Corrigendum: Case Report: Chorea-Acanthocytosis Presents as Epilepsy in a Consanguineous Family With a Nonsense Mutation of in VPS13A
 Fang-Mei Luo
Fang-Mei Luo Ming-Xing Deng3†
Ming-Xing Deng3† Liang-Liang Fan
Liang-Liang Fan- 1Department of Respiratory Medicine, Diagnosis and Treatment Center of Respiratory Disease, The Second XiangYa Hospital of Central South University, Changsha, China
- 2Department of Cell Biology, The School of Life Sciences, Central South University, Changsha, China
- 3Department of Dermatology, Loudi Central Hospital, Loudi, China
- 4Department of Anesthesiology, The Second Xiangya Hospital, Central South University, Changsha, China
- 5Hunan Key Laboratory of Animal Models for Human Diseases, Changsha, China
Chorea-Acanthocytosis (ChAc), a rare autosomal recessive inherited neurological disorder, originated from variants in Vacuolar Protein Sorting 13 homolog A (VPS13A) gene. The main symptoms of ChAc contain hyperkinetic movements, seizures, cognitive impairment, neuropsychiatric symptoms, elevated serum biochemical indicators, and acanthocytes detection in peripheral blood smear. Recently, researchers found that epilepsy may be a presenting and prominent symptom of ChAc. Here, we enrolled a consanguineous family with epilepsy and non-coordinated movement. Whole exome sequencing was employed to explore the genetic lesion of the family. After data filtering, co-separation analysis was performed by Sanger sequencing and bioinformatics analysis, the homozygous nonsense variant (NM_033305.2: c.8282C>G, p.S2761X) of VPS13A were identified which could be genetic factor of the patient. No other meaningful mutations were detected. This mutation (p.S2761X) led to a truncated protein in exon 60 of the VPS13A gene, was simultaneously absent in our 200 local control participants. The homozygous mutation (NM_033305.2: c.8282C>G, p.S2761X) of VPS13A may be the first time be identified in ChAc patient with epilepsy. Our study assisted to the diagnosis of ChAc in this patient and contributed to the genetic diagnosis and counseling of families with ChAc presented as epilepsy. Moreover, we further indicated that epilepsy was a crucial phenotype in ChAc patients caused by VPS13A mutations.
Introduction
Chorea-acanthocytosis (ChAc, OMIM #200150) is a primary neurological disorder characterized by repetitive movements of various parts of the body and abnormal red blood cell shape (Velayos Baeza et al., 1993; Liu et al., 2018; Roulis et al., 2018). As fewer than 1000 cases have been reported worldwide, researchers commonly consider ChAc to be a rare genetic disease (Peikert et al., 2018). Previous studies have revealed that autosomal-recessive mutations in the Vacuolar Protein Sorting 13 homolog A (VPS13A) gene may be the genetic lesion of ChAc (Velayos Baeza et al., 1993). Epilepsy is also a central nervous system disorder that is described as “a common brain condition that causes repeated seizures.” In addition to recurrent seizures, other symptoms of epilepsy include temporary confusion, staring spells, uncontrollable jerking movements of the arms and legs, and loss of consciousness or awareness (Yuen et al., 2018; Thijs et al., 2019). A recent study revealed that the prevalence of epilepsy was 3.5–6.5 per 1,000 children and 10.8 per 1,000 elderly individuals worldwide (Abramovici and Bagic, 2016). In a recent summary of 5 years of research in patients with VPS13A mutations, ~45% of patients have a history of epileptic seizures, which is consistent with previous studies in ChAc claiming that ~42% of ChAc patients have at least one seizure at some point in their clinical course (Benninger et al., 2016).
In the past decade, the rapidly growing number of genes underlying genetic epilepsy has been attributed to the application of high-throughput sequencing technology. According to the EpilepsyGene database (http://www.wzgenomics.cn/EpilepsyGene/index.php), ~500 genes may be associated with epilepsy, and ~2% of idiopathic epilepsy patients are thought to be monogenic (Ran et al., 2015). Mutations in Sodium Voltage-Gated Channel Alpha Subunit 1 (SCN1A), Potassium Voltage-Gated Channel Subfamily Q Member 2 (KCNQ2), Cyclin Dependent Kinase Like 5 (CDKL5), Protocadherin 19 (PCDH19), and Proline Rich Transmembrane Protein 2 (PRRT2) were the most common genetic lesions of epilepsy (Wang J. et al., 2017; Kang et al., 2019). In addition, recent studies revealed that epilepsy may be a presenting and prominent symptom of ChAc caused by VPS13A mutations (Benninger et al., 2016; Weber et al., 2018).
Here, a patient with simultaneous epilepsy and non-coordinated movement was enrolled. Family history indicated that the parents were in a consanguineous marriage. Whole-exome sequencing and Sanger sequencing were applied to explore the candidate genes of the family.
Case Presentation
All 13 family members were investigated in this study (Figure 1A). The peripheral blood samples of one patient (IV-3) and three unaffected family members (IV-2, IV-4, and V-1) were collected. Clinical data, such as magnetic resonance imaging (MRI) and electroencephalogram (EEG), were recorded carefully. In addition, 200 unrelated, ethnically matched healthy controls were used as internal controls to exclude SNPs in local individuals. These healthy controls (male/female: 100/100, age 36.7 ± 8.6 years) lacked ChAc diagnostic features. Each participant underwent thorough examination for clinical diagnosis or exclusion, including general examination, such as EEG and other behavioral testing.
FIGURE 1
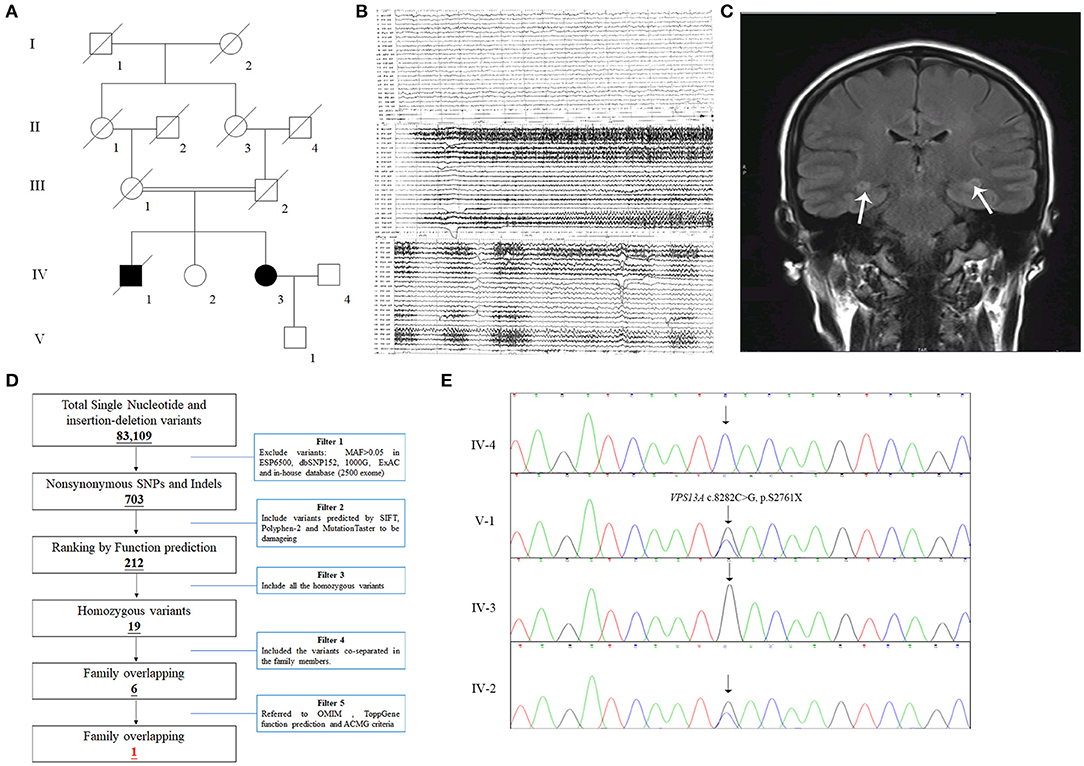
Figure 1. The clinical and genetic data of the family. (A) Pedigree of the family with epilepsy. Double lines indicate consanguineous unions. Square denotes male family member, circle represents female family member, slashed symbol indicates deceased family member, fully shaded symbol shows patient with epilepsy, and open symbol presents no symptoms member. (B) EEG testing of the IV-3 showed frequent abnormal epileptiform discharges in the right temple, and a small amount of epileptiform discharges in the left; (C) MRI scanning of the IV-3 revealed that T2 and T2 flair signals of bilateral hippocampus were slightly higher (the arrows), the hippocampal neuron may be damaged. (D) Schematic representation of the filter strategies employed in this study. (E) Sanger sequencing validated that the patient IV-3 carried a homozygous mutation (NM_033305.2: c.8282C>G, p.S2761X) of VPS13A and her son (V-1) and sister (IV-2) carried the heterozygous mutation.
The patient, a 43-year-old female, was admitted to the hospital due to lung infection. However, the patient presented recurrent seizures during hospitalization. EEG testing showed frequent abnormal epileptiform discharges in the right temple and a small amount of epileptiform discharges in the left temple (Figure 1B). MRI scanning revealed that T2 and T2 flair signals of the bilateral hippocampus were slightly higher, and hippocampal neurons may be damaged (Figure 1C). Serum creatine kinase, l-lactate dehydrogenase and alpha hydroxybutyrate dehydrogenase levels were all within normal ranges (Table 1). Medical history revealed that the patient presented mild non-coordinated mandibular movement and upper limb movements for seven months. A family history survey indicated that her mother and father were intermarried, and her brother (IV-1) also presented non-coordinated mandibular movement and upper limb movements at 37 years old according to the family members' descriptions. No other family members presented similar symptoms.
TABLE 1
Table 1. The clinical summary of the patient.
Genetic Analysis
Genomic DNA was extracted from peripheral blood lymphocytes of four family members with PureLink™ Genomic DNA Mini Kit (Invitrogen, the USA). We selected the proband's DNA to perform the whole exome sequencing. Whole exome sequencing services were provided by the Novogene Bioinformatics Institute (Beijing, China). Exomes were captured by Agilent SureSelect Human All Exon V6 kits, and next-generation sequencing was conducted with an Illumina HiSeq X-10 system as we previous described (Yu et al., 2017; Wang et al., 2020).
The strategies of data filtering are as follows (Figure 1D): (a) Non-synonymous SNPs or frameshift-causing INDELs with an alternative allele frequency > 0.05 in the NHLBI Exome Sequencing Project Exome Variant Server (ESP6500), dbSNP152 (http://www.ncbi.nlm.nih.gov/projects/SNP/index.html), the 1000 Genomes project (http://www.1000genomes.org/), the ExAC database (http://exac.broadinstitute.org) or in-house exome databases of Novogene (2500 exomes) were excluded; (b) the filtered SNVs and INDELs, predicted by SIFT (http://sift.jcvi.org/), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (http://www.mutationtaster.org/) to be damaging, were remained; (c) all the homozygous mutations were remained; (d) Co-segregation analysis was conducted in the family.
Here, 99.3% coverage of the target regions and 97.70 × sequencing depth were achieved for the proband. Approximately 83,109 variants were found in the proband. Via the abovementioned filtering method, six homozygous mutations were detected (Table 2). Further Sanger sequencing and bioinformatics analysis indicated that only the nonsense mutation (NM_033305.2: c.8282C>G, p.S2761X) in the VPS13A gene may be the genetic lesion of the patient (Figure 1E). No other potential pathogenic mutations for epilepsy diseases were found. The novel mutation, generating a truncated protein in exon 60 of the VPS13A gene, was absent in our 200 local control participants. Bioinformatics programs predicted that this mutation (NM_033305.2: c.8282C>G, p. S2761X) is a pathogenic mutation located in an evolutionarily conserved site of the VPS13A protein. According to ACMG guidelines (Richards et al., 2015), this mutation belongs to pathogenic (PVS1+PM2+PM3).
TABLE 2
Table 2. The filtered homozygous mutations of whole exome sequencing.
Discussion
After data filtering, six homozygous mutations remained, including the mutation (NM_033305.2: c.8282C>G, p.S2761X) of VPS13A. In the other five homozygous mutations, previous studies have revealed that mutations in NDUFAF7 may lead to pathologic myopia (Wang B. et al., 2017), and mutations in DSP may be linked to cardiomyopathy (Singh et al., 2018). Hence, we exclude these two mutations (NM_144736.5: c.581T>A and NM_004415.4: c.1_2insC). The GDPD4 gene may be related to obesity (Kaewsutthi et al., 2016), and RPUSD1 may be related to the methylome at birth and adolescence (Alfano et al., 2019). Hence, mutations in both GDPD4 and RPUSD1 may have extremely low effects on epilepsy. Few studies on the LMF2 gene are available, and ToppGene predicted that this gene may be related to digestion of dietary lipids. In our candidate mutations, the VPS13A mutation was a nonsense mutation, which has been reported in ChAc. In recent years, an increasing number of studies have revealed that epilepsy may be a presenting and prominent symptom of ChAc caused by VPS13A mutations (Benninger et al., 2016; Weber et al., 2018). In addition, according to ACMG criteria, only VPS13A and DSP mutations belong to the pathogenic level. Synthetic judgments indicated that the mutation (NM_033305.3: c.8282C>G) of VPS13A was the genetic lesion of the family.
The human VPS13A gene encoding a vacuolar protein sorting-associated protein named chorein is located on chromosome 9q21.2 and consists of 72 exons spanning ~244.2 kilobases (kb) (Velayos-Baeza et al., 2004). Chorein controls steps in the cycling of proteins through the trans-Golgi network to endosomes, lysosomes and the plasma membrane (Kumar et al., 2018; Yeshaw et al., 2019). In 2001, Rampoldi et al. identified 16 different mutations of VPS13A in 11 unrelated families with ChAc, indicating that VPS13A mutations might be involved in the etiology of this autosomal recessive disorder (Rampoldi et al., 2001). Since then, more than 100 VPS13A gene mutations have been identified in ChAc patients (Shen et al., 2017). However, in recent years, an increasing number of studies have revealed that ChAc caused by VPS13A mutations may present a prominent symptom of epilepsy or even present only an isolated epilepsy phenotype (Al-Asmi et al., 2005; Benninger et al., 2016; Weber et al., 2018). In this study, we identified a homozygous nonsense variant (NM_033305.2: c.8282C>G, p.S2761X) of VPS13A in a consanguineous family with epilepsy, which further demonstrated that VPS13A mutation may exhibit an epilepsy phenotype.
The human VPS13A protein contains several conserved domains, including the N-terminal chorein domain (aa 3-119), SHR-binding domain and APT1 domain (aa 2522-2856) (Rzepnikowska et al., 2017; Kumar et al., 2018; Soczewka et al., 2019). Previous studies have indicated that phosphatidylinositol 3-phosphate (PI3P) can regulate the functioning of VPS13A, both in protein trafficking and actin cytoskeleton organization (Rzepnikowska et al., 2017). VPS13A can interact with actin and regulate the level of phosphatidylinositol 4-phosphate (PI4P) in the membranes of neuronal cells (Rzepnikowska et al., 2017). In our study, the nonsense mutation (NM_033305.2: c.8282C>G, p. S2761X) may generate a truncated protein without an integrated APT1 domain, which can disrupt the structure and function of the VPS13A protein and affect the level of phosphatidylinositol 4-phosphate (PI4P) in neuronal cell membranes, disrupt the structure and function of neurons, and finally present T2 and T2 flair signals in the bilateral hippocampus that increase on MRI, which indicates damage to hippocampal neurons.
Simultaneously, recent studies have also indicated that the VPS13A protein may also regulate the storage and release of calcium (Yu et al., 2016; Pelzl et al., 2017a,b). VPS13A can interact with and foster stimulation of phosphoinositide-3-kinase (PI3K), which may further activate NF-κB with subsequent upregulation of the Ca2+ channel (Sukkar et al., 2018). Studies have indicated that the functional connection between VPS13A and calcium signaling is a possible target for chemical intervention in ChAc (Soczewka et al., 2019). Lithium treatment can reverse the enhanced neuronal apoptosis of ChAc fibroblasts, which is caused by decreased SOCE, a Ca2+-channel accomplishing store-operated Ca2+ entry (Pelzl et al., 2017b; Sukkar et al., 2018). In our study, the mutation (NM_033305.2: c.8282C>G, p. S2761X) changed the structure of VPS13A and may affect the interaction between VPS13A and PI3K, disrupting the release of calcium signals in neurons and leading to epilepsy and one calcium disorder (Steinlein, 2014), presenting frequent abnormal epileptiform discharges in EEG.
The mutation (NM_033305.2: c.8282C>G, p. S2761X) of VPS13A was first identified in a ChAc patient who carried the heterozygous mutation p. S2761X and did not present epilepsy phenotypes (Shen et al., 2017). Here, we may be the first detect the homozygous mutation (NM_033305.2: c.8282C>G, p. S2761X) of VPS13A in a Chinese consanguineous family member who presented epilepsy phenotypes. Recently, an increasing number of studies have revealed that epilepsy is a crucial presentation in patients with VPS13A mutations (Benninger et al., 2016; Weber et al., 2018). Therefore, our study provided a new epilepsy case caused by a VPS13A mutation, which may further confirm the epilepsy phenotype in ChAc and expand the phenotype of the mutation (NM_033305.2: c.8282C>G, p.S2761X) of VPS13A. Some limitations in our study should be noted. For instance, we only enrolled one patient with ChAc who presented with an epilepsy phenotype. The data filtering of whole-exome sequencing was insufficient to exclude unknown mutations. Additional functional analysis of the VPS13A protein with this mutation is recommended and may result in additional information about the pathogenetic mechanism of ChAc presented as epilepsy.
Conclusions
In summary, a homozygous missense mutation (NM_033305.2: c.8282C>G, p. S2761X) in the VPS13A gene was identified in a consanguineous Chinese family with epilepsy. To our knowledge, this is the first report of the homozygous VPS13A c.8282C>G mutation in a ChAc patient. Our study further confirmed that epilepsy was an important phenotype in patients with mutated VPS13A, which may extend the genotype-phenotype relationship between mutations in the VPS13A gene and clinical findings of ChAc and help us understand how the different mutations could contribute to different symptoms of the same disease. In addition, our study may be helpful in the genetic counseling of patients with VPS13A mutations.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Second Xiangya Hospital of Central South University, Changsha, China. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
F-ML and M-XD enrolled the family members. M-XD performed DNA isolation and Sanger sequencing. F-ML and RY performed genetic analysis. F-ML and LL wrote the manuscript. L-LF supported the project. All authors reviewed the manuscript.
Funding
This study was supported by National Natural Science Foundation of China (81800220, 82000079, and 82000427) and National Natural Science Foundation of Hunan province (2020JJ5785).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all subjects for participating in this study. We thank Dr. Shuai Guo from University of Texas MD Anderson Cancer Center, the USA for editing the language.
References
Abramovici, S., and Bagic, A. (2016). Epidemiology of epilepsy. Handb. Clin. Neurol. 138, 159–171. doi: 10.1016/B978-0-12-802973-2.00010-0
Al-Asmi, A., Jansen, A. C., Badhwar, A., Dubeau, F., Tampieri, D., Shustik, C., et al. (2005). Familial temporal lobe epilepsy as a presenting feature of choreoacanthocytosis. Epilepsia 46, 1256–1263. doi: 10.1111/j.1528-1167.2005.65804.x
Alfano, R., Guida, F., Galobardes, B., Chadeau-Hyam, M., Delpierre, C., Ghantous, A., et al. (2019). Socioeconomic position during pregnancy and DNA methylation signatures at three stages across early life: epigenome-wide association studies in the ALSPAC birth cohort. Int. J. Epidemiol. 48, 30–44. doi: 10.1093/ije/dyy259
Benninger, F., Afawi, Z., Korczyn, A. D., Oliver, K. L., Pendziwiat, M., Nakamura, M., et al. (2016). Seizures as presenting and prominent symptom in chorea-acanthocytosis with c.2343del VPS13A gene mutation. Epilepsia 57, 549–556. doi: 10.1111/epi.13318
Kaewsutthi, S., Santiprabhob, J., Phonrat, B., Tungtrongchitr, A., Lertrit, P., and Tungtrongchitr, R. (2016). Exome sequencing in Thai patients with familial obesity. Genet. Mol. Res. 15:gmr8311. doi: 10.4238/gmr.15028311
Kang, K. W., Kim, W., Cho, Y. W., Lee, S. K., Jung, K. Y., Shin, W., et al. (2019). Genetic characteristics of non-familial epilepsy. PeerJ. 7:e8278. doi: 10.7717/peerj.8278
Kumar, N., Leonzino, M., Hancock-Cerutti, W., Horenkamp, F. A., Li, P., Lees, J. A., et al. (2018). VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 217, 3625–3639. doi: 10.1083/jcb.201807019
Liu, Y., Liu, Z. Y., Wan, X. H., and Guo, Y. (2018). Progress in the diagnosis and management of chorea-acanthocytosis. Chin. Med. Sci. J. 33, 53–59. doi: 10.1016/S1001-9294(10)60021-1
Peikert, K., Danek, A., and Hermann, A. (2018). Current state of knowledge in chorea-acanthocytosis as core neuroacanthocytosis syndrome. Eur. J. Med. Genet. 61, 699–705. doi: 10.1016/j.ejmg.2017.12.007
Pelzl, L., Elsir, B., Sahu, I., Bissinger, R., Singh, Y., Sukkar, B., et al. (2017a). Lithium sensitivity of store operated Ca2+ entry and survival of fibroblasts isolated from chorea-acanthocytosis patients. Cell. Physiol. Biochem. 42, 2066–2077. doi: 10.1159/000479901
Pelzl, L., Hauser, S., Elsir, B., Sukkar, B., Sahu, I., Singh, Y., et al. (2017b). Lithium sensitive ORAI1 expression, store operated Ca(2+) entry, and suicidal death of neurons in chorea-acanthocytosis. Sci. Rep. 7:6457. doi: 10.1038/s41598-017-06451-1
Rampoldi, L., Dobson-Stone, C., Rubio, J. P., Danek, A., Chalmers, R. M., Wood, N. W., et al. (2001). A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat. Genet. 28, 119–120. doi: 10.1038/88821
Ran, X., Li, J., Shao, Q., Chen, H., Lin, Z., Sun, Z. S., et al. (2015). EpilepsyGene: a genetic resource for genes and mutations related to epilepsy. Nucleic Acids Res. 43, D893–D899. doi: 10.1093/nar/gku943
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Roulis, E., Hyland, C., Flower, R., Gassner, C., Jung, H. H., and Frey, B. M. (2018). Molecular basis and clinical overview of mcleod syndrome compared with other neuroacanthocytosis syndromes: a review. JAMA Neurol. 75, 1554–1562. doi: 10.1001/jamaneurol.2018.2166
Rzepnikowska, W., Flis, K., Kaminska, J., Grynberg, M., Urbanek, A., Ayscough, K. R., et al. (2017). Amino acid substitution equivalent to human chorea-acanthocytosis I2771R in yeast Vps13 protein affects its binding to phosphatidylinositol 3-phosphate. Hum. Mol. Genet. 26, 1497–1510. doi: 10.1093/hmg/ddx054
Shen, Y., Liu, X., Long, X., Han, C., Wan, F., Fan, W., et al. (2017). Novel VPS13A gene mutations identified in patients diagnosed with chorea-acanthocytosis (ChAc): case presentation and literature review. Front. Aging Neurosci. 9:95. doi: 10.3389/fnagi.2017.00095
Singh, S. M., Casey, S. A., Berg, A. A., Abdelhadi, R. H., Katsiyiannis, W. T., Bennett, M. K., et al. (2018). Autosomal-dominant biventricular arrhythmogenic cardiomyopathy in a large family with a novel in-frame DSP nonsense mutation. Am. J. Med. Genet. A 176, 1622–1626. doi: 10.1002/ajmg.a.38719
Soczewka, P., Kolakowski, D., Smaczynska-de Rooij, I., Rzepnikowska, W., Ayscough, K. R., Kaminska, J., et al. (2019). Yeast-model-based study identified myosin- and calcium-dependent calmodulin signalling as a potential target for drug intervention in chorea-acanthocytosis. Dis. Model. Mech. 12:dmm036830. doi: 10.1242/dmm.036830
Steinlein, O. K. (2014). Calcium signaling and epilepsy. Cell Tissue Res. 357, 385–393. doi: 10.1007/s00441-014-1849-1
Sukkar, B., Hauser, S., Pelzl, L., Hosseinzadeh, Z., Sahu, I., Al-Maghout, T., et al. (2018). Inhibition of lithium sensitive orai1/STIM1 expression and store operated Ca2+ entry in chorea-acanthocytosis neurons by NF-kappaB inhibitor wogonin. Cell. Physiol. Biochem. 51, 278–289. doi: 10.1159/000495229
Thijs, R. D., Surges, R., O'Brien, T. J., and Sander, J. W. (2019). Epilepsy in adults. Lancet 393, 689–701. doi: 10.1016/S0140-6736(18)32596-0
Velayos Baeza, A., Dobson-Stone, C., Rampoldi, L., Bader, B., Walker, R. H., Danek, A., et al. (1993). “Chorea-Acanthocytosis,” in GeneReviews(R), eds. M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, and A. Amemiya (Seattle, WA: University of Washington).
Velayos-Baeza, A., Vettori, A., Copley, R. R., Dobson-Stone, C., and Monaco, A. P. (2004). Analysis of the human VPS13 gene family. Genomics 84, 536–549. doi: 10.1016/j.ygeno.2004.04.012
Wang, B., Liu, Y., Chen, S., Wu, Y., Lin, S., Duan, Y., et al. (2017). A novel potentially causative variant of NDUFAF7 revealed by mutation screening in a Chinese family with pathologic myopia. Invest. Ophthalmol. Vis. Sci. 58, 4182–4192. doi: 10.1167/iovs.16-20941
Wang, C. Y., Chen, Y. Q., Jin, J. Y., Du, R., Fan, L. L., and Xiang, R. (2020). A novel nonsense mutation of ABCA8 in a han-Chinese family with ASCVD leads to the reduction of HDL-c levels. Front. Genet. 11:755. doi: 10.3389/fgene.2020.00755
Wang, J., Lin, Z. J., Liu, L., Xu, H. Q., Shi, Y. W., Yi, Y. H., et al. (2017). Epilepsy-associated genes. Seizure 44, 11–20. doi: 10.1016/j.seizure.2016.11.030
Weber, J., Frings, L., Rijntjes, M., Urbach, H., Fischer, J., Weiller, C., et al. (2018). Chorea-acanthocytosis presenting as autosomal recessive epilepsy in a family with a novel VPS13A mutation. Front. Neurol. 9:1168. doi: 10.3389/fneur.2018.01168
Yeshaw, W. M., van der Zwaag, M., Pinto, F., Lahaye, L. L., Faber, A. I., Gomez-Sanchez, R., et al. (2019). Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. Elife 8:e43561. doi: 10.7554/eLife.43561.038
Yu, R., Liu, L., Chen, C., and Shen, J. M. (2017). Exome sequencing identifies a novel DES mutation (R227C) in a Chinese dilated cardiomyopathy family. Cardiology 137, 78–82. doi: 10.1159/000455181
Yu, W., Honisch, S., Schmidt, S., Yan, J., Schmid, E., Alkahtani, S., et al. (2016). Chorein sensitive orai1 expression and store operated Ca2+ entry in rhabdomyosarcoma cells. Cell. Physiol. Biochem. 40, 1141–1152. doi: 10.1159/000453168
Keywords: chorea-acanthocytosis, epilepsy, VPS13A, homozygous variant, CHAC
Citation: Luo F-M, Deng M-X, Yu R, Liu L and Fan L-L (2021) Case Report: Chorea-Acanthocytosis Presents as Epilepsy in a Consanguineous Family With a Nonsense Mutation of in VPS13A. Front. Neurosci. 15:604715. doi: 10.3389/fnins.2021.604715
Received: 10 September 2020; Accepted: 21 January 2021;
Published: 10 February 2021.
Edited by:
Ole A. Andreassen, University of Oslo, NorwayReviewed by:
William Hennah, Orion Corporation, FinlandIda Sønderby, Oslo University Hospital, Norway
Copyright © 2021 Luo, Deng, Yu, Liu and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lv Liu, ZG9jbGl1bHYmI3gwMDA0MDtjc3UuZWR1LmNu; Liang-Liang Fan, c3dmYW5saWFuZ2xpYW5nJiN4MDAwNDA7Y3N1LmVkdS5jbg==
†These authors have contributed equally to this work