 Zhilian Jia
Zhilian Jia Qiang Wu
Qiang Wu- Center for Comparative Biomedicine, MOE Key Laboratory of Systems Biomedicine, State Key Laboratory of Oncogenes and Related Genes, School of Life Sciences and Biotechnology, Institute of Systems Biomedicine, Shanghai Jiao Tong University, Shanghai, China
The clustered protocadherins (cPcdhs) are a subfamily of type I single-pass transmembrane cell adhesion molecules predominantly expressed in the brain. Their stochastic and combinatorial expression patterns encode highly diverse neural identity codes which are central for neuronal self-avoidance and non-self discrimination in brain circuit formation. In this review, we first briefly outline mechanisms for generating a tremendous diversity of cPcdh cell-surface assemblies. We then summarize the biological functions of cPcdhs in a wide variety of neurodevelopmental processes, such as neuronal migration and survival, dendritic arborization and self-avoidance, axonal tiling and even spacing, and synaptogenesis. We focus on genetic, epigenetic, and 3D genomic dysregulations of cPcdhs that are associated with various neuropsychiatric and neurodevelopmental diseases. A deeper understanding of regulatory mechanisms and physiological functions of cPcdhs should provide significant insights into the pathogenesis of mental disorders and facilitate development of novel diagnostic and therapeutic strategies.
Introduction
The assembly of complex neural circuits, which are essential for accurate processing of sensory and cognitive information, requires individual neurons to discriminate self from non-self. This neuronal discrimination is thought to be accomplished by cell identity codes – neural recognition addresses determined by specific cell-surface receptors (Zipursky and Grueber, 2013). Given that there are billions of neurons each forming up to twenty thousand synapses in the human brain (Pakkenberg and Gundersen, 1997; DeFelipe et al., 1999), it’s fascinating how neurons use limited number of genes to generate such seemingly unlimited diversities. Failure in generating diversified neuronal codes causes deficits in neural circuit formation and may be related to pathogenesis of brain disorders.
Vertebrates adopt several mechanisms to achieve molecular diversity in different systems. For example, in the immune system, individual B and T cells express distinct repertoires of immunoglobulin (Ig) and T-cell receptor (TCR) genes through stochastic V(D)J rearrangements in the Ig and TCR gene clusters, respectively (Tonegawa, 1983; Wu et al., 2020). In the olfactory system, each olfactory sensory neuron expresses a single olfactory receptor (OR) out of more than a thousand OR genes. In this case, interchromosomal interactions bring 63 OR enhancers (Greek islands) together to activate one and only one OR gene in each neuron (Monahan et al., 2019). In the nervous system, more than 1,000 distinct isoforms of neurexins are generated by alternative splicing (Ullrich et al., 1995). In addition, the clustered protocadherins (cPcdhs), the largest subfamily of cadherin superfamily proteins, endow each individual neuron with a unique identity code (Wu and Maniatis, 1999). The molecular diversity of cPcdhs is generated by stochastic promoter choice and combinatorial expression regulated by the genome architectural protein CTCF (CCCTC-binding factor) (Golan-Mashiach et al., 2012; Guo et al., 2012, 2015; Monahan et al., 2012; Jia et al., 2020).
The expressed Pcdh isoforms are proposed to form repertoires of dimers on the cell membrane through promiscuous cis-interactions, as observed in crystal structures (Rubinstein et al., 2015; Goodman et al., 2016b, 2017; Brasch et al., 2019). When contacting with opposed neurites expressing the same combinations, the cis-dimer repertoires are thought to assemble into a large zipper lattice through stringent homophilic trans-interactions (Schreiner and Weiner, 2010; Thu et al., 2014; Brasch et al., 2019). Thus, the cPcdhs mediate specific cell-cell adhesion through these trans-interactions and are strong candidates for cell-surface neuronal identity codes (Wu and Maniatis, 1999; Thu et al., 2014; Brasch et al., 2019). In addition to cPcdhs, non-clustered Pcdhs also mediate cell adhesion through homophilic trans-interactions (Honig and Shapiro, 2020). Clustered and non-clustered Pcdhs may have cooperative functions in brain development.
Consistent with their prominent expression in the nervous system, cPcdh proteins function in many processes during brain development and are thought to be important for the construction of neuronal connectivity leading to the adult brain (Wang et al., 2002b; Weiner et al., 2005; Garrett and Weiner, 2009; Lefebvre et al., 2012; Katori et al., 2017; Mountoufaris et al., 2017; Fan et al., 2018). Emerging evidence suggests that genomic variants, epigenetic alterations, and 3D genome architectural dysregulation of the cPcdhs are associated with a wide variety of brain disorders in both animal models and human patients. In this review, we specifically focus on recent progress of the roles of cPcdhs in brain development and their dysregulation in brain disorders. We propose that epigenetic dysregulation of cPcdhs underlies many mental disorders. We refer interested readers to other recent reviews describing various aspects of cPcdhs (Flaherty and Maniatis, 2020; Honig and Shapiro, 2020; Pancho et al., 2020; Sanes and Zipursky, 2020; Wu and Jia, 2020).
Molecular Mechanisms for Generating Diversified Neural Identity Codes
Promoter Choice of cPcdhs in the Cell Nucleus
The unique genome organization of cPcdh genes has the potential to generate unparalleled diversities for neuron identity codes. The cPcdh locus contains three closely-linked gene clusters (Pcdhα, Pcdhβ, and Pcdhγ) encompassing nearly one million base pairs at the human chromosome 5q31 region (Figure 1A) (Wu and Maniatis, 1999). Both the Pcdhα and Pcdhγ clusters contain tandem arrays of large variable exons, each of which is transcribed from its own promoter and then spliced to the common set of three downstream constant exons within the respective cluster (Figure 1A) (Wu and Maniatis, 1999; Tasic et al., 2002; Wang et al., 2002a). The Pcdhβ cluster has no constant region and thus contains only variable exons (Figure 1A) (Wu and Maniatis, 1999; Wu et al., 2001). Each Pcdh α, β, and γ variable exon encodes an extracellular domain with 6 ectodomains (ECs), a transmembrane domain (TM), and a juxtamembrane variable cytoplasmic domain (VCD). The three constant exons encode a common membrane-distal cytoplasmic domain (αCD and γCD) (Figures 1B,C) (Wu and Maniatis, 1999). Based on phylogenetic analyses, the 53 human cPcdh genes are divided into five groups: Pcdhα (α1 – α13), Pcdhβ (β1 – β16), PcdhγA (γa1 – a12), PcdhγB (γb1 – γb7), and C-type (αc1, αc2, γc3, γc4, γc5) (Wu and Maniatis, 1999; Wu et al., 2001). Interestingly, different species, even between the closest relatives such as human and chimpanzee, do not have exact the same number of cPcdh genes, suggesting that their absolute numbers are not important once reached a certain threshold (Wu, 2005).
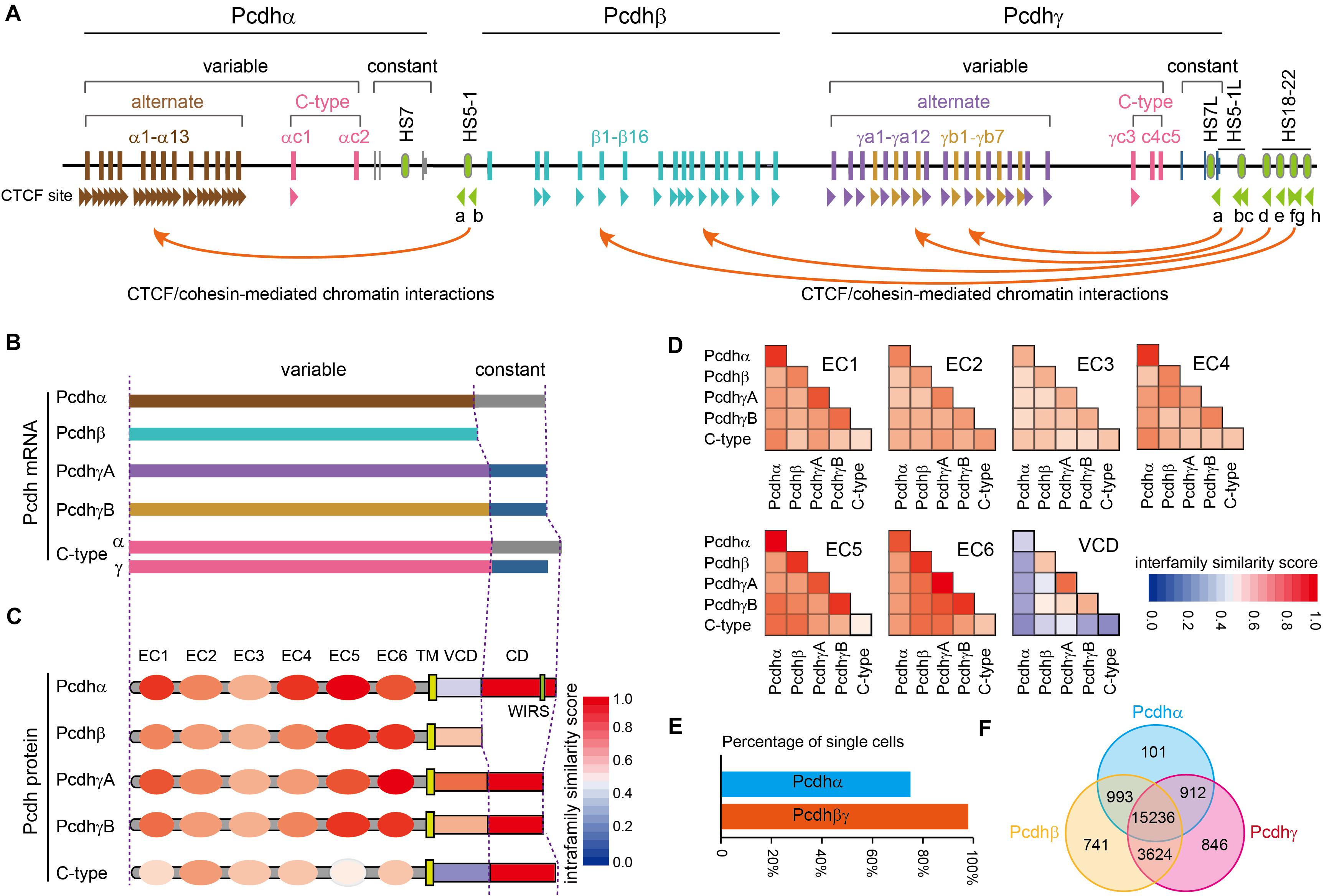
Figure 1. Characteristics of the human clustered Pcdhs. (A) The unique genomic organization of the three closely-linked human Pcdh gene clusters. The Pcdhα and Pcdhγ clusters are organized into variable and constant regions while the Pcdhβ cluster contains only variable region with no constant region. The Pcdhα cluster contains 15 variable exons (13 alternate exons of α1-α13 and 2 C-type exons of αc1 and αc2) each independently spliced to a single set of three downstream constant exons. Each Pcdhα variable exon is associated with its own promoter. Each Pcdhα alternate promoter carries two forward CTCF sites, Pcdhαc1 carries one forward CTCF site, and Pcdhαc2 carries no CTCF site. These forward CTCF sites function as topological insulators and form directional chromatin looping with the two reverse CTCF sites of the downstream HS5-1 enhancer. The Pcdhβ cluster contains 16 variable exons of β1–β16 each encoding a single Pcdhβ protein. Each Pcdhβ variable exon is associated with its own promoter. Each Pcdhβ promoter carries a forward CTCF site except β1. The Pcdhγ cluster contains 22 variable exons (19 alternate exons of 12 A-type: γa1–γa12; 7 B-type: γb1–γb7; 3 C-type exons: γc3–γc5) each independently spliced to a single set of three downstream constant exons. Each Pcdhγ variable exon is associated with a promoter. Each Pcdhγ promoter carries a forward CTCF site except γc4 and γc5. These forward Pcdhβ and Pcdhγ CTCF sites function as topological insulators and form directional chromatin looping with a tandem array of reverse CTCF sites of the downstream super-enhancer. Enhancer and super-enhancer are marked with green ovals. The location of CTCF sites and their orientation are shown as arrowheads. (B). Diverse cPcdh mRNAs, which can be divided into five groups, are generated by a combination of stochastic promoter activation and alternative splicing. (C). Shown is the domain organization of the encoded cPcdh proteins. Each variable exon of the Pcdhα, Pcdhβ, and Pcdhγ clusters encodes an extracellular domain with a signal peptide and 6 ectodomains (ECs), a transmembrane domain (TM), and a variable cytoplasmic domain (VCD). The constant exons encode an intracellular common domain (CD) shared by all members of the Pcdhα or Pcdhγ cluster. The color in each domain represents the amino-acid sequence similarity score between isoforms of the same group. WIRS, Wiskott-Aldrich syndrome family verprolin homologous protein (WAVE) interacting receptor sequence. (D) The amino-acid sequence similarity of each domain between different groups. Note that the EC2/EC3 are the most diversified ectodomains and EC5/EC6 are the most conserved ectodomains among all six ectodomains. VCDs are the least conserved. (E) Percentage of cortical cells expressing Pcdhα or Pcdhβγ. The Pcdhα isoforms are expressed in 74.5% of while the Pcdhβγ isoforms are expressed in nearly all of cortical cells. (F) Venn diagram of populations of single cells expressing Pcdhαβγ.
The three Pcdh clusters contain tandem arrays of CTCF binding sites (CBS) in both gene promoters and enhancers (Figure 1A) (Golan-Mashiach et al., 2012; Guo et al., 2012; Monahan et al., 2012). CTCF/cohesin-mediated long-range chromatin interactions between these CBS elements determine the stochastic and combinatorial expression of diverse cPcdh repertoires on the cell surface (Wu and Jia, 2020). Differential expression of cPcdh genes in individual neurons is regulated by epigenetic modifications, such as DNA methylation and H3K9me3 histone modification in the promoter region (Guo et al., 2012; Toyoda et al., 2014; Chen et al., 2015; Jiang et al., 2017; Wada et al., 2018). In particular, DNA methylation precludes the binding of CTCF proteins (Wu et al., 2001; Guo et al., 2012; Yin et al., 2017). Without CTCF binding, cPcdh promoters cannot form long-range chromatin contacts with remote enhancers and thus cannot be activated (Guo et al., 2012; Canzio et al., 2019). It is this higher-order chromatin architecture that determines the Pcdh promoter choice in the cell nucleus (Wu and Jia, 2020).
Cell-Surface Delivery of cPcdhs Through Cytoplasm
Significant progress has been made on cell-surface delivery mechanisms of cPcdhs. In particular, Pcdhγ proteins are initially detected in the cytoplasm as a mobile pool and their cell-surface delivery corresponds to the maturation of synapses (Fernandez-Monreal et al., 2009, 2010; LaMassa et al., 2020). For example, Pcdhγa3 and Pcdhγb2 form juxtanuclear membrane tubules in the cytoplasm when overexpressed (Hanson et al., 2010). In contrast, Pcdhγc4 accumulates as continuous sheets but does not form tubules (Hanson et al., 2010). Intracellular trafficking and tubulation of Pcdhγa3 are determined by a 26-amino-acid VCD motif (O’Leary et al., 2011). Consistently, despite that the VCD domain is the most diverse region of cPcdhs (Figures 1C,D), there are several highly conserved residues within the VCD motif (Wu and Maniatis, 1999; Shonubi et al., 2015).
Pcdhαc2 and isoforms of the Pcdh β and γ families (except γc4) can be delivered to the cell surface by themselves; by contrast, surface localization of Pcdhα (except αc2) and Pcdhγc4 requires the co-expression with other isoforms of Pcdh β or γ, known as carrier isoforms (Murata et al., 2004; Bonn et al., 2007; Thu et al., 2014; Goodman et al., 2016b). The mechanism by which carrier isoforms facilitate the delivery of members of Pcdhα and Pcdhγc4 to the cell surface is not known but may require cis-dimerization on the ER membrane between cPcdh isoforms. This hypothesis is consistent with evidence of cis-dimerization or multimerization between cPcdhs in transfection experiments (Schalm et al., 2010; Schreiner and Weiner, 2010; Biswas et al., 2012; Thu et al., 2014; Rubinstein et al., 2015). Finally, analyses of single-cell RNA-seq data of 23,178 cortical cells (Tasic et al., 2018) revealed that almost every cell (97.0%) expresses isoforms of Pcdh β or γ but only 74.5% cells express members of Pcdhα (Figure 1E). Moreover, every Pcdhα-expressing cell (99.4%) co-expresses isoforms of either Pcdh β or γ, while only 76.7% of Pcdh β- or γ-expressing cells co-express members of Pcdhα, consistent with the requirement of Pcdh β or γ for the cell-surface delivery of Pcdhα (Figure 1F).
Self-Recognition and Non-self Discrimination on the Cell Surface
The cis-dimers of cPcdhs can engage in highly stringent homophilic trans-interactions to generate cell-recognition specificities (Thu et al., 2014). The trans-interactions of cPcdhs are mediated through EC1–EC4 domains, especially the most-divergent EC2 and EC3 domains, in an anti-parallel manner (Figures 1C,D, 2) (Wu, 2005; Schreiner and Weiner, 2010; Nicoludis et al., 2015, 2016, 2019; Rubinstein et al., 2015; Goodman et al., 2016a; Brasch et al., 2019). Remarkably, liposome-tethered ectodomains spontaneously assemble into a zipper-like lattice structure (Brasch et al., 2019). Thus, when neurites bearing the same cPcdh isoforms contact with each other, the cPcdhs are thought to form a zipper lattice through homophilic trans-interactions and trigger an intracellular signaling pathway that eventually leads to adhesion or repulsion (e.g., self-avoidance) through cytoskeletal remodeling (Molumby et al., 2016; Fan et al., 2018; Brasch et al., 2019).
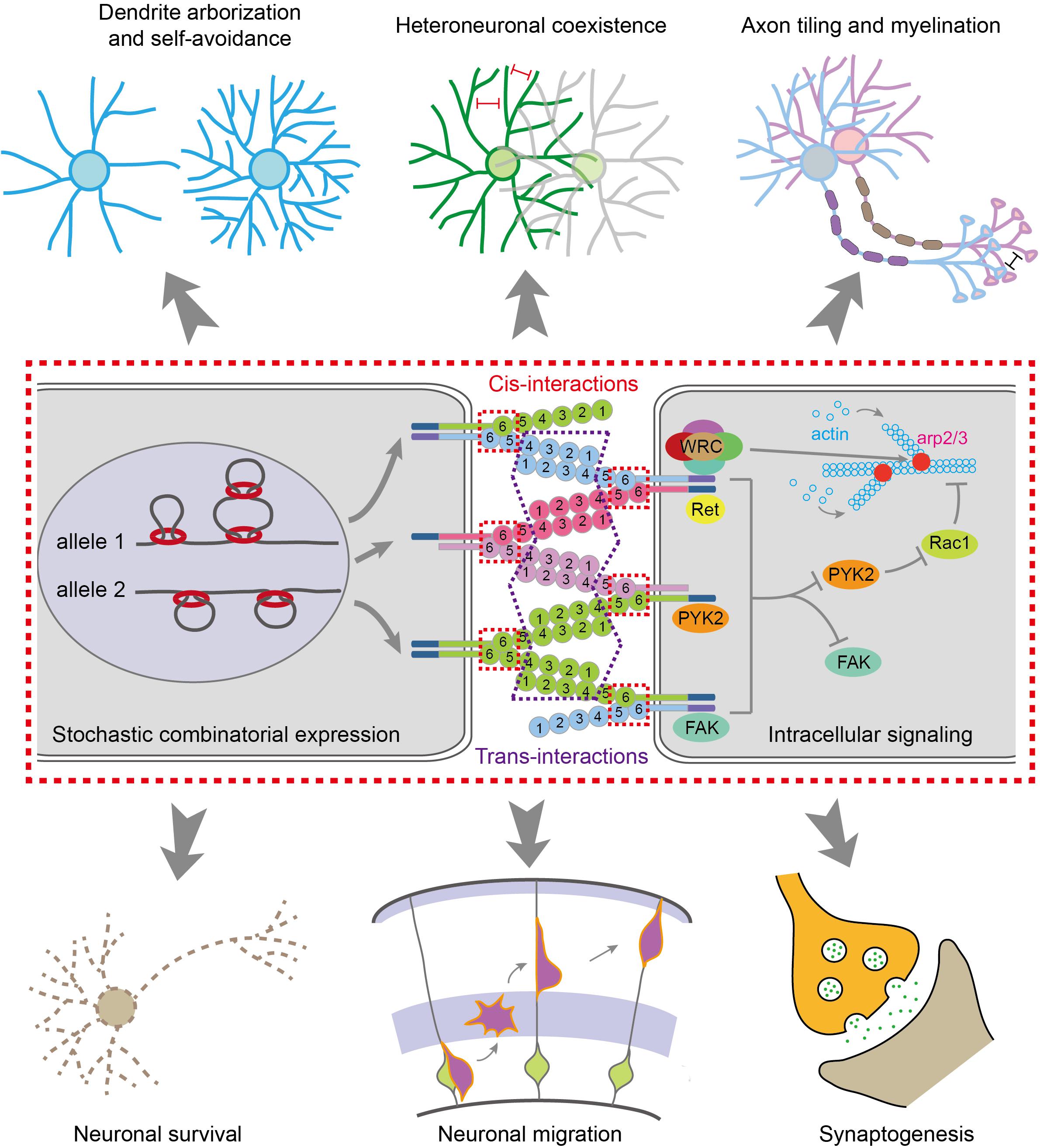
Figure 2. Molecular mechanisms for generating diverse neural identity codes. The stochastic and combinatorial expression of cPcdhs by CTCF/cohesin-mediated chromatin interactions generates vast repertoires of cell surface proteins. Most cPcdh genes are expressed monoallelically. The cPcdh proteins form cis-dimers and engage in specific homophilic trans-interactions. Through cytoplasmic common effectors of WRC, Pyk2, FAK, Ret and Rac1, cPcdhs regulate actin dynamics and cytoskeletal remodeling, eventually leading to various cellular functions, such as dendritic arborization and self avoidance, heteroneuronal neurite coexistence, axon tiling and myelination, neuronal survival and migration, and synaptogenesis.
Isoforms of Pcdhα and Pcdhγ could be cleaved by the metalloproteinase ADAM10 and γ-secretase to release the intracellular domain which translocates into the nucleus to regulate gene expression (Haas et al., 2005; Hambsch et al., 2005; Reiss et al., 2006; Bonn et al., 2007; Buchanan et al., 2010). Thus, diverse extracellular signals can converge on the same intracellular signaling pathway through the common CD shared by members of Pcdhα or Pcdhγ (Wu and Maniatis, 1999; Wu et al., 2001; Schalm et al., 2010; Fan et al., 2018) (reviewed in Wu and Jia, 2020). In addition, the intracellular domains of Pcdhα and Pcdhγ can bind to several kinases including FAK (focal adhesion kinase), Pyk2 (proline-rich tyrosine kinase 2), and Ret (receptor tyrosine kinase rearranged during transformation) (Figure 2) (Chen et al., 2009; Schalm et al., 2010; Garrett et al., 2012; Suo et al., 2012; Keeler et al., 2015). Finally, Pyk2 inhibits the activities of Rac1, which regulates lamellipodial dynamics and actin cytoskeletal remodeling (Figure 2) (Suo et al., 2012; Fan et al., 2018).
Biological Functions of Clustered Pcdhs
Homophilic Adhesion-Induced Repulsion Is Required for Dendrite Self-Avoidance
During development, sister neurites avoid each other in innervating the receptive field, a phenomenon known as self-avoidance. Neurite self-avoidance requires neurons to discriminate self from non-self (Grueber and Sagasti, 2010; Zipursky and Grueber, 2013). In Drosophila, self-avoidance is mediated through diversified Dscam1 (Down syndrome cell adhesion molecule 1) generated by alternative splicing (Zipursky and Grueber, 2013). In vertebrates, it is thought to be achieved by diverse cPcdhs as supported by a growing body of evidence. In particular, knockout of Pcdhγ causes collapses of dendrites, a defect of self-avoidance, of the same starburst amacrine cells (Figure 3A) (Lefebvre et al., 2012; Kostadinov and Sanes, 2015). When the entire Pcdhγ cluster is replaced with a single Pcdhγ isoform, self-avoidance defects are rescued. However, these cells recognize heteroneuronal dendrites as ‘self’ since neighboring starburst cells also express the same cPcdh isoform (Lefebvre et al., 2012). Similar to starburst cells, dendritic self-avoidance defects were also observed in cerebellar Purkinje cells in the absence of Pcdhγ (Figure 3B) (Lefebvre et al., 2012; Toyoda et al., 2014). Subsequent studies demonstrated that Pcdhα and Pcdhγ function together to mediate dendritic self-avoidance of starburst and Purkinje cells (Ing-Esteves et al., 2018).
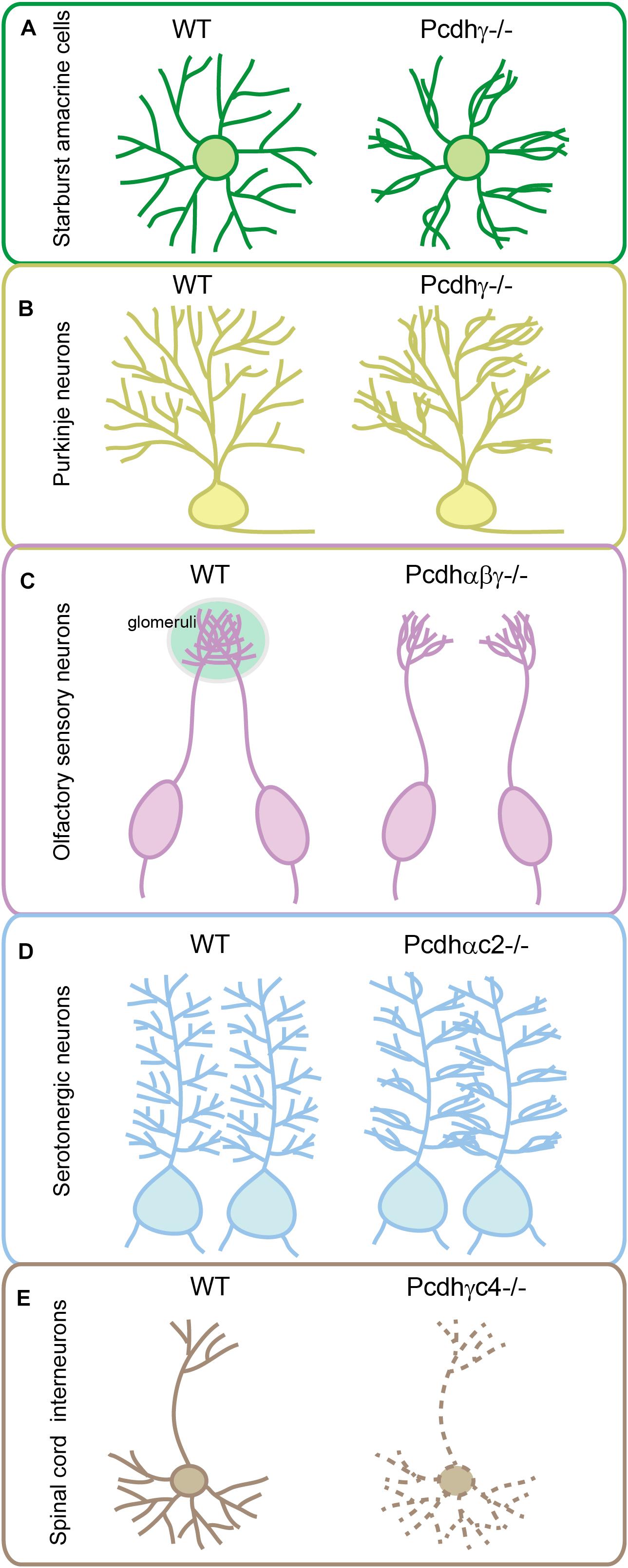
Figure 3. Genetic evidence for cPcdh functions in neuronal wiring. Pcdhγ has critical function in sister dendrite self-avoidance of the retinal starburst amacrine cells (A) and cerebellar Purkinje neurons (B). Tri-cluster Pcdhs are required for olfactory sensory neurons to form glomeruli in the olfactory bulb (C). Pcdhαc2 is essential for the even spacing and tiling of serotonergic axons (D). Pcdhγc4 is the sole necessary and sufficient isoform for interneuron survival in the spinal cord (E).
Isoform Diversity Is Required for Neurite Coexistence
In addition to a critical role in dendritic self-avoidance, cPcdh diversity plays a crucial role in dendrite coexistence (Lefebvre et al., 2012). Specifically, reducing the diversity of Pcdhγ disrupts dendrite coexistence in retinal starburst amacrine cells (Lefebvre et al., 2012). The cPcdh proteins also function in patterning of axon terminals. For example, olfactory sensory neurons (OSNs) expressing identical OR genes project their axons to the same location in the olfactory bulb and converge to form a glomerulus (Mombaerts et al., 1996). Mechanistically, alternate cPcdh isoforms from all three Pcdh clusters are stochastically expressed in single OSNs, ensuring that axons from different OSNs do not share the same profile and thus can coexist in the same glomeruli of the olfactory bulb (Mountoufaris et al., 2017). Genetic deletion of the entire Pcdhα cluster in mice results in abnormal sorting of OSN axons leading to the formation of ectopic small glomeruli (Hasegawa et al., 2008; Mountoufaris et al., 2017). Deletion of Pcdhβ or Pcdhγ also affects OSN wiring (Hasegawa et al., 2016; Mountoufaris et al., 2017). Finally, deletion of all three Pcdh clusters in mice causes the most severe phenotype with axons from the same OR-expressing OSNs branching profusely, reminiscent of the axonal self-avoidance defects (Figure 3C) (Mountoufaris et al., 2017).
Gain-of-function experiments provide additional insights into the role of cPcdhs in OSN wiring. Overexpression of the same three cPcdh isoforms (one isoform from each cluster) in different OSNs, which overrides the endogenous cPcdh diversity and makes every OSN express the same set of cPcdh isoforms, results in their axons repelling each other and projecting diffusely (Mountoufaris et al., 2017). These axons cannot converge in the same location of the olfactory bulb to form proper glomeruli. The severity of convergence defects correlates with numbers of neurons overexpressing cPcdhs (Mountoufaris et al., 2017).
Pcdhαc2 Regulates Axonal Tiling of Serotonergic Neurons
Serotonin is a critical neurotransmitter that regulates diverse higher-order brain functions including mood, cognition, reward, learning and memory. Although there are only 300,000 serotonergic neurons in the human brain, serotonergic axon terminals innervate the entire brain with even spacing. This tilling pattern is required for the maintenance of constant serotonin levels since serotonin molecules can only function over a short distance from their releasing sites. Genetic evidence suggests that Pcdhα participates in the even spacing of serotonergic axons in mice (Chen et al., 2017; Katori et al., 2017). Specifically, deletion of the Pcdhα cluster, but not double deletion of the Pcdh β and γ clusters, causes serotonergic axons to form clumps throughout the brain and results in insufficient terminal arborization in mice (Chen et al., 2017; Katori et al., 2017). In addition, labeling individual serotonergic neurons with brainbow technologies revealed that serotonergic axons from different neurons have tiling and self-avoidance defects upon constitutive Pcdhα deletion (Chen et al., 2017). Finally, these Pcdhα-deleted mice displayed depression-like behaviors, including increased immobility time in tail-suspension, forced-swimming, and contextual fear conditioning tests (Chen et al., 2017).
Conditional knockout of the entire Pcdhα cluster in serotonergic neurons or their target fields revealed cell-autonomous functions of Pcdhα in serotonergic neurons (Chen et al., 2017; Katori et al., 2017). However, deletion of α1-α12 of the Pcdhα cluster results in no obvious defect in serotonergic wiring, suggesting an important role of C-type Pcdhα genes in serotonin circuit formation. Consistently, deletion of both Pcdhαc1 and Pcdhαc2, or deletion of Pcdhαc2 alone, causes the clumping of serotonergic axons similar to deletion of the entire Pcdhα cluster (Chen et al., 2017; Katori et al., 2017). In addition, Pcdhαc2 is predominately expressed in serotonergic neurons (Chen et al., 2017; Katori et al., 2017). Thus, among all members of the Pcdhα cluster, only Pcdhαc2 has been shown to be required for serotonergic axon wiring (Figure 3D) (Chen et al., 2017; Katori et al., 2017). This is a striking example that a neuronal cell type uses a single cPcdh isoform to achieve axonal tiling or even spacing in all of its projecting fields. Therefore, in contrast to the diversity generated by multi-cPcdh isoforms for cell–cell recognition of starburst cells, Purkinje cells, and OSNs, the single-isoform-mediated repulsion is a remarkable strategy adopted by serotonergic axons to tile brain regions with even spacing. However, since Pcdhγ c4 and c5 are also expressed but at lower levels in serotonergic neurons (Chen et al., 2017), it is difficult to rule out their partially redundant contribution in patterning the serotonergic axons. Combinatorial C-type gene deletion and rescue experiments are needed to fully address this important question.
Pcdhγc4 Is Required for Interneuron Survival
Experiments of in situ hybridization and immunohistochemistry showed that Pcdhγc4 is expressed in many brain regions as well as in spinal cord (Zou et al., 2007; Miralles et al., 2020). Detailed analyses of constitutive or conditional mutant mice lacking all Pcdhγ genes revealed the increased apoptosis of interneurons in the spinal cord, retina, cerebral and cerebellar cortex (Wang et al., 2002b; Lefebvre et al., 2008; Prasad et al., 2008; Chen et al., 2012; Hasegawa et al., 2016; Carriere et al., 2020; Mancia Leon et al., 2020). For example, in retina, Pcdhγ deletion results in interneuron apoptosis but does not affect survival of neighboring WT interneurons, suggesting a cell-autonomous effect (Lefebvre et al., 2008). In the spinal cord, however, the survival of Pcdhγ-deficient interneurons can be enhanced if they are surrounded by neurons expressing Pcdhγ (Prasad et al., 2008). This non-cell autonomous effect on survival was also observed for cortical interneurons (Mancia Leon et al., 2020). Taken together, these studies suggest that the effect of Pcdhγ on interneuron survival is cell- or tissue-type dependent.
Knockout of the three C-type isoforms (Pcdhγc3-c5) of the Pcdhγ cluster results in neuronal apoptosis that is indistinguishable from that observed in mice with deletion of the entire Pcdhγ cluster (Chen et al., 2012; Carriere et al., 2020; Mancia Leon et al., 2020). Moreover, deletion of only Pcdhγc4 fully recapitulates interneuron apoptosis caused by deletion of the entire Pcdhγ cluster, suggesting that Pcdhγc4 is the only crucial isoform required for neuronal survival (Figure 3E) (Garrett et al., 2019). Although genetic evidence suggested that Pcdhγc4 contributes to interneuron survival, analyses of double or tri-cluster deletion mice revealed that Pcdh α and β clusters also have cooperative roles in neuronal survival. For example, cell apoptosis in the spinal cord of the Pcdhβγ double-cluster or tri-cluster deletion mice is much more severe than that of the Pcdhγ-cluster deletion mice, although deletion of Pcdhα or Pcdhβ themselves reveals no apoptosis (Hasegawa et al., 2016). In addition, double deletion of Pcdhα and Pcdhγ also leads to more severe apoptosis of retina cells than the Pcdhγ deletion alone (Ing-Esteves et al., 2018). It’s possible that members of Pcdhβ and Pcdhγ contribute to neuronal survival by acting as carrier isoforms to facilitate Pcdhγc4 to be delivered to the cell surface.
A Role of Pcdhα in Myelination
Pcdhα has been observed to be strongly expressed in the developing, but not mature, axons at the late embryonic and early postnatal stages in mice. Specifically, Pcdhα expression is negatively correlated with the increasing expression levels of myelination markers during development (Morishita et al., 2004). In addition, deletion of the Pcdhα cluster in mice results in delayed oligodendrocyte maturation and defects in myelination (Yu et al., 2012). Moreover, imaging by transmission electron microscopy revealed the reduced ratio of myelinated nerve fibers and abnormal myelin sheaths in the Pcdhα deletion mice (Lu et al., 2018). Consistently, patients with a deletion in the 5q31.3 region, which covers the Pcdhα cluster, show delayed myelination (Shimojima et al., 2011). Further investigation of how cPcdhs participate in myelination processes is urgently needed.
Clustered Pcdhs Regulate Spine Morphogenesis and Synaptogenesis
The establishment of proper synaptic connections in the brain is central to information processing. Subcellular fractionation and microscopic imaging studies have shown that isoforms of Pcdhα and Pcdhγ are located at synaptic junctions (Kohmura et al., 1998; Wang et al., 2002b; Phillips et al., 2003; Lefebvre et al., 2008; Garrett and Weiner, 2009). In addition, two isoforms of Pcdhβ, β16 and β22, have been found to be enriched in subsets of synapses in the retina and cerebellum (Junghans et al., 2008; Puller and Haverkamp, 2011; Nuhn and Fuerst, 2014). Finally, isoforms of Pcdhγ are found to accumulate at axodendritic and dendrodendritic synapses (Fernandez-Monreal et al., 2009). These expression patterns suggest a role of cPcdhs in synaptogenesis.
Deletion of Pcdhγ leads to a significant loss of synapses in the mouse spinal cord (Wang et al., 2002b). To specifically investigate the role of cPcdhs in synapse development, apoptosis is blocked by BAX mutation in the Pcdhγ deletion mice. In this case, the spinal cord still displays decreased synaptic density, suggesting a direct role of Pcdhγ in synaptogenesis (Weiner et al., 2005). In contrast, blocking apoptosis in Pcdhγ-deficient retina by mutating BAX reveals normal synaptic numbers (Lefebvre et al., 2008). In addition, the decreased VGAT+ and increased VGLUT1+ synapses seen in the Pcdhγ-deficient spinal cord are secondary to interneuron apoptosis (Chen et al., 2012). Another study showed that synapse development requires Pcdhγ-mediated contacts between astrocytes and neurons (Garrett and Weiner, 2009). Specifically, deletion of Pcdhγ in astrocytes in vivo reduces both excitatory and inhibitory synapses in a contact-dependent mechanism (Garrett and Weiner, 2009). Finally, reciprocal synaptic connections between sister neurons derived from the same neural stem cell are impaired by deletion of the three Pcdh clusters (Tarusawa et al., 2016).
In the cortical neurons of Pcdhγ knockout mice, only thin spines are slightly increased (Molumby et al., 2017). In addition, overexpression of a single Pcdhγ gene, γa1, significantly decreases spine density through repressing the postsynaptic cell adhesion molecule neuroligin-1 (Molumby et al., 2017). However, conditional knockout of Pcdhγ in the olfactory granule cells decreases dendritic spines (Ledderose et al., 2013). In addition, in Pcdhα deletion mice, a significant decrease of spine density was observed in hippocampal Golgi staining in vivo and cultured hippocampal neurons in vitro (Suo et al., 2012). Similar phenotype was found in Pcdhγ knockdown hippocampal neurons (Suo et al., 2012). In summary, cPcdhs are implicated in various aspects of synapse development in a cell-type dependent manner.
A Signaling Pathway Linking Clustered Pcdhs to Lamellipodial Formation and Actin Cytoskeletal Dynamics
A growing body of evidence supports the vital roles of cPcdhs in neuronal survival and migration, dendritic arborization and self-avoidance, spine morphogenesis and maturation, axonal targeting and tiling, as well as synaptogenesis (Figure 2) (Wang et al., 2002b; Weiner et al., 2005; Chen et al., 2012, 2017; Garrett et al., 2012; Lefebvre et al., 2012; Suo et al., 2012; Katori et al., 2017; Mountoufaris et al., 2017; Fan et al., 2018; Ing-Esteves et al., 2018; reviewed in Mountoufaris et al., 2018). These diverse functions of cPcdhs are all subserved by the intracellular signaling pathway via their conserved CDs. Interestingly, Pcdhα cluster contains a WIRS (Wiskott-Aldrich syndrome family verprolin homologous protein (WAVE) interacting receptor sequence) motif in CD which can directly bind WRC (WAVE regulatory complex), linking to actin filament branching via the Arp2/3 complex, to regulate lamellipodial dynamics and cytoskeletal remodeling (Fan et al., 2018).
Pcdhα also indirectly modulates actin dynamics through inhibiting Pyk2, which is a synaptic non-receptor tyrosine kinase. Pyk2, a known risk factor in neuropsychiatric diseases (Lambert et al., 2013), then regulates conformational changes of WRC via Rac1 (Fan et al., 2018). The Pcdh β and γ proteins, on the other hand, do not contain the WIRS motif. However, they may also modulate actin dynamics through forming cis-heterodimers with Pcdhα (Thu et al., 2014; Rubinstein et al., 2015; Goodman et al., 2017), or through the shared downstream effector Pyk2 (Chen et al., 2009; Suo et al., 2012) (reviewed in Wu and Jia, 2020). Thus, this actin cytoskeletal dynamic regulation via Pyk2 and WRC is a general underlying mechanism for various functions of cPcdhs (Fan et al., 2018). It is known that dysregulation of actin dynamics and synaptic structural plasticity is closely related to neuropsychiatric diseases (Forrest et al., 2018). In the following four sections, we discuss the genetic, epigenetic, 3D genomic, and environmental dysregulations of cPcdhs in neuropsychiatric diseases.
Genetic Variants of Clustered Pcdhs in Mental Disorders
Patients with the 5q31.3 deletion, which includes the three Pcdh clusters, show severe neurodevelopmental delay, encephalopathy associated with myelination defects, and hypotonia (Table 1) (Shimojima et al., 2011; Brown et al., 2013). In addition, human genetic studies revealed that deletion of a 5q31.3 fragment covering the Pcdhα cluster is associated with poor music perception, suggesting its role in human higher cognitive functions (Table 1) (Ukkola-Vuoti et al., 2013).
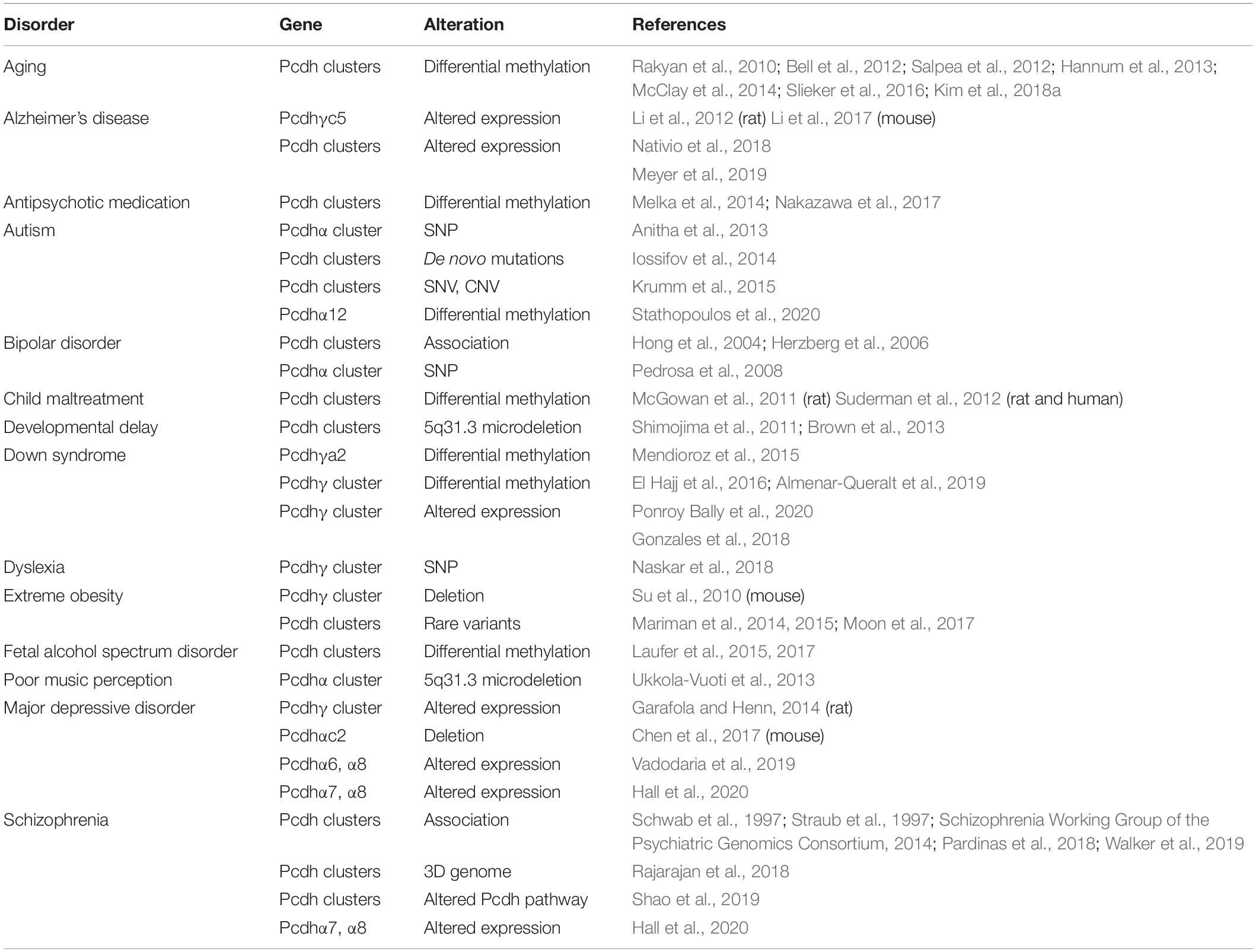
Table 1. Genetic/epigenetic dysregulation of the three Pcdh clusters causes various brain disorders.
Clustered Pcdh Genes in Autism Spectrum Disorders (ASD)
Autism spectrum disorders (ASD) are common neurodevelopment diseases with social, communicational, and behavioral impairments (Betancur et al., 2009). Genetic studies have identified cPcdhs as candidate genes for ASD. First, whole exome sequencing identified de novo mutation of Pcdhβ4 D555H in sporadic autism probands (O’Roak et al., 2012). Second, five single-nucleotide polymorphisms (SNPs) of Pcdhα are reported to have significant genetic associations with autism in a large cohort of 841 families (Anitha et al., 2013). Third, whole exome sequencing revealed that single nucleotide variants (SNVs) within the Pcdh clusters are associated with ASD (Iossifov et al., 2014; Krumm et al., 2015). Finally, abnormal CpG methylation patterns of Pcdhα12 are found in a South African ASD cohort (Stathopoulos et al., 2020).
Clustered Pcdh Genes in Bipolar Disorders
The 5q31 region is found to be associated with bipolar disorders (Hong et al., 2004; Herzberg et al., 2006). Specifically, patients with bipolar disorders showed a striking increase in homozygosity of a minor allele of the Pcdhα cluster, in which a SNP within the HS5-1 enhancer is found to be associated with aberrant Pcdhα expression (Pedrosa et al., 2008).
Clustered Pcdh Genes in Dyslexia
A set of SNPs within the Pcdhγ cluster is dominantly inherited in a family with dyslexia, a neurodevelopmental disorder characterized by reading and writing difficulties (Naskar et al., 2018). Two of them alter amino acids within EC2 and EC3 domains of Pcdh γa3 and γa4, respectively (Naskar et al., 2018). These missense SNPs located in the two critical ectodomains required for homophilic interactions, which might alter cell recognition and contribute to aberrant neuronal circuits of dyslexia.
Clustered Pcdh Genes in Extreme Obesity with Behavioral Abnormality
In patients with extreme obesity, cPcdhs have a significantly higher frequency of rare variants than in general population, indicating an association between rare variants of the Pcdh clusters and extreme obesity (Table 1) (Mariman et al., 2014, 2015). In addition, copy number variations (CNVs) of two Pcdh genes, Pcdhβ7 and Pcdhβ8, are associated with increased body mass index (Moon et al., 2017). In mice, Pcdhγ regulates hypothalamic feeding circuitry (Su et al., 2010), which is known to be involved in extreme obesity. In particular, conditional knockout of Pcdhγ in mouse hypothalamic neurons causes excessive feeding behaviors and defects in energy homeostasis, leading to extreme obesity (Su et al., 2010). These studies suggest that cPcdhs may play an important role in regulating neural circuits controlling feeding behavior, resulting in extreme obesity.
Epigenetic Dysregulations of Clustered Pcdhs in Mental Disorders
Clustered Pcdhs in Down Syndrome
Down syndrome (DS) is a genetic disorder with intellectual disabilities caused by an extra copy of chromosome 21. In the developing DS brain tissues, 5′ alternate isoforms of the Pcdhγ cluster are significantly hypermethylated (Table 1) (Mendioroz et al., 2015; El Hajj et al., 2016). In addition, in postmortem DS brains, 3′ alternate isoforms of the Pcdhγ cluster retain the fetal-like methylation state (Almenar-Queralt et al., 2019). Consequently, expression levels of these isoforms remain high in adult brain tissues, similar to that of the fetal stage, supporting a model of delayed brain maturation in the DS patients (Almenar-Queralt et al., 2019). Moreover, human DS iPSC-derived astrocytes show a transcriptional profile between fetal and mature stages, further supporting the delayed maturation model of DS (Ponroy Bally et al., 2020). Furthermore, these DS astrocytes show reduced levels of the Pcdhγ expression and have selective impairments in Pcdhγ-mediated cell adhesion (Ponroy Bally et al., 2020). Finally, nearly all of the cPcdh genes are downregulated in trisomic iPSCs comparing to the genetically edited disomic iPSCs derived from the same DS patient (Gonzales et al., 2018). Considering the important functions of cPcdhs in brain development, it’s possible that dysregulated cPcdhs may be related to defective synaptogenesis and neurite growth seen in DS brains.
Clustered Pcdhs in Alzheimer’s Disease (AD)
In humans, the expression of cPcdh genes is frequently found to be altered in AD. For example, Pcdhαc2 and Pcdhγc5 are decreased while subsets of alternate Pcdhγ isoforms are increased in the lateral temporal lobe of postmortem AD brains (Nativio et al., 2018). Interestingly, APOE4, a prominent genetic risk allele for late-onset AD, causes cPcdh gene upregulation in iPSC-derived cerebral organoids (Meyer et al., 2019).
In mice, loss of excitatory or inhibitory synapses has been implicated in the pathogenesis of AD. For example, Pcdhγc5 specifically interacts with GABA receptors through its cytoplasmic domain to stabilize and promote inhibitory GABAergic synaptic transmission (Li et al., 2012). The expression levels of Pcdhγc5 in GABAergic synapses are increased in hyperexcitatory conditions. The increased Pcdhγc5 enhances synaptic inhibition and elevates GABAergic protein levels, which may contribute to pathogenic excitatory/inhibitory imbalance in an AD mouse model (Table 1) (Li et al., 2017). As a downstream effector of cPcdhs, the cell-adhesion kinase Pyk2 has been identified as a major susceptibility gene for late-onset AD (Lambert et al., 2013). Consistently, overexpression of Pyk2 results in synapse loss and learning deficit (Lee et al., 2019; Salazar et al., 2019). Therefore, dysregulation of cPcdhs or disruption of its intracellular signaling pathway may contribute to AD pathogenesis.
Clustered Pcdhs in Major Depressive Disorders (MDD)
Reduced serotonin levels have been implicated in the pathogenesis of MDD and selective serotonin reuptake inhibitors (SSRIs) are the most commonly used antidepressants (Belmaker and Agam, 2008). In humans, altered expression patterns of members of the Pcdhα cluster are detected in serotonergic neurons derived from patients of SSRI-resistant MDD (Vadodaria et al., 2019). In addition, low expression levels of Pcdhα7 and Pcdhα8 in the prenatal brain are associated with MDD (Hall et al., 2020). Moreover, in rats with learned helplessness, a model of depression, members of the Pcdhγ cluster are upregulated in CA1 neurons (Garafola and Henn, 2014). Finally, in mice Pcdhαc2 is specifically expressed in serotonergic neurons and knockout of the Pcdhα cluster leads to depressive-like behaviors (Chen et al., 2017). In conjunction with the important role of cPcdhs in serotonergic axon wiring (Chen et al., 2017), these findings suggest that uneven distribution of serotonergic fibers by cPcdh dysregulation could lead to reduced serotonin signaling in various brain regions.
Clustered Pcdhs in Schizophrenia
Clustered Pcdhs have been identified as susceptible loci for schizophrenia (Schwab et al., 1997; Straub et al., 1997; Pardinas et al., 2018; Walker et al., 2019). In particular, large-scale genome-wide association studies have revealed that the cPcdh locus is associated with schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). In addition, high expression levels of members of the Pcdhα cluster, Pcdhα7 and Pcdhα8, are associated with schizophrenia (Hall et al., 2020). Finally, cPcdhs and their downstream effector PKC are dysregulated in hiPSC-derived cortical interneurons from schizophrenic patients (Shao et al., 2019).
Epigenetic modifications, especially DNA methylation of gene promoters, play a major role in cPcdh regulation (reviewed in Wu and Jia, 2020). Antisense transcription of long non-coding RNAs leads to demethylation of cPcdh promoters (Canzio et al., 2019). CTCF then binds the unmethylated cPcdh promoters and brings them in close contacts with remote enhancers via long-distance chromatin interactions to activate cPcdh transcription (Guo et al., 2012). Collectively, these studies suggest that epigenetic dysregulation of cPcdhs is related to various mental disorders. Since epigenetic and higher-order chromatin regulations are a general phenomenon, it is very likely that these regulations also affect other cell-surface neural receptors.
3D Genome Dysregulations of Clustered Pcdhs in Mental Disorders
The proper expression of the cPcdh genes requires specific long-range enhancer-promoter contacts in the 3D nuclear space. Mutations in genes encoding 3D genome architectural proteins, such as CTCF, cohesin, MeCP2, SETDB1, and WIZ, dysregulate cPcdh genes through altering higher-order chromatin contacts (Figure 4). Therefore, it’s interesting to note that an increasing list of mutations in genes encoding 3D genome architectural proteins is linked to brain disorders. We suggest that many complex neuropsychiatric diseases of mutations of 3D genome regulators are ‘actually’ resulted from cPcdh dysregulations.
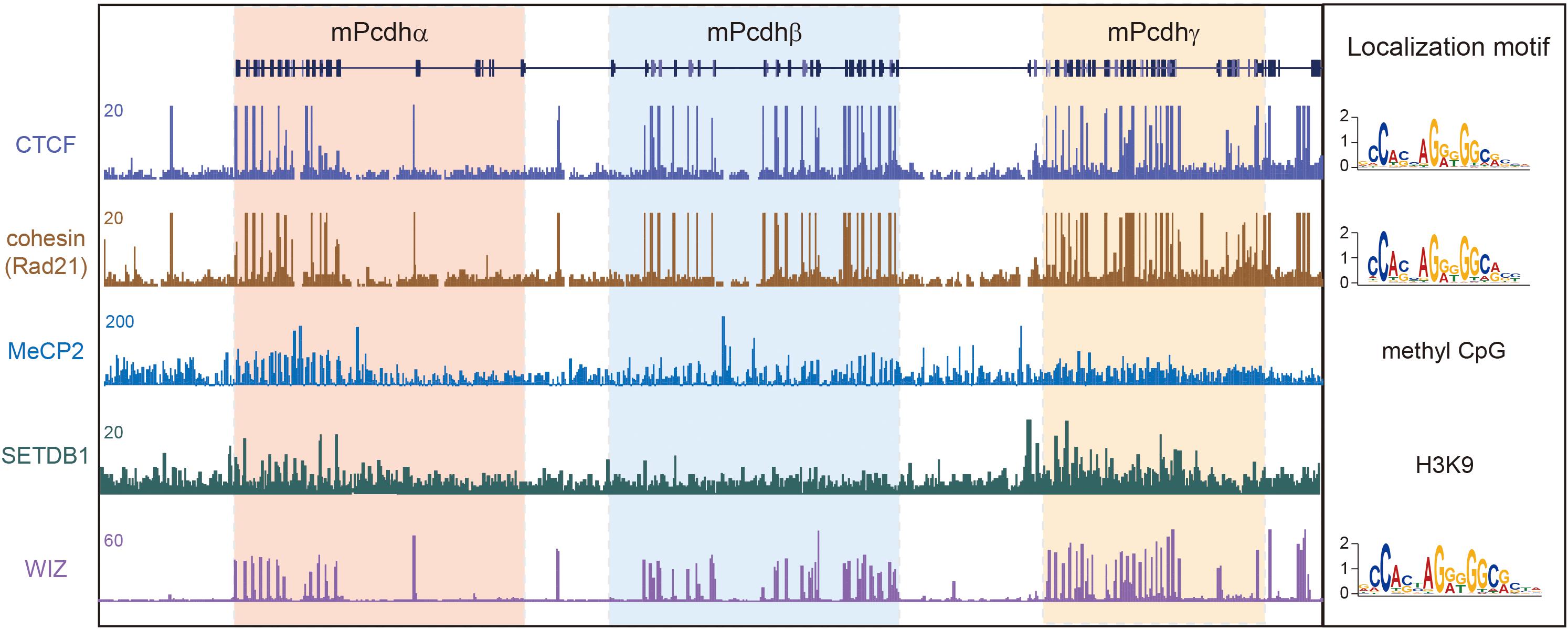
Figure 4. 3D genome architectural proteins regulate cPcdh gene expression. Shown are schematic binding patterns of architectural proteins in the three Pcdh clusters.
3D Genome Dysregulation of Clustered Pcdhs by CTCF
De novo mutations of CTCF in humans causes the type 21 of autosomal dominant mental retardation (MRD21) (Gregor et al., 2013; Bastaki et al., 2017; Hori et al., 2017; Chen et al., 2019; Konrad et al., 2019). This rare condition is first described in four individuals with intellectual disability, microcephaly, and growth retardation. Genetic studies revealed one individual with CTCF missense mutation, two with CTCF frameshift mutations, and one with CTCF deletion (Gregor et al., 2013). A subsequent study reported an additional individual of MRD21 with a different de novo CTCF frameshift mutation (Bastaki et al., 2017). Recent studies independently reported 2, 3, and 39 additional individuals of MRD21 with pathogenic variants of CTCF (Hori et al., 2017; Chen et al., 2019; Konrad et al., 2019). Finally, a human genetic study showed strong associations between CTCF SNPs and schizophrenia in multiple cohorts (Juraeva et al., 2014). Given that CTCF mutations or variants dysregulate cPcdh genes, it’s likely that this cPcdh dysregulation is responsible for complex brain disorders caused by CTCF mutations or variants.
In mice, conditional knockout of CTCF in cortical neurons downregulates nearly all of the cPcdh genes. These mice display abnormal behavior and growth retardation (Hirayama et al., 2012). Strikingly, every member of the cPcdh genes carrying CBS elements is downregulated, suggesting that long-distance chromatin looping between enhancers and promoters is impaired. Histological analyses revealed the loss of the functional somatosensory circuits and the reduction of dendritic arborizations in CTCF knockout mice, reminiscent of the phenotypes of cPcdh deletion (Hirayama et al., 2012). In addition, conditional CTCF knockout in excitatory or inhibitory neurons impairs long term memory and cortical synaptic plasticity (Kim et al., 2018b). Finally, RNA-seq experiments revealed a predominant dysregulation of the cPcdh genes (Kim et al., 2018b). In conjunction with the known role of CTCF in the cPcdh 3D regulation, it is possible that neuropsychiatric symptoms in human CTCF mutations are related to the dysregulation of cPcdh genes.
3D Genome Dysregulation of Clustered Pcdhs by Cohesin and its Regulators
The cohesin complex, which mediates chromatin loop extrusion and sister chromatid cohesion, is central for 3D genome folding. Mutations of cohesin or of its regulators have been shown to be associated with a large set of complex human diseases, collectively called cohesinopathies (Liu and Krantz, 2008). The symptoms of cohesinopathies vary in different mutations but share the same manifestations such as cognitive retardation and intellectual disability. Cornelia de Lange syndrome (CdLS), the best characterized cohesinopathy, is caused by mutations of cohesin regulators, such as NIPBL and HDAC8, or of cohesin subunits, including SMC1A, SMC3, and RAD21 (Bose and Gerton, 2010). Roberts syndrome, which is phenotypically related to CdLS, is caused by mutations of the cohesin regulator ESCO2 (Liu and Krantz, 2008). Similar to CTCF, knockdown of cohesin in cell lines significantly downregulates the expression of cPcdh genes (Guo et al., 2012). Interestingly, only the SA1 but not SA2 subunit of cohesin is located at cPcdh promoters and regulates their activities (Remeseiro et al., 2012). Collectively, cognitive impairments in cohesinopathies with mutations of cohesin subunits or of its regulators may result from their 3D genome dysregulations of the cPcdh genes.
3D Genome Dysregulation of Clustered Pcdhs by MeCP2
The Rett syndrome is a rare, severe type of ASDs caused by mutations in the X-linked MeCP2 (methyl-CpG binding protein 2) gene (Amir et al., 1999). In addition, Pcdhβ1 is upregulated in both the MeCP2 mutant mice and postmortem brains of Rett syndrome patients (Miyake et al., 2011). Moreover, in MeCP2 mutant mice, several members of the Pcdh clusters are dysregulated (Chahrour et al., 2008). Finally, in the human cPcdh locus, each of the 15 Pcdhα, 16 Pcdhβ, and 22 Pcdhγ genes is associated with a CpG island (Wu et al., 2001) and methylation of a CpG dinucleotide within the cPcdh promoter CTCF sites precludes CTCF binding (Guo et al., 2012). Given the known role of MeCP2 in gene regulation via the binding to methylated CpG islands, MeCP2 and CTCF may bind mutual exclusively to the cPcdh promoters. Thus, the dysregulation of cPcdhs by MeCP2 mutations may result from altering 3D genome configuration via CTCF.
3D Genome Dysregulation of Clustered Pcdhs by SETDB1
Deletion of SETDB1 (SET domain bifurcated 1), which encodes a histone H3K9 methyltransferase, results in a five-hundred-fold increase of cPcdh transcripts in the mouse cortex compared to the rest of the genome (Jiang et al., 2017). SETDB1 protects the cPcdh locus from excessive CTCF binding to maintain the cPcdh superTAD structure (Jiang et al., 2017). Interestingly, the 5’ boundary of the cPcdh superTAD, which enriches with H3K9me3 histone modifications (Figure 4), contains a haplotype significantly associated with schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Jiang et al., 2017). This region regulates the expression of cPcdh genes through long-range chromatin loops across hundreds of kilobases (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Rajarajan et al., 2018). Within this region a schizophrenia-risk SNP (rs111896713) significantly affects the expression of Pcdhα genes in a dosage-dependent manner (Fromer et al., 2016; Rajarajan et al., 2018).
Mutations of SETDB1 itself are also associated with psychiatric disorders such as autism (Cukier et al., 2012). In addition, the expression of SETDB1 is markedly increased in patients with Huntington’s disease (HD) (Ryu et al., 2006). In the mouse model of HD, 37 out of 58 cPcdh genes are striking dysregulated as the size of huntingtin CAG repeats increases (Langfelder et al., 2016). Together, cognitive defects associated with SETDB1 mutations may result from the dysregulation of cPcdhs.
3D Genome Dysregulation of Clustered Pcdhs by WIZ
WIZ (Widely interspaced zinc finger-containing protein) forms a heteromeric H3K9 methyltransferase complex with G9a and GLP. In autism patients, exon resequencing identified non-synonymous variants in WIZ and its partners G9a/GLP (Balan et al., 2014). Haploinsufficiency of GLP causes the Kleefstra syndrome, a multi-system syndrome associated with intellectual disability, neurodevelopmental delay, and neuropsychiatric diseases (Willemsen et al., 2012). WIZ is a modulator of chromatin loops and its genomic binding sites overlap with CTCF (Figure 4) (Isbel et al., 2016; Justice et al., 2020). Remarkably, only members of the Pcdhβ cluster are dysregulated in WIZ mutant mice (Isbel et al., 2016). Interestingly, these mice display an anxiety-like phenotype (Isbel et al., 2016), consistent with the requirement for cPcdhs in neuronal development and connectivity. All in all, these human and mouse genetic studies suggest that mutations of genome architectural proteins may be involved in pathogenesis of complex neural diseases through dysregulation of cPcdh genes (Figure 4).
Dysregulations of Clustered Pcdhs in Mental Disorders by Environmental Factors
Clustered Pcdhs and Child Maltreatment
Early life experiences are known to have long-term effects on mental health and behavior through epigenetic reprogramming, including alterations in stress responses. cPcdhs display differential methylations in response to maternal cares. In children suffered from maltreatment or abuse, increased methylation levels are seen in promoters across the three Pcdh clusters, especially in Pcdhα (Table 1) (Suderman et al., 2012). In rat pups who received less licking and grooming from mothers, cPcdh promoters are hypermethylated and their expression levels are downregulated (McGowan et al., 2011; Suderman et al., 2012). These studies suggest that early life experience affects cPcdh expression through epigenetic modification of their promoters, which may contribute to the psychological abnormalities in abused children.
Clustered Pcdhs and Fetal Alcohol Spectrum Disorders (FASD)
Similar to early life experiences, prenatal alcohol exposure during pregnancy, which can cause lifelong alterations in cognition and behavior, also influences the methylation states of the three Pcdh clusters (Laufer et al., 2015, 2017). In mouse models of FASDs and in children suffered from prenatal alcohol exposure, there are similar methylation changes in the cPcdh genes (Laufer et al., 2015, 2017).
Clustered Pcdhs and Antipsychotic Medication
Olanzapine, an antipsychotic medication commonly used for treating bipolar disorders and schizophrenia, causes methylation changes in the cortex in a rat model. Interestingly, the promoter regions of cPcdhs showed prominent methylation alterations in the cerebrum and hippocampus (Table 1) (Melka et al., 2014). In addition, clozapine, the most effective medication for treatment-resistant schizophrenia, causes differential expression of cPcdh genes in iPSC-derived neurons (Nakazawa et al., 2017). Thus, antipsychotic drugs may exert their therapeutic effects through altering the regulation of cPcdhs.
Clustered Pcdhs and Brain Aging
Epigenetic alteration is a hallmark of aging. Interestingly, many age-related differentially methylated regions are found to be located within the three Pcdh clusters (Rakyan et al., 2010; Bell et al., 2012; Hannum et al., 2013; Slieker et al., 2016). In addition, Pcdhα is prominently differentially methylated during aging (McClay et al., 2014). Moreover, epigenetic modifications in the three Pcdh clusters are reprogrammed during aging (Salpea et al., 2012). Finally, a 134-twin aging study revealed that methylation states of cPcdh promoters correlate with biological ages (Kim et al., 2018a). Altogether, various environmental factors can influence methylation states of the Pcdh clusters, leading to their abnormal expression patterns, which may be related to mental disorders and behavioral abnormalities.
Concluding Remarks
Clustered Pcdhs are thought to function as molecular identity codes for individual neurons to discriminate self from non-self (Lefebvre et al., 2012; Mountoufaris et al., 2018; Honig and Shapiro, 2020; Sanes and Zipursky, 2020; Wu and Jia, 2020). The tremendous diversity afforded by unique variable and constant genomic organization (Figure 1), the stochastic promoter choice realized by topological chromatin looping and balanced enhancer contacts (Figure 1A), and amazing structural recognition evolved through promiscuous cis-interactions and stringent homophilic trans-interactions (Figure 2) enable billions of neurons to achieve neurite self-avoidance and to establish proper connectivity in the brain. A key unresolved question is how cPcdh homophilic adhesions are transformed into neurite repulsion. This is likely achieved through common intracellular signaling transduction pathways with Pyk2, FAK, Ret, and WRC complex, eventually leading to actin dynamics and cytoskeletal remodeling (Figure 2). Shedding the extracellular domain by metalloproteinase may be essential for separating the homophilic-interaction-bridged plasma membranes. Finally, genetic studies provide strong evidence that cPcdh genes are central for neurodevelopment and neurite morphogenesis in the brain (Figure 3).
The diverse roles of cPcdh genes in neural circuit formation suggest that their mutations or dysregulations may be involved in complex brain disorders. Indeed, numerous studies reported mutations of cPcdh genes in Alzheimer’s disease, ASDs, bipolar disorders, major depressive disorders, schizophrenia, behavioral abnormalities, and several neurodevelopmental disorders with cognitive impairments (Table 1). In addition, mutations of 3D genome architectural proteins or regulators of cPcdh genes such as CTCF, cohesin, MeCP2, SETDB1, and WIZ cause a wide variety of brain disorders (Figure 4). These overwhelming data suggest that mutations or dysregulations of cPcdh genes play an important role in the pathogenesis of brain disorders.
In summary, growing lines of evidence suggest the important roles of cPcdh genes in complex mental or brain disorders. However, most brain and mind diseases have complex etiology and pathogenesis. How these diseases are initiated and progressed through genetic mutations and epigenetic dysregulations of cPcdh genes remain largely unknown. A deeper understanding of regulatory mechanisms and biological functions of cPcdh genes may be the key to understand the etiology and pathogenesis and to facilitate developing rational therapeutic strategies in the future.
Author Contributions
Both authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31630039 and 31700666), the Ministry of Science and Technology of China (2017YFA0504203 and 2018YFC1004504), and the Science and Technology Commission of Shanghai Municipality (19JC1412500).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of our lab for extensive discussions and constructive comments. We apologize to our colleagues whose important contributions could not be cited due to space limitations.
References
Almenar-Queralt, A., Merkurjev, D., Kim, H. S., Navarro, M., Ma, Q., Chaves, R. S., et al. (2019). Chromatin establishes an immature version of neuronal protocadherin selection during the naive-to-primed conversion of pluripotent stem cells. Nat. Genet. 51, 1691–1701. doi: 10.1038/s41588-019-0526-4
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Anitha, A., Thanseem, I., Nakamura, K., Yamada, K., Iwayama, Y., Toyota, T., et al. (2013). Protocadherin alpha (PCDHA) as a novel susceptibility gene for autism. J. Psychiatry Neurosci. 38, 192–198. doi: 10.1503/jpn.120058
Balan, S., Iwayama, Y., Maekawa, M., Toyota, T., Ohnishi, T., Toyoshima, M., et al. (2014). Exon resequencing of H3K9 methyltransferase complex genes, EHMT1, EHTM2 and WIZ, in Japanese autism subjects. Mol. Autism 5:49. doi: 10.1186/2040-2392-5-49
Bastaki, F., Nair, P., Mohamed, M., Malik, E. M., Helmi, M., and Al-Ali, M. T. (2017). Identification of a novel CTCF mutation responsible for syndromic intellectual disability – a case report. BMC Med. Genet. 18:68. doi: 10.1186/s12881-017-0429-0
Bell, J. T., Tsai, P. C., Yang, T. P., Pidsley, R., Nisbet, J., Glass, D., et al. (2012). Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 8:e1002629. doi: 10.1371/journal.pgen.1002629
Belmaker, R. H., and Agam, G. (2008). Major depressive disorder. N. Engl. J. Med. 358, 55–68. doi: 10.1056/NEJMra073096
Betancur, C., Sakurai, T., and Buxbaum, J. D. (2009). The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 32, 402–412. doi: 10.1016/j.tins.2009.04.003
Biswas, S., Emond, M. R., and Jontes, J. D. (2012). The clustered protocadherins Pcdhalpha and Pcdhgamma form a heteromeric complex in zebrafish. Neuroscience 219, 280–289. doi: 10.1016/j.neuroscience.2012.05.058
Bonn, S., Seeburg, P. H., and Schwarz, M. K. (2007). Combinatorial expression of alpha- and gamma-protocadherins alters their presenilin-dependent processing. Mol. Cell. Biol. 27, 4121–4132. doi: 10.1128/MCB.01708-06
Bose, T., and Gerton, J. L. (2010). Cohesinopathies, gene expression, and chromatin organization. J. Cell Biol. 189, 201–210. doi: 10.1083/jcb.200912129
Brasch, J., Goodman, K. M., Noble, A. J., Rapp, M., Mannepalli, S., Bahna, F., et al. (2019). Visualization of clustered protocadherin neuronal self-recognition complexes. Nature 569, 280–283. doi: 10.1038/s41586-019-1089-3
Brown, N., Burgess, T., Forbes, R., McGillivray, G., Kornberg, A., Mandelstam, S., et al. (2013). 5q31.3 Microdeletion syndrome: clinical and molecular characterization of two further cases. Am. J. Med. Genet. A 161A, 2604–2608. doi: 10.1002/ajmg.a.36108
Buchanan, S. M., Schalm, S. S., and Maniatis, T. (2010). Proteolytic processing of protocadherin proteins requires endocytosis. Proc. Natl. Acad. Sci. U.S.A. 107, 17774–17779. doi: 10.1073/pnas.1013105107
Canzio, D., Nwakeze, C. L., Horta, A., Rajkumar, S. M., Coffey, E. L., Duffy, E. E., et al. (2019). Antisense lncRNA transcription mediates DNA demethylation to drive stochastic protocadherin alpha promoter choice. Cell 177, 639–653.e15. doi: 10.1016/j.cell.2019.03.008
Carriere, C. H., Wang, W. X., Sing, A. D., Fekete, A., Jones, B. E., Yee, Y., et al. (2020). The gamma-Protocadherins regulate the survival of GABAergic interneurons during developmental cell death. J. Neurosci. doi: 10.1523/JNEUROSCI.1636-20.2020 [Epub ahead of print].
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chen, F., Yuan, H., Wu, W., Chen, S., Yang, Q., Wang, J., et al. (2019). Three additional de novo CTCF mutations in Chinese patients help to define an emerging neurodevelopmental disorder. Am. J. Med. Genet. C Semin. Med. Genet. 181, 218–225. doi: 10.1002/ajmg.c.31698
Chen, J., Lu, Y., Meng, S., Han, M. H., Lin, and Wang, X. (2009). alpha- and gamma-Protocadherins negatively regulate PYK2. J. Biol. Chem. 284, 2880–2890. doi: 10.1074/jbc.M807417200
Chen, K., Hu, J., Moore, D. L., Liu, R., Kessans, S. A., Breslin, K., et al. (2015). Genome-wide binding and mechanistic analyses of Smchd1-mediated epigenetic regulation. Proc. Natl. Acad. Sci. U.S.A. 112, E3535–E3544. doi: 10.1073/pnas.1504232112
Chen, W. V., Alvarez, F. J., Lefebvre, J. L., Friedman, B., Nwakeze, C., Geiman, E., et al. (2012). Functional significance of isoform diversification in the protocadherin gamma gene cluster. Neuron 75, 402–409. doi: 10.1016/j.neuron.2012.06.039
Chen, W. V., Nwakeze, C. L., Denny, C. A., O’Keeffe, S., Rieger, M. A., Mountoufaris, G., et al. (2017). Pcdhalphac2 is required for axonal tiling and assembly of serotonergic circuitries in mice. Science 356, 406–411. doi: 10.1126/science.aal3231
Cukier, H. N., Lee, J. M., Ma, D., Young, J. I., Mayo, V., Butler, B. L., et al. (2012). The expanding role of MBD genes in autism: identification of a MECP2 duplication and novel alterations in MBD5, MBD6, and SETDB1. Autism Res. 5, 385–397. doi: 10.1002/aur.1251
DeFelipe, J., Marco, P., Busturia, I., and Merchan-Perez, A. (1999). Estimation of the number of synapses in the cerebral cortex: methodological considerations. Cereb. Cortex 9, 722–732. doi: 10.1093/cercor/9.7.722
El Hajj, N., Dittrich, M., Bock, J., Kraus, T. F., Nanda, I., Muller, T., et al. (2016). Epigenetic dysregulation in the developing Down syndrome cortex. Epigenetics 11, 563–578. doi: 10.1080/15592294.2016.1192736
Fan, L., Lu, Y., Shen, X., Shao, H., Suo, L., and Wu, Q. (2018). Alpha protocadherins and Pyk2 kinase regulate cortical neuron migration and cytoskeletal dynamics via Rac1 GTPase and WAVE complex in mice. eLife 7:e35242. doi: 10.7554/eLife.35242
Fernandez-Monreal, M., Kang, S., and Phillips, G. R. (2009). Gamma-protocadherin homophilic interaction and intracellular trafficking is controlled by the cytoplasmic domain in neurons. Mol. Cell. Neurosci. 40, 344–353. doi: 10.1016/j.mcn.2008.12.002
Fernandez-Monreal, M., Oung, T., Hanson, H. H., O’Leary, R., Janssen, W. G., Dolios, G., et al. (2010). gamma-protocadherins are enriched and transported in specialized vesicles associated with the secretory pathway in neurons. Eur. J. Neurosci. 32, 921–931. doi: 10.1111/j.1460-9568.2010.07386.x
Flaherty, E., and Maniatis, T. (2020). The role of clustered protocadherins in neurodevelopment and neuropsychiatric diseases. Curr. Opin. Genet. Dev. 65, 144–150. doi: 10.1016/j.gde.2020.05.041
Forrest, M. P., Parnell, E., and Penzes, P. (2018). Dendritic structural plasticity and neuropsychiatric disease. Nat. Rev. Neurosci. 19, 215–234. doi: 10.1038/nrn.2018.16
Fromer, M., Roussos, P., Sieberts, S. K., Johnson, J. S., Kavanagh, D. H., Perumal, T. M., et al. (2016). Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 19, 1442–1453. doi: 10.1038/nn.4399
Garafola, C. S., and Henn, F. A. (2014). A change in hippocampal protocadherin gamma expression in a learned helpless rat. Brain Res. 1593, 55–64. doi: 10.1016/j.brainres.2014.08.071
Garrett, A. M., Bosch, P. J., Steffen, D. M., Fuller, L. C., Marcucci, C. G., Koch, A. A., et al. (2019). CRISPR/Cas9 interrogation of the mouse Pcdhg gene cluster reveals a crucial isoform-specific role for Pcdhgc4. PLoS Genet. 15:e1008554. doi: 10.1371/journal.pgen.1008554
Garrett, A. M., Schreiner, D., Lobas, M. A., and Weiner, J. A. (2012). gamma-protocadherins control cortical dendrite arborization by regulating the activity of a FAK/PKC/MARCKS signaling pathway. Neuron 74, 269–276. doi: 10.1016/j.neuron.2012.01.028
Garrett, A. M., and Weiner, J. A. (2009). Control of CNS synapse development by {gamma}-protocadherin-mediated astrocyte-neuron contact. J. Neurosci. 29, 11723–11731. doi: 10.1523/JNEUROSCI.2818-09.2009
Golan-Mashiach, M., Grunspan, M., Emmanuel, R., Gibbs-Bar, L., Dikstein, R., and Shapiro, E. (2012). Identification of CTCF as a master regulator of the clustered protocadherin genes. Nucleic Acids Res. 40, 3378–3391. doi: 10.1093/nar/gkr1260
Gonzales, P. K., Roberts, C. M., Fonte, V., Jacobsen, C., Stein, G. H., and Link, C. D. (2018). Transcriptome analysis of genetically matched human induced pluripotent stem cells disomic or trisomic for chromosome 21. PLoS One 13:e0194581. doi: 10.1371/journal.pone.0194581
Goodman, K. M., Rubinstein, R., Dan, H., Bahna, F., Mannepalli, S., Ahlsen, G., et al. (2017). Protocadherin cis-dimer architecture and recognition unit diversity. Proc. Natl. Acad. Sci. U.S.A. 114, E9829–E9837. doi: 10.1073/pnas.1713449114
Goodman, K. M., Rubinstein, R., Thu, C. A., Bahna, F., Mannepalli, S., Ahlsen, G., et al. (2016a). Structural basis of diverse homophilic recognition by clustered alpha- and beta-protocadherins. Neuron 90, 709–723. doi: 10.1016/j.neuron.2016.04.004
Goodman, K. M., Rubinstein, R., Thu, C. A., Mannepalli, S., Bahna, F., Ahlsen, G., et al. (2016b). gamma-Protocadherin structural diversity and functional implications. eLife 5:e20930. doi: 10.7554/eLife.20930
Gregor, A., Oti, M., Kouwenhoven, E. N., Hoyer, J., Sticht, H., Ekici, A. B., et al. (2013). De novo mutations in the genome organizer CTCF cause intellectual disability. Am. J. Hum. Genet. 93, 124–131. doi: 10.1016/j.ajhg.2013.05.007
Grueber, W. B., and Sagasti, A. (2010). Self-avoidance and tiling: mechanisms of dendrite and axon spacing. Cold Spring Harb. Perspect. Biol. 2:a001750. doi: 10.1101/cshperspect.a001750
Guo, Y., Monahan, K., Wu, H., Gertz, J., Varley, K. E., Li, W., et al. (2012). CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proc. Natl. Acad. Sci. U.S.A. 109, 21081–21086. doi: 10.1073/pnas.1219280110
Guo, Y., Xu, Q., Canzio, D., Shou, J., Li, J., Gorkin, D. U., et al. (2015). CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell 162, 900–910. doi: 10.1016/j.cell.2015.07.038
Haas, I. G., Frank, M., Veron, N., and Kemler, R. (2005). Presenilin-dependent processing and nuclear function of gamma-protocadherins. J. Biol. Chem. 280, 9313–9319. doi: 10.1074/jbc.M412909200
Hall, L. S., Pain, O., O’Brien, H. E., Anney, R., Walters, J. T. R., Owen, M. J., et al. (2020). Cis-effects on gene expression in the human prenatal brain associated with genetic risk for neuropsychiatric disorders. Mol. Psychiatry. doi: 10.1038/s41380-020-0743-3 [Epub ahead of print].
Hambsch, B., Grinevich, V., Seeburg, P. H., and Schwarz, M. K. (2005). {gamma}-Protocadherins, presenilin-mediated release of C-terminal fragment promotes locus expression. J. Biol. Chem. 280, 15888–15897. doi: 10.1074/jbc.M414359200
Hannum, G., Guinney, J., Zhao, L., Zhang, L., Hughes, G., Sadda, S., et al. (2013). Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367. doi: 10.1016/j.molcel.2012.10.016
Hanson, H. H., Kang, S., Fernandez-Monreal, M., Oung, T., Yildirim, M., Lee, R., et al. (2010). LC3-dependent intracellular membrane tubules induced by gamma-protocadherins A3 and B2: a role for intraluminal interactions. J. Biol. Chem. 285, 20982–20992. doi: 10.1074/jbc.M109.092031
Hasegawa, S., Hamada, S., Kumode, Y., Esumi, S., Katori, S., Fukuda, E., et al. (2008). The protocadherin-alpha family is involved in axonal coalescence of olfactory sensory neurons into glomeruli of the olfactory bulb in mouse. Mol. Cell. Neurosci. 38, 66–79. doi: 10.1016/j.mcn.2008.01.016
Hasegawa, S., Kumagai, M., Hagihara, M., Nishimaru, H., Hirano, K., Kaneko, R., et al. (2016). Distinct and cooperative functions for the protocadherin-alpha, -beta and -gamma clusters in neuronal survival and axon targeting. Front. Mol. Neurosci. 9:155. doi: 10.3389/fnmol.2016.00155
Herzberg, I., Jasinska, A., Garcia, J., Jawaheer, D., Service, S., Kremeyer, B., et al. (2006). Convergent linkage evidence from two Latin-American population isolates supports the presence of a susceptibility locus for bipolar disorder in 5q31-34. Hum. Mol. Genet. 15, 3146–3153. doi: 10.1093/hmg/ddl254
Hirayama, T., Tarusawa, E., Yoshimura, Y., Galjart, N., and Yagi, T. (2012). CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell Rep. 2, 345–357. doi: 10.1016/j.celrep.2012.06.014
Hong, K. S., McInnes, L. A., Service, S. K., Song, T., Lucas, J., Silva, S., et al. (2004). Genetic mapping using haplotype and model-free linkage analysis supports previous evidence for a locus predisposing to severe bipolar disorder at 5q31-33. Am. J. Med. Genet. B Neuropsychiatr. Genet. 125B, 83–86. doi: 10.1002/ajmg.b.20091
Honig, B., and Shapiro, L. (2020). Adhesion protein structure, molecular affinities, and principles of cell-cell recognition. Cell 181, 520–535. doi: 10.1016/j.cell.2020.04.010
Hori, I., Kawamura, R., Nakabayashi, K., Watanabe, H., Higashimoto, K., Tomikawa, J., et al. (2017). CTCF deletion syndrome: clinical features and epigenetic delineation. J. Med. Genet. 54, 836–842. doi: 10.1136/jmedgenet-2017-104854
Ing-Esteves, S., Kostadinov, D., Marocha, J., Sing, A. D., Joseph, K. S., Laboulaye, M. A., et al. (2018). Combinatorial effects of alpha- and gamma-protocadherins on neuronal survival and dendritic self-avoidance. J. Neurosci. 38, 2713–2729. doi: 10.1523/JNEUROSCI.3035-17.2018
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Isbel, L., Prokopuk, L., Wu, H., Daxinger, L., Oey, H., Spurling, A., et al. (2016). Wiz binds active promoters and CTCF-binding sites and is required for normal behaviour in the mouse. eLife 5:e15082. doi: 10.7554/eLife.15082
Jia, Z., Li, J., Ge, X., Wu, Y., Guo, Y., and Wu, Q. (2020). Tandem CTCF sites function as insulators to balance spatial chromatin contacts and topological enhancer-promoter selection. Genome Biol. 21:75. doi: 10.1186/s13059-020-01984-7
Jiang, Y., Loh, Y. E., Rajarajan, P., Hirayama, T., Liao, W., Kassim, B. S., et al. (2017). The methyltransferase SETDB1 regulates a large neuron-specific topological chromatin domain. Nat. Genet. 49, 1239–1250. doi: 10.1038/ng.3906
Junghans, D., Heidenreich, M., Hack, I., Taylor, V., Frotscher, M., and Kemler, R. (2008). Postsynaptic and differential localization to neuronal subtypes of protocadherin beta16 in the mammalian central nervous system. Eur. J. Neurosci. 27, 559–571. doi: 10.1111/j.1460-9568.2008.06052.x
Juraeva, D., Haenisch, B., Zapatka, M., Frank, J., Witt, S. H., Muhleisen, T. W., et al. (2014). Integrated pathway-based approach identifies association between genomic regions at CTCF and CACNB2 and schizophrenia. PLoS Genet. 10:e1004345. doi: 10.1371/journal.pgen.1004345
Justice, M., Carico, Z. M., Stefan, H. C., and Dowen, J. M. (2020). A WIZ/cohesin/CTCF complex anchors DNA loops to define gene expression and cell identity. Cell Rep. 31:107503. doi: 10.1016/j.celrep.2020.03.067
Katori, S., Noguchi-Katori, Y., Okayama, A., Kawamura, Y., Luo, W., Sakimura, K., et al. (2017). Protocadherin-alphaC2 is required for diffuse projections of serotonergic axons. Sci. Rep. 7:15908. doi: 10.1038/s41598-017-16120-y
Keeler, A. B., Schreiner, D., and Weiner, J. A. (2015). Protein Kinase C phosphorylation of a gamma-protocadherin c-terminal lipid binding domain regulates focal adhesion kinase inhibition and dendrite arborization. J. Biol. Chem. 290, 20674–20686. doi: 10.1074/jbc.M115.642306
Kim, S., Wyckoff, J., Morris, A. T., Succop, A., Avery, A., Duncan, G. E., et al. (2018a). DNA methylation associated with healthy aging of elderly twins. Geroscience 40, 469–484. doi: 10.1007/s11357-018-0040-0
Kim, S., Yu, N. K., Shim, K. W., Kim, J. I., Kim, H., Han, D. H., et al. (2018b). Remote memory and cortical synaptic plasticity require neuronal CCCTC-binding factor (CTCF). J. Neurosci. 38, 5042–5052. doi: 10.1523/JNEUROSCI.2738-17.2018
Kohmura, N., Senzaki, K., Hamada, S., Kai, N., Yasuda, R., Watanabe, M., et al. (1998). Diversity revealed by a novel family of cadherins expressed in neurons at a synaptic complex. Neuron 20, 1137–1151.
Konrad, E. D. H., Nardini, N., Caliebe, A., Nagel, I., Young, D., Horvath, G., et al. (2019). CTCF variants in 39 individuals with a variable neurodevelopmental disorder broaden the mutational and clinical spectrum. Genet. Med. 21, 2723–2733. doi: 10.1038/s41436-019-0585-z
Kostadinov, D., and Sanes, J. R. (2015). Protocadherin-dependent dendritic self-avoidance regulates neural connectivity and circuit function. eLife 4:e08964. doi: 10.7554/eLife.08964
Krumm, N., Turner, T. N., Baker, C., Vives, L., Mohajeri, K., Witherspoon, K., et al. (2015). Excess of rare, inherited truncating mutations in autism. Nat. Genet. 47, 582–588. doi: 10.1038/ng.3303
LaMassa, N., Sverdlov, H., Mambetalieva, A., Shapiro, S., Bucaro, M., Fernandez-Monreal, M., et al. (2020). Gamma-protocadherin localization at the synapse corresponds to parameters of synaptic maturation. bioRxiv [Preprint]. doi: 10.1101/760041
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Langfelder, P., Cantle, J. P., Chatzopoulou, D., Wang, N., Gao, F., Al-Ramahi, I., et al. (2016). Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci. 19, 623–633. doi: 10.1038/nn.4256
Laufer, B. I., Chater-Diehl, E. J., Kapalanga, J., and Singh, S. M. (2017). Long-term alterations to DNA methylation as a biomarker of prenatal alcohol exposure: from mouse models to human children with fetal alcohol spectrum disorders. Alcohol 60, 67–75. doi: 10.1016/j.alcohol.2016.11.009
Laufer, B. I., Kapalanga, J., Castellani, C. A., Diehl, E. J., Yan, L., and Singh, S. M. (2015). Associative DNA methylation changes in children with prenatal alcohol exposure. Epigenomics 7, 1259–1274. doi: 10.2217/epi.15.60
Ledderose, J., Dieter, S., and Schwarz, M. K. (2013). Maturation of postnatally generated olfactory bulb granule cells depends on functional gamma-protocadherin expression. Sci. Rep. 3:1514. doi: 10.1038/srep01514
Lee, S., Salazar, S. V., Cox, T. O., and Strittmatter, S. M. (2019). Pyk2 signaling through Graf1 and RhoA GTPase is required for amyloid-beta oligomer-triggered synapse loss. J. Neurosci. 39, 1910–1929. doi: 10.1523/JNEUROSCI.2983-18.2018
Lefebvre, J. L., Kostadinov, D., Chen, W. V., Maniatis, T., and Sanes, J. R. (2012). Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature 488, 517–521. doi: 10.1038/nature11305
Lefebvre, J. L., Zhang, Y., Meister, M., Wang, X., and Sanes, J. R. (2008). gamma-Protocadherins regulate neuronal survival but are dispensable for circuit formation in retina. Development 135, 4141–4151. doi: 10.1242/dev.027912
Li, Y., Chen, Z., Gao, Y., Pan, G., Zheng, H., Zhang, Y., et al. (2017). Synaptic adhesion molecule Pcdh-gammaC5 mediates synaptic dysfunction in Alzheimer’s disease. J. Neurosci. 37, 9259–9268. doi: 10.1523/JNEUROSCI.1051-17.2017
Li, Y., Xiao, H., Chiou, T. T., Jin, H., Bonhomme, B., Miralles, C. P., et al. (2012). Molecular and functional interaction between protocadherin-gammaC5 and GABAA receptors. J. Neurosci. 32, 11780–11797. doi: 10.1523/JNEUROSCI.0969-12.2012
Liu, J., and Krantz, I. D. (2008). Cohesin and human disease. Annu. Rev. Genomics Hum. Genet. 9, 303–320. doi: 10.1146/annurev.genom.9.081307.164211
Lu, W. C., Zhou, Y. X., Qiao, P., Zheng, J., Wu, Q., and Shen, Q. (2018). The protocadherin alpha cluster is required for axon extension and myelination in the developing central nervous system. Neural. Regen. Res. 13, 427–433. doi: 10.4103/1673-5374.228724
Mancia Leon, W. R., Spatazza, J., Rakela, B., Chatterjee, A., Pande, V., Maniatis, T., et al. (2020). Clustered gamma-protocadherins regulate cortical interneuron programmed cell death. eLife 9:e55374. doi: 10.7554/eLife.55374
Mariman, E. C., Bouwman, F. G., Aller, E. E., van Baak, M. A., and Wang, P. (2014). High frequency of rare variants with a moderate-to-high predicted biological effect in protocadherin genes of extremely obese. Genes Nutr. 9:399. doi: 10.1007/s12263-014-0399-1
Mariman, E. C., Szklarczyk, R., Bouwman, F. G., Aller, E. E., van Baak, M. A., and Wang, P. (2015). Olfactory receptor genes cooperate with protocadherin genes in human extreme obesity. Genes Nutr. 10:465. doi: 10.1007/s12263-015-0465-3
McClay, J. L., Aberg, K. A., Clark, S. L., Nerella, S., Kumar, G., Xie, L. Y., et al. (2014). A methylome-wide study of aging using massively parallel sequencing of the methyl-CpG-enriched genomic fraction from blood in over 700 subjects. Hum. Mol. Genet. 23, 1175–1185. doi: 10.1093/hmg/ddt511
McGowan, P. O., Suderman, M., Sasaki, A., Huang, T. C., Hallett, M., Meaney, M. J., et al. (2011). Broad epigenetic signature of maternal care in the brain of adult rats. PLoS One 6:e14739. doi: 10.1371/journal.pone.0014739
Melka, M. G., Castellani, C. A., Rajakumar, N., O’Reilly, R., and Singh, S. M. (2014). Olanzapine-induced methylation alters cadherin gene families and associated pathways implicated in psychosis. BMC Neurosci. 15:112. doi: 10.1186/1471-2202-15-112
Mendioroz, M., Do, C., Jiang, X., Liu, C., Darbary, H. K., Lang, C. F., et al. (2015). Trans effects of chromosome aneuploidies on DNA methylation patterns in human Down syndrome and mouse models. Genome Biol. 16:263. doi: 10.1186/s13059-015-0827-6
Meyer, K., Feldman, H. M., Lu, T., Drake, D., Lim, E. T., Ling, K. H., et al. (2019). REST and neural gene network dysregulation in iPSC models of Alzheimer’s disease. Cell Rep. 26:e1119. doi: 10.1016/j.celrep.2019.01.023
Miralles, C. P., Taylor, M. J., Bear, J. Jr., Fekete, C. D., George, S., et al. (2020). Expression of protocadherin-gammaC4 protein in the rat brain. J. Comp. Neurol. 528, 840–864. doi: 10.1002/cne.24783
Miyake, K., Hirasawa, T., Soutome, M., Itoh, M., Goto, Y., Endoh, K., et al. (2011). The protocadherins, PCDHB1 and PCDH7, are regulated by MeCP2 in neuronal cells and brain tissues: implication for pathogenesis of Rett syndrome. BMC Neurosci. 12:81. doi: 10.1186/1471-2202-12-81
Molumby, M. J., Anderson, R. M., Newbold, D. J., Koblesky, N. K., Garrett, A. M., Schreiner, D., et al. (2017). gamma-Protocadherins interact with Neuroligin-1 and negatively regulate dendritic spine morphogenesis. Cell Rep. 18, 2702–2714. doi: 10.1016/j.celrep.2017.02.060
Molumby, M. J., Keeler, A. B., and Weiner, J. A. (2016). Homophilic protocadherin cell-cell interactions promote dendrite complexity. Cell Rep. 15, 1037–1050. doi: 10.1016/j.celrep.2016.03.093
Mombaerts, P., Wang, F., Dulac, C., Chao, S. K., Nemes, A., Mendelsohn, M., et al. (1996). Visualizing an olfactory sensory map. Cell 87, 675–686. doi: 10.1016/s0092-8674(00)81387-2
Monahan, K., Horta, A., and Lomvardas, S. (2019). LHX2- and LDB1-mediated trans interactions regulate olfactory receptor choice. Nature 565, 448–453. doi: 10.1038/s41586-018-0845-0
Monahan, K., Rudnick, N. D., Kehayova, P. D., Pauli, F., Newberry, K. M., Myers, R. M., et al. (2012). Role of CCCTC binding factor (CTCF) and cohesin in the generation of single-cell diversity of protocadherin-alpha gene expression. Proc. Natl. Acad. Sci. U.S.A. 109, 9125–9130. doi: 10.1073/pnas.1205074109
Moon, S., Hwang, M. Y., Jang, H. B., Han, S., Kim, Y. J., Hwang, J. Y., et al. (2017). Whole-exome sequencing study reveals common copy number variants in protocadherin genes associated with childhood obesity in Koreans. Int. J. Obes. 41, 660–663. doi: 10.1038/ijo.2017.12
Morishita, H., Kawaguchi, M., Murata, Y., Seiwa, C., Hamada, S., Asou, H., et al. (2004). Myelination triggers local loss of axonal CNR/protocadherin alpha family protein expression. Eur. J. Neurosci. 20, 2843–2847. doi: 10.1111/j.1460-9568.2004.03803.x
Mountoufaris, G., Canzio, D., Nwakeze, C. L., Chen, W. V., and Maniatis, T. (2018). Writing, reading, and translating the clustered protocadherin cell surface recognition code for neural circuit assembly. Annu. Rev. Cell Dev. Biol. 34, 471–493. doi: 10.1146/annurev-cellbio-100616-060701
Mountoufaris, G., Chen, W. V., Hirabayashi, Y., O’Keeffe, S., Chevee, M., Nwakeze, C. L., et al. (2017). Multicluster Pcdh diversity is required for mouse olfactory neural circuit assembly. Science 356, 411–414. doi: 10.1126/science.aai8801
Murata, Y., Hamada, S., Morishita, H., Mutoh, T., and Yagi, T. (2004). Interaction with protocadherin-gamma regulates the cell surface expression of protocadherin-alpha. J. Biol. Chem. 279, 49508–49516. doi: 10.1074/jbc.M408771200
Nakazawa, T., Kikuchi, M., Ishikawa, M., Yamamori, H., Nagayasu, K., Matsumoto, T., et al. (2017). Differential gene expression profiles in neurons generated from lymphoblastoid B-cell line-derived iPS cells from monozygotic twin cases with treatment-resistant schizophrenia and discordant responses to clozapine. Schizophr. Res. 181, 75–82. doi: 10.1016/j.schres.2016.10.012
Naskar, T., Faruq, M., Banerjee, P., Khan, M., Midha, R., Kumari, R., et al. (2018). Ancestral variations of the PCDHG gene cluster predispose to dyslexia in a multiplex family. EBioMedicine 28, 168–179. doi: 10.1016/j.ebiom.2017.12.031
Nativio, R., Donahue, G., Berson, A., Lan, Y., Amlie-Wolf, A., Tuzer, F., et al. (2018). Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 21, 497–505. doi: 10.1038/s41593-018-0101-9
Nicoludis, J. M., Green, A. G., Walujkar, S., May, E. J., Sotomayor, M., Marks, D. S., et al. (2019). Interaction specificity of clustered protocadherins inferred from sequence covariation and structural analysis. Proc. Natl. Acad. Sci. U.S.A. 116, 17825–17830. doi: 10.1073/pnas.1821063116
Nicoludis, J. M., Lau, S. Y., Scharfe, C. P., Marks, D. S., Weihofen, W. A., and Gaudet, R. (2015). Structure and sequence analyses of clustered protocadherins reveal antiparallel interactions that mediate homophilic specificity. Structure 23, 2087–2098. doi: 10.1016/j.str.2015.09.005
Nicoludis, J. M., Vogt, B. E., Green, A. G., Scharfe, C. P., Marks, D. S., and Gaudet, R. (2016). Antiparallel protocadherin homodimers use distinct affinity- and specificity-mediating regions in cadherin repeats 1-4. eLife 5:e18449. doi: 10.7554/eLife.18449
Nuhn, J. S., and Fuerst, P. G. (2014). Developmental localization of adhesion and scaffolding proteins at the cone synapse. Gene Expr. Patterns 16, 36–50. doi: 10.1016/j.gep.2014.07.003
O’Leary, R., Reilly, J. E., Hanson, H. H., Kang, S., Lou, N., and Phillips, G. R. (2011). A variable cytoplasmic domain segment is necessary for gamma-protocadherin trafficking and tubulation in the endosome/lysosome pathway. Mol. Biol. Cell 22, 4362–4372. doi: 10.1091/mbc.E11-04-0283
O’Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Pakkenberg, B., and Gundersen, H. J. (1997). Neocortical neuron number in humans: effect of sex and age. J. Comp. Neurol. 384, 312–320.
Pancho, A., Aerts, T., Mitsogiannis, M. D., and Seuntjens, E. (2020). Protocadherins at the crossroad of signaling pathways. Front. Mol. Neurosci. 13:117. doi: 10.3389/fnmol.2020.00117
Pardinas, A. F., Holmans, P., Pocklington, A. J., Escott-Price, V., Ripke, S., Carrera, N., et al. (2018). Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 50, 381–389. doi: 10.1038/s41588-018-0059-2
Pedrosa, E., Stefanescu, R., Margolis, B., Petruolo, O., Lo, Y., Nolan, K., et al. (2008). Analysis of protocadherin alpha gene enhancer polymorphism in bipolar disorder and schizophrenia. Schizophr. Res. 102, 210–219. doi: 10.1016/j.schres.2008.04.013
Phillips, G. R., Tanaka, H., Frank, M., Elste, A., Fidler, L., Benson, D. L., et al. (2003). Gamma-protocadherins are targeted to subsets of synapses and intracellular organelles in neurons. J. Neurosci. 23, 5096–5104.
Ponroy Bally, B., Farmer, W. T., Jones, E. V., Jessa, S., Kacerovsky, J. B., Mayran, A., et al. (2020). Human iPSC-derived Down syndrome astrocytes display genome-wide perturbations in gene expression, an altered adhesion profile, and increased cellular dynamics. Hum. Mol. Genet. 29, 785–802. doi: 10.1093/hmg/ddaa003
Prasad, T., Wang, X., Gray, P. A., and Weiner, J. A. (2008). A differential developmental pattern of spinal interneuron apoptosis during synaptogenesis: insights from genetic analyses of the protocadherin-gamma gene cluster. Development 135, 4153–4164. doi: 10.1242/dev.026807
Puller, C., and Haverkamp, S. (2011). Cell-type-specific localization of protocadherin beta16 at AMPA and AMPA/Kainate receptor-containing synapses in the primate retina. J. Comp. Neurol. 519, 467–479. doi: 10.1002/cne.22528
Rajarajan, P., Borrman, T., Liao, W., Schrode, N., Flaherty, E., Casino, C., et al. (2018). Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 362:eaat4311. doi: 10.1126/science.aat4311
Rakyan, V. K., Down, T. A., Maslau, S., Andrew, T., Yang, T. P., Beyan, H., et al. (2010). Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 20, 434–439. doi: 10.1101/gr.103101.109
Reiss, K., Maretzky, T., Haas, I. G., Schulte, M., Ludwig, A., Frank, M., et al. (2006). Regulated ADAM10-dependent ectodomain shedding of gamma-protocadherin C3 modulates cell-cell adhesion. J. Biol. Chem. 281, 21735–21744. doi: 10.1074/jbc.M602663200
Remeseiro, S., Cuadrado, A., Gomez-Lopez, G., Pisano, D. G., and Losada, A. (2012). A unique role of cohesin-SA1 in gene regulation and development. EMBO J. 31, 2090–2102.
Rubinstein, R., Thu, C. A., Goodman, K. M., Wolcott, H. N., Bahna, F., Mannepalli, S., et al. (2015). Molecular logic of neuronal self-recognition through protocadherin domain interactions. Cell 163, 629–642. doi: 10.1016/j.cell.2015.09.026
Ryu, H., Lee, J., Hagerty, S. W., Soh, B. Y., McAlpin, S. E., Cormier, K. A., et al. (2006). ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 103, 19176–19181. doi: 10.1073/pnas.0606373103
Salazar, S. V., Cox, T. O., Lee, S., Brody, A. H., Chyung, A. S., Haas, L. T., et al. (2019). Alzheimer’s disease risk factor Pyk2 mediates amyloid-beta-induced synaptic dysfunction and loss. J. Neurosci. 39, 758–772. doi: 10.1523/JNEUROSCI.1873-18.2018
Salpea, P., Russanova, V. R., Hirai, T. H., Sourlingas, T. G., Sekeri-Pataryas, K. E., Romero, R., et al. (2012). Postnatal development- and age-related changes in DNA-methylation patterns in the human genome. Nucleic Acids Res. 40, 6477–6494. doi: 10.1093/nar/gks312
Sanes, J. R., and Zipursky, S. L. (2020). Synaptic specificity, recognition molecules, and assembly of neural circuits. Cell 181, 536–556. doi: 10.1016/j.cell.2020.04.008
Schalm, S. S., Ballif, B. A., Buchanan, S. M., Phillips, G. R., and Maniatis, T. (2010). Phosphorylation of protocadherin proteins by the receptor tyrosine kinase Ret. Proc. Natl. Acad. Sci. U.S.A. 107, 13894–13899. doi: 10.1073/pnas.1007182107
Schreiner, D., and Weiner, J. A. (2010). Combinatorial homophilic interaction between gamma-protocadherin multimers greatly expands the molecular diversity of cell adhesion. Proc. Natl. Acad. Sci. U.S.A. 107, 14893–14898. doi: 10.1073/pnas.1004526107
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Schwab, S. G., Eckstein, G. N., Hallmayer, J., Lerer, B., Albus, M., Borrmann, M., et al. (1997). Evidence suggestive of a locus on chromosome 5q31 contributing to susceptibility for schizophrenia in German and Israeli families by multipoint affected sib-pair linkage analysis. Mol. Psychiatry 2, 156–160. doi: 10.1038/sj.mp.4000263
Shao, Z., Noh, H., Bin Kim, W., Ni, P., Nguyen, C., Cote, S. E., et al. (2019). Dysregulated protocadherin-pathway activity as an intrinsic defect in induced pluripotent stem cell-derived cortical interneurons from subjects with schizophrenia. Nat. Neurosci. 22, 229–242. doi: 10.1038/s41593-018-0313-z
Shimojima, K., Isidor, B., Le Caignec, C., Kondo, A., Sakata, S., Ohno, K., et al. (2011). A new microdeletion syndrome of 5q31.3 characterized by severe developmental delays, distinctive facial features, and delayed myelination. Am. J. Med. Genet. A 155A, 732–736. doi: 10.1002/ajmg.a.33891
Shonubi, A., Roman, C., and Phillips, G. R. (2015). The clustered protocadherin endolysosomal trafficking motif mediates cytoplasmic association. BMC Cell Biol. 16:28. doi: 10.1186/s12860-015-0074-4
Slieker, R. C., van Iterson, M., Luijk, R., Beekman, M., Zhernakova, D. V., Moed, M. H., et al. (2016). Age-related accrual of methylomic variability is linked to fundamental ageing mechanisms. Genome Biol. 17:191. doi: 10.1186/s13059-016-1053-6
Stathopoulos, S., Gaujoux, R., Lindeque, Z., Mahony, C., Van Der Colff, R., Van Der Westhuizen, F., et al. (2020). DNA methylation associated with mitochondrial dysfunction in a South African autism spectrum disorder cohort. Autism Res. 13, 1079–1093. doi: 10.1002/aur.2310
Straub, R. E., MacLean, C. J., O’Neill, F. A., Walsh, D., and Kendler, K. S. (1997). Support for a possible schizophrenia vulnerability locus in region 5q22-31 in Irish families. Mol. Psychiatry 2, 148–155. doi: 10.1038/sj.mp.4000258
Su, H., Marcheva, B., Meng, S., Liang, F. A., Kohsaka, A., Kobayashi, Y., et al. (2010). Gamma-protocadherins regulate the functional integrity of hypothalamic feeding circuitry in mice. Dev. Biol. 339, 38–50. doi: 10.1016/j.ydbio.2009.12.010
Suderman, M., McGowan, P. O., Sasaki, A., Huang, T. C., Hallett, M. T., Meaney, M. J., et al. (2012). Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc. Natl. Acad. Sci. U.S.A. 109, (Suppl. 2), 17266–17272. doi: 10.1073/pnas.1121260109
Suo, L., Lu, H., Ying, G., Capecchi, M. R., and Wu, Q. (2012). Protocadherin clusters and cell adhesion kinase regulate dendrite complexity through Rho GTPase. J. Mol. Cell Biol. 4, 362–376. doi: 10.1093/jmcb/mjs034
Tarusawa, E., Sanbo, M., Okayama, A., Miyashita, T., Kitsukawa, T., Hirayama, T., et al. (2016). Establishment of high reciprocal connectivity between clonal cortical neurons is regulated by the Dnmt3b DNA methyltransferase and clustered protocadherins. BMC Biol. 14:103. doi: 10.1186/s12915-016-0326-6
Tasic, B., Nabholz, C. E., Baldwin, K. K., Kim, Y., Rueckert, E. H., Ribich, S. A., et al. (2002). Promoter choice determines splice site selection in protocadherin alpha and gamma pre-mRNA splicing. Mol. Cell 10, 21–33. doi: 10.1016/s1097-2765(02)00578-6
Tasic, B., Yao, Z., Graybuck, L. T., Smith, K. A., Nguyen, T. N., Bertagnolli, D., et al. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78. doi: 10.1038/s41586-018-0654-5
Thu, C. A., Chen, W. V., Rubinstein, R., Chevee, M., Wolcott, H. N., Felsovalyi, K. O., et al. (2014). Single-cell identity generated by combinatorial homophilic interactions between alpha, beta, and gamma protocadherins. Cell 158, 1045–1059. doi: 10.1016/j.cell.2014.07.012
Tonegawa, S. (1983). Somatic generation of antibody diversity. Nature 302, 575–581. doi: 10.1038/302575a0
Toyoda, S., Kawaguchi, M., Kobayashi, T., Tarusawa, E., Toyama, T., Okano, M., et al. (2014). Developmental epigenetic modification regulates stochastic expression of clustered protocadherin genes, generating single neuron diversity. Neuron 82, 94–108. doi: 10.1016/j.neuron.2014.02.005
Ukkola-Vuoti, L., Kanduri, C., Oikkonen, J., Buck, G., Blancher, C., Raijas, P., et al. (2013). Genome-wide copy number variation analysis in extended families and unrelated individuals characterized for musical aptitude and creativity in music. PLoS One 8:e56356. doi: 10.1371/journal.pone.0056356
Ullrich, B., Ushkaryov, Y. A., and Sudhof, T. C. (1995). Cartography of neurexins: more than 1000 isoforms generated by alternative splicing and expressed in distinct subsets of neurons. Neuron 14, 497–507. doi: 10.1016/0896-6273(95)90306-2
Vadodaria, K. C., Ji, Y., Skime, M., Paquola, A. C., Nelson, T., Hall-Flavin, D., et al. (2019). Altered serotonergic circuitry in SSRI-resistant major depressive disorder patient-derived neurons. Mol. Psychiatry 24, 808–818. doi: 10.1038/s41380-019-0377-5
Wada, T., Wallerich, S., and Becskei, A. (2018). Stochastic gene choice during cellular differentiation. Cell Rep. 24, 3503–3512. doi: 10.1016/j.celrep.2018.08.074
Walker, R. L., Ramaswami, G., Hartl, C., Mancuso, N., Gandal, M. J., de la Torre-Ubieta, L., et al. (2019). Genetic control of expression and splicing in developing human brain informs disease mechanisms. Cell 179, 750–771.e22. doi: 10.1016/j.cell.2019.09.021
Wang, X., Su, H., and Bradley, A. (2002a). Molecular mechanisms governing Pcdh-gamma gene expression: evidence for a multiple promoter and cis-alternative splicing model. Genes Dev. 16, 1890–1905. doi: 10.1101/gad.1004802
Wang, X., Weiner, J. A., Levi, S., Craig, A. M., Bradley, A., and Sanes, J. R. (2002b). Gamma protocadherins are required for survival of spinal interneurons. Neuron 36, 843–854. doi: 10.1016/s0896-6273(02)01090-5
Weiner, J. A., Wang, X., Tapia, J. C., and Sanes, J. R. (2005). Gamma protocadherins are required for synaptic development in the spinal cord. Proc. Natl. Acad. Sci. U.S.A. 102, 8–14. doi: 10.1073/pnas.0407931101
Willemsen, M. H., Vulto-van Silfhout, A. T., Nillesen, W. M., Wissink-Lindhout, W. M., van Bokhoven, H., Philip, N., et al. (2012). Update on Kleefstra syndrome. Mol. Syndromol. 2, 202–212. doi: 10.1159/000335648
Wu, Q. (2005). Comparative genomics and diversifying selection of the clustered vertebrate protocadherin genes. Genetics 169, 2179–2188. doi: 10.1534/genetics.104.037606
Wu, Q., and Jia, Z. (2020). Wiring the brain by clustered protocadherin neural codes. Neurosci. Bull. doi: 10.1007/s12264-020-00578-4 [Epub ahead of print].
Wu, Q., Liu, P., and Wang, L. (2020). Many facades of CTCF unified by its coding for three dimensional genome architecture. J. Genet. Genomics. doi: 10.1016/j.jgg.2020.06.008 [Epub ahead of print].
Wu, Q., and Maniatis, T. (1999). A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell 97, 779–790. doi: 10.1016/s0092-8674(00)80789-8
Wu, Q., Zhang, T., Cheng, J. F., Kim, Y., Grimwood, J., Schmutz, J., et al. (2001). Comparative DNA sequence analysis of mouse and human protocadherin gene clusters. Genome Res. 11, 389–404. doi: 10.1101/gr.167301
Yin, M., Wang, J., Wang, M., Li, X., Zhang, M., Wu, Q., et al. (2017). Molecular mechanism of directional CTCF recognition of a diverse range of genomic sites. Cell Res. 27, 1365–1377. doi: 10.1038/cr.2017.131
Yu, Y., Suo, L., and Wu, Q. (2012). Protocadherin alpha gene cluster is required for myelination and oligodendrocyte development. Dongwuxue Yanjiu 33, 362–366. doi: 10.3724/SP.J.1141.2012.04362
Zipursky, S. L., and Grueber, W. B. (2013). The molecular basis of self-avoidance. Annu. Rev. Neurosci. 36, 547–568. doi: 10.1146/annurev-neuro-062111-150414
Keywords: axonal tiling and myelination, neuronal self-recognition and dendrite self-avoidance, cell adhesion, neuropsychiatric diseases, neuronal connectivity, gene dysregulation, 3D genome architecture, clustered protocadherins
Citation: Jia Z and Wu Q (2020) Clustered Protocadherins Emerge as Novel Susceptibility Loci for Mental Disorders. Front. Neurosci. 14:587819. doi: 10.3389/fnins.2020.587819
Received: 27 July 2020; Accepted: 26 October 2020;
Published: 12 November 2020.
Edited by:
Yan Jiang, Fudan University, ChinaReviewed by:
Julie L. Lefebvre, Hospital for Sick Children, CanadaYanfang Li, Xiamen University, China
Schahram Akbarian, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2020 Jia and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Wu, cWlhbmd3dUBzanR1LmVkdS5jbg==