 Xiuqi Chen
Xiuqi Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 24 May 2021
Sec. Neurodegeneration
Volume 15 - 2021 | https://doi.org/10.3389/fnins.2021.614107
As a type of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases, sirtuin 2 (SIRT2) is predominantly found in the cytoplasm of cells in the central nervous system (CNS), suggesting its potential role in neurological disorders. Though SIRT2 is generally acknowledged to accelerate the development of neurological pathologies, it protects the brain from deterioration in certain circumstances. This review summarized the complex roles SIRT2 plays in the pathophysiology of diverse neurological disorders, compared and analyzed the discrete roles of SIRT2 in different conditions, and provided possible explanations for its paradoxical functions. In the future, the rapid growth in SIRT2 research may clarify its impacts on neurological disorders and develop therapeutic strategies targeting this protein.
The mammalian sirtuin family consists of seven members, sirtuins 1 to 7 (SIRT1–SIRT7), which are classified by their highly conserved central nicotinamide adenine dinucleotide (NAD+)-binding and catalytic domains. Their diverse functions are based on different enzymatic activities, unique binding partners, substrates, and distinct subcellular localization and expression patterns (Haigis and Sinclair, 2010). Each sirtuin resides in its preferable cellular compartment. SIRT1, SIRT6, and SIRT7 are chiefly found in the nucleus, whereas SIRT3, SIRT4, and SIRT5 are primarily found in the mitochondria (Haigis and Sinclair, 2010). SIRT2 is dominantly localized in the cytoplasm and is able to enter the nucleus to interact with histone H4 Lys 16 (H4K16) during mitosis (Vaquero et al., 2006), suggesting its essential role in chromatin condensation. α-Tubulin, a key component of the skeleton of microtubes, is also an important substrate of SIRT2. Acetylation of α-tubulin by SIRT2 is closely related to brain aging and neurological disorders (Li et al., 2007). SIRT2 is abundantly expressed in the brain by both neurons and oligodendrocytes, confined to the cytoplasm of these postmitotic cells (Harting and Knöll, 2010). Therefore, it is reasonable to assume that SIRT2 plays a vital role in neurological disorders.
Previous studies have corroborated that specific SIRT2 single-nucleotide polymorphisms (SNPs) correlate to the risk of a number of neurodegenerative diseases, like Alzheimer’s disease (AD) (Polito et al., 2013; Porcelli et al., 2013; Xia et al., 2014; Cacabelos et al., 2019; Shen et al., 2020) and Parkinson’s disease (PD) (Wang et al., 2018; Chen et al., 2019) under certain conditions. Recent studies demonstrated that SIRT2 participates in even more pathological processes such as frontotemporal dementia (FTD) (Spires-Jones et al., 2012), stroke (Shu et al., 2019), and brain injury (Wang et al., 2016). Although these diseases have different etiologies, they share some common pathological characteristics: neuroinflammation, synaptic dysfunction, metabolism abnormality, oxidative stress, etc., all of which can be affected by SIRT2. Some characteristics are only observed in specific diseases, such as amyloid beta (Aβ) aggregation and tau protein phosphorylation.
This review not only emphasized and compared the shared characteristics impacted by SIRT2 but also discussed unique SIRT2 functions in each diseased condition. First, we introduced the biochemistry and physiology of SIRT2, then described the mechanisms SIRT2 plays in different neurological disorders, analyzed why SIRT2 has paradoxical impacts on similar pathological processes, and discussed the possibility of developing drugs targeting SIRT2.
Sirtuins are lysine deacetylases (KDACs) and previously classified as class III histone deacetylases (HDACs). Unlike classes I, II, and IV that are dependent on Zn2+, they take NAD+ as a cofactor and herein manifest catalytic activities rather than deacetylation (Schiedel et al., 2018). Sirtuins are also known to catalyze ADP-ribosylation (Frye, 1999), which is especially prominent for SIRT2.
The chemical structure of SIRT2 is fully revealed in 2001, when Finnin et al. (2001) discovered that SIRT2 has a 304-amino acid catalytic core and a 19-residue N-terminal helical extension. The SIRT2 catalytic core has two domains: the larger one is a variant of the Rossmann fold, and the smaller one consists of the zinc-binding and the helical modules (Finnin et al., 2001). At the interface of the two domains, there is a large grove, which includes the NAD binding site and presents the HDAC activity.
In general, there are three explanations for the deacetylation mechanism, among which the ADPR peptidyl-imidate one is well accepted with substantial and solid evidence to support it. This mechanism shows that the process of deacetylation begins with the release of nicotinamide from NAD+ and the formation of a C1′-O-alkylamidate intermediate from the nucleophilic addition of the acetyl oxygen to the 1′-carbon of the nicotinamide ribose. The N-ribose 2′-hydroxyl group activated by a conserved catalytic His residue then attacks the previous intermediate carbon to produce a 1′2′-cyclic intermediate, which will eventually be resolved by a base-activated water molecule to generate the 2′-O-acetyl-ADP-ribose and deacetylated lysine (Sauve, 2010; Moniot et al., 2013). The characteristics of the structure and biological reaction of SIRT2 lay foundations for the design and synthesis of SIRT2 modulators.
SIRT2 is mainly present in the cytoplasm but enriched in the nucleus during the cell cycle (North and Verdin, 2007a), implicating its close relationship with cell cycle and cell proliferation. SIRT2 and its yeast counterpart HDAC Hst2 (Hst2) have preferences for H4K16Ac, which peaks at the S phase and dramatically drops in the G2 phase during the cell cycle. Loss of SIRT2 produces a high level of H4K16Ac during mitosis, a delay in S-phase/M transition and abnormal levels of H4K16Ac in heterochromatic foci (Vaquero et al., 2006). The deacetylation of H4K16 by SIRT2 leads to a series of downstream reactions. For example, SIRT2 suppresses the expression of NEDD4 E3 ubiquitin protein ligase (NEDD4), and the latter targets Myc oncoproteins for ubiquitination and degradation. Consequently, the inhibitory effect of NEDD4 on oncogenesis is reduced (Liu et al., 2013). In another study, SIRT2 regulates monomethylation of H4K20 (H4K20mel) and determines the levels of H4K20me2/H4K20me3 throughout the cell cycle. SIRT2-deficient animals exhibit genomic instability and chromosomal aberrations and are prone to tumorigenesis (Serrano et al., 2013), reflecting opposite roles of SIRT2 in cell proliferation and tumorigenesis. This paradox has been reported in other studies as well: the pharmacological blockade of SIRT2 activity or downregulation of SIRT2 expression with small interfering RNA (siRNA) counteracted the inhibitory effect of resveratrol on cell proliferation (Sayd et al., 2014), while another study demonstrated that the SIRT2 inhibitor AGK2 suppressed cell growth and resulted in G1-phase arrest by inhibiting the expression of cyclin-dependent kinase 6 (CDK6) and/or cyclin-dependent kinase 4 (CDK4) (Kim et al., 2016). This suggests that SIRT2 is involved in a complex network regulating cell cycle and exerts discrete functions under different conditions.
SIRT2 correlates to cell cycle checkpoints as well. It has been proved that SIRT2 curbs microtubule poison-induced hyperploid cell formation by blocking the entry to chromosome condensation (Inoue et al., 2007). SIRT2 deficiency contributes to fragility to replication stress, spontaneous accumulation of replication protein A to foci and the chromatin, and a G2/M checkpoint deficit by regulating the activity of cyclin-dependent kinase 9 (CDK9) (Zhang et al., 2013). Another study corroborated that the mutation of serine 368 in SIRT2 reduces hyperploidy in cells under mitotic stress in the presence of microtubule poisons, while SIRT2 overexpression mediates a delay in cellular proliferation by enhancing the level of serine 368 phosphorylation (North and Verdin, 2007b). SIRT2 also affects mitosis by triggering Golgi fragmentation and impairing Golgi assembly (Zhang et al., 2019). SIRT2 deficiency or overexpression results in impairments in nuclear envelope reassembly and acetylation as well as phosphorylation of the ankyrin repeat and LEM domain containing 2 (ANKLE2) throughout the cell cycle (Kaufmann et al., 2016). Apart from cell proliferation and cell cycle, SIRT2 also induces cell detachment and halts migration of mouse embryonic fibroblasts (MEFs) possibly by changing the stability of microtubules due to differential acetylation (Pandithage et al., 2008). The same conclusion is also drawn from another study which showed that SIRT2 hampers cell motility by curbing actin polymerization through heat shock protein 90 (HSP90) destabilization and subsequent repression of the LIM domain kinase 1 (LIMK1)/cofilin pathway (Min et al., 2018). However, Kim et al. (2016) proved that SIRT2 inhibition by AGK2 suppresses migration by compromising the stability of the heat shock transcription factor 1 (HSF1) protein. Dispute on SIRT2 in cell migration has encouraged further research.
The relationship between calorie restriction (CR) and aging dates back to 1986 when Weindruch et al. (1986) discovered the antiaging effects of dietary restriction. Some people attribute this phenomenon to low reactive oxygen species (ROS), and others assume that the radical shift in the metabolic strategy in cells may account for it (Guarente, 2000). To elucidate the detailed mechanism, Guarente (2000) proposed that Sir2 proteins may link the metabolic rate to the pace of aging by sensing NAD levels and generating the mandated level of chromatin silencing. This assumption has been corroborated later. Only yeast that expresses Sir2 and nicotinate phosphoribosyltransferase (NPT1) show life span extension under the CR condition (Lin et al., 2000), suggesting that Sir2p and NAD are necessary for life span extension induced by CR. The authors assumed that this may be linked to the shunting of carbon metabolism toward the mitochondrial tricarboxylic acid cycle and the concomitant increase in respiration (Lin et al., 2002). Another study verified the above findings and demonstrated that the overexpression of Sir2 and deletion of the fork barrier binding protein (Fob1) contribute to increased longevity induced by CR. The authors interpreted this as an outcome of differences in approaches to achieve CR and yeast strain background (Kaeberlein et al., 2004). Though Sir2 synergistically increases CR-induced replicative life span extension, a study found that Sir2 exerts the opposite effect on the chronological life span of yeast. In this study, lack of Sir2 along with CR showed a dramatic chronological life span extension (Fabrizio et al., 2005). The potential mechanism underlying this inconsistency was discussed in another study which stated that the regulatory function of Sir2 in chronological aging responds to nutrient cues including glucose, possibly through Sir2-dependent modification of chromatin or deacetylation of a non-histone protein (McCleary and Rine, 2017).
In addition to studies on yeast, more experiments were conducted on species like Drosophila and other eukaryotes. Drosophila with low-calorie intake exhibits high spontaneous activities, which are dependent on the presence of dSir2 (Parashar and Rogina, 2009). Another study concluded that Sir2 in the body is a key regulator of organismal energy homeostasis required for maintaining the metabolic regulatory network across tissues (Banerjee et al., 2013). In a mouse model, SIRT2 regulates insulin-mediated glucose uptake by deacetylating the TUG (encoded by the Aspscr1 gene) peptide (Belman et al., 2015).
Though it is widely accepted that Sir2 and its homolog present synergistic functions in CR-mediated longevity, some researchers showed results that CR-mediated longevity is not dependent on Sir2 (Tsuchiya et al., 2006; Mei and Brenner, 2015), which needs further validation.
Neuroinflammation is ubiquitously present in neurological disorders. It directly impacts neuronal apoptosis, which results in acute neural damage or neuron death and accelerates long-term neurodegeneration (Lyman et al., 2014). Glial cells, including astrocytes and microglia, along with chemokines and cytokines, play a vital role in neuroinflammation (Perry et al., 2010; Ransohoff, 2016). Neuroinflammation is closely associated with oxidative stress (Akbar et al., 2016). The latter can be triggered by protein aggregates, impair mitochondrial function, and injure neurons, leading to DNA damage as well as structural alteration of proteins and lipids (Zhang et al., 2020).
The relationship between SIRT2 and neuroinflammation has been inconclusive for a long time. SIRT2 inhibition by a high concentration of AGK2 was reported to block Aβ-triggered inflammation in vitro (Scuderi et al., 2014). SIRT2-knockout mice manifest less inflammatory response triggered by lipopolysaccharides (LPS) (Lee et al., 2014), mainly due to inhibition of nuclear factor kappa-B (NF-κB) activation by deficiency of SIRT2. Consequently, the phosphorylation and degradation of IκBα was decreased, resulting from decreased p65 phosphorylation and nuclear translocation (Lee et al., 2014). Application of AGK2 inhibits inflammatory signals triggered by LPS as well (Wang et al., 2016; Harrison et al., 2018), which is possibly attributed to decreased nuclear translocation of NF-κB (Wang et al., 2016). Interestingly, another study showed similar results in BV2 microglia cells (Chen et al., 2015), but altered nuclear translocation of NF-κB was not observed. This discrepancy may be the result of different experimental conditions or tissue specificity, with one study selecting bone-derived macrophage (Lee et al., 2014) and another selecting BV2 microglia (Chen et al., 2015). Apart from its direct regulating role, SIRT2 is also able to curtail the anti-inflammatory effect of Treg cells by modulating the expression of immunosuppression-associated molecules including forkhead box P3 (Foxp3), indirectly ameliorating neuroinflammation in the middle cerebral artery occlusion (MCAO) mouse model (Shu et al., 2019). Though the majority of studies support the anti-neuroinflammation role of SIRT2, conflicting results were also reported. In the controlled cortical impact (CCI) injury model mimicking brain injury, inhibition of SIRT2 by AK-7 exacerbated inflammatory response, which may result from elevated NF-κB activation and increased acetylation and activation of the p65 subunit (Yuan et al., 2016). The following factors may explain the conflicting results: (1) difference in the animal model used (mainly LPS-induced inflammation models or the CCI injury model); (2) the endurance of the trigger applied to the animal model; (3) approaches to silence SIRT2; and (4) tissue specificity.
Studies have shown that targets of SIRT2 deacetylation have important roles in antioxidant- and redox-mediated cellular homeostasis both in vitro and in vivo (Gomes et al., 2015). In SH-SY5Y cells, elevated SIRT2 protected cells from rotenone- or diquat-induced cell death through increasing the expression of antioxidant substances such as superoxide dismutase 2 (SOD2) (Singh et al., 2017). In vivo, 13-month-old mice lacking SIRT2 exhibit energy failure due to mitochondrial depletion, and redox dyshomeostasis (Fourcade et al., 2017). Overall, SIRT2 acts as a protectant for cells to combat against oxidative stress (Singh et al., 2018). However, another study claimed that inhibiting SIRT2 by AK-7 may protect 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mice from redox dysfunction (Guan et al., 2016). Therefore, more comprehensive studies are required to clarify the exact role of SIRT2 in oxidative stress.
Synaptic changes and axonal dysfunction directly contribute to cognitive impairment and behavior changes. SIRT2 is one of the key factors modulating hippocampus function such as cell proliferation, memory formation, and neuroblast differentiation (Yoo et al., 2015; Jung et al., 2016). Nicotinamide is confirmed to be protective in the slow Wallerian degeneration (Wlds) mouse model, and its function does not relate to SIRT1 (Wang et al., 2005). Though participation of SIRT2 was not tested, it was expected that inhibition of SIRT2 is related to axonal degeneration. This hypothesis was verified by showing that SIRT2 overexpression abrogates microtubule hyperacetylation and resistance to axonal degeneration, while SIRT2 silencing by siRNA enhances microtubule acetylation and resistance to axonal degeneration in WldS mice (Suzuki and Koike, 2007). Though it is known that inhibition of SIRT2 protects axons, middle-aged mice lacking SIRT2 exhibit axonal degeneration and locomotor dysfunction (Fourcade et al., 2017). The opposite roles of SIRT2 may arise from the difference in the choice of mouse models or the approaches to induce axonal dysfunction. Sirtuin-knockout mice exhibit axonal dysfunction (Fourcade et al., 2017) or less voluntary differentiation of dopaminergic neurons due to knockout of the sirtuin gene (Szego et al., 2017), whereas the axonal degeneration in WldS mice is due to the transection of axons (Suzuki and Koike, 2007). We assume that SIRT2 may be necessary for normal axonal formation but functions differently after that.
As for SIRT2 on synaptic plasticity, SIRT2 functions as an AMPA receptor (AMPAR) deacetylase (Wang et al., 2017). It interacts with GluA1, a subunit of AMPAR. The downregulation or absence of SIRT2 increases GluA1 acetylation by competitively decreasing GluA1 ubiquitination, through which AMPAR trafficking and proteostasis are regulated and learning and memory capabilities are influenced. There are few studies focusing on SIRT2 and synaptic plasticity, and more investigations are in demand.
SIRT2 influences autophagy and apoptosis mainly by regulating microtubule-related proteins and alleviating the toxicity of misfolded proteins. Pharmacological inhibition of SIRT2 prevents microtubules from impairment by selectively enhancing α-tubulin acetylation in sPD cells, and SIRT2-knockout mice also maintain stable microtubule assembly after being exposed to MPP +. The stability of microtubules is essential for the clearance of misfolded proteins and normal autophagic flux (Esteves et al., 2018). α-syn, AβPP, and tau protein can lead to mitophagy and autophagy, and SIRT2 inhibition attenuates the toxicity of these misfolded proteins, indirectly participating in the regulation of autophagy. Deletion of Sir2 in the yeast abolishes autophagy and mitophagy and consequently rescues cells (Sampaio-Marques et al., 2012). In microglial BV2 cells, SIRT2-induced autophagy was also found as a result of AGK2-induced ATP decline (Li et al., 2013). A similar phenomenon was observed in mammalian models (de Oliveira et al., 2017). In addition, SIRT2 has been shown to enhance apoptosis in the MPTP model of PD through deacetylating Foxo3a and increasing levels of Bim RNA and protein, exacerbating MPTP-induced nigrostriatal damage (Liu et al., 2014). Inhibition of SIRT2 either by AK-1 or by SIRT2 knockout restored microtubule stability and improved autophagy, favoring cell survival through eliminating toxic Aβ oligomers (Silva et al., 2017). In addition to clearing Aβ, SIRT2 is able to impact the phosphorylation of tau and autophagic influx in AD (Esteves et al., 2019). The neuroprotective effects of SIRT2 inhibition have also been found in ischemic stroke, which is mediated by the downregulation of AKT/FOXO3a and mitogen-activated protein kinase (MAPK) pathways (She et al., 2018).
Accumulating evidence has shown that necrosis is also a programmed process (Vandenabeele et al., 2010). SIRT2 regulates this process in ischemic stroke. Deletion or knockdown of SIRT2 blocks cellular necrosis induced by tumor necrosis factor α (TNF-α) through preventing the formation of the receptor-interacting protein 1 (RIP1)–receptor-interacting protein 3 (RIP3) complex (Narayan et al., 2012). There are relatively few studies available in this field, and future research is required to confirm the role of SIRT2 in necrosis.
The concomitant presence of Aβ and tau is still the classical characteristics of AD (Scheltens et al., 2016). SIRT2 influences the amyloid beta-protein precursor (APP) transformation process and tau protein phosphorylation. Inhibition of SIRT2 by AGK2 in H4-SW cells reduced levels of Aβ40 and Aβ42 as well as soluble AβPPβ (sAβPPβ), whereas inhibition of SIRT2 by AK-7 in both APP23 and 3xTg transgenic mice increased the level of soluble AβPPα (sAβPPα), indicating that SIRT2 may regulate the activity or quantity of α and β secretases (Biella et al., 2016). Intriguingly, in vivo experiment did not find significant changes in Aβ40 and Aβ42 after inhibiting SIRT2, which might be attributed to environmental differences between in vivo and in vitro conditions (Biella et al., 2016). Apart from the direct impacts of SIRT2 inhibition on Aβ, SIRT2 loss of function indirectly promoted the clearance of Aβ by restoring autophagy (Silva et al., 2017). Though inhibition of SIRT2 improved memory in murine models (Biella et al., 2016), clinical evidence did not show correlations between the level of SIRT2 mRNA and cognitive performance of patients, and no difference in the level of SIRT2 mRNA in the periphery between normal senior people and AD patients was found (Wongchitrat et al., 2019). This inconsistency between animal experiments and clinical trials implies the complicated relationship between AD and SIRT2.
When investigating the role of SIRT2 in AD pathogenesis, ideal animal models ensure the significance of research findings and the possibility of clinical translation. One of the animal models that researchers usually use is the 3xTg mouse model. Though this mouse model is not perfect for examining the relationship between SIRT2 and cognitive impairment because of the presence of Aβ deposit in the 3xTg model, AD models with sole tau pathology are hard to find. Transgenic tau models like rTg4510 and PS19 are not applicable to AD research for no tau mutations relate to AD. Though humanized tau (htau) models almost perfectly recapitulate the characteristics of tau pathology of AD, their behavior deficits and pathological phenotypes are very mild, which limits their application (Jankowsky and Zheng, 2017).
Inhibition of SIRT2 by AK-7 increased the steady-state level of tau protein, which assisted in stabilizing microtubules in a healthy neuron, which may account for alleviated neurodegeneration in AD models. But this research failed to measure the level of phosphorylated tau due to the relatively young age of mice (Biella et al., 2016). Another study found that nicotinamide, a competitive inhibitor of the sirtuins, can restore cognitive function by selectively reducing a specific phospho-species of tau (Thr231) (Green et al., 2008); SIRT2 inhibition by AK-1 is corroborated to decrease phospho-tau as well (Esteves et al., 2019). Interestingly, it has been demonstrated in this study that the effect of nicotinamide on cognition is not related to changes of Aβ (Green et al., 2008). Discrepancy in findings by Biella et al. (2016) and Green et al. (2008) may be partially due to the selection of different inhibitors of SIRT2. It is known that NAD is not a specific SIRT2 inhibitor and that the involvement of other sirtuin family members cannot be excluded. In the study of Green et al. (2008), quantities of APP and its metabolites were measured, but the distribution of Aβ deposit was not tested, and this may directly contribute to cognitive impairment or indirectly participate in tau pathology and cognitive impairment.
SIRT2 not only influences AD by intervening with the process of Aβ aggregation and tau phosphorylation through regulating the Aβ/tau-mediated autophagy but also impacts AD by increasing the clearance of misfolded proteins. Inhibition of SIRT2 either by AK-1 or by SIRT2 knockout restored microtubule stabilization and improved autophagy, favoring cell survival through eliminating toxic Aβ oligomers (Silva et al., 2017). In addition to Aβ, SIRT2 was shown to impact the phosphorylation of tau and autophagic influx in AD (Esteves et al., 2019).
Given that AD may benefit from SIRT2 inhibition, scientists have been testing if a selective SIRT2 inhibitor may be a promising drug target for AD. Diaz-Perdigon et al. (2020) found that early SIRT2 inhibition by a selective SIRT2 inhibitor 33i might be beneficial for preventing age-related cognitive deficits, neuroinflammation, and AD progression in the senescence-accelerated mouse prone-8 (SAMP8) model, which provided new insights into AD therapy.
PD is characterized by progressive loss of dopaminergic neurons and accumulating Lewy bodies (LBs) in the substantia nigra (SN). The pathologically misfolded protein α-syn is the main component of LBs (Ghosh et al., 2017). α-Syn is acetylated on lysines 6 and 10, and these residues can be deacetylated by SIRT2, implying the potential role of SIRT2 in α-syn acetylation, aggregation, and autophagy (de Oliveira et al., 2017). SIRT2 inhibition increased the level of acetylated α-syn, decreased the aggregation and toxicity of α-syn, and elevated the activity of alkaline phosphatase (ALP), which led to clearance of α-syn aggregates (de Oliveira et al., 2017). Pharmacological inhibition of SIRT2 by AGK2 or AK-1 rescued α-syn-mediated toxicity by transforming toxic, submicroscopic a-syn oligomers into larger inclusions, but why SIRT2 inhibition affected α-syn aggregation is not clear. Though researchers assume that an increase in acetylated α-tubulin by SIRT2 may account for it, they have not validated this hypothesis (Outeiro et al., 2007). A later study found that the absence of Sir2, a homologous-to-mammalian sirtuin protein, can alleviate α-syn toxicity by inhibiting mitophagy/autophagy (Sampaio-Marques et al., 2012). In this study, it was shown that α-syn can induce detrimental autophagy/mitophagy and superoxide anion accumulation in the presence of ATG11/ATG32 (two scaffold proteins closely related to autophagy/mitophagy) (Sampaio-Marques et al., 2012). Therefore, it is reasonable to speculate that absence of Sir2 may be related to oxidative stress, and this was confirmed by Guan et al. (2016) who showed that SIRT2 inhibition by AK-7 improved MPTP-induced neurochemical and behavioral deficits by ameliorating dysfunction of the redox network. In contrast, another study demonstrated that SIRT2 inhibition exacerbated α-syn aggregation under the condition of oxidative stress caused by diquat/rotenone (Singh et al., 2017). Though the majority of studies claimed the protective role of SIRT2 inhibition in α-synucleinopathy, the detailed mechanism is still unclear, which demands more investigations.
SIRT2 reduces the toxicity of misfolded proteins by accelerating the process of clearance and regulating the process of autophagy. Pharmacological inhibition of SIRT2 prevented microtubules from being impaired by selectively enhancing α-tubulin acetylation in sPD cells, and SIRT2-knockout mice also maintained stable microtubule assembly after being exposed to MPP +. The stability of microtubules is essential for the clearance of misfolded proteins and normal autophagic flux (Esteves et al., 2018). Though normal autophagic flux is protective, excessive autophagy is detrimental. Deletion of Sir2 in the yeast abolishes autophagy and mitophagy and consequently rescues cells (Sampaio-Marques et al., 2012), and a similar phenomenon was observed in mammalian models (de Oliveira et al., 2017). However, SIRT2 is not always protective. A study reported that SIRT2 aggravated MPTP-induced nigrostriatal damage by enhancing apoptosis in the MPTP model (Liu et al., 2014). Therefore, it is necessary to investigate the exact role of SIRT2 in autophagy of PD.
Brain injury consists of penetrating and non-penetrating injuries. The main pathophysiological changes include autoregulatory disturbances, abnormal glutaminergic and GABAergic functions, massive accumulation of K+ in the extracellular space, the influx of Na+ and Ca2+ through glutamate receptor-gated ion channels, and uniform decline in oxygen and glucose metabolism (McGinn and Povlishock, 2016). Given that SIRT2 is closely related to inflammation and autophagy, both of which are present in brain injury, SIRT2 may be involved in the process of brain injury. A study has discovered that inhibition of SIRT2 by AK-7 led to exacerbation of brain edema and neuroinflammation in an experimental traumatic brain injury (TBI) model CCI (Yuan et al., 2016), whereas another study has reported conflicting results. It was found that administration of the SIRT2 inhibitor AGK2 significantly decreased LPS-induced increase in CD11b, TNF-α, and IL-6 and blocked LPS-induced nuclear translocation of NF-κB and signaling pathways related to activation of autophagy (Wang et al., 2016).
Infarction of the central nervous system refers to cell death of the brain, cell death of the spinal cord, or cell death due to ischemia, and diagnosis is based on imaging, clinical, and other objective evidence (Sacco et al., 2013). It is classified into ischemic and hemorrhagic stroke, and ischemic stroke is more common. In stroke, SIRT2 presents contradictory functions as well. SIRT2 impairs the anti-inflammatory effect of Treg cells and indirectly impacts the process of stroke in the MCAO model (Shu et al., 2019). Inhibition of SIRT2 by AK-7 rescued neurological function and decreased stroke volume via p38 activation. SIRT2 has also been verified to regulate programmed necrosis in ischemic stroke. Deletion or knockdown of SIRT2 blocked cellular necrosis induced by TNF-α through preventing the formation of the RIP1–RIP3 complex (Narayan et al., 2012). However, the neuroprotective effect of SIRT2 inhibition on ischemic stroke has also been reported, which is mediated by downregulation of AKT/FOXO3a and MAPK pathways (She et al., 2018).
FTD is classified as a neurodegenerative disease. It mainly manifests by progressive decline in behavior and language caused by focal degeneration of frontal and anterior temporal lobes (Shinagawa, 2013). FTD is also defined as a tau-associated disease. Given that SIRT2 can influence the process of tau pathology, it is reasonable to deduce that SIRT2 may also play an important role in FTD. SIRT2 inhibition by AK-1 is reported to be non-toxic and to prevent neurodegeneration in the rTg4510 brain by decreasing neuronal loss (Spires-Jones et al., 2012). There are few studies focusing on SIRT2 and FTD, and further investigations are in demand.
Overexpression or inhibition/knockout/silencing of SIRT2 can lead to changes in different phenotypes. These phenotypes interact with each other under certain circumstances as well. Figure 1 and Table 1 have concluded the sophisticated relationship between SIRT2 and various phenotypes.
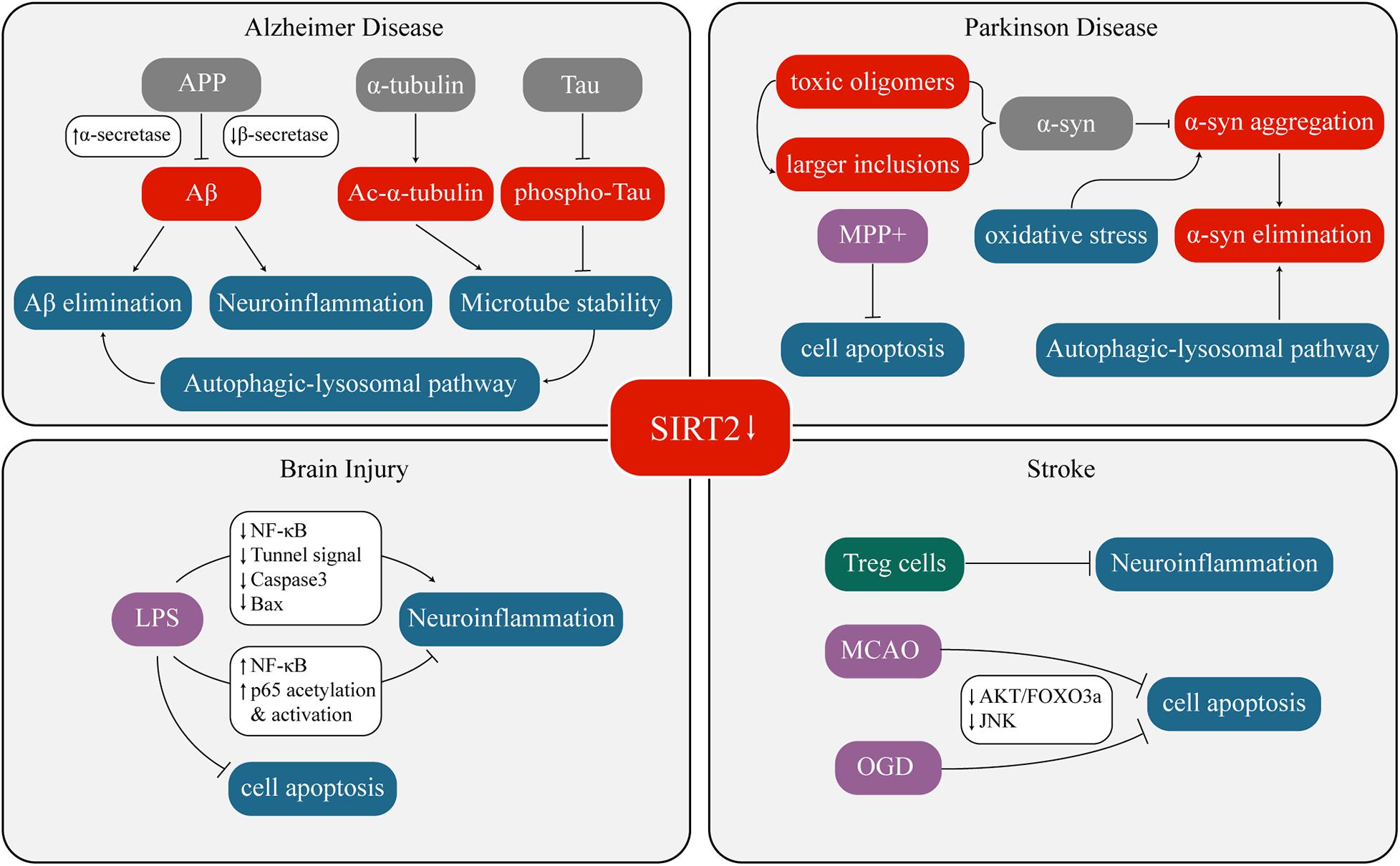
Figure 1. Crosstalk between SIRT2 and neurological diseases.
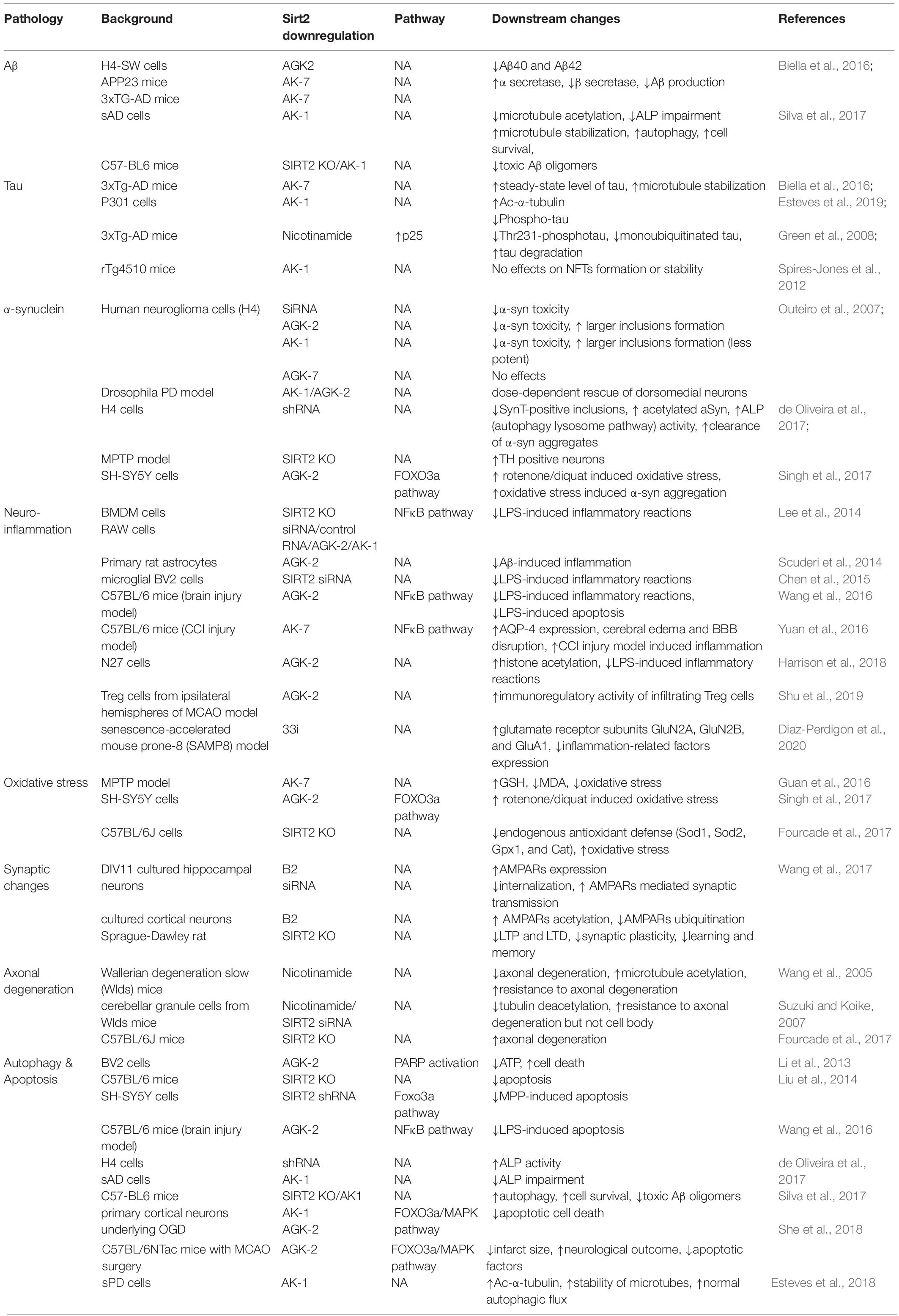
Table 1. Relationship between SIRT2 and phenotypes.
As stated above, SIRT2 might play a negative role in neurological diseases, and it is a hot spot to search for disease-modifying SIRT2 modulators. This section mainly focused on SIRT2 modulators targeting neurological diseases.
The formation of a C1’-O-alkylamidate intermediate is important in the deacetylation process of SIRT2. Nicotinamide is a potent inhibitor of this reaction because it can bind to and attack the intermediate (Alcaín and Villalba, 2009). Green et al. (2008) verified the effect of nicotinamide in the 3xTg AD mouse model: nicotinamide decreased the level of phosphorylated tau protein and increased its degradation. Nicotinamide is also protective in axonal degeneration by elevating the acetylation of microtubules (Wang et al., 2005) or reducing the deacetylation of tubulin (Suzuki and Koike, 2007).
As previously discussed, SIRT2 modulators AGK2, AK-1, and AK-7 have been widely explored in different neurological models (Table 1). AK-1 has been reported to stabilize microtubes, improve cell survival, alleviate the toxicity of Aβ oligomers (Silva et al., 2017), and increase the level of acetylated α-tubulin as well as decrease tau phosphorylation (Esteves et al., 2019) in AD. However, it has been proven to be ineffective on formation of neurofibrillary tangles (NFTs) or stability in FTD though it possesses the ability to prevent neurodegeneration in general (Spires-Jones et al., 2012). AGK2 and AK-7 are also effective in improving cognitive performance in AD by regulating the metabolism of AβPP (Biella et al., 2016). In PD, AK-1 and AGK2 were demonstrated to modify the morphology of inclusion bodies, rescue α-syn toxicity, and dose-dependently rescue dorsomedial neurons, whereas AK-7 did not have such protective effects (Outeiro et al., 2007). Conflicting results were also reported. A study showed that AGK2 aggravated oxidative stress and the resulting α-syn aggregation in PD (Singh et al., 2017). Another study demonstrated that AK-7 improved neurological functions by alleviating oxidative stress (Guan et al., 2016). In brain injury and stroke, SIRT2 modulators usually take their effect through regulating neuroinflammation and autophagy, but a definite conclusion on their efficacy has not been reached (Wang et al., 2016; Yuan et al., 2016; She et al., 2018; Shu et al., 2019), mainly due to lack of substantial data.
Besides nicotinamide, AK-1, AK-7, and AGK2, new highly selective SIRT2 inhibitors have emerged. 3-((2-Methoxynaphthalen-1-yl)methyl)-7-((pyridin-3- ylmethyl) amino)-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(3H)-one (ICL-SIRT078) showed a neuroprotective effect on parkinsonian neuronal cell death in the lactacystin-induced N27 cell model (Di Fruscia et al., 2015). Another new class of SIRT2 inhibitors, 5-((3-amidobenzyl)oxy)nicotinamides, is corroborated to protect SH-SY5Y cells from α-synuclein aggregation-induced cytotoxicity (Ai et al., 2016).
When it comes to clinical translation, findings in these studies are contradictory, and most of them focused on the relationship between SIRT2 and phenotypes but neglected what roles the SIRT2 modulators eventually play in cognitive/behavioral improvement and neuroimaging changes. Each disease has a series of phenotypes, and improvement in one or two phenotypes does not extrapolate to state improvement in all phenotypes of this disease. Further studies should focus on in vivo experiments and take cognitive and/or behavioral improvement and neuroimaging changes as indicators for evaluating the efficacy of SIRT2 modulators.
In summary, SIRT2 exerts a detrimental effect on majority of neurological diseases, inhibiting SIRT2 by chemical inhibitors, SIRT2 knockout, or siRNA silencing protects the brain. Though SIRT2 inhibitors are promising in curing neurological diseases, we cannot dismiss the paradoxical role of SIRT2 in neuroinflammation, axonal dysfunction, and autophagy. The continuous growth of SIRT2 research will help clarify functions of SIRT2 and accelerate translation of SIRT2 research findings.
XC searched literatures, drafted the manuscript, and drew the figure and tables. All authors contributed to the revision of the manuscript and approved the submitted version.
This study was supported by a grant from Shanghai Committee of Science and Technology (Grant No. 20S31904400) and Shanghai Fifth People’s Hospital (Grant No. 2018WYZD01).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thanked Duanlu Hou for critical reading and helpful advice.
Ai, T., Wilson, D. J., More, S. S., Xie, J., and Chen, L. (2016). 5-((3-Amidobenzyl)oxy)nicotinamides as Sirtuin 2 Inhibitors. J. Med. Chem. 59, 2928–2941. doi: 10.1021/acs.jmedchem.5b01376
Akbar, M., Essa, M. M., Daradkeh, G., Abdelmegeed, M. A., Choi, Y., Mahmood, L., et al. (2016). Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res. 1637, 34–55. doi: 10.1016/j.brainres.2016.02.016
Alcaín, F. J., and Villalba, J. M. (2009). Sirtuin inhibitors. Exp. Opin. Ther. Pat. 19, 283–294. doi: 10.1517/13543770902755111
Banerjee, K. K., Ayyub, C., Sengupta, S., and Kolthur-Seetharam, U. (2013). Fat body dSir2 regulates muscle mitochondrial physiology and energy homeostasis nonautonomously and mimics the autonomous functions of dSir2 in muscles. Mol. Cell Biol. 33, 252–264. doi: 10.1128/mcb.00976-12
Belman, J. P., Bian, R. R., Habtemichael, E. N., Li, D. T., Jurczak, M. J., Alcázar-Román, A., et al. (2015). Acetylation of TUG protein promotes the accumulation of GLUT4 glucose transporters in an insulin-responsive intracellular compartment. J. Biol. Chem. 290, 4447–4463. doi: 10.1074/jbc.M114.603977
Biella, G., Fusco, F., Nardo, E., Bernocchi, O., Colombo, A., Lichtenthaler, S. F., et al. (2016). Sirtuin 2 Inhibition Improves Cognitive Performance and Acts on Amyloid-β Protein Precursor Processing in Two Alzheimer’s Disease Mouse Models. J. Alzheimers Dis. 53, 1193–1207. doi: 10.3233/jad-151135
Cacabelos, R., Carril, J. C., Cacabelos, N., Kazantsev, A. G., Vostrov, A. V., Corzo, L., et al. (2019). Sirtuins in Alzheimer’s Disease: SIRT2-Related GenoPhenotypes and Implications for PharmacoEpiGenetics. Int. J. Mol. Sci. 20:20051249. doi: 10.3390/ijms20051249
Chen, H., Wu, D., Ding, X., and Ying, W. (2015). SIRT2 is required for lipopolysaccharide-induced activation of BV2 microglia. Neuroreport 26, 88–93. doi: 10.1097/wnr.0000000000000305
Chen, X., Mai, H., Chen, X., Cai, Y., Cheng, Q., Chen, X., et al. (2019). Rs2015 Polymorphism in miRNA Target Site of Sirtuin2 Gene Is Associated with the Risk of Parkinson’s Disease in Chinese Han Population. Biomed. Res. Int. 2019:1498034. doi: 10.1155/2019/1498034
de Oliveira, R. M., Vicente, M. H., Francelle, L., Pinho, R., Szegö, ÉM., Martinho, R., et al. (2017). The mechanism of sirtuin 2-mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol. 15:e2000374. doi: 10.1371/journal.pbio.2000374
Di Fruscia, P., Zacharioudakis, E., Liu, C., Moniot, S., Laohasinnarong, S., Khongkow, M., et al. (2015). The discovery of a highly selective 5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(3H)-one SIRT2 inhibitor that is neuroprotective in an in vitro Parkinson’s disease model. Chem. Med. Chem. 10, 69–82. doi: 10.1002/cmdc.201402431
Diaz-Perdigon, T., Belloch, F. B., Ricobaraza, A., Elboray, E. E., Suzuki, T., Tordera, R. M., et al. (2020). Early sirtuin 2 inhibition prevents age-related cognitive decline in a senescence-accelerated mouse model. Neuropsycho. Pharm. 45, 347–357. doi: 10.1038/s41386-019-0503-8
Esteves, A. R., Arduino, D. M., Silva, D. F., Viana, S. D., Pereira, F. C., and Cardoso, S. M. (2018). Mitochondrial Metabolism Regulates Microtubule Acetylome and Autophagy Trough Sirtuin-2: Impact for Parkinson’s Disease. Mol. Neurobiol. 55, 1440–1462. doi: 10.1007/s12035-017-0420-y
Esteves, A. R., Palma, A. M., Gomes, R., Santos, D., Silva, D. F., and Cardoso, S. M. (2019). Acetylation as a major determinant to microtubule-dependent autophagy: Relevance to Alzheimer’s and Parkinson disease pathology. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 2008–2023. doi: 10.1016/j.bbadis.2018.11.014
Fabrizio, P., Gattazzo, C., Battistella, L., Wei, M., Cheng, C., McGrew, K., et al. (2005). Sir2 blocks extreme life-span extension. Cell 123, 655–667. doi: 10.1016/j.cell.2005.08.042
Finnin, M. S., Donigian, J. R., and Pavletich, N. P. (2001). Structure of the histone deacetylase SIRT2. Nat. Struct. Biol. 8, 621–625. doi: 10.1038/89668
Fourcade, S., Morato, L., Parameswaran, J., Ruiz, M., Ruiz-Cortes, T., Jove, M., et al. (2017). Loss of SIRT2 leads to axonal degeneration and locomotor disability associated with redox and energy imbalance. Aging Cell 16, 1404–1413. doi: 10.1111/acel.12682
Frye, R. A. (1999). Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 260, 273–279. doi: 10.1006/bbrc.1999.0897
Ghosh, D., Mehra, S., Sahay, S., Singh, P. K., and Maji, S. K. (2017). α-synuclein aggregation and its modulation. Int. J. Biol. Macromol. 100, 37–54. doi: 10.1016/j.ijbiomac.2016.10.021
Gomes, P., Fleming Outeiro, T., and Cavadas, C. (2015). Emerging Role of Sirtuin 2 in the Regulation of Mammalian Metabolism. Trends Pharmacol. Sci. 36, 756–768. doi: 10.1016/j.tips.2015.08.001
Green, K. N., Steffan, J. S., Martinez-Coria, H., Sun, X., Schreiber, S. S., Thompson, L. M., et al. (2008). Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 28, 11500–11510. doi: 10.1523/jneurosci.3203-08.2008
Guan, Q., Wang, M., Chen, H., Yang, L., Yan, Z., and Wang, X. (2016). Aging-related 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurochemial and behavioral deficits and redox dysfunction: improvement by AK-7. Exp. Gerontol. 82, 19–29. doi: 10.1016/j.exger.2016.05.011
Guarente, L. (2000). Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 14, 1021–1026.
Haigis, M. C., and Sinclair, D. A. (2010). Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295. doi: 10.1146/annurev.pathol.4.110807.092250
Harrison, I. F., Smith, A. D., and Dexter, D. T. (2018). Pathological histone acetylation in Parkinson’s disease: Neuroprotection and inhibition of microglial activation through SIRT 2 inhibition. Neurosci. Lett. 666, 48–57. doi: 10.1016/j.neulet.2017.12.037
Harting, K., and Knöll, B. (2010). SIRT2-mediated protein deacetylation: An emerging key regulator in brain physiology and pathology. Eur. J. Cell Biol. 89, 262–269. doi: 10.1016/j.ejcb.2009.11.006
Inoue, T., Hiratsuka, M., Osaki, M., Yamada, H., Kishimoto, I., Yamaguchi, S., et al. (2007). SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 26, 945–957. doi: 10.1038/sj.onc.1209857
Jankowsky, J. L., and Zheng, H. (2017). Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 12:89. doi: 10.1186/s13024-017-0231-7
Jung, H. Y., Yoo, D. Y., Kim, J. W., Kim, D. W., Choi, J. H., Chung, J. Y., et al. (2016). Sirtuin-2 inhibition affects hippocampal functions and sodium butyrate ameliorates the reduction in novel object memory, cell proliferation, and neuroblast differentiation. Lab. Anim. Res. 32, 224–230. doi: 10.5625/lar.2016.32.4.224
Kaeberlein, M., Kirkland, K. T., Fields, S., and Kennedy, B. K. (2004). Sir2-independent life span extension by calorie restriction in yeast. PLoS Biol. 2:E296. doi: 10.1371/journal.pbio.0020296
Kaufmann, T., Kukolj, E., Brachner, A., Beltzung, E., Bruno, M., Kostrhon, S., et al. (2016). SIRT2 regulates nuclear envelope reassembly through ANKLE2 deacetylation. J. Cell Sci. 129, 4607–4621. doi: 10.1242/jcs.192633
Kim, H. W., Kim, S. A., and Ahn, S. G. (2016). Sirtuin inhibitors, EX527 and AGK2, suppress cell migration by inhibiting HSF1 protein stability. Oncol. Rep. 35, 235–242. doi: 10.3892/or.2015.4381
Lee, A. S., Jung, Y. J., Kim, D., Nguyen-Thanh, T., Kang, K. P., Lee, S., et al. (2014). SIRT2 ameliorates lipopolysaccharide-induced inflammation in macrophages. Biochem. Biophys. Res. Commun. 450, 1363–1369. doi: 10.1016/j.bbrc.2014.06.135
Li, W., Zhang, B., Tang, J., Cao, Q., Wu, Y., Wu, C., et al. (2007). Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha-tubulin. J. Neurosci. 27, 2606–2616. doi: 10.1523/jneurosci.4181-06.2007
Li, Y., Nie, H., Wu, D., Zhang, J., Wei, X., and Ying, W. (2013). Poly(ADP-ribose) polymerase mediates both cell death and ATP decreases in SIRT2 inhibitor AGK2-treated microglial BV2 cells. Neurosci. Lett. 544, 36–40. doi: 10.1016/j.neulet.2013.03.032
Lin, S. J., Defossez, P. A., and Guarente, L. (2000). Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126–2128. doi: 10.1126/science.289.5487.2126
Lin, S. J., Kaeberlein, M., Andalis, A. A., Sturtz, L. A., Defossez, P. A., Culotta, V. C., et al. (2002). Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418, 344–348. doi: 10.1038/nature00829
Liu, L., Arun, A., Ellis, L., Peritore, C., and Donmez, G. (2014). SIRT2 enhances 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced nigrostriatal damage via apoptotic pathway. Front. Aging Neurosci. 6:184. doi: 10.3389/fnagi.2014.00184
Liu, P. Y., Xu, N., Malyukova, A., Scarlett, C. J., Sun, Y. T., Zhang, X. D., et al. (2013). The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ. 20, 503–514. doi: 10.1038/cdd.2012.147
Lyman, M., Lloyd, D. G., Ji, X., Vizcaychipi, M. P., and Ma, D. (2014). Neuroinflammation: the role and consequences. Neurosci. Res. 79, 1–12. doi: 10.1016/j.neures.2013.10.004
McCleary, D. F., and Rine, J. (2017). Nutritional Control of Chronological Aging and Heterochromatin in Saccharomyces cerevisiae. Genetics 205, 1179–1193. doi: 10.1534/genetics.116.196485
McGinn, M. J., and Povlishock, J. T. (2016). Pathophysiology of Traumatic Brain Injury. Neurosurg. Clin. N. Am. 27, 397–407. doi: 10.1016/j.nec.2016.06.002
Mei, S. C., and Brenner, C. (2015). Calorie restriction-mediated replicative lifespan extension in yeast is non-cell autonomous. PLoS Biol. 13:e1002048. doi: 10.1371/journal.pbio.1002048
Min, J. S., Kim, J. C., Kim, J. A., Kang, I., and Ahn, J. K. (2018). SIRT2 reduces actin polymerization and cell migration through deacetylation and degradation of HSP90. Biochim. Biophys. Acta Mol. Cell Res. 1865, 1230–1238. doi: 10.1016/j.bbamcr.2018.06.005
Moniot, S., Schutkowski, M., and Steegborn, C. (2013). Crystal structure analysis of human Sirt2 and its ADP-ribose complex. J. Struct. Biol. 182, 136–143. doi: 10.1016/j.jsb.2013.02.012
Narayan, N., Lee, I. H., Borenstein, R., Sun, J., Wong, R., Tong, G., et al. (2012). The NAD-dependent deacetylase SIRT2 is required for programmed necrosis. Nature 492, 199–204. doi: 10.1038/nature11700
North, B. J., and Verdin, E. (2007a). Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS One 2:e784. doi: 10.1371/journal.pone.0000784
North, B. J., and Verdin, E. (2007b). Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J. Biol. Chem. 282, 19546–19555. doi: 10.1074/jbc.M702990200
Outeiro, T. F., Kontopoulos, E., Altmann, S. M., Kufareva, I., Strathearn, K. E., Amore, A. M., et al. (2007). Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317, 516–519. doi: 10.1126/science.1143780
Pandithage, R., Lilischkis, R., Harting, K., Wolf, A., Jedamzik, B., Lüscher-Firzlaff, J., et al. (2008). The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J. Cell Biol. 180, 915–929. doi: 10.1083/jcb.200707126
Parashar, V., and Rogina, B. (2009). dSir2 mediates the increased spontaneous physical activity in flies on calorie restriction. Aging 1, 529–541. doi: 10.18632/aging.100061
Perry, V. H., Nicoll, J. A., and Holmes, C. (2010). Microglia in neurodegenerative disease. Nat. Rev. Neurol. 6, 193–201. doi: 10.1038/nrneurol.2010.17
Polito, L., Kehoe, P. G., Davin, A., Benussi, L., Ghidoni, R., Binetti, G., et al. (2013). The SIRT2 polymorphism rs10410544 and risk of Alzheimer’s disease in two Caucasian case-control cohorts. Alzheimers Dement 9, 392–399. doi: 10.1016/j.jalz.2012.02.003
Porcelli, S., Salfi, R., Politis, A., Atti, A. R., Albani, D., Chierchia, A., et al. (2013). Association between Sirtuin 2 gene rs10410544 polymorphism and depression in Alzheimer’s disease in two independent European samples. J. Neural. Transm. 120, 1709–1715. doi: 10.1007/s00702-013-1045-6
Ransohoff, R. M. (2016). How neuroinflammation contributes to neurodegeneration. Science 353, 777–783. doi: 10.1126/science.aag2590
Sacco, R. L., Kasner, S. E., Broderick, J. P., Caplan, L. R., Connors, J. J., Culebras, A., et al. (2013). An updated definition of stroke for the 21st century: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 44, 2064–2089. doi: 10.1161/STR.0b013e318296aeca
Sampaio-Marques, B., Felgueiras, C., Silva, A., Rodrigues, M., Tenreiro, S., Franssens, V., et al. (2012). SNCA (alpha-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy 8, 1494–1509. doi: 10.4161/auto.21275
Sauve, A. A. (2010). Sirtuin chemical mechanisms. Biochim. Biophys. Acta 1804, 1591–1603. doi: 10.1016/j.bbapap.2010.01.021
Sayd, S., Thirant, C., El-Habr, E. A., Lipecka, J., Dubois, L. G., Bogeas, A., et al. (2014). Sirtuin-2 activity is required for glioma stem cell proliferation arrest but not necrosis induced by resveratrol. Stem. Cell Rev. Rep. 10, 103–113. doi: 10.1007/s12015-013-9465-0
Scheltens, P., Blennow, K., Breteler, M. M., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517. doi: 10.1016/s0140-6736(15)01124-1
Schiedel, M., Robaa, D., Rumpf, T., Sippl, W., and Jung, M. (2018). The Current State of NAD(+) -Dependent Histone Deacetylases (Sirtuins) as Novel Therapeutic Targets. Med. Res. Rev. 38, 147–200. doi: 10.1002/med.21436
Scuderi, C., Stecca, C., Bronzuoli, M. R., Rotili, D., Valente, S., Mai, A., et al. (2014). Sirtuin modulators control reactive gliosis in an in vitro model of Alzheimer’s disease. Front. Pharmacol. 5:89. doi: 10.3389/fphar.2014.00089
Serrano, L., Martínez-Redondo, P., Marazuela-Duque, A., Vazquez, B. N., Dooley, S. J., Voigt, P., et al. (2013). The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 27, 639–653. doi: 10.1101/gad.211342.112
She, D. T., Wong, L. J., Baik, S. H., and Arumugam, T. V. (2018). SIRT2 Inhibition Confers Neuroprotection by Downregulation of FOXO3a and MAPK Signaling Pathways in Ischemic Stroke. Mol. Neurobiol. 55, 9188–9203. doi: 10.1007/s12035-018-1058-0
Shen, Y., Chen, L., Zhang, S., and Xie, L. (2020). Correlation Between SIRT2 3’UTR Gene Polymorphism and the Susceptibility to Alzheimer’s Disease. J. Mol. Neurosci. 70, 878–886. doi: 10.1007/s12031-020-01513-y
Shinagawa, S. (2013). Phenotypic variety in the presentation of frontotemporal lobar degeneration. Int. Rev. Psychiatr. 25, 138–144. doi: 10.3109/09540261.2012.743877
Shu, L., Xu, C. Q., Yan, Z. Y., Yan, Y., Jiang, S. Z., and Wang, Y. R. (2019). Post-Stroke Microglia Induce Sirtuin2 Expression to Suppress the Anti-inflammatory Function of Infiltrating Regulatory T Cells. Inflammation 42, 1968–1979. doi: 10.1007/s10753-019-01057-3
Silva, D. F., Esteves, A. R., Oliveira, C. R., and Cardoso, S. M. (2017). Mitochondrial Metabolism Power SIRT2-Dependent Deficient Traffic Causing Alzheimer’s-Disease Related Pathology. Mol. Neurobiol. 54, 4021–4040. doi: 10.1007/s12035-016-9951-x
Singh, C. K., Chhabra, G., Ndiaye, M. A., Garcia-Peterson, L. M., Mack, N. J., and Ahmad, N. (2018). The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox. Signal 28, 643–661. doi: 10.1089/ars.2017.7290
Singh, P., Hanson, P. S., and Morris, C. M. (2017). Sirtuin-2 Protects Neural Cells from Oxidative Stress and Is Elevated in Neurodegeneration. Parkinsons Dis. 2017:2643587. doi: 10.1155/2017/2643587
Spires-Jones, T. L., Fox, L. M., Rozkalne, A., Pitstick, R., Carlson, G. A., and Kazantsev, A. G. (2012). Inhibition of Sirtuin 2 with Sulfobenzoic Acid Derivative AK1 is Non-Toxic and Potentially Neuroprotective in a Mouse Model of Frontotemporal Dementia. Front. Pharmacol. 3:42. doi: 10.3389/fphar.2012.00042
Suzuki, K., and Koike, T. (2007). Mammalian Sir2-related protein (SIRT) 2-mediated modulation of resistance to axonal degeneration in slow Wallerian degeneration mice: a crucial role of tubulin deacetylation. Neuroscience 147, 599–612. doi: 10.1016/j.neuroscience.2007.04.059
Szego, E. M., Gerhardt, E., and Outeiro, T. F. (2017). Sirtuin 2 enhances dopaminergic differentiation via the AKT/GSK-3beta/beta-catenin pathway. Neurobiol. Aging 56, 7–16. doi: 10.1016/j.neurobiolaging.2017.04.001
Tsuchiya, M., Dang, N., Kerr, E. O., Hu, D., Steffen, K. K., Oakes, J. A., et al. (2006). Sirtuin-independent effects of nicotinamide on lifespan extension from calorie restriction in yeast. Aging Cell 5, 505–514. doi: 10.1111/j.1474-9726.2006.00240.x
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., and Kroemer, G. (2010). Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714. doi: 10.1038/nrm2970
Vaquero, A., Scher, M. B., Lee, D. H., Sutton, A., Cheng, H. L., Alt, F. W., et al. (2006). SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 20, 1256–1261. doi: 10.1101/gad.1412706
Wang, B., Zhang, Y., Cao, W., Wei, X., Chen, J., and Ying, W. (2016). SIRT2 Plays Significant Roles in Lipopolysaccharides-Induced Neuroinflammation and Brain Injury in Mice. Neurochem. Res. 41, 2490–2500. doi: 10.1007/s11064-016-1981-2
Wang, G., Li, S., Gilbert, J., Gritton, H.J., Wang, Z., Li, Z., et al. (2017). Crucial Roles for SIRT2 and AMPA Receptor Acetylation in Synaptic Plasticity and Memory. Cell Rep. 20, 1335–1347. doi: 10.1016/j.celrep.2017.07.030
Wang, J., Zhai, Q., Chen, Y., Lin, E., Gu, W., McBurney, M. W., et al. (2005). A local mechanism mediates NAD-dependent protection of axon degeneration. J. Cell Biol. 170, 349–355. doi: 10.1083/jcb.200504028
Wang, Y., Cai, Y., Huang, H., Chen, X., Chen, X., Chen, X., et al. (2018). miR-486-3p Influences the Neurotoxicity of a-Synuclein by Targeting the SIRT2 Gene and the Polymorphisms at Target Sites Contributing to Parkinson’s Disease. Cell Physiol. Biochem. 51, 2732–2745. doi: 10.1159/000495963
Weindruch, R., Walford, R. L., Fligiel, S., and Guthrie, D. (1986). The retardation of aging in mice by dietary restriction: longevity, cancer, immunity and lifetime energy intake. J. Nutr. 116, 641–654. doi: 10.1093/jn/116.4.641
Wongchitrat, P., Pakpian, N., Kitidee, K., Phopin, K., Dharmasaroja, P. A., and Govitrapong, P. (2019). Alterations in the Expression of Amyloid Precursor Protein Cleaving Enzymes mRNA in Alzheimer Peripheral Blood. Curr. Alzheimer. Res. 16, 29–38. doi: 10.2174/1567205015666181109103742
Xia, M., Yu, J. T., Miao, D., Lu, R. C., Zheng, X. P., and Tan, L. (2014). SIRT2 polymorphism rs10410544 is associated with Alzheimer’s disease in a Han Chinese population. J. Neurol. Sci. 336, 48–51. doi: 10.1016/j.jns.2013.10.001
Yoo, D. Y., Kim, D. W., Kim, M. J., Choi, J. H., Jung, H. Y., Nam, S. M., et al. (2015). Sodium butyrate, a histone deacetylase Inhibitor, ameliorates SIRT2-induced memory impairment, reduction of cell proliferation, and neuroblast differentiation in the dentate gyrus. Neurol. Res. 37, 69–76. doi: 10.1179/1743132814y.0000000416
Yuan, F., Xu, Z. M., Lu, L. Y., Nie, H., Ding, J., Ying, W. H., et al. (2016). SIRT2 inhibition exacerbates neuroinflammation and blood-brain barrier disruption in experimental traumatic brain injury by enhancing NF-kappaB p65 acetylation and activation. J. Neurochem. 136, 581–593. doi: 10.1111/jnc.13423
Zhang, H., Park, S. H., Pantazides, B. G., Karpiuk, O., Warren, M. D., Hardy, C. W., et al. (2013). SIRT2 directs the replication stress response through CDK9 deacetylation. Proc. Natl. Acad. Sci. U S A 110, 13546–13551. doi: 10.1073/pnas.1301463110
Zhang, X., Brachner, A., Kukolj, E., Slade, D., and Wang, Y. (2019). SIRT2 deacetylates GRASP55 to facilitate post-mitotic Golgi assembly. J. Cell Sci. 132:232389. doi: 10.1242/jcs.232389
Keywords: SIRT2, neuroinflammation, oxidative stress, synaptic change, axonal degeneration, autophagy, apoptosis, programmed necrosis
Citation: Chen X, Lu W and Wu D (2021) Sirtuin 2 (SIRT2): Confusing Roles in the Pathophysiology of Neurological Disorders. Front. Neurosci. 15:614107. doi: 10.3389/fnins.2021.614107
Received: 08 October 2020; Accepted: 12 April 2021;
Published: 24 May 2021.
Edited by:
Arumugam R. Jayakumar, Miami VA Healthcare System, United StatesReviewed by:
Abdeljalil Elgot, Hassan Premier University, MoroccoCopyright © 2021 Chen, Lu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danhong Wu, danhongwu@fudan.edu.cn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.