2000; Klein et al., 2004; Buch, 2005; Gehrs et al., 2006; Katta et al., 2009). Thus, it is not surprising that proteins isolated from RPE cells are heavily modified by oxidative stress markers, including malondialdehyde, 4-hydroxynonenal, AGE, and RAGE modifications (Schutt et al., 2002, 2003). Degradation-resistant aggregates of lipofuscin consisting of cross-linked, oxidized proteins and lipids also accumulate in the brain and in the eye of AD and AMD in association with the occurrence of pro-oxidant environmental conditions, especially those caused by light stimulation (Kaarniranta et al., 2011). Damage of mtDNA, which is more vulnerable than nuclear DNA to damage from oxidation and blue light, is proven to accumulate during aging in the retina along with changes in morphology and function of respiration-competent mitochondria (Barreau et al., 1996; Ballinger et al., 1999; Barron et al., 2001; Liang and Godley, 2003; King et al., 2004; Godley et al., 2005; Feher et al., 2006). Correspondingly, mitochondrial dysfunction and oxidative injury marked by peroxidation, nitration, reactive carbonyls, and nucleic acid oxidation are early and prominent into selectively damaged hippocampal and cortical neuronal populations in AD brain (Nunomura et al., 2001; Ganguly et al., 2021; Ke et al., 2021). The autophagic, lysosomal, and proteasomal signal transduction pathways, the main protein quality control mechanisms endowed with the capacity of repairing oxidative stress-induced damages, are also altered both in AMD eyes and AD brain (Kaarniranta et al., 2011). This imbalance in clearance systems likely causes an intracellular buildup in misfolded/damaged proteins, including Aβ and phospho-tau (ptau; Loeffler et al., 1993; Johnson et al., 2002; Ohno-Matsui, 2011), which ends up in the formation of detrimental insoluble aggregates in the brain and in the eye (Kaarniranta et al., 2010; Bruni et al., 2020), as largely shared both in these neurodegenerative diseases. Impaired or insufficient autophagy activity is implicated in numerous age-related degenerative diseases, including AMD (Kaarniranta et al., 2011) and AD (Rami, 2009; Wong and Cuervo, 2010), since postmitotic, terminally differentiated neurons are largely sensitive to stress in degradative intracellular pathways leading to proteostasis. The neurotoxic intracellular Aβ, present in late endosomes and lysosome compartments of retinal neurons from aged familial AD mice models showing AMD pathology, is more likely to undergo release into cytosolic compartment by provoking destabilization/leaking of the lysosome membrane (Park et al., 2017; Habiba et al., 2020). This further corroborates the finding that an impaired autophagy is involved in Aβ-dependent eye neurodegeneration (Golestaneh et al., 2017). Markers of activated autophagy colocalize with Aβ deposits, especially in aged human RPE cells and specimens from postmortem AMD subjects, and clearly display impairment of an autophagic pathway in the retina (Mitter et al., 2014). In transgenic animal models expressing E693Δ mutation (referred to as the “Osaka” mutation) of amyloid precursor protein (APP), the age-dependent accumulation of intraneuronal Aβ oligomers causes leakage of cathepsin D from endosomes/lysosomes into the cytoplasm, cytochrome c release from the mitochondria, and activation of caspase-3 in the hippocampi of 18-month-old mice (Umeda et al., 2011) as well. Neuroinflammation is considered to crucially take part in neuronal loss and deposition of extracellular aggregates both in AD retina and brain. In particular, microglial cells, which are phagocytes of the CNS, have functional similarities with RPE cells, macrophages, or dendritic cells in AMD, and when activated, all of them secrete similar inflammatory mediators. The phagocytic activity of reactive microglial cells engulfing and degrading Aβ is progressively lost in close proximity to the fibrillary plaque into the brains of AD patients, while chronically activated neuroglia start to release damaging chemokines and cytokines – notably IL-1, IL-6, and tumor necrosis factor α – which further contribute to affect the protein degradation systems and neuronal viability. Likewise, under pathological conditions, microglia migrating into the subretinal space from the inner retina is early activated by Aβ deposited in the drusen with the production of different types of neurotoxic cytokines which drive both the protein aggregation and cell death during development and progression of AMD (Ohno-Matsui, 2011). Astrocyte activation is also present around drusens and in the subretinal space and senile plaques in AMD and AD specimens (Sivak, 2013). In addition to chronic oxidative stress, complement activation is a central mechanism in both pathologies with classical and alternative pathways preferentially involved in AD and AMD, respectively (McGeer et al., 2005; McGeer and McGeer, 2010). Colocalization of Aβ and iC3b immunoreactivity have been detected in the amyloid vesicles within the drusens with markers of chronic complement activation, such as C3d and C4 being elevated in the plasma of both AMD and AD patients (Ohno-Matsui, 2011). Proteins of the acute phase can be found both in the drusens and senile plaques (McGeer and McGeer, 2004; Donoso et al., 2006). Due to oxidative stress and inflammation, secondary neovascularization specifically manifests in exudative AMD with new vessels sprouting from the choroidal capillaries through the Bruch’s membrane into the sub-RPE space or into the retinal layer (Miller, 2013; Wang and Mao, 2021). Aβ accumulation, alterations of blood flow dynamics, and an increased endothelial cell apoptosis have been detected in the retina and choroidal vessels of complement factor H knockout mice [Cfh(–/–)], suggested as a valuable model for AMD mice (Aboelnour et al., 2016). Similarly, angiogenesis hallmark is featured in the hippocampus of postmortem human brain tissues from patients with AD by an elevated immunoreactivity of integrin aVb3, a dimeric glycoprotein expressed on the endothelial cell surface markedly increased on angiogenic vessels (Boscolo et al., 2007; Desai et al., 2009). Nevertheless, even though many parallel pathophysiological aspects are shared by both AMD and AD, several concerns have challenged the development of common biomarkers and effective therapies for their clinical management. For instance, the genetic background is completely different between AMD and AD. Besides, even though Aβ is deposited both in the brain and in drusens, the phenotypic traits characterizing AD and AMD are quite dissimilar. Extracellular Aβ-containing deposits are observed as drusens into sub-RPE space in AMD and as senile plaques into the hippocampus and cortex (brain), ganglion cell layer (retina), and around retinal vessels in AD. The main cell types affected in AMD are the RPE cells, whereas in AD, the hippocampal neural cells are primarily damaged. Again, Aβ accumulation occurs outside the outer blood–retinal barrier in AMD and inside the blood–brain barrier (BBB) and blood–cerebrospinal fluid (CSF) barrier in AD. Collectively, these findings suggest that AMD should be better considered as a subtype of “amyloid disease” with several similarities and differences with AD (Ohno-Matsui, 2011; Wang and Mao, 2021).
Neurotrophin Impairment in AD and AMD: The Neuroprotective and Regenerative Actions of NGF Delivery in the Brain and Eye
NGF is part, and indeed the pioneer, of neurotrophin family including brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT-3), and neurotrophin 4/5 (NT-4/5) (Huang and Reichardt, 2001; Calissano et al., 2010a, b; Cattaneo and Calissano, 2012; Aloe et al., 2015) expressed in mammalian brain and in the neural retina (Garcia et al., 2017). NGF is a pleiotropic factor that promotes the survival, growth, and differentiation of neuronal cells of the CNS and peripheral nervous system (PNS) during development and adulthood. Its “trophic” prosurvival activity has been more recently documented to consist of an anti-amyloidogenic action keeping under control the apoptotic program (Calissano et al., 2010a, b). NGF also exerts a modulatory role by acting on specific non-neuronal cells of the neuro-immuno-endocrine system (Aloe and Chaldakov, 2013). NGF and its cognate receptors (the high-affinity tyrosine kinase receptor TrkA and the low-affinity pan-receptor p75), in addition to their classical distribution in the cholinergic basal forebrain nuclei and in the cortical and hippocampal regions of the CNS, are also expressed in different ocular fluids and tissues, such as aqueous/vitreous humor, posterior lens, retina, and optic nerve (Micera et al., 2007). An imbalance between the NGF-dependent, TrkA-mediated survival, the growth actions, and the p75NTR-mediated activation of apoptosis/growth inhibitory pathway is causally associated with the onset and/or development of several neurodegenerative diseases of both CNS and PNS (Micera et al., 2004; Cuello et al., 2007, 2010; Garcia et al., 2017), including AD (Sofroniew et al., 2001; Schulte-Herbruggen et al., 2008; Cattaneo and Calissano, 2012) and AMD (Lambiase et al., 2009; Telegina et al., 2019; Esposito et al., 2021). In view of its strong neuroprotective and regenerative actions both in vitro and in vivo, delivery of NGF to the brain and eye has been always explored with therapeutic purposes for cerebral and extracerebral (retinal) neurodegenerative diseases, including AD and AMD (Gupta et al., 2016). Nevertheless, the clinical benefits of NGF therapy to the brain are restricted by its inability of bypassing the BBB to reach the CSF via the brain ventricles, poor pharmacokinetic profile, and nociceptive unwanted side effects due to the broad distribution of its receptors along the intrathecal space (Eriksdotter Jönhagen et al., 1998). All these pharmacological aspects of NGF drastically reduce its efficacy and safety when administered in vivo. In order to achieve a cerebral long-term delivery of NGF in biologically active therapeutic doses, multiple approaches have been employed to allow a sustained release/targeting of NGF both in the brain and eye. However, starting from the initial attempts of direct injection, the classical gene- and cell-based routes along with the more innovative nanotechnological strategies – including carrier-mediated release, polymer-based and encapsulated cell (EC) delivery system, nanoparticles, and quantum dots – have encountered several hurdles prior to becoming suitable treatments of cerebral and extracerebral symptoms of patients suffering from AD and, potentially, from AMD. The main therapeutic approaches for NGF delivery will be discussed in the following sections.
Direct Intracerebral Infusion of NGF
To date, local and direct intracerebroventricular (ICV) injections of NGF in close proximity to the cholinergic neuronal soma located in the CNS basal forebrain have been carried out by implantation of fibroblasts or neuronal progenitors genetically modified to secrete NGF or by delivery of NGF-expressing adeno-associated virus both in animal models and in early clinical trials (Fischer et al., 1987; Olson et al., 1992; Seiger et al., 1993; Eriksdotter Jönhagen et al., 1998). Adult cholinergic neurons are successfully rescued from degeneration by NGF ICV delivery, and mnemonic recovery is greatly sustained in vivo following infusion of this neurotrophin as well (Tuszynski et al., 1990, 2005, 2015; Markowska et al., 1994, 1996; Martínez-Serrano et al., 1995; Castel-Barthe et al., 1996; Klein et al., 2000; Bishop et al., 2008). An intraparenchymal application of recombinant NGF protein into sites adjacent to degenerating cholinergic cell bodies of rodents is effective and well-tolerated at least up to a 2-week period of treatment (Tuszynski, 2000; Pizzo and Thal, 2004).
Peripheral Administration of NGF Using Nasal and Intraocular Delivery
Even though the initial improvement of cholinergic functions following the direct intracerebral delivery of NGF in vivo turned out to be encouraging, the manifestations of an undesirable back pain and weight loss along with a low diffusivity of the drug within the brain have prompted researchers and companies to further discontinue this route of administration for therapeutic reasons (Faustino et al., 2017; Mitra et al., 2019). Thus, peripheral intranasal administration (INS) has provided an alternative approach to target via the olfactory path a sizeable amount of NGF directly to the brain, especially in a rodent model (Cattaneo et al., 2008). By taking advantage of the unique anatomic connections of the olfactory and trigeminal nerves linking the nasal mucosa and the CNS, INS delivery provides a high-vascularized absorption area favoring rapid penetration of NGF, but it does not overcome the occurrence of its adverse effects. Moreover, long-term INS of a drug causes damage to the nasal mucosa by altering the mucociliar activity (Malerba et al., 2011; Zhu et al., 2012). On the other hand, more recent studies have underscored the interesting possibility that the ocularly administered exogenous NGF (oNGF) is able to exert neuroprotective and/or regenerative properties on the retina and its brain projections, thus representing an innovative, safe, and non-invasive option for the cure of affected patients in the eye–brain system (Di Fausto et al., 2007; Lambiase et al., 2007; Tirassa et al., 2018). Furthermore, comparative studies carried out on adult and aged anti-NGF AD11 transgenic mice – an animal model characterized by AD-like neuropathology following chronic NGF antibody-mediated deprivation – have clearly demonstrated that ocular administration is less effective than intranasal delivery to protect cholinergic neurons and prevent behavioral deficits, even if used at higher doses (Capsoni et al., 2009; Covaceuszach et al., 2009). Likewise, intranasal delivery of NGF significantly improves the clinical outcome in children with neurological impairment following traumatic brain injury (TBI; Chiaretti et al., 2017).
Gene- and Cell-Mediated NGF Delivery
Different approaches aimed at achieving a direct administration of exogenous NGF to the brain with beneficial outcomes have been recently provided by gene- and cell-based therapies (Faustino et al., 2017; Mitra et al., 2019). In this regard, preclinical and clinical studies have proved that the stereotactic surgical delivery of CERE-110 – an AAV serotype 2-based vector encoding for the human NGF – to the nucleus basalis of Meynert is a reliable and accurate neuroprotective strategy, being largely neurorestorative for a large part of cholinergic neurons localized in the rat fimbria-fornix lesion, in aged monkey models, and in human patients (Bishop et al., 2008; Mandel, 2010). Unfortunately, preliminary clinical studies designed to evaluate the actual safety and efficacy of the long-term administration of CERE-110 in subjects suffering from early-moderate AD (Rafii et al., 2014) are not followed by encouraging results. As reported in a recently published phase II clinical trial, no significant clinical rescue was detected by using AAV vectors expressing human NGF on 49 enrolled AD-diagnosed patients who underwent intracerebral injections of AAV-NGF or sham surgery, respectively (Rafii et al., 2018). In parallel with these findings, following 1-year lentivirus NGF gene delivery to the cholinergic basal forebrain, neither the systemic leakage of NGF and the occurrence of anti-NGF antibodies nor the activation of detrimental neuroinflammatory response in the brain with back pain or weight loss has been detected in aged non-human primates (Nagahara et al., 2009). Although the gene therapy procedures exploit recombinant AAV in which the viral genes for self-replication and incorporation of genetic material into chromosomal DNA have been deleted, the impossibility of modifying the dosage and/or interrupting the treatment once started along with potential toxicity and immunogenicity has raised concerns regarding the practicability, suitability, and usefulness of its application. To overcome the limitations of direct viral vector-mediated gene delivery, ex vivo gene therapy has been also evaluated. Neuronal stem cells (NSCs) transduced with human NGF by using a AAV2 vector, when grafted into the cerebral cortex of cognitively impaired rats undergoing chronic ICV infusion of the serine/threonine protein phosphatase (PP) inhibitor okadaic acid, are proven to successfully integrate into the host brain and to improve cognitive performance after transplantation (Wu et al., 2008). The NSC line overexpressing the human choline acetyltransferase (ChAT) gene recovers the learning/memory deficits and elevates the levels of acetylcholine (ACh) in CSF when transplanted into rat brain of an experimentally induced AD model, in which the application of ethylcholine mustard aziridinium ion (AF64A) specifically inactivates the cholinergic nerves. Interestingly, transplanted ChAT human NSCs migrate to different brain areas including the cerebral cortex, hippocampus, striatum, and septum of AF64A-cholinotoxin-lesioned AD rat model and successfully differentiate into viable and functional neurons and astrocytes as well (Park et al., 2012).
Carrier-Mediated Sustained Release of NGF
Encapsulated cell biodelivery is an innovative strategy involving an in vitro genetically engineered human cell line which grows on a polymer scaffold behind a semipermeable filter and continuously releases a low but sufficient amount of therapeutic protein directly into a localized region of brain cells. This method combines the potency of gene therapy with the advantage offered by an implantable and retrievable device, acting as an actual biological micropump. In this regard, grafts of polymer-ECs modified to release NGF delay and/or prevent the degeneration of axotomized cholinergic neurons in the basal forebrain of rodents as well as of aged monkeys (Hoffman et al., 1993; Emerich et al., 1994; Kordower et al., 1994; Lindner et al., 1996). Implantation of encapsulated NsG0202, a clinical device which houses an NGF-secreting cell line (NGC-0295) derived from a human RPE cell line, is well-tolerated into the basal forebrain of Göttingen minipigs up to 12 months leading to an increase of NGF levels in the surrounding tissue (Fjord-Larsen et al., 2010; Wahlberg et al., 2012). EC-NGF biodelivery devices targeting the basal forebrain of implanted AD patients (n = 6) are safe in a 12-month study, leading to improvement in cognition assessed by clinical rating scales, EEG, MRI, and positron emission tomography (PET) (Eriksdotter-Jönhagen et al., 2012). Nevertheless, several drawbacks, such as the invasive nature of the procedures requiring hospitalization and neurosurgical intervention and their high costs, have discouraged the pursuing of both gene- and cell-based therapies as interventions to achieve a sustained and controlled release of NGF into the brain. An advancement in the field of NGF therapeutic strategies has been made with the contemporary development of intracerebral implants composed of biocompatible, synthetic, and natural polymers, including poly(ethylene co-vinyl acetate) (EVAc), poly(D,L-lactide-co-glycolic acid) (PLGA), polyanions (e.g., heparin, dextran sulfate, gelatin), and polycationic or polyanionic hydrogels which are endowed with different shapes and surface areas to provide high and localized doses of this neurotrophin into the brain (Hoffman et al., 1990; Powell et al., 1990; Johnson et al., 2008; Yang et al., 2009; Song B. et al., 2012; Hines and Kaplan, 2013). Nanoparticles have been more recently explored due to their multiple advantages, including small size (nanoscale dimensions), high solubility, multifunctionality, improved transport properties and pharmacokinetic profile, enhanced permeability, and retention. These parameters significantly enhance the penetrance of a drug into tissues through capillaries and also enhance its efficient delivery to target sites, thus providing a more powerful medicament for patients. Liposomes, polymeric micelles, dendrimers, magnetic nanoparticles, and quantum dots functionalized with NGF effectively deliver this neurotrophin both in neuronal cell culture and/or in animal model of AD leading to neurite outgrowth, diminution of cerebral Aβ accumulation, rescue in cholinergic functions, improved viability, and neuroprotection (Angelova et al., 2013; Zhao et al., 2016; Faustino et al., 2017; Marcus et al., 2018; Mitra et al., 2019; Tosi et al., 2020).
In summary, the pivotal role of NGF in the development, growth, maintenance, and plasticity of the CNS and PNS, both in normal and diseased conditions, represents a strong rationale and incentive of advancing innovative techniques to provide its localized and finely modulated delivery to target tissues.
Ocular Administration of NGF as a Promising Therapeutic Approach to Counteract Degeneration of the Eye and Brain Both in AMD and AD Pathologies
The independent trophic and neuroprotective roles of NGF on the retina, optic nerve, and brain visual area and its direct and indirect projections to limbic structures, including the cholinergic hippocampus and septum, have been largely documented in different animal models and in paradigms of retinal injury and disease (Micera et al., 2004; Cattaneo and Calissano, 2012; Aloe et al., 2015; Garcia et al., 2017; Esposito et al., 2021).
In particular, under oxidative stress and inflammatory conditions similar to those early occurring both in AMD and AD, the activity of matrix metalloproteinase-7 protease is altered in the eye causing a local accumulation of proNGF and concomitant reduction of mature NGF followed by changes in the expression and function of its receptors (i.e., the survival TrkA receptor and the neurotrophin p75NTR receptor). Thus, even though it is still unclear whether ocular neuronal death is associated with the lack of neurotrophic support or impairment of its signaling events regardless of the level of the growth factor itself, ophthalmological clinical trials have taken advantage of the well-known anti-apoptotic action of NGF to slow and/or stop sight loss resulting from death of injured RGC or photoreceptors (Pardue and Allen, 2018; Hill et al., 2021). Compelling experimental evidence has documented the regenerative role of NGF on retinal ganglion cell survival in different animal models of optic nerve transection, ischemic injury, ocular hypertension, and glaucoma (Roberti et al., 2014). The impressive pharmacological potential of this neurotrophin in ophthalmic diseases has been recently brought to light by Guo et al. (2020) reporting that topical application of recombinant human NGF (rh-NGF) significantly reduces the RGC apoptosis in vivo following partial optic nerve transection (pONT) by protecting their soma and axons likely via the retrobulbar route (Guo et al., 2020). Intravitreal administration of rhNGF counteracts RGC degeneration occurring within 2 weeks following optic nerve crush (ONC) by reducing p75NTR and proNGF and by enhancing phosphorylation of TrkA and its intracellular signals at the retina level (Mesentier-Louro et al., 2019), in line with previous findings on another animal model of retinal disease, such as inherited retinitis pigmentosa (RP) (Sacchetti et al., 2017). The beneficial effect of NGF on injured eye has been also validated in RP experimental paradigm following retrobulbar injection (Lenzi et al., 2005) or in affected patients after eye drop administration (Falsini et al., 2016b). A randomized double-blind phase II clinical trial, aimed at testing the vision functions in 18 patients aged 2–23 years with stable disease and severe blindness caused by glioma, has proven that ocular instillation of NGF protects the optic pathway glioma-related visual impairment in the absence of any apparent side effects (Falsini et al., 2016a).
More interestingly, a growing body of experimental and clinical studies have shown that administration of NGF in different ocular sites can be a feasible means to convey NGF and/or activate its downstream signals into the brain (Eftimiadi et al., 2021). Thus, the increase of the endogenous NGF availability, following its intraocular (intravitreal or periocular) exogenous injection and/or topic instillation (Figure 2), is currently exploited as a safe, non-invasive approach to successfully counteract retina and even brain neurodegeneration underlying clinical signs of AMD and AD phenotypes (Tirassa et al., 2018; Mitra et al., 2019). Consistently, when administered topically in animal models, NGF becomes available to the retina and optic nerve and exerts neuroprotective and/or regenerative properties on projections of the eye–brain system, including the nucleus basalis and septum as well (Lambiase et al., 2005, 2007). The local application of NGF drops using the ocular surface way induces an upregulation in the expression of NGF receptors and ChAT immunoreactivity (cholinergic markers) in chemically injured basal forebrain neurons of adult rats, indicating that eye NGF application somehow affects brain cells (Di Fausto et al., 2007; Lambiase et al., 2007). Along this line, the finding that a single ocular administration of NGF solution is sufficient to enhance the distribution of Ki67-positive cells (marker for proliferative cells) also expressing p75 neurotrophin receptors in the proliferating layer of the subventricular zone (SVZ) further supports the feasibility of ocular application of NGF in upregulating the development and maturation of neuronal progenitors in the CNS (Tirassa, 2011). Immunohistochemical studies have reported that an instillation of NGF as eye drops increases in a time-dependent manner the immunoreactivity of c-fos (markers for neuronal activation) in several areas of the limbic system and in primary visual centers of rats as well (Calza et al., 2011). Furthermore, an improvement in visual acuity and electrofunctional parameters has been found in a 94-year-old female affected with AMD 3 months after initiation of treatment with NGF and in the absence of any side effects up to 5 years of follow-up (Lambiase et al., 2009). Thus, despite intrinsic limitations due to proper physiology of the eye (tear turnover, nasolachrymal drainage, reflex blinking, ocular static, and dynamic barriers) which makes difficult a deeper drug penetration, the chance of exploiting in common clinical practice the ocular route to achieve a non-invasive distribution of several medicaments, including NGF, in therapeutic concentrations for the brain is gaining increasing interest to ocular scientists (Patel et al., 2013; Esposito et al., 2021). The advent of innovative nanotechnologies, new techniques, and devices (nanoparticles, nanosuspensions, liposomes, dendrimers, in situ gelling systems, intraocular implants, and microneedles) designed to overcome ocular barriers and side effects linked with conventional topical drops is currently allowing a more sustained drug release and an improved target specificity (Patel et al., 2013). Furthermore, the recent development of a genetically engineered NGF variant endowed with biological activity and reduced pain effect (painless NGF) further opens the expanding panorama of its applications via eye drop administration (Malerba et al., 2015; Testa et al., 2021).
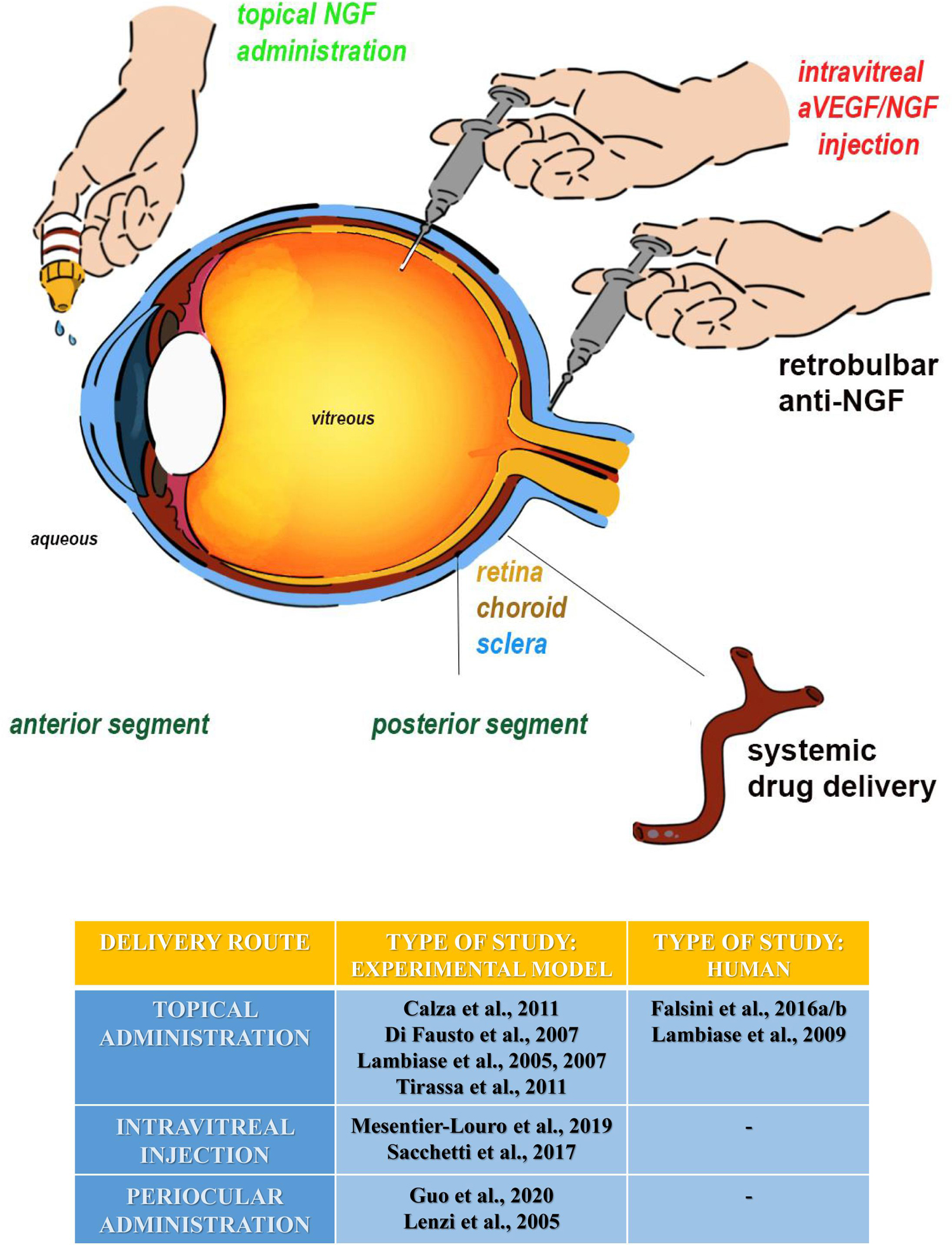
Figure 2. All ocular sites tested for experimental NGF delivery are summarized as follows: (1) the topical administration (eye drop instillation and emulsion/gel application) (green labeled), (2) the intravitreal injections (ophthalmic solutions/suspentions, implants, and nanotecnology), and (3) the periocular administration (implants or injections) at the posterior segment. The periocular route includes the subconjunctival, subtenon, peribulbar, and retrobulbar sites of injection. Except for the systemic route, all the others have been tested for NGF and anti-NGF at both experimental and clinical levels. The topical administration is actually a routine procedure for specific eye disorders. Highlighted in red: our future directions on experimental animals, comprising the anti-VEGF/NGF combined intravitreal injection or the use of NGF nanoparticles layered in the vitreal chamber. The table summarizes the different routes of NGF administration in the eye, in an experimental model and human beings.
Collectively, these recent and promising in vivo results underline that the protective and reparative actions of NGF used as eye drops are not only confined to the primary visual areas, but are also extended to other retinal central targets, including the forebrain structure (Calza et al., 2011; Tirassa, 2011). Moreover, these studies encourage investigations on the clinical effects of NGF therapy for the treatment of neurodegenerative diseases characterized by an imbalance in NGF/TrkA signal transduction pathway in the eye and in the brain, including AMD and AD, respectively (Mufson et al., 2008; Aloe et al., 2015; Canu et al., 2017; Esposito et al., 2021).
Essential Trace Metals: Diagnostic Perspectives in AD and AMD
The common physiopathological aspects linking AD and AMD not only have important therapeutic implications but also can give additional insights into the development of novel biomarkers for both diseases. Besides, since retinal degenerative alterations manifest before the corresponding changes detected in the brain, quantification of ocular biomarkers will facilitate (i) the diagnosis of AD as well as AMD at their earliest stages when therapy is more likely to be effective in halting/slowing down symptomatology and (ii) a non-invasive monitoring of disease progression (Micera et al., 2019; Ong et al., 2019). In this context, a growing body of evidence has shown that dyshomeostasis of essential trace metals, namely, both depletion and excess of copper (Cu), iron (Fe), and zinc (Zn), causes severe damage to neurons and is causally associated with various neurodegenerative and retinal diseases, including AD and AMD (Bush, 2013; Faller et al., 2013; Micera et al., 2019).
In AD, a Cu imbalance is evident and associated with the misplacement of metal from cellular and tissue compartments: meta-analytic studies reported that Cu levels are decreased in the AD brain while increased in the periphery, suggesting a breakdown of Cu homeostasis control in the disease (Mathys and White, 2017). Elevated values of Cu in serum and decreased Cu values in the brain are associated with increased levels of Cu not bound to proteins and primarily to ceruloplasmin (the main Cu protein in serum) in general circulation: this Cu component, called non-ceruloplasmin Cu, also known as “free” Cu, is an established marker of Wilson disease, the paradigmatic disease of Cu toxicosis/accumulation. Non-ceruloplasmin Cu is an exchangeable, small molecular weight and filterable Cu component in the bloodstream that is toxic above a certain cutoff and can pass the BBB and distribute to brain parenchyma (Choi and Zheng, 2009; Squitti, 2012; Kepp and Squitti, 2019). Aβ40 and Aβ42 are flexible small α-helix and β-sheet structures representing the majority of Aβ forms in the AD brain. They can bind Cu(I/II) with relevant effects in initiating Aβ aggregation processes (Squitti, 2012; Mathys and White, 2017; Kepp and Squitti, 2019). Cu binding confers redox activity to Aβ (Squitti et al., 2017) that affects neuron viability (White et al., 1999). It has been recently hypothesized that a Cu dysfunction in the aging human brain might evolve as a gradual displacement of Cu from pools tightly bound to proteins to pools of loosely bound metal ions, involved in gain-of-function oxidative stress associated with Aβ peptides, a shift that might be provoked by chemical aging (Kepp and Squitti, 2019). The loss of Cu tightly bound to proteins may be associated with the loss of energy production and antioxidant function within neuronal cells, two important drivers of AD neurodegeneration (Kepp and Squitti, 2019). Non-ceruloplasmin Cu fits well in this construct since it is a loosely bound specie of Cu in general circulation and has been demonstrated to be a potential prognostic biomarker for conversion from mild cognitive impairment (MCI) to symptomatic AD, typified by copper imbalance (Squitti et al., 2014; Rozzini et al., 2018; Sensi et al., 2018). In AD, non-ceruloplasmin Cu reaches values similar to Wilson disease (Squitti et al., 2018) and has demonstrated a fair sensitivity in detecting which individuals with MCI will convert to Cu-AD (Squitti et al., 2014) and high specificity (95%) in detecting patients affected with Cu-AD due to copper exposure (Squitti et al., 2017), in line with preclinical models of the disease (Sparks and Schreurs, 2003; Singh et al., 2013; Yu et al., 2018; Hsu et al., 2019). Recently, several studies have shown that non-ceruloplasmin Cu might serve as a stratification biomarker for a subset of AD patients (Kepp and Squitti, 2019). The feature, called Cu-AD (Squitti et al., 2017), appears specific to a subset (50%–60%) of AD patients and is characterized by being a carrier of selective ATP7B gene variants (Squitti et al., 2013) and by having peculiar cortical activity and neuroimaging deficits (Kepp and Squitti, 2019).
Fe dysfunction has been strongly associated with AD pathogenesis in recent years. It is known that Fe imbalance and Aβ metabolism are synergistically regulated in neurodegeneration and can facilitate cellular processes which may result in AD development, pointing out to a central role of Fe dyshomeostasis in AD pathology. Recently, potential biomarkers associated with Fe imbalance have been individuated: ferritin, either in plasma or in CSF, has been shown to have potential in discriminating AD patients in their preclinical stage, categorized into low and high neocortical amyloid-β load, prior to cognitive impairment (Goozee et al., 2018). High ferritin levels suggest an increase of Fe abundance in the CSF and brain which can be associated with ferroptosis, an iron-dependent cell death that results from a buildup of lipid peroxides and is regulated by glutathione (GSH) peroxidase 4 (Acevedo et al., 2019). A recent study provided a very interesting theoretical significance of abnormalities of Fe associated with AD onset and progression (Ayton et al., 2021). Fe and Cu metabolism are intertwined in physiology, and they are certainly tied in AD (Squitti et al., 2010). Ceruloplasmin exemplifies the crosstalk protein between Cu and Fe balance because it is the main Cu protein in the body, and it controls Fe oxidative state: imbalance in one of the two metals has effects on the other (Siotto et al., 2016). Consistent with both Cu and Fe balance abnormalities, ceruloplasmin in the CSF has been shown to predict cognitive decline and brain atrophy in people with underlying Aβ pathology (Diouf et al., 2020). High levels of ceruloplasmin and the activation of the ceruloplasmin:transferrin (Cp:Tf) system, one of the main antioxidant systems acting in general circulation, were associated with worse cognitive performances and a severe medial temporal lobe atrophy, supporting the view that local iron accumulation in brain areas critical for AD might be strictly associated with Fe systemic alterations (Squitti et al., 2010). Recently, another point of conversion between Cu and Fe metabolism has been provided by the evidence of the loss in GSH levels in AD. GSH is one of the main endogenous free radical scavenger systems in the brain, acting as a co-substrate in GSH peroxidase-catalyzed reactions (Pinnen et al., 2011). GSH loss in the brain is linked to cognitive dysfunction (Currais and Maher, 2013) and is associated with Cu imbalance. This has been mainly stressed in preclinical models of AD induced by Cu exposure: high Cu and cholesterol levels trigger Aβ aggregation and plaque formation in the hippocampus and temporal cortex and impair learning and memory, thus recapitulating deficits featuring mental dementia (Yao et al., 2018).
Zinc homeostatic imbalance has been advocated as a driver of neurodegenerative processes, primarily linked to oxidative stress and the aging brain (Capasso et al., 2005; Frazzini et al., 2006). Somewhat reduced levels of zinc have been shown in AD through meta-analyses (da Silva et al., 2014; Ventriglia et al., 2015; Wang et al., 2015). The hypothetical role of this metal in AD pathology has been connected to neurotoxicity induced by mitochondrial production of ROS and by disruption of metabolic enzymatic activity (i.e., Cu, Zn superoxide dismutase activity) eventually leading to apoptosis and/or neurodegeneration (Li et al., 2017; Kabir et al., 2021). Zinc therapy, used in Wilson disease, effectively reduces non-ceruloplasmin Cu levels: this suggests potential beneficial effects in slowing the progression of cognitive decline in subsets of individuals who show both cognitive disturbances and signs of Cu imbalance (Squitti et al., 2020).
Concerning AMD, there is evidence that heavy metals might be involved in the pathogenesis of AMD (Ugarte et al., 2013; Micera et al., 2019). Beyond the deposition of Fe, Cu, and Zn metal ions in extracellular deposits of AMD, recent studies have shown accumulation of heavy intoxicant metals in eye tissues and in general circulation of AMD patients (Aberami et al., 2019). Major metal changes involve the accumulation of loosely bound Fe in human RPE, in Bruch’s membrane, and in photoreceptors located in the macula (Biesemeier et al., 2015; Dalvi et al., 2019). Fe plays a pivotal role in retina physiology as a cofactor of several processes of visual transduction: the expression of transferrin, ferritin, and ferroportin is upregulated in AMD macula (Baumann et al., 2017). A tight Fe control can avoid the accumulation of pro-oxidant and proinflammatory loosely bound species of the metal (Courtois et al., 2020). To contrast the accumulation of Fe in the aged retina of AMD patients, Fe chelation therapy has been proposed (Shu and Dunaief, 2018). Indeed, treatment with the Fe chelator deferiprone has been shown to be useful to prevent retinal degeneration (Song D. et al., 2012; Cui et al., 2020). However, several chelators (deferoxamine B) have been shown to cause adverse events (Micera et al., 2019). Recently, intravitreal injection of transferrin, a natural Fe chelator, has been demonstrated to have neuroprotective effects against oxidative stress in retinal degeneration: after injection in the vitreous, transferrin spans rapidly within the retina and accumulates in photoreceptors and in RPE, protecting retinal function from loosely bound Fe (Picard et al., 2015). Consistent with previous evidence (Micera et al., 2019), new findings show that Cu is increased in choroid-RPE of AMD. In contrast to Fe and Cu trends, Zn appears decreased in the aged retina and Zn supplementation has been proposed for AMD treatment (Gilbert et al., 2019). Zn supplementation has beneficial effects in reducing the progression of AMD according to the Age-Related Eye Disease Study (AREDS; Age-Related Eye Disease Study Research Group (AREDSRG), 2000; Chew et al., 2015; Seddon et al., 2016): daily supplementation with a formulation consisting of 500 mg vitamin C, 400 IU vitamin E, 25 mg zinc, 2 mg copper, 10 mg lutein, and 2 mg zeaxanthin (AREDS and AREDS2) was effective for slowing AMD progression (Kim et al., 2016). Cochrane reviews support the use of Zn for the delay of AMD progression and vision loss (Evans and Lawrenson, 2017; Micera et al., 2019). Beyond studies on essential trace metals, an increasing number of studies have been performed on circulating or local concentrations of intoxicant heavy metals, mainly linked to environmental or lifestyle risk factors. It is acknowledged that environmental and lifestyle factors affect the balance of bodily trace metals, and in particular, this occurs in the retina mainly through oxidative stress that originated from exposure of macular tissues to sunlight and local or systemic exposure to oxidative stressors, including smoke. In this line of research, important results have been gained mainly for cadmium (Cd) that, in most of these studies, has been found to be elevated in AMD patients, especially within the smoking population (Kim et al., 2016; Wu et al., 2014; Güngör et al., 2018). These findings are coherent with the results from histology studies, showing Cd accumulation in the retina and in the RPE, particularly in smokers (Wills et al., 2008). The proposed mechanisms of Cd uptake in the retina involve zinc transporters (ZIP4 and ZIP8) with high Cd affinity (Girijashanker et al., 2008). Studies on circulating intoxicant heavy metals have shown also increased levels of blood lead (Pb) (Güngör et al., 2018). Higher blood Pb and Cd levels in AMD (Güngör et al., 2018; Heesterbeek et al., 2020) together with increased levels of barium (Ba) have been found in neovascular AMD (236 patients with neovascular AMD compared with 236 age-matched controls). Conversely, chromium (Cr) concentration decreased. Interestingly, higher Cd concentrations have been found mostly in smokers. On this basis, preventive strategies targeting decreased Cd exposure have been postulated to reduce the burden of AMD, primarily directed to skip smoking (Heesterbeek et al., 2020).
Conclusion
Alzheimer’s disease and AMD are brain and retinal degenerative diseases whose incidence is increasing worldwide due to lengthening of life expectancy. AMD and AD share several clinical and pathological aspects, including Aβ accumulation and aggregation, oxidative stress, inflammation, and alterations in local supportive/regulatory actions of NGF. Based on these similarities and in view of the strong evidence that NGF is endowed with regenerative actions on both the brain and the retina, a growing number of opportunities can be offered by the administration of this neurotrophin as a common therapeutic agent and neuroprotective strategy for AD and AMD. Currently, great efforts are being made to enhance the effectiveness of NGF-based therapy by exploring novel, safe, and reliable routes of its delivery to the brain and eye, ultimately assessed in preclinical and clinical trials. Alterations in the systemic and ocular levels of essential trace metals are currently evaluated as diagnostic and/or even predictive biomarkers for future precision medicine of both AD and AMD.
Author Contributions
GA, RS, and AM designed and wrote the manuscript. VL, BB, MV, and PC contributed to the data, text, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported (in part) by Fondo Ordinario Enti (FOE D.M 865/2019) funds in the framework of a collaboration agreement between the Italian National Research Council and EBRI (2019–2021) and Regione Lazio, POR FESR Lazio 2014-2020, “Progetti di Gruppi di Ricerca 2020” (Determinazione dirigenziale n.G08487 del 19 luglio 2020) to GA. VL is supported by a postdoctoral fellowship by Operatori Sanitari Associati (OSA). AM is supported partially by the Italian Ministry of Health (RC2765949) and 5xMille 2016 to Fondazione Bietti. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
BB and AM are grateful to Fondazione Roma (Italy) for the continuous support. Many thanks to Angelica Napoli for drawing Figure 2. We are also grateful to Dott. Egidio Stigliano for critical reading of the manuscript and suggestions.
References
Aberami, S., Nikhalashree, S., Bharathselvi, M., Biswas, J., Sulochana, K. N., and Coral, K. (2019). Elemental concentrations in Choroid-RPE and retina of human eyes with age-related macular degeneration. Exp. Eye Res. 186:107718. doi: 10.1016/j.exer.2019.107718
Aboelnour, A., Kam, J. H., Elnasharty, M. A., Sayed-Ahmed, A., and Jeffery, G. (2016). Amyloid beta deposition and phosphorylated tau accumulation are key features in aged choroidal vessels in the complement factor H knock out model of retinal degeneration. Exp. Eye Res. 147, 138–143. doi: 10.1016/j.exer.2016.05.015
Acevedo, K., Shashank, M., Opazo, C. M., and Bush, A. I. (2019). Redox active metals in neurodegenerative diseases. J. Biol. Inorg. Chem. 24, 1141–1157. doi: 10.1007/s00775-019-01731-9
Age-Related Eye Disease Study Research Group (AREDSRG) (2000). Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: age-Related Eye Disease Study Report Number 3. Ophthalmology 107, 2224–2232. doi: 10.1016/S0161-6420(00)00409-7
Aloe, L., and Chaldakov, G. N. (2013). The multiple life of nerve growth factor: tribute to Rita Levi-Montalcini (1909–2012). Balkan Med. J. 30, 4–7. doi: 10.5152/balkanmedj.2013.003
Aloe, L., Rocco, M. L., Balzamino, B. O., and Micera, A. (2015). Nerve Growth Factor: a Focus on Neuroscience and Therapy. Curr. Neuropharmacol. 13, 294–303. doi: 10.2174/1570159x13666150403231920
Ambati, J., and Fowler, B. J. (2012). Mechanisms of age-related macular degeneration. Neuron 75, 26–39. doi: 10.1016/j.neuron.2012.06.018
Anderson, D. H., Talaga, K. C., Rivest, A. J., Barron, E., Hageman, G. S., and Johnson, L. V. (2004). Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp. Eye Res. 78, 243–256. doi: 10.1016/j.exer.2003.10.011
Angelova, A., Angelov, B., Drechsler, M., and Lesieur, S. (2013). Neurotrophin delivery using nanotechnology. Drug Discov. Today 18, 1263–1271. doi: 10.1016/j.drudis.2013.07.010
Ashok, A., Singh, N., Chaudhary, S., Bellamkonda, V., Kritikos, A. E., Wise, A. S., et al. (2020). Retinal Degeneration and Alzheimer’s Disease: an Evolving Link. Int. J. Mol. Sci. 21:7290. doi: 10.3390/ijms21197290
Atwood, C. S., Martins, R. N., Smith, M. A., and Perry, G. (2002). Senile plaque composition and posttranslational modification of amyloid-beta peptide and associated proteins. Peptides 23, 1343–1350. doi: 10.1016/S0196-9781(02)00070-0
Ayton, S., Portbury, S., Kalinowski, P., Agarwal, P., Diouf, I., Schneider, J. A., et al. (2021). Regional brain iron associated with deterioration in Alzheimer’s disease: a large cohort study and theoretical significance. Alzheimers Dement. 17, 1244–1256. doi: 10.1002/alz.12282
Ballinger, S. W., Van Houten, B., Jin, G. F., Conklin, C. A., and Godley, B. F. (1999). Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells. Exp. Eye Res. 68, 765–772. doi: 10.1006/exer.1998.0661
Barreau, E., Brossas, J. Y., Courtois, Y., and Treton, J. A. (1996). Accumulation of mitochondrial DNA deletions in human retina during aging. Invest. Ophthalmol. Vis. Sci. 37, 384–391.
Barron, M. J., Johnson, M. A., Andrews, R. M., Clarke, M. P., Griffiths, P. G., Bristow, E., et al. (2001). Mitochondrial abnormalities in ageing macular photoreceptors. Invest. Ophthalmol. Vis. Sci. 42, 3016–3022.
Baumann, B., Sterling, J., Song, Y., Song, D., Fruttiger, M., Gillies, M., et al. (2017). Conditional Müller Cell Ablation Leads to Retinal Iron Accumulation. Invest. Ophthalmol. Vis. Sci. 58, 4223–4234. doi: 10.1167/iovs.17-21743
Baumgart, M., Snyder, H. M., Carrillo, M. C., Fazio, S., Kim, H., and Johns, H. (2015). Summary of the evidence on modifiable risk factors for cognitive decline and dementia: a population-based perspective. Alzheimers Dement. 11, 718–726. doi: 10.1016/j.jalz.2015.05.016
Beatty, S., Koh, H., Phil, M., Henson, D., and Boulton, M. (2000). The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 45, 115–134. doi: 10.1016/s0039-6257(00)00140-5
Biesemeier, A., Yoeruek, E., Eibl, O., and Schraermeyer, U. (2015). Iron accumulation in Bruch’s membrane and melanosomes of donor eyes with age-related macular degeneration. Exp. Eye Res. 137, 39–49. doi: 10.1016/j.exer.2015.05.019
Biscetti, L., Luchetti, E., Vergaro, A., Menduno, P., Cagini, C., and Parnetti, L. (2017). Associations of Alzheimer’s disease with macular degeneration. Front. Biosci. (Elite Ed.) 9, 174–191. doi: 10.2741/e794
Bishop, K. M., Hofer, E. K., Mehta, A., Ramirez, A., Sun, L., Tuszynski, M., et al. (2008). Therapeutic potential of CERE-110 (AAV2-NGF): targeted, stable, and sustained NGFdelivery and trophic activity on rodent basal forebrain cholinergic neurons. Exp. Neurol. 211, 574–584. doi: 10.1016/j.expneurol.2008.03.004
Blasiak, J. (2020). Senescence in the pathogenesis of age-related macular degeneration. Cell. Mol. Life Sci. 77, 789–805. doi: 10.1007/s00018-019-03420-x
Boscolo, E., Folin, M., Nico, B., Grandi, C., Mangieri, D., Longo, V., et al. (2007). Beta amyloid angiogenic activity in vitro and in vivo. Int. J. Mol. Med. 19, 581–587.
Brookmeyer, R., Johnson, E., Ziegler-Graham, K., and Arrighi, H. M. (2007). Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 3, 186–191. doi: 10.1016/j.jalz.2007.04.381
Bruni, A. C., Bernardi, L., and Gabelli, C. (2020). From beta amyloid to altered proteostasis in Alzheimer’s disease. Ageing Res. Rev. 64:101126. doi: 10.1016/j.arr.2020.101126
Buch, H. (2005). Fourteen-year incidence of age-related maculopathy and cause-specific prevalence of visual impairment and blindness in a Caucasian population: the Copenhagen City Eye Study. Acta Ophthalmol. Scand. 83, 400–401. doi: 10.1111/j.1600-0420.2005.thesis.x
Bush, A. I. (2013). The metal theory of Alzheimer’s disease. J. Alzheimers Dis. 33, S277–S281. doi: 10.3233/JAD-2012-129011
Calissano, P., Amadoro, G., Matrone, C., Ciafrè, S., Marolda, R., Corsetti, V., et al. (2010a). Does the term ‘trophic’ actually mean anti-amyloidogenic? The case of NGF. Cell Death Differ. 17, 1126–1133. doi: 10.1038/cdd.2010.38
Calissano, P., Matrone, C., and Amadoro, G. (2010b). Nerve growth factor as a paradigm of neurotrophins related to Alzheimer’s disease. Dev. Neurobiol. 70, 372–383. doi: 10.1002/dneu.20759
Calza, A., Florenzano, F., Pellegrini, D., and Tirassa, P. (2011). Time-dependent activation of c-fos in limbic brain areas by ocular administration of nerve growth factor in adult rats. J. Ocul. Pharmacol. Ther. 27, 209–218. doi: 10.1089/jop.2010.0139
Canu, N., Amadoro, G., Triaca, V., Latina, V., Sposato, V., Corsetti, V., et al. (2017). The Intersection of NGF/TrkA Signaling and Amyloid Precursor Protein Processing in Alzheimer’s Disease Neuropathology. Int. J. Mol. Sci. 18:1319. doi: 10.3390/ijms18061319
Capasso, M., Jeng, J. M., Malavolta, M., Mocchegiani, E., and Sensi, S. L. (2005). Zinc dyshomeostasis: a key modulator of neuronal injury. J. Alzheimers Dis. 8, 93–108. doi: 10.3233/JAD-2005-8202
Capsoni, S., Covaceuszach, S., Ugolini, G., Spirito, F., Vignone, D., Stefanini, B., et al. (2009). Delivery of NGF to the brain: intranasal versus ocular administration in anti-NGF transgenic mice. J. Alzheimers Dis. 16, 371–388. doi: 10.3233/JAD-2009-0953
Castel-Barthe, M. N., Jazat-Poindessous, F., Barneoud, P., Vigne, E., Revah, F., Mallet, J., et al. (1996). Direct intracerebral nerve growth factor gene transfer using a recombinant adenovirus: effect on basal forebrain cholinergic neurons during aging. Neurobiol. Dis. 3, 76–86. doi: 10.1006/nbdi.1996.0008
Cattaneo, A., and Calissano, P. (2012). Nerve growth factor and Alzheimer’s disease: new facts for an old hypothesis. Mol. Neurobiol. 46, 588–604. doi: 10.1007/s12035-012-8310-9
Cattaneo, A., Capsoni, S., and Paoletti, F. (2008). Towards non invasive nerve growth factor therapies for Alzheimer’s disease. J. Alzheimers Dis. 15, 255–283. doi: 10.3233/jad-2008-15210
Çerman, E., Eraslan, M., and Çekiç, O. (2015). Age-related macular degeneration and Alzheimer disease. Turk. J. Med. Sci. 45, 1004–1009. doi: 10.3906/sag-1406-146
Chew, E. Y., Clemons, T. E., Agrón, E., Launer, L. J., Grodstein, F., Bernstein, P. S., et al. (2015). Effect of Omega-3 Fatty Acids. Lutein/Zeaxanthin, or Other Nutrient Supplementation on Cognitive Function: the AREDS2 Randomized Clinical Trial. JAMA 314, 791–801. doi: 10.1001/jama.2015.9677
Chiaretti, C., Conti, G., Falsini, B., Buonsenso, D., Crasti, M., Manni, L., et al. (2017). Intranasal Nerve Growth Factor administration improves cerebral functions in a child with severe traumatic brain injury: a case report. Brain Inj. 31, 1538–1547. doi: 10.1080/02699052.2017.1376760
Choi, B. S., and Zheng, W. (2009). Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 1248, 14–21. doi: 10.1016/j.brainres.2008.10.056
Cobley, N., Fiorello, M. L., and Bailey, D. M. (2018). 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 15, 490–503. doi: 10.1016/j.redox.2018.01.008
Coleman, H. R., Chan, C. C., Ferris, F. L., and Chew, E. Y. (2008). Age-related macular degeneration. Lancet 372, 1835–1845. doi: 10.1016/S0140-6736(08)61759-6
Courtois, Y., Behar-Cohen, J. Y. F., and Picard, E. (2020). Iron and age-related macular degeneration: a new track. Med. Sci. (Paris) 36, 616–625. doi: 10.1051/medsci/2020096
Covaceuszach, S., Capsoni, S., Ugolini, G., Spirito, F., Vignone, D., and Cattaneo, A. (2009). Development of a non invasive NGF-based therapy for Alzheimer’s disease. Curr. Alzheimer Res. 6, 158–170. doi: 10.2174/156720509787602870
Crabb, J. W., Miyagi, M., Gu, X., Shadrach, K., West, K. A., Sakaguchi, H., et al. (2002). Drusen proteome analysis:an approach to the etiology of age-related maculardegeneration. Proc. Natl. Acad. Sci. U S A. 99, 14682–14687. doi: 10.1073/pnas.222551899
Cuello, A. C., Bruno, M. A., Allard, S., Leon, W., and Iulita, M. F. (2010). Cholinergic involvement in Alzheimer’s disease. A link with NGF maturation and degradation. J. Mol. Neurosci. 40, 230–235. doi: 10.1007/s12031-009-9238-z
Cuello, A. C., Bruno, M. A., and Bell, K. F. (2007). NGF-cholinergic dependency in brain aging. MCI and Alzheimer’s disease. Curr. Alzheimer Res. 4, 351–358. doi: 10.2174/156720507781788774
Cui, Q. N., Bargoud, A. R., Ross, A. G., Song, Y., and Dunaief, J. L. (2020). Oral administration of the iron chelator deferiprone protects against loss of retinal ganglion cells in a mouse model of glaucoma. Exp. Eye Res. 193:107961. doi: 10.1016/j.exer.2020.107961
Currais, A., and Maher, P. (2013). Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid. Redox Signal. 19, 813–822. doi: 10.1089/ars.2012.4996
da Silva, S. L., Vellas, B., Elemans, S., Luchsinger, J., Kamphuis, P., Yaffe, K., et al. (2014). Plasma nutrient status of patients with Alzheimer’s disease: systematic review and meta-analysis. Alzheimers Dement. 10, 485–502. doi: 10.1016/j.jalz.2013.05.1771
Dalvi, S., Galloway, C. A., Winschel, L., Hashim, A., Soto, C., Tang, C., et al. (2019). Environmental stress impairs photoreceptor outer segment (POS) phagocytosis and degradation and induces autofluorescent material accumulation in hiPSC-RPE cells. Cell Death Discov. 5:96. doi: 10.1038/s41420-019-0171-9
Desai, B. S., Schneider, J. A., Li, J. L., Carvey, P. M., and Hendey, B. (2009). Evidence of angiogenic vessels in Alzheimer’s disease. J. Neural Transm. 116, 587–597. doi: 10.1007/s00702-009-0226-9
Di Fausto, V., Fiore, M., Tirassa, P., Lambiase, A., and Aloe, L. (2007). Eye drop NGF administration promotes the recovery of chemically injured cholinergic neurons of adult mouse forebrain. Eur. J. Neurosci. 26, 2473–2480. doi: 10.1111/j.1460-9568.2007.05883.x
Ding, J. D., Johnson, L. V., Herrmann, R., Farsiu, S., Smith, S. G., Groelle, M., et al. (2011). Anti-amyloid therapy protects against retinal pigmented epithelium damage and vision loss in a model of age-related macular degeneration. Proc. Natl. Acad. Sci. U S A. 108, E279–E287. doi: 10.1073/pnas.1100901108
Ding, J. D., Lin, J., Mace, B. E., Herrmann, R., Sullivan, P., and Bowes Rickman, C. (2008). Targeting age-related macular degeneration with Alzheimer’s disease based immunotherapies: anti-amyloid-beta antibody attenuates pathologies in an agerelated macular degeneration mouse model. Vision Res. 48, 339–345. doi: 10.1016/j.visres.2007.07.025
Ding, X., Patel, M., and Chan, C. C. (2009). Molecular pathology of age-related macular degeneration. Prog. Retin Eye Res. 28, 1–18. doi: 10.1016/j.preteyeres.2008.10.001
Diouf, I., Bush, A. I., Ayton, S., and Alzheimer’s disease Neuroimaging Initiative. (2020). Cerebrospinal fluid ceruloplasmin levels predict cognitive decline and brain atrophy in people with underlying β-amyloid pathology. Neurobiol. Dis. 139:104810. doi: 10.1016/j.nbd.2020.104810
Donoso, L. A., Kim, D., Frost, A., Callahan, A., and Hageman, G. (2006). The role of inflammation in the pathogenesis ofage-related macular degeneration. Surv. Ophthalmol. 51, 137–152. doi: 10.1016/j.survophthal.2005.12.001
Eftimiadi, G., Soligo, M., Manni, L., Di Giuda, D., Calcagni, M. L., and Chiaretti, A. (2021). Topical delivery of nerve growth factor for treatment of ocular and brain disorders. Neural Regen. Res. 16, 1740–1750. doi: 10.4103/1673-5374.306062
Emerich, D. F., Winn, S. R., Harper, J., Hammang, J. P., Baetge, E. E., and Kordower, J. H. (1994). Implants of polymer-encapsulated human NGF-secreting cells in the nonhumanprimate: rescue and sprouting of degenerating cholinergic basal forebrain neurons. J. Comp. Neurol. 349, 148–164. doi: 10.1002/cne.903490110
Eriksdotter Jönhagen, M., Nordberg, A., Amberla, K., Bäckman, L., Ebendal, T., Meyerson, B., et al. (1998). Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 9, 246–257. doi: 10.1159/000017069
Eriksdotter-Jönhagen, M., Linderoth, B., Lind, G., Aladellie, L., Almkvist, O., Andreasen, N., et al. (2012). Encapsulated cell biodelivery of nerve growth factor to the basal forebrain in patients with Azheimer’s disease. Dement. Geriatr. Cogn. Disord. 33, 18–28. doi: 10.1159/000336051
Erskine, L., and Herrera, E. (2014). Connecting the retina to the brain. ASN Neuro. 6:1759091414562107. doi: 10.1177/1759091414562107
Esposito, G., Balzamino, B. O., Bruno, L., Cacciamani, A., and Micera, A. (2021). NGF in Inflammatory and Neurodegenerative Diseases of the Eye: new Findings Supporting Neuroprotection and Proper Tissue Remodeling in Vitreoretinal Disorders,” in Recent Advances in NGF and Related Molecules, Advances in Experimental Medicine and Biology, eds L. Calzà, L. Aloe, and L. Giardino. (New York: Springer International Publishing).
Evans, J. R., and Lawrenson, J. G. (2017). Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst. Rev. 11:CD000254. doi: 10.1002/14651858.CD000254.pub4
Faller, P., Hureau, C., and Berthoumieu, O. (2013). Role of metal ions in the selfassembly of the Alzheimer’s amyloid-β peptide. Inorg. Chem. 52, 12193–12206. doi: 10.1021/ic4003059
Falsini, B., Chiaretti, A., Rizzo, D., Piccardi, M., Ruggiero, A., Manni, L., et al. (2016a). Nerve growth factor improves visual loss in childhood optic gliomas: a randomized, double-blind, phase II clinical trial. Brain 139, 404–414. doi: 10.1093/brain/awv366
Falsini, B., Iarossi, G., Chiaretti, A., Ruggiero, A., Manni, L., Galli-Resta, L., et al. (2016b). NGF eye-drops topical administration in patients with retinitis pigmentosa, a pilot study. J. Transl. Med. 14:8. doi: 10.1186/s12967-015-0750-3
Faustino, C., Patrícia Rijo, P., and Reis, C. P. (2017). Nanotechnological strategies for nerve growth factor delivery: therapeutic implications in Alzheimer’s disease. Pharmacol. Res. 120, 68–87. doi: 10.1016/j.phrs.2017.03.020
Feher, J., Kovacs, I., Artico, M., Cavallotti, C., Papale, A., and Balacco Gabrieli, C. (2006). Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 27, 983–993. doi: 10.1016/j.neurobiolaging.2005.05.012
Fine, S. L., Berger, J. W., Maguire, M. G., and Ho, A. C. (2000). Age related macular degeneration. N. Engl. J. Med. 342, 483–492. doi: 10.1056/NEJM200002173420707
Fischer, W., Wictorin, K., Björklund, A., Williams, L. R., Varon, S., and Gage, F. H. (1987). Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature 329, 65–68. doi: 10.1038/329065a0
Fjord-Larsen, L., Kusk, P., Tornoe, J., Juliusson, B., Torp, M., Bjarkam, C. R., et al. (2010). Long-term delivery of nerve growth factor by encapsulated cell biodelivery in the Göttingen minipig basal forebrain. Mol. Ther. 18, 2164–2172. doi: 10.1038/mt.2010.154
Frazzini, V., Rockabrand, E., Mocchegiani, E., and Sensi, S. L. (2006). Oxidative stress and brain aging: is zinc the link? Biogerontology 7, 307–314. doi: 10.1007/s10522-006-9045-7
Frost, S., Guymer, R., Aung, K. Z., Macaulay, S. L., Sohrabi, H. R., Bourgeat, P., et al. (2016). Alzheimer’s Disease and the Early Signs of Age-Related Macular Degeneration. Curr. Alzheimer Res. 13, 1259–1266. doi: 10.2174/1567205013666160603003800
Ganguly, U., Kaur, U., Chakrabarti, S. S., Sharma, P., Agrawal, B. K., Saso, L., et al. (2021). Oxidative Stress, Neuroinflammation, and NADPH Oxidase: implications in the Pathogenesis and Treatment of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2021:7086512. doi: 10.1155/2021/7086512
Garcia, T. B., Hollborn, M., and Bringmann, A. (2017). Expression and signaling of NGF in the healthy and injured retina. Cytokine Growth Factor Rev. 34, 43–57. doi: 10.1016/j.cytogfr.2016.11.005
Gehrs, K. M., Anderson, D. H., Johnson, L. V., and Hageman, G. S. (2006). Age-related macular degeneration – emerging pathogenetic and therapeutic concepts. Ann. Med. 38, 450–471. doi: 10.1080/07853890600946724
Gilbert, R., Peto, T., Lengyel, I., and Emri, E. (2019). Zinc Nutrition and Inflammation in the Aging Retina. Mol. Nutr. Food Res. 63:e1801049. doi: 10.1002/mnfr.201801049
Girijashanker, K., He, L., Soleimani, M., Reed, J. M., Li, H., Liu, Z., et al. (2008). Slc39a14 gene encodes ZIP14, a metal/bicarbonate symporter: similarities to the ZIP8 transporter. Mol. Pharmacol. 73, 1413–1423. doi: 10.1124/mol.107.043588
Godley, B. F., Shamsi, F. A., Liang, F. Q., Jarrett, S. G., Davies, S., and Boulton, M. (2005). Blue light induces mitochondrial damage and free radical production in epithelial cells. J. Biol. Chem. 280, 21061–21066. doi: 10.1074/jbc.M502194200
Golestaneh, N., Chu, Y., Xiao, Y. Y., Stoleru, G. L., and Theos, A. C. (2017). Dysfunctional autophagy in RPE, a contributing factor in age-relatedmacular degeneration. Cell Death. Dis. 8:e2537. doi: 10.1038/cddis.2016.453
Goozee, K., Chatterjee, P., James, I., Shen, K., Sohrabi, H. R., Asih, P. R., et al. (2018). Elevated plasma ferritin in elderly individuals with high neocortical amyloid-β load. Mol. Psychiatry 23, 1807–1812. doi: 10.1038/mp.2017.146
Güngör, E. D., Yülek, F., Serkant, U., Toklu, Y., Hocaoğlu, A., and S̨imsek, S. (2018). Blood lead and cadmium in age related macular degeneration in a Turkish urban population. J. Trace Elem. Med. Biol. 48, 16–19. doi: 10.1016/j.jtemb.2018.02.019
Guo, L., Davis, B. M., Ravindran, N., Galvao, J., Kapoor, N., Haamedi, N., et al. (2020). Topical recombinant human Nerve growth factor (rh-NGF) is neuroprotective to retinal ganglion cells by targeting secondary degeneration. Sci Rep. 10:3375. doi: 10.1038/s41598-020-60427-2
Gupta, V., Gupta, V. B., Chitranshi, N., Gangoda, S., Vander Wall, R., Abbasi, M., et al. (2016). One protein, multiple pathologies: multifaceted involvement of amyloid β in neurodegenerative disorders of the brain and retina. Cell. Mol. Life Sci. 73, 4279–4297. doi: 10.1007/s00018-016-2295-x
Habiba, U., Merlin, S., Lim, J. K. H., Wong, V. H. Y., Nguyen, C. T. O., Morley, J. W., et al. (2020). Age-Specific Retinal and Cerebral Immunodetection of Amyloid-beta Plaques and Oligomers in a Rodent Model of Alzheimer’s Disease. J. Alzheimers Dis. 76, 1135–1150. doi: 10.3233/JAD-191346
Heesterbeek, T. J., Rouhi-Parkouhi, M., Church, S. J., Lechanteur, Y. T., Lorés-Motta, L., Kouvatsos, N., et al. (2020). Association of plasma trace element levels with neovascular age-related macular degeneration. Exp. Eye Res. 201:108324. doi: 10.1016/j.exer.2020.108324
Hernandez-Zimbron, L. F., Zamora-Alvarado, R., Ochoa-De la Paz, L., Velez-Montoya, R., Zenteno, E., Gulias-Canizo, R., et al. (2018). Age-Related Macular Degeneration: new Paradigms for Treatment and Management of AMD. Oxid. Med. Cell Longev 2018:8374647. doi: 10.1155/2018/8374647
Hill, D., Compagnoni, C., and Cordeiro, M. F. (2021). Investigational neuroprotective compounds in clinical trials for retinal disease. Expert Opin. Investig. Drugs 30, 571–577. doi: 10.1080/13543784.2021.1896701
Hines, D. J., and Kaplan, D. L. (2013). Poly(lactic-co-glycolic acid) controlled release systems: experimental and modeling insights. Crit. Rev. Ther. Drug Carrier Syst. 30, 257–276. doi: 10.1615/critrevtherdrugcarriersyst.2013006475
Hoffman, D., Breakefield, X. O., Short, M. P., and Aebischer, P. (1993). Transplantation of apolymer-encapsulated cell line genetically engineered to release NGF. Exp. Neurol. 122, 100–106. doi: 10.1006/exnr.1993.1111
Hoffman, D., Wahlberg, L., and Aebischer, P. (1990). NGF released from a polymer matrix prevents loss of ChAT expression in basal forebrain neurons following a fimbria-fornix lesion. Exp. Neurol. 110, 39–44. doi: 10.1016/0014-4886(90)90049-x
Hsu, H. W., Rodriguez-Ortiz, C. J., Lim, S. L., Zumkehr, J., Kilian, J. G., Vidal, J., et al. (2019). Copper-Induced Upregulation of MicroRNAs Directs the Suppression of Endothelial LRP1 in Alzheimer’s Disease Model. Toxicol. Sci. 170, 144–156. doi: 10.1093/toxsci/kfz084
Huang, E. J., and Reichardt, L. F. (2001). Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736. doi: 10.1146/annurev.neuro.24.1.677
Isas, J. M., Luibl, V., Johnson, L. V., Kayed, R., Wetzel, R., Glabe, C. G., et al. (2010). Soluble and mature amyloid fibrilsin drusen deposits. Invest. Ophthalmol. Vis. Sci. 51, 1304–1310. doi: 10.1167/iovs.09-4207
Jager, R. D., Mieler, W. F., and Miller, J. W. (2008). Age-related macular degeneration. N. Engl. J. Med. 358, 2606–2617. doi: 10.1056/NEJMra0801537
Jindal, V. (2015). Interconnection Between Brain and Retinal Neurodegenerations. Mol. Neurobiol. 51, 885–892. doi: 10.1007/s12035-014-8733-6
Johnson, L. V., Leitner, W. P., Rivest, A. J., Staples, M. K., Radeke, M. J., and Anderson, D. H. (2002). The Alzheimer’s A beta -peptide is deposited at sites of complementactivation in pathologic deposits associated with aging and age-related macular degeneration. Proc. Natl. Acad. Sci. U. S. A. 99, 11830–11835. doi: 10.1073/pnas.192203399
Johnson, P. J., Skornia, S. L., Stabenfeldt, S. E., and Willits, R. K. (2008). Maintaining bioactivity of NGF for controlled release from PLGA using PEG. J. Biomed. Mater. Res. A. 86, 420–427. doi: 10.1002/jbm.a.31635
Kaarniranta, K., Hyttinen, J., Ryhanen, T., Viiri, J., Paimela, T., Toropainen, E., et al. (2010). Mechanisms of protein aggregation in the retinal pigment epithelial cells. Front. Biosci. (Elite Ed.) 2, 1374–1384. doi: 10.2741/e198
Kaarniranta, K., Salminen, A., Haapasalo, A., Soininen, H., and Hiltunen, M. (2011). Age-related macular degeneration (AMD): Alzheimer’s disease in the eye? J. Alzheimers Dis. 24, 615–631. doi: 10.3233/JAD-2011-101908
Kabir, M. T., Uddin, M. S., Zaman, S., Begum, Y., Ashraf, G. M., Bin-Jumah, M. N., et al. (2021). Molecular Mechanisms of Metal Toxicity in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 58, 1–20. doi: 10.1007/s12035-020-02096-w
Katta, S., Kaur, I., and Chakrabarti, S. (2009). The molecular genetic basis of age-related macular degeneration: an overview. J. Genet. 88, 425–449. doi: 10.1007/s12041-009-0064-4
Ke, J., Tian, Q., Xu, Q., Fu, Z., and Fu, Q. (2021). Mitochondrial dysfunction: a potential target for Alzheimer’s disease intervention and treatment. Drug Discov. Today doi: 10.1016/j.drudis.2021.04.025 [Epub Online ahead of Print].
Kepp, K. P., and Squitti, R. (2019). Copper imbalance in Alzheimer’s disease: convergence of the chemistry and the clinic Coordination. Chemistry 397, 168–187. doi: 10.1016/j.ccr.2019.06.018
Kim, M. H., Zhao, D., Cho, J., and Guallar, E. (2016). Cadmium exposure and age-related macular degeneration. J. Expo. Sci. Environ. Epidemiol. 26, 214–218. doi: 10.1038/jes.2014.75
King, A., Gottlieb, E., Brooks, D. G., Murphy, M. P., and Dunaief, J. L. (2004). Mitochondria-derived reactive oxygen species mediate blue light-induced death of retinal pigment epithelial cells. Photochem. Photobiol. 79, 470–475. doi: 10.1562/le-03-17.1
Kivipelto, M., Helkala, E. L., Laakso, M. P., Hanninen, T., Hallikainen, M., Alhainen, K., et al. (2001). Midlife vascular risk factors andAlzheimer’s disease in later life: longitudinal, population based study. BMJ 322, 1447–1451. doi: 10.1136/bmj.322.7300.1447
Kivipelto, M., Helkala, E. L., Laakso, M. P., Hanninen, T., Hallikainen, M., Alhainen, K., et al. (2002). Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann. Intern. Med. 137, 149–155. doi: 10.7326/0003-4819-137-3-200208060-00006
Kivipelto, M., Ngandu, T., Fratiglioni, L., Viitanen, M., Kareholt, I., Winblad, B., et al. (2005). Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 62, 1556–1560. doi: 10.1001/archneur.62.10.1556
Klein, R. L., Hirko, A. C., Meyers, C. A., Grimes, J. R., Muzyczka, N., and Meyer, E. M. (2000). NGF gene transfer to intrinsic basal forebrain neurons increases cholinergic cell size and protects from age-related, spatial memory deficits in middle-aged rats. Brain Res. 875, 144–151. doi: 10.1016/s0006-8993(00)02634-2
Klein, R., Peto, T., Bird, A., and Vannewkirk, M. R. (2004). The epidemiology of age-related macular degeneration. Am. J. Ophthalmol. 137, 486–495. doi: 10.1016/j.ajo.2003.11.069
Kordower, J. H., Winn, S. R., Liu, Y. T., Mufson, E. J., Sladek, J. R. Jr., Hammang, J. P., et al. (1994). The aged monkey basal forebrain: rescue and sprouting of axotomized basal forebrain neurons after grafts of encapsulated cells secreting human nerve growth factor. Proc. Natl. Acad. Sci. U. S. A. 91, 10898–109902. doi: 10.1073/pnas.91.23.10898
Koronyo-Hamaoui, M., Koronyo, Y., Ljubimov, A. V., Miller, C. A., Ko, M. K., Black, K. L., et al. (2011). Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 54, S204–S217. doi: 10.1016/j.neuroimage.2010.06.020
Lambiase, A., Coassin, M., Tirassa, P., Mantelli, F., and Aloe, L. (2009). Nerve growth factor eye drops improve visual acuity and electrofunctional activity in age-related macular degeneration: a case report. Ann. Ist. Super. Sanita. 45, 439–442. doi: 10.1590/s0021-25712009000400014
Lambiase, A., Pagani, L., Di Fausto, V., Sposato, V., Coassin, M., Bonini, S., et al. (2007). Nerve growth factor eye drop administrated on the ocular surface of rodents affects the nucleus basalis and septum: biochemical and structural evidence. Brain Res. 1127, 45–51. doi: 10.1016/j.brainres.2006.09.102
Lambiase, A., Tirassa, P., Micera, A., Aloe, L., and Bonini, S. (2005). Pharmacokinetics of conjunctivally applied nerve growth factor in the retina and optic nerve of adult rats. Invest. Ophthalmol. Vis. Sci. 46, 3800–3806. doi: 10.1167/iovs.05-0301
Lenzi, L., Coassin, M., Lambiase, A., Bonini, S., Amendola, T., and Aloe, L. (2005). Effect of exogenous administration of nerve growth factor in the retina of rats with inherited retinitis pigmentosa. Vision Res. 45, 1491–1500. doi: 10.1016/j.visres.2004.12.020
Li, Y., Jiao, Q., Xu, H., Du, X., Shi, L., Jia, F., et al. (2017). Biometal Dyshomeostasis and Toxic Metal Accumulations in the Development of Alzheimer’s Disease. Front. Mol. Neurosci. 10:339. doi: 10.3389/fnmol.2017.00339
Liang, F. Q., and Godley, B. F. (2003). Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp. Eye Res. 76, 397–403. doi: 10.1016/s0014-4835(03)00023-x
Lindner, M. D., Kearns, C. E., Winn, S. R., Frydel, B., and Emerich, D. F. (1996). Effects ofintraventricular encapsulated hNGF-secreting fibroblasts in aged rats. Cell Transplant. 5, 205–223. doi: 10.1016/0963-6897(95)02029-2
Loeffler, K. U., Edward, D. P., and Tso, M. O. (1993). Tau-2 immunoreactivity of corpora amylacea in the human retina and optic nerve. Invest. Ophthalmol. Vis. Sci. 34, 2600–2603.
Logue, M. W., Schu, M., Vardarajan, B. N., Farrell, J., Lunetta, K. L., Jun, G., et al. (2014). Search for age-related macular degeneration risk variants in Alzheimer disease genes and pathways. Neurobiol. Aging 35, 1510.e7–18. doi: 10.1016/j.neurobiolaging.2013.12.007
Luibl, V., Isas, J. M., Kayed, R., Glabe, C. G., Langen, R., and Chen, J. (2006). Drusen deposits associated with aging and agerelatedmacular degeneration contain nonfibrillar amyloidoligomers. J. Clin. Invest. 116, 378–385. doi: 10.1172/JCI25843
Malerba, F., Paoletti, F., Bruni Ercole, B., Materazzi, S., Nassini, R., Coppi, E., et al. (2015). Functional Characterization of Human ProNGF and NGF Mutants: identification of NGF P61SR100E as a “Painless” Lead Investigational Candidate for Therapeutic Applications. PLoS One 10:e0136425. doi: 10.1371/journal.pone.0136425
Malerba, F., Paoletti, F., Capsoni, S., and Cattaneo, A. (2011). Intranasal delivery of therapeutic proteins for neurological diseases. Expert. Opin. Drug Deliv. 8, 1277–1296. doi: 10.1517/17425247.2011.588204
Mandel, R. J. (2010). CERE-110, an adeno-associated virus-based gene delivery vector expressing human nerve growth factor for the treatment of Alzheimer’s disease. Curr. Opin. Mol. Ther. 12, 240–247.
Marcus, M., Smith, A., Maswadeh, A., Shemesh, Z., Zak, I., Motiei, M., et al. (2018). Magnetic Targeting of Growth Factors Using Iron Oxide Nanoparticles. Nanomaterials (Basel) 8:707. doi: 10.3390/nano8090707
Markowska, A. L., Koliatsos, V. E., Breckler, S. J., Price, D. L., and Olton, D. S. (1994). Human nerve growth factor improves spatial memory in aged but not in young rats. J. Neurosci. 14, 4815–4824. doi: 10.1523/JNEUROSCI.14-08-04815.1994
Markowska, A. L., Price, D., and Koliatsos, V. E. (1996). Selective effects of nerve growth factor on spatial recent memory as assessed by a delayed nonmatching-to-position task in the water maze. J. Neurosci. 16, 3541–3548. doi: 10.1523/JNEUROSCI.16-10-03541.1996
Martínez-Serrano, A., Fischer, W., and Björklund, W. A. (1995). Reversal of age-dependent cognitive impairments and cholinergic neuron atrophy by NGF-secreting neural progenitors grafted to the basal forebrain. Neuron 15, 473–484. doi: 10.1016/0896-6273(95)90051-9
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., and Cummings, J. L. (2015). Alzheimer’s disease. Nat. Rev. Dis. Prim. 1:15056. doi: 10.1038/nrdp.2015.56
Masuzzo, A., Dinet, V., Cavanagh, C., Mascarelli, F., and Krantic, S. (2016). Amyloidosis in Retinal Neurodegenerative Diseases. Front. Neurol. 7:127. doi: 10.3389/fneur.2016.00127
Mathys, Z. K., and White, A. R. (2017). Copper and Alzheimer’s Disease. Adv. Neurobiol. 18, 199–216. doi: 10.1007/978-3-319-60189-2_10
Mayeux, R., and Stern, Y. (2012). Epidemiology of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2:a006239. doi: 10.1101/cshperspect.a006239
McGeer, E. G., and McGeer, P. L. (2010). Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J. Alzheimers Dis. 19, 355–361. doi: 10.3233/JAD-2010-1219
McGeer, E. G., Klegeris, A., and McGeer, P. L. (2005). Inflammation, the complement system and the diseases of aging. Neurobiol. Aging 26, 94–97. doi: 10.1016/j.neurobiolaging.2005.08.008
McGeer, P. L., and McGeer, E. G. (2004). Inflammation and the degenerative diseases of aging. Ann. NY. Acad. Sci. 1035, 104–116. doi: 10.1196/annals.1332.007
Mesentier-Louro, L. A., Rosso, P., Carito, V., Mendez-Otero, R., Santiago, M. F., Rama, P., et al. (2019). Nerve Growth Factor Role on Retinal Ganglion Cell Survival and Axon Regrowth: effects of Ocular Administration in Experimental Model of Optic Nerve Injury. Mol Neurobiol. 56, 1056–1069. doi: 10.1007/s12035-018-1154-1
Micera, A., Bruno, L., Cacciamani, A., Rongioletti, M., and Squitti, R. (2019). Alzheimer’s Disease and Retinal Degeneration: a Glimpse at Essential Trace Metals in Ocular Fluids and Tissues. Curr. Alzheimer Res. 16, 1073–1083. doi: 10.2174/1567205016666191023114015
Micera, A., Lambiase, A., Aloe, L., Bonini, S., Levi-Schaffer, F., and Bonini, S. (2004). Nerve growth factor involvement in the visual system: implications in allergic and neurodegenerative diseases. Cytokine Growth Factor Rev. 15, 411–417. doi: 10.1016/j.cytogfr.2004.09.003
Micera, A., Lambiase, A., Stampachiacchiere, B., Bonini, S., Bonini, S., and Levi-Schaffer, F. (2007). Nerve growth factor and tissue repair remodeling: trkA (NGFR) and p75(NTR), two receptors one fate. Cytokine Growth Factor Rev. 18, 245–256. doi: 10.1016/j.cytogfr.2007.04.004
Miller, J. W. (2013). Age-related macular degeneration revisited - Piecing the puzzle: the LXIX edward jackson memorial lecture. Am. J. Ophthalmol. 155, 1–35. doi: 10.1016/j.ajo.2012.10.018
Mitra, S., Behbahani, H., and Eriksdotter, M. (2019). Innovative Therapy for Alzheimer’s Disease-With Focus on Biodelivery of NGF. Front. Neurosci. 13:38. doi: 10.3389/fnins.2019.00038
Mitter, S. K., Song, C., Qi, X., Mao, H., Rao, H., Akin, D., et al. (2014). Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 10, 1989–2005. doi: 10.4161/auto.36184
Mufson, E. J., Counts, S. E., Ginsberg, S. D., Mahady, L., Perez, S. E., Massa, S. M., et al. (2019). Nerve Growth Factor Pathobiology During the Progression of Alzheimer’s Disease. Front. Neurosci. 13:533. doi: 10.3389/fnins.2019.00533
Mufson, E. J., Counts, S. E., Perez, S. E., and Ginsberg, S. D. (2008). Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert. Rev. Neurother. 8, 1703–1718. doi: 10.1586/14737175.8.11.1703
Mullins, R. F., Russell, S. R., Anderson, D. H., and Hageman, G. S. (2000). Drusen associated with aging and age-related maculardegeneration contain proteins common to extracellulardeposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 14, 835–846.
Nagahara, A. H., Bernot, T., Moseanko, R., Brignolo, L., Blesch, A., Conner, J. M., et al. (2009). Long-term reversal of cholinergic neuronal decline in aged nonhuman primates by lentiviral NGF gene delivery. Exp. Neurol. 215, 153–159. doi: 10.1016/j.expneurol.2008.10.004
Newman, A. B., Fitzpatrick, A. L., Lopez, O., Jackson, S., Lyketsos, C., Jagust, W., et al. (2005). Dementia and Alzheimer’s disease incidence in relationship to cardiovascular disease in the Cardiovascular Health Study cohort. J. Am. Geriatr. Soc. 53, 1101–1107. doi: 10.1111/j.1532-5415.2005.53360.x
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767. doi: 10.1093/jnen/60.8.759
Ohno-Matsui, K. (2011). Parallel findings in age-related macular degeneration and Alzheimer’s disease. Prog. Retin. Eye Res. 30, 217–238. doi: 10.1016/j.preteyeres.2011.02.004
Olson, L., Nordberg, A., von Holst, H., Backman, L., Ebendal, T., Alafuzoff, I., et al. (1992). Nerve growth factor affects 11C-nicotine binding, blood flow, EEG, andverbal episodic memory in an Alzheimer patient (case report). J. Neural Transm. Park. Dis. Dement. Sect. 4, 79–95. doi: 10.1007/BF02257624
Ong, S. S., Proia, A. D., Whitson, H. E., Farsiu, S., Doraiswamy, P. M., and Lad, E. M. (2019). Ocular amyloid imaging at the crossroad of Alzheimer’s disease and age-related macular degeneration: implications for diagnosis and therapy. J. Neurol. 266, 1566–1577. doi: 10.1007/s00415-018-9028-z
Pardue, M. T., and Allen, R. S. (2018). Neuroprotective strategies for retinal disease. Prog. Retin. Eye Res. 65, 50–76. doi: 10.1016/j.preteyeres.2018.02.002
Park, D., Joo, S. S., Kim, T. K., Lee, S. H., Kang, H., Lee, H. J., et al. (2012). Human neural stem cells overexpressing choline acetyltransferase restore cognitive function of kainic acid-induced learning and memory deficit animals. Cell. Transplant. 21, 365–371. doi: 10.3727/096368911X586765
Park, S. W., Im, S., Jun, H. O., Lee, K., Park, Y. J., Kim, J. H., et al. (2017). Dry age-related macular degeneration like pathology in aged 5XFAD mice: ultrastructure and microarray analysis. Oncotarget 8, 40006–40018. doi: 10.18632/oncotarget.16967
Patel, A., Cholkar, K., Agrahari, V., and Mitra, A. K. (2013). Ocular drug delivery systems: an overview. World J. Pharmacol. 2, 47–64. doi: 10.5497/wjp.v2.i2.47
Picard, E., Le Rouzic, Q., Oudar, A., Berdugo, M., El Sanharawi, M., Andrieu-Soler, C., et al. (2015). Targeting iron-mediated retinal degeneration by local delivery of transferrin. Free Radic. Biol. Med. 89, 1105–11021. doi: 10.1016/j.freeradbiomed.2015.08.018
Pinnen, F., Sozio, P., Cacciatore, I., Cornacchia, C., Mollica, A., Iannitelli, A., et al. (2011). Ibuprofen and glutathione conjugate as a potential therapeutic agent for treating Alzheimer’s disease. Arch. Pharm. (Weinheim) 344, 139–148. doi: 10.1002/ardp.201000209
Pizzo, D. P., and Thal, L. J. (2004). Intraparenchymal nerve growth factor improves behavioral deficits while minimizing the adverse effects of intraventricular delivery. Neuroscience 124, 743–755. doi: 10.1016/j.neuroscience.2003.12.041
Polidori, M. C., Marvardi, M., Cherubini, A., Senin, U., and Mecocci, P. (2001). Heart disease and vascular risk factors in the cognitively impaired elderly: implications for Alzheimer’s dementia. Aging (Milano) 13, 231–239. doi: 10.1007/bf03351481
Powell, E. M., Sobarzo, M. R., and Saltzman, W. M. (1990). Controlled release of nerve growth factor from a polymeric implant. Brain Res. 515, 309–311. doi: 10.1016/0006-8993(90)90612-f
Prince, M., Guerchet, M., and Prina, M. (2013). The Global Impact of Dementia 2013-2050. London: Alzheimer’s Disease International.
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Rafii, M. S., Baumann, T. L., Bakay, R. A., Ostrove, J. M., Siffert, J., Fleisher, A. S., et al. (2014). A phase 1study of stereotactic gene delivery of AAV2-NGF for Alzheimer’s disease. Alzheimers Dement. 10, 571–581. doi: 10.1016/j.jalz.2013.09.004
Rafii, M. S., Tuszynski, M. H., Thomas, R. G., Barba, D., Brewer, J. B., Rissman, R. A., et al. (2018). Adeno-associated viral vector (serotype 2)-nerve growth factor for patients with Alzheimer disease: a randomized clinical trial. JAMA Neurol. 75, 834–841. doi: 10.1001/jamaneurol.2018.0233
Rami, A. (2009). Autophagy in neurodegeneration: firefighter and/or incendiarist? Neuropathol. Appl. Neurobiol. 35, 449–461. doi: 10.1111/j.1365-2990.2009.01034.x
Reitz, C., and Mayeux, R. (2014). Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 88, 640–651. doi: 10.1016/j.bcp.2013.12.024
Roberti, G., Mantelli, F., Macchi, I., Massaro-Giordano, M., and Centofanti, M. (2014). Nerve growth factor modulation of retinal ganglion cell physiology. J. Cell Physiol. 229, 1130–1133. doi: 10.1002/jcp.24573
Rozzini, L., Lanfranchi, F., Pilotto, A., Catalani, S., Gilberti, M. E., Paganelli, M., et al. (2018). Serum Non-Ceruloplasmin Non-Albumin Copper Elevation in Mild Cognitive Impairment and Dementia due to Alzheimer’s Disease: a Case Control Study. J. Alzheimers Dis. 61, 907–912. doi: 10.3233/JAD-170552
Rudnicka, A. R., Jarrar, Z., Wormald, R., Cook, D. G., Fletcher, A., and Owen, C. G. (2012). Age and gender variations in age-related macular degeneration prevalence in populations of European ancestry: a meta-analysis. Ophthalmology 119, 571–580. doi: 10.1016/j.ophtha.2011.09.027
Ryhänen, T., Hyttinen, J. M., Kopitz, J., Rilla, K., Kuusisto, E., Mannermaa, E., et al. (2009). Crosstalk between Hsp70 molecular chaperone, lysosomes and proteasomes in autophagy mediatedproteolysis in human retinal pigment epithelial cells. J. Cell. Mol. Med. 13, 3616–3631. doi: 10.1111/j.1582-4934.2008.00577.x
Sacchetti, M., Mantelli, F., Rocco, M. L., Micera, A., Brandolini, L., Focareta, L., et al. (2017). Recombinant Human Nerve Growth Factor Treatment Promotes Photoreceptor Survival in the Retinas of Rats with Retinitis Pigmentosa. Curr Eye Res. 42, 1064–1068. doi: 10.1080/02713683.2017.1279634
Schulte-Herbruggen, O., Jockers-Scherubl, M. C., and Hellweg, R. (2008). Neurotrophins: from pathophysiology to treatment in Alzheimer’s disease. Curr. Alzheimer Res. 5, 38–44. doi: 10.2174/156720508783884620
Schutt, F., Bergmann, M., Holz, F. G., and Kopitz, J. (2003). Proteins modified by malondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 44, 3663–3668.
Schutt, F., Ueberle, B., Schnolzer, M., Holz, F. G., and Kopitz, J. (2002). Proteome analysis of lipofuscin in human retinal pigment epithelial cells. FEBS Lett. 528, 217–221. doi: 10.1016/s0014-5793(02)03312-4
Seddon, J. M., Cote, J., Page, W. F., Aggen, S. H., and Neale, M. C. (2005). The US twin study of age-related macular degeneration: relative roles of genetic and environmental influences. Arch. Ophthalmol. 123, 321–327. doi: 10.1001/archopht.123.3.321
Seddon, J. M., Silver, R. E., and Rosner, B. (2016). Response to AREDS supplements according to genetic factors: survival analysis approach using the eye as the unit of analysis. Br. J. Ophthalmol. 100, 1731–1737. doi: 10.1136/bjophthalmol-2016-308624
Seiger, A., Nordberg, A., von Holst, H., Bäckman, L., Ebendal, T., Alafuzoff, I., et al. (1993). Intracranial infusion of purified nerve growth factor to an Alzheimer patient: the first attempt of a possible future treatment strategy. Behav. Brain Res. 57, 255–261. doi: 10.1016/0166-4328(93)90141-c
Sensi, S. L., Granzotto, A., Siotto, M., and Squitti, R. (2018). Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends. Pharmacol. Sci. 39, 1049–1063. doi: 10.1016/j.tips.2018.10.001
Shu, W., and Dunaief, J. L. (2018). Potential treatment of retinal diseases with iron chelators. Pharmaceuticals (Basel) 11:112. doi: 10.3390/ph11040112
Singh, I., Sagare, A. P., Coma, M., Perlmutter, D., Gelein, R., Bell, R. D., et al. (2013). Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. U. S. A. 110, 14771–14776. doi: 10.1073/pnas.1302212110
Siotto, M., Simonelli, I., Pasqualetti, P., Mariani, S., Caprara, D., Bucossi, S., et al. (2016). Association Between Serum Ceruloplasmin Specific Activity and Risk of Alzheimer’s Disease. J. Alzheimers Dis. 50, 1181–1189. doi: 10.3233/JAD-150611
Sivak, J. M. (2013). The aging eye: common degenerative mechanisms between the Alzheimer’s brain and retinal disease. Invest. Ophthalmol. Vis. Sci. 54, 871–880. doi: 10.1167/iovs.12-10827
Sofroniew, M. V., Howe, C. L., and Mobley, W. C. (2001). Nerve growth factor signaling, neuroprotection, and neural repair. Annu. Rev. Neurosci. 24, 1217–1281. doi: 10.1146/annurev.neuro.24.1.1217
Song, B., Song, J., Zhang, S., Anderson, M. A., Ao, Y., Yang, C. Y., et al. (2012). Sustained local delivery of bioactive nerve growth factor in the central nervous system via tunable diblock copolypeptide hydrogel depots. Biomaterials 33, 9105–9116. doi: 10.1016/j.biomaterials.2012.08.060
Song, D., Song, Y., Hadziahmetovic, M., Zhong, Y., and Dunaief, J. L. (2012). Systemic administration of the iron chelator deferiprone protects against light-induced photoreceptor degeneration in the mouse retina. Free Radic. Biol. Med. 53, 64–71. doi: 10.1016/j.freeradbiomed.2012.04.020
Sparks, D. L., and Schreurs, B. G. (2003). Trace amounts of copper in water induce β-amyloid plaques and learning deficits in a rabbit model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 100, 11065–11069. doi: 10.1073/pnas.1832769100
Spires-Jones, T. L., and Hyman, B. T. (2014). The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 82, 756–771. doi: 10.1016/j.neuron.2014.05.004
Squitti, R. (2012). Copper dysfunction in Alzheimer’s disease: from meta-analysis of biochemical studies to new insight into genetics. J. Trace Elem. Med. Biol. 26, 93–96. doi: 10.1016/j.jtemb.2012.04.012
Squitti, R., Ghidoni, R., Simonelli, I., Ivanova, I. D., Colabufo, N. A., Zuin, M., et al. (2018). Copper dyshomeostasis in Wilson disease and Alzheimer’s disease as shown by serum and urine copper indicators. J. Trace Elem. Med. Biol. 45, 181–188. doi: 10.1016/j.jtemb.2017.11.005
Squitti, R., Ghidoni, R., Siotto, M., Ventriglia, M., Benussi, L., Paterlini, A., et al. (2014). Value of serum nonceruloplasmin copper for prediction of mild cognitive impairment conversion to Alzheimer disease. Ann. Neurol. 75, 574–580. doi: 10.1002/ana.24136
Squitti, R., Pal, A., Picozza, M., Avan, A., Ventriglia, M., Rongioletti, M. C., et al. (2020). Zinc Therapy in Early Alzheimer’s Disease: safety and Potential Therapeutic Efficacy. Biomolecules 10:1164. doi: 10.3390/biom10081164
Squitti, R., Polimanti, R., Bucossi, S., Ventriglia, M., Mariani, S., Manfellotto, D., et al. (2013). Linkage disequilibrium and haplotype analysis of the ATP7B gene in Alzheimer’s disease. Rejuvenation Res. 16, 3–10. doi: 10.1089/rej.2012.1357
Squitti, R., Salustri, C., Siotto, M., Ventriglia, M., Vernieri, F., Lupoi, D., et al. (2010). Ceruloplasmin/Transferrin ratio changes in Alzheimer’s disease. Int. J. Alzheimers Dis. 2011:231595. doi: 10.4061/2011/231595
Squitti, R., Siotto, M., Cassetta, E., Imane Ghafir El Idrissi, and Colabufo, N. A. (2017). Measurements of serum non-ceruloplasmin copper by a direct fluorescent method specific to Cu(II). Clin. Chem. Lab. Med. 55, 1360–1367. doi: 10.1515/cclm-2016-0843
Telegina, D. V., Kolosova, N. G., and Kozhevnikova, O. S. (2019). Immunohistochemical localization of NGF. BDNF, and their receptors in a normal and AMD-like rat retina. BMC Med. Genomics 12:48. doi: 10.1186/s12920-019-0493-8
Testa, G., Cattaneo, A., and Capsoni, S. (2021). Understanding pain perception through genetic painlessness diseases: the role of NGF and proNGF. Pharmacol. Res. 169:105662. doi: 10.1016/j.phrs.2021.105662
Tirassa, P. (2011). The nerve growth factor administrated as eye drops activates mature and precursor cells in subventricular zone of adult rats. Arch. Ital. Biol. 149, 205–213. doi: 10.4449/aib.v149i1.1359
Tirassa, P., Rosso, P., and Iannitelli, A. (2018). Ocular Nerve Growth Factor (NGF) and NGF Eye Drop Application as Paradigms to Investigate NGF Neuroprotective and Reparative Actions Methods. Mol. Biol. 1727, 19–38. doi: 10.1007/978-1-4939-7571-6_2
Tosi, G., Duskey, J. T., and Kreuter, J. (2020). Nanoparticles as carriers for drug delivery of macromolecules across the blood-brain barrier. Expert Opin. Drug Deliv. 17, 23–32. doi: 10.1080/17425247.2020.1698544
Tsiloulis, A. N., Zacharaki, F., Kotoula, M. G., Chatzoulis, D. Z., Morrison, M. A., Mayne, K., et al. (2016). Genetic variants in complement pathway and ARMS2/HTRA1 genes and risk of age-related macular degeneration in a homogeneous population from central Greece. Ophthalmic Genet. 37, 339–344. doi: 10.3109/13816810.2015.1045525
Tuszynski, M. H. (2000). Intraparenchymal NGF infusions rescue degenerating cholinergic neurons. Cell Transplant. 9, 629–636. doi: 10.1177/096368970000900508
Tuszynski, M. H., Thal, L., Pay, M., Salmon, D. P., U, H. S., Bakay, R., et al. (2005). A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 11, 551–555. doi: 10.1038/nm1239
Tuszynski, M. H., U, H. S., Amaral, D. G., and Gage, F. H. (1990). Nerve growth factor infusion in the primate brain reduces lesion-induced cholinergic neuronal degeneration. J. Neurosci. 10, 3604–3614.
Tuszynski, M. H., Yang, J. H., Barba, D., U, H. S., Bakay, R. A., Pay, M. M., et al. (2015). Nerve growth factor gene therapy: activation of neuronal responses in Alzheimer disease. JAMA Neurol. 72, 1139–1147.
Ugarte, M., Osborne, N. N., Brown, L. A., and Bishop, P. N. (2013). Iron, zinc, and copper in retinal physiology and disease. Surv. Ophthalmol. 58, 585–609. doi: 10.1016/j.survophthal.2012.12.002
Umeda, T., Tomiyama, T., Sakama, N., Tanaka, S., Lambert, M. P., Klein, W. L., et al. (2011). Intraneuronal amyloid β oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J. Neurosci. Res. 89, 1031–1042. doi: 10.1002/jnr.22640
Usui, H., Nishiwaki, A., Landiev, L., Kacza, J., Eichler, W., Wako, R., et al. (2018). In Vitro drusen model—Three-dimensional spheroid culture of retinal pigment epithelial cells. J. Cell Sci. 132:jcs215798. doi: 10.1242/jcs.215798
van Lookeren Campagne, M., LeCouter, J., Yaspan, B. L., and Ye, W. (2014). Mechanisms of age-related macular degeneration and therapeutic opportunities. J. Pathol. 232, 151–164. doi: 10.1002/path.4266
Ventriglia, M., Brewer, G. J., Simonelli, I., Mariani, S., Siotto, M., Bucossi, S., et al. (2015). Zinc in Alzheimer’s disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J. Alzheimers Dis. 46, 75–87. doi: 10.3233/JAD-141296
Wahlberg, L. U., Lind, G., Almqvist, P. M., Kusk, P., Tornoe, J., Juliusson, B., et al. (2012). Targeted delivery of nerve growth factor via encapsulated cell biodelivery in Alzheimer disease: atechnology platform for restorative neurosurgery. J. Neurosurg. 117, 340–347. doi: 10.3171/2012.2.JNS11714
Walsh, D. M., and Selkoe, D. J. (2004). Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44, 181–193. doi: 10.1016/j.neuron.2004.09.010
Wang, L., and Mao, X. (2021). Role of Retinal Amyloid-beta in Neurodegenerative Diseases: overlapping Mechanisms and Emerging Clinical Applications. Int. J. Mol. Sci. 22:2360. doi: 10.3390/ijms22052360
Wang, L., Clark, M. E., Crossman, D. K., Kojima, K., Messinger, J. D., Mobley, J. A., et al. (2010). Abundant Lipid and Protein Components of Drusen. PLoS One 5:e10329. doi: 10.1371/journal.pone.0010329
Wang, Z. X., Tan, L., Wang, H. F., Ma, J., Liu, J., Tan, M.-S., et al. (2015). Serum Iron, Zinc, and Copper Levels in Patients with Alzheimer’s Disease: a Replication Study and Meta-Analyses. J. Alzheimers Dis. 47, 565–581. doi: 10.3233/JAD-143108
Wen, L. Y., Wan, L., Lai, J. N., Chen, C. S., Chen, J. J. Y., Wu, M. Y., et al. (2021). Increased risk of Alzheimer’s disease among patients with age-related macular degeneration: a nationwide population-based study. PLoS One 16:e0250440. doi: 10.1371/journal.pone.0250440
White, A. R., Multhaup, G., Maher, F., Bellingham, S., Camakaris, J., Zheng, H., et al. (1999). The Alzheimer’s disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J. Neurosci. 19, 9170–9179. doi: 10.1523/JNEUROSCI.19-21-09170
Williams, M. A., Silvestri, V., Craig, D., Passmore, A. P., and Silvestri, G. (2014). The prevalence of age-related macular degeneration in Alzheimer’s disease. J. Alzheimers Dis. 42, 909–914. doi: 10.3233/JAD-140243
Wills, N. K., Ramanujam, V. M. S., Chang, J., Kalariya, N., Lewis, J. R., Weng, T. X., et al. (2008). Cadmium accumulation in the human retina: effects of age, gender, and cellular toxicity. Exp. Eye Res. 86, 41–51. doi: 10.1016/j.exer.2007.09.005
Wong, E., and Cuervo, A. M. (2010). Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 13, 805–811. doi: 10.1038/nn.2575
Wong, W. L., Su, X., Li, X., Cheung, C. M. G., Klein, R., Cheng, C. Y., et al. (2014). Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob. Health 2, e106–16. doi: 10.1016/S2214-109X(13)70145-1
Woo, S. J., Ahn, J., Morrison, M. A., Ahn, S. Y., Lee, J., Kim, K. W., et al. (2015). Analysis of genetic and environmental risk factors and their interactions in Korean patients with age-related macular degeneration. PLoS One 10:e0132771. doi: 10.1371/journal.pone.0132771
Wu, E. W., Schaumberg, D. A., and Park, S. K. (2014). Environmental cadmium and lead exposures and age-related macular degeneration in U.S. adults: the National Health and Nutrition Examination Survey 2005 to 2008. Environ. Res. 133, 178–184. doi: 10.1016/j.envres.2014.05.023
Wu, S., Sasaki, A., Yoshimoto, R., Kawahara, Y., Manabe, T., Kataoka, K., et al. (2008). Neural stem cells improve learning and memory in rats with Alzheimer’s disease. Pathobiology 75, 186–194. doi: 10.1159/000124979
Xu, Q., Cao, S., Rajapakse, S., and Matsubara, J. A. (2018). Understanding AMD by analogy: systematic review of lipid-related common pathogenic mechanisms in AMD, AD, AS and GN. Lipids Health Dis. 17:3. doi: 10.1186/s12944-017-0647-7
Yang, C. Y., Song, B., Ao, Y., Nowak, A. P., Abelowitz, R. B., Korsak, R. A., et al. (2009). Biocompatibilityof amphiphilic diblock copolypeptide hydrogels in the central nervous system. Biomaterials 30, 2881–2898. doi: 10.1016/j.biomaterials.2009.01.056
Yao, D., Jing, T., Niu, L., Huang, X., Wang, Y., Deng, X., et al. (2018). Amyloidogenesis induced by diet cholesterol and copper in a model mouse for Alzheimer’s disease and protection effects of zinc and fluvastatin. Brain Res. Bull. 143, 1–8. doi: 10.1016/j.brainresbull.2018.09.003
Yu, H., Jiang, X., Lin, X., Zhang, Z., Wu, D., Zhou, L., et al. (2018). Hippocampal Subcellular Organelle Proteomic Alteration of Copper-Treated Mice. Toxicol. Sci. 164, 250–263. doi: 10.1093/toxsci/kfy082
Zhao, Y. Z., Jiang, X., Xiao, J., Lin, Q., Yu, W. Z., Tian, F. R., et al. (2016). Using NGF heparin-poloxamer thermosensitive hydrogels to enhance the nerve regeneration for spinal cord injury. Acta Biomater. 29, 71–80. doi: 10.1016/j.actbio.2015.10.014
Keywords: neuroprotection, brain degeneration, trace metals, Alzheimer’s disease, age-related macular degeneration, nerve growth factor, retinal degeneration, biomarkers
Citation: Amadoro G, Latina V, Balzamino BO, Squitti R, Varano M, Calissano P and Micera A (2021) Nerve Growth Factor-Based Therapy in Alzheimer’s Disease and Age-Related Macular Degeneration. Front. Neurosci. 15:735928. doi: 10.3389/fnins.2021.735928
Received: 03 July 2021; Accepted: 10 August 2021;
Published: 09 September 2021.
Edited by:
Bruno Pietro Imbimbo, Chiesi Farmaceutici, ItalyReviewed by:
Benedetto Falsini, Catholic University of the Sacred Heart, ItalyVittorio Porciatti, University of Miami, United States
Giancarlo Iarossi, Bambino Gesù Children Hospital (IRCCS), Italy
Copyright © 2021 Amadoro, Latina, Balzamino, Squitti, Varano, Calissano and Micera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppina Amadoro, Zy5hbWFkb3JvQGlubW0uY25yLml0, orcid.org/0000-0003-2080-2951; Alessandra Micera, YWxlc3NhbmRyYS5taWNlcmFAZm9uZGF6aW9uZWJpZXR0aS5pdA==