 Grant Richter
Grant Richter Vivian W. Y. Liao
Vivian W. Y. Liao Philip K. Ahring
Philip K. Ahring Mary Chebib
Mary Chebib
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurosci., 19 November 2020
Sec. Neuropharmacology
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.599812
This article is part of the Research TopicPharmacological Aspects of Ligand-gated Ion Channels as Targets of Natural and Synthetic AgentsView all 18 articles
γ-Aminobutyric-acid type A (GABAA) receptors expressing the γ1 or γ3 subunit are only found within a few regions of the brain, some of which are involved in sleep. No known compounds have been reported to selectively target γ1- or γ3-containing GABAA receptors. Pharmacological assessments of this are conflicting, possibly due to differences in experimental models, conditions, and exact protocols when reporting efficacies and potencies. In this study, we evaluated the modulatory properties of five non-benzodiazepine Z-drugs (zaleplon, indiplon, eszopiclone, zolpidem, and alpidem) used in sleep management and the benzodiazepine, diazepam on human α1β2γ receptors using all three γ subtypes. This was accomplished using concatenated GABAA pentamers expressed in Xenopus laevis oocytes and measured via two-electrode voltage clamp. This approach removes the potential for single subunits to form erroneous receptors that could contribute to the pharmacological assessment of these compounds. No compound tested had significant effects on γ1-containing receptors below 10 μM. Interestingly, zaleplon and indiplon were found to modulate γ3-containing receptors equally as efficacious as γ2-containing receptors. Furthermore, zaleplon had a higher potency for γ3- than for γ2-containing receptors, indicating certain therapeutic effects could occur via these γ3-containing receptors. Eszopiclone modulated γ3-containing receptors with reduced efficacy but no reduction in potency. These data demonstrate that the imidazopyridines zaleplon and indiplon are well suited to further investigate potential γ3 effects on sleep in vivo.
γ-Aminobutyric-acid type A (GABAA) receptors are ligand-gated ion channels that mediate most inhibitory responses in the brain. These receptors are made up of five building block subunits, and in mammals, there are nineteen identified subunits, α1-6, β1-3, γ1-3, δ, ε, θ, π, and ρ1-3 (Sieghart and Savic, 2018). Receptors typically form from two α, two β, and one of either γ or δ with the most widely expressed combination made from two α1, two β2/3, and one γ2, denoted as α1β2/3γ2 (Olsen and Sieghart, 2008). Distinctive GABAA receptor subtypes are found based on their cellular and anatomical locations and behave differently in response to agonists and modulating compounds.
Each GABAA subunit contains a principal side (+) and a complimentary side (−). GABA binding within the β(+) and α(−) interface induces a conformational change in the receptor channel allowing Cl– ions to pass into the cell to hyperpolarize neurons and make action potentials less likely (Figure 1). Benzodiazepines and Z-drugs allosterically modulate GABAA receptors making the frequency of Cl– channel opening more likely. These drugs bind to the interface within the α(+) and γ(−) (Sigel and Buhr, 1997; Zhu et al., 2018) to reduce the brain’s excitability and thus are primarily prescribed for their effects as anxiolytics, hypnotics, anti-epileptics, and muscle relaxants. Z-drugs are the most commonly prescribed treatment for insomnia and compared with benzodiazepines they more closely induce normal physiological sleep (Klimm et al., 1987; Fleming et al., 1988). However, it is still not precisely characterized which regions Z-drugs act on to induce sleep.
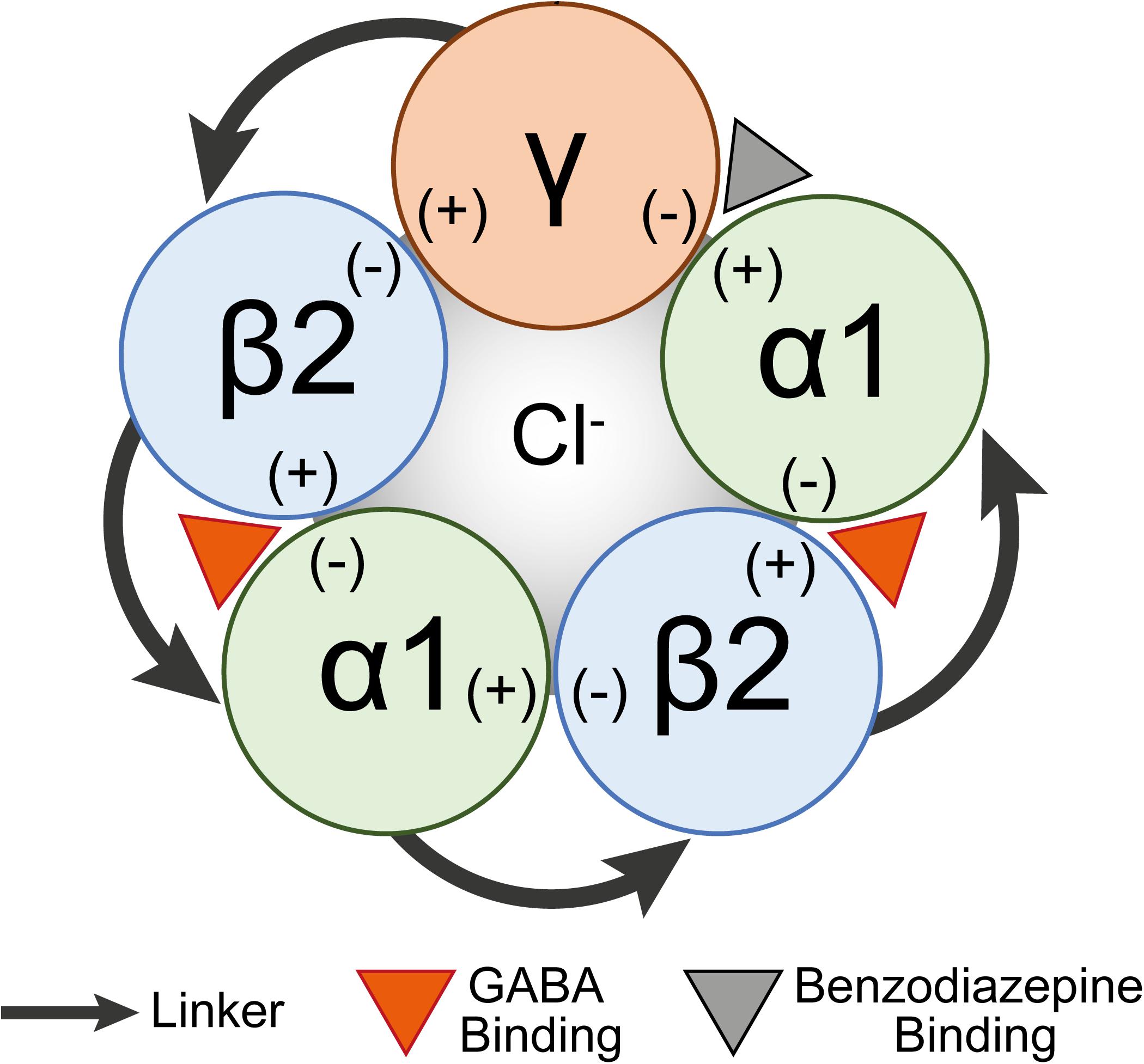
Figure 1. A schematic diagram of a concatenated pentamer GABAA receptor construct. Linkers concatenating subunits are shown as arrows. The GABA binding site (orange arrowhead) is shown between the β2(+) and α(–) interfaces and the benzodiazepine binding site (gray arrowhead) is between the α1(+) and γ(–) interfaces.
While three γ subunits exist, the actions of benzodiazepines and Z-drugs have typically been associated with the γ2 subunit with little information available for γ1 and γ3 subunits. The γ1 or γ3-subunits are found in at most 10 or 15% of GABAA receptors, respectively (Quirk et al., 1994; Benke et al., 1996; Sieghart and Sperk, 2002). Temporally, the γ2 subunit is expressed throughout all stages of development, while γ1 subunit expression peaks around birth and γ3 subunit expression peaks in 2-week old animals (Laurie et al., 1992; Allen Institute for Brain Science, 2008) GABAA receptors with a γ1 subunit have been detected mainly in the amygdala, basal ganglia, hypothalamus, thalamus, and in astrocytes, while receptors with γ3 subunits show some expression in the basal ganglia, thalamus, and midbrain (Bovolin et al., 1992; Quirk et al., 1994; Pirker et al., 2000; Sieghart and Sperk, 2002; Hertz and Chen, 2010). The thalamus and hypothalamus regions are intricately involved in the maintenance of the sleep-wake cycle (Gent et al., 2018) and Z-drugs have been shown to affect clusters of nuclei in these regions (Jia et al., 2009; Kumar et al., 2011; Uygun et al., 2016). Hence, it is a genuine possibility that γ1- or γ3-containing receptors could also play a role in the hypnotic effects of Z-drugs. Indeed the interface between α(+)/γ(−) is believed to be sensitive to benzodiazepine binding in both γ1 and γ3 containing receptors, though some ligands might have lower potencies and/or efficacies because of amino acid sequence differences (Knoflach et al., 1991; Sieghart, 1995; Khom et al., 2006).
Although some Z-drugs have been evaluated on γ1 or γ3-containing GABAA receptors, it is difficult to conclude any clear effects mediated from these subunits as there is conflicting literary evidence of the modulative ability of Z-drugs. This may be due to differences in experimental models, conditions, and exact protocols reported for efficacy and potencies of these compounds. Furthermore, studies that utilize Xenopus laevis oocytes to investigate the pharmacology of Z-drugs have conflicting results potentially due to using single subunit cRNAs to express recombinant receptors. Using single subunit cRNAs in a heterologous expression system can potentially result in a mix of receptor populations. For example, if unlinked cRNAs for α1, β2, and γ subunits are injected into a cell, there is potential for GABAA receptors to assemble from only α1 and β2 with two different stoichiometries [i.e., (α1)2(β2)3 or (α1)3(β2)2], potentially confounding results. Therefore, our group has recently optimized receptor concatenation technology to ensure a single receptor subtype population with assembly in the correct orientation (Liao et al., 2019).
In the present study, we systematically evaluated the pharmacology of five Z-drugs including the pyrazolopyrimidines (zaleplon and indiplon), cyclopyrrolones (zopiclone and its isolated S-enantiomer eszopiclone), and imidazopyridines (zolpidem and alpidem), along with diazepam on γ1, γ2, and γ3 concatenated pentameric GABAA receptors (Figure 2). We found that zaleplon, indiplon, and eszopiclone show comparable efficacy and potency on γ3 as γ2-containing receptors. Furthermore, zolpidem and alpidem modulate γ2 receptors with exclusive selectivity at concentrations below 10 μM. These data clarify conflicting observations and provide further insight into the receptor subtype populations targeted by Z-drugs, and identifies zaleplon, indiplon, and possibly eszopiclone as useful tools for further studies that understand the role γ3-containing receptors in sleep.
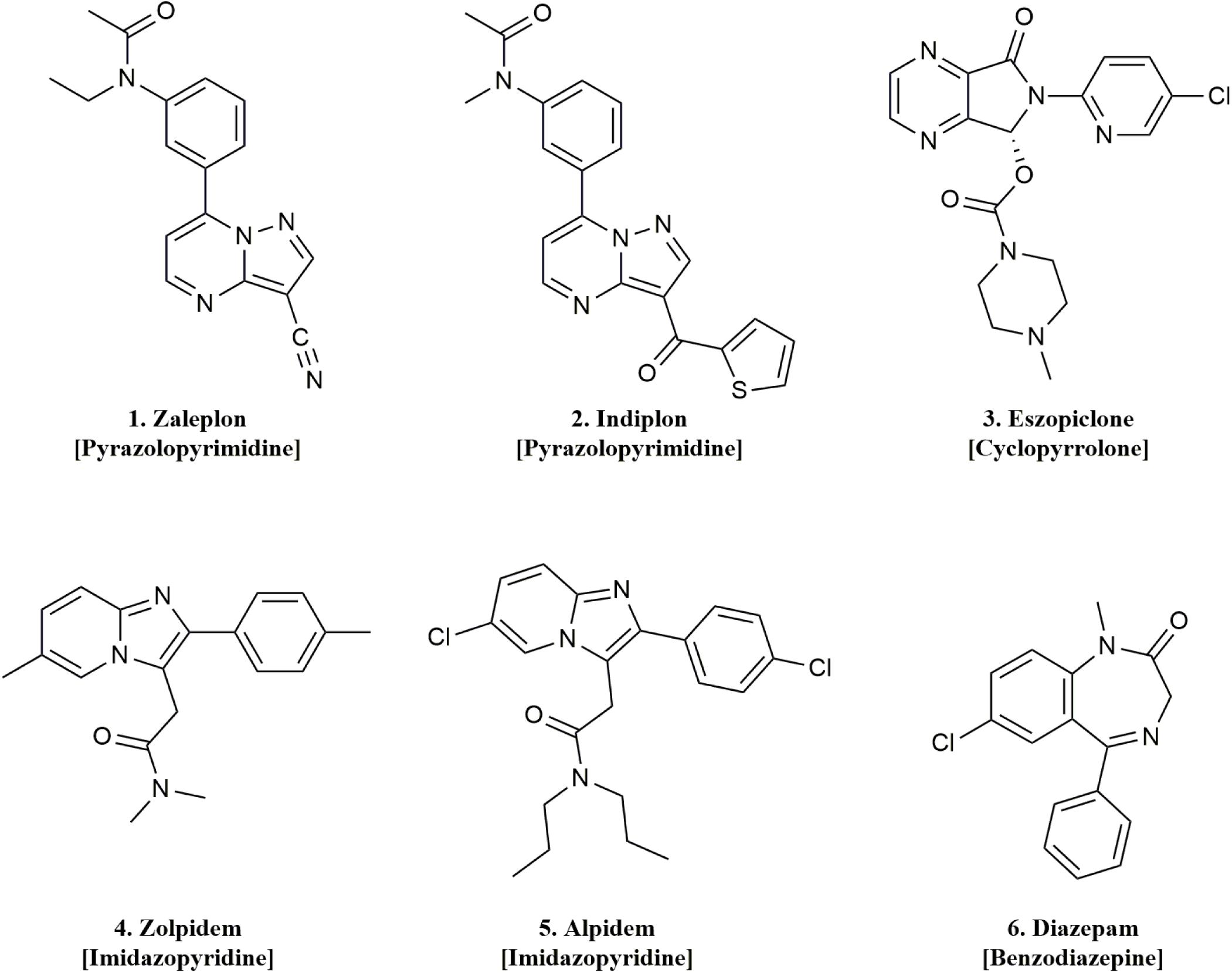
Figure 2. Chemical structures and classes of the drugs used in this study.
GABA, diazepam, alpidem, and all salts and chemicals not specifically mentioned were purchased from Sigma-Aldrich. Zolpidem was purchased from Chemieliva (Yubei District, Chongqing, China), zaleplon was purchased from Alomone Labs (Jerusalem, Israel), eszopiclone was purchased from Clearsynth (NJ, United States), and indiplon was purchased from Tocris (VIC, Australia). Human cDNA for α1 β2, γ1,2,3 GABAA receptor subunits were gifts from Saniona A/S. Oligonucleotides were purchased from Sigma-Aldrich. Restriction enzymes, Q5 polymerase, T4 DNA ligase, and 10-beta competent Escherichia coli were from New England Biolabs (Ipswich, MA, United States). Collagenase A was purchased from Roche (Basel, Switzerland). DNA purification kits were from Qiagen (Hilden, Germany). The QuickChange II Site-Directed Mutagenesis Kit was from Agilent Technologies (Santa Clara, CA, United States). The mMessage mMachine T7 transcription kit–were purchased from Thermo Fisher Scientific (Waltham, MA, United States).
To ensure homogenous receptor populations and subunit orientation, we used concatenated receptors expressed in X. laevis oocytes. Concatenated pentameric constructs were created using the subunits γx-β2-α1-β2-α1 (where x = 1, 2, or 3). A detailed description of the creation of concatenated receptor constructs has been previously described (Liao et al., 2019). Briefly, natural restriction sites BamHI, HindIII, and KpnI restriction sites in the γ1, 2, 3, β2, or α1 subunits were removed through silent mutations using site-directed mutagenesis. Linker sequences of 13 amino acids inserted between the natural C-terminal in the transmembrane segment 4 of the γ subunit and the N-terminal leucine anchor of the β subunit through standard PCR reactions and subunit cDNA ligated together (corresponding to a total linker length of 28 total amino acids between subunits) This was found to be an optimal length for relatively pure receptor expression and orientation without compromising function (Liao et al., 2019). Inserted linker lengths are as follows γx-13a-β2-27a-α1-18a-β2-27a-α1. E. coli bacteria were hosts for plasmid amplification and plasmid purification was performed using standard kits. RNA was produced from DNA using the mMessage mMachine T7 Transcription kit (Thermo Fisher Scientific, Waltham, MA, United States), but due to the size of the pentameric constructs (>10 kb), guanosine triphosphate concentration was increased to give a final cap analog to guanosine triphosphate ratio of 2:1.
The collection and preparation of oocytes were done as previously described (Ahring et al., 2016). Briefly, ovarian lobes were removed from anesthetized adult X. laevis following protocol approval by the Animal Ethics Committee of The University of Sydney (AEC No. 2016/970) in accordance with the National Health and Medical Research Council of Australia code for the care and use of animals. Oocytes were prepared by slicing lobes into small pieces and defolliculated through collagenase A treatment. Stage V and VI oocytes were injected with around 50 nL of 0.5 ng/nL RNA for each concatenated construct or α1/β2 subunits in a 1:1 ratio and incubated for 3–4 days at 18°C in modified Barth’s solution (96 mM NaCl, 2.0 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 5 mM HEPES, 2.5 mM sodium pyruvate, 0.5 mM theophylline, and 100 μg/mL gentamicin; pH 7.4).
This technique was performed as previously described (Ahring et al., 2016, 2018; Liao et al., 2019). Briefly, oocytes sit in a custom-built chamber and continuously perfused with a saline solution, ‘ND96’ (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, and 10 mM HEPES; pH 7.4). Glass electrode pipettes were filled with 3 M KCl, with resistances ranging from 0.4 to 2 MΩ. Oocytes were clamped to −60 mV using an Axon GeneClamp 500B amplifier (Molecular Devices). Currents were filtered at 20 Hz with a four-pole low pass Bessel filter (Axon GeneClamp 500 B) and digitized by a Digidata 1440A (Molecular Devices). Sampling was taken at 200 Hz and analyzed using pClamp 10.2 suite (Molecular Devices).
Stock solutions of 3.16 M GABA in ultrapure water and drug solutions of 100 mM in DMSO were stored at −20°C and aliquoted to avoid repeated freeze-thaw cycles. Each recording day, a fresh stock was used to prepare dilutions. The maximal concentration of DMSO in final drug ND96 solutions was <0.1%.
GABA concentration-response curves were determined for each construct as follows. To ensure RNA expression and reproducibility, a set of control applications were first applied consisting of three applications of 40 μM GABA, one 316 μM application, and three more 40 μM applications. After this, ten solutions of GABA each increasing in concentration by a factor of 3.16 were used starting with 100 nM and ending with 3.16 mM. Applications lasted for 30 s and were followed by 2–5 min of washout. EC50 and EC10 were calculated from this curve.
The drug modulation experiments were done as follows. Like the GABA dose-response curves, first, a set of three control applications were run consisting of GABA EC10, then a maximal response of GABA 3.16 mM, followed by three more GABA EC10. Before the application of modulators, EC10 was confirmed by comparing the ratio of the current of the last control application to the maximal response current. For each drug, 6 concentrations increasing by a factor of 10, ranging from 0.1 nM to 10 μM, were co-applied with GABA EC10 for 30 s followed by 2–5 min of washout.
The final dataset was from a minimum of four experiments and a minimum of two different X. laevis donors. Raw traces were analyzed using pClamp 10.2. Episodic traces for each application were overlaid and the baseline was subtracted. Peak current amplitude was quantified by measuring maximum inward current for each response. Peak current amplitudes (I) were fitted to the Hill equation and normalized to the maximal fitted response (Imax). The calculated Emax response is expressed as a percentage of the current obtained through GABA EC10 (actual GABA control percentage for each experiment is listed in Table 1). The Emax response and EC50 values were calculated by using non-linear regression to fit the data to the Hill equation in a monophasic model with three variables (top, bottom, EC50) using GraphPad Prism 8. Efficacy at infinitely low compound concentration was set to 0, and the slope was constrained to 1. For GABA concentration-response curves, the slope was unconstrained and listed in Table 1. Means are reported ± one SD. To compare differences in Emax response, EC50 values within drug groups and across γ3 and γ2 receptors, or to compare γ1/3 receptor responses at 10 μM with binary α1β2 receptors, one-way ANOVAs were run with Sidak multiple comparisons test. F tests, respectively, are [F (7, 48) = 32.77, p < 0.0001] and [F (7, 48) = 75.69, p < 0.0001], and [F (14,57) = 48.68, p < 0.0001]. All reported statistically significant comparisons within the results section are p < 0.01.
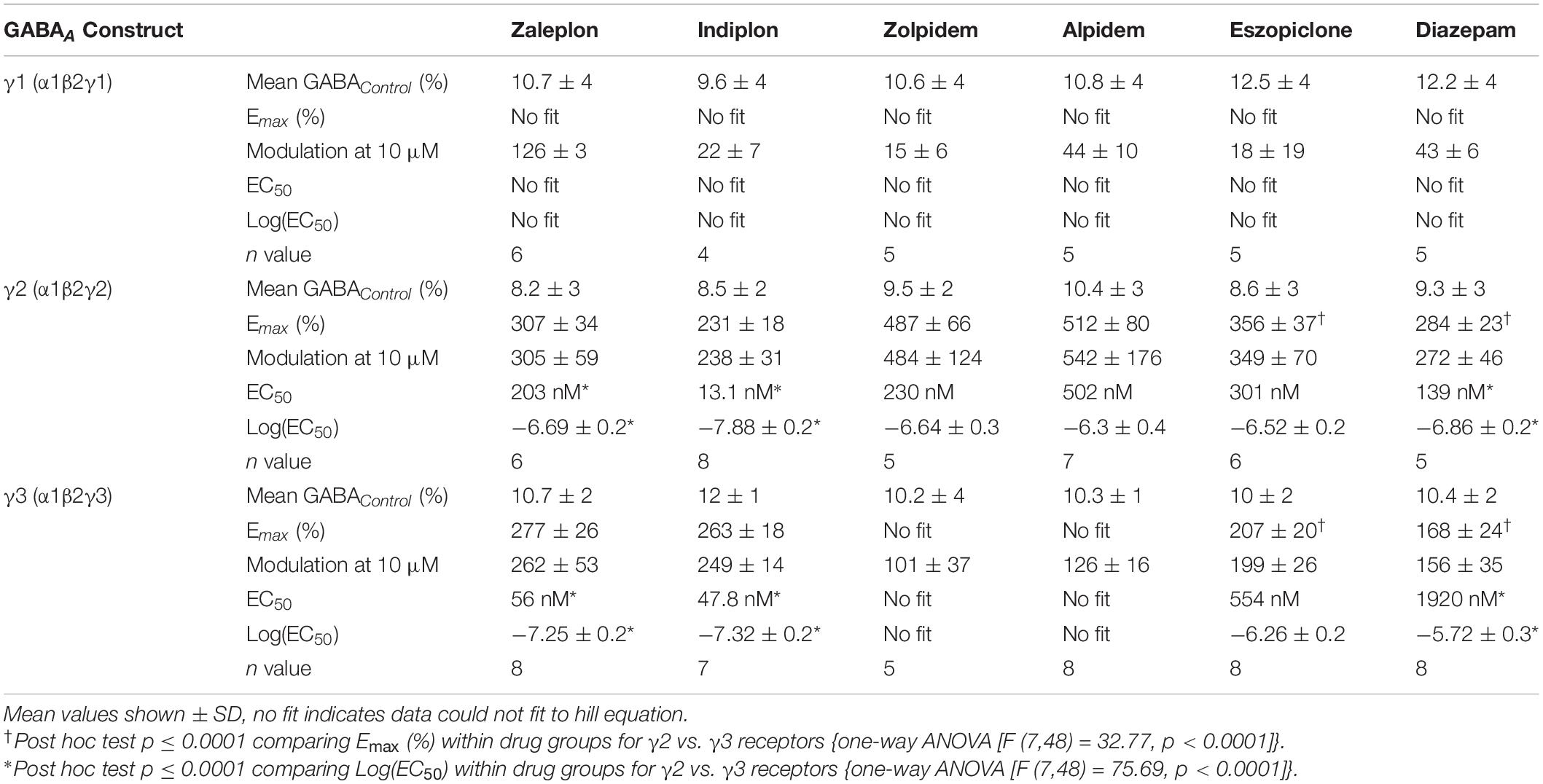
Table 1. Potency and efficacy of Z-drugs on GABAA receptors with varying γ subunits.
To ensure homogenous receptor populations and subunit orientation, we used concatenated receptors expressed in X. laevis oocytes. Concatenated pentameric constructs were created using the subunits γ-β2-α1-β2-α1 (where γ = γ1, γ2, or γ3). Subunits were linked with artificial linker sequences optimized to give relatively pure receptor expression and orientation without compromising function (Liao et al., 2019).
We first measured the concentration-response for GABA on each construct (Figure 3A). Upon visual inspection of representative traces (Figure 3B), γ1 and γ2 receptors presented similar current decay profiles at the highest GABA concentrations while the γ3 receptor showed a shorter current decay time. This could indicate that γ3 receptors undergo a higher degree of desensitization upon prolonged GABA exposure than the γ1 and γ2 receptor counterparts.
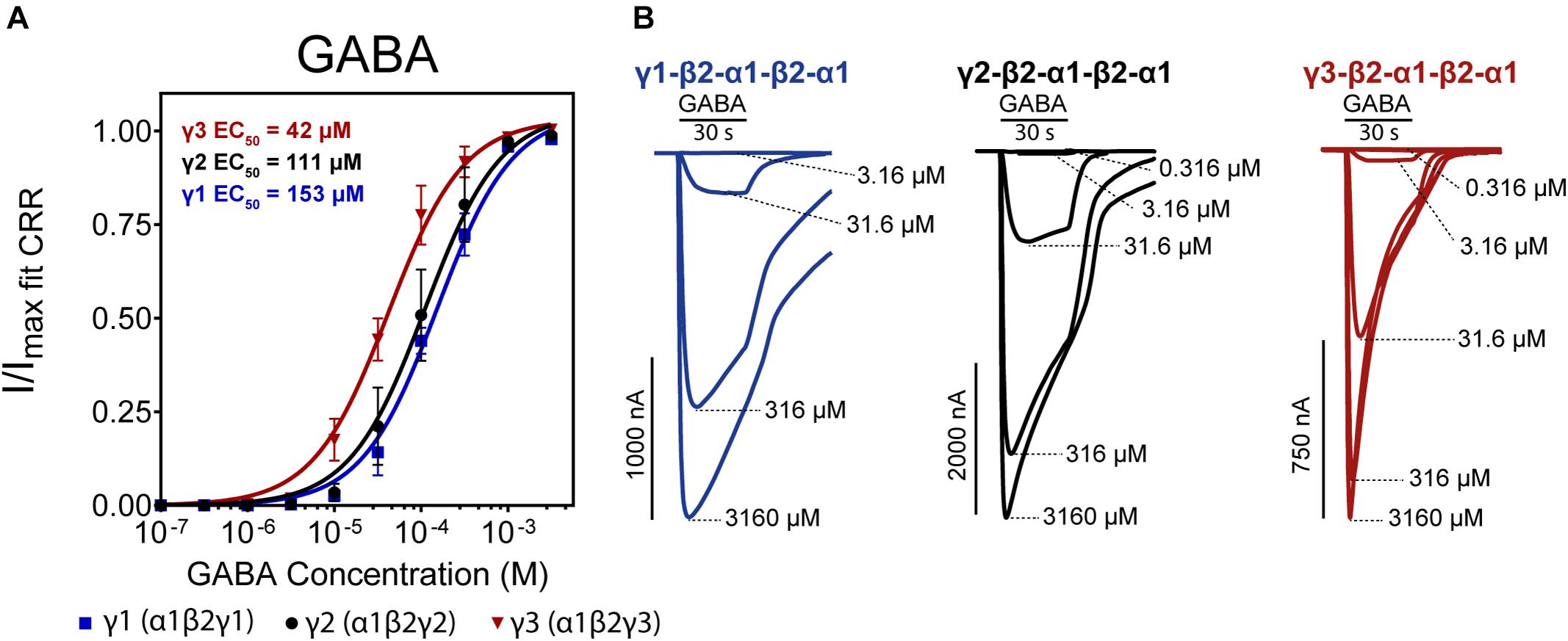
Figure 3. (A) Normalized GABA concentration-response curves of α1β2γx receptors (x = 1,2,3) expressed as concatenated pentamers in Xenopus laevis oocytes measured via two-electrode voltage clamp. Oocytes were injected with 50 nL of 0.5 ng/nL cRNA for each concatenated construct. Datapoints are depicted as means ± SD (n = 12–14). Data were fitted by non-linear regression to the Hill equation with an unconstrained Hill slope. Log(EC50) and Hill slope parameters are as follows; γ1 Log(EC50) = –3.82 ± 0.07 Hill slope = 1.22 ± 0.32, γ2 Log(EC50) = –3.96 ± 0.10 Hill slope = 1.19 ± 0.55, γ3 Log(EC50) = –4.38 ± 0.07 Hill slope = 1.36 ± 0.63. (B) Representative traces of each construct with indicated concatenated subunit combination. Application bars designate 30 s application time and concentrations of GABA are indicated at the peak of each trace.
The three receptor subtypes presented EC50 values in the range of 42–153 μM with γ3 being the most sensitive and γ1 being the least sensitive to GABA. The value for the γ2-containing concatenated receptor (EC50 of 111 μM) is in good agreement with Liao et al. (2019). Previously reported GABA EC50 values using single subunit injections of GABAA γ1, 2, 3 cRNAs in X. laevis oocytes show substantial variations in obtained GABA potencies ranging from 5–100 μM, but generally, γ3 receptors appear more sensitive to GABA (Knoflach et al., 1991; Wafford et al., 1993; Ebert et al., 1994; Khom et al., 2006; Esmaeili et al., 2009).
Positive allosteric modulators work by increasing the open-state probability of a receptor in the presence of an endogenous ligand (GABA). If the receptor is already at its maximal open-state probability, then the modulator will have no additional effect. For all γ-containing GABAA receptors, applications of high concentrations of GABA (>1 mM) are typically able to reach activation levels close to the maximal open-state probability, hence, modulators show no efficacy under conditions with high GABA concentrations. Whereas allosteric modulators, by definition, should not gate the receptor in the absence of GABA, substantial modulatory efficacies can be observed as GABA concentrations are lowered toward zero. Therefore, any efficacy of modulators described in percent will depend entirely on the selected concentration of the endogenous ligand. Low concentrations of GABA co-applied with modulators will yield large modulatory percent changes. Conversely, higher concentrations of GABA co-applied with modulators give small percent changes.
For our experiments, we selected to co-apply modulators with a GABAcontrol concentration that yields 10% of the maximum response (EC10) at the given receptor. Modulator efficacy is reported as a percent change of evoked current amplitude relative to the GABAcontrol application alone. To directly compare modulator efficacy across different receptors, it was critical that each experiment is run as close as possible to the EC10 of that receptor subtype. Due to GABA potency variations both between batches of oocytes and between individual oocytes, each experiment began with a full GABA concentration-response to determine EC10. Then for each oocyte, a set of 3 control applications at EC10 followed by a max GABA application, followed by three more EC10 applications were applied to confirm that the chosen GABAcontrol concentration yielded ∼10% of the maximum response. Any oocytes responding outside this narrow range (10% ± 5) were discarded before continuing with modulator experiments. GABAcontrol variation is reported in Table 1.
We examined the modulatory effects of the non-benzodiazepines ‘Z-drugs’ (zaleplon, indiplon, eszopiclone, zolpidem, and alpidem) and the benzodiazepine, diazepam on GABAA receptors with varying γ subunits (Figures 4A–C). Representative traces for each compound and receptor subtype are shown in Figures 4D–F. Concentrations ranging from 10–10 to 10–5 M were co-applied with GABA EC10. Full experimental results with Log(EC50) ± SD are listed in Table 1. Unless stated otherwise, all reported statistically significant comparisons have a p < 0.01.
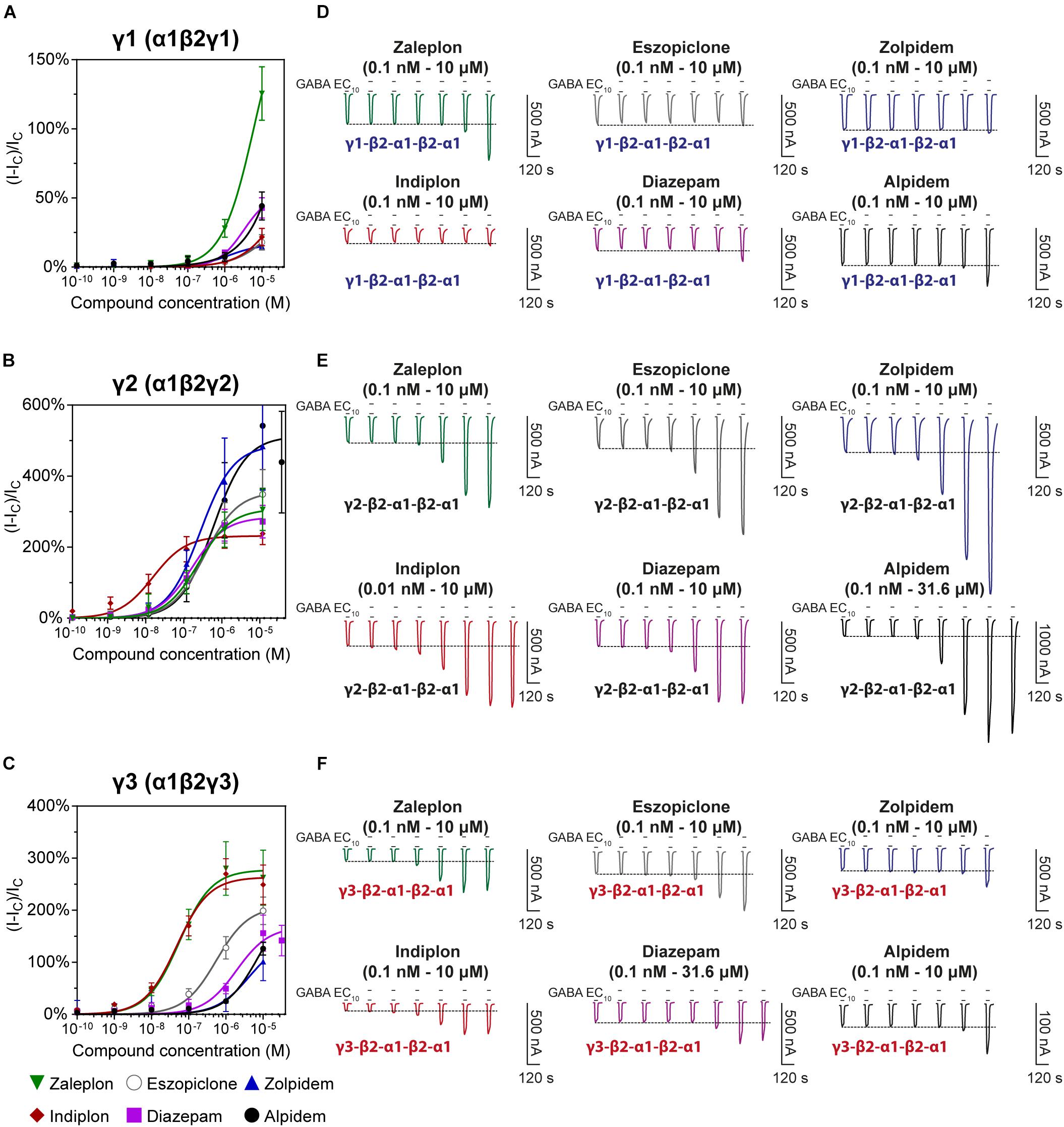
Figure 4. Modulatory actions of zaleplon, indiplon, eszopiclone, diazepam, zolpidem, and alpidem, on GABA evoked Cl– currents measured in human (A) α1β2γ1, (B) α1β2γ2, and (C) α1β2γ3 GABAA receptors expressed in Xenopus laevis oocytes measured via two-electrode voltage-clamp. The data are expressed as a percentage potentiation of GABA EC10 and are means ± SD (n = 4–8 from at least 2 separate Xenopus laevis donors). Data points were fitted to the Hill equation with bottom set to 0 and slope constrained to 1. (D–F) Representative traces illustrating modulator concentration-response experiments.
The pyrazolopyrimidines, zaleplon and indiplon, showed a reverse potency preference for γ2 and γ3 receptors (Figures 5A,B). Zaleplon had a ∼4-fold greater potency at γ3 receptors compared with γ2 (EC50 of approximately 50 vs. 200 nM), while indiplon had a ∼4-fold greater potency for γ2 vs. γ3 (10 vs. 45 nM). Neither compound had statistically significant different efficacies when γ2 was replaced by γ3 with Emax both in the range of ∼250–300%. On γ1 receptors, neither compound showed sufficient potency to enable fitting to the Hill equation within the concentration range tested. At the highest concentration applied (10 μM), zaleplon elicited a modulatory response of 125% and indiplon, 20%.
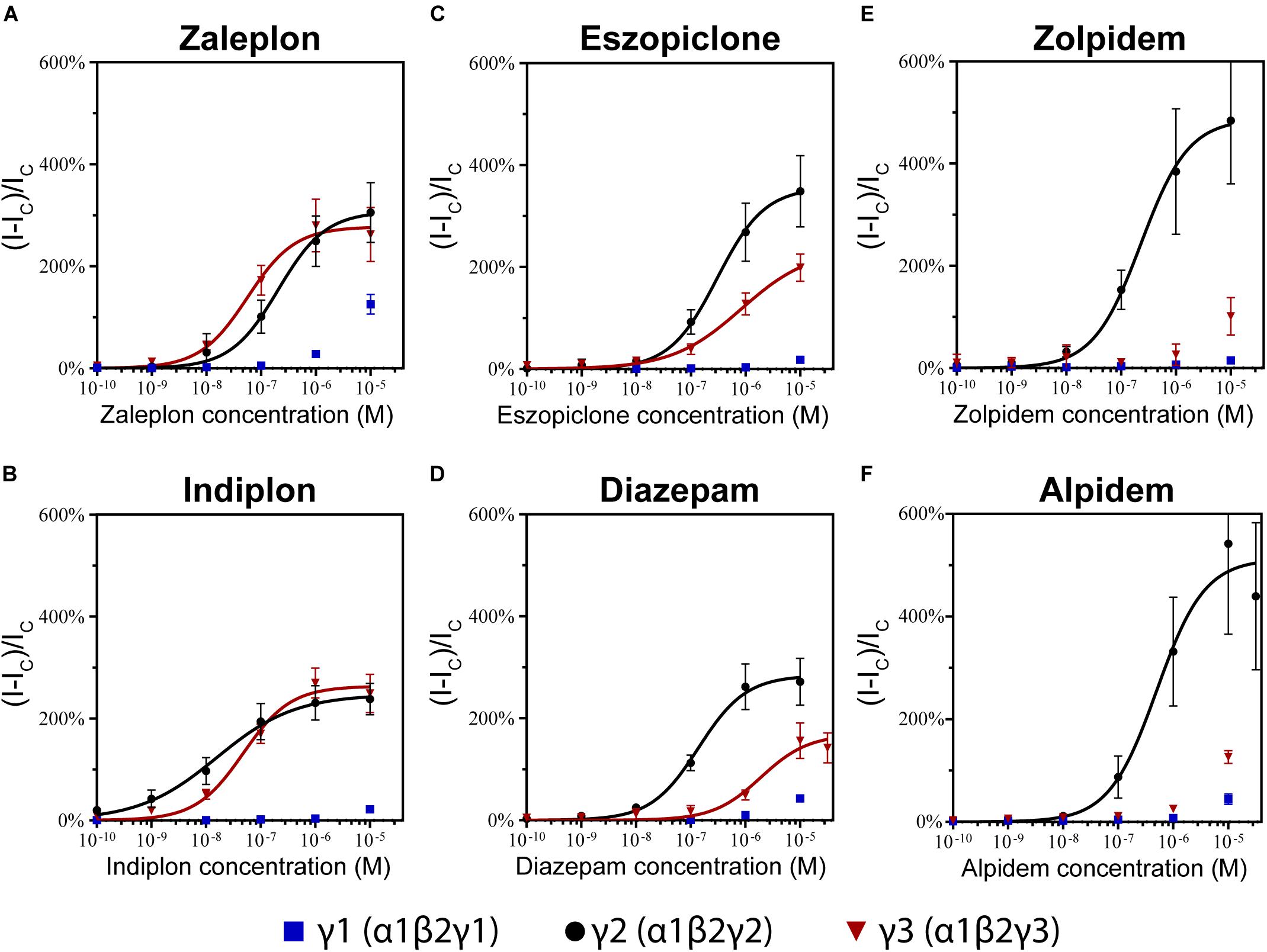
Figure 5. Modulatory actions of (A) zaleplon, (B) indiplon, (C) eszopiclone, (D) diazepam, (E) zolpidem, and (F) alpidem, on GABA evoked Cl– currents measured in human α1β2γ1, α1β2γ2, and α1β2γ3 GABAA receptors expressed in Xenopus laevis oocytes measured via two-electrode voltage-clamp. The data are expressed as a percentage potentiation of GABA EC10 and are means ± SD (n = 4–8 from at least 2 separate Xenopus laevis donors). Data points were fitted to the Hill equation with bottom set to 0 and slope constrained to 1.
In support of these findings, previous competitive binding studies using Ro15-4513 have suggested that zaleplon binds to γ3 GABAA receptors with an eightfold higher affinity than when γ2 is present (Dämgen and Lüddens, 1999). However, efficacy and potency have only been studied for the α2β2γ3 receptor which shows 10-fold less potency than what we have seen on α1β2γ3 with an EC50 of ∼500 nM.
The cyclopyrrolone, eszopiclone, and the benzodiazepine, diazepam modulated both γ2 and γ3 containing receptors with varying potency and efficacies and did not significantly modulate γ1 containing receptors (Figures 5C,D). Substituting the γ3 receptor for γ2 had no statistically significant difference on eszopiclone’s potency (in the range of 300–500 nM), but diazepam had a ∼15-fold reduction (1900 vs. 150 nM). Both compounds had ∼1.5-fold reductions in Emax when γ3 replaced γ2 (300 vs. 200%). At 10 μM, eszopiclone modulated γ1 receptors by 20% and diazepam by 40% above GABA EC10.
No literary data are available for eszopiclone, however, the racemic mixture zopiclone has been investigated. In a competitive binding study from Dämgen and Lüddens (1999), a marginal reduction in binding affinity was observed for zopiclone when γ3 was replaced by γ2. Yet, in another study zopiclone was observed to modulate α1β2γ3 receptors with comparable efficacy and potency to that of α1β2γ2 (Davies et al., 2000). Hence, eszopiclone and zopiclone seem to behave in a similar fashion at γ2- and γ3 containing receptors A previous study of diazepam on recombinant α1β2γ3 GABAA receptors shows good agreement for the potency (EC50 of 1.95 μM), but they observed no reduction in Emax comparing α1β2γ2 vs. α1β2γ3 receptors (Lippa et al., 2005).
The imidazopyridines, zolpidem, and alpidem were selective for the γ2 subunit, not showing significant potencies to be able to estimate an EC50 from fitting to the Hill equation for γ1 and γ3 receptors (Figures 5E,F). Zolpidem and alpidem had Emax on γ2 receptors ranging from 475–550%. Zolpidem’s EC50 on γ2 receptors was 230 nM and alpidem’s 500 nM. On γ3 receptors, both compounds had a measured response at concentrations of 10 μM of near 125% of GABA EC10. Neither compound showed robust efficacy on γ1 containing receptors. At 10 μM, zolpidem elicited a response of 15% and alpidem 40% above GABAcontrol. Overall this data indicates that zolpidem’s pharmacological activity is likely to be related only to the γ2 subunit.
Zolpidem’s selectivity for the γ2 subunit below 10 μM correlates with previous studies both on the binding for the γ1 (Benke et al., 1996) and γ3 subunit (Herb et al., 1992; Lüddens et al., 1994; Tögel et al., 1994; Hadingham et al., 1995; Sieghart, 1995; Dämgen and Lüddens, 1999), and with measurements in oocytes showing 20% or less efficacy (Wafford et al., 1993; Mckernan et al., 1995; Khom et al., 2006). These observations contrast with studies using HEK293 cells expressing α1βγ1 receptors observing zolpidem potentiating near 50–75% (Puia et al., 1991) and with an EC50 around 200 nM (Esmaeili et al., 2009).
To investigate whether the modulation observed at high compound concentrations on γ1 or γ3 receptors was specific to the γ subunit, 10 μM of each compound was applied to α1β2 binary receptors. Potentiation values are depicted along with the respective values at the γ-containing receptors in Figure 6. All 6 tested compounds elicited small responses on α1β2 receptors, with mean values ranging from 10–35%. Zaleplon was the only compound to show significantly higher γ1 receptor modulation above the value seen for α1/β2 receptors (p < 0.01) indicating that potentiation observed is specific to the γ1 subunit. Importantly, all tested compounds showed significantly higher potentiation values at γ3 receptors compared with α1β2 receptors (p < 0.01) indicating that modulation is specific to γ3.
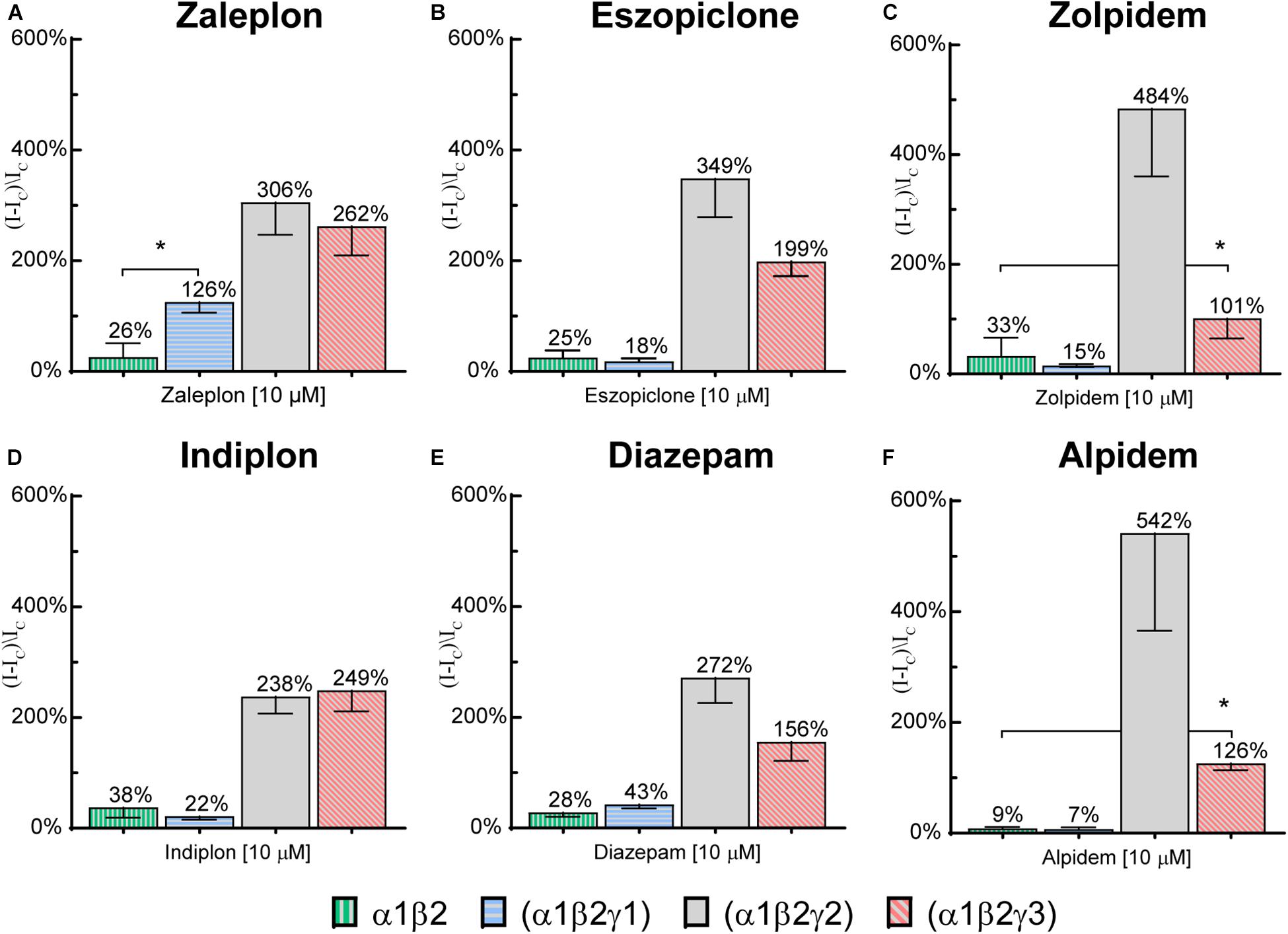
Figure 6. Modulatory actions of 10 μM (A) zaleplon, (B) indiplon, (C) eszopiclone, (D) diazepam, (E) zolpidem, and (F) alpidem, on GABA evoked Cl– currents measured in human α1β2γ1, α1β2γ2, α1β2γ3, and binary α1β2 GABAA receptors expressed in Xenopus laevis oocytes measured via two-electrode voltage-clamp. Data are expressed as percentage potentiation of GABA EC10 and are means ± SD (n = 3–8 from at least 2 separate Xenopus laevis donors). One-way ANOVA with post hoc Sidak multiple comparisons test were calculated between all compounds on γ1 vs. α1β2 and between zolpidem and alpidem on γ3 vs. α1β2; *p < 0.01.
In this study, we examined the effectiveness of the Z-drugs (zaleplon, indiplon, eszopiclone, zolpidem, and alpidem) and the benzodiazepine, diazepam on GABAA receptors containing γ1, γ2, or γ3 subunits under highly controlled experimental conditions. We used concatenated pentamers expressed in Xenopus laevis oocytes to reduce the potential of confounding mixed receptor populations arising when single subunits are injected (Boileau et al., 2002; Sigel et al., 2006; Ahring et al., 2016; Liao et al., 2019). Furthermore, all experiments were performed identically for each oocyte. Modulators were co-applied with a GABAcontrol concentration eliciting 10% of the maximum response.
All the tested drugs are efficient and potent modulators of γ2 receptors. Maximum efficacies ranged from 250–500% with the most and least efficacious being alpidem and indiplon, respectively. Potencies ranged from 10–500 nM with the most and least potent being indiplon and alpidem, respectively. In general, our results are within the range of variation from previous studies (Puia et al., 1991; Davies et al., 2000; Sanna et al., 2002; Petroski et al., 2006).
None of the Z-drugs exhibited sufficient potency within the tested concentration range to allow reliable fitting of the data to the Hill equation at γ1 receptors. At the highest tested concentration (10 μM) zaleplon had an efficacy of 125%. This contrasts the structurally similar compound, indiplon which at the same concentration did not affect α1β2γ1 receptors. Notably, zaleplon’s modulation was likely specific to the γ1 subunit, as the same concentration applied to α1/β2 receptors only elicited 25% above GABAcontrol. It remains a possibility that even higher concentrations of zaleplon could reveal further robust modulation at γ1-containing receptors. However, we generally chose to limit the concentration range tested to a maximum of 10 μM to avoid issues with compound solubility and potential interfering efficacies from binding to secondary modulatory sites as previously described for diazepam (Walters et al., 2000; Sieghart, 2015; Masiulis et al., 2019).
There is some discrepancy regarding γ1-containing receptors and zolpidem in the literature. Several studies observed that zolpidem displays no binding (Benke et al., 1996), or low maximum efficacies below 20% in α1βγ1 (Khom et al., 2006) and α2βγ1 receptors (Wafford et al., 1993; Mckernan et al., 1995) expressed in X. laevis oocytes. In contrast, other studies using HEK293 cells expressing α1βγ1 receptors observe zolpidem potentiations of near 50–75% (Puia et al., 1991) with an EC50 around 200 nM (Esmaeili et al., 2009). Differences in the experimental protocol, expression systems, assembling receptor populations, or chosen GABA control concentration may account for some of these divergences. Nevertheless, our data showing that zolpidem and the structurally similar compound, alpidem have negligible effects on concatenated pentameric α1β2γ1 receptors align with the findings that these compounds do not modulate γ1-containing receptors.
While definitive high-resolution crystal or Cryo-EM structures of GABAA receptors with bound diazepam exist (Zhu et al., 2018), they are still lacking for Z-drugs. Mutational studies and molecular modeling have provided insights into the nature of the important amino acids determining Z-drugs’ binding within the α1(+)–γ2(−) interface. The necessary His101 residue on the α1(+) interface is a well-characterized component of the benzodiazepine binding site (Wieland et al., 1992; Benson et al., 1998; Mckernan et al., 2000), but there are also important residues on the γ2(−) side. The amino acids Met130 and Phe77 have interactions with zolpidem, and mutating one or more of these abolishes binding (Buhr and Sigel, 1997; Wingrove et al., 1997). These residues are not present on the γ1 subunit, yet introducing them into the γ1 subunit does not fully restore zolpidem binding (Wingrove et al., 1997). Furthermore, the γ2 Phe77 mutation when expressed in the mouse eliminated zolpidem (but not flurazepam) dependent sedation and decreases motor exploration (Cope et al., 2004). Overall, our data pose the question of whether any of the tested drugs bind efficiently to α1-γ1 interfaces within the tested concentration range.
Zaleplon, indiplon, and eszopiclone modulate γ3-containing GABAA receptors at therapeutically relevant doses while diazepam, zolpidem, and alpidem do not. On α1β2γ3 receptors, zaleplon has equal efficacy compared with α1β2γ2, and a four-fold increase in potency. The structurally similar indiplon was also equally as efficacious on γ3- as γ2 containing receptors, but with reduced potency indicating that small differences in pyrazolopyrimidines can alter selectivity preferences between γ3- and γ2-containing GABAA subunits. Eszopiclone potentiates α1β2γ3 receptors with equal potency to α1β2γ2, but with a 1.5-fold reduction in efficacy. Overall these data indicate that even though classes of Z-drugs are quite similar, the arylamide moiety located at C4 of the pyrazolopyrimidines may be important for binding to the γ3 subunit.
Interestingly, high concentrations of zolpidem and alpidem potentiated GABA at γ3 receptors. This effect is specific to the γ3 subunit, as the same concentration applied to α1/β2 receptors elicited little response. Previous competitive binding studies using high-affinity benzodiazepine site ligands such as flunitrazepam or Ro-154513 have indicated that zolpidem has no binding to the classical γ3 receptor benzodiazepine site (Herb et al., 1992; Lüddens et al., 1994; Tögel et al., 1994; Hadingham et al., 1995; Sieghart, 1995; Dämgen and Lüddens, 1999).
While clinical studies observing the pharmacokinetics and pharmacodynamics of Z-drug mediated sleep are extensive, there have been relatively few studies comparing how hypnotic drugs target specific brain areas to induce sleep. Within the thalamus and hypothalamus are clusters of nuclei that relay information from subcortical structures to the cortex and both these regions are important for sleep-wake maintenance. The thalamic reticular nucleus generates characteristic sleep EEG firing rhythms, and the lateral hypothalamus is part of an ascending pathway stimulating cortical activity and wakefulness (Saper et al., 2001; Gent et al., 2018). Interestingly, eszopiclone but not zolpidem modulates GABAergic postsynaptic potentials in the thalamic reticular nucleus (Jia et al., 2009) and suppresses activity in the lateral hypothalamus (Kumar et al., 2011) to bring about sleep. Both of these regions contain a wider variety of GABAA subunits including the γ3 subunit (Pirker et al., 2000) which may, in part, account for the differences. Compared to zolpidem, eszopiclone has a faster sleep onset, more time spent in the restorative non-rapid eye movement stage, and a differing EEG signature (Xi and Chase, 2008).
There is a need to understand how hypnotics mediate their effect to aid in future drug development. Z-drugs were designed well before our detailed understanding of GABAA receptor subtypes (Bardone et al., 1978; Arbilla et al., 1985; Beer et al., 1997), and different GABAA subunit preferences contribute to differences in drug action along with pharmacokinetic factors like plasma concentration and drug half-life. In this study, we limited receptors to only contain α1 in combination with γ1, γ2, or γ3. While Z-drugs preferentially modulate α1 receptors at low concentrations, at moderate to high concentrations they also modulate receptors with α2 and α3 subunits (Petroski et al., 2006; Nutt and Stahl, 2010; Ramerstorfer et al., 2010; Sieghart and Savic, 2018), and these subunits may also play a role in sleep generation (Kopp et al., 2004). In addition to α subunit preference variations, we provide evidence here that there are also differences in how Z-drugs modulate GABAA receptors with γ3 subunits, but the significance of this in vivo is still unknown. In addition, future studies should characterize receptors with γ3 subunits in combination with α2/3.
The γ2 Phe77 mutation which abolishes zolpidem binding has been used as an in vivo pharmacogenetic model to explore zolpidem’s effects in particular brain regions (Wulff et al., 2007). This approach revealed that zolpidem specifically prolongs postsynaptic potentials within the hypothalamic tuberomammillary nucleus, reducing histamine levels across the brain sufficiently to induce sleep (Uygun et al., 2016). Because γ3-containing receptors are expressed within the same networks controlling sleep, elucidating any potential role they play would be important for the development of better hypnotics. Utilizing the approach of expressing the γ2 Phe77 mutation may reveal residual non-γ2 mediated behavioral effects related to zaleplon, indiplon, or eszopiclone administration. Moreover, because indiplon is efficacious on γ3, but not γ1-containing receptors, it would be well suited to specifically target γ3-containing receptors.
In conclusion, the approach taken of using concatenated GABAA receptors has overcome issues of forming unexpected receptor populations when using single subunit cRNAs to express recombinant receptors in X. laevis oocytes. We used this strategy to clarify inconsistencies within the literature on what effects Z-drugs have on γ1- and γ3-containing GABAA receptors. Using this strategy, we have shown that zaleplon, indiplon, and eszopiclone modulate γ3-containing GABAA receptors with no effects on γ1-containing GABAA receptors below 10 μM. Zolpidem and alpidem show no significant modulation on γ1 or γ3 subunits below 10 μM indicating that their pharmacological effects are likely limited to GABAA receptors with γ2 subunits. Gaining a complete picture of the GABAA receptor subtypes targeted by Z-drugs will help in the understanding of hypnotics and aid in developing drugs that more closely replicate physiological sleep with less adverse side effects.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by The University of Sydney Animal Ethics Committee.
GR, VL, PA, and MC conceptualized the study and designed the experiments. GR, VL, and PA collected and analyzed the data. GR wrote the manuscript and prepared the figures. VL, PA, and MC reviewed the manuscript. All authors contributed to the article and approved the submitted version.
We acknowledge the financial support of the National Health and Medical Research Council of Australia Project grant APP1124567 and the Australian Research Council Linkage grant LP160100560.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
GR acknowledges the award of a University of Sydney Deputy Vice Chancellor Postgraduate Scholarship. All authors acknowledge the support of the technical staff at the University of Sydney Live Animal Services.
Ahring, P. K., Bang, L. H., Jensen, M. L., Strøbæk, D., Hartiadi, L. Y., Chebib, M., et al. (2016). A pharmacological assessment of agonists and modulators at α4β2γ2 and α4β2δ GABAA receptors: the challenge in comparing apples with oranges. Pharmacol. Res. 111, 563–576. doi: 10.1016/j.phrs.2016.05.014
Ahring, P. K., Liao, V. W. Y., and Balle, T. (2018). Concatenated nicotinic acetylcholine receptors: a gift or a curse? J. Gen. Physiol. 150, 453–473. doi: 10.1085/jgp.201711846
Allen Institute for Brain Science (2008). Allen Developing Mouse Brain Atlas [Online]. Available online at: http://developingmouse.brain-map.org/ (accessed October, 2020).
Arbilla, S., Depoortere, H., George, P., and Langer, S. Z. (1985). Pharmacological profile of the imidazopyridine zolpidem at benzodiazepine receptors and electrocorticogram in rats. Naunyn Schmied. Arch. Pharmacol. 330, 248–251. doi: 10.1007/bf00572441
Bardone, M., Ducrot, R., Garret, C., and Julou, L. (1978). Benzodiazepine-Like Central Effects of RP 27267, A Dihydro-7-Oxo-5H-Pyrrolo [3, 4-b] Pyrazine Derivative. London: Pergamon Press.
Beer, B., Clody, D., Mangano, R., Levner, M., Mayer, P., and Barrett, J. (1997). A review of the preclinical development of zaleplon, a novel non−benzodiazepine hypnotic for the treatment of insomnia. CNS Drug Rev. 3, 207–224. doi: 10.1111/j.1527-3458.1997.tb00324.x
Benke, D., Honer, M., Michel, C., and Mohler, H. (1996). GABAA receptor subtypes differentiated by their γ-subunit variants: prevalence, pharmacology and subunit architecture. Neuropharmacology 35, 1413–1423. doi: 10.1016/s0028-3908(96)00068-8
Benson, J. A., Low, K., Keist, R., Mohler, H., and Rudolph, U. (1998). Pharmacology of recombinant gamma-aminobutyric acidA receptors rendered diazepam-insensitive by point-mutated alpha-subunits. FEBS Lett. 431, 400–404. doi: 10.1016/s0014-5793(98)00803-5
Boileau, A., Baur, R., Sharkey, L., Sigel, E., and Czajkowski, C. (2002). The relative amount of cRNA coding for γ2 subunits affects stimulation by benzodiazepines in GABAA receptors expressed in Xenopus oocytes. Neuropharmacology 43, 695–700. doi: 10.1016/s0028-3908(02)00036-9
Bovolin, P., Santi, M. R., Puia, G., Costa, E., and Grayson, D. (1992). Expression patterns of gamma-aminobutyric acid type A receptor subunit mRNAs in primary cultures of granule neurons and astrocytes from neonatal rat cerebella. Proc. Natl. Acad. Sci. U.S.A. 89, 9344–9348. doi: 10.1073/pnas.89.19.9344
Buhr, A., and Sigel, E. (1997). A point mutation in the γ2 subunit of γ-aminobutyric acid type A receptors results in altered benzodiazepine binding site specificity. Proc. Natl. Acad. Sci. U.S.A. 94, 8824–8829. doi: 10.1073/pnas.94.16.8824
Cope, D. W., Wulff, P., Oberto, A., Aller, M. I., Capogna, M., Ferraguti, F., et al. (2004). Abolition of zolpidem sensitivity in mice with a point mutation in the GABAA receptor γ2 subunit. Neuropharmacology 47, 17–34. doi: 10.1016/j.neuropharm.2004.03.007
Dämgen, K., and Lüddens, H. (1999). Zaleplon displays a selectivity to recombinant GABAA receptors different from zolipdem, zopiclone and benzodiazepines. Neurosci. Res. Commun. 25, 139–148. doi: 10.1002/(sici)1520-6769(199911/12)25:3<139::aid-nrc3>3.0.co;2-w
Davies, M., Newell, J. G., Derry, J. M. C., Martin, I. L., and Dunn, S. M. J. (2000). Characterization of the Interaction of zopiclone with γ-aminobutyric acid type a receptors. Mol. Pharmacol. 58, 756–762. doi: 10.1124/mol.58.4.756
Ebert, B., Wafford, K. A., Whiting, P. J., Krogsgaard-Larsen, P., and Kemp, J. A. (1994). Molecular pharmacology of gamma-aminobutyric acid type A receptor agonists and partial agonists in oocytes injected with different alpha, beta, and gamma receptor subunit combinations. Mol. Pharmacol. 46, 957–963.
Esmaeili, A., Lynch, J. W., and Sah, P. (2009). GABAA receptors containing gamma1 subunits contribute to inhibitory transmission in the central amygdala. J. Neurophysiol. 101, 341–349. doi: 10.1152/jn.90991.2008
Fleming, J. A., Bourgouin, J., and Hamilton, P. (1988). A sleep laboratory evaluation of the long-term efficacy of zopiclone. Can. J. Psychiatry 33, 103–107. doi: 10.1177/070674378803300206
Gent, T. C., Bassetti, C. L., and Adamantidis, A. R. (2018). Sleep-wake control and the thalamus. Curr. Opin. Neurobiol. 52, 188–197. doi: 10.1016/j.conb.2018.08.002
Hadingham, K. L., Wafford, K. A., Thompson, S. A., Palmer, K. J., and Whiting, P. J. (1995). Expression and pharmacology of human GABAA receptors containing γ3 subunits. Eur. J. Pharmacol. Mol. Pharmacol. 291, 301–309. doi: 10.1016/0922-4106(95)90070-5
Herb, A., Wisden, W., Lüddens, H., Puia, G., Vicini, S., and Seeburg, P. H. (1992). The third gamma subunit of the gamma-aminobutyric acid type A receptor family. Proc. Nat. Acad. Sci. 89, 1433–1437. doi: 10.1073/pnas.89.4.1433
Hertz, L., and Chen, Y. (2010). The astrocytic GABAA/benzodiazepinelike receptor: identifying the joker receptor for benzodiazepinemimetic drugs? Front. CNS Drug Discov. 1, 342–361. doi: 10.2174/978160805159511001010342
Jia, F., Goldstein, P. A., and Harrison, N. L. (2009). The modulation of synaptic GABAA receptors in the thalamus by eszopiclone and zolpidem. J. Pharmacol. Exp. Therap. 328:1000. doi: 10.1124/jpet.108.146084
Khom, S., Baburin, I., Timin, E. N., Hohaus, A., Sieghart, W., and Hering, S. (2006). Pharmacological properties of GABAA receptors containing γ1 subunits. Mol. Pharmacol. 69, 640–649. doi: 10.1124/mol.105.017236
Klimm, H. D., Dreyfus, J. F., and Delmotte, M. (1987). Zopiclone versus nitrazepam: a double-blind comparative study of efficacy and tolerance in elderly patients with chronic insomnia. Sleep 10(Suppl. 1) 73–78. doi: 10.1093/sleep/10.suppl_1.73
Knoflach, F., Rhyner, T., Villa, M., Kellenberger, S., Drescher, U., Malherbe, P., et al. (1991). The γ3-subunit of the GABAA-receptor confers sensitivity to benzodiazepine receptor ligands. FEBS Lett. 293, 191–194. doi: 10.1016/0014-5793(91)81184-a
Kopp, C., Rudolph, U., and Tobler, I. (2004). Sleep EEG changes after zolpidem in mice. Neuroreport 15, 2299–2302. doi: 10.1097/00001756-200410050-00031
Kumar, S., Alam, M., Rai, S., Bashir, T., Mcginty, D., and Szymusiak, R. (2011). Central nervous system sites of the sleep promoting effects of eszopiclone in rats. Neuroscience 181, 67–78. doi: 10.1016/j.neuroscience.2011.03.006
Laurie, D., Wisden, W., and Seeburg, P. (1992). The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J. Neurosci. 12, 4151–4172. doi: 10.1523/jneurosci.12-11-04151.1992
Liao, V. W. Y., Chua, H. C., Kowal, N. M., Chebib, M., Balle, T., and Ahring, P. K. (2019). Concatenated gamma-aminobutyric acid type A receptors revisited: finding order in chaos. J. Gen. Physiol. 151, 798–819. doi: 10.1085/jgp.201812133
Lippa, A., Czobor, P., Stark, J., Beer, B., Kostakis, E., Gravielle, M., et al. (2005). Selective anxiolysis produced by ocinaplon, a GABAA receptor modulator. Proc. Natl. Acad. Sci. U.S.A. 102, 7380–7385. doi: 10.1073/pnas.0502579102
Lüddens, H., Seeburg, P. H., and Korpi, E. R. (1994). Impact of beta and gamma variants on ligand-binding properties of gamma-aminobutyric acid type A receptors. Mol. Pharmacol. 45, 810–814.
Masiulis, S., Desai, R., Uchañski, T., Martin, I. S., Laverty, D., Karia, D., et al. (2019). GABA A receptor signalling mechanisms revealed by structural pharmacology. Nature 565, 454–459. doi: 10.1038/s41586-018-0832-5
Mckernan, R., Wafford, K., Quirk, K., Hadingham, K., Harley, E., Ragan, C., et al. (1995). The pharmacology of the benzodiazepine site of the GABA-A receptor is dependent on the type of γ-subunit present. J. Rec. Signal Trans. 15, 173–183. doi: 10.3109/10799899509045215
Mckernan, R. M., Rosahl, T. W., Reynolds, D. S., Sur, C., Wafford, K. A., Atack, J. R., et al. (2000). Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nat. Neurosci. 3, 587–592.
Nutt, D. J., and Stahl, S. M. (2010). Searching for perfect sleep: the continuing evolution of GABAA receptor modulators as hypnotics. J. Psychopharmacol. 24, 1601–1612. doi: 10.1177/0269881109106927
Olsen, R. W., and Sieghart, W. (2008). International union of pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev. 60, 243–260. doi: 10.1124/pr.108.00505
Petroski, R. E., Pomeroy, J. E., Das, R., Bowman, H., Yang, W., Chen, A. P., et al. (2006). Indiplon is a high-affinity positive allosteric modulator with selectivity for α1 subunit-containing GABAA receptors. J. Pharmacol. Exp. Therap. 317, 369–377. doi: 10.1124/jpet.105.096701
Pirker, S., Schwarzer, C., Wieselthaler, A., Sieghart, W., and Sperk, G. (2000). GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 101, 815–850. doi: 10.1016/s0306-4522(00)00442-5
Puia, G., Vicini, S., Seeburg, P. H., and Costa, E. (1991). Influence of recombinant gamma-aminobutyric acid-A receptor subunit composition on the action of allosteric modulators of gamma-aminobutyric acid-gated Cl- currents. Mol. Pharmacol. 39, 691–696.
Quirk, K., Gillard, N. P., Ragan, C., Whiting, P. J., and Mckernan, R. M. (1994). gamma-Aminobutyric acid type A receptors in the rat brain can contain both gamma 2 and gamma 3 subunits, but gamma 1 does not exist in combination with another gamma subunit. Mol. Pharmacol. 45, 1061–1070.
Ramerstorfer, J., Furtmüller, R., Vogel, E., Huck, S., and Sieghart, W. (2010). The point mutation γ2F77I changes the potency and efficacy of benzodiazepine site ligands in different GABAA receptor subtypes. Eur. J. Pharmacol. 636, 18–27. doi: 10.1016/j.ejphar.2010.03.015
Sanna, E., Busonero, F., Talani, G., Carta, M., Massa, F., Peis, M., et al. (2002). Comparison of the effects of zaleplon, zolpidem, and triazolam at various GABAA receptor subtypes. Eur. J. Pharmacol. 451, 103–110. doi: 10.1016/s0014-2999(02)02191-x
Saper, C. B., Chou, T. C., and Scammell, T. E. (2001). The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 24, 726–731. doi: 10.1016/s0166-2236(00)02002-6
Sieghart, W. (1995). Structure and pharmacology of gamma-amino-butyric acid A receptor subtypes. Pharmacol. Rev. 47, 181–234.
Sieghart, W. (2015). Allosteric modulation of GABAA receptors via multiple drug-binding sites. Adv. Pharmacol. 72, 53–96. doi: 10.1016/bs.apha.2014.10.002
Sieghart, W., and Savic, M. M. (2018). International union of basic and clinical pharmacology. CVI: GABAA receptor subtype- and function-selective ligands: key issues in translation to humans. Pharmacol. Rev. 70, 836–878. doi: 10.1124/pr.117.014449
Sieghart, W., and Sperk, G. (2002). Subunit composition, distribution and function of GABA-A receptor subtypes. Curr. Top. Med. Chem. 2, 795–816. doi: 10.2174/1568026023393507
Sigel, E., Baur, R., Boulineau, N., and Minier, F. (2006). Impact of Subunit Positioning on GABAA Receptor Function. London: Portland Press Ltd.
Sigel, E., and Buhr, A. (1997). The benzodiazepine binding site of GABAA receptors. Trends Pharmacol. Sci. 18, 425–429. doi: 10.1016/s0165-6147(97)90675-1
Tögel, M., Mossier, B., Fuchs, K., and Sieghart, W. (1994). γ-Aminobutyric acidA receptors displaying association of gamma 3-subunits with β2/3 and different α-subunits exhibit unique pharmacological properties. J. Biol. Chem. 269, 12993–12998.
Uygun, D. S., Ye, Z., Zecharia, A. Y., Harding, E. C., Yu, X., Yustos, R., et al. (2016). Bottom-up versus top-down induction of sleep by zolpidem acting on histaminergic and neocortex neurons. J. Neurosci. 36, 11171–11184. doi: 10.1523/jneurosci.3714-15.2016
Wafford, K. A., Bain, C. J., Whiting, P. J., and Kemp, J. A. (1993). Functional comparison of the role of gamma subunits in recombinant human gamma-aminobutyric acidA/benzodiazepine receptors. Mol. Pharmacol. 44, 437–442.
Walters, R. J., Hadley, S. H., Morris, K. D. W., and Amin, J. (2000). Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nat. Neurosci. 3, 1274–1281. doi: 10.1038/81800
Wieland, H. A., Lüddens, H., and Seeburg, P. H. (1992). A single histidine in GABAA receptors is essential for benzodiazepine agonist binding. J. Biol. Chem. 267, 1426–1429.
Wingrove, P. B., Thompson, S. A., Wafford, K. A., and Whiting, P. J. (1997). Key amino acids in the γ subunit of the γ-aminobutyric acidA receptor that determine ligand binding and modulation at the benzodiazepine site. Mol. Pharmacol. 52, 874–881. doi: 10.1124/mol.52.5.874
Wulff, P., Goetz, T., Leppä, E., Linden, A.-M., Renzi, M., Swinny, J. D., et al. (2007). From synapse to behavior: rapid modulation of defined neuronal types with engineered GABAA receptors. Nat. Neurosci. 10, 923–929. doi: 10.1038/nn1927
Xi, M., and Chase, M. H. (2008). Effects of eszopiclone and zolpidem on sleep and waking states in the adult guinea pig. Sleep 31, 1043–1051.
Keywords: GABAA receptors, Z-drugs, modulators, γ1 subunit, γ3 subunit, zolpidem, zaleplon, eszopiclone
Citation: Richter G, Liao VWY, Ahring PK and Chebib M (2020) The Z-Drugs Zolpidem, Zaleplon, and Eszopiclone Have Varying Actions on Human GABAA Receptors Containing γ1, γ2, and γ3 Subunits. Front. Neurosci. 14:599812. doi: 10.3389/fnins.2020.599812
Received: 28 August 2020; Accepted: 26 October 2020;
Published: 19 November 2020.
Edited by:
Christian Legros, Université d’Angers, FranceReviewed by:
Petra Scholze, Medical University of Vienna, AustriaCopyright © 2020 Richter, Liao, Ahring and Chebib. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Grant Richter, Z3JhbnQucmljaHRlckBzeWRuZXkuZWR1LmF1; Mary Chebib, bWFyeS5jb2xsaW5zQHN5ZG5leS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.