 A Young Sim1,2†
A Young Sim1,2† Jong Youl Kim
Jong Youl Kim Yong-ho Lee
Yong-ho Lee Jong Eun Lee
Jong Eun Lee
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 11 August 2021
Sec. Neurodegeneration
Volume 15 - 2021 | https://doi.org/10.3389/fnins.2021.708547
This article is part of the Research Topic Neurodegenerative Diseases: Looking Beyond the Boundaries of the Brain View all 12 articles
Alzheimer’s disease (AD) is characterized by memory loss and cognitive decline. Additionally, abnormal extracellular amyloid plaques accumulation and nerve damage caused by intracellular neurofibrillary tangles, and tau protein are characteristic of AD. Furthermore, AD is associated with oxidative stress, impaired mitochondrial structure and function, denormalization, and inflammatory responses. Recently, besides the amyloid β hypothesis, another hypothesis linking AD to systemic diseases has been put forth by multiple studies as a probable cause for AD. Particularly, type 2 diabetes mellitus (T2DM) and its features, including hyperinsulinemia, and chronic hyperglycemia with an inflammatory response, have been shown to be closely related to AD through insulin resistance. The brain cannot synthesize or store glucose, but it does require glucose, and the use of glucose in the brain is higher than that in any other organ in the mammalian body. One of the therapeutic drugs for T2DM, dipeptidyl peptidase-4 (DPP-4) inhibitor, suppresses the degradation of incretins, glucagon-like peptides and glucose-dependent insulinotropic peptide. Sodium-glucose cotransporter 2 (SGLT2) inhibitors, recently used in T2DM treatment, have a unique mechanism of action via inhibition of renal glucose reabsorption, and which is different from the mechanisms of previously used medications. This manuscript reviews the pathophysiological relationship between the two diseases, AD and T2DM, and the pharmacological effects of therapeutic T2DM drugs, especially DPP-4 inhibitors, and SGLT2 inhibitors.
Alzheimer’s disease (AD) and type 2 diabetes mellitus (T2DM) are two of the most common disorders affecting older adults. AD occurs in 60–80% of the elderly population as the most common neurocognitive disorder and type of dementia. Clinically, AD is characterized by progressive memory loss and decreased cognitive function, leading to premature death several years after diagnosis. The most common pathological features of AD are the abnormal accumulation of amyloid plaques due to the aggregation of amyloid β (Aβ) peptides and neurofibrillary tangles (NFT) consisting of hyperphosphorylated tau protein. Recent studies have revealed that AD is associated with extracellular amyloid plaques, intracellular NFT, neuronal loss, and cellular damage. It is caused by oxidative stress, abnormal mitochondrial structure and function, inflammation, and aging (Kandimalla et al., 2017). This injury has also been associated with conditions related to insulin resistance, including hyperinsulinemia, chronic hyperglycemia, inflammation, and vascular changes. It was also confirmed that approximately 80% of AD patients are affected by insulin resistance or T2DM (de la Monte, 2014). This allows the mechanical relationship between T2DM and AD to be better understood. Research results have shown that AD can be regarded as a metabolic disorder with impaired brain glucose uptake and energy production. Therefore, studies on the causes of AD, based on the potential neuroprotective effects of anti-diabetic drugs and their direct and indirect mechanisms of action, are ongoing (Boccardi et al., 2019). In this review, we provide a summary of the mechanisms that link AD and T2DM; thereafter, we focus on the principal drugs of T2DM and explore their potential as suitable candidates for the treatment of AD.
The worldwide incidence of diabetes, which is a chronic metabolic disorder, is rapidly increasing. Diabetes can be classified as types 1 and 2. T2DM, which accounts for 95% of all cases of diabetes, is characterized by hyperinsulinemia and insulin resistance (Taylor, 2012). Another feature of T2DM is the formation of amyloid polypeptides, which induce pancreatic β cell dysfunction (Marzban et al., 2003). Insulin resistance and amyloid peptides reduce the absorption of blood glucose and ultimately induce chronic hyperglycemia, one of the pathological features of T2DM (Chatterjee and Mudher, 2018). Maintaining the insulin secretory function and ameliorating insulin resistance are important in the management of T2DM.
Insulin is a hormone secreted by the β cells of the pancreas in response to high glucose levels. The binding of insulin to the insulin receptor (IR) initiates insulin signaling.
During insulin binding, IR auto-phosphorylates tyrosine residues in the intracellular portion of the receptor and then rapidly phosphorylates the tyrosine residues of C substrates 1 to 4 (IRS1-4; Lavan et al., 1997). IRS converts insulin via several pathways, the most well-known of which is the phosphoinositide 3-kinase (PIK3)/protein kinase B (AKT)/mechanistic target of rapamycin (mTOR) pathway of IRS1. When the serine residues of IRS1 and IRS2 are phosphorylated, these get separated from the IR, tyrosine phosphorylation of IRS is reduced, and the downward regulation of insulin signal is inhibited (Mothe and Van Obberghen, 1996).
In addition, insulin maintains glucose homeostasis by suppressing glycogen synthase kinase-3 β (GSK3-β), which regulates glucose production and glucose consumption by muscle and adipose tissue after passing through the glucose transporter type 4 (GLUT4; Guo, 2014; Roberts et al., 2014).
Insulin resistance indicates a reduced function of insulin in target tissues, such as the liver, muscles, and adipose tissue. The ability of IRS to get activated and transmit downstream signals is diminished in insulin-responsive tissues, leading to impaired insulin secretion and insulin dysfunction, which are major causes of diabetes (Leng et al., 2004; Guo, 2014).
Insulin resistance in skeletal muscle reduces glucose intake, making it difficult to regulate muscle glycogen synthesis (Hunter and Garvey, 1998). This is considered to occur due to the suppression of GLUT4 gene by excessive free fatty acids. High saturated fatty acid levels, which can suppress normal IRS1 tyrosine phosphorylation and induce insulin resistance in skeletal muscle, show a correlation with skeletal muscle insulin activity (Roden et al., 1996; Pan et al., 1997). Recent studies investigating the deformation of O-linked-β-N-acetylglucosamine (OGlcNAc) protein found that OGlcNAc transferase and β-anomalous variants of N-acetylglucosamine [mediated by O-GlcNAcase (OGA)] are IRS Ser/Thr residues. An important function of the liver is to produce and store glycogen in a glucose reservoir that is readily available to the body (Ma and Hart, 2013). Glucose production during normal postprandial state with glycogenolysis is sufficient to meet the energy needs of the brain and other body tissues. However, insulin resistance results in a systemic insulin resistance phenomenon (Beck-Nielsen et al., 2002; Bugianesi et al., 2005) which causes the body tissues to be deprived of glucose.
Insulin receptors are expressed in all brain cells, but the differences in expression levels vary considerably from region to region and are most visible in the cerebral cortex, striatum, and cerebellum. This suggests that insulin signaling is important in the brain and plays various roles (Arnold et al., 2018). Insulin and insulin-like growth factor (IGF) signaling mechanisms in the brain are important for maintaining synaptic plasticity, and function (Boucher et al., 2014). When insulin binds to an IR, multiple tyrosine residues are auto-phosphorylated to activate IRS1 and IRS2, which mediate downstream signaling through PIK3. This PIK3 activates AKT and suppresses activity at serine 9 residues via GSK3-β phosphorylation, leading to glycogen synthesis (Avila et al., 2012). The PIK/AKT pathway stimulates excitatory and inhibitory cell membrane receptors to regulate synaptic plasticity, enhance N-Methyl-D-aspartic acid (NMDA) receptor-mediated long-term potentiation and neurotransmitter activity, and is important for learning and memory. Additionally, the PIK/AKT pathway increases cortical glucose metabolism (Farrar et al., 2005; Bradley et al., 2012). However, in an abnormal state, GSK3-β is overactivated and tau is phosphorylated; this hyperphosphorylated tau gets aggregated and entangled in nerve fibers (Avila et al., 2010). GSK3-β also acts as a mediator of cell death, increasing the production of Aβ (Qu et al., 2014; Meng et al., 2020).
Insulin activates the mitogen-activated protein kinase (MAPK) pathway, leading to Ras activation and activation of rapidly accelerated fibrosarcoma (Raf), MAPK/ERK kinase (MEK), and extracellular signal-regulated kinase (ERK) in the protoplasmic membrane (Zhang et al., 2011). Although the direct role of the MAPK pathway components that mediate AD pathology has not yet been clarified, recent studies have reported that ERK plays an important role in synaptogenesis, learning, and memory function and has neuroprotective functions (Thiels and Klann, 2001).
Epidemiological studies and neuroimaging studies of the brain have indicated that insulin and IGF signaling pathways are important for the preservation and maintenance of learning and memory processes, and it can be confirmed that the function of learning and memory is improved in AD patients with intranasal insulin injection (Benedict et al., 2007).
Alzheimer’s disease can be classified into two clinical subtypes: familial AD (fAD) and sporadic AD (sAD). Although the two types of disease (fAD and sAD) exhibit similar pathological phenotypes such as presence of plaques, tangles, synaptic damage, and neuronal loss, the factors that induce the neurodegenerative process are completely different. Pathological accumulation in fAD occurs due to the presence of autosomal dominant mutations in one of the three genes: amyloid protein precursor (APP), presenilin-1, or presenilin-2 (Querfurth and LaFerla, 2010). However, the cause of sAD, which accounts for the majority of AD cases, is complex and multifactorial based on the combination of genetic factors, epigenetic factors, and lifestyle-related factors. Moreover, most sAD patients are elderly individuals with various comorbidities (e.g., stroke, stress, diabetes, seizures, osteoporosis, and kidney disease) that can significantly increase the complexity underlying the pathogenesis of sAD (Doraiswamy et al., 2002; Magaki et al., 2014; Aubert et al., 2015).
Many recent studies have confirmed that insulin signaling impaired due to insulin resistance also occurs in AD (Talbot et al., 2012). In fluorodeoxyglucose (FDG)–positron emission tomography (PET) studies of the brains of patients with “early-stage” AD, AD was referred to as “type 3 diabetes” because of reduced glucose intake (de la Monte and Wands, 2008).
The important role of insulin in the peripheral system is well known and has been widely studied, but studies on insulin function in the central nervous system are currently underway. Previously, it was believed that due to the size of insulin, insulin could not pass through the brain-blood barrier (BBB), and the brain was considered to be insulin-independent; however, some studies have shown that IRs are expressed in the brain, and there are several mechanisms to support the presence of insulin (Banks, 2004; Gray et al., 2014).
Recent studies have established that insulin is transported through the BBB via carrier-mediated, saturated, and temperature-sensitive active processes. All types of brain cells, including neurons, have insulin signaling pathways, and insulin regulates the concentration of neurotransmitters such as acetylcholine, recovery, differentiation, proliferation, regeneration, and neuronal cell death (de la Monte et al., 2003; Goberdhan and Wilson, 2003; Russo et al., 2005).
Studies have shown that ICR mice on a long-term high-fat diet (HFD) developed T2DM with insulin resistance in both the body and brain, along with Alzheimer’s pathologies such as cognitive deficits, Aβ accumulation, and hyperphosphorylated tau. Aβ oligomers remove IRs in the protoplasmic membrane, and insulin also affects Aβ accumulation and systemic tau phosphorylation (Zhao et al., 2008).
The insulin-degrading enzyme (IDE) is also known to degrade other substrates such as Aβ (Farris et al., 2003). When insulin levels increase, IDE expression is activated, and it inhibits long-term insulin activity. However, in an insulin-resistant state, because IDE is used to remove insulin, senile plaques are formed in which IDE cannot lower Aβ (Shiiki et al., 2004).
GSK3-β, which is the most widely studied tau kinase, is also involved in Aβ production (Jeon et al., 2015). A study showed that GSK3-β, which is a multifunctional Ser/Thr kinase affected by tau phosphorylation and aggregation inhibition, improved learning and memory and reduced tau phosphorylation in an AD transgenic mouse model (Farr et al., 2016). In addition, GSK3-β expression was suppressed in an AD-pathology mouse model. In particular, intranasal insulin injection has been shown to help improve memory by maintaining serum insulin and glucose levels (Benedict et al., 2004), suggesting that insulin is a therapeutic target for AD.
In addition to PI3K/AKT and Ras/Raf/MAPK insulin signaling pathways, mTOR and its downstream targets that regulate neuronal survival and nutrient sensing play roles in AD pathogenesis; however, these roles are not well-defined. mTOR regulates protein synthesis by phosphorylating the key substrates of the translational machinery, namely, the eukaryotic initiation factor 4E-binding protein and p70S6 kinase. Rapamycin inhibits mTOR in vivo and halts cellular growth and proliferation (Showkat et al., 2014). Also, genetic inhibition of mTOR reduces the level of memory loss, improves cognitive function and reduces tan and Aβ deposits (Kaeberlein and Galvan, 2019). It is hypothesized that in an insulin-resistant state, these downstream signaling pathways are compromised, leading to increased levels of Aβ oligomers and hyperphosphorylated tau. These increased levels of Aβ oligomers and hyperphosphorylated tau occur not only due to a dysregulation of downstream kinases but also due to an impairment of autophagic clearance that arises as a result of an imbalance of the mTOR and autophagy pathways. Autophagic dysfunction which is recently gaining attention feature of AD causes the progressive accumulation of toxic proteins and eventually leads to neuronal death (Orr and Oddo, 2013).
Alzheimer’s disease, a degenerative brain disease, is the most common cause of dementia, with clinical features including gradual decline of cognitive function, amnesia, behavioral and personality changes, and pathological features including extracellular Aβ plaques and intracellular NFT intraneuronal deposition, tau protein degeneration, and severe neuronal loss in the brain tissue (Saxena, 2010). Most AD treatments have focused on Aβ but have failed to look at AD from various perspectives, for example, relating AD to obesity and T2DM (Kang et al., 2017). Considering the relationship between AD and T2DM, it might be possible to treat AD with T2DM drugs. For instance, Thiazolidinedlones (TZDs), such as pioglitazone and rosiglitazone, are PPARγ agonists used as anti-diabetic drugs, that induce a decrease in plasma free fatty acid concentration and fasting hyperglycemia through an insulin-reducing effect. A recent pioglitazone-related study found that it may be of therapeutic benefit, showing a significant reduction in Aβ and tau pathology measured in cerebral blood flow from patients with early-stage and mild to moderate AD patients (Pérez and Quintanilla, 2015).
According to the Mayo Clinic Alzheimer Disease Patient Registry, 80% of patients with AD have impaired glucose tolerance or diabetes (Janson et al., 2004). Epidemiological studies have shown that T2DM induces cognitive impairment and that T2DM patients are 1.5−2 times more likely to be diagnosed with dementia than healthy individuals are (Biessels et al., 2014). There is also evidence of cellular insulin resistance or insulin deficiency in the brains of patients with AD, including non-diabetic patients (Vijan, 2015).
Type 2 diabetes mellitus is a chronic metabolic disorder that can damage blood vessels, nerves, eyes, and kidneys and causes serious complications. Typical symptoms of T2DM associated with insulin dysfunction, including hyperglycemia, insulin resistance, and relative insulin deficiency, also induce the accumulation of Aβ in the brain, contributing to AD pathogenesis (Ramos-Rodriguez et al., 2017).
Several pathogenic mechanisms overlap the two diseases, including dysregulation of glucose and insulin signals, increased inflammation, Aβ deposition, mitochondrial dysfunction, and oxidative stress (Liu et al., 2011). Insulin resistance and deficiency are increased by abnormally creating insulin signaling through PI3K/Akt/GSK3-β signals; GSK3-β activation is an important component of NFT and can lead to hyperphosphorylated tau (Zheng et al., 2015). In addition, IDE associated with insulin signaling plays an important role in insulin and Aβ clearance, so that impaired IDE function can cause AD and T2DM (Ramos-Rodriguez et al., 2017). IRS1 plays an important role in transferring insulin and IGF-1 receptor signals to signal adapter proteins and the intracellular pathway. PI3K/AKT kinase pathway and IRS1 dysfunction causes AD and T2DM (illustrated in Figure 1).
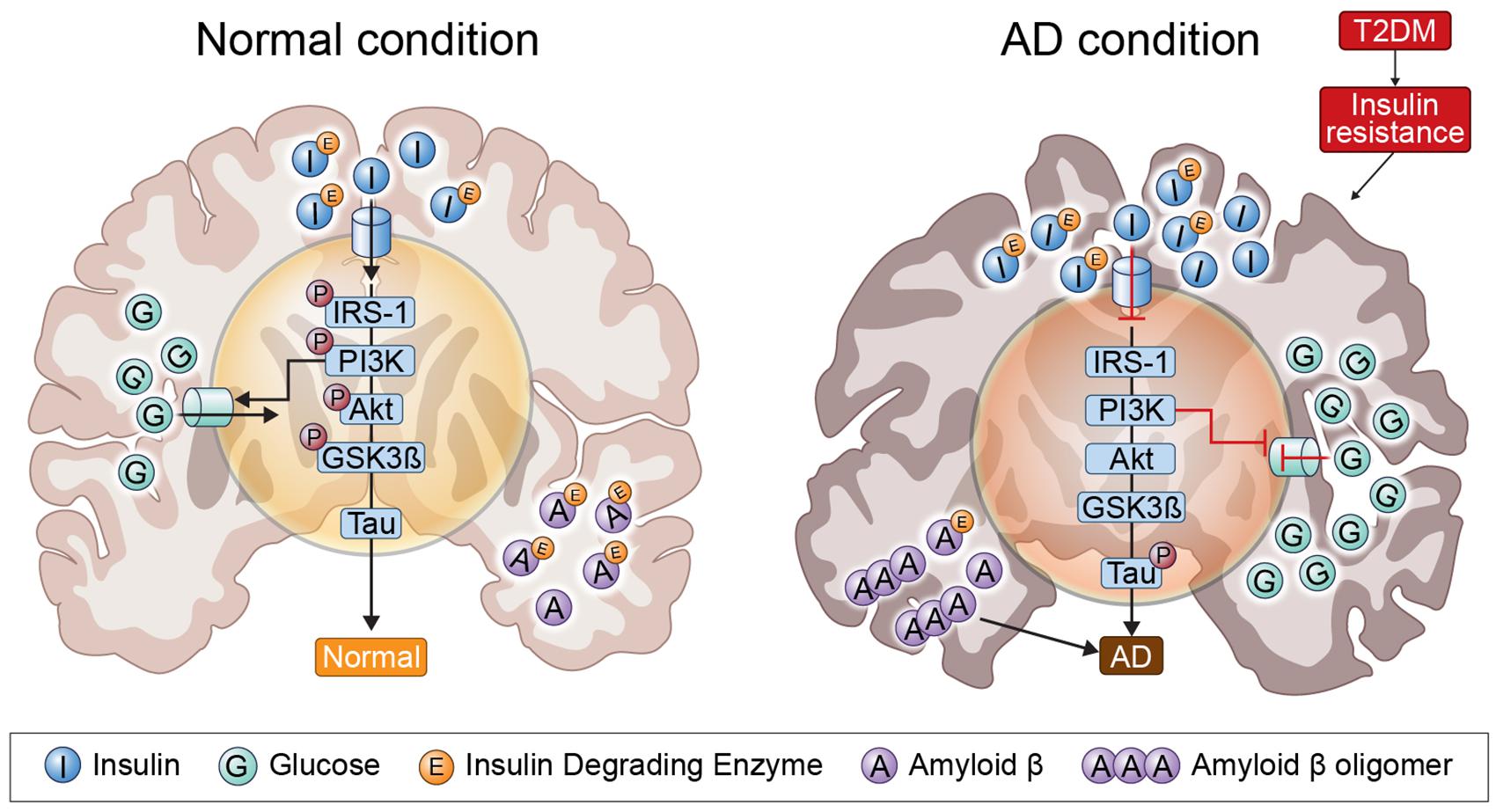
Figure 1. Insulin signaling in the normal condition brain and AD brain. In insulin-resistant state induced by T2DM, as insulin signaling is impaired, insulin is unable to bind to an insulin receptor, and multiple tyrosine residues are not auto-phosphorylated to activate insulin receptor substrate 1, which mediates downstream signaling through phosphoinositide 3-kinase. Therefore, GSK3-β is overactivated and tau is phosphorylated.
When food is ingested, a series of hormones are secreted by epithelial cells of the small intestine to increase insulin secretion. When glucose is orally administered, insulin secretion from the pancreas increases, and this phenomenon is called the incretin effect. A typical incretin is an endogenous peptide that is mainly synthesized and secreted by enteroendocrine L cells into the gastrointestinal peptide hormone (GLP-1). Physiologically, it promotes β cell proliferation, improves β cell function, decreases β cell apoptosis, increases insulin secretion, and regulates glucose homeostasis (Graaf et al., 2016; Keshava et al., 2017). Glucagon-like peptide-1 receptor (GLP-1R) is widely expressed in the hippocampus, hypothalamus, cortex, nucleus basalis of the Meynert, choroidal plexus, and nucleus of the solitary tract (Erreger et al., 2012; Yildirim Simsir et al., 2018). This factor, which is overexpressed in the hippocampus of mice, also affects neurite growth, learning, and memory (McClean et al., 2011). In addition, GLP-1 analogs and GLP-1R agonists administered peripherally or centrally reduce Aβ deposition, prevent tau and NFT protein hyperphosphorylation, and have a neuroprotective effect against rodent AD-like neurodegeneration (Hölscher, 2018). There have also been reported to be effective for maintaining synaptic plasticity and learning and memory (McClean et al., 2011; Li et al., 2012; Xiong et al., 2013; McClean and Hölscher, 2014; Candeias et al., 2015; Wang et al., 2016).
However, GLP-1 has a short duration of reaction time as it is rapidly degraded by dipeptidyl peptidase-4 (DPP-4) present in plasma and other body fluids, such as cerebrospinal fluid (Gong et al., 2014).
Dipeptidyl peptidase-4 is a type 2 transmembrane glycoprotein with various functions. Usually, the substrates of DPP-4 are peptides with a size of 80 amino acids or less, and there are more than 35 neuropeptides and chemokines, including GLP-1, GLP, neuropeptide Y, peptide YY, substance P, and stromal cell-derived factor 1, that serve as substrates for this peptide. It is found in epithelial cells, immune cells, including T lymphocytes, various cells, such as vascular endothelial cells, and almost all tissues, including kidney, liver, adrenal gland, skeletal muscle, pancreas, lung, small intestine, bone marrow, and spleen (Chen et al., 2019).
Usually, 180 g of glucose per day is filtered by the kidneys and reabsorbed in the proximal tubules. Glucose reabsorption occurs via sodium-glucose cotransporter 2 (SGLT2) present mainly in the kidney, which comprises SGLT2 located in front of the proximal tubule and sodium-glucose cotransporter 1 (SGLT1) in the latter half. In a normal blood glucose level state, SGLT2 is responsible for approximately 97% of the reabsorption of filtered glucose, whereas and SGLT1 is responsible for approximately 3%.
Glucose reabsorption begins with active transfer of Na+ the extracellular region by Na+/K+ ATPase in the proximal tubule. The electrochemical force generated while moving Na+ extracellularly causes Na+ and glucose to move intracellularly via SGLT. One Na+ and glucose molecule move together through SGLT2, and two Na+ and glucose molecules move together through SGLT1. When the glucose concentration increases by the glucose transferred into the cell, glucose is reabsorbed into the bloodstream via the glucose transporter based on the difference in glucose concentration between the cell and epilepsy.
Recently, there have been increasing reports of the presence of SGLTs in the mammalian central nervous system (Yu et al., 2010, 2013). The receptors for SGLT1 are expressed in CA1, CA3 (regions 1 and 3 of hippocampal cornu ammonis), and the dentate gyrus hippocampal subfields, and SGLT2 has been reported to be expressed in the hippocampus, cerebellum, and blood-hippocampal barrier endothelial cells (Poppe et al., 1997; Enerson and Drewes, 2006; Shah et al., 2012; Jurcovicova, 2014).
Dipeptidyl peptidase-4 inhibitors (DPP-4is) include sitagliptin, vildagliptin, saxagliptin, linagliptin, alogliptin, and gemigliptin. As hormones that increase insulin secretion are degraded by DPP-4, DPP-4is can be used to suppress this hormone degradation. In other words, the principle of DPP-4 is the increase in insulin secretion following food intake and the period of insulin secretion time can be improved, and blood glucose levels can be additionally improved by suppressing glucagon secretion without inducing hypoglycemia.
The SGLT2 inhibitors (SGLT2is) include dapagliflozin, canagliflozin, empagliflozin, ipragliflozin, tofogliflozin, luseogliflozin, and ertugliflozin. These inhibitors reduce glycated hemoglobin level by 0.3–0.9% and fasting blood glucose levels by 18–36 mg/dl, regardless of use of other drugs, and decrease body weight as well as blood pressures due to drug effects on glucosuria and natriuresis. In addition, since these have an insulin-independent hypoglycemic effect, SGLT2is can reduce the blood glucose level even in an environment where the insulin secretory capacity is decreased. By increasing the excretion of glucose into the urine, insulin resistance can be improved, and by improving glucose toxicity, the function of pancreatic β cells can be maintained. These are diabetic treatment agents with a low risk of hypoglycemia because they facilitate the excretion of glucose in a hyperglycemic state without affecting insulin secretion.
Glucagon-like peptide-1 signaling in the brain regulates glucose metabolism. Inhibitors of DPP-4 improve neuronal insulin resistance by restoring insulin-induced phosphorylation of neuronal IR, IRS1 phosphorylation, and AKT/PKB-Ser phosphorylation, resulting in the brain mitochondrial dysfunction. Previous studies on diabetes-related AD rat models have demonstrated that GLP-1 positively affects learning and memory (Chen et al., 2012). In addition, a recent study has shown that DPP4is can increase the levels of active GLP-1 in the brain and improve memory behaviors in AD mice models (D’Amico et al., 2010). These have also been shown to improve spatial learning and memory ability and protect synaptic proteins by increasing GLP-1 and GLP-1R expression levels in the hippocampus and cortex of AD mice (Pipatpiboon et al., 2013). Cognitive function was improved as a result of the administration of DPP-4is and quercetin (3,3′,4′,5,7-pentahydroxyflavone) found in vegetables and fruits which is one of the major groups of polyphenols with effects on inflammation, diabetes and the nervous system in a study (Babaei et al., 2018; Li et al., 2019), and DPP-4is was shown to ameliorate memory impairment, increase GLP-1 levels in the brain which acts as a neuroprotective agent (Holst et al., 2011) and could lead to improved brain and hippocampal mitochondrial function and reduced brain MDA (Pintana et al., 2013), and significantly reduce nitrosative stress, inflammation hallmarks, and Aβ deposits (D’Amico et al., 2010; Kosaraju et al., 2013a, 2017). These inhibitors showed a time-dependent improvement in memory retention and dose-dependent attenuation of Aβ, tau phosphorylation, and inflammatory markers, and AD-associated proteins were decreased in the hippocampus following DPP-4is administration (Kosaraju et al., 2013b; Ma et al., 2018). The combination of DPP-4i and memantine could reduce the expression of APP and phosphorylated tau protein (Khalaf et al., 2019). Inhibitors of DPP-4 alleviated cognitive deficits in 3xTG AD mice. These improve incretin levels in the brain and reduce Aβ deposition, tau phosphorylation, and neuroinflammation (Thomas et al., 2008) and can significantly protect against Aβ-induced cytotoxicity, and inhibit the activation of GSK3-β and tau hyperphosphorylation by restoring downstream insulin signaling. Inhibitors of DPP-4 ameliorated Aβ-induced mitochondrial dysfunction and intracellular reactive oxygen species (ROS) generation and upregulated Sirt1 expression (Kosaraju et al., 2017). HFD rats had brain mitochondrial dysfunction as shown by increased ROS production, mitochondrial depolarization, and mitochondrial swelling. In the mitochondria, it has been shown that increased levels of ROS could cause the opening of the inner membrane anion channel (IMAC), thus leading to mitochondrial membrane depolarization (Zorov et al., 2006). The depolarization of mitochondria could also lead to the dysfunction of mitochondria to produce ATP synthesis (Aon et al., 2006). Furthermore, increased ROS levels could play a role in the cognitive decline observed in HFD rats (Table 1 for a summary, illustrated in Figure 2).

Table 1. Research of DPP-4 inhibitors in AD models.
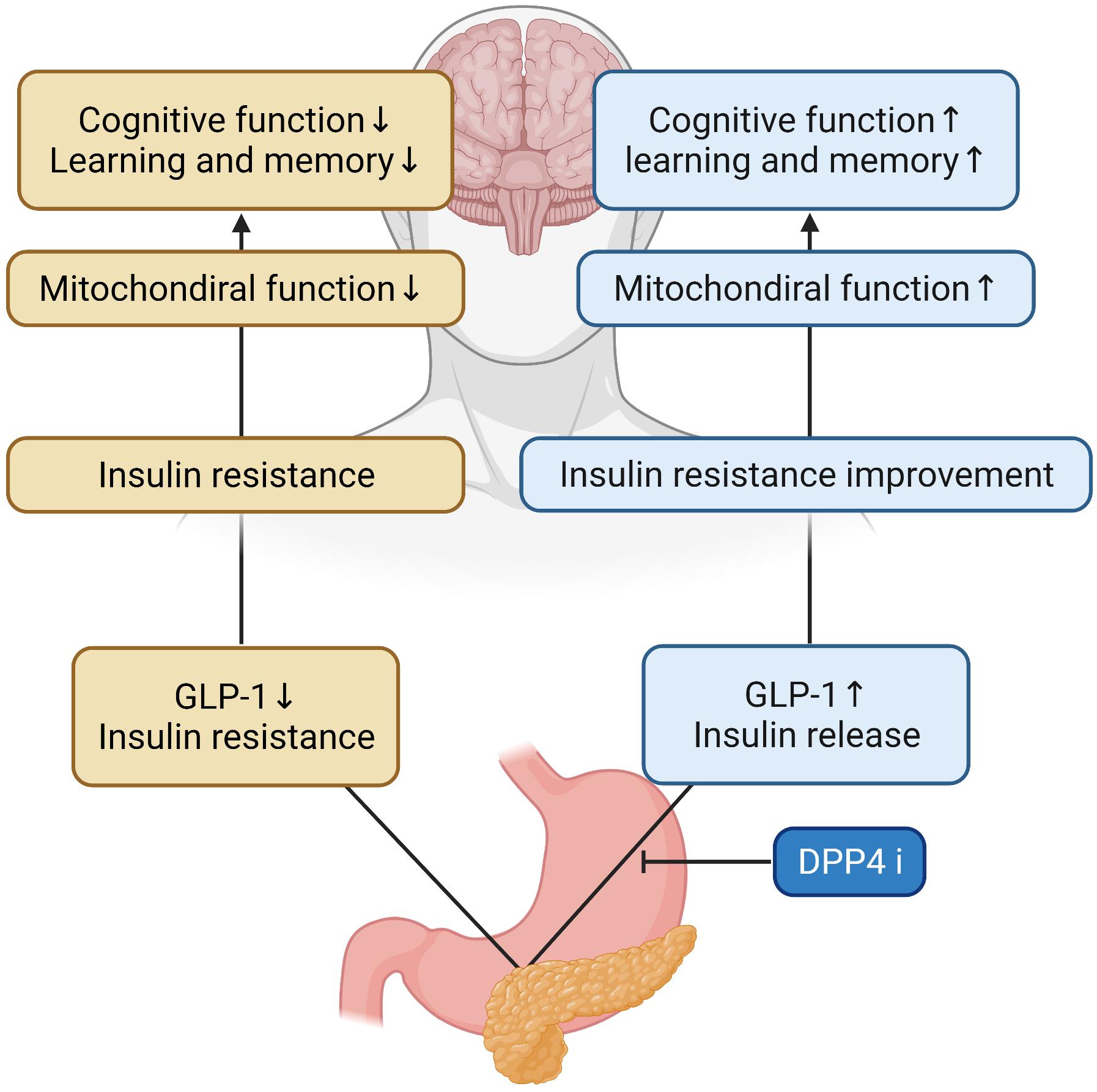
Figure 2. Effects of DPP-4 inhibitor in the AD brain. During DPP-4 inhibitor treatment, GLP-1 increases and insulin is secreted, which improves insulin resistance and mitochondrial function in the brain. Therefore, cognitive function and learning and memory ability are improved.
Inhibitors of SGLT2 not only improve peripheral insulin sensitivity and reduce body weight (Xu et al., 2017) but also improve brain mitochondrial function and insulin signaling, and reduce cell death. Furthermore, SGLT2is prevent cognitive decline and protect synaptic plasticity in the hippocampus (Sa-Nguanmoo et al., 2017). Inhibitors of SGLT2 reduced the accumulation of Aβ in the cortical region of AD-T2DM mice (APP/PS1xdb/db mice) which is a genetically diabetic model of T2DM and showed the same effect on the amount of tau induced pathological cerebral atrophy (Wiciński et al., 2020). SGLT2is-mediated mTOR inhibition, through continuous loss of glucose in the urine, routinely restores a reliable, overnight catabolic-fasted state in older, inactive individuals and re-establishes the benefits associated wit circadian catabolic/anabolic metabolism (e.g., reactivation of the endo-lysosomal pathway through inhibition of mTOR), removal and replacement of dysfunctional organelles/proteins, and lowering of blood pressure through mTOR-mediated modulation of sympathetic tone. Unrestrained chronic mTOR activation may be responsible for sustaining metabolic and mitochondrial dysfunction in AD, driving the breakdown of the BBB via endothelial cell dysfunction, as well as driving the hyperphosphorylation of tau, and formation of amyloid plaques in the brain (Mueed et al., 2018). These inhibitors can restore mTOR signaling through mTOR inhibition and prevent the progression of AD pathology (Esterline et al., 2020). In addition, SGLT2is physiologically elevates blood ketone bodies such as β-hydroxybutyrate (Kim et al., 2019), which can modulate NLRP3 inflammasome-IL-1β signaling (Kim et al., 2020), a key pathologic pathway in AD (Heneka et al., 2013). Decreased blood glucose levels were seen in db/db mice after 10 weeks of treatment with SGLT2is for T2DM. These inhibitors not only ameliorated albuminuria and glomerular injury in db/db mice but also significantly prevented the impairment of cognitive function, which was associated with the attenuation of cerebral oxidative stress and increase in cerebral brain-derived neurotrophic factor level (Lin et al., 2014). SGLT2is treatment significantly attenuated cerebral oxidative stress and DNA oxidative damage in db/db mice, as shown by the reduction of cerebral superoxide and 8-OHdG, and this attenuation of cerebral oxidative stress was associated with the reduction of cerebral NADPH oxidase subunit. Therefore, the improvement of cognitive function by SGLT2is seems to be attributed to the attenuation of oxidative stress. Moreover, the effect of SGLT2is treatment on cerebral BDNF, since BDNF, a key protein promoting memory and survival of neurons, is significantly reduced in diabetic patients, and diabetic animals including db/db mice and the decrease in cerebral BDNF is shown to be associated with cognitive decline (Lin et al., 2014; Table 2 for a summary, illustrated in Figure 3).

Table 2. Research of SGLT2 inhibitors in AD models.
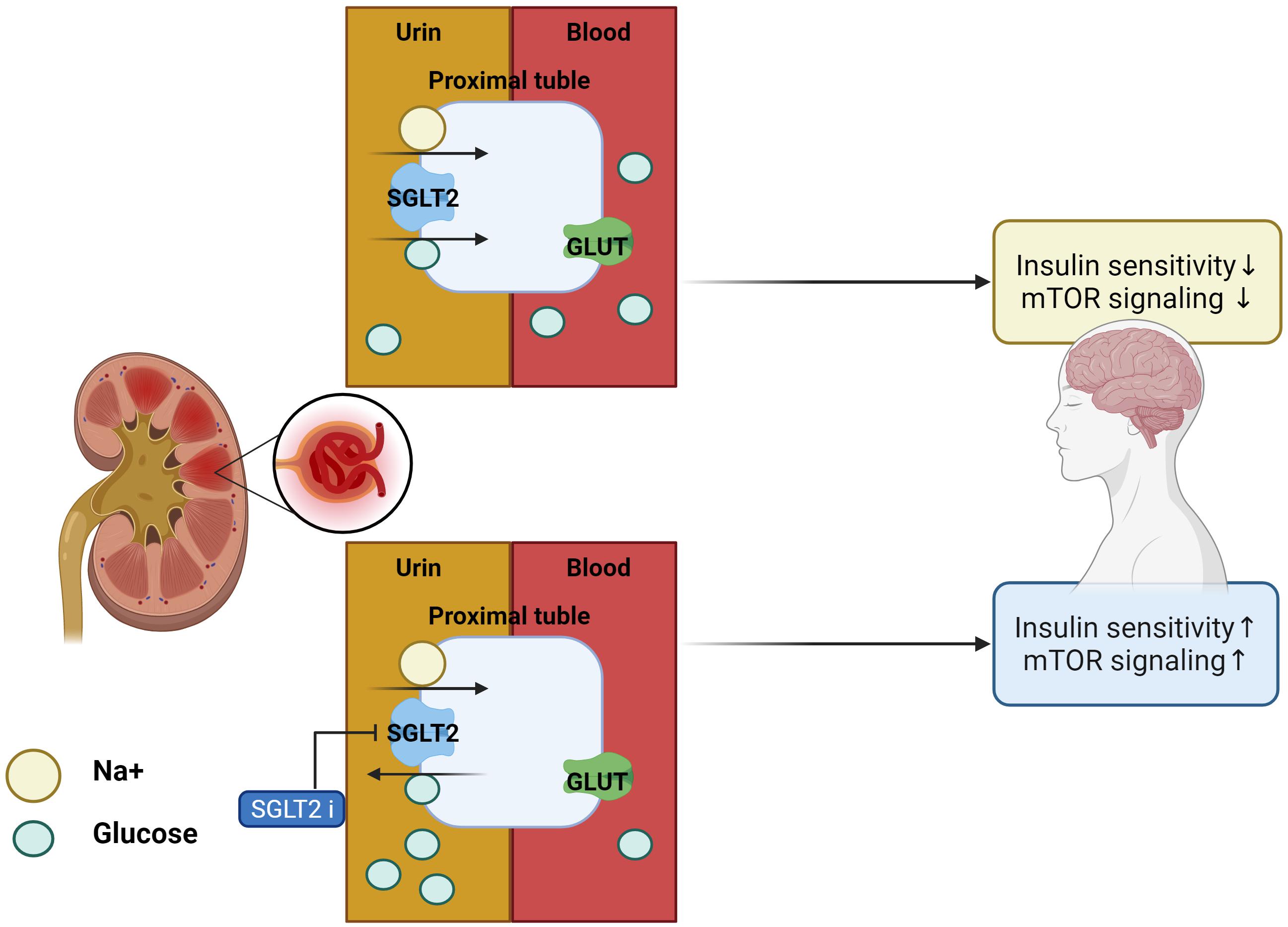
Figure 3. Effects of SGLT2 inhibitor in the AD brain. With SGLT2 inhibitor, insulin sensitivity and mTOR signaling in the brain are improved, as excess glucose from insulin resistance is filtered out by the kidneys.
Majority of reports regarding the effects of anti-diabetic agents on dementia have been investigated from retrospective studies. In 240 elderly patients with T2DM affected by mild cognitive impairment (MCI), 2 years treatment group of DPP-4i significantly improve cognitive functions measured by mini-mental state examination (MMSE), compared to the sulfonylurea which increases endogenous release of insulin from pancreatic β cells group (Rizzo et al., 2014). A prospective, non-randomized study showed that sitagliptin therapy prevented from the decline of MMSE during 6 months in old T2DM (Isik et al., 2017). Using a health insurance claim database in Korea, DPP-4i use demonstrated a significant 46% decrease in AD development among elderly T2DM (Kim et al., 2018). Although there are few data on SGLT2i, one randomized clinical trial reported no changes in MMSE after 12-month treatment of incretins vs. SGLT2i (Perna et al., 2018). Further larger and well-designed clinical studies are needed to evaluate the neuroprotective effects of DPP-4i and SGLT2i.
Recent studies have identified the key mechanisms by which the brain becomes resistant to insulin in AD and how impaired insulin signaling in AD is linked to memory impairment (Vieira et al., 2018). In this review, we describe the connections between AD and T2DM. Although the reason why many T2DM patients develop AD is not clear, the two diseases are associated with insulin resistance. Significant effort is required to identify the common pathological and molecular mechanisms between AD and T2DM, which will help better understand the onset and development of both diseases. Therefore, novel approaches to identify biomarkers for detecting early-stage of AD will likely increase the efficacy of anti-diabetic agents and allow treatment before severe neuronal dysfunction occurs in the AD brains.
AYS and SB wrote the manuscript. AYS, SB, JYK, Y-HL, and JEL participated in the discussion and revision. JEL designed and edited the final manuscript. All authors read and approved the final manuscript.
This study was supported by the National Research Foundation of Korea (NRF), funded by the Korea government (NRF-2021R1A2C2008034) to JEL.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank Medical Illustration and Design, part of the Medical Research Support Services of Yonsei University College of Medicine, for all artistic support related to this work.
Aon, M. A., Cortassa, S., Akar, F. G., and O’Rourke, B. (2006). Mitochondrial criticality: a new concept at the turning point of life or death. Biochim. Biophys. Acta 1762, 232–240. doi: 10.1016/j.bbadis.2005.06.008
Arnold, S. E., Arvanitakis, Z., Macauley-Rambach, S. L., Koenig, A. M., Wang, H.-Y., Ahima, R. S., et al. (2018). Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat. Rev. Neurol. 14:168. doi: 10.1038/nrneurol.2017.185
Aubert, L., Pichierri, S., Hommet, C., Camus, V., Berrut, G., and de Decker, L. (2015). Association between comorbidity burden and rapid cognitive decline in individuals with mild to moderate Alzheimer’s disease. J. Am. Geriatr. Soc. 63, 543–547. doi: 10.1111/jgs.13314
Avila, J., León-Espinosa, G., García, E., García-Escudero, V., Hernández, F., and Defelipe, J. (2012). Tau phosphorylation by GSK3 in different conditions. Int. J. Alzheimers Dis. 2012:578373. doi: 10.1155/2012/578373
Avila, J., Wandosell, F., and Hernández, F. (2010). Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Exp. Rev. Neurother. 10, 703–710. doi: 10.1586/ern.10.40
Babaei, F., Mirzababaei, M., and Nassiri-Asl, M. (2018). Quercetin in food: possible mechanisms of its effect on memory. J. Food Sci. 83, 2280–2287. doi: 10.1111/1750-3841.14317
Banks, W. A. (2004). The source of cerebral insulin. Eur. J. Pharmacol. 490, 5–12. doi: 10.1016/j.ejphar.2004.02.040
Beck-Nielsen, H., Hother-Nielsen, O., and Staehr, P. (2002). Is hepatic glucose production increased in type 2 diabetes mellitus? Curr. Diab. Rep. 2, 231–236. doi: 10.1007/s11892-002-0088-0
Benedict, C., Hallschmid, M., Hatke, A., Schultes, B., Fehm, H. L., Born, J., et al. (2004). Intranasal insulin improves memory in humans. Psychoneuroendocrinology 29, 1326–1334. doi: 10.1016/j.psyneuen.2004.04.003
Benedict, C., Hallschmid, M., Schultes, B., Born, J., and Kern, W. (2007). Intranasal insulin to improve memory function in humans. Neuroendocrinology 86, 136–142. doi: 10.1159/000106378
Biessels, G. J., Strachan, M. W., Visseren, F. L., Kappelle, L. J., and Whitmer, R. A. (2014). Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. Lancet Diabet. Endocrinol. 2, 246–255. doi: 10.1016/s2213-8587(13)70088-3
Boccardi, V., Murasecco, I., and Mecocci, P. (2019). Diabetes drugs in the fight against Alzheimer’s disease. Ageing Res. Rev. 54:100936. doi: 10.1016/j.arr.2019.100936
Boucher, J., Kleinridders, A., and Kahn, C. R. (2014). Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 6:9191. doi: 10.1101/cshperspect.a009191
Bradley, C. A., Peineau, S., Taghibiglou, C., Nicolas, C. S., Whitcomb, D. J., Bortolotto, Z. A., et al. (2012). A pivotal role of GSK-3 in synaptic plasticity. Front. Mol. Neurosci. 5:13. doi: 10.3389/fnmol.2012.00013
Bugianesi, E., McCullough, A. J., and Marchesini, G. (2005). Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology 42, 987–1000. doi: 10.1002/hep.20920
Candeias, E. M., Sebastião, I. C., Cardoso, S. M., Correia, S. C., Carvalho, C. I., Plácido, A. I., et al. (2015). Gut-brain connection: The neuroprotective effects of the anti-diabetic drug liraglutide. World J. Diabetes 6, 807–827. doi: 10.4239/wjd.v6.i6.807
Chatterjee, S., and Mudher, A. (2018). Alzheimer’s disease and type 2 diabetes: a critical assessment of the shared pathological traits. Front. Neurosci. 12:383. doi: 10.3389/fnins.2018.00383
Chen, S., Liu, A. R., An, F. M., Yao, W. B., and Gao, X. D. (2012). Amelioration of neurodegenerative changes in cellular and rat models of diabetes-related Alzheimer’s disease by exendin-4. Age (Dordr.) 34, 1211–1224. doi: 10.1007/s11357-011-9303-8
Chen, S., Zhou, M., Sun, J., Guo, A., Fernando, R. L., Chen, Y., et al. (2019). DPP-4 inhibitor improves learning and memory deficits and AD-like neurodegeneration by modulating the GLP-1 signaling. Neuropharmacology 157:107668. doi: 10.1016/j.neuropharm.2019.107668
D’Amico, M., Di Filippo, C., Marfella, R., Abbatecola, A. M., Ferraraccio, F., Rossi, F., et al. (2010). Long-term inhibition of dipeptidyl peptidase-4 in Alzheimer’s prone mice. Exp. Gerontol. 45, 202–207. doi: 10.1016/j.exger.2009.12.004
de la Monte, S. M. (2014). Type 3 diabetes is sporadic Alzheimer’s disease: mini-review. Eur. Neuropsychopharmacol. 24, 1954–1960. doi: 10.1016/j.euroneuro.2014.06.008
de la Monte, S. M., and Wands, J. R. (2008). Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2, 1101–1113. doi: 10.1177/193229680800200619
de la Monte, S. M., Chen, G. J., Rivera, E., and Wands, J. R. (2003). Neuronal thread protein regulation and interaction with microtubule-associated proteins in SH-Sy5y neuronal cells. Cell Mol. Life Sci. 60, 2679–2691. doi: 10.1007/s00018-003-3305-3
Doraiswamy, P. M., Leon, J., Cummings, J. L., Marin, D., and Neumann, P. J. (2002). Prevalence and impact of medical comorbidity in Alzheimer’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 57, M173–M177. doi: 10.1093/gerona/57.3.m173
Enerson, B. E., and Drewes, L. R. (2006). The rat blood-brain barrier transcriptome. J. Cereb. Blood Flow Metab. 26, 959–973. doi: 10.1038/sj.jcbfm.9600249
Erreger, K., Davis, A. R., Poe, A. M., Greig, N. H., Stanwood, G. D., and Galli, A. (2012). Exendin-4 decreases amphetamine-induced locomotor activity. Physiol. Behav. 106, 574–578. doi: 10.1016/j.physbeh.2012.03.014
Esterline, R., Oscarsson, J., and Burns, J. (2020). A role for sodium glucose cotransporter 2 inhibitors (SGLT2is) in the treatment of Alzheimer’s disease? Int. Rev. Neurobiol. 155, 113–140. doi: 10.1016/bs.irn.2020.03.018
Farr, S. A., Sandoval, K. E., Niehoff, M. L., Witt, K. A., Kumar, V. B., and Morley, J. E. (2016). Peripheral administration of GSK-3β antisense oligonucleotide improves learning and memory in SAMP8 and Tg2576 mouse models of Alzheimer’s disease. J. Alzheimers Dis. 54, 1339–1348. doi: 10.3233/jad-160416
Farrar, C., Houser, C. R., and Clarke, S. (2005). Activation of the PI3K/Akt signal transduction pathway and increased levels of insulin receptor in protein repair-deficient mice. Aging Cell 4, 1–12. doi: 10.1111/j.1474-9728.2004.00136.x
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 4162–4167. doi: 10.1073/pnas.0230450100
Goberdhan, D. C., and Wilson, C. (2003). The functions of insulin signaling: size isn’t everything, even in Drosophila. Differentiation 71, 375–397. doi: 10.1046/j.1432-0436.2003.7107001.x
Gong, N., Xiao, Q., Zhu, B., Zhang, C. Y., Wang, Y. C., Fan, H., et al. (2014). Activation of spinal glucagon-like peptide-1 receptors specifically suppresses pain hypersensitivity. J. Neurosci. 34, 5322–5334. doi: 10.1523/jneurosci.4703-13.2014
Graaf, C., Donnelly, D., Wootten, D., Lau, J., Sexton, P. M., Miller, L. J., et al. (2016). Glucagon-like peptide-1 and its class B G protein-coupled receptors: a long march to therapeutic successes. Pharmacol. Rev. 68, 954–1013. doi: 10.1124/pr.115.011395
Gray, S. M., Meijer, R. I., and Barrett, E. J. (2014). Insulin regulates brain function, but how does it get there? Diabetes 63, 3992–3997. doi: 10.2337/db14-0340
Guo, S. (2014). Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J. Endocrinol. 220, T1–T23. doi: 10.1530/joe-13-0327
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Hölscher, C. (2018). Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology 136, 251–259. doi: 10.1016/j.neuropharm.2018.01.040
Holst, J. J., Burcelin, R., and Nathanson, E. (2011). Neuroprotective properties of GLP-1: theoretical and practical applications. Curr. Med. Res. Opin. 27, 547–558. doi: 10.1185/03007995.2010.549466
Hunter, S. J., and Garvey, W. T. (1998). Insulin action and insulin resistance: diseases involving defects in insulin receptors, signal transduction, and the glucose transport effector system. Am. J. Med. 105, 331–345. doi: 10.1016/s0002-9343(98)00300-3
Isik, A. T., Soysal, P., Yay, A., and Usarel, C. (2017). The effects of sitagliptin, a DPP-4 inhibitor, on cognitive functions in elderly diabetic patients with or without Alzheimer’s disease. Diabet. Res. Clin. Pract. 123, 192–198. doi: 10.1016/j.diabres.2016.12.010
Janson, J., Laedtke, T., Parisi, J. E., O’Brien, P., Petersen, R. C., and Butler, P. C. (2004). Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53, 474–481. doi: 10.2337/diabetes.53.2.474
Jeon, S., Park, J. E., Lee, J., Liu, Q. F., Jeong, H. J., Pak, S. C., et al. (2015). Illite improves memory impairment and reduces Aβ level in the Tg-APPswe/PS1dE9 mouse model of Alzheimer’s disease through Akt/CREB and GSK-3β phosphorylation in the brain. J. Ethnopharmacol. 160, 69–77. doi: 10.1016/j.jep.2014.11.029
Jurcovicova, J. (2014). Glucose transport in brain–effect of inflammation. Endocr. Regul. 48, 35–48. doi: 10.4149/endo_2014_01_35
Kaeberlein, M., and Galvan, V. (2019). Rapamycin and Alzheimer’s disease: time for a clinical trial? Sci. Transl. Med. 11:4289. doi: 10.1126/scitranslmed.aar4289
Kandimalla, R., Thirumala, V., and Reddy, P. H. (2017). Is Alzheimer’s disease a Type 3 diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1078–1089. doi: 10.1016/j.bbadis.2016.08.018
Kang, S., Lee, Y. H., and Lee, J. E. (2017). Metabolism-centric overview of the pathogenesis of Alzheimer’s disease. Yonsei. Med. J. 58, 479–488. doi: 10.3349/ymj.2017.58.3.479
Keshava, H. B., Mowla, A., Heinberg, L. J., Schauer, P. R., Brethauer, S. A., and Aminian, A. (2017). Bariatric surgery may reduce the risk of Alzheimer’s diseases through GLP-1 mediated neuroprotective effects. Med. Hypoth. 104, 4–9. doi: 10.1016/j.mehy.2017.05.002
Khalaf, S. S., Hafez, M. M., Mehanna, E. T., Mesbah, N. M., and Abo-Elmatty, D. M. (2019). Combined vildagliptin and memantine treatment downregulates expression of amyloid precursor protein, and total and phosphorylated tau in a rat model of combined Alzheimer’s disease and type 2 diabetes. Naunyn. Schmiedeb. Arch Pharmacol. 392, 685–695. doi: 10.1007/s00210-019-01616-3
Kim, J. H., Lee, M., Kim, S. H., Kim, S. R., Lee, B. W., Kang, E. S., et al. (2019). Sodium-glucose cotransporter 2 inhibitors regulate ketone body metabolism via inter-organ crosstalk. Diabetes Obes. Metab. 21, 801–811. doi: 10.1111/dom.13577
Kim, S. R., Lee, S. G., Kim, S. H., Kim, J. H., Choi, E., Cho, W., et al. (2020). SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 11:2127. doi: 10.1038/s41467-020-15983-6
Kim, Y. G., Jeon, J. Y., Kim, H. J., Kim, D. J., Lee, K. W., Moon, S. Y., et al. (2018). Risk of dementia in older patients with type 2 diabetes on Dipeptidyl-Peptidase iv inhibitors versus sulfonylureas: a real-world population-based cohort study. J. Clin. Med. 8:28. doi: 10.3390/jcm8010028
Kosaraju, J., Gali, C. C., Khatwal, R. B., Dubala, A., Chinni, S., Holsinger, R. M., et al. (2013a). Saxagliptin: a dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology 72, 291–300. doi: 10.1016/j.neuropharm.2013.04.008
Kosaraju, J., Holsinger, R. M. D., Guo, L., and Tam, K. Y. (2017). Linagliptin, a Dipeptidyl peptidase-4 inhibitor, mitigates cognitive deficits and pathology in the 3xTg-AD mouse model of Alzheimer’s disease. Mol. Neurobiol. 54, 6074–6084. doi: 10.1007/s12035-016-0125-7
Kosaraju, J., Murthy, V., Khatwal, R. B., Dubala, A., Chinni, S., Muthureddy Nataraj, S. K., et al. (2013b). Vildagliptin: an anti-diabetes agent ameliorates cognitive deficits and pathology observed in streptozotocin-induced Alzheimer’s disease. J. Pharm. Pharmacol. 65, 1773–1784. doi: 10.1111/jphp.12148
Lavan, B. E., Fantin, V. R., Chang, E. T., Lane, W. S., Keller, S. R., and Lienhard, G. E. (1997). A novel 160-kDa phosphotyrosine protein in insulin-treated embryonic kidney cells is a new member of the insulin receptor substrate family. J. Biol. Chem. 272, 21403–21407. doi: 10.1074/jbc.272.34.21403
Leng, Y., Karlsson, H. K., and Zierath, J. R. (2004). Insulin signaling defects in type 2 diabetes. Rev. Endocr. Metab. Disord. 5, 111–117. doi: 10.1023/B:REMD.0000021432.84588.f6
Li, L., Zhang, Z. F., Holscher, C., Gao, C., Jiang, Y. H., and Liu, Y. Z. (2012). (Val8) glucagon-like peptide-1 prevents tau hyperphosphorylation, impairment of spatial learning and ultra-structural cellular damage induced by streptozotocin in rat brains. Eur. J. Pharmacol. 674, 280–286. doi: 10.1016/j.ejphar.2011.11.005
Li, Y., Tian, Q., Li, Z., Dang, M., Lin, Y., and Hou, X. (2019). Activation of Nrf2 signaling by sitagliptin and quercetin combination against β-amyloid induced Alzheimer’s disease in rats. Drug Dev. Res. 80, 837–845. doi: 10.1002/ddr.21567
Lin, B., Koibuchi, N., Hasegawa, Y., Sueta, D., Toyama, K., Uekawa, K., et al. (2014). Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc. Diabetol. 13:148. doi: 10.1186/s12933-014-0148-1
Liu, Y., Liu, F., Grundke-Iqbal, I., Iqbal, K., and Gong, C. X. (2011). Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 225, 54–62. doi: 10.1002/path.2912
Ma, J., and Hart, G. W. (2013). Protein O-GlcNAcylation in diabetes and diabetic complications. Exp. Rev. Proteom. 10, 365–380. doi: 10.1586/14789450.2013.820536
Ma, Q. H., Jiang, L. F., Mao, J. L., Xu, W. X., and Huang, M. (2018). Vildagliptin prevents cognitive deficits and neuronal apoptosis in a rat model of Alzheimer’s disease. Mol. Med. Rep. 17, 4113–4119. doi: 10.3892/mmr.2017.8289
Magaki, S., Yong, W. H., Khanlou, N., Tung, S., and Vinters, H. V. (2014). Comorbidity in dementia: update of an ongoing autopsy study. J. Am. Geriatr. Soc. 62, 1722–1728. doi: 10.1111/jgs.12977
Marzban, L., Park, K., and Verchere, C. B. (2003). Islet amyloid polypeptide and type 2 diabetes. Exp. Gerontol. 38, 347–351. doi: 10.1016/s0531-5565(03)00004-4
McClean, P. L., and Hölscher, C. (2014). Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer’s disease. Neuropharmacology 86, 241–258. doi: 10.1016/j.neuropharm.2014.07.015
McClean, P. L., Parthsarathy, V., Faivre, E., and Hölscher, C. (2011). The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. 31, 6587–6594. doi: 10.1523/jneurosci.0529-11.2011
Meng, L., Li, X. Y., Shen, L., and Ji, H. F. (2020). Type 2 diabetes mellitus drugs for Alzheimer’s disease: current evidence and therapeutic opportunities. Trends. Mol. Med. 26, 597–614. doi: 10.1016/j.molmed.2020.02.002
Mothe, I., and Van Obberghen, E. (1996). Phosphorylation of insulin receptor substrate-1 on multiple serine residues, 612, 632, 662, and 731, modulates insulin action. J. Biol. Chem. 271, 11222–11227. doi: 10.1074/jbc.271.19.11222
Mueed, Z., Tandon, P., Maurya, S. K., Deval, R., Kamal, M. A., and Poddar, N. K. (2018). Tau and mTOR: the hotspots for multifarious diseases in Alzheimer’s development. Front. Neurosci. 12:1017. doi: 10.3389/fnins.2018.01017
Orr, M. E., and Oddo, S. (2013). Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res. Ther. 5:53. doi: 10.1186/alzrt217
Pan, D. A., Lillioja, S., Kriketos, A. D., Milner, M. R., Baur, L. A., Bogardus, C., et al. (1997). Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 46, 983–988. doi: 10.2337/diab.46.6.983
Pérez, M. J., and Quintanilla, R. A. (2015). Therapeutic actions of the thiazolidinediones in Alzheimer’s disease. PPAR Res. 2015:957248. doi: 10.1155/2015/957248
Perna, S., Mainardi, M., Astrone, P., Gozzer, C., Biava, A., Bacchio, R., et al. (2018). 12-month effects of incretins versus SGLT2-inhibitors on cognitive performance and metabolic profile. A randomized clinical trial in the elderly with Type-2 diabetes mellitus. Clin. Pharmacol. 10, 141–151. doi: 10.2147/cpaa.S164785
Pintana, H., Apaijai, N., Chattipakorn, N., and Chattipakorn, S. C. (2013). DPP-4 inhibitors improve cognition and brain mitochondrial function of insulin-resistant rats. J. Endocrinol. 218, 1–11. doi: 10.1530/joe-12-0521
Pipatpiboon, N., Pintana, H., Pratchayasakul, W., Chattipakorn, N., and Chattipakorn, S. C. (2013). DPP4-inhibitor improves neuronal insulin receptor function, brain mitochondrial function and cognitive function in rats with insulin resistance induced by high-fat diet consumption. Eur. J. Neurosci. 37, 839–849. doi: 10.1111/ejn.12088
Poppe, R., Karbach, U., Gambaryan, S., Wiesinger, H., Lutzenburg, M., Kraemer, M., et al. (1997). Expression of the Na+-D-glucose cotransporter SGLT1 in neurons. J. Neurochem. 69, 84–94. doi: 10.1046/j.1471-4159.1997.69010084.x
Qu, Z. S., Li, L., Sun, X. J., Zhao, Y. W., Zhang, J., Geng, Z., et al. (2014). Glycogen synthase kinase-3 regulates production of amyloid-β peptides and tau phosphorylation in diabetic rat brain. ScientificWorldJournal 2014:878123. doi: 10.1155/2014/878123
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Ramos-Rodriguez, J. J., Spires-Jones, T., Pooler, A. M., Lechuga-Sancho, A. M., Bacskai, B. J., and Garcia-Alloza, M. (2017). Progressive neuronal pathology and synaptic loss induced by prediabetes and Type 2 diabetes in a mouse model of Alzheimer’s disease. Mol. Neurobiol. 54, 3428–3438. doi: 10.1007/s12035-016-9921-3
Rizzo, M. R., Barbieri, M., Boccardi, V., Angellotti, E., Marfella, R., and Paolisso, G. (2014). Dipeptidyl peptidase-4 inhibitors have protective effect on cognitive impairment in aged diabetic patients with mild cognitive impairment. J. Gerontol. A Biol. Sci. Med. Sci. 69, 1122–1131. doi: 10.1093/gerona/glu032
Roberts, L. D., Koulman, A., and Griffin, J. L. (2014). Towards metabolic biomarkers of insulin resistance and type 2 diabetes: progress from the metabolome. Lancet Diab. Endocrinol. 2, 65–75. doi: 10.1016/s2213-8587(13)70143-8
Roden, M., Price, T. B., Perseghin, G., Petersen, K. F., Rothman, D. L., Cline, G. W., et al. (1996). Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Invest. 97, 2859–2865. doi: 10.1172/jci118742
Russo, V. C., Gluckman, P. D., Feldman, E. L., and Werther, G. A. (2005). The insulin-like growth factor system and its pleiotropic functions in brain. Endocr. Rev. 26, 916–943. doi: 10.1210/er.2004-0024
Sa-Nguanmoo, P., Tanajak, P., Kerdphoo, S., Jaiwongkam, T., Pratchayasakul, W., Chattipakorn, N., et al. (2017). SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuating mitochondrial dysfunction, insulin resistance, inflammation, and apoptosis in HFD-induced obese rats. Toxicol. Appl. Pharmacol. 333, 43–50. doi: 10.1016/j.taap.2017.08.005
Saxena, U. (2010). Alzheimer’s disease amyloid hypothesis at crossroads: where do we go from here? Exp. Opin. Ther. Targets 14, 1273–1277. doi: 10.1517/14728222.2010.528285
Shah, K., Desilva, S., and Abbruscato, T. (2012). The role of glucose transporters in brain disease: diabetes and Alzheimer’s Disease. Int. J. Mol. Sci. 13, 12629–12655. doi: 10.3390/ijms131012629
Shiiki, T., Ohtsuki, S., Kurihara, A., Naganuma, H., Nishimura, K., Tachikawa, M., et al. (2004). Brain insulin impairs amyloid-beta(1-40) clearance from the brain. J. Neurosci. 24, 9632–9637. doi: 10.1523/jneurosci.2236-04.2004
Showkat, M., Beigh, M. A., and Andrabi, K. I. (2014). mTOR signaling in protein translation regulation: implications in cancer genesis and therapeutic interventions. Mol. Biol. Int. 2014:686984. doi: 10.1155/2014/686984
Talbot, K., Wang, H. Y., Kazi, H., Han, L. Y., Bakshi, K. P., Stucky, A., et al. (2012). Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 122, 1316–1338. doi: 10.1172/jci59903
Taylor, R. (2012). Insulin resistance and type 2 diabetes. Diabetes 61, 778–779. doi: 10.2337/db12-0073
Thiels, E., and Klann, E. (2001). Extracellular signal-regulated kinase, synaptic plasticity, and memory. Rev. Neurosci. 12, 327–345. doi: 10.1515/revneuro.2001.12.4.327
Thomas, L., Eckhardt, M., Langkopf, E., Tadayyon, M., Himmelsbach, F., and Mark, M. (2008). (R)-8-(3-amino-piperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (BI 1356), a novel xanthine-based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase-4 inhibitors. J. Pharmacol. Exp. Ther. 325, 175–182. doi: 10.1124/jpet.107.135723
Vieira, M. N. N., Lima-Filho, R. A. S., and De Felice, F. G. (2018). Connecting Alzheimer’s disease to diabetes: underlying mechanisms and potential therapeutic targets. Neuropharmacology 136, 160–171. doi: 10.1016/j.neuropharm.2017.11.014
Vijan, S. (2015). In the clinic. Type 2 diabetes. Ann. Intern. Med. 162, Itc1–Itc16. doi: 10.7326/aitc201503030
Wang, X., Wang, L., Jiang, R., Xu, Y., Zhao, X., and Li, Y. (2016). Exendin-4 antagonizes Aβ1-42-induced attenuation of spatial learning and memory ability. Exp. Ther. Med. 12, 2885–2892. doi: 10.3892/etm.2016.3742
Wiciński, M., Wódkiewicz, E., Górski, K., Walczak, M., and Malinowski, B. (2020). Perspective of SGLT2 inhibition in treatment of conditions connected to neuronal loss: focus on Alzheimer’s disease and ischemia-related brain injury. Pharmaceuticals (Basel) 13:79. doi: 10.3390/ph13110379
Xiong, H., Zheng, C., Wang, J., Song, J., Zhao, G., Shen, H., et al. (2013). The neuroprotection of liraglutide on Alzheimer-like learning and memory impairment by modulating the hyperphosphorylation of tau and neurofilament proteins and insulin signaling pathways in mice. J. Alzheimers Dis. 37, 623–635. doi: 10.3233/jad-130584
Xu, L., Nagata, N., Nagashimada, M., Zhuge, F., Ni, Y., Chen, G., et al. (2017). SGLT2 Inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine 20, 137–149. doi: 10.1016/j.ebiom.2017.05.028
Yildirim Simsir, I., Soyaltin, U. E., and Cetinkalp, S. (2018). Glucagon like peptide-1 (GLP-1) likes Alzheimer’s disease. Diabetes Metab. Syndr. 12, 469–475. doi: 10.1016/j.dsx.2018.03.002
Yu, A. S., Hirayama, B. A., Timbol, G., Liu, J., Basarah, E., Kepe, V., et al. (2010). Functional expression of SGLTs in rat brain. Am. J. Physiol. Cell Physiol. 299, C1277–C1284. doi: 10.1152/ajpcell.00296.2010
Yu, A. S., Hirayama, B. A., Timbol, G., Liu, J., Diez-Sampedro, A., Kepe, V., et al. (2013). Regional distribution of SGLT activity in rat brain in vivo. Am. J. Physiol. Cell Physiol. 304, C240–C247. doi: 10.1152/ajpcell.00317.2012
Zhang, W., Thompson, B. J., Hietakangas, V., and Cohen, S. M. (2011). MAPK/ERK signaling regulates insulin sensitivity to control glucose metabolism in Drosophila. PLoS Genet. 7:e1002429. doi: 10.1371/journal.pgen.1002429
Zhao, W. Q., De Felice, F. G., Fernandez, S., Chen, H., Lambert, M. P., Quon, M. J., et al. (2008). Amyloid beta oligomers induce impairment of neuronal insulin receptors. Faseb J. 22, 246–260. doi: 10.1096/fj.06-7703com
Zheng, T., Yang, X., Wu, D., Xing, S., Bian, F., Li, W., et al. (2015). Salidroside ameliorates insulin resistance through activation of a mitochondria-associated AMPK/PI3K/Akt/GSK3β pathway. Br. J. Pharmacol. 172, 3284–3301. doi: 10.1111/bph.1312s0
Keywords: insulin signaling, insulin resistance, Alzheimer’s disease, type 2 diabetes mellitus, DPP-4 inhibitor, SGLT2 inhibitor
Citation: Sim AY, Barua S, Kim JY, Lee Y-h and Lee JE (2021) Role of DPP-4 and SGLT2 Inhibitors Connected to Alzheimer Disease in Type 2 Diabetes Mellitus. Front. Neurosci. 15:708547. doi: 10.3389/fnins.2021.708547
Received: 12 May 2021; Accepted: 12 July 2021;
Published: 11 August 2021.
Edited by:
Elena Zenaro, University of Verona, ItalyReviewed by:
Habib Yaribeygi, Semnan University of Medical Sciences, IranCopyright © 2021 Sim, Barua, Kim, Lee and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jong Eun Lee, amVsZWVAeXVocy5hYw==; orcid.org/0000-0001-6203-7413
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.