 Meng Zhang
Meng Zhang Zhigang Bian
Zhigang Bian
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurosci. , 24 May 2021
Sec. Neurodegeneration
Volume 15 - 2021 | https://doi.org/10.3389/fnins.2021.687973
Alzheimer’s disease (AD) is a common neurodegenerative disease in the elderly and is the most common type of dementia. AD is mostly gradual onset, and involves slow, progressive mental decline, accompanied by personality changes; the incidence of AD gradually increases with age. The etiology of AD is unknown, although it is currently believed to be related to abnormal deposition of amyloid β-protein (Aβ) in the brain, hyperphosphorylation of microtubule-associated protein tau, and the release of various cytokines, complements, activators and chemokines by cells. MicroRNAs (miRNAs) are a class of highly conserved non-coding RNAs that regulate gene expression at the post-transcriptional level, and manipulate the functions of intracellular proteins and physiological processes. Emerging studies have shown that miRNA plays an important role in regulating AD-related genes. MiR-132 is known as “NeurimmiR” due to its involvement in numerous neurophysiological and pathological processes. Accumulating pre-clinical results suggest that miR-132 may be involved in the progression of Aβ and tau pathology. Moreover, clinical studies have indicated that decreased circulating miR-132 levels could be used a potential diagnostic biomarker in AD. Here, we review the pathogenic role of miR-132 activity in AD, and the potential of targeting miR-132 for developing future therapeutic strategies.
Alzheimer’s disease is one of the most common neurodegenerative disease in the elderly; the main neuropathological hallmarks of AD are senile plaques (SP), neurofibrillary tangles (NFT), and loss of neurons (Iqbal et al., 2010; Bloom, 2014). AD is the most common type of senile dementia, and is characterized by recessive onset, slow progressive mental decline, and personality changes (Cerovic et al., 2019). The incidence of AD increases gradually with age (Herrera-Espejo et al., 2019). The etiology of AD is believed to involve abnormal deposition of amyloid β-protein (Aβ) in the brain (Chakravorty et al., 2019). The Aβ in AD is mainly produced by degradation of β-site amyloid precursor protein-cleaving enzyme 1 (BACE1). An increase in concentration and activity of BACE1 in the brain indicates over-expression of the BACE1 gene (Mazdeh et al., 2018). Intracellular NFT are mainly composed of paired helical fibers (PHF). It has been found that hyperphosphorylation of microtubule-associated protein tau results in the formation of PHF, which form NFT, damaging the stability of the cytoskeleton and causing neurotoxicity (Sorrentino and Bonavita, 2007). A variety of cytokines, complements, their activators, and chemokines, such as COX-2 and complement factor H (CFH), are involved in the inflammatory response, leading to non-specific inflammatory cell infiltration, contributing to the pathogenesis of AD (Rivas-Arancibia et al., 2015; Williams et al., 2015). Currently, as the specific pathogenesis of AD is unknown in clinical practice, it is difficult to have a simple and effective method for early diagnosis (Zeng et al., 2019). The main therapeutic drugs for AD in clinical practice are cholinesterase inhibitors (CHEI) and selective antagonists of N-methyl-D-aspartic acid (NMDA) receptors (Reisberg et al., 2003; Wattmo and Wallin, 2017). However, due to the unclear pathogenesis of AD, the currently marketed therapeutic drugs can only delay the progression of the disease or slightly improve it, and there are no effective drugs that can reverse or prevent the progression of AD. Therefore, it is crucial to identify the molecular mechanism of AD pathogenesis and develop new and effective treatment methods.
MicroRNAs are a class of small non-coding single-stranded RNA molecules about 21–25 nt in length, which are usually involved in regulating transcription and expression of target genes at the post-mRNA transcription level. Single miRNAs can regulate up to 200 mRNAs, thus playing important roles in many key biological metabolic processes, such as cell growth, tissue differentiation, cell proliferation, embryonic development, and apoptosis (Dickson et al., 2013; Dong et al., 2015). Therefore, miRNA mutations, changes in miRNA synthesis, and dysfunction of miRNA and its target sites may provoke the development of diseases. In addition, some miRNAs are widely distributed in the central nervous system (CNS) and play important regulatory roles in neural development, differentiation and maturation. Deregulated expression of these miRNAs may lead to the development of various neurological diseases, including AD (Song and Lee, 2015; Maffioletti et al., 2019).
More specifically, significant reduction in miR-29a/b-1 expression was found in patients with AD, indicating an abnormal increase in BACE1 (Hébert et al., 2008). In addition, Peter et al., found that miR-107 levels in the temporal lobe were lower in patients with early AD, and there was a negative correlation between miR-107 and BACE1 mRNA levels (Nelson and Wang, 2010). In particular, miR-146a is upregulated in AD brains, causing upregulation of immune and inflammatory signals through IRAK1 and TRAF6, suggesting that miR-146a may be dysregulated and lead to the inflammatory response in AD (Ferguson-Chanowitz et al., 1990; Wang et al., 2012).
Importantly, one of the most abundant miRNAs in the brain is miR-132 (Klein et al., 2007) which was first discovered in mouse nerve tissue by Lagos-Quintana et al. (2002). Mature miR-132 is 22 bp in length and is processed from a precursor sequence of 66 bp. Human miR-132 is composed of two homologous miRNAs, hsa-miR-132-5p, and hsa-miR-132-3p. MiR-132 is evolutionary conserved and has the same sequence and structure in humans, rats, mice, apes and other species. MiR-132 has tissue specificity and is highly expressed in nerve-related tissues. MiR-132 is transcribed by the activity-dependent transcription factor cAMP-response element binding protein (CREB), and regulates axon, dendritic and spinal maturation in response to multiple signaling pathways (Klein et al., 2007; Magill et al., 2010). Sirtuin 1, encoded in humans by SIRT1, is best recognized as associated with longevity and aging (Jeong et al., 2011), miR-132 is shown to target SIRT1 (Strum et al., 2009). Deletion of miR-132 is shown to induce tau aggregation and impair mouse cognitive skills (Smith et al., 2015; Hansen et al., 2016). In mammals, the IGF-1 pathway affects the phenotype of aging (Kenyon et al., 1993). FOXO1 is one of the major components of IGF-1 axis (Pöllänen et al., 2010), and it is also a potential regulatory target gene of miR-132 (Budzinska et al., 2016). Pathologically, various studies have indicated that miR-132 is the most common downregulated miRNA in the postmortem AD brain, and is involved in the progression of Aβ and tau pathology (Lau et al., 2013; Patrick et al., 2017; Pichler et al., 2017).
In this review, we will discuss preclinical and clinical data on the novel role of miR-132 in AD pathophysiology, with the aim of gaining a deeper understanding of the molecular mechanisms of AD and developing novel therapeutic strategies (Figure 1).
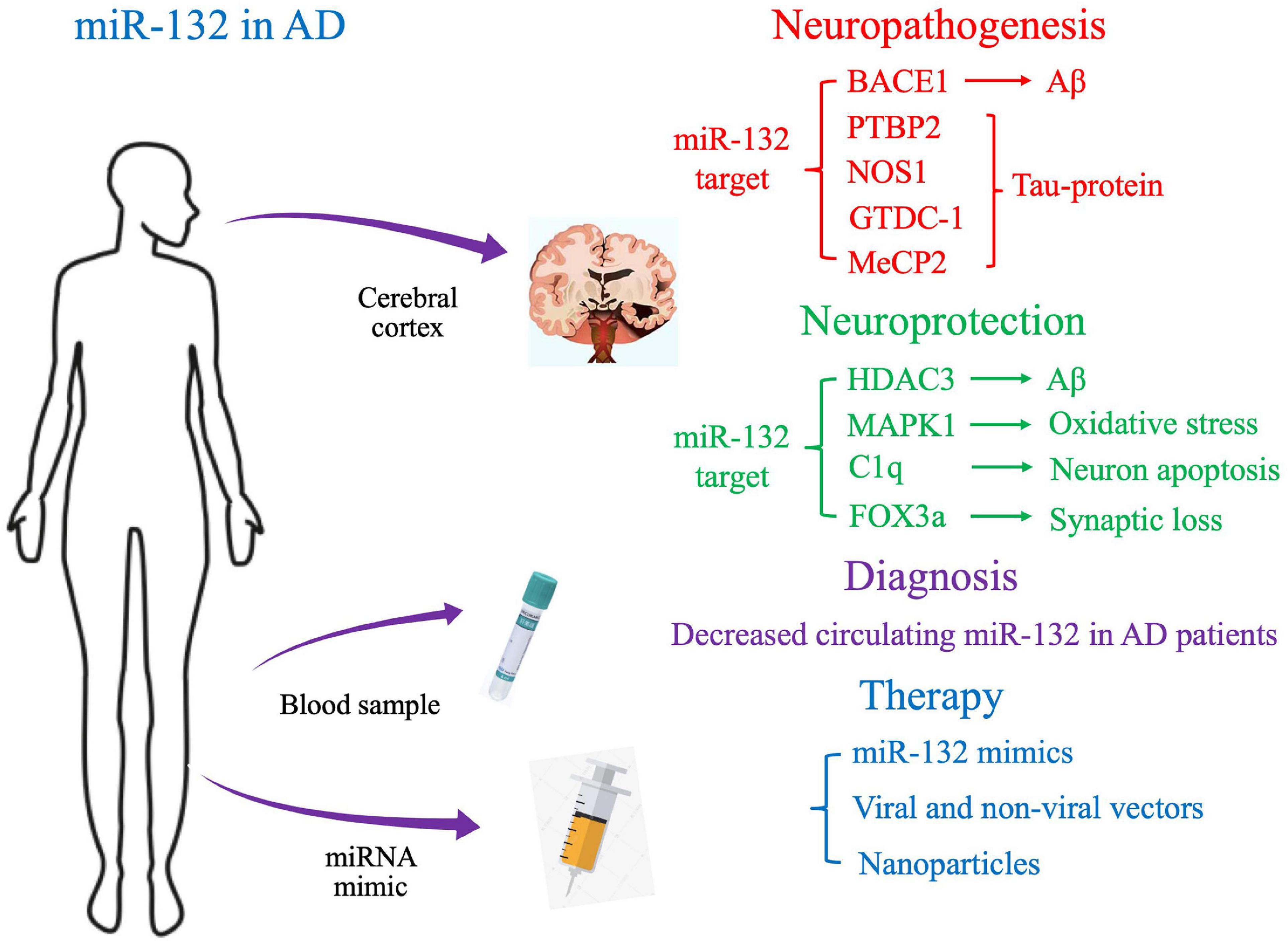
Figure 1. Proposed molecular targets and clinical application of miR-132 in Alzheimer’s disease.
Downregulation of miR-132 has been shown to be involved in the pathogenesis of AD both in vivo and in vitro AD models, therefore, regulating miR-132 expression by different mechanisms may provide a new therapeutic strategy for AD treatment.
Soluble Aβ has been shown to decrease miR-132 expression and increase histone deacetylase (HDAC) levels in cultured primary neurons. In addition, Wei et al. (2020) indicated that there was a direct regulatory relationship between miR-132 and HDAC3. Upregulating miR-132 expression or downregulating HDAC3 levels could enhance hippocampal long-term potentiation and prevent hippocampal impairment by Aβ in AD mice. They also concluded that upregulated miR-132 expression or reduced HDAC3 signaling may counteract synaptotoxicity and lessen the progressive effects of Aβ accumulation during human brain aging (Wei et al., 2020).
Mitogen-activated protein kinases (MAPKs) are serine-threonine kinases that are highly expressed in the CNS. MAPK1 is involved in the increase of inflammation and apoptosis of neurons. Neuron apoptosis during AD is closely associated with the MAPK pathway (Li et al., 2019). In the study by Deng et al. (2020) miR-132 expression was significantly downregulated in an AD rat model, and MAPK1 expression was significantly upregulated; they suggested that miR-132 and MAPK1 had specific binding sites, and that miR-132 could inhibit MAPK1 expression. They concluded that miR-132 can inhibit hippocampal oxidative stress and iNOS expression by inhibiting MAPK1 expression to improve the cognitive function of AD rats (Deng et al., 2020).
Synaptic loss in the hippocampus and neocortex occurs throughout the pathological process of AD. In the early stage of AD (Zhang et al., 2014), Aβ-induced synaptic loss is the main cause, while in the late stage, accumulation of tau protein promotes synaptic degeneration, which is the key factor that leads to dementia (Shankar et al., 2008; Marciniak et al., 2017). Furthermore, C1q is a major protein in the classical complement cascade, and is highly expressed in synaptic regions of the CNS in AD patients (Bialas and Stevens, 2013). It has been reported that, in mouse models of AD, C1q levels were increased and associated with synaptic loss before apparent plaque deposition (Hong et al., 2016), and inhibition of C1q expression reduced the number of phagocytic microglia, and may be involved in early synaptic loss (Stevens et al., 2007). Xu et al. (2019) showed that miR-132 was significantly reduced in the brains of AD patients; they also found that upregulated miR-132 could increase expression levels of synaptic proteins in the temporal cortex of mice, and the same effect could also be obtained by suppressing C1q levels. Subsequently, they verified that C1q was genuinely regulated by miR-132 in vivo, and suggested that miR-132 could prevent synaptic loss by regulating C1q expression, which may provide new strategies for treating AD (Xu et al., 2019).
Samandari-Bahraseman et al. (2019) found that miR-132 decreased in the brain cells of AD patients when studying the mechanism of Eremostachys labiosiformis in the treatment of AD. This downregulation of miR-132 expression led to increased neuronal cell apoptosis. Accordingly, upregulating miR-132 expression could prevent neuronal apoptosis by directly regulating the expression of FOX3a, PTEN and P300 as the main components of the APK pathway (Samandari-Bahraseman et al., 2019).
Furthermore, El Fatimy et al. (2018) demonstrated that miR-132 exerted neuroprotective effects for AD through multiple signaling pathways; miR-132 could eliminate multiple forms of tau protein implicated in tauopathies. In addition, miR-132 could attenuate phospho-tau pathology and enhance long-term potentiation in P301S tau transgenic mice. Moreover, miR-132 protected neurons against Aβ and glutamate excitotoxicity; El Fatimy et al. (2018) indicated that miR-132 over-expression preserved cell body clusters and neurite integrity in PS19 neurons treated with Aβ. Collectively, these results demonstrated that miR-132 could regulate tau homeostasis, as well as protect neurons against Aβ deposition, suggesting that miR-132 supplementation could be therapeutically beneficial for treating AD (El Fatimy et al., 2018; Table 1).
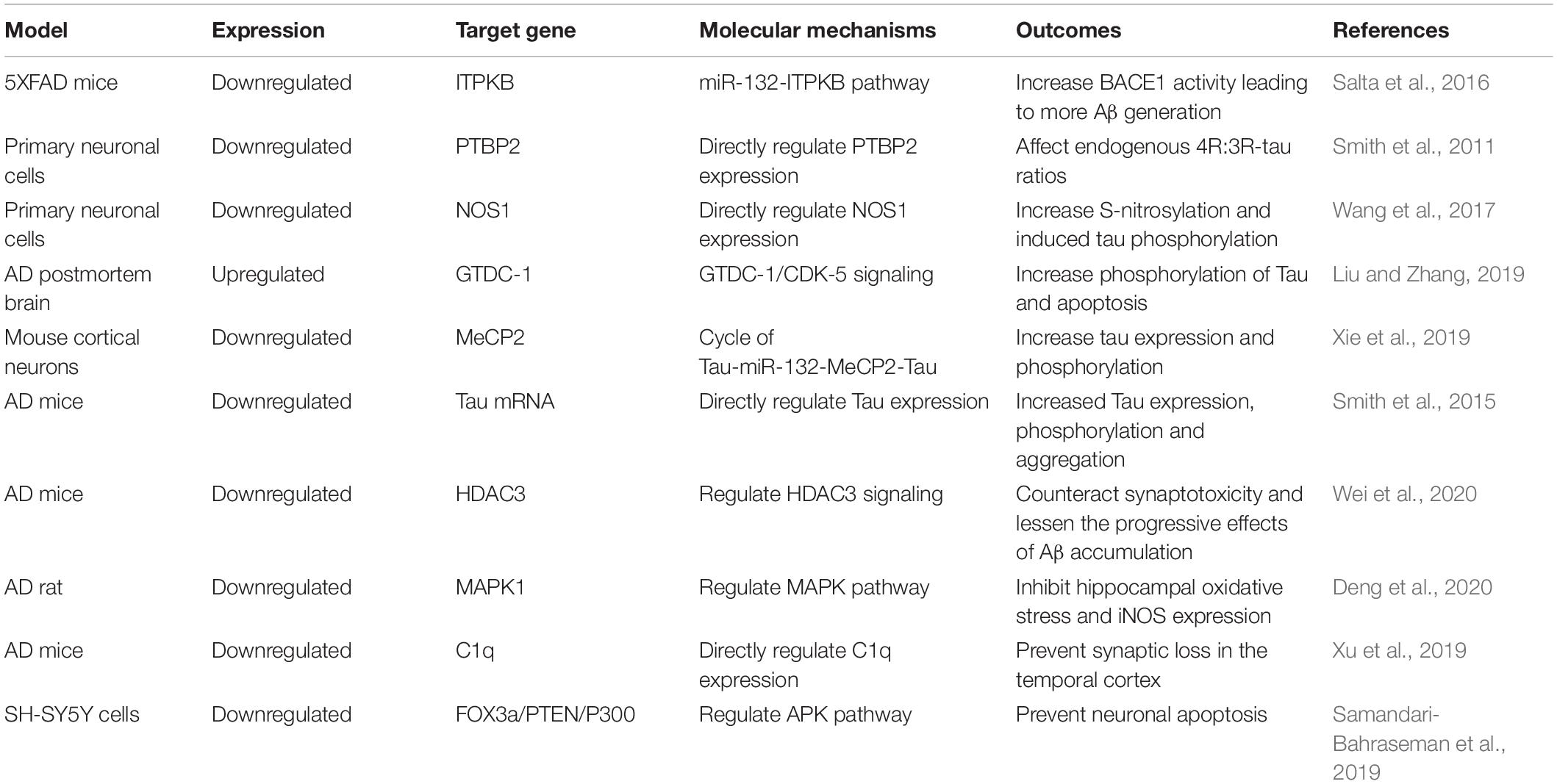
Table 1. The role of miR-132 in Alzheimer’s disease: Evidence from in vitro and in vivo studies.
Previous studies have found that several lncRNAs are specifically expressed in brain tissue, and are involved in many important neurological functions (Smalheiser, 2014; Barry et al., 2015; Lee et al., 2015; He et al., 2017; Yang et al., 2017). β-amyloid cleaving enzyme 1 antisense RNA (BACE1-AS) can promote tau phosphorylation, which is related to another important pathological sign of AD-NFTs. Ge et al. (2020) showed that miR-132-3p was a direct target of BACE1-AS, and was negatively regulated by BACE1-AS in neuronal cells. Depletion of BACE1-AS upregulated miR-132-3p expression and attenuated the neuronal damage induced by Aβ25–35, indicating that modulating the BACE1-AS/miR-132-3p axis could provide new insights into AD treatment (Ge et al., 2020). Wang et al. (2018) indicated that miR-132 was a target of XIST, and XIST could relieve Aβ25–35 induced toxicity, oxidative stress, and apoptosis in rat hippocampal neurons by upregulation of miR-132 expression, suggesting the potential of manipulating XIST in AD treatment.
In conclusion, these mechanisms may be applicable in the case of AD, and studying these potential miR-132 targets in AD may provide a promising area of research.
It is believed that Aβ is the initiating factor of AD, and Aβ is hydrolyzed by β-amyloid precursor protein (APP); BACE1, as a lytic enzyme, plays an important role in the pathogenesis of AD. It has been shown that miR-132 is involved in regulating Aβ and BACE1 expression. Autopsy results have confirmed that BACE1 expression in the AD brain was upregulated at the protein level, but not at the mRNA level, further confirming that miRNA regulates gene expression after transcription rather than at the mRNA level (Ji et al., 2019).
Furthermore, Hernandez-Rapp et al. (2016) found that the amount of endogenous soluble Aβ42 in the hippocampus was significantly increased in 18-month-old triple transgenic AD (3xTg-AD) mice (with miR-132 knockout) compared to controls (without miR-132 knockout), suggesting that lack of miR-132 in mice can promote Aβ production, aggregation and deposition. In addition, regarding data from the Religious Orders Study (ROS), the researchers found that miR-132 levels were lower in mild cognitive impairment (MCI) and AD patients compared to healthy controls, and there was a negative correlation between miR-132 levels and memory deficits; miR-132 and insoluble Aβ42 levels were also significantly correlated (Hernandez-Rapp et al., 2016). To further verify the connection between miR-132 and Aβ, miR-132 mimics were established in Neuro2a and HEK293 APPSwe cell lines stably expressing (human) Aβ. In both cell lines, miR-132 significantly downregulated (soluble) human Aβ40 and Aβ42 levels (determined by ELISA), suggesting that miR-132 deficiency enhanced Aβ production (Hernandez-Rapp et al., 2016).
Abnormal aggregation of tau protein is involved in the pathogenesis of AD, and the most common pathological changes in AD patients are tau protein phosphorylation, abnormal precipitation and aggregation of tau protein, resulting in the formation of NFTs (Congdon and Sigurdsson, 2018). A large number of specific miRNAs are reduced during AD development, which may be involved in the pathogenesis of AD by affecting the abnormal aggregation of tau protein (Fitzpatrick et al., 2017; Nisbet and Götz, 2018). MiR-132 directly targeted the neuronal splicing factor polypyrimidine tract-binding protein 2 (PTBP2); thus, miR-132 over-expression or knockdown of PTBP2 could affect endogenous 4R:3R-tau ratios in neuronal cells (Smith et al., 2011). Wang et al., suggested that miR-132 can directly regulate neuronal nitric oxide synthase (NOS1) expression through primate-specific binding sites. Inhibiting miR-132 expression in neural cells led to increased levels of NOS1 and triggered excessive production of nitric oxide, followed by abnormal S-nitrosylation of specific proteins related to tau pathology. This indicated that downregulation of miR-132 can disturb the balance of S-nitrosylation and induce tau phosphorylation in a NOS1-dependent manner, which may be involved in the pathogenesis of AD (Wang et al., 2017). Furthermore, Liu et al., showed that miR-132 expression was significantly higher in postmortem brain specimens of AD patients compared to normal controls. In addition, miR-132 could induce neuron apoptosis by affecting the expression of cell apoptosis-related factors, such as Bax and Bcl-2. Cyclic-dependent kinase-5 (CDK-5) is a critical kinase associated with tau phosphorylation. Over-expression of miR-132 was shown to promote CDK5 expression and accumulation of p35/p25. Glycosyltransferase-Like domain containing-1 (GTDC-1) was a direct target of miR-132, and over-expression of miR-132 could suppress GTDC-1 expression, inducing apoptosis of neuronal cells. In addition, GTDC-1 decreased Bax expression and increased Bcl-2 expression. Furthermore, GTDC-1 markedly inhibited tau phosphorylation. Therefore, miR-132 plays an important role in the pathogenesis of AD by regulating apoptosis and GTDC-1/CDK-5/tau phosphorylation signaling mechanisms (Liu and Zhang, 2019). Notably, in human tau-overexpressing neurons, methyl-CpG-binding protein 2 (MeCP2) levels were increased, while miR-132 was decreased, and miR-132 was found to negatively regulate MeCP2 expression both in vitro and in vivo (Xie et al., 2019). Xie et al., found that miR-132 deficiency led to increased tau expression, phosphorylation, and aggregation in mice. In addition, miR-132 regulated tau expression and phosphorylation, and thus contributed to tauopathy in AD by regulating MeCP2 levels, suggesting a vicious cycle of tau-miR-132-MeCP2-tau abnormalities in AD (Xie et al., 2019). Smith et al., found that miR-132 deficiency led to increased tau expression, phosphorylation and aggregation in mice, as well as demonstrating that miR-132 directly targeted tau mRNA to regulate its expression, by using reporter assays and cell-based studies. In addition, miR-132 mimics could partly restore memory function and tau metabolism in AD mice. Moreover, miR-132 levels were associated with insoluble tau and cognitive impairment in humans. MiR-132 could eliminate multiple forms of tau protein implicated in tauopathies, including the cleaved, phosphorylated and acetylated forms, and promote the extension and branching of neurites and reduce neuronal death. MiR-132 also directly regulated the tau modifiers acetyltransferase EP300, kinase GSK3β, RNA-binding protein Rbfox1, Caspases 3/7 and proteases Calpain 2; miR-132 could attenuate phospho-tau pathology and enhance long-term potentiation in P301S tau transgenic mice. These results support a role for miR-132 in the regulation of tau pathology in mice and humans, and may provide new strategies for therapeutic development (Smith et al., 2015).
The etiology and pathogenesis of AD are not completely clear, and there are no effective means for early diagnosis and treatment (Cordell et al., 2013). As diagnostic biomarkers, both Aβ and tau protein are mainly expressed in cerebrospinal fluid (CSF), but collection of CSFs is invasive and inconvenient, making clinical development difficult. If reliable diagnostic biomarkers can be found in peripheral blood, it would be greatly significant for early screening, diagnosis and treatment of AD. MiRNAs are secreted extracellularly in the form of small vesicles or non-vesicles (such as peripheral blood, serum, plasma, saliva, and urine) and can bind to proteins or other compounds. For example, miRNAs in plasma can bind to high-density lipoproteins to form a stable structure and thus exist in peripheral blood (Noren Hooten et al., 2013; Turchinovich et al., 2013). Currently, miRNAs in the peripheral circulation are on the way to becoming important biomarkers for evaluating diseases (Schwarzenbach et al., 2014; Wang and Zhang, 2020).
Given the important role of miR-132 in the pathogenesis of AD, it is reasonable to suggest that it could serve as a potential biomarker for the diagnosis and progression of AD. A recent study of 24 AD patients and 45 controls showed that miR-132 levels in lymphoblastoid cell lines (LCLs) were dramatically decreased in AD patients compared with the controls. In addition, miR-132 expression levels were negatively correlated with Braak stage scores in AD olfactory bulb tissues (Hadar et al., 2018). Another study measured miR-132-3p levels in neurally derived plasma exosomes from 16 early stage AD patients, 16 MCI individuals and 31 healthy controls; Cha et al. (2019) indicated that miR-132-3p levels were lower in AD patients and MCI individuals compared to controls, thus demonstrating preferable sensitivity and specificity to diagnose AD. However, the results failed to separate AD patients from MCI individuals (Cha et al., 2019). Furthermore, miR-132 may also contribute to discriminate early cognitive dysfunction. Xie et al., performed a cross-sectional cohort study, which included 66 MCI patients and 76 healthy controls. They found that serum miR-132 levels were significantly increased in MCI patients compared with healthy controls. These results preliminarily suggested that circulating miR-132 was upregulated in MCI patients and could be potentially a biomarker for diagnosing MCI (Xie et al., 2015).
However, it is worth mentioning that, so far, there are relatively few studies that have investigated various circulating miRNA levels in AD patients, and all published data indicate that the overlap between detected miRNA targets is particularly limited. Moreover, the sample sizes of these studies were relatively limited, and the results were inconclusive. Most studies reported that miR-132 levels were downregulated in AD patients, and this abnormal expression appeared in the cerebral cortex and hippocampus of AD patients. In LCLs, miR-132 gradually decreased with age in patients with cognitive impairment. However, Xie et al., found serum miR-132 levels were significantly increased in MCI. Therefore, future studies are required to confirm these results, as well as the standardization of application methods, cohort verification from multiple centers, larger patient samples and ideal postmortem diagnosis of AD to obtain clinically meaningful results.
Therefore, miR-132 may be used as a diagnostic biomarker for AD, but in order to obtain clinically meaningful results, additional large sample studies are needed. In view of the importance of miR-132 in the pathogenesis of AD, even in the initial stage mentioned above, we could speculate that it may also be used as a preclinical biomarker in the pre-symptomatic stage, especially for high-risk groups.
Currently, AD treatment still has no efficient strategy to prevent the disease from progressing (Ayaz et al., 2017). Therefore, there is an urgent need to find new and effective therapeutic targets, especially regarding the early stages of the disease.
MicroRNAs have been shown to play an important role in biological processes such as cell differentiation, proliferation, apoptosis and other cell activities. In addition, a large number of experimental results have shown that miRNAs play a critical role in the development of neurons, muscles, and striatum (Follert et al., 2014). In recent years, studies have shown that miR-132, as an important regulator of neural development, is overexpressed in neural progenitor and mature neurons and can promote neural development (Chen D. et al., 2018; Jia et al., 2019). Currently, many miRNA-based treatment strategies have been developed, such as miRNA antisense technology and alternative therapies (Srivastava et al., 2016; Titze-de-Almeida et al., 2020). Antisense technology has been widely used to inhibit the expression of miRNAs. When there is over-expression of miRNAs in the body, which promotes disease development, antisense technology, namely miRNA inhibition therapy, can be used to block the expression of relevant proteins and thus inhibit miRNAs (Lima et al., 2018). Inhibitors of miRNA include anti-miRNA oligonucleotides (AMOs), which can be designed to complement and bind to miRNAs to prevent mRNA degradation (Lennox et al., 2017). It has been found that AMOs can enhance the selective hybridization of endogenous miRNA and achieve effective inhibition through chemical modification (Baumann and Winkler, 2014). Conversely, the development of miRNA mimics with the ability to bind to RNA-induced silencing complex and target mRNA as miRNA replacement therapy can further reduce mRNA expression levels (Lykhmus et al., 2016). These miRNAs mimics contain sequences consistent with those of mature endogenous miRNAs.
Gene therapy has received widespread attention as a way to treat certain diseases, such as cancer. miRNA therapy can be divided into two categories according to the expression status of the target miRNA: (1) miRNA inhibition therapy when miRNA is overexpressed; (2) miRNA replacement therapy when miRNA is inhibited (Roitbak, 2018). However, miRNA therapies still face a number of challenges, including achieving effective cell uptake and tissue-specific delivery, as well as issues of toxicity minimization and off-target effects. Therefore, miRNA-based therapies are highly dependent on utilizing vector-mediated delivery mechanisms. To improve the efficiency of miRNA delivery, viral and non-viral vectors have been developed (Wang et al., 2015; Labatut and Mattheolabakis, 2018). Virus-based delivery systems have high transduction efficiency and can effectively deliver genetic material to target cells. However, the disadvantages of viral vectors (e.g., inflammatory/immunogenicity response) limit their application in gene delivery and make it difficult to achieve large-scale manufacturing and quality control (Itaka and Kataoka, 2009; Chen Y. H. et al., 2018). Compared with viral vectors, non-viral delivery systems (polymer-based systems, liposomes and inorganic nanoparticles) are diverse and relatively safe, thus effectively avoiding the problems posed by viral vectors through reasonable design and appropriate modifications. Therefore, non-viral delivery systems can be widely used in clinical studies (Khalil et al., 2019; Zhang et al., 2019). Salta et al. (2016) showed downregulation of miR-132 in three distinct human AD patient cohorts, and showed that inositol 1,4,5-trisphosphate 3-kinase B (ITPKB) was a direct target gene of miR-132, which affected BACE1 activity, increasing Aβ expression in 5XFAD mice. This indicated the pathological relevance of the miR-132-ITPKB pathway in AD; considering the versatility of miRNAs as therapeutic drugs and the ever-increasing technical level, it was speculated that miR-132 mimics have potential applications in alleviating the progressive neurodegeneration of AD patients (Salta et al., 2016). In addition, Su et al. (2020) built wheat germ agglutinin (WGA)-nanoparticle-miR132 intranasally to treat AD mice, and found that nasal administration can enable the blood-brain barrier to be crossed, which increases drug bioavailability in the brain; moreover, synaptic protein expression levels were increased significantly after administration. Thus, proposal of the nasal delivery of WGA-nanoparticle-miR132 was an interesting novel therapeutic approach for AD treatment (Su et al., 2020).
However, as miR-132 may target various genes and be involved in multiple pathways in a cell-type-specific manner, its biological effects of miR-132-based therapy should be fully studied before they can be translated into clinical practice. Importantly, although emerging studies have investigated the potential role of various miRNAs in human disease, the most challenging part of the field is selecting the “correct” miRNA from a great number of potential candidates (Godlewski et al., 2019). Based on the above evidence, miR-132 is expected to become a target for AD therapy and thus merits further study.
In conclusion, miR-132 is involved in the core pathophysiological mechanism of AD and may serve as a potential diagnostic biomarker. However, more studies are needed to successfully clarify its biological and clinical values. Although the effective delivery of miR-132 in the CNS has practical limitations, developing new strategies currently under study can pave the way for the clinical application of miR-132 for treating AD.
MZ and ZB drafted and revised the manuscript. ZB drafted and modified the figures. Both authors approved the final version of the manuscript and agreed to be accountable for all aspects of the work to ensure that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ayaz, M., Sadiq, A., Junaid, M., Ullah, F., Subhan, F., and Ahmed, J. (2017). Neuroprotective and anti-aging potentials of essential oils from aromatic and medicinal plants. Front. Aging Neurosci. 9:168. doi: 10.3389/fnagi.2017.00168
Barry, G., Guennewig, B., Fung, S., Kaczorowski, D., and Weickert, C. S. (2015). Long non-coding RNA expression during aging in the human subependymal zone. Front. Neurol. 6:45. doi: 10.3389/fneur.2015.00045
Baumann, V., and Winkler, J. (2014). miRNA-based therapies: strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med. Chem. 6, 1967–1984. doi: 10.4155/fmc.14.116
Bialas, A. R., and Stevens, B. (2013). TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat. Neurosci. 16, 1773–1782. doi: 10.1038/nn.3560
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Budzinska, M., Owczarz, M., Pawlik-Pachucka, E., Roszkowska-Gancarz, M., Slusarczyk, P., and Puzianowska-Kuznicka, M. (2016). miR-96, miR-145 and miR-9 expression increases, and IGF-1R and FOXO1 expression decreases in peripheral blood mononuclear cells of aging humans. BMC Geriatr. 16:200. doi: 10.1186/s12877-016-0379-y
Cerovic, M., Forloni, G., and Balducci, C. (2019). Neuroinflammation and the gut microbiota: possible alternative therapeutic targets to counteract Alzheimer’s disease? Front. Aging Neurosci. 11:284. doi: 10.3389/fnagi.2019.00284
Cha, D. J., Mengel, D., Mustapic, M., Liu, W., Selkoe, D. J., Kapogiannis, D., et al. (2019). miR-212 and miR-132 are downregulated in neurally derived plasma exosomes of Alzheimer’s patients. Front. Neurosci. 13:1208. doi: 10.3389/fnins.2019.01208
Chakravorty, A., Jetto, C. T., and Manjithaya, R. (2019). Dysfunctional mitochondria and mitophagy as drivers of Alzheimer’s disease pathogenesis. Front. Aging Neurosci. 11:311. doi: 10.3389/fnagi.2019.00311
Chen, D., Hu, S., Wu, Z., Liu, J., and Li, S. (2018). The role of miR-132 in regulating neural stem cell proliferation, differentiation and neuronal maturation. Cell. Physiol. Biochem. 47, 2319–2330. doi: 10.1159/000491543
Chen, Y. H., Keiser, M. S., and Davidson, B. L. (2018). Viral vectors for gene transfer. Curr. Protoc. Mouse Biol. 8:e58.
Congdon, E. E., and Sigurdsson, E. M. (2018). Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 14, 399–415. doi: 10.1038/s41582-018-0013-z
Cordell, C. B., Borson, S., Boustani, M., Chodosh, J., Reuben, D., Verghese, J., et al. (2013). Alzheimer’s Association recommendations for operationalizing the detection of cognitive impairment during the Medicare Annual Wellness Visit in a primary care setting. Alzheimers Dement. 9, 141–150. doi: 10.1016/j.jalz.2012.09.011
Deng, Y., Zhang, J., Sun, X., Ma, G., Luo, G., Miao, Z., et al. (2020). miR-132 improves the cognitive function of rats with Alzheimer’s disease by inhibiting the MAPK1 signal pathway. Exp. Ther. Med. 20:159.
Dickson, J. R., Kruse, C., Montagna, D. R., Finsen, B., and Wolfe, M. S. (2013). Alternative polyadenylation and miR-34 family members regulate tau expression. J. Neurochem. 127, 739–749. doi: 10.1111/jnc.12437
Dong, H., Li, J., Huang, L., Chen, X., Li, D., Wang, T., et al. (2015). Serum microRNA profiles serve as novel biomarkers for the diagnosis of Alzheimer’s disease. Dis. Markers 2015:625659.
El Fatimy, R., Li, S., Chen, Z., Mushannen, T., Gongala, S., Wei, Z., et al. (2018). MicroRNA-132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathol. 136, 537–555. doi: 10.1007/s00401-018-1880-5
Ferguson-Chanowitz, K. M., Katocs, A. S. Jr., Pickett, W. C., Kaplan, J. B., Sass, P. M., Oronsky, A. L., et al. (1990). Platelet-activating factor or a platelet-activating factor antagonist decreases tumor necrosis factor-alpha in the plasma of mice treated with endotoxin. J. Infect. Dis. 162, 1081–1086. doi: 10.1093/infdis/162.5.1081
Fitzpatrick, A. W. P., Falcon, B., He, S., Murzin, A. G., Murshudov, G., Garringer, H. J., et al. (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190.
Follert, P., Cremer, H., and Béclin, C. (2014). MicroRNAs in brain development and function: a matter of flexibility and stability. Front. Mol. Neurosci. 7:5. doi: 10.3389/fnmol.2014.00005
Ge, Y., Song, X., Liu, J., Liu, C., and Xu, C. (2020). The combined therapy of berberine treatment with lncRNA BACE1-AS depletion attenuates Aβ(25-35) induced neuronal injury through regulating the expression of miR-132-3p in neuronal cells. Neurochem. Res. 45, 741–751. doi: 10.1007/s11064-019-02947-6
Godlewski, J., Lenart, J., and Salinska, E. (2019). MicroRNA in brain pathology: neurodegeneration the other side of the brain cancer. Noncoding RNA 5:20. doi: 10.3390/ncrna5010020
Hadar, A., Milanesi, E., Walczak, M., Puzianowska-Kuźnicka, M., Kuźnicki, J., Squassina, A., et al. (2018). SIRT1, miR-132 and miR-212 link human longevity to Alzheimer’s Disease. Sci. Rep. 8:8465.
Hansen, K. F., Sakamoto, K., Aten, S., Snider, K. H., Loeser, J., Hesse, A. M., et al. (2016). Targeted deletion of miR-132/-212 impairs memory and alters the hippocampal transcriptome. Learn. Mem. 23, 61–71. doi: 10.1101/lm.039578.115
He, D., Wang, J., Lu, Y., Deng, Y., Zhao, C., Xu, L., et al. (2017). lncRNA functional networks in oligodendrocytes reveal stage-specific myelination control by an lncOL1/Suz12 complex in the CNS. Neuron 93, 362–378. doi: 10.1016/j.neuron.2016.11.044
Hébert, S. S., Horré, K., Nicolaï, L., Papadopoulou, A. S., Mandemakers, W., Silahtaroglu, A. N., et al. (2008). Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. U.S.A. 105, 6415–6420. doi: 10.1073/pnas.0710263105
Hernandez-Rapp, J., Rainone, S., Goupil, C., Dorval, V., Smith, P. Y., Saint-Pierre, M., et al. (2016). microRNA-132/212 deficiency enhances Aβ production and senile plaque deposition in Alzheimer’s disease triple transgenic mice. Sci. Rep. 6:30953.
Herrera-Espejo, S., Santos-Zorrozua, B., Álvarez-González, P., Lopez-Lopez, E., and Garcia-Orad, Á (2019). A systematic review of microRNA expression as biomarker of late-onset Alzheimer’s disease. Mol. Neurobiol. 56, 8376–8391. doi: 10.1007/s12035-019-01676-9
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Iqbal, K., Liu, F., Gong, C. X., and Grundke-Iqbal, I. (2010). Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7, 656–664. doi: 10.2174/156720510793611592
Itaka, K., and Kataoka, K. (2009). Recent development of nonviral gene delivery systems with virus-like structures and mechanisms. Eur. J. Pharm. Biopharm. 71, 475–483. doi: 10.1016/j.ejpb.2008.09.019
Jeong, H., Cohen, D. E., Cui, L., Supinski, A., Savas, J. N., Mazzulli, J. R., et al. (2011). Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med. 18, 159–165. doi: 10.1038/nm.2559
Ji, Y., Wang, D., Zhang, B., and Lu, H. (2019). MiR-361-3p inhibits β-amyloid accumulation and attenuates cognitive deficits through targeting BACE1 in Alzheimer’s disease. J. Integr. Neurosci. 18, 285–291. doi: 10.31083/j.jin.2019.03.1136
Jia, M., Wang, X., Zhang, H., Ye, C., Ma, H., Yang, M., et al. (2019). MicroRNA-132 in the adult dentate gyrus is involved in opioid addiction via modifying the differentiation of neural stem cells. Neurosci. Bull. 35, 486–496. doi: 10.1007/s12264-019-00338-z
Kenyon, C., Chang, J., Gensch, E., Rudner, A., and Tabtiang, R. (1993). A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464. doi: 10.1038/366461a0
Khalil, I. A., Sato, Y., and Harashima, H. (2019). Recent advances in the targeting of systemically administered non-viral gene delivery systems. Expert Opin. Drug Deliv. 16, 1037–1050. doi: 10.1080/17425247.2019.1656196
Klein, M. E., Lioy, D. T., Ma, L., Impey, S., Mandel, G., and Goodman, R. H. (2007). Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 10, 1513–1514. doi: 10.1038/nn2010
Labatut, A. E., and Mattheolabakis, G. (2018). Non-viral based miR delivery and recent developments. Eur. J. Pharm. Biopharm. 128, 82–90. doi: 10.1016/j.ejpb.2018.04.018
Lagos-Quintana, M., Rauhut, R., Yalcin, A., Meyer, J., Lendeckel, W., and Tuschl, T. (2002). Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12, 735–739. doi: 10.1016/s0960-9822(02)00809-6
Lau, P., Bossers, K., Janky, R., Salta, E., Frigerio, C. S., Barbash, S., et al. (2013). Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol. Med. 5, 1613–1634.
Lee, D. Y., Moon, J., Lee, S. T., Jung, K. H., Park, D. K., Yoo, J. S., et al. (2015). Distinct expression of long non-coding RNAs in an Alzheimer’s disease model. J. Alzheimers Dis. 45, 837–849.
Lennox, K. A., Vakulskas, C. A., and Behlke, M. A. (2017). Non-nucleotide modification of Anti-miRNA oligonucleotides. Methods Mol. Biol. 1517, 51–69. doi: 10.1007/978-1-4939-6563-2_3
Li, Y., Ba, M., Du, Y., Xia, C., Tan, S., Ng, K. P., et al. (2019). Aβ1-42 increases the expression of neural KATP subunits Kir6.2/SUR1 via the NF-κB, p38 MAPK and PKC signal pathways in rat primary cholinergic neurons. Hum. Exp. Toxicol. 38, 665–674. doi: 10.1177/0960327119833742
Lima, J. F., Cerqueira, L., Figueiredo, C., Oliveira, C., and Azevedo, N. F. (2018). Anti-miRNA oligonucleotides: a comprehensive guide for design. RNA Biol. 15, 338–352. doi: 10.1080/15476286.2018.1445959
Liu, D. Y., and Zhang, L. (2019). MicroRNA-132 promotes neurons cell apoptosis and activates Tau phosphorylation by targeting GTDC-1 in Alzheimer’s disease. Eur. Rev. Med. Pharmacol. Sci. 23, 8523–8532.
Lykhmus, O., Mishra, N., Koval, L., Kalashnyk, O., Gergalova, G., Uspenska, K., et al. (2016). Molecular mechanisms regulating LPS-induced inflammation in the brain. Front. Mol. Neurosci. 9:19. doi: 10.3389/fnmol.2016.00019
Maffioletti, E., Milanesi, E., Ansari, A., Zanetti, O., Galluzzi, S., Geroldi, C., et al. (2019). miR-146a plasma levels are not altered in Alzheimer’s disease but correlate with age and illness severity. Front. Aging Neurosci. 11:366. doi: 10.3389/fnagi.2019.00366
Magill, S. T., Cambronne, X. A., Luikart, B. W., Lioy, D. T., Leighton, B. H., Westbrook, G. L., et al. (2010). microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc. Natl. Acad. Sci. U.S.A. 107, 20382–20387. doi: 10.1073/pnas.1015691107
Marciniak, E., Leboucher, A., Caron, E., Ahmed, T., Tailleux, A., Dumont, J., et al. (2017). Tau deletion promotes brain insulin resistance. J. Exp. Med. 214, 2257–2269. doi: 10.1084/jem.20161731
Mazdeh, M., Komaki, A., Omrani, M. D., Gharzi, V., Sayad, A., Taheri, M., et al. (2018). Expression analysis of beta-secretase 1 (BACE1) and its naturally occurring antisense (BACE1-AS) in blood of epileptic patients. Neurol. Sci. 39, 1565–1569. doi: 10.1007/s10072-018-3458-3
Nelson, P. T., and Wang, W. X. (2010). MiR-107 is reduced in Alzheimer’s disease brain neocortex: validation study. J. Alzheimers Dis. 21, 75–79. doi: 10.3233/jad-2010-091603
Nisbet, R. M., and Götz, J. (2018). Amyloid-β and Tau in Alzheimer’s disease: novel pathomechanisms and non-pharmacological treatment strategies. J. Alzheimers Dis. 64, S517–S527.
Noren Hooten, N., Fitzpatrick, M., Wood, W. H. III, De, S., Ejiogu, N., Zhang, Y., et al. (2013). Age-related changes in microRNA levels in serum. Aging 5, 725–740. doi: 10.18632/aging.100603
Patrick, E., Rajagopal, S., Wong, H. A., Mccabe, C., Xu, J., Tang, A., et al. (2017). Dissecting the role of non-coding RNAs in the accumulation of amyloid and tau neuropathologies in Alzheimer’s disease. Mol. Neurodegener. 12:51.
Pichler, S., Gu, W., Hartl, D., Gasparoni, G., Leidinger, P., Keller, A., et al. (2017). The miRNome of Alzheimer’s disease: consistent downregulation of the miR-132/212 cluster. Neurobiol. Aging 50, 167.e1–167.e10.
Pöllänen, E., Ronkainen, P. H., Horttanainen, M., Takala, T., Puolakka, J., Suominen, H., et al. (2010). Effects of combined hormone replacement therapy or its effective agents on the IGF-1 pathway in skeletal muscle. Growth Horm. IGF Res. 20, 372–379. doi: 10.1016/j.ghir.2010.07.003
Reisberg, B., Doody, R., Stöffler, A., Schmitt, F., Ferris, S., and Möbius, H. J. (2003). Memantine in moderate-to-severe Alzheimer’s disease. N. Engl. J. Med. 348, 1333–1341.
Rivas-Arancibia, S., Zimbrón, L. F., Rodríguez-Martínez, E., Maldonado, P. D., Borgonio Pérez, G., and Sepúlveda-Parada, M. (2015). Oxidative stress-dependent changes in immune responses and cell death in the substantia nigra after ozone exposure in rat. Front. Aging Neurosci. 7:65. doi: 10.3389/fnagi.2015.00065
Roitbak, T. (2018). Silencing a multifunctional microRNA is beneficial for stroke recovery. Front. Mol. Neurosci. 11:58. doi: 10.3389/fnmol.2018.00058
Salta, E., Sierksma, A., Vanden Eynden, E., and De Strooper, B. (2016). miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol. Med. 8, 1005–1018. doi: 10.15252/emmm.201606520
Samandari-Bahraseman, M. R., Jahanshahi, M., Asadi Barbariha, S., and Elyasi, L. (2019). Altered micro-RNA regulation and neuroprotection activity of Eremostachys labiosiformis in Alzheimer’s disease model. Ann. Neurosci. 25, 160–165. doi: 10.1159/000489551
Schwarzenbach, H., Nishida, N., Calin, G. A., and Pantel, K. (2014). Clinical relevance of circulating cell-free microRNAs in cancer. Nat. Rev. Clin. Oncol. 11, 145–156. doi: 10.1038/nrclinonc.2014.5
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Smalheiser, N. R. (2014). The RNA-centred view of the synapse: non-coding RNAs and synaptic plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130504. doi: 10.1098/rstb.2013.0504
Smith, P. Y., Delay, C., Girard, J., Papon, M. A., Planel, E., Sergeant, N., et al. (2011). MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Hum. Mol. Genet. 20, 4016–4024. doi: 10.1093/hmg/ddr330
Smith, P. Y., Hernandez-Rapp, J., Jolivette, F., Lecours, C., Bisht, K., Goupil, C., et al. (2015). miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum. Mol. Genet. 24, 6721–6735. doi: 10.1093/hmg/ddv377
Song, J., and Lee, J. E. (2015). miR-155 is involved in Alzheimer’s disease by regulating T lymphocyte function. Front. Aging Neurosci. 7:61. doi: 10.3389/fnagi.2015.00061
Sorrentino, G., and Bonavita, V. (2007). Neurodegeneration and Alzheimer’s disease: the lesson from tauopathies. Neurol. Sci. 28, 63–71. doi: 10.1007/s10072-007-0789-x
Srivastava, A., Dixit, A. B., Banerjee, J., Tripathi, M., and Sarat Chandra, P. (2016). Role of inflammation and its miRNA based regulation in epilepsy: implications for therapy. Clin. Chim. Acta 452, 1–9. doi: 10.1016/j.cca.2015.10.023
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Strum, J. C., Johnson, J. H., Ward, J., Xie, H., Feild, J., Hester, A., et al. (2009). MicroRNA 132 regulates nutritional stress-induced chemokine production through repression of SirT1. Mol. Endocrinol. 23, 1876–1884. doi: 10.1210/me.2009-0117
Su, Y., Sun, B., Gao, X., Dong, X., Fu, L., Zhang, Y., et al. (2020). Intranasal delivery of targeted nanoparticles loaded with miR-132 to brain for the treatment of neurodegenerative diseases. Front. Pharmacol. 11:1165. doi: 10.3389/fphar.2020.01165
Titze-de-Almeida, S. S., Soto-Sánchez, C., Fernandez, E., Koprich, J. B., Brotchie, J. M., and Titze-de-Almeida, R. (2020). The promise and challenges of developing miRNA-based therapeutics for Parkinson’s disease. Cells 9:841. doi: 10.3390/cells9040841
Turchinovich, A., Samatov, T. R., Tonevitsky, A. G., and Burwinkel, B. (2013). Circulating miRNAs: cell-cell communication function? Front. Genet. 4:119. doi: 10.3389/fgene.2013.00119
Wang, H., Jiang, Y., Peng, H., Chen, Y., Zhu, P., and Huang, Y. (2015). Recent progress in microRNA delivery for cancer therapy by non-viral synthetic vectors. Adv. Drug Deliv. Rev. 81, 142–160. doi: 10.1016/j.addr.2014.10.031
Wang, L., and Zhang, L. (2020). Circulating exosomal miRNA as diagnostic biomarkers of neurodegenerative diseases. Front. Mol. Neurosci. 13:53. doi: 10.3389/fnmol.2020.00053
Wang, L. L., Huang, Y., Wang, G., and Chen, S. D. (2012). The potential role of microRNA-146 in Alzheimer’s disease: biomarker or therapeutic target? Med. Hypotheses 78, 398–401. doi: 10.1016/j.mehy.2011.11.019
Wang, X., Wang, C., Geng, C., and Zhao, K. (2018). LncRNA XIST knockdown attenuates Aβ(25-35)-induced toxicity, oxidative stress, and apoptosis in primary cultured rat hippocampal neurons by targeting miR-132. Int. J. Clin. Exp. Pathol. 11, 3915–3924.
Wang, Y., Veremeyko, T., Wong, A. H., El Fatimy, R., Wei, Z., Cai, W., et al. (2017). Downregulation of miR-132/212 impairs S-nitrosylation balance and induces tau phosphorylation in Alzheimer’s disease. Neurobiol. Aging 51, 156–166. doi: 10.1016/j.neurobiolaging.2016.12.015
Wattmo, C., and Wallin, ÅK. (2017). Early- versus late-onset Alzheimer’s disease in clinical practice: cognitive and global outcomes over 3 years. Alzheimers Res. Ther. 9:70.
Wei, Z., Meng, X., El Fatimy, R., Sun, B., Mai, D., Zhang, J., et al. (2020). Environmental enrichment prevents Aβ oligomer-induced synaptic dysfunction through mirna-132 and hdac3 signaling pathways. Neurobiol. Dis. 134:104617. doi: 10.1016/j.nbd.2019.104617
Williams, M. A., Haughton, D., Stevenson, M., Craig, D., Passmore, A. P., and Silvestri, G. (2015). Plasma complement factor H in Alzheimer’s disease. J. Alzheimers Dis. 45, 369–372. doi: 10.3233/jad-142742
Xie, A. J., Hou, T. Y., Xiong, W., Huang, H. Z., Zheng, J., Li, K., et al. (2019). Tau overexpression impairs neuronal endocytosis by decreasing the GTPase dynamin 1 through the miR-132/MeCP2 pathway. Aging Cell 18:e12929. doi: 10.1111/acel.12929
Xie, B., Zhou, H., Zhang, R., Song, M., Yu, L., Wang, L., et al. (2015). Serum miR-206 and miR-132 as potential circulating biomarkers for mild cognitive impairment. J. Alzheimers Dis. 45, 721–731. doi: 10.3233/jad-142847
Xu, N., Li, A. D., Ji, L. L., Ye, Y., Wang, Z. Y., and Tong, L. (2019). miR-132 regulates the expression of synaptic proteins in APP/PS1 transgenic mice through C1q. Eur. J. Histochem. 63:3008.
Yang, B., Xia, Z. A., Zhong, B., Xiong, X., Sheng, C., Wang, Y., et al. (2017). Distinct hippocampal expression profiles of long non-coding RNAs in an Alzheimer’s disease model. Mol. Neurobiol. 54, 4833–4846. doi: 10.1007/s12035-016-0038-5
Zeng, F., Li, Y., Xu, Y., Yang, J., Liu, Z., Li, X., et al. (2019). Strategies targeting soluble β-amyloid oligomers and their application to early diagnosis of Alzheimer’s disease. Curr. Alzheimer Res. 16, 1132–1142. doi: 10.2174/1567205016666191031163504
Zhang, N., Chin, J. S., and Chew, S. Y. (2019). Localised non-viral delivery of nucleic acids for nerve regeneration in injured nervous systems. Exp. Neurol. 319:112820. doi: 10.1016/j.expneurol.2018.09.003
Keywords: miR-132, Alzheimer’s disease, pathogenesis, biomarker, neuroprotection
Citation: Zhang M and Bian Z (2021) Alzheimer’s Disease and microRNA-132: A Widespread Pathological Factor and Potential Therapeutic Target. Front. Neurosci. 15:687973. doi: 10.3389/fnins.2021.687973
Received: 30 March 2021; Accepted: 30 April 2021;
Published: 24 May 2021.
Edited by:
Cristina Lanni, University of Pavia, ItalyReviewed by:
Maria Giulia Bacalini, University of Bologna, ItalyCopyright © 2021 Zhang and Bian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhigang Bian, YmlhbnpnMDgxMEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.