 Shu Zhao
Shu Zhao Zhiwei Bao1
Zhiwei Bao1 Ming D. Li
Ming D. Li Zhongli Yang
Zhongli Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurosci. , 18 June 2021
Sec. Neurogenomics
Volume 15 - 2021 | https://doi.org/10.3389/fnins.2021.645998
Background: Major depressive disorder (MDD) is a global health challenge that impacts the quality of patients’ lives severely. The disorder can manifest in many forms with different combinations of symptoms, which makes its clinical diagnosis difficult. Robust biomarkers are greatly needed to improve diagnosis and to understand the etiology of the disease. The main purpose of this study was to create a predictive model for MDD diagnosis based on peripheral blood transcriptomes.
Materials and Methods: We collected nine RNA expression datasets for MDD patients and healthy samples from the Gene Expression Omnibus database. After a series of quality control and heterogeneity tests, 302 samples from six studies were deemed suitable for the study. R package “MetaOmics” was applied for systematic meta-analysis of genome-wide expression data. Receiver operating characteristic (ROC) curve analysis was used to evaluate the diagnostic effectiveness of individual genes. To obtain a better diagnostic model, we also adopted the support vector machine (SVM), random forest (RF), k-nearest neighbors (kNN), and naive Bayesian (NB) tools for modeling, with the RF method being used for feature selection.
Results: Our analysis revealed six differentially expressed genes (AKR1C3, ARG1, KLRB1, MAFG, TPST1, and WWC3) with a false discovery rate (FDR) < 0.05 between MDD patients and control subjects. We then evaluated the diagnostic ability of these genes individually. With single gene prediction, we achieved a corresponding area under the curve (AUC) value of 0.63 ± 0.04, 0.67 ± 0.07, 0.70 ± 0.11, 0.64 ± 0.08, 0.68 ± 0.07, and 0.62 ± 0.09, respectively, for these genes. Next, we constructed the classifiers of SVM, RF, kNN, and NB with an AUC of 0.84 ± 0.09, 0.81 ± 0.10, 0.73 ± 0.11, and 0.83 ± 0.09, respectively, in validation datasets, suggesting that the SVM classifier might be superior for constructing an MDD diagnostic model. The final SVM classifier including 70 feature genes was capable of distinguishing MDD samples from healthy controls and yielded an AUC of 0.78 in an independent dataset.
Conclusion: This study provides new insights into potential biomarkers through meta-analysis of GEO data. Constructing different machine learning models based on these biomarkers could be a valuable approach for diagnosing MDD in clinical practice.
From 1990 to 2016, major depressive disorder (MDD) was one of the five leading causes of years lived with disability (GBD 2016 Disease and Injury Incidence and Prevalence Collaborators, 2017). Patients with MDD have a higher risk of diabetes, stroke, cardiovascular disease, obesity, cancer, cognitive impairment, and Alzheimer’s disease (Otte et al., 2016). Moreover, MDD is one of the most common disorders associated with suicidal behavior. It has been estimated that the risk of suicide in MDD patients is increased substantially (greater than 10 times) compared with the general population (Chesney et al., 2014).
Early diagnosis and appropriate treatment would undoubtedly reduce the incidence and mortality rate of MDD patients. However, like many other affective disorders, the complex etiology of MDD and the inevitable need for clinical judgment based on an individual’s medical history may cause a lack of reliability in diagnosis. More objective diagnostic methods thus are required. Previous studies have explored molecular biomarkers of MDD based on genomic, epigenetic, transcriptomic, and proteomic sources (Gururajan et al., 2016). Several types of molecules have been revealed with these approaches, which include mitochondrial DNA (Cai et al., 2015), small non-coding RNAs (Bocchio-Chiavetto et al., 2013), neurotransmitters (Belzeaux et al., 2010), neurotrophic and growth factors (Iga et al., 2007; Cattaneo et al., 2013), HPA axis-related molecules (Austin et al., 2003), and mediators of neuroinflammation (Cattaneo et al., 2013; Iacob et al., 2013). For example, several studies (Cattaneo et al., 2013; Iacob et al., 2013, 2014) reported increased expression of peripheral mRNAs for the pro-inflammatory cytokines interleukin (IL)-1α, IL-1β, IL-6, IL-8, IL-10, interferon (IFN)-γ, migration inhibitory factor (MIF), and tumor necrosis factor (TNF)-α in MDD patients compared with healthy control subjects. In addition, neuroimaging approaches, such as magnetic resonance imaging (MRI), electroencephalography (EEG), diffusion tensor imaging (DTI), near-infrared spectroscopy (NIRS), and molecular imaging (i.e., PET and SPECT) (Kang and Cho, 2020) have been used to discover biomarkers for diagnosis and treatment of MDD.
This study focused on the peripheral transcriptomic biomarkers, which have been described as “sentinels of disease” (Liew et al., 2006). Because of the complicated and heterogeneous pathogenesis of MDD, there existed some limitations in the study of relevant transcriptomic biomarkers. For example, studies on brain-derived neurotrophic factor (BDNF) had inconsistent results. The studies of Karege et al. (2005) and Piccinni et al. (2008) reported a reduction in BDNF in depressed patients compared with healthy persons, but in the study of Serra-Millàs et al. (2011), MDD patients showed higher plasma BDNF concentrations. However, in another study, researchers found no significant difference in plasma BDNF concentrations between MDD patients and control subjects (Bocchio-Chiavetto et al., 2010). Based on these facts, it appears that identification of reliable biomarkers for predicting diagnosis and treatment of MDD remains a challenge.
Therefore, in this study, meta-analysis was first performed to identify consistent biomarkers from different large-sample datasets. Although there are various meta-analyses of microarray data, they generally focus on one or a few genes; few have been developed for systematic integration of multiple microarray datasets (Sun et al., 2017; Li et al., 2018). The current commonly used meta-analysis method was proposed by Choi et al. (2003) and facilitates the detection of small but consistent expression changes and increases sensitivity and reliability. With this method, Chen et al. (2019) acquired gene expression data from eight commonly used in vitro macrophage models to perform a meta-analysis and identified consistently differentially expressed genes (DEGs) that have been implicated in inflammatory and metabolic processes. Forero et al. (2017) found MDD-related DEGs for blood, amygdala, cerebellum, anterior cingulate cortex, and prefrontal cortex regions based on GWES using meta-analysis. However, whether these DEGs would be useful as biomarkers has not been evaluated yet.
MDD is influenced by both genetics and environment where the transcriptome feature patterns or feature function patterns may represent disease subtypes, outcome prognosis, drug benefit prediction, or specific biological process. The machine learning (Anttila et al., 2018) approach has an advantage in recognizing subtle patterns in large and noisy datasets, which is particularly useful in the study of complex transcriptome data. For example, Xu et al. (2012) employed the SVM-RFE approach to select genes for prediction of breast cancer prognosis and discovered a 50-gene signature that yielded significantly higher accuracy than the widely used 70-gene signature (van ‘t Veer et al., 2002). By adopting fuzzy forests of transcriptome data, Ciobanu et al. (2020) found that the downregulated TFRC (transferrin receptor) can predict recurrent MDD with an accuracy of 63%.
The aim of this study was to identify potential transcriptional biosignatures that might be used for the diagnosis of MDD. Here, we applied meta-analysis to discover DEGs differing between MDD patients and healthy controls. Six significant DEGs with FDRs < 0.05 were investigated for their diagnostic capability. To obtain better diagnostic efficacy, we compared four ML models. Finally, an SVM prediction model consisting of 70 feature genes was constructed and validated by a reserved independent gene expression dataset.
MDD-related keywords were searched in the Medical Subject Headings(MeSH) library1. Then we conducted a systematic search in the GEO repository2 using the following search sentence: ((((((((MDD) OR major depressive disorders) OR depressive disorders) OR depressive syndromes) OR depression)) AND (((blood) OR peripheral blood) OR PB)) AND Homo sapiens [Organism]) AND Expression profiling by array [Filter]. A report was included in the analysis if the following criteria were satisfied: (1) used a case-control design; (2) patients did not have any diseases other than MDD; and (3) the patients were medication free. We finally obtained a total of 9 datasets (Figure 1).
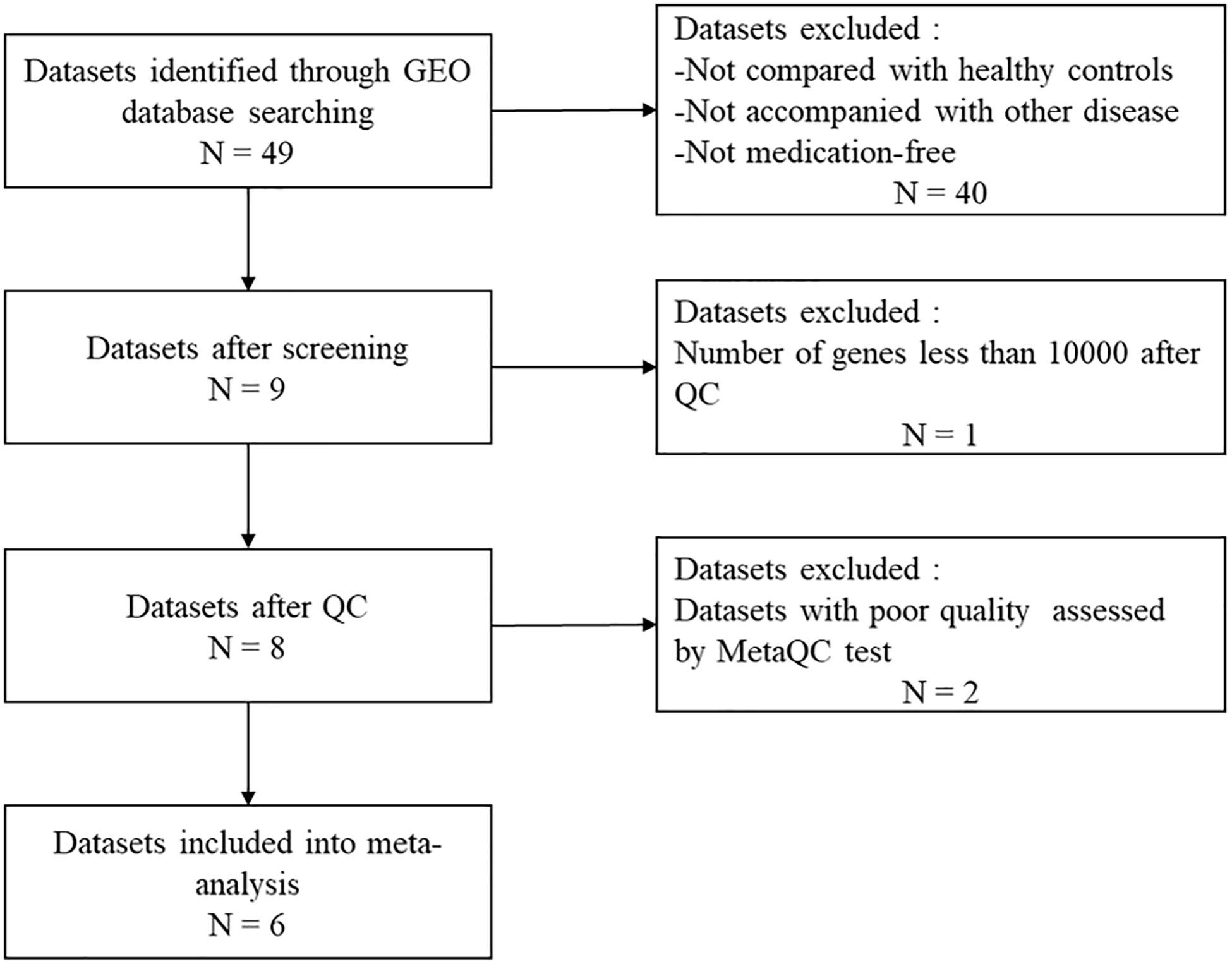
Figure 1. Workflow of data processing. GEO, Gene Expression Omnibus; QC, quality control.
Nine microarray datasets were retrieved from the GEO database: GSE98793, GSE19738, GSE38206, GSE52790, GSE39653, GSE76826, GSE58430, GSE32280, and GSE46743, with a sample size of 128, 67, 18, 22, 45, 22, 12, 16, and 160, respectively (Table 1).
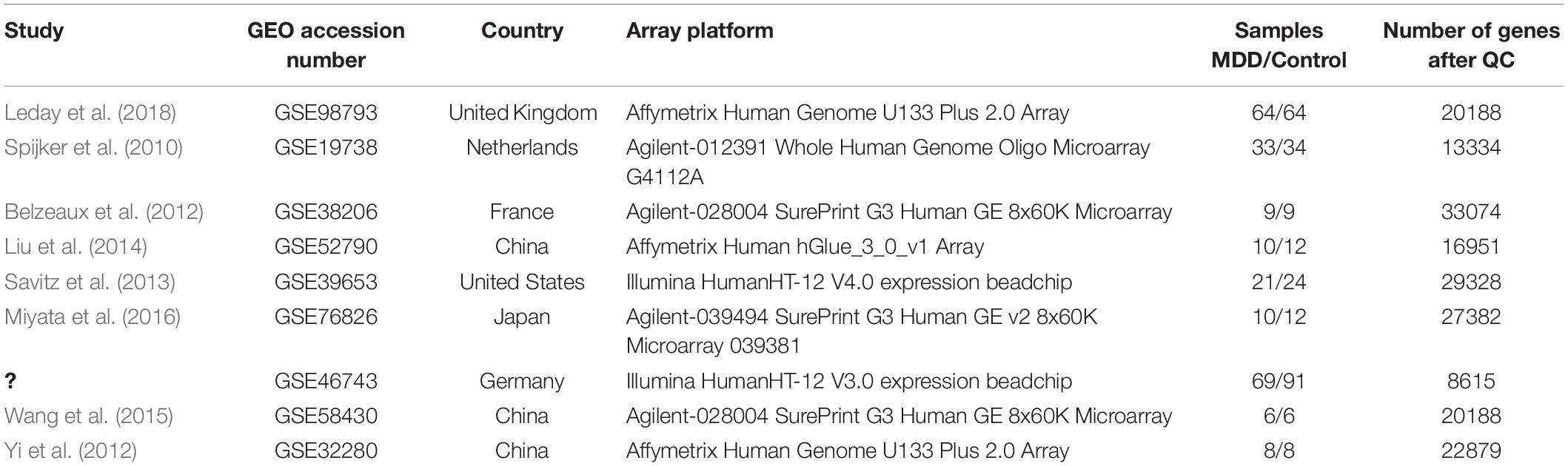
Table 1. Basic information of collected microarray datasets.
For the GSE98793, GSE52790, and GSE32280 datasets, based on the Affymetrix platform (Thermo Fisher Scientific, Inc., Waltham, MA, United States), the raw CEL data were downloaded; and the Robust Multi-Array Average (RMA) method and the ‘‘Oligo’’ package from BioConductor3 were used to normalize the data and annotate the probe information. For the GSE19738, GSE38206, GSE58430, and GSE76826 data, based on the Agilent platform (Agilent Technologies, Inc., Santa Clara, CA, United States), the quantile method was used to normalize the data. Annotation of the probe information was based on Agilent platform information. For the GSE39653 and GSE46743 datasets based on the Illumina platform (Illumina Inc., San Diego, CA, United States), the quantile method was used to normalize the data, and annotation of the probe information was based on Illumina platform information.
Normalized signal intensity data were imported into BRB-Array Tools (v. 4.5)4 for initial processing. We excluded those genes with more than 50% of the data missing. The most variable probe measured by inter-quartile range (IQR) was used to handle redundant probe sets that correspond to the same gene.
Meta-analysis of microarray data was carried out in “MetaOmics” based on R language (Ma et al., 2019), which includes three packages: MetaQC, MetaDE, and MetaPath. MetaQC (Kang et al., 2012) was used for the quality control of datasets before meta-analysis, and the MetaDE (Wang et al., 2012) package was used to identifying differentially expressed genes.
We used the following six quantitative quality control indexes to assess heterogeneity across different studies: internal homogeneity of co-expression structure among studies (IQC), external consistency of co-expression pattern with pathway database (EQC), accuracy of biomarker detection (AQCg), accuracy of enriched pathway detection (AQCp), consistency of differentially expressed genes (CQCg), and consistency of enriched pathway ranking (CQCp). Each QC index was defined as the minus log-transformed p-value from formal hypothesis testing in each QC criterion (Wang et al., 2012). Finally, standardized mean rank (SMR) was generated to assist decision making. In this study, datasets with SMR values > 5 were excluded from analysis (Esmaeili et al., 2020).
MetaDE was used for identifying differentially expressed genes, and the meta-analysis method used in the current study was developed by Choi et al. (2003). The change of gene expression was represented as “effect size,” a standardized index measuring the magnitude of a treatment or covariate effect. The effect sizes of different studies were combined to obtain an estimate of the overall mean. Herein, we applied the random effects model, and FDR correction was used to control for multiple testing. Finally, genes were considered significant at FDR < 0.05.
A total of 217,249 pairs of FIs were downloaded from Reactome (v. 20145; Croft et al., 2011). These pairwise relations were derived from datasets of protein–protein interactions in BioGrid (Chatr-Aryamontri et al., 2015), the Database of Interacting Proteins (Salwinski et al., 2004), the Human Protein Reference Database (Keshava Prasad et al., 2009), I2D (Brown and Jurisica, 2007), IntACT (Orchard et al., 2014), and MINT (Licata et al., 2012), as well as from gene co-expression data derived from multiple high-throughput techniques, including yeast two-hybrid assays, mass spectrometry pull-down experiments, and DNA microarrays (Wu et al., 2010). The above interaction information was imported into Cytoscape software (v. 3.2.16) to construct the FI network (Shannon et al., 2003). A spectral partition-based network clustering (Newman, 2006) was used to search for modules based on the FI network. A KEGG pathway enrichment analysis was used to analyze functions for each individual network module. We selected a size cutoff of 2 to filter out small network modules. An FDR value of < 0.05 was considered to represent significantly enriched processes or signaling pathways. Co-expression patterns of the genes in the same module were analyzed by using the Pearson correlation test.
Supplementary Figure 3 shows an overview of the proposed ML method involving feature extraction, selection, classification, and validation. R package ‘‘caret’’ (v. 6.0-847) was applied in the following steps.
The six datasets included in this study were divided into two parts: GSE98793 (Leday et al., 2018), GSE19738 (Spijker et al., 2010), GSE39653 (Savitz et al., 2013), GSE52790 (Liu et al., 2014), and GSE76826 (Miyata et al., 2016) were used as discovery datasets and GSE38206 (Belzeaux et al., 2012) as an independent validation dataset. We performed meta-analysis on discovery sets to identify DGEs between MDD patients and healthy controls. DEGs with p < 0.01 were considered potential biomarkers for further feature selection. Then, the dataset GSE98793 with the largest sample size was used as the training dataset in both feature screening and ML modeling. The other four datasets in discovery sets were used for internal validation.
We applied an RF algorithm to reduce the number of feature genes, with the following steps: (1) ranked genes in descending order based on their importance; (2) eliminated feature genes one by one according to the importance of each feature with the goal of producing a new feature set; and (3) repeated the above process with the new feature set. We used 10-fold cross-validation for verification and calculated the average accuracy value to assess the classification capability. Finally, the feature set with the highest average accuracy was selected for model construction.
The following ML algorithms, SVM (Cortes and Vapnik, 1995), RF (Tin Kam, 1998), kNN (Altman, 1992), and NB (Friedman et al., 1997), were used to build prediction models for gene expression data.
The aim of SVM algorithm is to identify a decision hyperplane that make the distance between the hyperplane and the instances that are closest to boundary is maximized. By introducing the concept of “soft margin” and using “kernel trick,” SVM performs well with linear indivisibility data (Boser et al., 1996). SVM with the radial basis function (RBF) kernel was used in this study and parameters “C” and “sigma” were returned.
Random Forest is an ensembles-learning algorithm forming with a series of decision trees. Each tree is developed from a subset from the training data. The class of the new instance is determined by using the majority vote of individual trees in the forest (Tin Kam, 1998). In this study, R package “randomForest” was used in the modeling and we tuned the parameter “mtry.”
In kNN, classification is based on the distance between the instances and an object is classified according to the status of its k nearest neighbors. R package “kknn” was used in the modeling, where we set distance = 2 (Euclidean Distance), and tuned the parameter “kmax.”
Naive Bayes is a statistical classification algorithm based on theBayes theorem. The core idea of Naive Bayes is, if the probability ofinstance x belonging to A is greater than the probability of belonging to B under some attribute conditions, it is said that instance x belongs to A. R package “klaR” was used in the modeling and output parameter “usekernel.”
Default values were used for other parameters in each model. To evaluate the overall performance of each model, leave-one-out cross-validation was performed. Details about the tested parameters and their corresponding test values for each model are provided in Supplementary Table 6.
We used the average AUC to assess the classification capability of each model. Finally, a model with the highest average AUC in validation sets was chosen.
To facilitate clinical application, we attempted to construct a model with fewer genes without affecting the accuracy of classification efficiency. We compared the classification ability of the model based on the average AUC value of discovery set. The criterion for determining the final model was that the model achieved the optimal average AUC value in the discovery sets.
The feature genes in the final determined SVM classifier were used to perform the supervised clustering of samples and extents of expression. The clustering results were visualized using a heatmap (Gu et al., 2016).
The information in the datasets is shown inTable 1. Dataset GSE46743 (Miyata et al., 2016) has a number of genes < 10,000 after QC was excluded. Eight eligible datasets were finally used for the following analysis, which consisted of a total of 161 MDD and 169 control samples.
The quality of the eight datasets was assessed utilizing “MetaQC.” Among the eight microarray datasets, six were included in the further meta-analysis for DEGs (Supplementary Figure 1 and Supplementary Table 1), and the other two studies (Yi et al., 2012; Wang et al., 2015) were excluded because of their lower quantitative quality control scores (SMR < 5). We then combined the other six datasets and obtained a total of 9,263 common genes that were used as input for the meta-analysis, which revealed 137 DEGs with p < 0.01. Of them, 66 were upregulated and 71 downregulated in MDD (Figure 2). A detailed list of these DEGs is given in Supplementary Table 2. Figure 2 shows the six most significant DEGs with FDRs < 0.05; they are tyrosylprotein sulfotransferase 1 (TPST1), arginase 1 (ARG1), killer cell lectin-like receptor B1 (KLRB1), WWC family member 3 (WWC3), aldo-keto reductase family 1 member C3 (AKR1C3), and MAF bZIP transcription factor G (MAFG). The forest plots of the six genes’ expression in different datasets are shown in Figure 3. These six genes were associated with immune process, inflammatory response, and hormonal metabolic process (Supplementary Table 3). In short, these results implied that these six biomarkers might play important roles in MDD.
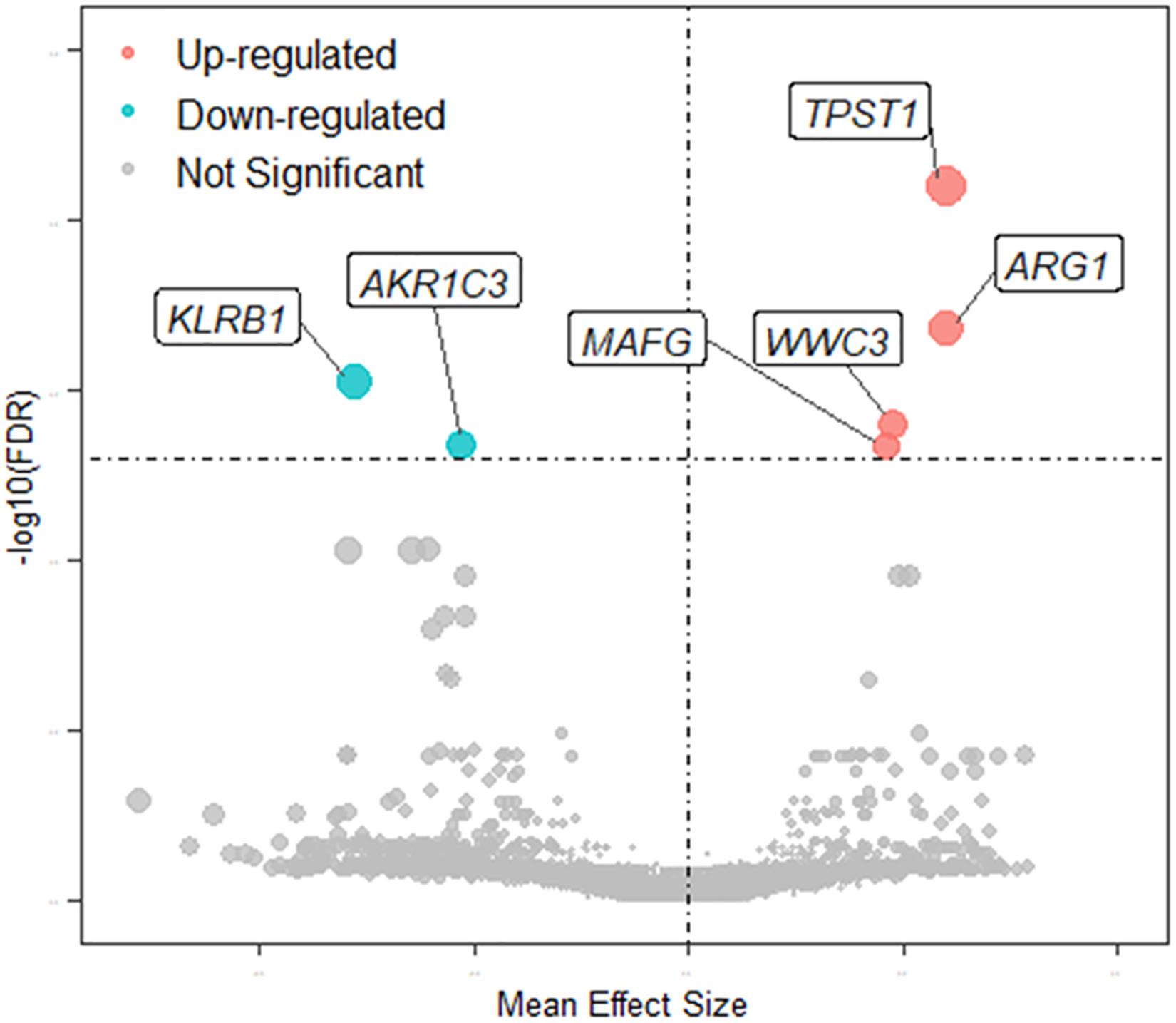
Figure 2. Volcano plot of MDD-related DEGs. Node colors define change direction in DEGs: red for upregulated genes, green for downregulated genes, and gray for not significant genes. Node size combines the effect size and FDR value: a larger node indicates that mean effect size of gene is large and FDR value is small.
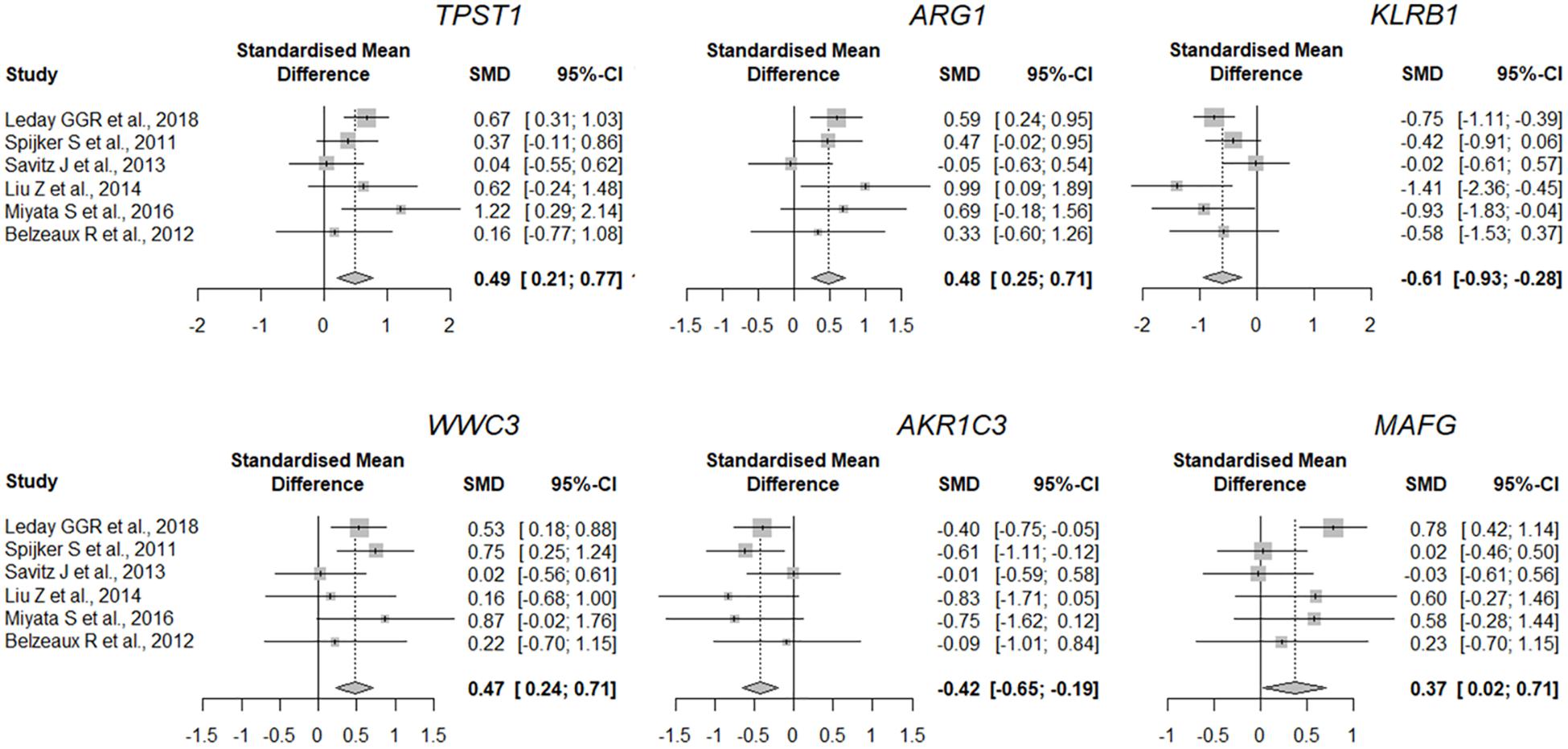
Figure 3. Expression of most significant DEGs between MDD and control group (a random-effects model) in different studies. Lines indicate 95% confidence intervals (CI), and the midpoint of each line is denoted by a square indicating the standardized mean difference (SMD) for each study. Diamond indicates overall SMD and 95% confidence interval.
By mapping 137 MDD-related DEGs to the FI data, we constructed an MDD-related FI network comprising 137 nodes, of which 103 were isolated and 34 were classified into seven clusters (Supplementary Figure 2A). A topographical analysis of the FI network revealed five modules ranging in size from three to eight genes (Supplementary Figure 2B). We next explored the potential co-expression of the DEGs in each module. There was a moderate to high positive correlation among the expression of most genes in Modules 0, 3, and 4 (Supplementary Figure 2C). In Module 1, there was a moderate to high positive correlation among MLKL, CEP63, and CSNK1E, a moderate negative correlation among DNAJC7, MLKL, and CSNK1E (Supplementary Figure 2C). Most genes in Module 2 showed a low correlation between each other. To understand how the 26 genes of the five modules were related to the molecular mechanisms of MDD, we performed a functional enrichment analysis of these modules based on pathway annotation (Supplementary Table 4). The enriched pathways of Modules 0 and 2 were related to some elements and events in transcription and translation, such as the ribosome, spliceosome, RNA degradation, or mRNA surveillance pathway. Similarly, genes in Module 3 were involved in transcriptional mis-regulation in cancers. Module 1 was related mainly to signaling pathways associated with the immune response, for example, antigen processing and presentation and the IL-17 signaling pathway. Neurodegenerative disease-related pathways in KEGG were enriched in Module 4, which included genes involved in Alzheimer, Huntington, and Parkinson diseases. In addition, metabolic pathways were enriched in Module 4. Together, this identified MDD-related FI network of dysregulated pathways could serve as a pool of novel functional module genes for future investigation in the diagnosis of MDD.
Next, we constructed single gene models to distinguish MDD patientsfrom healthy control subjects. We chose the six most significant DEGsand calculated the diagnostic ability of these genes with ROC curveanalysis. Table 2 shows the predicted resultsof single gene model in different datasets, represented by AUC values. The gene with the most significant expression difference, TPST1, had an average AUC value of 0.68 and a predictive ability of 0.82 in the dataset of Miyata et al. (2016), but only 0.62 in the dataset of Belzeaux et al. (2012). The gene with the better predictive potency was KLRB1, with an average AUC value of 0.70 and an SD of 0.11. The performance of WWC3 was the worst, with an average AUC of 0.63 and an SD of 0.04. These results indicated that the model developed with individual genes was not effective for diagnosis of MDD in clinics.
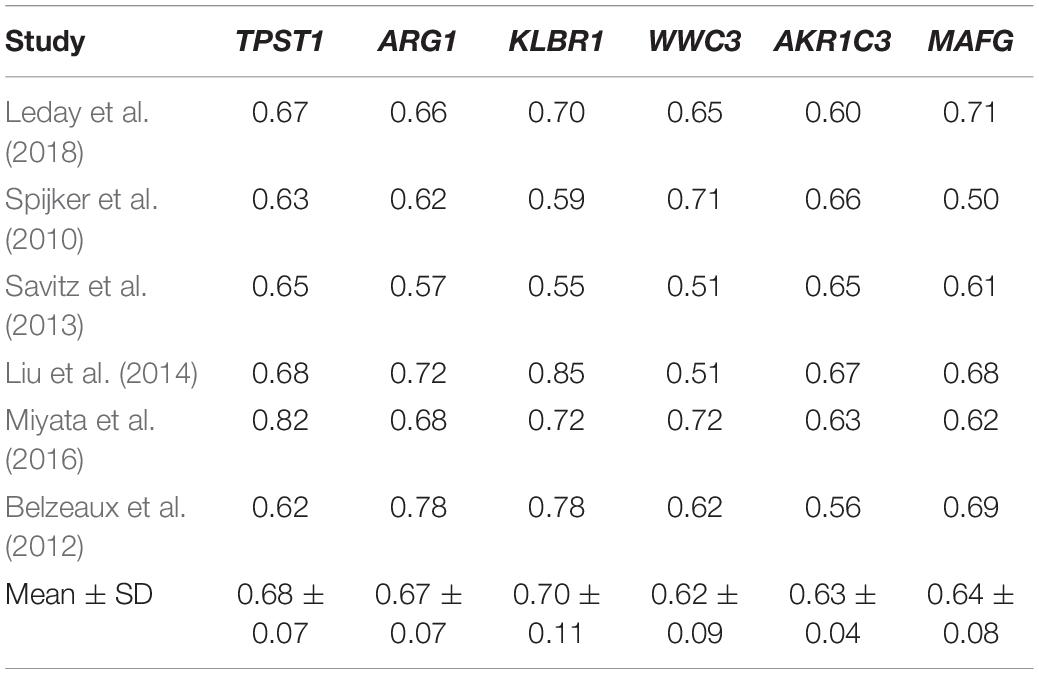
Table 2. AUC of single gene models for MDD diagnosis.
The above analyses indicated that the average AUCs of most single gene models were less than 0.70, suggesting that more efficient diagnostic models were necessary. We used the transcriptome data from the discovery sets for meta-analysis and obtained 114 DEGs (Supplementary Table 5) with p < 0.01 as input for feature screening. First, the feature set containing 108 genes was selected as it had the highest accuracy in the training dataset (Supplementary Figure 4A). Based on this feature set, four ML classification methods were used for modeling and the parameters of each model were shown in Supplementary Table 6. All models yielded an average AUC > 0.7 in validation datasets (Table 3), with SVM producing the highest average AUC (0.84). We thus chose SVM as the final diagnostic model.
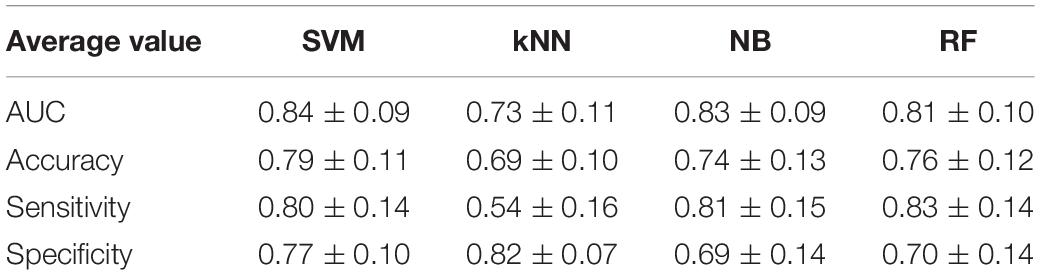
Table 3. Comparison of different models in the validation sets.
To facilitate clinical application, we attempted to construct a modelwith fewer genes. Feature genes ranked with an average AUC ofdiscovery datasets were picked at 10 intervals from the top 10 to number 108. As shown in Supplementary Figure 4B, the accuracy of the SVM classifier was improved with an increasing number of genes, and the average accuracy reached the top at 0.84 once 70 genes were selected. The SVM classifier was still able to distinguish MDD samples from the healthy controls in test datasets with an average AUC of 0.82, accuracy of 0.75, sensitivity of 0.78, and specificity of 0.74 (Table 4). In the independent dataset, the classifier achieved an AUC of 0.78 (Table 4), which was greatly better than the model with randomly selected 70 genes (Supplementary Table 9). Besides, we calculated positive predictive value (0.74 ± 0.06) and Matthews correlation coefficient (0.52 ± 0.14) in training and test data sets which also reflect a great performance of our model (Supplementary Table 8). The top six most significant genes mentioned above were all included in this SVM model (Supplementary Figure 5 and Supplementary Table 7). Compared with a single gene, the SVM model had better predictive performance (Figure 4).
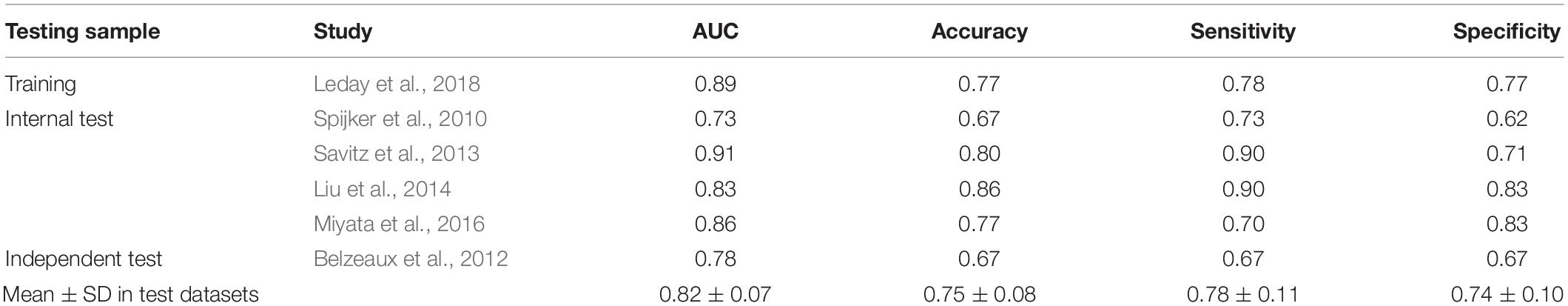
Table 4. Evaluation of classification effect of the SVM model.
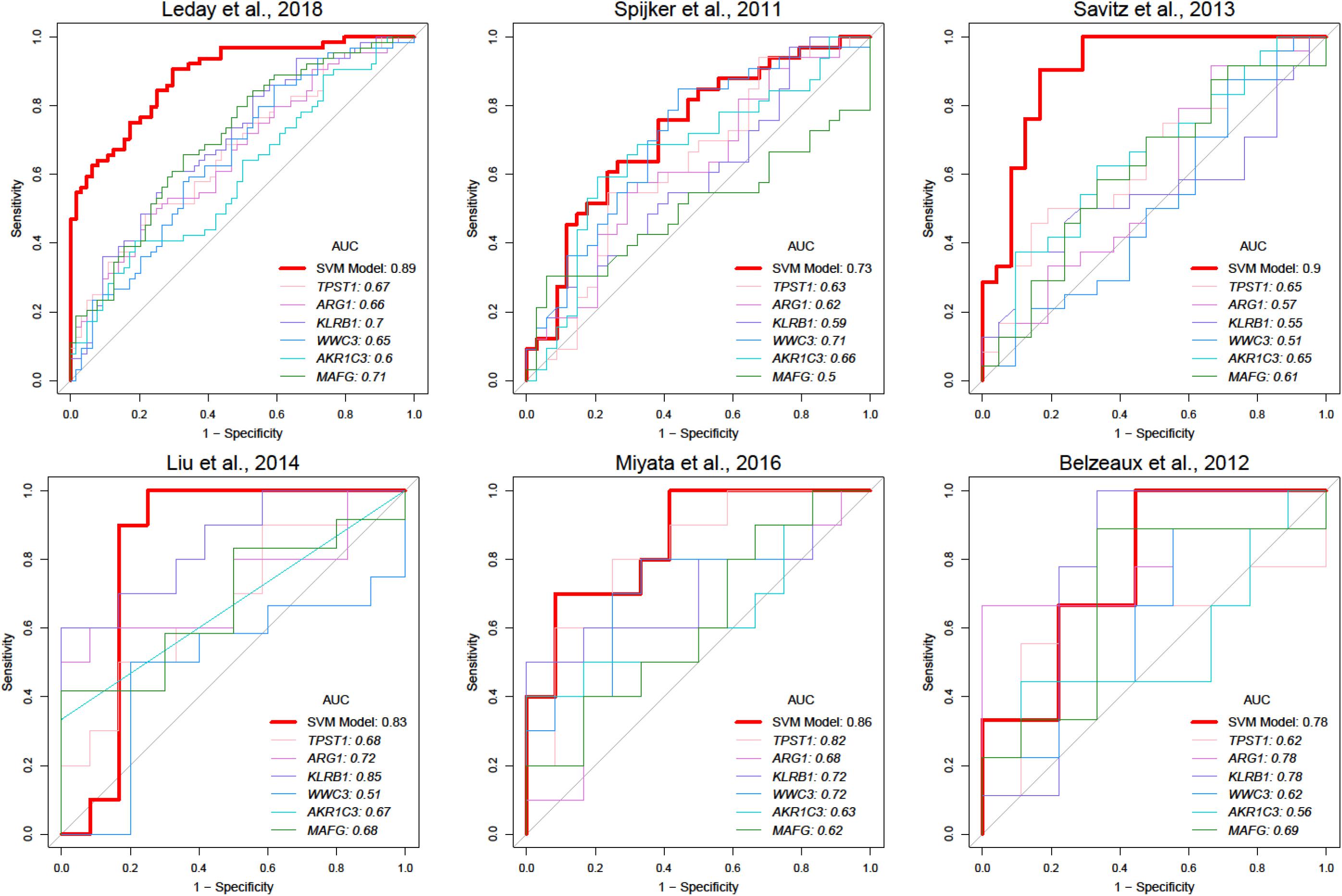
Figure 4. Comparison of prediction performance between SVM and single-gene models. Red lines represent ROC curves of SVM model in different studies, and lines with other colors represent ROC curves of various single gene models.
Although there have been numerous reports analyzing DEGs in MDD patients and healthy individuals, an exploration of diagnosis and etiology of MDD remains a challenge. To identify effective diagnostic biomarkers of MDD, in this study, we first conducted a meta-analysis of six studies, which revealed 137 DEGs with a p < 0.01. Then we identified functional module genes showing that these DEGs were involved in the processes of transcription and translation, inflammation, immune-related pathways, and neurodegenerative diseases. Six DEGs with FDR < 0.05 were investigated by ROC curve analysis for their potential to distinguish MDD patients from healthy controls. To improve predictive power, we applied four ML methods with RF being used for feature selection. Finally, we constructed an SVM model containing 70 feature genes and showed that it was superior to the single gene prediction model.
In the first part of the present study, we used meta-analysis to identify reliable DEGs as biomarkers. Six differentially expressed genes with FDRs < 0.05 were identified in MDD and healthy control subjects and used to calculate classification efficiency. Among them, the functions of KLRB1, ARG1, and TPST1 were associated with immune and inflammatory responses (Supplementary Table 2). It is known that inflammation and immunity play an important role in the etiology of depression (Liu et al., 2019). For example, individuals with autoimmune diseases and severe infections are more likely to have depression (Malhi and Mann, 2018). Peripheral cytokine concentrations have been linked to brain function, wellbeing, and cognition (Bollen et al., 2017). Therefore, we speculate that KLRB1, ARG1, and TPST1 influence the occurrence and progression of MDD by participating in the immune or inflammation pathways. AKR1C3 has the activity of aldo-keto reductase (nicotinamide adenine nucleotide phosphate; NADP), which plays an important role in interconversion of androgens, estrogens, and progestins to their cognate inactive metabolites (Supplementary Table 2). Over the past several years, both clinical and preclinical studies have established a strong link between sex hormones and depression (Thériault and Perreault, 2019). For example, in women, the prevalence of depression correlates with changes in hormonal fluctuations, such as puberty, prior to menstruation, during the postpartum period, and after the onset of menopause (Thériault and Perreault, 2019). Even if these differentially expressed genes are biologically related to the MDD process, ROC analysis showed that the diagnostic AUC value of a single gene was generally < 0.7, and, importantly, there existed great differences in the effectiveness of a diagnostic model developed on the basis of individual genes among different datasets or studies. These results strongly indicated that including only the top DEGs in a diagnostic model might lack a reliable distinguishing effect in all datasets, which has been one of the reasons transcriptome DEGs are currently difficult to use as MDD biomarkers.
Further in this study, four ML classifiers of screened feature genes were constructed. Among them, SVM produced better classification results. We then reduced the model size to 70 feature genes and found that the SVM model of these genes displayed acceptable performance in distinguishing MDD from control samples of discovery sets. The verification on an independent dataset exhibited an AUC of 0.78 and an accuracy of 0.67. The results showed that the predictive power of the model was superior to that of a single gene as an indicator of classification. The application of ML in some large-scale omics data is a popular undertaking, such as in cancer genomics (Zhou et al., 2018) and radiomics (Huang et al., 2018; Ding et al., 2019). In the depression-related studies, ML algorithms also have been used to find radiomics (Rubin-Falcone et al., 2018) and video- (Schultebraucks et al., 2020) and audio-based markers (Schultebraucks et al., 2020) for diagnosis or medication prediction. However, unlike cancers, in some studies of peripheral transcriptome biomarkers of MDD, it was difficult to find a relatively credible database such as TCGA (The Cancer Genome Atlas) as an independent validation. For example, one study constructed an elastic net model using immune-inflammatory signature to classify MDD and BD; it achieved high accuracy (AUC = 97%), but the result lacked the independent validation to acquire a true diagnostic effect of these biomarkers.
In contrast to other studies on MDD biomarkers, the blood transcriptomic data used for modeling here were from multiple studies, and the validation data were completely independent from the training data. Besides, we identified feature biomarkers by using meta-analysis followed by RF. Therefore, our strategy could help in identifying consistent and reliable biomarkers from different studies and was more conducive to the evaluation of the generalization ability of the model.
This study has several limitations. First, although existing data of MDD and healthy control subjects were used, it is unknown at this stage how much data are required to establish a reliable predictive model, which can be answered through empirical investigation. Second, the biomarkers used in this study were based on statistical significance, although the biological conception of these genes could also be considered. For example, our subsequent research can build a diagnostic model to refer to genes in the module of the PPI network. Third, because as many data as possible were used for modeling to improve the accuracy, independent validation data are needed in the future.
MDD occurs in a heterogeneous patient population, which makes accurate diagnosis a challenge. To address this challenge, we conducted meta-analysis of six datasets and found significant DEGs. The TPST1, ARG1, KLRB1, WWC3, AKR1C3, MAF, and MAFG genes were highlighted as potential feature genes influencing MDD. In addition, we constructed four ML models and chose SVM as a diagnostic model for MDD. We finally obtained an SVM diagnostic model containing 70 feature genes with an average AUC of 0.83, whose diagnostic effectiveness was superior to that of a single gene. Together, this study provided some markers that may be prospective precise diagnosis targets for MDD. Besides, this study provides new insight into how meta-analysis and ML can be used to find relatively objective transcriptional markers for complex mental diseases.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
SZ, ZB, XZ, and MX collected the data, conducted the analysis, and drafted the manuscript. ML provided resources and revised the manuscript. ZY administered project and revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the China Precision Medicine Initiative (2016YFC0906300), Research Center for Air Pollution and Health of Zhejiang University and State Key Laboratory for Diagnosis and Treatment of Infectious Diseases of the First Affiliated Hospital of Zhejiang University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank David Bronson and Judith Gunn Bronson for their editing of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2021.645998/full#supplementary-material
Supplementary Figure 1 | Quality control results of merged datasets from eight microarray profiles. SMR, standardized mean rank.
Supplementary Figure 2 | Biological functional of protein–protein interaction network constructed using MDD-related DEGs. (A) Functional interaction (FI) network. Node sizes represent p-values of FI network, large for low value and small for high. Node colors define mean effect size in MDD-related DEGs, ranging from red for high expression to blue for low expression. (B) Five generated network modules incorporate 26 genes, which are shown in different colors in network modules. (C) Correlation heat map of genes in different modules.
Supplementary Figure 3 | Workflow of machine learning. TP, true positive; TN, true negative; FP, false positive; FN, false negative.
Supplementary Figure 4 | (A) The accuracy of gene selection procedure using RF. (B) Average AUC when modeling with different numbers of features. (C) ROC in model parameter calculation procedure using SVM.
Supplementary Figure 5 | Clustering heatmap of feature genes and samples in training dataset. Color gradient from purple to red represents changes in expression from low to high. Bars represent samples (orange refers to MDD samples, blue to control samples).
Altman, N. S. (1992). An introduction to kernel and nearest-neighbor nonparametric regression. Am. Stat. 46, 175–185. doi: 10.1080/00031305.1992.10475879
Anttila, V., Bulik-Sullivan, B., Finucane, H. K., Walters, R. K., Bras, J., Duncan, L., et al. (2018). Analysis of shared heritability in common disorders of the brain. Science 360:eaa8757. doi: 10.1126/science.aap8757
Arloth, J., Bogdan, R., Weber, P., Frishman, G., Menke, A., Wagner, K. V., et al. (2015). Genetic differences in the immediate transcriptome response to stress predict risk-related brain function and psychiatric disorders. Neuron. 86, 1189–1202. doi: 10.1016/j.neuron.2015.05.034
Austin, M. C., Janosky, J. E., and Murphy, H. A. (2003). Increased corticotropin-releasing hormone immunoreactivity in monoamine-containing pontine nuclei of depressed suicide men. Mol. Psychiatry 8, 324–332. doi: 10.1038/sj.mp.4001250
Belzeaux, R., Bergon, A., Jeanjean, V., Loriod, B., Formisano-Tréziny, C., Verrier, L., et al. (2012). Responder and nonresponder patients exhibit different peripheral transcriptional signatures during major depressive episode. Transl. Psychiatry 2:e185. doi: 10.1038/tp.2012.112
Belzeaux, R., Formisano-Tréziny, C., Loundou, A., Boyer, L., Gabert, J., Samuelian, J. C., et al. (2010). Clinical variations modulate patterns of gene expression and define blood biomarkers in major depression. J. Psychiatr. Res. 44, 1205–1213. doi: 10.1016/j.jpsychires.2010.04.011
Bocchio-Chiavetto, L., Bagnardi, V., Zanardini, R., Molteni, R., Nielsen, M. G., Placentino, A., et al. (2010). Serum and plasma BDNF levels in major depression: a replication study and meta-analyses. World J. Biol. Psychiatry 11, 763–773. doi: 10.3109/15622971003611319
Bocchio-Chiavetto, L., Maffioletti, E., Bettinsoli, P., Giovannini, C., Bignotti, S., Tardito, D., et al. (2013). Blood microRNA changes in depressed patients during antidepressant treatment. Eur. Neuropsychopharmacol. 23, 602–611. doi: 10.1016/j.euroneuro.2012.06.013
Bollen, J., Trick, L., Llewellyn, D., and Dickens, C. (2017). The effects of acute inflammation on cognitive functioning and emotional processing in humans: a systematic review of experimental studies. J. Psychosom. Res. 94, 47–55. doi: 10.1016/j.jpsychores.2017.01.002
Boser, B., Guyon, I., and Vapnik, V. (1996). “A Training Algorithm for Optimal Margin Classifier,” in Proceedings of the Fifth Annual Workshop on Computational Learning Theory, Pittsburgh.
Brown, K. R., and Jurisica, I. (2007). Unequal evolutionary conservation of human protein interactions in interologous networks. Genome Biol. 8:R95. doi: 10.1186/gb-2007-8-5-r95
Cai, N., Li, Y., Chang, S., Liang, J., Lin, C., Zhang, X., et al. (2015). Genetic control over mtDNA and its relationship to major depressive disorder. Curr. Biol. 25, 3170–3177. doi: 10.1016/j.cub.2015.10.065
Cattaneo, A., Gennarelli, M., Uher, R., Breen, G., Farmer, A., Aitchison, K. J., et al. (2013). Candidate genes expression profile associated with antidepressants response in the GENDEP study: differentiating between baseline ‘predictors’ and longitudinal ‘targets’. Neuropsychopharmacology 38, 377–385. doi: 10.1038/npp.2012.191
Chatr-Aryamontri, A., Breitkreutz, B. J., Oughtred, R., Boucher, L., Heinicke, S., Chen, D., et al. (2015). The BioGRID interaction database: 2015 update. Nucleic Acids Res. 43, D470–D478. doi: 10.1093/nar/gku1204
Chen, H. J., Li Yim, A. Y. F., Griffith, G. R., de Jonge, W. J., Mannens, M., Ferrero, E., et al. (2019). Meta-analysis of in vitro-differentiated macrophages identifies transcriptomic signatures that classify disease macrophages in vivo. Front. Immunol. 10:2887. doi: 10.3389/fimmu.2019.02887
Chesney, E., Goodwin, G. M., and Fazel, S. (2014). Risks of all-cause and suicide mortality in mental disorders: a meta-review. World Psychiatry 13, 153–160. doi: 10.1002/wps.20128
Choi, J. K., Yu, U., Kim, S., and Yoo, O. J. (2003). Combining multiple microarray studies and modeling interstudy variation. Bioinformatics 19(Suppl. 1), i84–i90. doi: 10.1093/bioinformatics/btg1010
Ciobanu, L. G., Sachdev, P. S., Trollor, J. N., Reppermund, S., Thalamuthu, A., Mather, K. A., et al. (2020). Downregulated transferrin receptor in the blood predicts recurrent MDD in the elderly cohort: a fuzzy forests approach. J. Affect. Disord. 267, 42–48. doi: 10.1016/j.jad.2020.02.001
Cortes, C., and Vapnik, V. (1995). Support-vector networks. Mach. Learn. 20, 273–297. doi: 10.1007/BF00994018
Croft, D., O’Kelly, G., Wu, G., Haw, R., Gillespie, M., Matthews, L., et al. (2011). Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 39, D691–D697. doi: 10.1093/nar/gkq1018
Ding, X., Yue, X., Zheng, R., Bi, C., Li, D., and Yao, G. (2019). Classifying major depression patients and healthy controls using EEG, eye tracking and galvanic skin response data. J. Affect. Disord. 251, 156–161. doi: 10.1016/j.jad.2019.03.058
Esmaeili, S., Mehrgou, A., Kakavandi, N., and Rahmati, Y. (2020). Exploring Kawasaki disease-specific hub genes revealing a striking similarity of expression profile to bacterial infections using weighted gene co-expression network analysis (WGCNA) and co-expression modules identification tool (CEMiTool): an integrated bioinformatics and experimental study. Immunobiology 225:151980. doi: 10.1016/j.imbio.2020.151980
Forero, D. A., Guio-Vega, G. P., and González-Giraldo, Y. (2017). A comprehensive regional analysis of genome-wide expression profiles for major depressive disorder. J. Affect. Disord. 218, 86–92. doi: 10.1016/j.jad.2017.04.061
Friedman, N., Geiger, D., and Goldszmidt, M. (1997). Bayesian network classifiers. Mach. Learn. 29, 131–163. doi: 10.1023/A:1007465528199
GBD 2016 Disease and Injury Incidence and Prevalence Collaborators (2017). Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1211–1259. doi: 10.1016/s0140-6736(17)32154-2
Gu, Z., Eils, R., and Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. doi: 10.1093/bioinformatics/btw313
Gururajan, A., Clarke, G., Dinan, T. G., and Cryan, J. F. (2016). Molecular biomarkers of depression. Neurosci. Biobehav. Rev. 64, 101–133. doi: 10.1016/j.neubiorev.2016.02.011
Huang, S., Cai, N., Pacheco, P. P., Narrandes, S., Wang, Y., and Xu, W. (2018). Applications of support vector machine (SVM) learning in cancer genomics. Cancer Genomics Proteomics 15, 41–51. doi: 10.21873/cgp.20063
Iacob, E., Light, K. C., Tadler, S. C., Weeks, H. R., White, A. T., Hughen, R. W., et al. (2013). Dysregulation of leukocyte gene expression in women with medication-refractory depression versus healthy non-depressed controls. BMC Psychiatry 13:273. doi: 10.1186/1471-244x-13-273
Iacob, E., Tadler, S. C., Light, K. C., Weeks, H. R., Smith, K. W., White, A. T., et al. (2014). Leukocyte gene expression in patients with medication refractory depression before and after treatment with ECT or isoflurane anesthesia: a pilot study. Depress. Res. Treat. 2014:582380. doi: 10.1155/2014/582380
Iga, J., Ueno, S., Yamauchi, K., Numata, S., Tayoshi-Shibuya, S., Kinouchi, S., et al. (2007). Gene expression and association analysis of vascular endothelial growth factor in major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 31, 658–663. doi: 10.1016/j.pnpbp.2006.12.011
Kang, D. D., Sibille, E., Kaminski, N., and Tseng, G. C. (2012). MetaQC: objective quality control and inclusion/exclusion criteria for genomic meta-analysis. Nucleic Acids Res. 40, e15. doi: 10.1093/nar/gkr1071
Kang, S. G., and Cho, S. E. (2020). Neuroimaging biomarkers for predicting treatment response and recurrence of major depressive disorder. Int. J. Mol. Sci. 21:2148. doi: 10.3390/ijms21062148
Karege, F., Bondolfi, G., Gervasoni, N., Schwald, M., Aubry, J. M., and Bertschy, G. (2005). Low brain-derived neurotrophic factor (BDNF) levels in serum of depressed patients probably results from lowered platelet BDNF release unrelated to platelet reactivity. Biol. Psychiatry 57, 1068–1072. doi: 10.1016/j.biopsych.2005.01.008
Keshava Prasad, T. S., Goel, R., Kandasamy, K., Keerthikumar, S., Kumar, S., Mathivanan, S., et al. (2009). Human protein reference database–2009 update. Nucleic Acids Res. 37, D767–D772. doi: 10.1093/nar/gkn892
Leday, G. G. R., Vértes, P. E., Richardson, S., Greene, J. R., Regan, T., Khan, S., et al. (2018). Replicable and coupled changes in innate and adaptive immune gene expression in two case-control studies of blood microarrays in major depressive disorder. Biol. Psychiatry 83, 70–80. doi: 10.1016/j.biopsych.2017.01.021
Li, Z., Liu, Z. M., and Xu, B. H. (2018). A meta-analysis of the effect of microRNA-34a on the progression and prognosis of gastric cancer. Eur. Rev. Med. Pharmacol. Sci. 22, 8281–8287. doi: 10.26355/eurrev_201812_16525
Licata, L., Briganti, L., Peluso, D., Perfetto, L., Iannuccelli, M., Galeota, E., et al. (2012). MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 40, D857–D861. doi: 10.1093/nar/gkr930
Liew, C. C., Ma, J., Tang, H. C., Zheng, R., and Dempsey, A. A. (2006). The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J. Lab. Clin. Med. 147, 126–132. doi: 10.1016/j.lab.2005.10.005
Liu, C. H., Zhang, G. Z., Li, B., Li, M., Woelfer, M., Walter, M., et al. (2019). Role of inflammation in depression relapse. J. Neuroinflammation 16:90. doi: 10.1186/s12974-019-1475-7
Liu, Z., Li, X., Sun, N., Xu, Y., Meng, Y., Yang, C., et al. (2014). Microarray profiling and co-expression network analysis of circulating lncRNAs and mRNAs associated with major depressive disorder. PLoS One 9:e93388. doi: 10.1371/journal.pone.0093388
Ma, T., Huo, Z., Kuo, A., Zhu, L., Fang, Z., Zeng, X., et al. (2019). MetaOmics: analysis pipeline and browser-based software suite for transcriptomic meta-analysis. Bioinformatics 35, 1597–1599. doi: 10.1093/bioinformatics/bty825
Malhi, G. S., and Mann, J. J. (2018). Depression. Lancet 392, 2299–2312. doi: 10.1016/s0140-6736(18)31948-2
Miyata, S., Kurachi, M., Okano, Y., Sakurai, N., Kobayashi, A., Harada, K., et al. (2016). Blood transcriptomic markers in patients with late-onset major depressive disorder. PLoS One 11:e0150262. doi: 10.1371/journal.pone.0150262
Newman, M. E. (2006). Modularity and community structure in networks. Proc. Natl. Acad. Sci. U.S.A. 103, 8577–8582. doi: 10.1073/pnas.0601602103
Orchard, S., Ammari, M., Aranda, B., Breuza, L., Briganti, L., Broackes-Carter, F., et al. (2014). The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 42, D358–D363. doi: 10.1093/nar/gkt1115
Otte, C., Gold, S. M., Penninx, B. W., Pariante, C. M., Etkin, A., Fava, M., et al. (2016). Major depressive disorder. Nat. Rev. Dis. Primers 2:16065. doi: 10.1038/nrdp.2016.65
Piccinni, A., Marazziti, D., Catena, M., Domenici, L., Del Debbio, A., Bianchi, C., et al. (2008). Plasma and serum brain-derived neurotrophic factor (BDNF) in depressed patients during 1 year of antidepressant treatments. J. Affect. Disord. 105, 279–283. doi: 10.1016/j.jad.2007.05.005
Rubin-Falcone, H., Zanderigo, F., Thapa-Chhetry, B., Lan, M., Miller, J. M., Sublette, M. E., et al. (2018). Pattern recognition of magnetic resonance imaging-based gray matter volume measurements classifies bipolar disorder and major depressive disorder. J. Affect. Disord. 227, 498–505. doi: 10.1016/j.jad.2017.11.043
Salwinski, L., Miller, C. S., Smith, A. J., Pettit, F. K., Bowie, J. U., and Eisenberg, D. (2004). The database of interacting proteins: 2004 update. Nucleic Acids Res. 32, D449–D451. doi: 10.1093/nar/gkh086
Savitz, J., Frank, M. B., Victor, T., Bebak, M., Marino, J. H., Bellgowan, P. S., et al. (2013). Inflammation and neurological disease-related genes are differentially expressed in depressed patients with mood disorders and correlate with morphometric and functional imaging abnormalities. Brain Behav. Immun. 31, 161–171. doi: 10.1016/j.bbi.2012.10.007
Schultebraucks, K., Yadav, V., Shalev, A. Y., Bonanno, G. A., and Galatzer-Levy, I. R. (2020). Deep learning-based classification of posttraumatic stress disorder and depression following trauma utilizing visual and auditory markers of arousal and mood. Psychol. Med. 1–11. doi: 10.1017/s0033291720002718 [Epub ahead of print],
Serra-Millàs, M., López-Vílchez, I., Navarro, V., Galán, A. M., Escolar, G., Penadés, R., et al. (2011). Changes in plasma and platelet BDNF levels induced by S-citalopram in major depression. Psychopharmacology (Berl.) 216, 1–8. doi: 10.1007/s00213-011-2180-0
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Spijker, S., Van Zanten, J. S., De Jong, S., Penninx, B. W., van Dyck, R., Zitman, F. G., et al. (2010). Stimulated gene expression profiles as a blood marker of major depressive disorder. Biol. Psychiatry 68, 179–186. doi: 10.1016/j.biopsych.2010.03.017
Sun, M., Song, H., Wang, S., Zhang, C., Zheng, L., Chen, F., et al. (2017). Integrated analysis identifies microRNA-195 as a suppressor of Hippo-YAP pathway in colorectal cancer. J. Hematol. Oncol. 10:79. doi: 10.1186/s13045-017-0445-8
Thériault, R. K., and Perreault, M. L. (2019). Hormonal regulation of circuit function: sex, systems and depression. Biol. Sex Differ. 10:12. doi: 10.1186/s13293-019-0226-x
Tin Kam, H. (1998). The random subspace method for constructing decision forests. IEEE Trans. Pattern Anal. Mach. Intell. 20, 832–844. doi: 10.1109/34.709601
van ‘t Veer, L. J., Dai, H., van de Vijver, M. J., He, Y. D., Hart, A. A., Mao, M., et al. (2002). Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536. doi: 10.1038/415530a
Wang, T., Ji, Y. L., Yang, Y. Y., Xiong, X. Y., Wang, I. M., Sandford, A. J., et al. (2015). Transcriptomic profiling of peripheral blood CD4? T-cells in asthmatics with and without depression. Gene 565, 282–287. doi: 10.1016/j.gene.2015.04.029
Wang, X., Kang, D. D., Shen, K., Song, C., Lu, S., Chang, L. C., et al. (2012). An R package suite for microarray meta-analysis in quality control, differentially expressed gene analysis and pathway enrichment detection. Bioinformatics 28, 2534–2536. doi: 10.1093/bioinformatics/bts485
Wu, G., Feng, X., and Stein, L. (2010). A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 11:R53. doi: 10.1186/gb-2010-11-5-r53
Xu, X., Zhang, Y., Zou, L., Wang, M., and Li, A. (2012). “A gene signature for breast cancer prognosis using support vector machine,” in Proceedings of the 2012 5th International Conference on BioMedical Engineering and Informatics, Chongqing, 928–931.
Yi, Z., Li, Z., Yu, S., Yuan, C., Hong, W., Wang, Z., et al. (2012). Blood-based gene expression profiles models for classification of subsyndromal symptomatic depression and major depressive disorder. PLoS One 7:e31283.
Keywords: biomarkers, depression, machine learning, major depressive disorder, meta-analysis
Citation: Zhao S, Bao Z, Zhao X, Xu M, Li MD and Yang Z (2021) Identification of Diagnostic Markers for Major Depressive Disorder Using Machine Learning Methods. Front. Neurosci. 15:645998. doi: 10.3389/fnins.2021.645998
Received: 24 December 2020; Accepted: 25 May 2021;
Published: 18 June 2021.
Edited by:
Yunpeng Wang, University of Oslo, NorwayCopyright © 2021 Zhao, Bao, Zhao, Xu, Li and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming D. Li, bWwya21Aemp1LmVkdS5jbg==; Zhongli Yang, enkzcEB6anUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.