 Chunxue Liu1†
Chunxue Liu1† Qiang Li
Qiang Li Xiu Xu
Xiu Xu- 1Department of Child Health Care, Children’s Hospital of Fudan University, National Children’s Medical Center, Shanghai, China
- 2Center for Translational Medicine, Institute of Pediatrics, Shanghai Key Laboratory of Birth Defects Prevention and Control, Children’s Hospital of Fudan University, National Children’s Medical Center, Shanghai, China
Mutations of the SHANK3 gene are found in some autism spectrum disorder (ASD) patients, and animal models harboring SHANK3 mutations exhibit a variety of ASD-like behaviors, presenting a unique opportunity to explore the underlying neuropathological mechanisms and potential pharmacological treatments. The histone deacetylase (HDAC) valproic acid (VPA) has demonstrated neuroprotective and neuroregenerative properties, suggesting possible therapeutic utility for ASD. Therefore, SHANK3-associated ASD-like symptoms present a convenient model to evaluate the potential benefits, therapeutic window, and optimal dose of VPA. We constructed a novel shank3-deficient (shank3ab–/–) zebrafish model through CRISPR/Cas9 editing and conducted comprehensive morphological and neurobehavioral evaluations, including of core ASD-like behaviors, as well as molecular analyses of synaptic proteins expression levels. Furthermore, different VPA doses and treatment durations were examined for effects on ASD-like phenotypes. Compared to wild types (WTs), shank3ab–/– zebrafish exhibited greater developmental mortality, more frequent abnormal tail bending, pervasive developmental delay, impaired social preference, repetitive swimming behaviors, and generally reduced locomotor activity. The expression levels of synaptic proteins were also dramatically reduced in shank3ab–/– zebrafish. These ASD-like behaviors were attenuated by low-dose (5 μM) VPA administered from 4 to 8 days post-fertilization (dpf), and the effects persisted to adulthood. In addition, the observed underexpression of grm5, encoding glutamate metabotropic receptor 5, was significantly improved in VPA-treated shank3ab–/– zebrafish. We report for the first time that low-dose VPA administered after neural tube closure has lasting beneficial effects on the social deficits and repetitive behavioral patterns in shank3-deficient ASD model zebrafish. These findings provide a promising strategy for ASD clinical drug development.
Introduction
Autism spectrum disorder (ASD) encompasses a group of neurodevelopmental syndromes characterized by deficits in social interaction and communication as well as repetitive behaviors and restricted interests (American Psychiatric Association, 2013). There is strong evidence for the involvement of inherited genetic factors in ASD (accounting for at least 80% of the variation in disease risk) (Jutla et al., 2019; Liu et al., 2021). Furthermore, mutations in numerous genes encoding synaptic proteins have been identified in patients with ASD and intellectual disability (Verpelli and Sala, 2012; Lai et al., 2014). According to a meta-analysis, monogenic mutations in SHANK3, which encodes the major postsynaptic density (PSD) scaffolding protein at excitatory glutamatergic synapses, are found in approximately 0.69% of ASD cases and up to 2.12% of all moderate to profound intellectual disability cases (Leblond et al., 2014). De novo mutations, interstitial deletions, and terminal deletions have been identified in ASD (Durand et al., 2007; Moessner et al., 2007; Gauthier et al., 2009; Boccuto et al., 2013; Leblond et al., 2014). Additionally, SHANK3 mutations underlie Phelan–McDermid syndrome (PMS, also known as 22q13.3 deletion syndrome), a rare autosomal dominant neurodevelopmental disorder characterized by autistic-like behaviors, absent to severely delayed speech, developmental delay, and moderate to profound intellectual disability as well as neonatal hypotonia and minor dysmorphic facial features (Phelan et al., 1993; Wilson et al., 2003; Phelan, 2008; Bonaglia et al., 2011; Phelan and McDermid, 2012). The genomic rearrangements in PMS are diverse, ranging from simple 22q13 deletions (72%), ring chromosomes (14%), and unbalanced translocations (7%) to interstitial deletions (9%), all leading to SHANK3 haploinsufficiency (Bonaglia et al., 2011). Although the severity of the developmental delay tends to vary with deletion size (Sarasua et al., 2011; Zwanenburg et al., 2016), individuals with the same size deletion may exhibit vastly different degrees of disability (Dhar et al., 2010). Thus, SHANK3 deficits appear to profoundly disrupt the neural circuitry required for social behavior, communication, and cognition.
The SHANK3 gene (also known as ProSAP2, at 22q13.33) is the best studied of the three SHANK family members, which encoding an extensive number of mRNA and protein isoforms via multiple intragenic promoters and alternative splicing (Durand et al., 2007; Wang et al., 2014b). In the brain, Shank3 mRNA is enriched in the cortex, thalamus, striatum, hippocampus, dentate gyrus, and cerebellar granule cells (Peca et al., 2011; Wang et al., 2016; Monteiro and Feng, 2017), suggesting important functions in synaptic plasticity underlying cortical organization, sensory processing, behavioral control, and cognition.
Owing to the strong genetic association between SHANK3 deficiency and ASD, many studies have focused on the neurodevelopmental functions of this particular gene. Numerous animal models of SHANK3 deficiency, including zebrafish, Drosophila, rat, mouse, and monkey models, demonstrate ASD-like behaviors, suggesting a causative role of SHANK3 deficiency in ASD (Peca et al., 2011; Wang et al., 2016; Monteiro and Feng, 2017). Wang et al. (2016) reported that homozygous Shank3 knockout mice displayed core behavioral features of ASD as well as impaired mGluR5–Homer association at the PSD, resulting in corticostriatal circuit abnormalities that may underlie learning deficits and ASD-like behaviors. In addition, monkey models also displayed core ASD features including impaired social interactions, repetitive behaviors, delayed vocalization, and reduced brain network activities (Tu et al., 2019; Zhou et al., 2019). The zebrafish genome harbors two homologs of human SHANK3, shank3a, and shank3b. In our previous studies, we generated the first shank3b loss-of-function mutation in zebrafish and reported prominent ASD-like behaviors (Liu et al., 2018). However, we did not examine the effects of shank3a and shank3b double mutant combinations, which would be more analogous to mammalian models harboring a single SHANK3 mutation, or assess potential pharmacological strategies to mitigate behavioral deficits.
Current treatment options for ASD are limited, especially pharmacotherapies (Wang et al., 2014a; Penagarikano et al., 2015). Evidence-based treatments for ASD children are restricted mainly to educational practices, and intensive behavioral interventions such as Treatment and Education of Autistic and Related Communication-Handicapped Children (TEACCH) and the Early Start Denver Model (ESDM) (Dawson et al., 2010; Hyman et al., 2020). Outcomes of these behavioral therapies vary markedly according to intervention intensity, disease severity, and a variety of other factors (Sandbank et al., 2020). Further, education and behavioral interventions do not target the underlying neurobiological mechanisms (Wang et al., 2014a; Weitlauf et al., 2014) and are costly both for educational institutions and primary caregivers (Lord et al., 2018, 2020). Similarly, current pharmacologic treatments address only the associated symptoms or comorbidities, including agitation and hyperactivity, rather than the core symptoms and underlying causes (Fung et al., 2016; Lord et al., 2018, 2020; Muhle et al., 2018). Risperidone and aripiprazole are approved by the United States Food and Drug Administration (FDA) to treat comorbidities common in ASD, including irritability, and agitation (Lord et al., 2018, 2020), but similar to behavioral interventions, these evidence-based pharmacologic treatments lack sufficient biological support.
Based on available evidence, behavioral interventions should be implemented as early and intensively as possible following ASD diagnosis to improve the cognitive and adaptive outcomes of preschoolers (Weitlauf et al., 2014; Muhle et al., 2018). The preschool years are critical for acquiring language and social skills, key areas of difficulty in ASD, as this period coincides with the temporal window of enhanced plasticity in relevant neural circuits (Franz and Dawson, 2019). Similar to early intervention, pharmaceutical treatments appear effective in animal models when administered early, and Muhle et al. (2018) even suggested greater emphasis on early drug treatment rather than strict adherence to the standard timeline of efficacy based on studies in adults and adolescents. For instance, early postnatal treatment improves social deficits in adult mice with mutations in the ASD risk gene cntnap2 (Franz and Dawson, 2019). Pharmacological inhibitors of histone deacetylase (HDAC) have garnered interest as possible ASD therapeutics due to demonstrated neuroprotective efficacy (Fischer et al., 2010). The class I HDAC inhibitor valproic acid (VPA) was found to reduce repetitive behaviors in a small randomized controlled trial involving 13 ASD children (Hollander et al., 2006). In addition, several studies have reported that VPA can attenuate irritability in young ASD patients (Hellings et al., 2005; DeFilippis and Wagner, 2016). Further, three daily VPA treatments transiently restored social preference deficits in adult Shank3-deficient mice, although the effect disappeared within a few days following treatment (Qin et al., 2018). Therefore, the effects of VPA on shank3 mutant models warrant further study.
Here we investigated the developmental characteristics of shank3-deficient zebrafish, neurobehavioral features relevant to ASD, and the effects of various VPA treatment regimens. We speculated that VPA administration in the early postnatal period would be more effective at reversing the core ASD-like deficits in shank3-deficient zebrafish than juvenile or adult treatment.
Materials and Methods
Zebrafish and Embryo Maintenance
Wild-type (WT) zebrafish of Tu strains were acquired from Children’s Hospital of Fudan University. They were raised and maintained under standard laboratory conditions at 28.5°C in “system water” under a 14 h light/10 h dark cycle according to standard protocols (Kalueff et al., 2014; Evans and Erickson, 2019). Freshly fertilized eggs were collected from multiple breeding tanks containing 25 females and 25 males. All animal experimental procedures were in compliance with local and international regulations, and approved by the institutional animal care committee of Children’s Hospital of Fudan University.
Generation of shank3a and shank3b Double Deficient Zebrafish Model
Zebrafish shank3a and shank3b genes and their exon/intron boundaries were identified by searching the NCBI database (gene ID: shank3b, NC_007115.7; shank3a, NC_007129.7). Mutations in shank3a and shank3b were generated using CRISPR/Cas9 editing as previously reported (Hwang et al., 2013; Mali et al., 2013). The CRISPR/Cas9 target of shank3a was 5′-GGACCCCAGCCCTCCTCCCGTGG-3′ and that of shank3b was 5′-GGGCGTGTTGTTGCCACGGCCGG-3′ (Liu et al., 2018; Supplementary Table 1). In vitro-transcribed RNA of the guide RNA (120 ng each) and Cas9 mRNA (500 pg) were microinjected into WT zebrafish embryos (F0) at the one-cell stage. The progeny were propagated via a series of out-crossings with WT zebrafish and genotyping of each generation. Eventually, these animals were in-crossed to obtain the homozygous knockouts shank3a–/– and shank3b–/–. The shank3ab–/– homozygous line was obtained by mutant crossing and subsequent genotyping.
Behavior Tests for Adult Zebrafish
All behavioral experiments were conducted on 2–3.5 month old male zebrafish between 10 a.m. and 4 p.m. Behaviors were recorded for 30 min after 1–2 min habituation period using a video camera (zebrabox) suspended above the test tank. Zebrafish were returned to their home tanks immediately after completion of the test. The raw data was analyzed using Viewpoint software.
Open Field Test
The free-swimming open field test was performed in novel tanks as previously described (Liu et al., 2018). Each tank was 30 cm × 30 cm × 30 cm, and its walls consisted of opaque partitions. Swim velocity was calculated as the total distance traveled divided by the total swim time.
For the danger awareness test, the tank was virtually divided into two equal areas, peripheral and center, and greater peripheral swimming distance relative to central swimming distance was measured as a metric of danger awareness.
For analyzing repetitive and stereotyped behaviors, we used a double-blind method to score the swimming pattern within each minute, and counted the number of the four stereotyped swimming pattern episodes separately.
Shoaling Test
The shoaling test assesses social cohesion in homogeneous groups of zebrafish (Figure 3A; Liu et al., 2018). A shoal refers to a loose aggregation of individuals who swim close to one another, whereas a school describes a group of fish exhibiting polarized, synchronized motion (Perathoner et al., 2016). The distance between each fish can reveal the degree of shoaling behavior (i.e., social cohesion). Six zebrafish were placed in a novel 30 cm × 30 cm × 30 cm tank with walls consisting of opaque partitions, and mean inter-individual distance was measured (Kim et al., 2017).
Social Preference Test
Sociability was evaluated as the difference score between the time spent in proximity to a conspecific sector and an empty sector (Busnelli et al., 2016). Briefly, social testing was conducted over 30 min in a standard mating tank of inner dimensions 21 cm × 10 cm × 7.5 cm. A transparent plexiglas divider was placed in the middle of the tank, which allowed sufficient visual presentation for forming a social preference, and a single shank3 mutant or WT zebrafish was placed on the left side while a group of six conspecific zebrafish (conspecific sector) was placed in the right side (Figure 3C). Social preference behavior was quantified as a distance distribution or as presence in a zone adjacent to the group of conspecifics. The distance ratio was calculated as the distance swam in the conspecific sector divided by the total distance.
Kin Preference Test
Another test was performed to assess preference for kinship using various colored variants. The duration and frequency of contact was compared between conspecifics and a phenotypically distinct strain (Figure 3E) in mating tanks with dimensions and configuration the same as those used in the “social preference test.” Briefly, two transparent separators divided the tank into three compartments, with a single test fish placed in the middle and Kin zebrafish placed on the right and non-Kin (red color) zebrafish placed on the left. Kin preference was represented by the ratio of the time spent in the Kin-sector to the total time.
VPA Treatment and Phenotypic Assessments
To assess the extent to which VPA exposure affects morphology, 4 dpf WT or shank3ab–/– larvae were reared in Petri dishes containing blue egg water alone or blue egg water containing 5, 10, 20, or 50 μM sodium valproate (Cat No. 4543-10G, Sigma-Aldrich). The egg water with or without VPA was changed daily. At 8 dpf, larvae were observed under a microscope for mortality and any morphologic abnormalities, including distended abdominal and thoracic regions, lordosis, yolk sac edema, and pericardial edema. Adverse effects including mortality and malformation rates were calculated to determine the optimal VPA concentration for subsequent experiments (Supplementary Figure 1).
To examine the effects of early postnatal low-dose VPA exposure on autism-like behaviors, WT or shank3ab–/– larvae were exposed to blue egg water with or without 5 μM VPA from 4 to 8 dpf. At 8 dpf, each larva was pipetted into fresh paramecium liquid, and raised to 2.5 months old (juvenile) or 3.5 months old (adult). The juveniles were then examined for 30 min using the 1 versus 6 social preference assay, while adults were subjected to social preference, repetitive behavior, locomotor activity, and thigmotaxis tests to comprehensively evaluate the effects of VPA on autism-like behaviors.
Real-Time Quantitative PCR
Total RNA was extracted from 15 WT, shank3a–/–, shank3b–/–, and shank3ab–/– larvae each at 3.5–4.5 months post-fertilization (mpf) using the RNA Extraction Kit from Takara, and reverse transcribed to cDNA using the PrimeScriptTM reagent Kit with gDNA Eraser (Takara) according to the manufacturer’s recommendations. The Cas9 target region of shank3a and shank3b were amplified in duplicate samples from shank3ab–/– zebrafish by real-time quantitative PCR (RT-qPCR) to confirm genotype (Figures 1C,D and Supplementary Table 1).
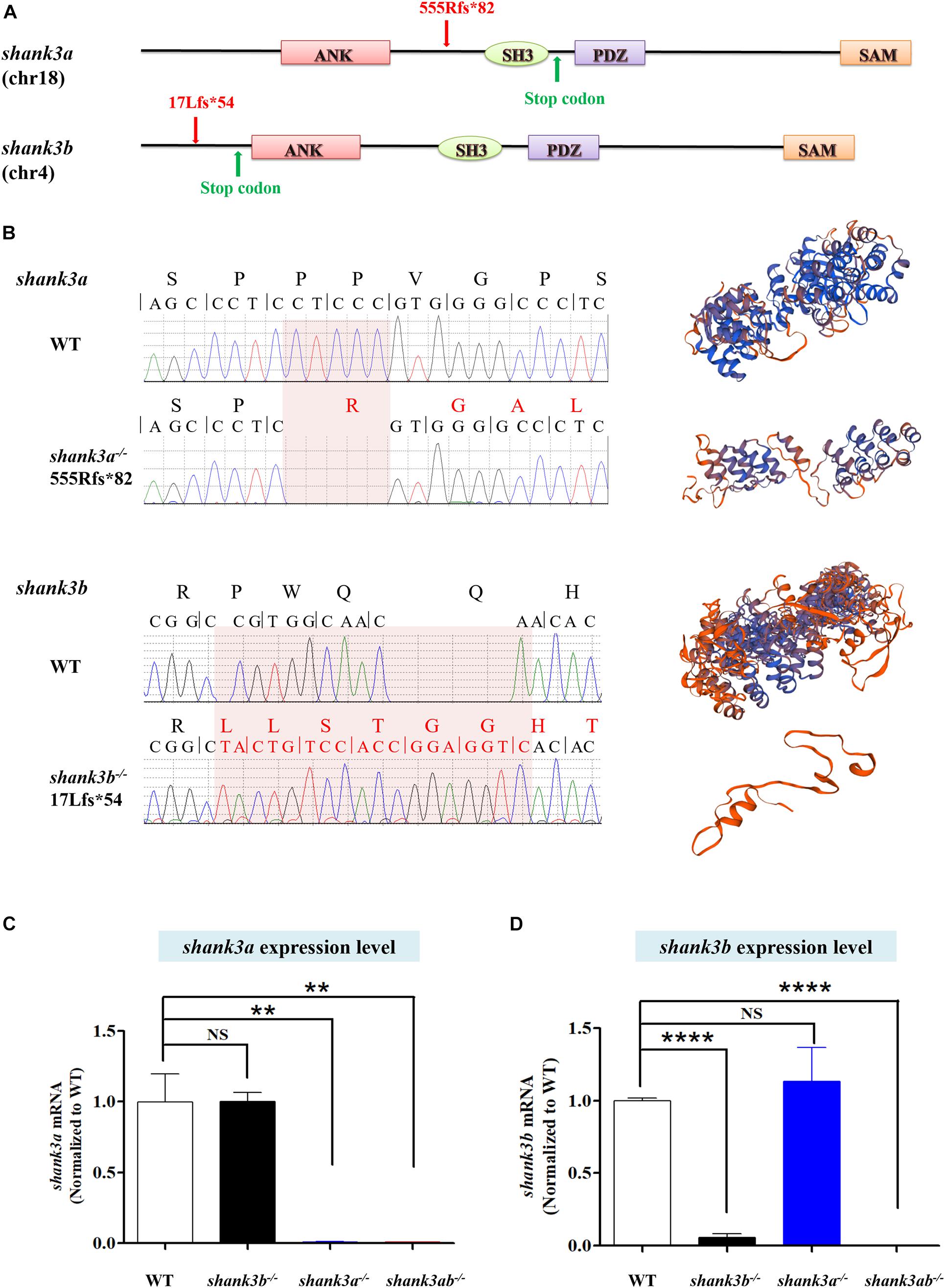
Figure 1. Generation of shank3a and shank3ab mutants in zebrafish by CRISPR-Cas9 gene editing. (A) Structures of zebrafish shank3a and shank3b (Liu et al., 2018) gene and protein domains (ANK, ankyrin repeat domain; SH3, Src homology 3 domain; PDZ, PSD-95/discs large/ZO-1 domain; SAM, sterile alpha motif domain). Exon 9 is the target for CRISPR/Cas9 gene editing in zebrafish shank3a and exon 2 is the target for shank3b. The CRISPR/Cas9 induced frameshift mutations in shank3a (5-base deletion) and in shank3b (5-base deletion and 13-base insertion) which led to the truncation of the protein (Liu et al., 2018). (B) The mutations of shank3a and shank3b (Liu et al., 2018) were verified by Sanger sequencing. The predictions of protein spatial structures were both suggested that shank3a and shank3b proteins were likely to turn into truncated proteins (https://www.swissmodel.expasy.org/interactive/wUdwQQ/models/). (C) Reduced expression level of shank3a mRNA in the brain of shank3a– /– and shank3ab– /– adult (4 mpf) male zebrafish analyzed by RT-qPCR, while shank3b– /– was not affected. (D) Reduced expression level of shank3b mRNA in the brain of shank3b– /– and shank3ab– /– adult (4 mpf) male zebrafish analyzed by RT-qPCR, while shank3a– /– was not affected. Data are shown as mean ± SEM; **P < 0.01. ****P < 0.0001.
To assess the effect of VPA exposure on synaptic genes and class I hdac genes (as VPA belongs to class I HDAC inhibitor), groups of ∼15 WT and shank3ab–/– zebrafish larvae were exposed to vehicle or VPA from 4 to 8 dpf, reared under normal conditions, then sacrificed for whole-brain total RNA isolation. The expression levels of the following genes analyzed by RT-qPCR: NMDAR subunits (grin1a, grin1b, grin2bb, grin2ca, grin2da, grin2aa), AMPAR subunits (gria1a, gria1b, gria2b), mGluR subunits (grm1a, grm1b, grm5a), and class I hdacs (hdac1, hdac3, hdac8). We selected β-actin or Rpl13α as internal controls because both are expressed in the brain throughout development. Primer sequence are shown in Supplementary Table 1.
Western Blotting
Proteins (50 μg per sample) were separated by SDS-PAGE and transferred on polyvinylidene difluoride membranes. After blocking the membrane at room temperature for 2 h with 5% BSA or 7% skim milk, and incubated with primary antibodies at 4°C overnight at the following concentrations: NeuN (Abcam, ab177487, 1:1500), homer1 (Aviva systems biology, ARP40181_P050, 1:1000), and synaptophysin (Abcam, ab32594, 1:1500). The blots were washed in TBS containing 0.1% Tween-20 (TBST) and incubated with HRP-conjugated secondary antibodies (1:5000) for 1.5 h at room temperature. Following six washes in TBST, the blots were incubated with ECL reagent (BeyoECL Plus, P0018M) and exposed to Kodak X-ray film (Tanon 5200). The gray values of proteins were analyzed by ImageJ software (NIH, Bethesda, MD, United States1), and normalized to that of corresponding internal controls, β-actin (1:2000) or Vinculin (1:5000).
Statistical Analysis
Values are presented as mean ± SEM. All data were analyzed using SPSS 20.0. In all experiments, WT and shank3-deficient zebrafish were compared by two-sided unpaired Student’s t-tests, while three or more groups were compared by analysis of variance (ANOVA). Genotypes within treatment groups were compared by one-way ANOVA. All experiments were conducted in triplicate using independently treated animals. A P < 0.05 was considered statistically significant for all tests.
Results
Generation of shank3a–/– and shank3ab–/– Zebrafish
To model shank3 deficiency in zebrafish, we generated a loss-of-function mutant using CRISPR/Cas9 mutagenesis technology (Hwang et al., 2013; Mali et al., 2013; Liu et al., 2018). Generation of the shank3b–/– line was described in our previous study (Liu et al., 2018). Briefly, shank3b–/– harbors an early stop codon (p. 17Lfs∗54) due to a deletion of 5 bases and insertion of 13 bases, resulting in a frameshift mutation and a 90-amino acid truncated protein (Figure 1B). The shank3a–/– line carries a deletion of five bases leading to protein truncation (p. 555Rfs∗82), including most of the shank3 domain (Figure 1A). The shank3ab–/– line was generated by crossing shank3b–/– and shank3a–/–, and individuals were selected by genotyping. RT-qPCR confirmed that shank3a and shank3b mRNA expression levels were significantly reduced in shank3a–/– and shank3b–/– zebrafish, respectively, and that both genes were downregulated in shank3ab–/– zebrafish (Figures 1C,D).
Aberrant Morphology and Swimming Patterns of shank3-Deficient Zebrafish
The developmental progression and morphological characteristics of shank3 mutants were evaluated on 1 day post-fertilization (1 dpf). All mutants demonstrated a higher rate of mortality compared to WTs (shank3a–/–: 6%, 15/266; shank3b–/–: 7%, 19/270; shank3ab–/–: 15%, 18/118; WT: 3%, 10/340) (Figure 2A). In addition, the rate of severe developmental delay was significantly greater in the shank3ab–/– group compared to all other genotypes (shank3ab–/–: 13%, 8/60; shank3a–/–: 7%, 4/60; shank3b–/–: 12%, 7/60; WT: 3%, 2/60). Abnormal tail bending was more frequent in shank3a–/– zebrafish than other genotypes (shank3a–/–: 35%, 21/60; shank3b–/–: 17%, 10/60; shank3ab–/–: 23%, 14/60; WT: 13%, 7/60).
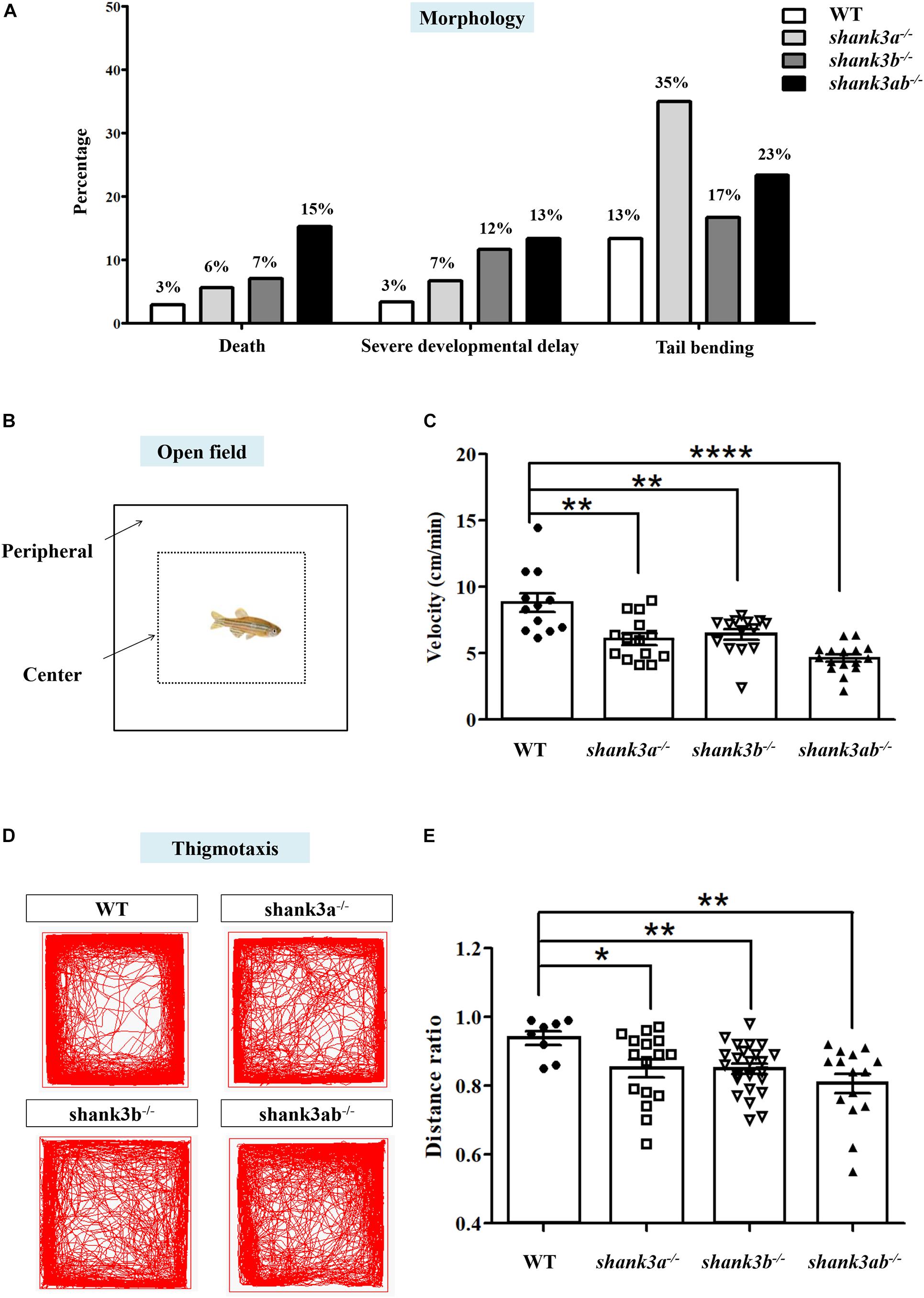
Figure 2. Morphological characteristics and locomotion activity alteration in shank3-deficient zebrafish. (A) Abnormal morphological changes in shank3a– /–, shank3b– /–, and shank3ab– /– larvae at ∼1 dpf, including death (WT, N = 340; shank3a– /–, N = 266; shank3b– /–, N = 270; shank3ab– /–, N = 118, severe developmental delay and tail bending (N = 60 each group). (B) Schematic diagram of the open field test and thigmotaxis test of adult male zebrafish (3.5 mpf). In the analysis of thigmotaxis test, the area of the peripheral zone is equal to the center zone (dotted line). (C) Compared with WT zebrafish (N = 12, 8.8 ± 2.4 cm/min), all shank3-deficient zebrafish displayed significantly decreased swimming velocity (shank3a– /–, N = 14, 6.1 ± 1.6 cm/min; shank3b– /–, N = 14, 6.4 ± 1.4 cm/min; shank3ab– /– zebrafish, N = 16, 4.6 ± 1.1 cm/min, respectively). (D) Representative traces of individual WT or shank3-deficient zebrafish in the thigmotaxis test. (E) Ratio for the distance traveled (periphery divided by the total zone) over 30 min in adult male zebrafish (3.5 mpf). WT, N = 8; shank3a– /–, N = 16; shank3b– /–, N = 24; shank3ab– /– zebrafish, N = 15. Data are shown as mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The remaining shank3a–/–, shank3b–/–, and shank3ab–/– zebrafish were viable and fertile into adulthood. The locomotor activities of adult shank3a–/–, shank3b–/–, and shank3ab–/– zebrafish were examined in the open home tank as previously described (Liu et al., 2018; Figure 2B). Compared to WT zebrafish, all shank3-deficient zebrafish displayed significantly decreased swimming velocity, with shank3ab–/– zebrafish demonstrating the slowest swimming speed (shank3ab–/–: 4.6 ± 1.1 cm/min; WT: 8.8 ± 2.4 cm/min; shank3a–/–: 6.1 ± 1.6 cm/min; shank3b–/–: 6.4 ± 1.4 cm/min) (Figure 2C).
Adult WT zebrafish typically avoid open areas near the water surface for protection against predation. To examine whether shank3 deficiency modulates these avoidance behaviors, the relative proportions of swim time and distance in the pool periphery (thigmotaxis) versus the center (dotted line in Figure 2B) were calculated in a novel square tank with opaque walls. All shank3-deficient genotypes spent a significantly greater proportion of total swim time and traveled longer distances in the center of the tank compared to WT zebrafish, and shank3ab–/– zebrafish exhibited the greatest peripheral to center distance ratio of the three mutant genotypes (Figures 2D,E). This behavior can be interpreted as reduced alertness or reduced danger awareness (Mathur and Guo, 2010).
Core ASD-Like Behaviors of shank3-Deficient Adult Zebrafish
Since ASD diagnosis is based on behavioral criteria, a valid zebrafish model should exhibit core behavioral symptoms, including impaired social interactions and repetitive and stereotyped behaviors. The shoaling test showed that adult WT zebrafish spent the majority of swimming time in compact schools, while all three shank3-deficient genotypes swam in looser and larger schools with more frequent deviation (leaving the group), resulting in a greater average inter-fish distance compared to WT zebrafish (Figure 3B). This social deficit was particularly strong among the shank3ab–/– zebrafish group.
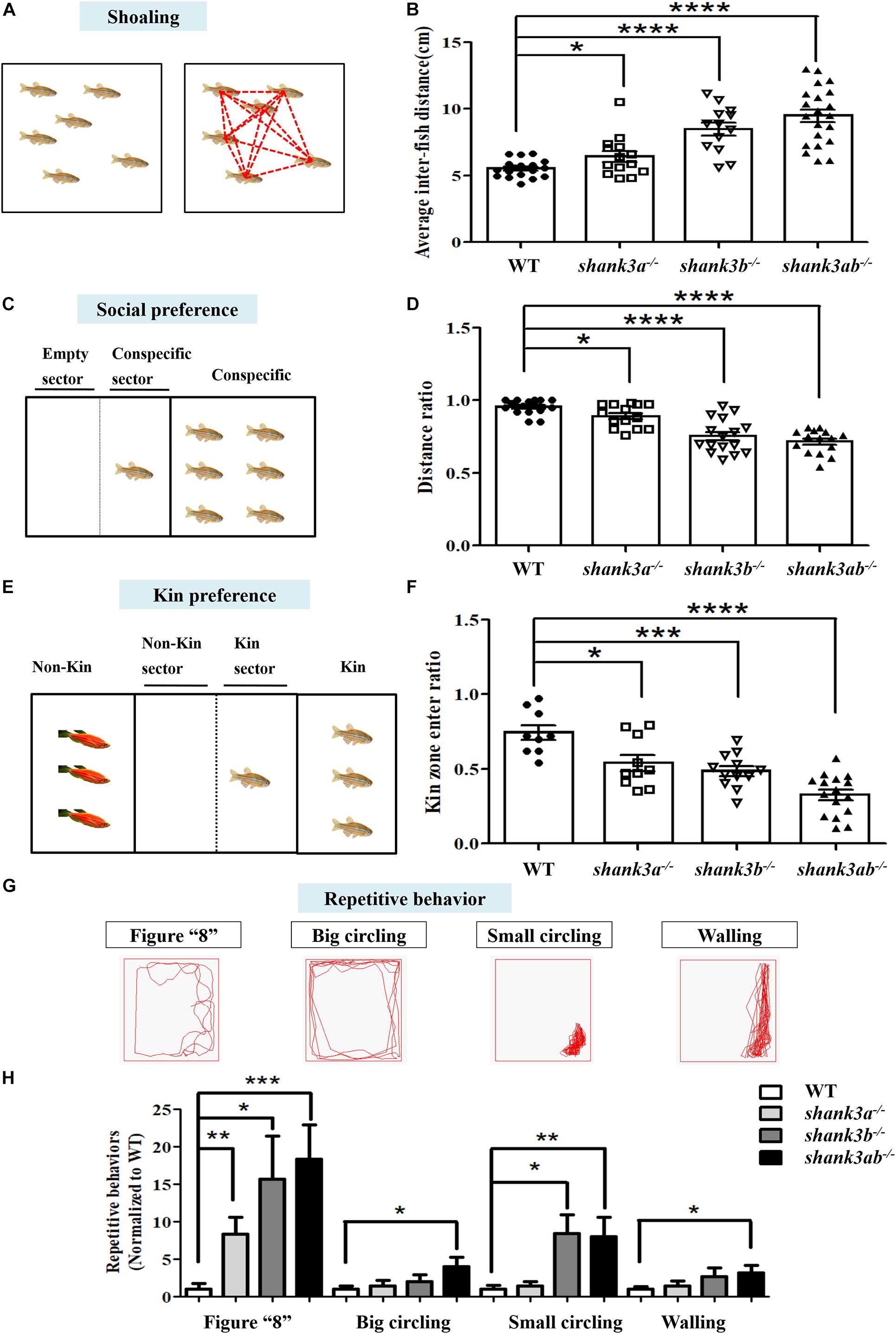
Figure 3. Core behavioral features of ASD-like displayed in shank3-deficient zebrafish. (A,B) The shoaling test showed significantly increased average inter-fish distance of adult male shank3-deficient zebrafish (3.5 mpf). WT, N = 18; shank3a– /–, N = 14; shank3b– /–, N = 13; shank3ab– /– zebrafish, N = 21. (C,D) The social preference test showed distance ratio in the conspecific sector were significantly reduced in shank3-deficient zebrafish compared to WT adult male zebrafish (3.5 mpf). WT, N = 16; shank3a– /–, N = 14; shank3b– /–, N = 16; shank3ab– /– zebrafish, N = 15. (E,F) The Kin recognition and preference test showed significantly reduced ratio of Kin zone entering in shank3-deficient zebrafish compared to WT adult male zebrafish (3.5 mpf). WT, N = 9; shank3a– /–, N = 10; shank3b– /–, N = 12; shank3ab– /– zebrafish, N = 16. (G) Representative trace of different types of stereotyped behaviors of shank3-deficient adult male zebrafish (3.5 mpf). (H) shank3-deficient zebrafish had a significantly higher proportion of stereotyped movements than WT zebrafish. WT, N = 12; shank3a– /–, N = 14; shank3b– /–, N = 14; shank3ab– /– zebrafish, N = 16. Data are presented as mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Social preference was further assessed by measuring conspecific proximity. WT zebrafish (3.5 mpf) maintain closer proximity with a conspecific group members on the right side (Figure 3C). In contrast, all shank3-deficient genotypes showed reduced frequency and duration of conspecific proximity, again with the shank3ab–/– genotype demonstrating the greatest average inter-conspecific distance (Figure 3D).
Wild type zebrafish also typically spend more time with a Kin group (conspecific and same color) than a non-Kin group in mixed populations. However, this Kin recognition and preference was markedly reduced in all shank3-deficient genotypes as measured by the proportion of time spent in close proximity with non-Kin (red-skinned) zebrafish among a mixed population (Figure 3F). Consistent with other social behavior tests, shank3ab–/– zebrafish spent the least amount of time in proximity to other conspecifics.
Repetitive and stereotyped behavior is another core symptom of ASD. Compared to adult WTs, shank3ab–/– zebrafish demonstrated greater behavioral perseveration, including repetitive stereotypic “figure 8” swimming, cycling behavior (swimming in circles), and other locomotor changes and patterns such as stereotyped “corner” or “wall” swimming (Figures 3G,H).
Dysregulation of Synapse-Related Protein Expression in shank3-Deficient Zebrafish
These behavioral abnormalities in shank3-deficient zebrafish suggest possible disruption of normal synaptic function, so we compared the expression levels of several important synaptic proteins among genotypes. Expression of the neuronal marker NeuN was reduced by 50% in the brains of adult shank3ab–/– zebrafish compared to age-matched WTs (Figures 4A,B). As SHANK3 is a major synaptic scaffolding protein enriched at the PSD of excitatory synapses (Jiang and Ehlers, 2013; Monteiro and Feng, 2017), we also examined expression of the postsynaptic marker homer1 and found an approximately 90% reduction in shank3ab–/– zebrafish relative to WTs (Figures 4C,D). It was reported that Shank3 deficiency in mice disrupts the presynaptic neurexin-neuroligin-mediated signaling pathway required for synapse targeting and development (Arons et al., 2012), so we further compared the expression levels of presynaptic proteins among genotypes, including the ubiquitous synaptic vesicle protein synaptophysin (Kwon and Chapman, 2011). Indeed, synaptophysin expression level was reduced by ∼64% in shank3ab–/– zebrafish compared to WTs (Figures 4E,F). Thus, shank3 deficiency reduced the expressions of several pre- and postsynaptic proteins which are likely important to protein transmitter signaling.
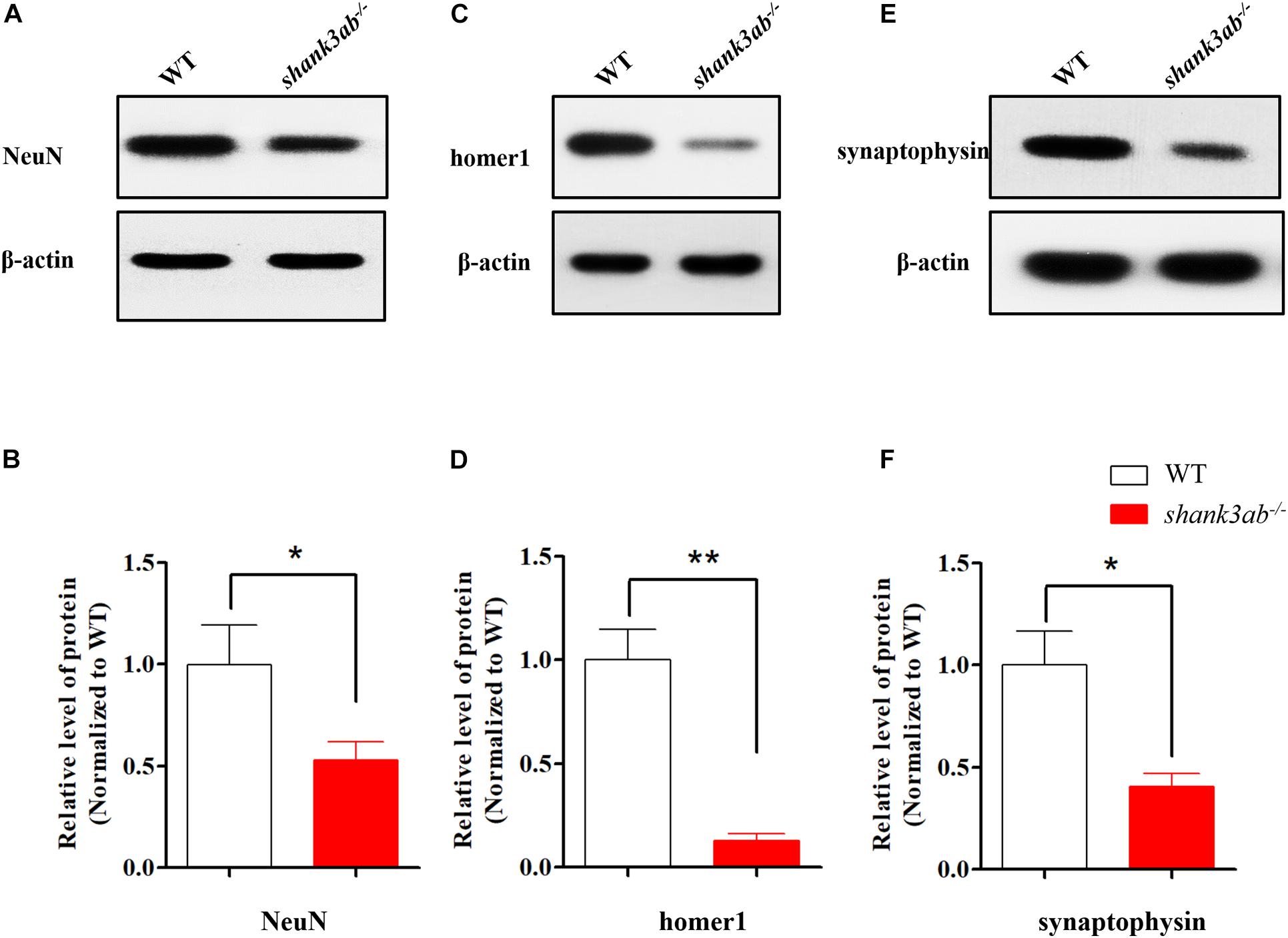
Figure 4. shank3 deficiency resulted in the reduction of synapse-related proteins in adult zebrafish brain. (A,B) Quantitative immunoblot blot analysis showed that the neuron protein NeuN was significantly decreased (50% of WT) in the shank3ab– /– male zebrafish brain relative to WT zebrafish (3.5 mpf). (C,D) The expression of post-synaptic homer1 protein was markedly reduced in shank3ab– /– male zebrafish brain compared with that of WT zebrafish (3.5 mpf, 10% of WT). (E,F) The expression of presynaptic synaptophysin protein was significantly reduced in shank3ab– /– male zebrafish brain compared with that of WT zebrafish (3.5 mpf, 36% of WT). N = 3 for each group. Data are presented as mean ± SEM; *P < 0.05, **P < 0.01.
Improved ASD Core Symptoms and grm5 Receptor Expression by Early VPA Treatment
We then examined the efficacy of VPA to mitigate autism-like symptoms in these zebrafish models. In WT zebrafish, both shank3a and shank3b expression levels increased gradually from 3 to 7 dpf, a period of intense synaptogenesis (Liu et al., 2016). Exposure regimens of 3–7 and 4–8 dpf were thus judged as potentially suitable for optimal therapeutic effect. Based on preliminary observations, 4–8 dpf was chosen as the optimal exposure regimen (Supplementary Figure 1), and exposure concentrations (5, 20, and 50 μM) were then evaluated to identify the safety. We found that 5 μM completely eliminated mortality of shank3ab–/– larvae at 8 dpf and almost completely eliminated morphological dysgenesis (1.04%, 1/96) with no adverse effects on WT larvae (Supplementary Figure 1).
Once-daily administration of 5 μM VPA from 4 to 8 dpf (Figure 5A) significantly improved the social preference behavior of shank3ab–/– zebrafish both in juvenile and adulthood. In addition, VPA-treated shank3ab–/– juvenile (2.5 mpf) zebrafish spent significantly more time exploring a tank section containing conspecifics compared to an empty zone as measured by the distance ratio (shank3ab–/–: 0.84 ± 0.02; shank3ab–/– + VPA: 0.91 ± 0.02; P = 0.013) (Figure 5B). Moreover, VPA had no statistically significant effect on the social preference of WT zebrafish as measured by distance ratio (WT: 0.94 ± 0.02; WT + VPA: 0.95 ± 0.01; P = 0.558). This improvement was also observed in adult shank3-deficient zebrafish (3.5 mpf) (shank3ab–/–: 0.79 ± 0.03; shank3ab–/– + VPA: 0.91 ± 0.02; P = 0.006) (Figure 5C). In fact, the deficit in social preference relative to WTs was completely reversed by postnatal VPA (WT: 0.94 ± 0.02; shank3ab–/– + VPA: 0.91 ± 0.02).
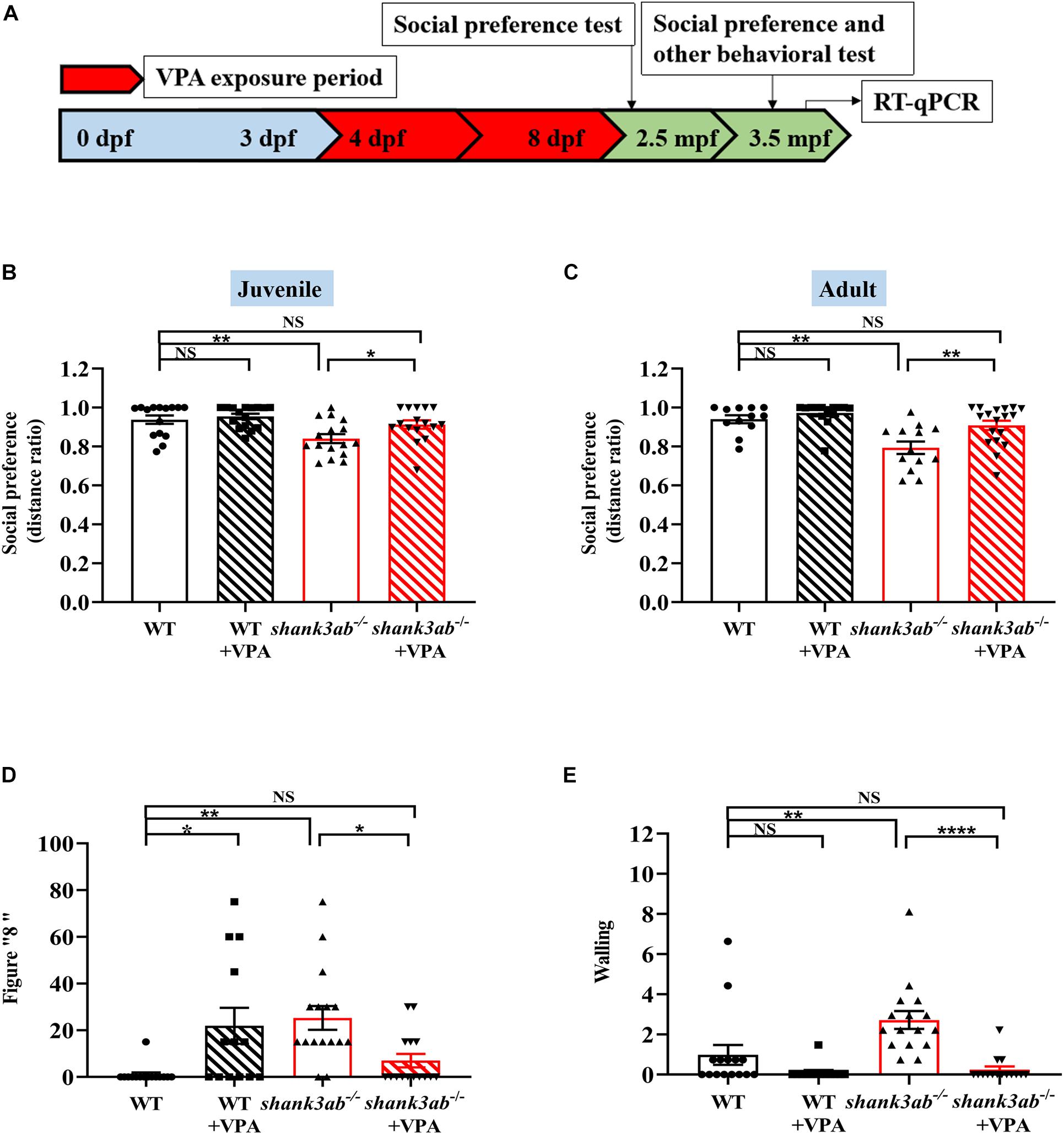
Figure 5. Improved ASD core symptoms in shank3-deficient zebrafish upon early VPA treatment. (A) Schematic overview of the protocol used for the VPA exposure period, the evaluation of behavioral tests at juvenile (2.5 mpf) and adult (3.5 mpf), and RT-qPCR analysis at 4.5 mpf. Red color indicates the VPA exposure phases. (B) Social preference index (distance ratio) of social test on juvenile zebrafish, WT, n = 15; WT-VPA, n = 16; shank3ab– /–, n = 16; shank3ab– /– -VPA, n = 16. (C) Social preference index (distance ratio) of social test on adult zebrafish, WT, n = 12; WT-VPA, n = 14; shank3ab– /–, n = 13; shank3ab– /– -VPA, n = 18. (D) The VPA treatment reduced abnormal proportion of stereotypic figure “8” swimming and (E) back and forth swimming (walling) in shank3ab– /– zebrafish, WT, n = 15; WT-VPA, n = 13; shank3ab– /–, n = 16; shank3ab– /– -VPA, n = 15. Data are shown as mean ± SEM. Statistical analyses: (B) one-way ANOVA with LSD correction for multiple testing. (C–E) One-way ANOVA with Bonferroni correction for multiple testing. Data are presented as mean ± SEM; *P < 0.05, **P < 0.01, ****P < 0.0001.
Compared to untreated shank3ab–/– zebrafish, those receiving early postnatal VPA treatment also showed reduced frequencies of stereotypic “figure 8” swim patterns (P = 0.035) (Figure 5D) and “wall” swimming (P < 0.0001) (Figure 5E). Taken together, these results suggest that early low-dose VPA treatment can induce sustained reversal of core ASD-like symptoms in shank3-deficient zebrafish. Conversely, VPA-treated WT zebrafish showed a greater frequency of stereotypic “figure 8” swimming (P = 0.018) (Figure 5D). Moreover, postnatal VPA treatment rescued the deficient avoidance behavior of shank3ab–/– adult zebrafish as evidenced by a significant increase in peripheral to central distance ratio (shank3ab–/–: 0.77 ± 0.02; shank3ab–/– + VPA: 0.85 ± 0.02; P = 0.032; Supplementary Figure 2A). In contrast, VPA had no effect on the distance ratio of WT zebrafish (WT: 0.91 ± 0.02; WT + VPA: 0.88 ± 0.03) or the slower swim velocity of shank3ab–/– zebrafish (shank3ab–/–: 6.38 ± 0.20 cm/s; shank3ab–/– + VPA: 5.71 ± 0.25 cm/s; P = 0.890) (Supplementary Figure 2B).
We also tested the effects of VPA treatment on synaptic proteins in shank3ab–/– zebrafish. As shown in Figures 6A–F, the expression levels of synaptic proteins (NeuN, homer1, and synaptophysin) were not significantly restored between WT and shank3ab–/– fish after exposed to VPA. To identify potential mechanisms underlying the amelioration of autism-like behaviors by early postnatal VPA, we first examined the expression levels of class I hdac genes (hdac1, hdac2, hdac3, and hdac8), as HDACs are the major known targets of this agent. Given the absence of hdac2 gene in the zebrafish genome (Ko et al., 2019), we detected the expression levels of the rest three genes. However, RT-qPCR analysis revealed no changes in mRNA expression levels of hdac1, hdac3, and hdac8 (Supplementary Figure 2C). We then examined the expression levels of glutamate receptors after cessation of treatment and found that VPA reversed the underexpression of grm5a observed in shank3ab–/– zebrafish (P < 0.05) (Figure 6G) but had no effects on the mRNA levels of AMPAR subunits (gria1a, gria1b, gria2b) (Supplementary Figure 2D), and NMDAR subunits (grin1a, grin1b, grin2bb, grin2ca, grin2da, grin2aa) (Supplementary Figure 2E) compared to untreated zebrafish.
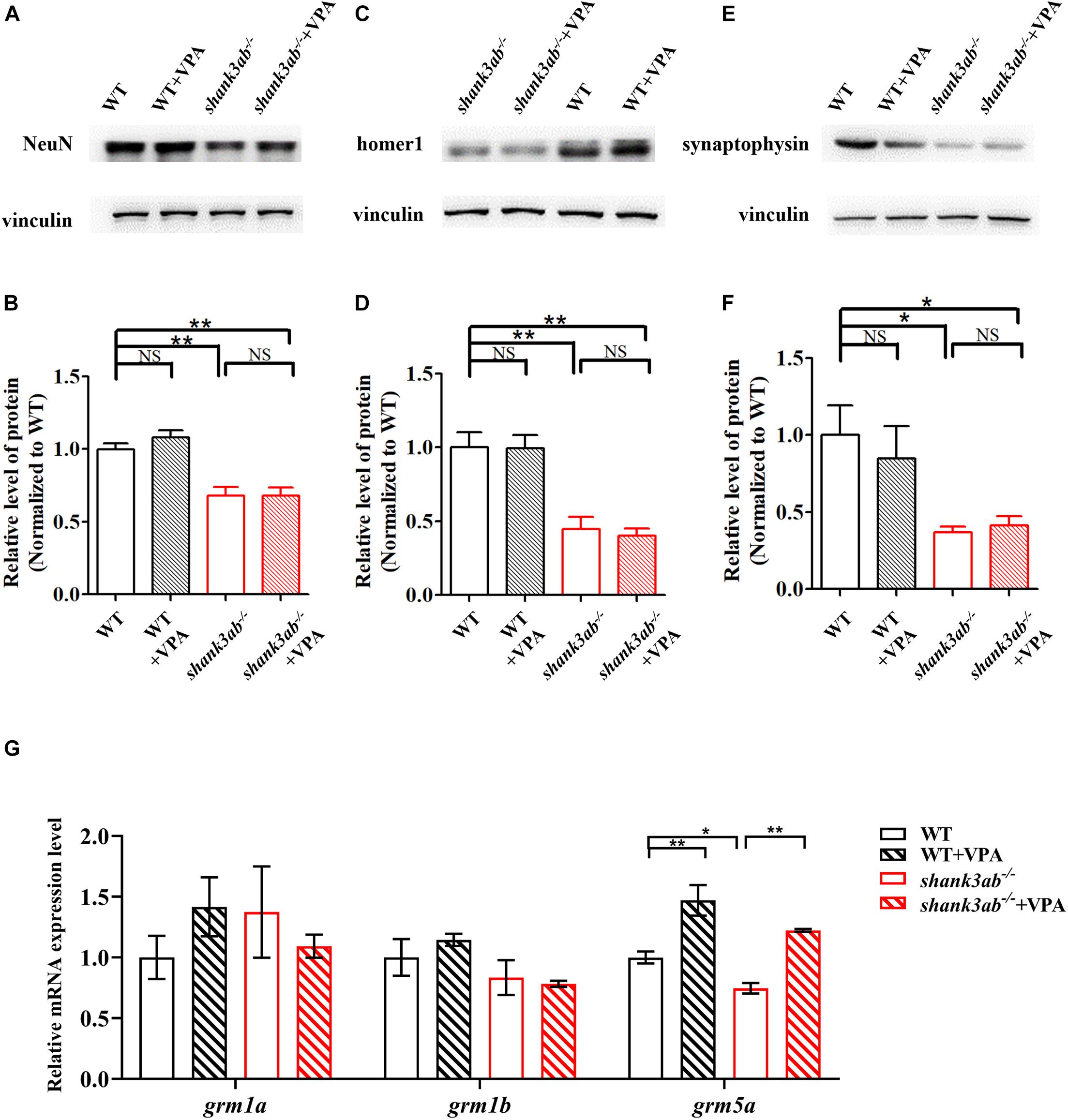
Figure 6. Increased grm5 expression level in shank3-deficient zebrafish upon early VPA treatment. (A,B) Quantitative immunoblot blot analysis showed that the expression level of neuron protein NeuN was not significantly restored in the brain of shank3ab– /– zebrafish treated with VPA relative to WT zebrafish (2 mpf). Similarly, the expressions of post-synaptic homer1 protein (C,D) and presynaptic synaptophysin protein (E,F) were not significantly increased in in the brain of shank3ab– /– zebrafish treated with VPA relative to WT zebrafish (2 mpf). (G) The relative mRNA expression levels of grm1a, grm1b, and grm5a at 4.5 mpf were detected. Each group n = 3. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01.
Discussion
We described a novel shank3-deficient zebrafish, shank3ab–/–, demonstrating stable autism-like behaviors from the juvenile stage through adulthood, including social deficits and stereotyped behaviors. These deficits were generally more severe than exhibited by either shank3a or shank3b mutants. All three mutants also exhibited higher postnatal mortality and rates of morphological dysgenesis than WTs, but adults were fertile. We also found there were decreases in several pre- and postsynaptic proteins in shank3ab–/– mutants. Low-dose VPA reversed some of these autism-like behaviors, consistent with the potential efficacy of this treatment strategy for ASD patients (Hellings et al., 2005; Hollander et al., 2006; DeFilippis and Wagner, 2016). Therefore, the shank3ab–/– zebrafish line is a robust model to explore the neurological mechanisms underlying ASD as well as potential pharmacological treatments.
Among molecular alterations, these shank3-deficient zebrafish exhibited a significant reduction in the expression levels of several synaptic proteins, including pre- and postsynaptic markers, which were consistent with previous mouse or Drosophila models. As a postsynaptic protein, change in postsynaptic homer1 protein was prominent in shank3ab–/– zebrafish, which was consistent with findings from other ASD mouse models (Tu et al., 1999; Wang et al., 2016). In addition, we also demonstrate reduced expression of the presynaptic protein synaptophysin, which was not previously detected in Shank3 deficient mouse models. This finding suggests that shank3 deficiency alters presynaptic formation and neurotransmission through direct or trans-synaptic mechanisms in zebrafish. The presynaptic functions of SHANK3 are not well characterized in comparison to postsynaptic functions. Several recent studies have suggested that SHANK3 is expressed in presynaptic terminals of rodent brain and dorsal root ganglion (DRG) neurons (Han et al., 2016). In a Drosophila Shank mutant (analogous to a SHANK3 mutation in humans), Shank was found in both axons and the presynaptic sites of neuromuscular junctions (NMJs) (Wu et al., 2017). Moreover, ultrastructural analysis of synaptic boutons at Shank mutant calyces showed disorganization of both presynaptic and postsynaptic components, and lack of synaptic clefts (Wu et al., 2017). This first demonstration of reduced synaptophysin in a vertebrate ASD model supports a presynaptic function for shank3 protein. The contribution of shank3-associated presynaptic deficits to ASD warrant further investigation. Generally, the overall morphology of the brain tissues were relatively normal in KO group as compared to WT group (Supplementary Figure 3). Moreover, the expression levels of synaptic proteins were not significantly restored between WT and shank3ab–/– fish after exposed to VPA (Figure 6). Similarly, in mouse model, neuronal morphology and density were not changed between WT and Shank3-deficient mice and also not altered by romidepsin (a highly potent class I inhibitor) treatment (Qin et al., 2018). Additionally, there were no apparent morphological changes in neurons treated with VPA compared with controls (Fujiki et al., 2013).
Postnatal low-dose VPA treatment profoundly and persistently improved social preference deficits, abnormal repetitive behaviors, and impaired thigmotaxis, suggesting activation of a compensatory mechanism under shank3 deficiency. Shank3 facilitates both synaptogenesis and the synaptic plasticity processes underlying social learning and cognition (Duffney et al., 2015). In addition, the behavioral abnormalities exhibited by Shank3-deficient animal models have been attributed to altered glutamatergic signaling (Peca et al., 2011; Wang et al., 2011; Bozdagi et al., 2013; Jiang and Ehlers, 2013), and VPA has been reported to increase synaptic transmission (Rinaldi et al., 2007; Akhtar et al., 2009).
We also found that postnatal low-dose VPA significantly reduced stereotyped swimming patterns (“figure 8” and “walling”). One possible explanation for this effect is improved transcription of grm5a, as Wang et al. (2016) demonstrated that disrupted mGluR5 scaffolding and abnormal mGluR5 signaling contribute to the excessive self-grooming and other behavioral and functional abnormalities of Shank3-defcient mice. Moreover, pharmacological enhancement of mGluR5 receptors rescued behavioral deficits, including repetitive behaviors and social deficits, in Shank3 knockout mice (Vicidomini et al., 2017). Collectively, an important inference from this study is that early VPA treatment can have long-lasting benefits on repetitive behaviors in shank3-deficient zebrafish, possibly by improving grm5a expression.
Valproic acid has anticonvulsant and mood stabilizing activities and are used to treat epilepsy and bipolar disorder. Generally, VPA is a HDAC inhibitor (Phiel et al., 2001), a GABA transaminase inhibitor, and a sodium channel blocker (Johannessen, 2000; Löscher, 2002; Owens and Nemeroff, 2003; Zanatta et al., 2019). Several studies confirmed the role of HDAC inhibitor of VPA. Fujiki et al. (2013) have reported that VPA, trichostatin A and sodium butyrate (all are HDAC inhibitors), but not valpromide, which is a structural analog of VPA having the same antiepileptic effect as VPA but lacking the HDAC inhibitor activity, have proapoptotic effects on neural progenitor cells (NPCs) of embryonic stem (ES) cell-derived glutamatergic neurons. In this study, we also have added a positive control – romidepsin, a highly potent class I HDAC inhibitor, to treat fish (Supplementary Table 2 and Figure 4). WT or shank3ab–/– larvae were exposed to blue egg water with or without 0.05 or 0.1 μM romidepsin from 4 to 8 dpf. At 8 dpf, each larva was pipetted into fresh paramecium liquid, and raised to 2 months old (juvenile). Compared to shank3ab–/– zebrafish, romidepsin treated (0.05 or 0.1 μM) juvenile shank3ab–/– zebrafish exhibited significantly elevated social preference behaviors, which was consistent with Shank3-deficient mice model (Qin et al., 2018).
Valproic acid is a broad-spectrum inhibitor against class I (HDAC1, HDAC2, HDAC3, and HDAC8) HDAC (Chelladurai et al., 2020). Qin et al. (2018) reported that Shank3-deficient mice exhibited an abnormally low level of histone acetylation resulting from HDAC2 upregulation in the prefrontal cortex (PFC) and β-catenin/HDAC2 played a causal role in social deficits of Shank3-deficiency mouse model and the therapeutic effect of romidepsin. While the levels of HDAC1, HDAC3, and HDAC8 mRNA were largely unchanged. Given the absence of hdac2 gene in the zebrafish genome (Ko et al., 2019), we detected the expression levels of the rest three genes. Similarly, RT-qPCR analysis revealed no changes in mRNA expression levels of hdac1, hdac3, and hdac8 (Supplementary Figure 2C). In the further study, a VPA analog that does not have the HDAC inhibitory activity should be used as a control to detect the effects of the HDAC inhibitory activity of VPA.
This study also supports previous studies demonstrating that the early postnatal period is a critical therapeutic time window for long-lasting effects on core autistic symptoms (Dinstein et al., 2011). Hensch (2005) reported that novel interventions should be applied before irreversible neural function changes coinciding with the end of these critical periods. However, clinical studies have demonstrated that VPA exposure before the neural tube is closed (20–24 days of gestation in humans) increases the incidence of neurodevelopmental disorders including ASD (Rice and Barone, 2000; Jentink et al., 2010; Meador et al., 2013). Moreover, prenatal VPA exposure is actually used to establish rodent ASD models. Animals exposed to VPA during neural tube closure [E12.5 in rats according to Kim et al. (2011) and E10.5 in mice according to Kim et al. (2014)] showed an increased incidence of autism-like symptoms. When VPA was administered earlier than this critical time point, embryonic malformation was very likely to occur (Kim et al., 2019). In contrast, VPA administered after neural tube closure did not cause embryonic lethality or autism-like phenotypes (Kim et al., 2019). Furthermore, long-term VPA therapy had no noticeable noxious effect on cognition and learning in school children (Calandre et al., 1990). Thus, the optimal postnatal time window appears essential for VPA treatment efficacy against ASD.
Dose was the other key factor for effective VPA treatment. In animal models, fetal VPA exposure impairs cognitive outcome and increases malformation rate in a dose-dependent manner (Meador et al., 2013; Kaplan et al., 2015). Nicolini and Fahnestock (2018) reported ASD-like deficits following exposure of rat embryos to 350-600 mg/kg or of mouse embryos to 300-800 mg/kg VPA, while exposure to 25 μM from 10 to 24 hpf elicited social deficits in zebrafish (Baronio et al., 2018). VPA can also induce neuronal apoptosis (Ikonomidou et al., 1999). Conversely, low-dose VPA induced a dramatic rescue of core autistic deficits in shank3-deficient zebrafish, and this dose had no detectable effects on WT animals.
This study provides a new genetic zebrafish model of shank3 deficiency which displayed distinctly abnormal social behaviors and increased stereotyped behaviors. Importantly, the autism-like behaviors could be improved by postnatal low-dose VPA treatment. These findings may suggest a path for further research to identify medicinal development and allow for more in-depth understandings of future clinical drug research.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by Research Ethics Board of Children’s Hospital of Fudan University.
Author Contributions
XX and QL conceived the study. CL and YW performed the experiments, analyzed the data, and wrote the initial draft of the manuscript. All authors contributed to the interpretation of the results, provided critical feedback, helped shape the analysis and manuscript, and approved the submitted manuscript.
Funding
This study was supported by grants from “Haiju” International Joint Laboratory of National Children’s Medical Center of Children’s Hospital of Fudan University (No. EK1125180106), Industry-University-Research High-tech Transformation Incubation Project of Fudan University (FDEKCXY08), and Shanghai Key Clinical Specialty Construction Project to XX. This study was also supported by grants from the Shanghai Sailing Program (No. 19YF1403800) and Clinical Research of Shanghai Municipal Health Commission (No. 20204Y0102) to CL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2021.682054/full#supplementary-material
Footnotes
References
American Psychiatric Association (2013). Diagnostic and Statistical Manual of Mental Disorders, 5th Edn. Washington, DC: American Psychiatric Association.
Akhtar, M. W., Raingo, J., Nelson, E. D., Montgomery, R. L., Olson, E. N., Kavalali, E. T., et al. (2009). Histone deacetylases 1 and 2 form a developmental switch that controls excitatory synapse maturation and function. J. Neurosci. 29, 8288–8297. doi: 10.1523/JNEUROSCI.009709.2009
Arons, M. H., Thynne, C. J., Grabrucker, A. M., Li, D., Schoen, M., Cheyne, J. E., et al. (2012). Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. J. Neurosci. 32, 14966–14978. doi: 10.1523/JNEUROSCI.2215-12.2012
Baronio, D., Puttonen, H., Sundvik, M., Semenova, S., and Lehtonen, E. (2018). Embryonic exposure to valproic acid affects the histaminergic system and the social behaviour of adult zebrafish (Danio rerio). Br. J. Pharmacol. 175, 797–809. doi: 10.1111/bph.14124
Bonaglia, M. C., Giorda, R., Beri, S., De Agostini, C., Novara, F., Fichera, M., et al. (2011). Molecular mechanisms generating and stabilizing terminal 22q13 deletions in 44 subjects with Phelan/McDermid syndrome. PLoS Genet 7:e1002173. doi: 10.1371/journal.pgen.1002173
Bozdagi, O., Tavassoli, T., and Buxbaum, J. D. (2013). Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Mol. Autism 4:9. doi: 10.1186/2040-2392-4-9
Boccuto, L., Lauri, M., Sarasua, S. M., Skinner, C. D., Buccella, D., Dwivedi, A., et al. (2013). Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur. J. Hum. Genet. 21, 310–316. doi: 10.1038/ejhg.2012.175
Busnelli, M., Kleinau, G., Muttenthaler, M., Stoev, S., Manning, M., Bibic, L., et al. (2016). Design and characterization of superpotent bivalent ligands targeting oxytocin receptor dimers via a channel-like structure. J. Med. Chem. 59, 7152–7166. doi: 10.1021/acs.jmedchem.6b00564
Calandre, E. P., Dominguez-Granados, R., Gomez-Rubio, M., and Molina-Font, J. A. (1990). Cognitive effects of long-term treatment with phenobarbital and valproic acid in school children. Acta Neurol. Scand. 81, 504–506. doi: 10.1111/j.1600-0404.1990.tb01008.x
Chelladurai, P., Dabral, S., Basineni, S. R., Chen, C., Schmoranzer, M., Bender, N., et al. (2020). Isoform-specific characterization of class I histone deacetylases and their therapeutic modulation in pulmonary hypertension. Sci. Rep. 10:12864. doi: 10.1038/s41598-020-69737-x
Dawson, G., Rogers, S., Munson, J., Smith, M., Winter, J., Greenson, J., et al. (2010). Randomized, controlled trial of an intervention for toddlers with autism: the Early Start Denver Model. Pediatrics 125, e17–e23. doi: 10.1542/peds.2009-2958
DeFilippis, M., and Wagner, K. D. (2016). Treatment of autism spectrum disorder in children and adolescents. Psychopharmacol. Bull. 46, 18–41.
Dhar, S. U., Del, G. D., German, J. R., Peters, S. U., Ou, Z., Bader, P. I., et al. (2010). 22q13.3 deletion syndrome: clinical and molecular analysis using array CGH. Am. J. Med. Genet A 152A, 573–581. doi: 10.1002/ajmg.a.33253
Dinstein, I., Pierce, K., Eyler, L., Solso, S., Malach, R., Behrmann, M., et al. (2011). Disrupted neural synchronization in toddlers with autism. Neuron 70, 1218–1225. doi: 10.1016/j.neuron.2011.04.018
Duffney, L. J., Zhong, P., Wei, J., Matas, E., Cheng, J., Qin, L., et al. (2015). Autism-like deficits in Shank3-deficient mice are rescued by targeting actin regulators. Cell Rep. 11, 1400–1413. doi: 10.1016/j.celrep.2015.04.064
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Evans, C. R., and Erickson, N. (2019). Intersectionality and depression in adolescence and early adulthood: a MAIHDA analysis of the national longitudinal study of adolescent to adult health, 1995-2008. Soc. Sci. Med. 220, 1–11. doi: 10.1016/j.socscimed.2018.10.019
Fischer, A., Sananbenesi, F., Mungenast, A., and Tsai, L. H. (2010). Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol. Sci. 31, 605–617. doi: 10.1016/j.tips.2010.09.003
Franz, L., and Dawson, G. (2019). Implementing early intervention for autism spectrum disorder: a global perspective. Pediatr. Med. 2:44. doi: 10.21037/pm.2019.07.09
Fujiki, R., Sato, A., Fujitani, M., and Yamashita, T. (2013). A proapoptotic effect of valproic acid on progenitors of embryonic stem cell-derived glutamatergic neurons. Cell Death Dis. 4:e677. doi: 10.1038/cddis.2013.205
Fung, L. K., Mahajan, R., Nozzolillo, A., Bernal, P., Krasner, A., Jo, B., et al. (2016). Pharmacologic treatment of severe irritability and problem behaviors in autism: a systematic review and meta-analysis. Pediatrics 137(Suppl. 2), S124–S135. doi: 10.1542/peds.2015-2851K
Gauthier, J., Spiegelman, D., Piton, A., Lafreniere, R. G., Laurent, S., St-Onge, J., et al. (2009). Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet B Neuropsychiatr. Genet 150B, 421–424. doi: 10.1002/ajmg.b.30822
Han, Q., Kim, Y. H., Wang, X., Liu, D., Zhang, Z. J., Bey, A. L., et al. (2016). SHANK3 deficiency impairs heat hyperalgesia and TRPV1 signaling in primary sensory neurons. Neuron 92, 1279–1293. doi: 10.1016/j.neuron.2016.11.007
Hellings, J. A., Weckbaugh, M., Nickel, E. J., Cain, S. E., Zarcone, J. R., Reese, R. M., et al. (2005). A double-blind, placebo-controlled study of valproate for aggression in youth with pervasive developmental disorders. J. Child Adolesc. Psychopharmacol. 15, 682–692. doi: 10.1089/cap.2005.15.682
Hensch, T. K. (2005). Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 6, 877–888. doi: 10.1038/nrn1787
Hollander, E., Soorya, L., Wasserman, S., Esposito, K., and Chaplin, W. (2006). Divalproex sodium vs. placebo in the treatment of repetitive behaviours in autism spectrum disorder. Int. J. Neuropsychopharmacol. 9, 209–213. doi: 10.1017/S1461145705005791
Hwang, W. Y., Fu, Y., Reyon, D., Maeder, M. L., Tsai, S. Q., Sander, J. D., et al. (2013). Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229. doi: 10.1038/nbt.2501
Hyman, S. L., Levy, S. E., and Myers, S. M. (2020). Identification, evaluation, and management of children with autism spectrum disorder. Pediatrics 145:e20193447. doi: 10.1542/peds.2019-3447
Ikonomidou, C., Bosch, F., Miksa, M., Bittigau, P., Vockler, J., Dikranian, K., et al. (1999). Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283, 70–74. doi: 10.1126/science.283.5398.70
Jentink, J., Loane, M. A., Dolk, H., Barisic, I., Garne, E., Morris, J. K., et al. (2010). Valproic acid monotherapy in pregnancy and major congenital malformations. N. Engl. J. Med. 362, 2185–2193. doi: 10.1056/NEJMoa0907328
Jiang, Y. H., and Ehlers, M. D. (2013). Modeling autism by SHANK gene mutations in mice. Neuron 78, 8–27. doi: 10.1016/j.neuron.2013.03.016
Jutla, A., Reed, H., and Veenstra-VanderWeele, J. (2019). The architecture of autism spectrum disorder risk: what do we know, and where do we go from here? JAMA Psychiatry 76, 1005–1006. doi: 10.1001/jamapsychiatry.2019.1375
Kalueff, A. V., Stewart, A. M., and Gerlai, R. (2014). Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol. Sci. 35, 63–75. doi: 10.1016/j.tips.2013.12.002
Kaplan, Y. C., Nulman, I., and Koren, G. (2015). Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Ther. Drug Monit. 37, 557–558. doi: 10.1097/FTD.0000000000000193
Kim, J. W., Park, K., Kang, R. J., Gonzales, E. L., Oh, H. A., Seung, H., et al. (2019). Gene-environment interaction counterbalances social impairment in mouse models of autism. Sci. Rep. 9:11490. doi: 10.1038/s41598-019-47680-w
Kim, J. W., Seung, H., Kwon, K. J., Ko, M. J., Lee, E. J., Oh, H. A., et al. (2014). Subchronic treatment of donepezil rescues impaired social, hyperactive, and stereotypic behavior in valproic acid-induced animal model of autism. PLoS One 9:e104927. doi: 10.1371/journal.pone.0104927
Kim, K. C., Kim, P., Go, H. S., Choi, C. S., Yang, S. I., Cheong, J. H., et al. (2011). The critical period of valproate exposure to induce autistic symptoms in Sprague-Dawley rats. Toxicol. Lett. 201, 137–142. doi: 10.1016/j.toxlet.2010.12.018
Kim, O. H., Cho, H. J., Han, E., Hong, T. I., Ariyasiri, K., Choi, J. H., et al. (2017). Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism. Mol. Autism 8:50. doi: 10.1186/s13229-017-0168-162
Ko, S., Russell, J. O., Tian, J., Gao, C., Kobayashi, M., Feng, R., et al. (2019). Hdac1 regulates differentiation of bipotent liver progenitor cells during regeneration via Sox9b and Cdk8. Gastroenterology 156, 187–202. doi: 10.1053/j.gastro.2018.09.039
Kwon, S. E., and Chapman, E. R. (2011). Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron 70, 847–854. doi: 10.1016/j.neuron.2011.04.001
Lai, M. C., Lombardo, M. V., and Baron-Cohen, S. (2014). Autism. Lancet 383, 896–910. doi: 10.1016/S0140-6736(13)61539-61531
Leblond, C. S., Nava, C., Polge, A., Gauthier, J., Huguet, G., Lumbroso, S., et al. (2014). Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet 10:e1004580. doi: 10.1371/journal.pgen.1004580
Liu, C. X., Li, C. Y., Hu, C. C., Wang, Y., Lin, J., Jiang, Y. H., et al. (2018). CRISPR/Cas9-induced shank3b mutant zebrafish display autism-like behaviors. Mol. Autism 9:23. doi: 10.1186/s13229-018-0204-x
Liu, C. X., Peng, X. L., Hu, C. C., Li, C. Y., and Li, Q. (2016). Developmental profiling of ASD-related shank3 transcripts and their differential regulation by valproic acid in zebrafish. Dev. Genes Evol. 226, 389–400. doi: 10.1007/s00427-016-0561-564
Liu, C. X., Li, D. Y., Yang, H. W., Li, H. P., Xu, Q., Zhou, B. R., et al. (2021). Altered striatum centered brain structures in SHANK3 deficient Chinese children with genotype and phenotype profiling. Prog. Neurobiol. 200:101985. doi: 10.1016/j.pneurobio.2020.101985
Lord, C., Brugha, T. S., Charman, T., Cusack, J., Dumas, G., Frazier, T., et al. (2020). Autism spectrum disorder. Nat. Rev. Dis. Primers 6:5. doi: 10.1038/s41572-019-0138-134
Lord, C., Elsabbagh, M., Baird, G., and Veenstra-Vanderweele, J. (2018). Autism spectrum disorder. Lancet 392, 508–520. doi: 10.1016/S0140-6736(18)31129-31122
Johannessen, C. U. (2000). Mechanisms of action of valproate: a commentatory. Neurochem. Int. 37, 103–110. doi: 10.1016/s0197-0186(00)00013-19
Löscher, W. (2002). Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs 16, 669–694. doi: 10.2165/00023210-200216100-00003
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., et al. (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. doi: 10.1126/science.1232033
Mathur, P., and Guo, S. (2010). Use of zebrafish as a model to understand mechanisms of addiction and complex neurobehavioral phenotypes. Neurobiol. Dis. 40, 66–72. doi: 10.1016/j.nbd.2010.05.016
Meador, K. J., Baker, G. A., Browning, N., Cohen, M. J., Bromley, R. L., Clayton-Smith, J., et al. (2013). Fetal antiepileptic drug exposure and cognitive outcomes at age 6 years (NEAD study): a prospective observational study. Lancet Neurol. 12, 244–252. doi: 10.1016/S1474-4422(12)70323-X
Moessner, R., Marshall, C. R., Sutcliffe, J. S., Skaug, J., Pinto, D., Vincent, J., et al. (2007). Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 81, 1289–1297. doi: 10.1086/522590
Monteiro, P., and Feng, G. (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 18, 147–157. doi: 10.1038/nrn.2016.183
Muhle, R. A., Reed, H. E., Stratigos, K. A., and Veenstra-VanderWeele, J. (2018). The emerging clinical neuroscience of autism spectrum disorder: a review. JAMA Psychiatry 75, 514–523. doi: 10.1001/jamapsychiatry.2017.4685
Nicolini, C., and Fahnestock, M. (2018). The valproic acid-induced rodent model of autism. Exp. Neurol. 299, 217–227. doi: 10.1016/j.expneurol.2017.04.017
Owens, M. J., and Nemeroff, C. B. (2003). Pharmacology of valproate. Psychopharmacol. Bull. 37(Suppl. 2), 17–24. doi: 10.2165/00023210-200216100-200216103
Peca, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N., et al. (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Penagarikano, O., Lazaro, M. T., Lu, X. H., Gordon, A., Dong, H., and Lam, H. A. (2015). Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci. Transl. Med. 7, 271r–278r. doi: 10.1126/scitranslmed.3010257
Perathoner, S., Cordero-Maldonado, M. L., and Crawford, A. D. (2016). Potential of zebrafish as a model for exploring the role of the amygdala in emotional memory and motivational behavior. J. Neurosci. Res. 94, 445–462. doi: 10.1002/jnr.23712
Phelan, K., and McDermid, H. E. (2012). The 22q13.3 deletion syndrome (Phelan-McDermid Syndrome). Mol. Syndromol. 2, 186–201. doi: 10.1159/000334260
Phelan, K., Rogers, R. C., and Boccuto, L. (1993). Phelan-McDermid Syndrome. Seattle (WA): University of Washington.
Phelan, M. C. (2008). Deletion 22q13.3 syndrome. Orphanet J. Rare Dis. 3:14. doi: 10.1186/1750-1172-3-14
Phiel, C. J., Zhang, F., Huang, E. Y., Guenther, M. G., Lazar, M. A., and Klein, P. S. (2001). Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 276, 36734–36741. doi: 10.1074/jbc.M101287200
Qin, L., Ma, K., Wang, Z. J., Hu, Z., Matas, E., Wei, J., et al. (2018). Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat. Neurosci. 21, 564–575. doi: 10.1038/s41593-018-0110-118
Rice, D., and Barone, S. J. (2000). Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ. Health Perspect 108(Suppl. 3), 511–533. doi: 10.1289/ehp.00108s3511
Rinaldi, T., Kulangara, K., Antoniello, K., and Markram, H. (2007). Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl. Acad. Sci. U S A. 104, 13501–13506. doi: 10.1073/pnas.0704391104
Sandbank, M., Bottema-Beutel, K., and Woynaroski, T. (2020). Intervention recommendations for children with autism in light of a changing evidence base. JAMA Pediatr. 175, 341–342. doi: 10.1001/jamapediatrics.2020.4730
Sarasua, S. M., Dwivedi, A., Boccuto, L., Rollins, J. D., Chen, C. F., Rogers, R. C., et al. (2011). Association between deletion size and important phenotypes expands the genomic region of interest in Phelan-McDermid syndrome (22q13 deletion syndrome). J. Med. Genet. 48, 761–766. doi: 10.1136/jmedgenet-2011-100225
Tu, J. C., Xiao, B., Naisbitt, S., Yuan, J. P., Petralia, R. S., Brakeman, P., et al. (1999). Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592. doi: 10.1016/s0896-6273(00)80810-80817
Tu, Z., Zhao, H., Li, B., Yan, S., Wang, L., Tang, Y., et al. (2019). CRISPR/Cas9-mediated disruption of SHANK3 in monkey leads to drug-treatable autism-like symptoms. Hum. Mol. Genet. 28, 561–571. doi: 10.1093/hmg/ddy367
Verpelli, C., and Sala, C. (2012). Molecular and synaptic defects in intellectual disability syndromes. Curr. Opin. Neurobiol. 22, 530–536. doi: 10.1016/j.conb.2011.09.007
Vicidomini, C., Ponzoni, L., Lim, D., Schmeisser, M. J., Reim, D., Morello, N., et al. (2017). Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 22:784. doi: 10.1038/mp.2016.70
Wang, X., Bey, A. L., Chung, L., Krystal, A. D., and Jiang, Y. H. (2014a). Therapeutic approaches for shankopathies. Dev. Neurobiol. 74, 123–135. doi: 10.1002/dneu.22084
Wang, X., Bey, A. L., Katz, B. M., Badea, A., Kim, N., David, L. K., et al. (2016). Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun. 7:11459. doi: 10.1038/ncomms11459
Wang, X., McCoy, P. A., Rodriguiz, R. M., Pan, Y., Je, H. S., Roberts, A. C., et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108. doi: 10.1093/hmg/ddr212
Wang, X., Xu, Q., Bey, A. L., Lee, Y., and Jiang, Y. H. (2014b). Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol. Autism 5:30. doi: 10.1186/2040-2392-5-30
Weitlauf, A. S., McPheeters, M. L., Peters, B., Sathe, N., Travis, R., Aiello, R., et al. (2014). Therapies for Children With Autism Spectrum Disorder: Behavioral Interventions Update. Rockville, MD: Agency for Healthcare Research and Quality.
Wilson, H. L., Wong, A. C., Shaw, S. R., Tse, W. Y., Stapleton, G. A., Phelan, M. C., et al. (2003). Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J. Med. Genet. 40, 575–584. doi: 10.1136/jmg.40.8.575
Wu, S., Gan, G., Zhang, Z., Sun, J., Wang, Q., Gao, Z., et al. (2017). A presynaptic function of Shank protein in Drosophila. J. Neurosci. 37, 11592–11604. doi: 10.1523/JNEUROSCI.0893-17.2017
Zanatta, G., Sula, A., Miles, A. J., Ng, L., Torella, R., Pryde, D. C., et al. (2019). Valproic acid interactions with the NavMs voltage-gated sodium channel. Proc. Natl. Acad. Sci. U S A. 116, 26549–26554. doi: 10.1073/pnas.1909696116
Zhou, Y., Sharma, J., Ke, Q., Landman, R., Yuan, J., Chen, H., et al. (2019). Atypical behaviour and connectivity in SHANK3-mutant macaques. Nature 570, 326–331. doi: 10.1038/s41586-019-1278-1270
Keywords: shank3, valproic acid, zebrafish model, drug treatment, autism spectrum disorder
Citation: Liu C, Wang Y, Deng J, Lin J, Hu C, Li Q and Xu X (2021) Social Deficits and Repetitive Behaviors Are Improved by Early Postnatal Low-Dose VPA Intervention in a Novel shank3-Deficient Zebrafish Model. Front. Neurosci. 15:682054. doi: 10.3389/fnins.2021.682054
Received: 17 March 2021; Accepted: 11 August 2021;
Published: 10 September 2021.
Edited by:
Xiaoyan Ke, Nanjing Brain Hospital Affiliated to Nanjing Medical University, ChinaReviewed by:
Sofia Lizarraga, University of South Carolina, United StatesJessica L. MacDonald, Syracuse University, United States
Copyright © 2021 Liu, Wang, Deng, Lin, Hu, Li and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Li, bGlxQGZ1ZGFuLmVkdS5jbg==; Xiu Xu, eHV4aXVAc2htdS5lZHUuY24=
†These authors have contributed equally to this work