 Bernice Frempong1,2
Bernice Frempong1,2 David R. Lynch
David R. Lynch
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Neurosci., 02 March 2021
Sec. Neurodegeneration
Volume 15 - 2021 | https://doi.org/10.3389/fnins.2021.653241
This article is part of the Research TopicThe Impact of Neurofilament Light Chain (NFL) Quantification in Serum and Cerebrospinal Fluid in Neurodegenerative DiseasesView all 17 articles
Friedreich Ataxia (FRDA) is a progressive neurological and systemic disorder that affects about one in 50,000 people worldwide (Strawser et al., 2017). It is caused by mutations, usually GAA repeat expansions (96%) but also point mutations or deletions (4%), in the FXN gene, resulting in decreased production of functional frataxin protein (Babady et al., 2007; Delatycki and Bidichandani, 2019). GAA length on the shorter allele inversely correlates with disease severity (Strawser et al., 2017). Frataxin is a small mitochondrial protein that functions in iron-sulfur-cluster biosynthesis (Colin et al., 2013). Its deficiency leads to difficulties in production of cellular ATP as well as sensitivity to reactive oxygen species in vitro (Rötig et al., 1997; Lodi et al., 2001; Pastore and Puccio, 2013; DeBrosse et al., 2016). These properties lead to neurological injury and clinical impairment, including ataxia, dysarthria, sensory loss, and weakness in FRDA patients. While most literature has focused on neurodegeneration in FRDA, the disorder also has a large developmental component (Koeppen et al., 2017a,b). In addition, individuals with FRDA develop cardiomyopathy, scoliosis and sometimes diabetes mellitus. The cardiomyopathy of FRDA is characterized by early hypertrophy, with later progression to fibrosis and systolic dysfunction, leading to death from end-stage heart failure (Tsou et al., 2011; Lynch et al., 2012; Strawser et al., 2017). Many agents are in development for FRDA, including some designed to ameliorate mitochondrial dysfunction and others that seek to increase levels of functional frataxin (Strawser et al., 2014; Li et al., 2015; Lynch et al., 2018, 2021; Piguet et al., 2018; Zesiewicz et al., 2018a,b; Belbella et al., 2019; Rodríguez-Pascau et al., 2021).
In many neurodegenerative diseases, the need for disease-modifying treatments is facilitated by identification of biomarkers to track disease progression. Such markers can capture subclinical changes in a rapid manner and show evidence of target engagement in clinical trials in slowly progressive neurological disorders. In other neurological disorders, including Multiple Sclerosis (MS), Alzheimer's disease (AD), and Parkinson's disease (PD), neurofilament light chain levels (NfL) in body fluids such as serum, plasma or CSF may provide a biomarker for tracking disease activity including progression (Bridel et al., 2019; Forgrave et al., 2019; Aktas et al., 2020; Del Prete et al., 2020; Milo et al., 2020; Thebault et al., 2020; Wang et al., 2020). Neurofilaments are cytoskeletal proteins located in both the peripheral and central nervous system, particularly in larger myelinated axons. They play a significant role in axonal growth and the determination of axonal caliber (Hsieh et al., 1994; Kurochkina et al., 2018; Bott and Winckler, 2020). Logically, as axons are damaged and die in neurodegenerative processes, NfL should leak into the interstitial space, then into CSF and plasma. Thus, concentrations of NfL should generally increase as neurodegenerative diseases progress and should reflect disease activity. For example, in progressive MS, NfL appears to track with neuronal and axonal death, the stage of disease, and treatment response. NfL concentration in the CSF of MS patients parallels T2 lesion changes on MRI. Similarly, plasma NfL concentration is higher in AD patients than in controls and is associated with greater cognitive deficit. Such findings suggest that NfL is a promising biomarker for determining the stage of disease, tracking progression and aiding in identification of disease-modifying treatments in neurological disorders. However, in stable MS, NfL levels may not track with clinical dysfunction, providing a reminder that changes in biomarkers must be interpreted in the context of clinical changes (Aktas et al., 2020).
In FRDA, data on NfL levels in serum is more complex. Most features of FRDA depend on genetic severity (GAA repeat length) and worsen over the course of time, thus correlating positively with disease duration or age (Strawser et al., 2017). Overall, the two main determinants of clinical severity in FRDA are genetic severity and disease duration. In the three studies evaluating serum NfL in FRDA patients, serum NfL is elevated in patients with FRDA when compared to controls and carriers (Zeitlberger et al., 2018; Clay et al., 2020; Hayer et al., 2020). This shows that serum NfL levels reflect a pathological process in FRDA. However, in these three studies NfL levels generally do not correlate with markers of clinical or genetic severity. Moreover, while NfL levels correlate positively with age in non-FRDA patients (controls and carriers) in cross sectional analysis, in FRDA patients, NfL levels are highest in young children and decrease with age as the disease progresses (Clay et al., 2020). Thus, serum NfL is paradoxically high in young individuals and far lower in older individuals with more severe disease. At later ages it even overlaps with control values. A greater genetic severity in early onset individuals could explain some of this paradox; however, NfL levels overall do not correlate with GAA repeat length after accounting for age, and they even appear to correlate inversely with GAA length in some of these studies. Accounting for age, individuals who are more severe genetically have lower levels of NfL. Interestingly, Nfl levels are relatively stable over 1–2 years; consequently, NfL levels could be used as an assessment of therapeutic response over the time used in most clinical trials. Still, while NfL may provide a biomarker of FRDA in some manner, the relationship of NfL to disease progression is complex suggesting its utility may be limited to certain situations.
Understanding the exact meaning of NfL levels in serum and how they reflect disease activity in FRDA would facilitate their use as a marker of FRDA. In most other disorders, NfL is viewed as a marker of neurodegeneration of either axons or other neuronal regions. Degeneration in FRDA, though, is complex, including both peripheral nerve degeneration (including very early degeneration of proprioceptive afferents) with later degeneration of central nervous system axons; loss of central nervous system elements controls most of the clinical progression of the disease (Selvadurai et al., 2016; Koeppen et al., 2017a; Strawser et al., 2017; Marty et al., 2019; Rezende et al., 2019; Harding et al., 2020; Naeije et al., 2020). Brain imaging studies are typically normal early in disease, with the exception of atrophy of the cervical spinal cord, with progressive loss of CNS pathways later (Selvadurai et al., 2016; Koeppen et al., 2017b; Marty et al., 2019; Rezende et al., 2019; Harding et al., 2020; Naeije et al., 2020). Thus, serum NfL levels in FRDA, with high values early in disease, are discrepant from the tangible loss of CNS axons by MRI and the loss of specific functional clinical systems (Figure 1). The present data on serum NfL levels could be explained by a relatively large early loss of peripheral axons that does not contribute to clinical progression. Similarly, the inverse correlation with GAA repeat length in early disease might lead to a large developmental deficit at presentation. This in turn might lead to lower serum NfL levels during neurodegeneration. This interpretation would be consistent with the prevailing concept of NfL levels reflecting a relatively passive leakage from dying neurons into surrounding fluids and eventually to the serum.
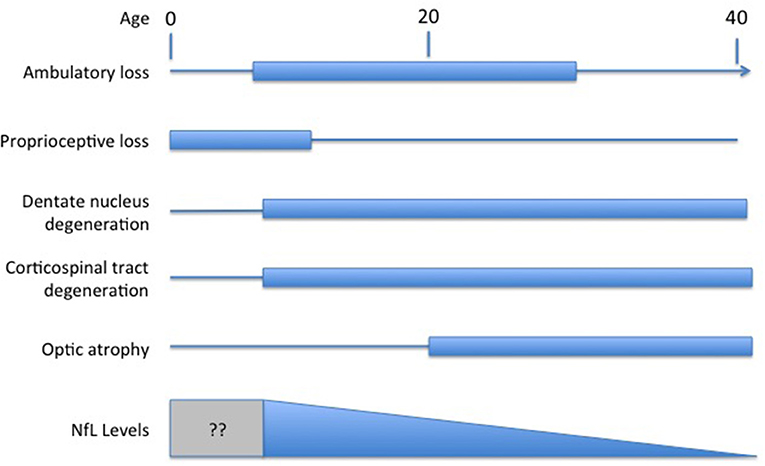
Figure 1. Temporal course of changes in FRDA. Diagram illustrating the contrasting temporal course of clinical changes in FRDA along with serum NfL levels over time (amalgamated from Bridel et al., 2019; Del Prete et al., 2020; Wang et al., 2020). Clinical changes are presented based on the clinical course of an early onset individual (onset between ages 5−10). At present, no study has measured NfL in the presymptomatic period before age five.
Alternatively, increased levels of NfL could reflect other components of the pathophysiology of FRDA in a manner not directly associated with cell death. FRDA is associated with abnormalities in lipid metabolism as well as lipid peroxidation (Navarro et al., 2010; Obis et al., 2014; Abeti et al., 2016; Chen et al., 2016; Tamarit et al., 2016; Cotticelli et al., 2019; Turchi et al., 2020). Both could lead to membranes that are inherently more permeable than normal, with consequent loss of NfL from the cell. Why these processes would decrease with age, however, is unclear.
Still, other processes might contribute to the paradox of elevated NfL levels early in FRDA. NfL levels must to some degree reflect its synthesis, as increased synthesis leads to higher levels of soluble NfL (before it is incorporated into neurofilaments) that should more readily efflux from neurons cells than NfL assembled into intact neurofilaments. Increased synthesis of structural proteins in axons occurs in response to injury and during neuronal regeneration (Pearson et al., 1988; Havton and Kellerth, 2001; Toth et al., 2008; Balaratnasingam et al., 2011; Yin et al., 2014; Liu et al., 2016), and neurofilaments play different roles in development than simply structural maintenance. The very high levels of NfL early in FRDA could reflect attempts at regeneration that become more impaired as the disease progresses, leading to falling levels of NfL later in the course of the disorder. In general, the plasticity of the nervous system decreases with aging, matching the falls in serum NfL over time in FRDA (Bouchard and Villeda, 2015). Thus, elevated levels of serum NfL early in the course of FRDA could be driven by enhanced synthesis of NFL during regeneration superimposed on increased membrane fragility.
Interestingly, cardiac troponin levels are elevated in FRDA serum during the period of hypertrophic disease, long before cardiomyocytes die and cardiac fibrosis develops (Friedman et al., 2013; Plehn et al., 2018; Legrand et al., 2020). Such elevated levels of troponin in early FRDA cardiomyopathy might result from similar mechanisms to the elevated levels of NfL early in FRDA (Thebault et al., 2020).
A final possibility is that both cell-loss and cell-repair mechanisms—and possibly still other mechanisms—mediate the changes in NfL in FRDA. Such interpretations may only be distinguishable over time with collection of long-term serial data, and with collection of data during the presymptomatic period. Furthermore, a more complete characterization of the features of immunoreactive NfL in FRDA serum may be helpful. While the assays used are specific for NfL, they do not assess whether it represents full-length protein. This does not change the observation that Nfl levels can serve as biomarkers of disease in FRDA. However, without understanding the reason for the unusual distribution of NfL values, it is difficult to provide precise interpretations for clinical trial results. Normalization of biomarker levels can provide evidence for benefit in the correct circumstances, but can also reflect impairment of compensatory mechanisms, thus being associated with deleterious effects. A deeper understanding of the mechanisms of NfL elevation in serum in FRDA is needed to make it a useful biomarker in FRDA.
BF created the first draft and edited the final work. RW and KS provided ideas, editing, and critical review. DL contributed to the first draft, performed critical review, ideas for the project, and designed the figure. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
FRDA, Friedreich Ataxia; NfL, Neurofilament light chain; GAA, Guanine-adenine-adenine; MS, Multiple sclerosis; PD, Parkinson's disease; CSF, Cerebrospinal fluid; AD, Alzheimer's disease; CNS, central nervous system.
Abeti, R., Parkinson, M. H., Hargreaves, I. P., Angelova, P. R., Sandi, C., Pook, M. A., et al. (2016). Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich's ataxia. Cell Death Dis. 7:e2237. doi: 10.1038/cddis.2016.111
Aktas, O., Renner, A., Huss, A., Filser, M., Baetge, S., Stute, N., et al. (2020). Serum neurofilament light chain: no clear relation to cognition and neuropsychiatric symptoms in stable, M. S. Neurol. Neuroimmunol. Neuroinflamm. 7:e885. doi: 10.1212/NXI.0000000000000885
Babady, N. E., Carelle, N., Wells, R. D., Rouault, T. A., Hirano, M., Lynch, D. R., et al. (2007). Advancements in the pathophysiology of Friedreich's Ataxia and new prospects for treatments. Mol. Genet. Metab. 92, 23–35. doi: 10.1016/j.ymgme.2007.05.009
Balaratnasingam, C., Morgan, W. H., Bass, L., Kang, M., Cringle, S. J., and Yu, D. Y. (2011). Axotomy-induced cytoskeleton changes in unmyelinated mammalian central nervous system axons. Neuroscience 177, 269–282. doi: 10.1016/j.neuroscience.2010.12.053
Belbella, B., Reutenauer, L., Monassier, L., and Puccio, H. (2019). Correction of half the cardiomyocytes fully rescue Friedreich ataxia mitochondrial cardiomyopathy through cell-autonomous mechanisms. Hum. Mol. Genet. 28, 1274–1285. doi: 10.1093/hmg/ddy427
Bott, C. J., and Winckler, B. (2020). Intermediate filaments in developing neurons: beyond structure. Cytoskeleton 77, 110–128. doi: 10.1002/cm.21597
Bouchard, J., and Villeda, S. A. (2015). Aging and brain rejuvenation as systemic events. J. Neurochem. 132, 5–19. doi: 10.1111/jnc.12969
Bridel, C., van Wieringen, W. N., Zetterberg, H., Tijms, B. M., Teunissen, CE., and the NFL Group. (2019). Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta-analysis. JAMA Neurol. 76, 1035–1048. doi: 10.1001/jamaneurol.2019.1534
Chen, K., Ho, T. S., Lin, G., Tan, K. L., Rasband, M. N., and Bellen, H. J. (2016). Loss of Frataxin activates the iron/sphingolipid/PDK1/Mef2 pathway in mammals. Elife 5:e20732. doi: 10.7554/eLife.20732
Clay, A., Obrochta, K. M., Soon, R. K. Jr., Russell, C. B., and Lynch, D. R. (2020). Neurofilament light chain as a potential biomarker of disease status in Friedreich ataxia. J. Neurol. 267, 2594–2598. doi: 10.1007/s00415-020-09868-3
Colin, F., Martelli, A., Clemancey, M., Latour, J. M., Gambarelli, S., Zeppieri, L., et al. (2013). Ollagnier de Choudens S. Mammalian frataxin controls sulfur production and iron entry during de novo Fe4S4 cluster assembly. J. Am. Chem. Soc. 135, 733–40. doi: 10.1021/ja308736e
Cotticelli, M. G., Xia, S., Lin, D., Lee, T., Terrab, L., Wipf, P., et al. (2019). Ferroptosis as a novel therapeutic target for friedreich's ataxia. J. Pharmacol. Exp. Ther. 369, 47–54. doi: 10.1124/jpet.118.252759
DeBrosse, C., Nanga, R. P. R., Wilson, N., D'Aquilla, K., Elliott, M., Hariharan, H., et al. (2016). Muscle oxidative phosphorylation quantitation using creatine chemical exchange saturation transfer (CrCEST) MRI in mitochondrial disorders. JCI Insight 1:e88207. doi: 10.1172/jci.insight.88207
Del Prete, E., Beatino, M. F., Campese, N., Giampietri, L., Siciliano, G., Ceravolo, R., et al. (2020). Fluid candidate biomarkers for Alzheimer's disease: a precision medicine approach. J. Pers Med. 10:221. doi: 10.3390/jpm10040221
Delatycki, M. B., and Bidichandani, S. I. (2019). Friedreich ataxia- pathogenesis and implications for therapies. Neurobiol. Dis. 132:104606. doi: 10.1016/j.nbd.2019.104606
Forgrave, L. M., Ma, M., Best, J. R., and DeMarco, M. L. (2019). The diagnostic performance of neurofilament light chain in CSF and blood for Alzheimer's disease, frontotemporal dementia, and amyotrophic lateral sclerosis: a systematic review and meta-analysis. Alzheimer's Dement. 11, 730–743. doi: 10.1016/j.dadm.2019.08.009
Friedman, L. S., Schadt, K. A., Regner, S. R., Mark, G. E., Lin, K. Y., Sciascia, T., et al. (2013). Elevation of serum cardiac troponin I in a cross-sectional cohort of asymptomatic subjects with Friedreich ataxia. Int. J. Cardiol. 167, 1622–1624. doi: 10.1016/j.ijcard.2012.04.159
Harding, I., Lynch, D. R., Koeppen, A., and Pandolfo, M. (2020). Central nervous system therapeutic targets in friedreich ataxia. Hum. Gene Ther. 31, 1226–1236. doi: 10.1089/hum.2020.264
Havton, L. A., and Kellerth, J. O. (2001). Neurofilamentous hypertrophy of intramedullary axonal arbors in intact spinal motoneurons undergoing peripheral sprouting. J. Neurocytol. 30, 917–926. doi: 10.1023/A:1020669201697
Hayer, S. N., Liepelt, I., Barro, C., Wilke, C., Kuhle, J., Martus, P., et al. (2020). NfL and pNfH are increased in Friedreich's ataxia. J. Neurol. 267, 1420–1430. doi: 10.1007/s00415-020-09722-6
Hsieh, S. T., Kidd, G. J., Crawford, T. O., Xu, Z., Lin, W. M., Trapp, B. D., et al. (1994). Regional modulation of neurofilament organization by myelination in normal axons. J. Neurosci. 14, 6392–6401. doi: 10.1523/JNEUROSCI.14-11-06392.1994
Koeppen, A. H., Becker, A. B., Qian, J., and Feustel, P. J. (2017b). Friedreich ataxia: hypoplasia of spinal cord and dorsal root ganglia. J. Neuropathol. Exp. Neurol. 76, 101–108. doi: 10.1093/jnen/nlw111
Koeppen, A. H., Becker, A. B., Qian, J., Gelman, B. B., and Mazurkiewicz, J. E. (2017a). Friedreich ataxia: developmental failure of the dorsal root entry zone. J. Neuropathol. Exp. Neurol. 76, 969–977. doi: 10.1093/jnen/nlx087
Kurochkina, N., Bhaskar, M., Yadav, S. P., and Pant, H. C. (2018). Phosphorylation, dephosphorylation, and multiprotein assemblies regulate dynamic behavior of neuronal cytoskeleton: a mini-review. Front. Mol. Neurosci. 11:373. doi: 10.3389/fnmol.2018.00373
Legrand, L., Maupain, C., Monin, M. L., Ewenczyk, C., Isnard, R., Alkouri, R., et al. (2020). Significance of NT-proBNP and high-sensitivity troponin in friedreich ataxia. J. Clin. Med. 9:1630. doi: 10.3390/jcm9061630
Li, Y., Polak, U., Bhalla, A. D., Lin, K., Shen, J., Farmer, J., et al. (2015). Excision of expanded GAA repeats alleviates the molecular phenotype of friedreich's ataxia. Mol. Ther. 23, 1055–1065. doi: 10.1038/mt.2015.41
Liu, D., Liu, Z., Liu, H., Li, H., Pan, X., and Li, Z. (2016). Brain-derived neurotrophic factor promotes vesicular glutamate transporter 3 expression and neurite outgrowth of dorsal root ganglion neurons through the activation of the transcription factors Etv4 and Etv5. Brain Res. Bull. 121, 215–226. doi: 10.1016/j.brainresbull.2016.02.010
Lodi, R., Hart, P. E., Rajagopalan, B., Taylor, D. J., Crilley, J. G., Bradley, J. L., et al. (2001). Antioxidant treatment improves in vivo cardiac and skeletal muscle bioenergetics in patients with Friedreich's ataxia. Ann. Neurol. 49, 590–596. doi: 10.1002/ana.1001
Lynch, D. R., Chin, M. P., Delatycki, M. B., Subramony, S. H., Corti, M., Hoyle, J. C., et al. (2021). Safety and efficacy of omaveloxolone in friedreich ataxia (MOXIe study). Ann. Neurol. 89, 212–225. doi: 10.1002/ana.25934
Lynch, D. R., Farmer, J., Hauser, L., Blair, I. A., Wang, Q. Q., Mesaros, C., et al. (2018). Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 6, 15–26. doi: 10.1002/acn3.660
Lynch, D. R., Regner, S. R., Schadt, K. A., Friedman, L. S., Lin, K. Y., and St John Sutton, M. G. (2012). Management and therapy for cardiomyopathy in Friedreich's ataxia. Expert. Rev. Cardiovasc. Ther. 10, 767–777. doi: 10.1586/erc.12.57
Marty, B., Naeije, G., Bourguignon, M., Wens, V., Jousmäki, V., Lynch, D. R., et al. (2019). Evidence for genetically determined degeneration of proprioceptive tracts in Friedreich ataxia. Neurology 93, e116–e124. doi: 10.1212/WNL.0000000000007750
Milo, R., Korczyn, A. D., Manouchehri, N., and Stüve, O. (2020). The temporal and causal relationship between inflammation and neurodegeneration in multiple sclerosis. Mult. Scler. 26, 876–886. doi: 10.1177/1352458519886943
Naeije, G., Bourguignon, M., Wens, V., Marty, B., Goldman, S., Hari, R., et al. (2020). Electrophysiological evidence for limited progression of the proprioceptive impairment in Friedreich ataxia. Clin. Neurophysiol. 131, 574–576. doi: 10.1016/j.clinph.2019.10.021
Navarro, J. A., Ohmann, E., Sanchez, D., Botella, J. A., Liebisch, G., Moltó, M. D., et al. (2010). Altered lipid metabolism in a Drosophila model of Friedreich's ataxia. Hum. Mol. Genet. 19, 2828–2840. doi: 10.1093/hmg/ddq183
Obis, È., Irazusta, V., Sanchís, D., Ros, J., and Tamarit, J. (2014). Frataxin deficiency in neonatal rat ventricular myocytes targets mitochondria and lipid metabolism. Free Radic. Biol. Med. 73, 21–33. doi: 10.1016/j.freeradbiomed.2014.04.016
Pastore, A., and Puccio, H. (2013). Frataxin: a protein in search for a function. J. Neurochem. 126(Suppl.1), 43–52. doi: 10.1111/jnc.12220
Pearson, R. C., Taylor, N., and Snyder, S. H. (1988). Tubulin messenger RNA: in situ hybridization reveals bilateral increases in hypoglossal and facial nuclei following nerve transection. Brain Res. 463, 245–249. doi: 10.1016/0006-8993(88)90396-4
Piguet, F., de Montigny, C., Vaucamps, N., Reutenauer, L., Eisenmann, A., and Puccio, H. (2018). Rapid and complete reversal of sensory ataxia by gene therapy in a novel model of friedreich ataxia. Mol. Ther. 26, 1940–1952. doi: 10.1016/j.ymthe.2018.05.006
Plehn, J. F., Hasbani, K., Ernst, I., Horton, K. D., Drinkard, B. E., and Di Prospero, N. A. (2018). The subclinical cardiomyopathy of friedreich's ataxia in a pediatric population. J. Card Fail. 24, 672–679. doi: 10.1016/j.cardfail.2017.09.012
Rezende, T. J. R., Martinez, A. R. M., Faber, I., Girotto Takazaki, K. A., Martins, M. P., de Lima, F. D., et al. (2019). Developmental and neurodegenerative damage in Friedreich's ataxia. Eur. J. Neurol. 26, 483–489. doi: 10.1111/ene.13843
Rodríguez-Pascau, L., Britti, E., Calap-Quintana, P., Dong, Y. N., Vergara, C., Delaspre, F., et al. (2021). PPAR gamma agonist leriglitazone improves frataxin-loss impairments in cellular and animal models of Friedreich Ataxia. Neurobiol. Dis. 148:105162. doi: 10.1016/j.nbd.2020.105162
Rötig, A., de Lonlay, P., Chretien, D., Foury, F., Koenig, M., Sidi, D., et al. (1997). Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 17, 215–217. doi: 10.1038/ng1097-215
Selvadurai, L. P., Harding, I. H., Corben, L. A., Stagnitti, M. R., Storey, E., Egan, G. F., et al. (2016). Cerebral and cerebellar grey matter atrophy in Friedreich ataxia: the IMAGE-FRDA study. J. Neurol. 263, 2215–2223. doi: 10.1007/s00415-016-8252-7
Strawser, C., Schadt, K., Hauser, L., McCormick, A., Wells, M., Larkindale, J., et al. (2017). Pharmacological therapeutics in Friedreich ataxia: the present state. Expert. Rev. Neurother. 17, 895–907. doi: 10.1080/14737175.2017.1356721
Strawser, C., Schadt, K., and Lynch, D. R. (2014). ataxia. Expert. Rev. Neurother. 14, 949–957. doi: 10.1586/14737175.2014.939173
Tamarit, J., Obis, È., and Ros, J. (2016). Oxidative stress and altered lipid metabolism in Friedreich ataxia. Free Radic. Biol. Med. 100, 138–146. doi: 10.1016/j.freeradbiomed.2016.06.007
Thebault, S., Booth, R. A., and Freedman, M. S. (2020). Blood neurofilament light chain: the neurologist's troponin? Biomedicines 8:523. doi: 10.3390/biomedicines8110523
Toth, C., Shim, S. Y., Wang, J., Jiang, Y., Neumayer, G., Belzil, C., et al. (2008). Ndel1 promotes axon regeneration via intermediate filaments. PLoS ONE 3:e2014. doi: 10.1371/journal.pone.0002014
Tsou, A. Y., Paulsen, E. K., Lagedrost, S. J., Perlman, S. L., Mathews, K. D., Wilmot, G. R., et al. (2011). Mortality in Friedreich ataxia. J. Neurol. Sci. 307, 46–913. doi: 10.1016/j.jns.2011.05.023
Turchi, R., Tortolici, F., Guidobaldi, G., Iacovelli, F., Falconi, M., Rufini, S., et al. (2020). Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 11:51. doi: 10.1038/s41419-020-2347-x
Wang, H., Wang, W., Shi, H., Han, L., and Pan, P. (2020). Cerebrospinal fluid and blood levels of neurofilament light chain in Parkinson disease: a protocol for systematic review and meta-analysis. Medicine. 99:e21458. doi: 10.1097/MD.0000000000021458
Yin, F., Men, C., Lu, R., Li, L., Zhang, Y., Chen, H., et al. (2014). Bone marrow mesenchymal stem cells repair spinal cord ischemia/reperfusion injury by promoting axonal growth and anti-autophagy. Neural. Regent Res. 9, 1665–1671. doi: 10.4103/1673-5374.141801
Zeitlberger, A. M., Thomas-Black, G., Garcia-Moreno, H., Foiani, M., Heslegrave, A. J., Zetterberg, H., et al. (2018). Plasma markers of neurodegeneration are raised in friedreich's ataxia. Front. Cell Neurosci. 12:366. doi: 10.3389/fncel.2018.00366
Zesiewicz, T., Heerinckx, F., De Jager, R., Omidvar, O., Kilpatrick, M., Shaw, J., et al. (2018a). Randomized, clinical trial of RT001: early signals of efficacy in Friedreich's ataxia. Mov. Disord. 33, 1000–1005. doi: 10.1002/mds.27353
Keywords: Ataxia, biomarker, neurodegenerative, clinical trial, axon
Citation: Frempong B, Wilson RB, Schadt K and Lynch DR (2021) The Role of Serum Levels of Neurofilament Light (NfL) Chain as a Biomarker in Friedreich Ataxia. Front. Neurosci. 15:653241. doi: 10.3389/fnins.2021.653241
Received: 14 January 2021; Accepted: 05 February 2021;
Published: 02 March 2021.
Edited by:
Isabella Zanella, University of Brescia, ItalyReviewed by:
Elisabetta Indelicato, Medizinische Universität Innsbruck, AustriaCopyright © 2021 Frempong, Wilson, Schadt and Lynch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David R. Lynch, bHluY2hkQHBlbm5tZWRpY2luZS51cGVubi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.