 Giovanna Morello1,2
Giovanna Morello1,2 Francesca Luisa Conforti
Francesca Luisa Conforti Sebastiano Cavallaro
Sebastiano Cavallaro
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 30 October 2020
Sec. Neurogenomics
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.577755
This article is part of the Research Topic Multifaceted Genes in Amyotrophic Lateral Sclerosis-Frontotemporal Dementia View all 11 articles
Amyotrophic lateral sclerosis (ALS) is a devastating and fatal neurodegenerative disorder, caused by the degeneration of upper and lower motor neurons for which there is no truly effective cure. The lack of successful treatments can be well explained by the complex and heterogeneous nature of ALS, with patients displaying widely distinct clinical features and progression patterns, and distinct molecular mechanisms underlying the phenotypic heterogeneity. Thus, stratifying ALS patients into consistent and clinically relevant subgroups can be of great value for the development of new precision diagnostics and targeted therapeutics for ALS patients. In the last years, the use and integration of high-throughput “omics” approaches have dramatically changed our thinking about ALS, improving our understanding of the complex molecular architecture of ALS, distinguishing distinct patient subtypes and providing a rational foundation for the discovery of biomarkers and new individualized treatments. In this review, we discuss the most significant contributions of omics technologies in unraveling the biological heterogeneity of ALS, highlighting how these approaches are revealing diagnostic, prognostic and therapeutic targets for future personalized interventions.
Amyotrophic lateral sclerosis (ALS) is a devastating and fatal neurodegenerative disease, characterized by the progressive deterioration of cortical and spinal motor neurons (MNs), leading invariably to progressive muscle weakness and paralysis. Death, often resulting from respiratory failure due to respiratory muscle weakness, generally occurs after 3–5 years from symptom onset, with only 5–10% of patients’ survival beyond 10 years (Brown and Al-Chalabi, 2017). ALS is the most common adult motor neuron disease with a worldwide annual incidence of about 2 per 100,000 persons and with an estimated prevalence of 5.4 per 100,000 individuals (Chiò et al., 2013). In most cases, mean age at onset is 50–60 years, while juvenile (before 25 years of age) and “young-onset” ALS cases (before 45 years), represent between∼1 and ∼10% of all patients, respectively (Artemiadis et al., 2016). No disease-modifying strategies are available so far, and therapies that can effectively stop or reverse the disease progression are urgently needed. The mainstay of treatment for ALS is mainly based on symptom management and respiratory support, with only two Food and Drug Administration (FDA)-approved treatments, riluzole, and edaravone, that appear to mildly slow disease progression and only in some patients (Bhandari et al., 2018; Dash et al., 2018; Jaiswal, 2019). The paucity of effective treatments has been attributed in part to the absence of complete knowledge of ALS pathogenesis, and in part to its heterogeneity with patients displaying widely distinct clinical features and progression patterns, together with a plurality of associated genes.
Over the last few years, the complexity of ALS has led to the concept of a spectrum of different disorders with different pathogenic mechanisms rather than a single disease. From a clinical point of view, in addition to typical or classic ALS (characterized by the simultaneous involvement of upper and lower motor neuron (UMN and LMN) at disease onset), several different phenotypic subtypes can be recognized based on the rate of progression, survival, age of onset, site of onset (bulbar vs. spinal) and prevalence of UMN or LMN motor signs (Brown and Al-Chalabi, 2017). Additionally, while ALS was historically judged as a pure motor neuron disease, it is now recognized that it represents a multi-systemic disorder affecting other brain regions, including frontotemporal, oculomotor, cerebellar, and/or sensory systems, and more rarely the basal ganglia and autonomic nervous system (Abrahams et al., 2014; Fang et al., 2017). To this regard, the most common alternative deficit observed in ALS patients is behavioral dysfunction and/or subtle cognitive impairment, which is also comorbid to ALS in about half of ALS individuals, and where a subset of ∼15% of patients receive the concomitant diagnosis of ALS with a frontotemporal dementia (FTD) syndrome (referred to as ALS-FTD or FTD-ALS patients) (Ferrari et al., 2011; Achi and Rudnicki, 2012; Chiò et al., 2019; Zucchi et al., 2019). The ALS-FTD relationship has been confirmed through genetic studies, suggesting these conditions can be viewed as divergent ends of the spectrum of a single clinically and etiologically heterogeneous condition (Ferrari et al., 2011).
Different clinical profiles are likely to reflect molecular heterogeneity in ALS. In fact, for example, the majority (∼90%) of ALS cases are sporadic (SALS), with unknown cause, while ∼10% of ALS patients show familiarity for the disease, usually transmitted according to an autosomal dominant inheritance (Ryan et al., 2018). However, this distinction is increasingly recognized to be artificial; FALS and SALS are, in fact, phenotypically indistinguishable and seem to show similar patterns of selective MN degeneration and vulnerability, and many mutations in one or more known FALS-associated genes have been found in SALS patients, suggesting the existence of common molecular mechanisms between these two disease forms (Renton et al., 2014; Kirby et al., 2016; Taylor et al., 2016). The complexity and heterogeneity of ALS also emerged from a pathophysiologic point of view, with a series of several biological and molecular pathways differently contributing to its development and progression. Despite the understanding of disease pathogenesis is far from exhaustive, numerous genetic and epidemiological risk factors have been identified, as well as various mechanisms have been suggested, including inflammatory and immune abnormalities, oxidative stress, mitochondrial dysfunction, glutamate excitotoxicity, proteasomal/autophagic impairment, defects in axonal transport and RNA metabolism (Taylor et al., 2016). With this in mind, it is clear that the current diagnostic classification criteria of ALS, primarily based on person’s signs and symptoms, are inadequate to characterize the complex and heterogeneous nature of ALS, as well as the use of a single compound to treat the patient population as a whole may hinder the identification of an effective therapy. Defining and stratification of ALS patients into disease subtypes cannot only provide important insights for diagnosis and prognosis but also for clinical trial planning and interpretation, thus achieving better care for ALS patients.
Advances in “omics” technologies (e.g., genome, transcriptome, proteome, epigenome, metabolome) and their correlation with the clinical phenotypes of the individual patient, are enabling medicine to move from a “one-size-fits-all” approach toward a “personalized” model, helping to clarify the molecular mechanisms underlying human disease and to provide both potential biomarkers and pharmacological targets for a more detailed patient stratification and personalized treatments (Figure 1). In this review, we discuss advances in the application of “-omics” to further our understanding of ALS, outline the evolving landscape of molecular classifications, and discuss how these techniques are contributing to reveal diagnostic and prognostic biomarkers and molecular targets for future personalized therapeutic interventions.
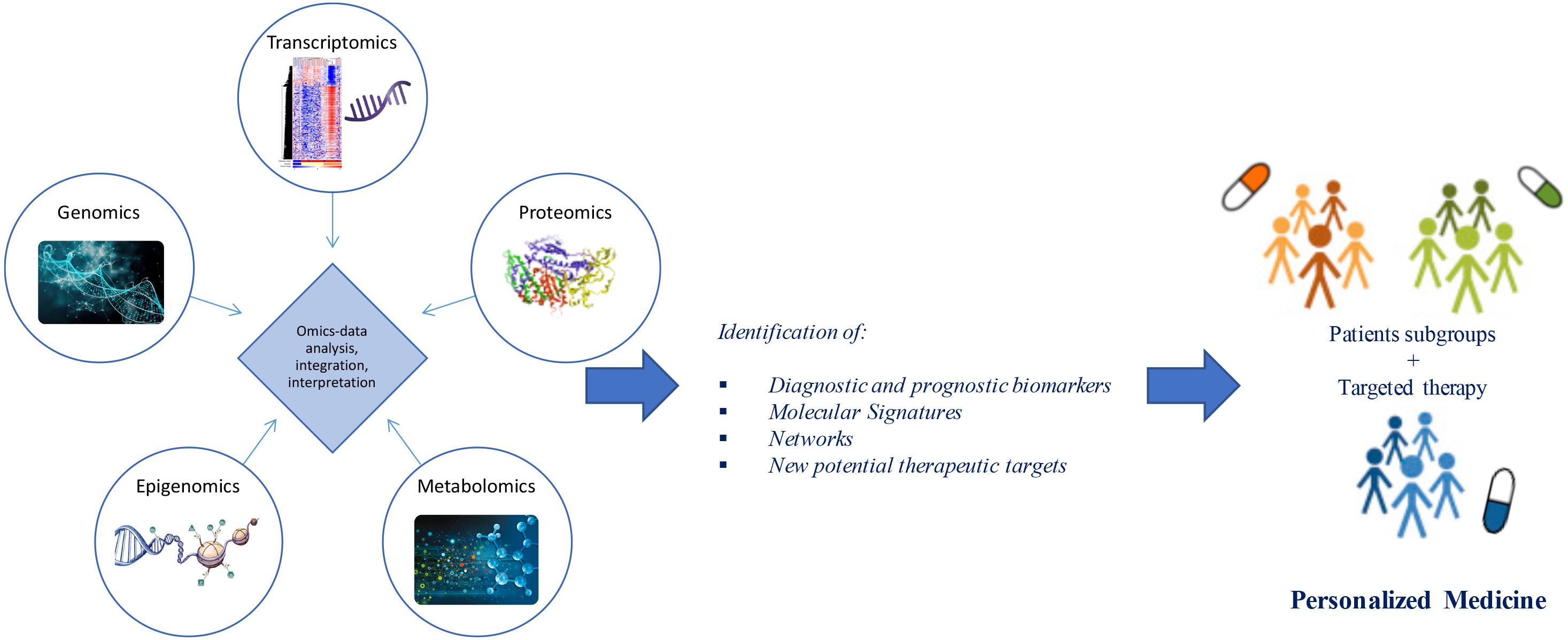
Figure 1. From omics technology to precision medicine in ALS. Multi-omics (e.g., genomics, transcriptomics, proteomics, epigenomics, metabolomics) data analysis and integration may allow patient stratification and targeted therapies. Through a “systems biology” approach, these technologies may move medicine from a “one-size-fits-all” toward a “personalized” model.
Applications of omics platforms range from the detection of genes (genomics), mRNA (transcriptomics), proteins (proteomics), epigenomic factors (epigenomics), and metabolites (metabolomics). Thanks to omics technologies, it is now possible to quantify the amount of particular molecules (genes, mRNA, protein levels, and metabolites) of a biological system, and observe massive interactomes describing their complex interconnections. For complex and multifactorial pathologies such as ALS, the analysis and integration of different omics layers are crucial for the full knowledge of the disease, opening the way to the development of personalized diagnostic and therapeutic tools. Several omics studies have suggested multiple pathologic mechanisms associated to ALS, providing new insights into molecular signatures/markers and moving toward molecular-based classifications and tailored interventions.
The genomic landscape of ALS has been extensively surveyed, contributing to our understanding of ALS biological and clinical complexity. Analysis at this level requires not only the study of DNA sequence variations, including single nucleotide polymorphisms (SNPs) or mutations, but also genomic alterations and chromosomal changes, with consequent protein dysfunction or differences in concentration levels. Detailed information regarding ALS-related genes is available via the Amyotrophic Lateral Sclerosis Online Database (ALSOD)1. After the identification of mutations in the SOD1 gene in 1993 (Rosen et al., 1993), more than 30 genes have been involved in the pathology, with the most common disease-causing variants in C9orf72, SOD1, FUS, and TARDBP. However, monogenic forms explain only a fraction of the diagnosed cases, suggesting ALS as a polygenic disease (McCann et al., 2017; Mejzini et al., 2019).
Thanks to the development of genome-wide association studies (GWAS) as well as the advances in massive parallel sequencing approaches, including whole-genome sequencing (WGS) and whole-exome sequencing (WES), enormous progress has been made in understanding genomics of ALS (Ramanan and Saykin, 2013; Cirulli et al., 2015; He et al., 2015; Butchbach, 2016; Van Rheenen et al., 2016; Little et al., 2017; Naruse et al., 2019; Bean et al., 2020). A growing number of causative and susceptibility genes have been identified so far in both familial and sporadic cases, the majority of which encode proteins implicated in cytoskeleton remodeling and axonal transport, mitochondrial metabolism and turnover, autophagy and proteostasis, membrane trafficking, RNA processing and DNA repair (Table 1; Ramanan and Saykin, 2013; Robberecht and Eykens, 2015; Maurel et al., 2018; Cook and Petrucelli, 2019; Mejzini et al., 2019; Gall et al., 2020). These genetic findings may guide patient stratification into different subgroups depending on which combination of pathways is deregulated, improving their recruitment for translational research and clinical trials (Vijayakumar et al., 2019; Volonté et al., 2020).
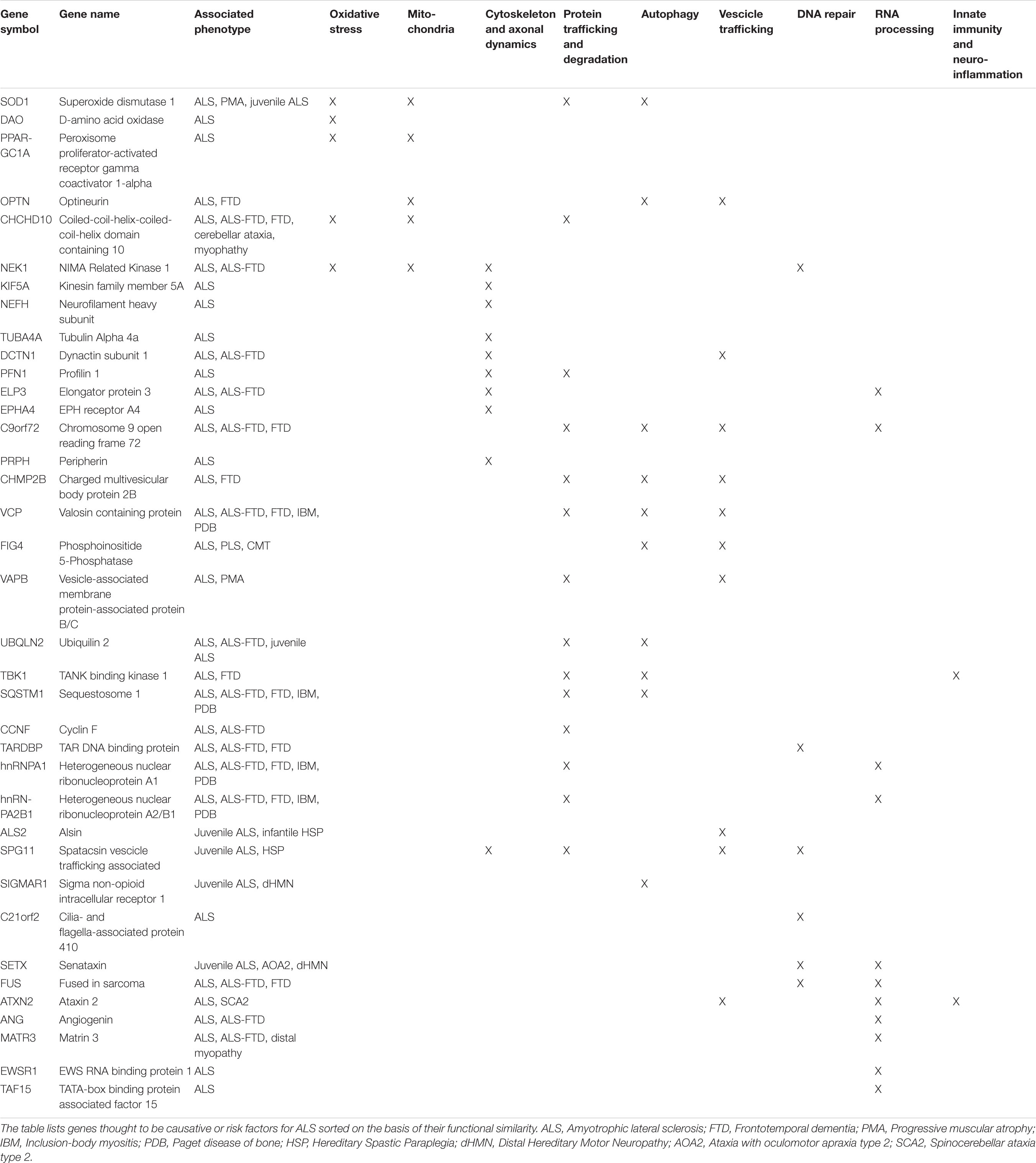
Table 1. Summary of the most known genes linked to ALS, their clinical phenotypes and affected pathway.
Another important factor increasing the complexity of phenotype-genotype correlations in ALS is the observation of a clinical pleiotropy for ALS genes. Although some mutations associate with very specific ALS clinical profiles (e.g., patients with the Ala4Val mutation in SOD1 usually have an aggressive form of ALS, whereas those with the homozygous Asp91Ala mutation tend to have a very slowly progressive disease with a generally ascending upper motor neuron phenotype), the majority of disease-causing genes show a high degree of phenotypic heterogeneity, with mutations in the same gene giving rise to different clinical entities, supporting a genetic basis for the observed clinical heterogeneity in ALS. A striking example of pleiotropy is due to C9orf72 hexanucleotide repeat expansion mutation, which is clearly linked to ALS and FTD but pathogenic expansions have been also observed in a small percentage of patients affected by Alzheimer’s (<1%), Huntington’s (1–5%), and Parkinson’s diseases (1%), as well as atypical parkinsonian syndromes, such as progressive supranuclear palsy (1–8%), corticobasal degeneration (3%), and Lewy body dementia (2%) (van Blitterswijk et al., 2014b; Al-Chalabi et al., 2017; Balendra and Isaacs, 2018; Bourinaris and Houlden, 2018; Foxe et al., 2018). Another interesting example regards a newly identified ALS gene, KIF5A. In fact, missense mutations in the N-terminal motor domain of this gene are known to cause hereditary spastic paraplegia and Charcot–Marie–Tooth disease type 2, while ALS-associated mutations are predominantly located at the C-terminal tail domain (Brenner et al., 2018; Nicolas et al., 2018). The possible existence of a common genetic background in neurodegeneration is also supported by the observation that mutations in ATXN2, SPAST, FIG4, SETX, DCTN1, MATR3, CHCHD10, SQSTM1, VAPB, HNRNPA1, VCP, APOE, and OPTN have been reported both ALS and other multisystem disorders, including FTD, spinocerebellar ataxias, parkinsonism and schizophrenia. Among these, APOE, the most prevalent genetic risk factor of AD, has been also studied both as a risk factor for ALS and as a modifier of various phenotypic aspects, including age at onset, site of onset, and duration of the disease. As already found for AD, inheritance of APOE alleles is associated with differences in the clinical course of ALS (with a protective role of E2 allele and a deleterious role of E4 allele) suggesting a potential implication of APOE genotype as a biomarker to discriminate clinical efficacy in ALS clinical trials (Moulard et al., 1996; Lacomblez et al., 2002; Li et al., 2004). Another genetic determinant of ALS is the trinucleotide repeat expansion occurring in the ATXN2 gene, with long-expanded repeats that are found to cause spinocerebellar ataxia 2 while intermediate-length polyQ expansion seems to increase the risk of developing ALS, significantly correlate to a spinal phenotype, and associate with shorter survival (Laffita-Mesa et al., 2013; van Blitterswijk et al., 2014a; Borghero et al., 2015; Chiò et al., 2015; Sproviero et al., 2017). As for mutant C9orf72 and other pathological repeats, ATXN2-mediated toxicity seems to involve the creation of small toxic homopolymeric proteins, called dipeptide repeats (DPRs), through a process known as repeat-associated non-ATG-initiated (RAN) translation, leading to an impairment of ribosomal biogenesis, nucleocytoplasmic transport, RNA metabolism and protein sequestration, that can cause neurodegeneration and behavioral deficits (Barker et al., 2017; Hutten and Dormann, 2019; Hergesheimer et al., 2020). Disease-modifying therapies designed or formulated to specifically target the ATXN2 gene, including the use of antisense oligonucleotides, are currently being studied as a promising therapeutic approach for ALS (Van Den Heuvel et al., 2014; Scoles and Pulst, 2018; Hergesheimer et al., 2020).
Besides clinical diagnosis and identification of risk variants and disease modifiers, the genomic analysis may be helpful for explaining the considerable differences in prognostic profiles of ALS patients, thus providing valuable information for designing new therapeutic strategies (Geyer et al., 2009; Tanaka et al., 2013; Su et al., 2014; Cappella et al., 2019; Chiò et al., 2020). In particular, mutations in SOD1, EPHA4, KIFAP3, and UNC13A seem to affect the progression of ALS disease or the survival of ALS patients (Landers et al., 2009). Loss-of-function mutations in EPHA4 results in significantly longer survival of ALS patients and pharmacological inhibition of EPHA4 signaling has demonstrated to improve functional performance and motor neuron survival in ALS animal models (Van Hoecke et al., 2012; Rué et al., 2019). Other genetic variants associated with ALS survival include Asp91Ala, one of the most common mutations in SOD1 that is associated with a long survival when the locus had homozygous genotype, while that of affected heterozygotes varies; and the rs12608932 located in intron 21 of the UNC13A gene that is associated with an increased risk and shorter survival of ALS patients (Daoud et al., 2010; Diekstra et al., 2012; Harms and Baloh, 2013; Cady et al., 2015; Gaastra et al., 2016; Yang et al., 2019).
In addition to genetic mutations, the screening of submicroscopic chromosomal changes, known as copy-number variations (CNVs), is potentially informative of genomic alterations related to disease phenotype through the modulation of the expression and function of genes. Several studies have investigated the involvement of these variants in ALS, demonstrating their involvement as risk factors, with multiple rare CNVs more important than common ones (Blauw et al., 2008, 2010; Wain et al., 2009; Uyan et al., 2013; Butchbach, 2016; Morello et al., 2018a; Vadgama et al., 2019). In particular, a large number of rare and novel ALS-specific CNV loci were identified in ALS patients, with the majority of these variants exerting a role in biochemical pathways relevant to ALS pathogenesis, including regulation of synaptic transmission and neuronal action potential, immune response and inflammation, cell adhesion, ion transport, transcriptional regulation and mRNA processing (Wain et al., 2009; Blauw et al., 2010; Morello et al., 2018a). One of the most interesting example is represented by the survival motor neuron (SMN) genes, whose copy number alterations seems to increase risk of developing SALS as well as other neurodegenerative disorders, including progressive muscular atrophy (PMA) (Blauw et al., 2012; Butchbach, 2016; Sangare et al., 2016; Morello et al., 2018a). However, other studies have not found any significant association between the deletion of either SMN1 or SMN2 in ALS, suggesting these conflicting results may be due, in part, to the existence of heterogeneous subgroups of ALS patients. The same ambiguous results are found for copy number changes affecting mitochondrial DNA (mtDNA), with some ALS patients characterized by an accumulation of deletions and other cases showing increased mtDNA copy numbers (Mawrin et al., 2004; Keeney and Bennett, 2010; Morello et al., 2018a). Other examples are heterozygous deletions of EPHA3, which seem to confer a protective role against the risk of developing ALS, and deletions in NEFL associated with a delayed disease onset and slowed disease progression (Uyan et al., 2013; Morello et al., 2018a).
Notwithstanding the increased knowledge of ALS from a genomic perspective, substantial dilemmas remain from a clinical perspective and large-scale NGS and GWAS projects are currently underway to fully unravel the underlying causes. Among these, of note is Project MinE, an international, large-scale research initiative devoted to discovering genetic causes of ALS by performing whole-genome sequencing of at least 15,000 ALS patients and 7,500 controls, resulting in an open-source genome database, in conjunction with the collection of skin samples to make patient induced pluripotent stem cell lines (iPSCs) (Van Rheenen et al., 2018; van der Spek et al., 2019). Future follow-up studies will be necessary to shed light on the biological drivers of disease and evaluate the direct effect of newly discovered genes on disease diagnosis and management, also determining if they could form candidates for novel gene therapies.
Changes in gene expression are widespread in ALS, as revealed by a large body of work on gene expression profiling of RNA samples from peripheral cells or post-mortem nervous tissue of ALS patients and animal models. These signature patterns of gene expression have started to provide a more detailed picture of molecular events implicated in ALS pathobiology (Dangond et al., 2004; Malaspina and de Belleroche, 2004; Jiang et al., 2005; Kirby et al., 2005; Pasinelli and Brown, 2006; Wang et al., 2006; Lederer et al., 2007; Malaspina et al., 2008; de Oliveira et al., 2013; Saris et al., 2013; Raman et al., 2015; Maria D’erchia et al., 2017; Krokidis and Vlamos, 2018; Recabarren-Leiva and Alarcón, 2018; Dickson et al., 2019; Rahman et al., 2019).
The advent of systems biology and development of high-throughput technologies, including RNA sequencing and high-density microarray platforms, is enabling us not only to discover and define mechanisms of pathogenesis in ALS, but also to differentiate ALS from the “ALS mimic syndromes” and healthy controls and stratify ALS patient into subgroups, facilitating the discovery of biomarkers and new individualized treatments for patients (Cooper-Knock et al., 2012; Heath et al., 2013; Krokidis and Vlamos, 2018; Recabarren-Leiva and Alarcón, 2018; Krokidis, 2020). In this regard, our research group, in the last years, has taken important steps toward the characterization of a biological and molecular heterogeneity of ALS based on transcriptional profiles. In particular, unsupervised hierarchical clustering of genome-wide transcriptomic profiles generated from post-mortem motor cortex samples from SALS patients has led to separate healthy controls and SALS patients and identify two distinct patient groups (SALS1 and SALS2) depending on the combinations of genes and pathways that were deregulated (Aronica et al., 2015). In particular, we observed that cell death, antigen processing and presentation and regulation of chemotaxis were the most representative subgroup-specific pathways in SALS1, while deregulated genes in SALS2 were associated with axonal guidance, oxidative and proteotoxic stress (Figure 2; Aronica et al., 2015; Morello et al., 2017a,b). Our analysis also showed that some of the deregulated genes in SALS patients were previously associated with FALS, further supporting the existence of common pathological events between two disease forms. Interestingly, we found the differential expression of a substantial number of genes encoding splicing factors in the motor cortex and spinal cord of the same SALS cohort (La Cognata et al., 2020). In particular, we observed transcriptional deregulation across the tissue types and/or disease states (SALS1, SALS2, controls), with expression changes that were more pronounced for the motor cortex regions than the spinal cord and revealing a significant trend of overexpression for the SALS1 group and a decreased trend in expression for SALS2 (La Cognata et al., 2020). Despite, taken together, our results provided a powerful means for revealing etiopathogenetic mechanisms that were not emerged by considering SALS as a single pathology, it is clear that to successfully translate this knowledge to the real-world clinical contexts, the number of biomarkers should be limited. For this purpose, we next asked if the transcriptome-based classification can be reproduced by utilizing just a list of 203 genes highly associated with an increased ALS susceptibility (Morello et al., 2017a,b). Our results showed that this restricted gene panel was sufficiently representative to separate control from SALS patients, reproducing our previous classification of these patients into molecularly defined and biologically meaningful subtypes and, consequently, facilitating the identification of promising cluster-specific biomarkers. Further studies will be necessary to investigate if peripheral tissues or easily accessible biological fluids (e.g., peripheral blood monocytes, cerebrospinal fluid, or muscle) can reproduce specific molecular patterns observed in brain regions of ALS patients, allowing for an effective mechanism-based selection of patients for clinical trials of molecular-targeted therapies. Emerging molecular heterogeneity of ALS lays the foundations for developing new therapeutic strategies, targeting disease pathogenesis as a complex system rather than at the level of the single protein molecule and that may have greater relevance to distinct sets of patients. In this regard, altered biological pathways emerged from our analysis provided a good number of potential subgroup-specific biomarkers and therapeutic targets, opening the way to the implementation of genomics-based personalized medicine (Morello and Cavallaro, 2015; Morello et al., 2015, 2017c). Of note, some of these target genes exhibit expression profiles similar to those observed in animal models of ALS, thus providing a rationale to ensure their preclinical trial success (Apolloni et al., 2017; Morello et al., 2017c; Apolloni et al., 2019).
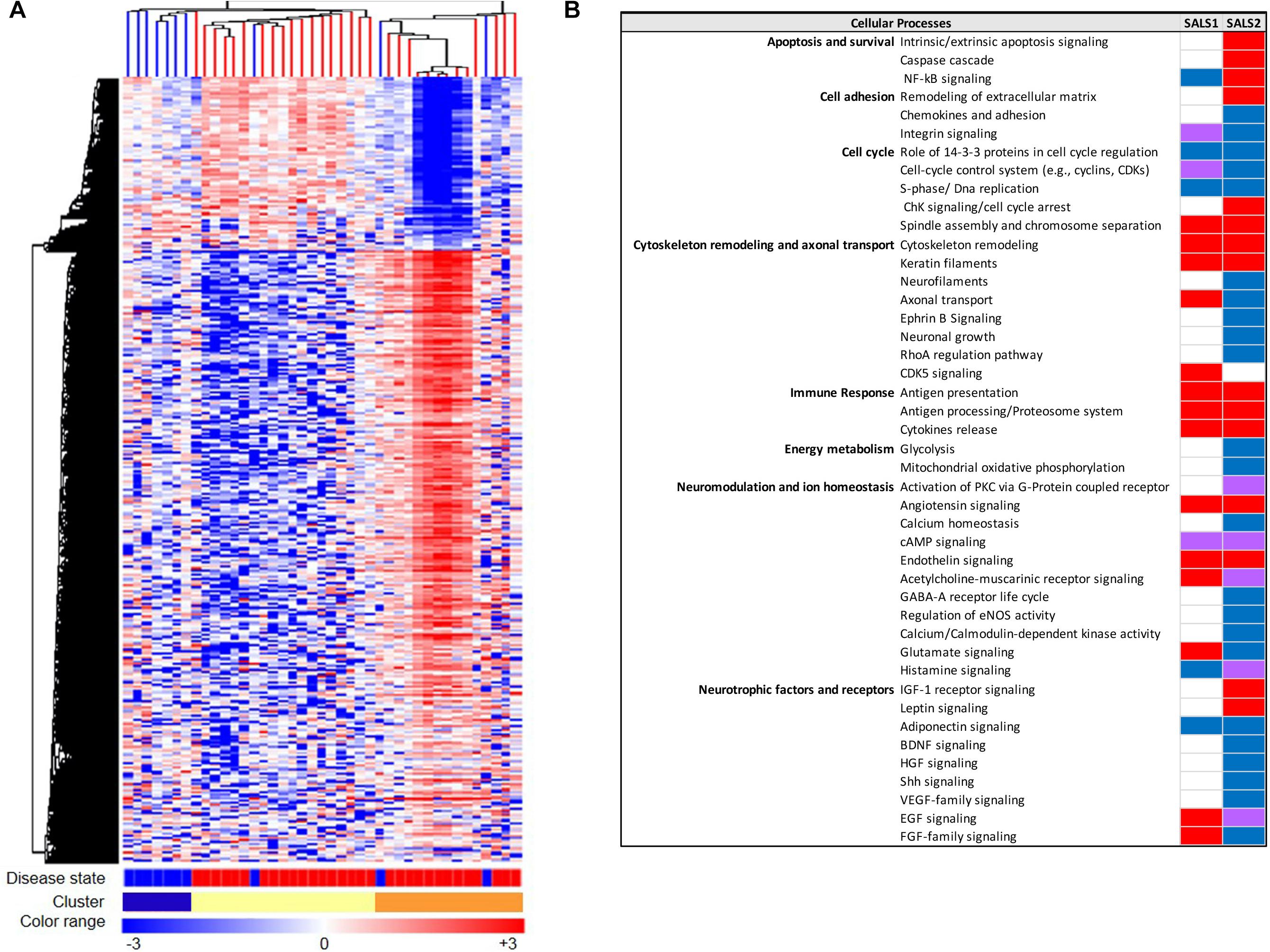
Figure 2. Molecular-based classification of SALS. (A) Unsupervised hierarchical clustering (similarity measure: Pearson centered; linkage rule: average) was used to cluster control (10 fresh-frozen motor cortex samples from non-neurological patients) and SALS patients (31 fresh-frozen motor cortex samples) on the basis of the similarity in expression profiles of the most “hypervariable genes” (9.646 genes with a standard deviation >1.5). The same clustering method clearly distinguished two SALS subgroups (SALS1 and SALS2), each associated to differentially expressed genes and pathways. In this two-dimensional presentation, each row represents a single gene and each column a motor cortex from control or SALS patients. As shown in the color bar, highly expressed genes are shown in orange, down-regulated genes in blue, no change in white. In the dendrograms shown (left and top), the length and the subdivision of the branches display the relatedness of the expression of the probes and the motor cortex (top). The disease state is marked as follows: controls patients are indicated by brown rectangles and SALS patients by red rectangles. In the cluster panel, red rectangle refers to control patients, brown to SALS1 and blue to SALS2 patients. For further details, the reader is referred to Aronica et al. (2015). (B) Functional pathways deregulated in clustered SALS patients. Orange boxes represent signaling pathways significantly up-regulated, blue bars down-regulated, green bars both up- and down-regulated, white bars indicate no significant change. Figure adapted from Morello and Cavallaro (2015).
Recently, a good number of studies investigated and confirmed the existence of distinct molecular-based clusters of ALS patients, calling attention to the need for better understanding their mechanistic underpinnings and developing treatments based on specific forms of ALS (Jones et al., 2015; Tam et al., 2019; Vijayakumar et al., 2019). In particular, Tam et al. (2019) were able to stratify the transcriptomes by RNA-seq of a largely sporadic set of ALS patients’ motor cortex samples into three distinct molecular subgroups, two of which overlapped the molecular signatures observed in our ALS patient samples (Tam et al., 2019). Another study compared brain transcriptome profiles in SALS cases carrying and not carrying the C9orf72 repeat expansion, revealing both shared and distinct transcriptome changes and pathways associated with these two subsets of ALS cases (Prudencio et al., 2015). A further interesting aspect is the possibility of separating rapid and slow ALS in earlier phases of drug development. To this regard, whole-genome expression analysis conducted by Nardo et al. (2013) in ALS animal models identified specific key genes and molecular pathways associated with fast or slow disease progression, highlighting their role as putative molecular targets for future therapeutic strategies (Prudencio et al., 2015).
The majority of the above-described studies assessed RNA samples from postmortem brain tissues. Although they provide essential elements in the pathophysiology of ALS that cannot be otherwise obtained through other approaches used in living patients, these studies reveal end-stage pathogenic mechanisms and do not clarify whether transcriptional differences that separate patient subtypes are a cause or a consequence of the disease process. In that context, the use of iPSC derived from patients suffering from ALS has provided important insights into disease pathophysiology, enabling researchers to explore molecular heterogeneity of ALS and follow the course of degeneration in the dish (Coatti et al., 2015; Bohl et al., 2016; Hedges et al., 2016; Myszczynska and Ferraiuolo, 2016; Sances et al., 2016; Csobonyeiova et al., 2017; Guo et al., 2017; Selvaraj et al., 2017; Centeno et al., 2018; Fujimori et al., 2018; Ghaffari et al., 2018; Lee et al., 2018; Halpern et al., 2019; Ziff and Patani, 2019; Chang et al., 2020; Hawrot et al., 2020). In addition, transcriptome studies on whole tissue (i.e., motor cortex and spinal cord) fail to capture dynamic changes and the complex heterogeneity of the nervous system, making it difficult to determine how gene expression changes disrupt functional interaction between motor neurons and non-neuronal cells (e.g., microglia, oligodendroglia, and astroglia) implicated in ALS pathology. Promising approaches, such as laser capture microdissection (LCM) coupled with RNA sequencing, offer a previously unavailable view of disease progression in ALS, enabling us to explore cell type-specific changes involved in the disease at a particular time point (Namboori et al., 2019; Liu et al., 2020). In a recent paper, Maniatis et al. (2019) used new RNA-seq based technologies, which they called “spatial transcriptomics,” for mapping gene expression changes occurring at different disease stages and in different regions of murine models of ALS and human postmortem spinal cords samples, providing important clues for identifying disease-associated pathways and establishing the key steps in motor neuron degeneration observed in ALS (Maniatis et al., 2019).
Detection of specific protein changes in affected brain tissue samples, cell cultures or body fluids such as CSF represents an important pillar in ALS. The discovery of protein biomarkers for ALS, in fact, may aid earlier diagnosis, measure disease progression, exclude other ALS-mimicking syndromes, discriminate between subtypes of ALS that may theoretically respond to different therapeutic strategies and monitoring drug efficacy during clinical trials (Ruegsegger and Saxena, 2016; Barschke et al., 2017; Webster et al., 2017; Chipika et al., 2019; Hedl et al., 2019; Yerbury et al., 2020). It is well established that the key neuropathological hallmark of the disease is the accumulation of misfolded cytoplasmic proteins in degenerating motor neurons and their non-neuronal neighbors (Perlson et al., 2010; Baloh, 2011; Prell et al., 2013; Tang, 2014; Navone et al., 2015; Parakh and Atkin, 2016; Nguyen et al., 2019; Malik and Wiedau, 2020). One of the main protein components of these protein aggregates is TDP-43, a nuclear RNA binding protein that under stress conditions or when mutated translocates to the cytoplasm where it is hyperphosphorylated and forms insoluble ubiquitin-positive aggregates (Baloh, 2011; Weskamp and Barmada, 2018). Such aggregates are present in almost all cases of ALS, including SALS and FALS patients with pathogenic variants of C9ORF72 (Mackenzie et al., 2007; Chew et al., 2015), as well as in other neurodegenerative disorders, including FTD, Parkinson’s and Alzheimer’s disease (Amador-Ortiz et al., 2007; Umoh et al., 2018). It is interesting to note that ALS and FTD have different forms of TDP-43 pathology, suggesting its utility for designing novel diagnostic procedures that could discriminate against these two diseases (Mackenzie and Rademakers, 2008). In addition, although controversial, several results reported that TDP-43 aggregates occur in the vast majority of SOD1- and FUS-negative FALS patients, but not in SOD1/FUS mutation carriers, suggesting that mutant TDP-43 may cause ALS through specific pathways of inclusion formation that are distinct from those that underlie SALS or other FALS-associated mutations, opening the way to the development of specific therapeutic approaches that take into account these selective modifications (Farrawell et al., 2015; Jeon et al., 2019).
Due to the complex and heterogeneous nature of ALS, it is plausible that a single biomarker could not detect or differentiate between disease subgroups and/or control subjects, sustaining the importance of developing biomarker panels for specific and sensitive diagnostic tests. Recent development of high-throughput Mass Spectrometry-based proteomic (MS) technologies has allowed the simultaneous analysis of multiple proteins, allowing for the definition of comprehensive lists of possible candidate ALS biomarkers (Ekegren et al., 2008; Krüger et al., 2013; Collins et al., 2015; Hedl et al., 2019). In this regard, due to its proximity to the central motor system, the cerebrospinal fluid (CSF) may most probably reflect disease-related alterations, including changes in protein expression, post-translational modification or biochemical turnover than in other body fluids (i.e., blood or urine) (Bowser et al., 2006; Pasinetti et al., 2006; Ryberg and Bowser, 2008; von Neuhoff et al., 2012; Krüger et al., 2013; Lehmer et al., 2017). Analyses of the CSF proteome of ALS patients revealed a panel of candidate biomarkers implicated in synaptic activity, extracellular matrix, inflammatory processes, glial response, axonal damage and apoptosis (Bowser et al., 2006; Pasinetti et al., 2006; Ryberg and Bowser, 2008; von Neuhoff et al., 2012; Krüger et al., 2013; Collins et al., 2015; Collins, 2016; Lehmer et al., 2017; Benatar et al., 2018; Zubiri et al., 2018; Online et al., 2019; Zhu et al., 2019; Oeckl et al., 2020). It is interesting to note that many candidate ALS protein biomarkers show subgroup-specific differential mRNA expression in SALS patients, suggesting their utility in patient stratification and personalized medicine (Table 2). Among the most extensively studied fluid biomarkers correlating with the survival of ALS patients, higher levels of neurofilament light chain (NF-L) and the phosphorylated form of neurofilament heavy chain (pNFH) in CSF and plasma samples, as well as their accumulation in brain tissue, have been correlated to shorter life expectancy and a more rapid disease progression and have demonstrated high sensitivity and specificity for separating ALS from ALS-mimic disorders (Gaiottino et al., 2013; Lu et al., 2015; Benatar et al., 2018; De Schaepdryver et al., 2018, 2019; Verde et al., 2019). Recent works also demonstrated the diagnostic utility of CSF pNFH levels in C9ORF72-ALS patients, revealing higher pNFH levels in ALS or ALS/FTD patients carrying C9ORF72 expansion compared with controls and other ALS or ALS/FTD patients (Balendra et al., 2017; Gendron et al., 2017; Floeter and Gendron, 2018). Several other proteins in CSF of ALS patients have demonstrated elevated sensitivity and specificity in distinguishing between ALS patients and neurological disease controls, including IL-10, IL-6, GM-CSF, IL-2, and IL-15 (Mitchell et al., 2009). Proteomic profiling of CSF also identified proteins with a potential prognostic value in ALS, including MIP-1α, wrCRP, HMGB, creatine kinase, granzyme B, and IL-8, whose increased levels have been correlated with more rapidly progressive disease; cystatin C protein levels were positively correlated with survival; increase in GPNMB and UCHL1 were specific for ALS patients showing a short survival time; bFGF increased in ALS patients with longer survival, whereas VGF levels correlated with progressing muscle weakness (Ranganathan et al., 2005; Barschke et al., 2017).
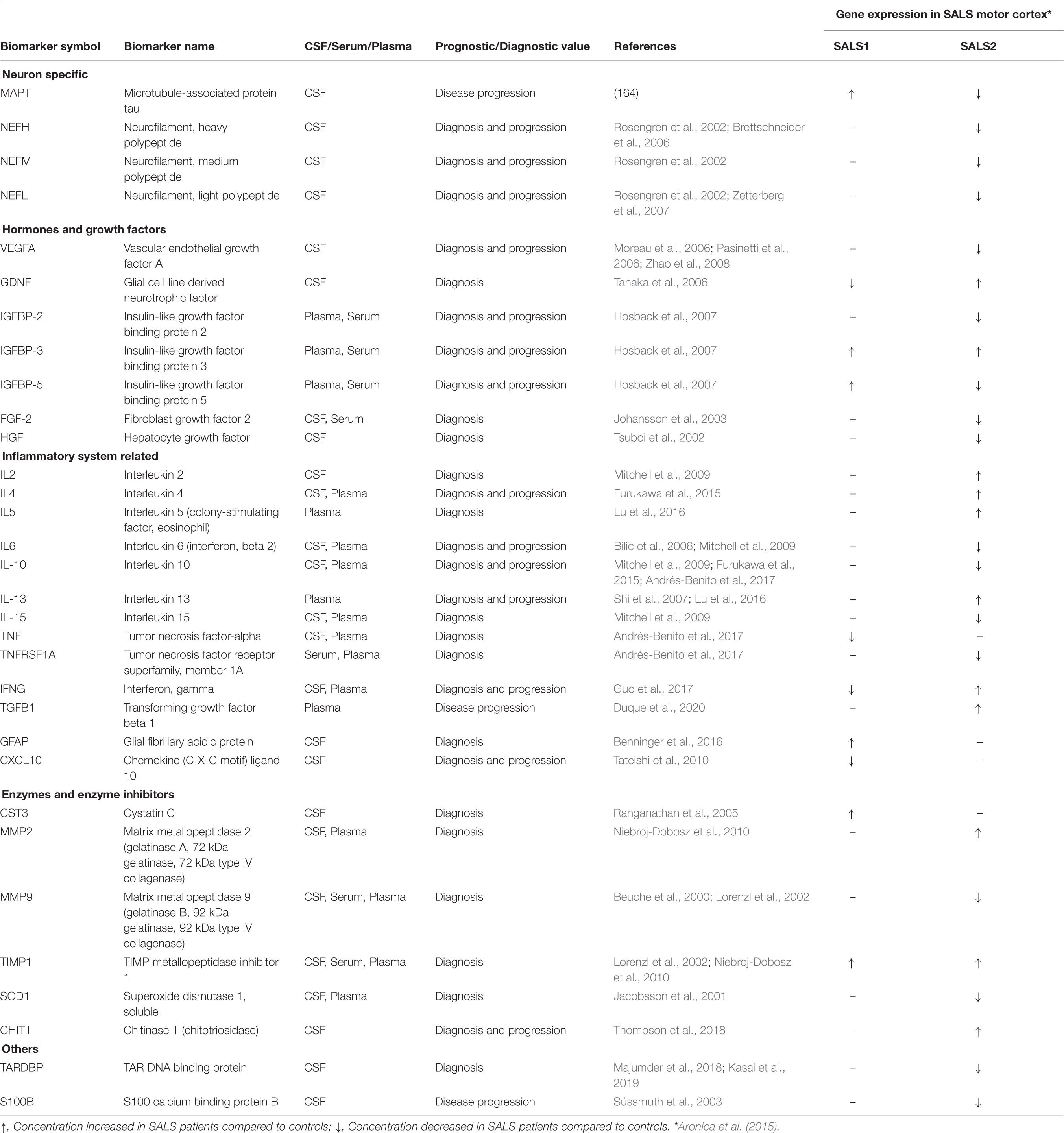
Table 2. Putative protein biomarkers and their differential expression in distinct SALS patient subgroups.
As for genomics studies, systems biology-oriented approaches in proteomics play a crucial role to reveal relevant biological knowledge on pathological mechanisms that trigger the onset and progression of ALS, providing a mechanistic rationale for stratification of ALS patients based on unique molecular profiles, and identification of disease biomarkers and targets for drug efficacy measurements. In this scenario, the analysis of protein-protein interaction (PPI) networks provides the possibility to group proteins that are interacting with each other’s in functional complexes and pathways, resulting critically important in helping us to comprehend complex processes, like ALS, and identify key signaling cascades, upstream regulatory components, interactome domains, and novel disease-associated protein candidates suitable for therapeutic intervention (Rao et al., 2014; Snider et al., 2015; Shurte, 2016; Mao et al., 2017; Vella et al., 2017). In this regard, an interesting example is represented by a recent study investigating modules of co-expressed genes or proteins altered in postmortem cortex samples from patients affected by ALS, FTD, ALS/FTD, and healthy disease controls. In this work, Umoh et al. (2018) identified co-expression modules (i.e., RNA binding proteins, synaptic transmission, inflammation) differing across the ALS-FTD disease spectrum that may be useful for identifying genes associated with different clinical phenotypes along the ALS-FTD disease spectrum (Umoh et al., 2018).
In addition to genomics, transcriptomics and proteomics, the exponential advances in technologies and informatics tools have stimulated an exponential growth of other areas of biomedical science (metabolomics, epigenomics, spliceomics), offering exciting new possibilities for ALS research. In this context, metabolomics, the scientific study of chemical processes involving metabolites (e.g., sugars, lipids, amino acids, organic acids), represents the downstream of systems biology that links the genome, transcriptome and proteome to patient phenotype, providing an important key tool for discovering potential markers in health or disease (Kumar et al., 2013; Blasco et al., 2016; Lanznaster et al., 2018, 2020; Germeys et al., 2019). In the last years, thanks to the development of high-throughput technologies (such as Mass Spectrometry Combined with Liquid and Gas Chromatography), metabolomics studies identified specific metabolic markers and signatures that can discriminate ALS from controls and non-ALS cases, as well as identify distinct subgroups of SALS patients, moving research toward the development of novel targeted personalized treatments (Gross et al., 2018; Lanznaster et al., 2018). In particular, Gross et al. (2018) recently identified two subgroups of SALS case fibroblasts displaying distinct metabotropic patterns that were also observed in plasma samples from the same patients, thus providing a basis for stratify SALS patients for appropriate targeted therapies (Gross et al., 2018). Other metabolite profiling-based studies revealed significantly different metabolic profiles among FALS, SALS and ALS patients carrying different mutations in disease-causing genes (i.e., C9ORF72, SOD1, TARDBP, and FUS), suggesting the existence of distinct neurodegenerative processes associated with different subtypes of ALS (Wuolikainen et al., 2012; Jääskeläinen et al., 2019; Lanznaster et al., 2020). It is interesting to note that changes in the metabolome as well as alterations in energy metabolism, such as an increase in resting energy expenditure, often precede the development of motor symptoms in ALS and correlates to disease progression. For instance, a lipid-specific metabolic abnormality is present at the pre-symptomatic stage of ALS animal models while increased serum levels of total cholesterol, LDL, LDL/HDL ratio, and triglycerides were associated with longer survival and slower disease progression in ALS patients and animal models (Dorst et al., 2011; De Aguilar, 2019; Germeys et al., 2019). However, the relationship between lipid levels and ALS is still rather controversial and poorly understood, and some follow-up observational studies of ALS did not observe any association between dyslipidemia and the incidence of ALS (Zufiría et al., 2016; De Aguilar, 2019; Zeng and Zhou, 2019). These conflicting results may be partly due to the relatively small sample sizes often employed in these observational studies and to the fact that lipid changes can be affected by a myriad of confounding factors, including genetic, nutritional, physical and pathological factors.
Analysis of metabolite profiles can be also used to identify metabolites and biochemical pathways in ALS patients that are modified before or after treatment exposure, giving rise to a new field called pharmacometabolomics (Rattray and Daouk, 2017; Blasco et al., 2018; Lanznaster et al., 2018). To this regard, an interesting example is represented by a study that analyzed changes in metabolites and lipids composition in the plasma of ALS patients enrolled in a phase III clinical trial for assessing the effects of TRO19622 (olesoxime), a compound with neuroprotective and neurodegenerative properties (Blasco et al., 2018). This study has permitted not only to identify distinct metabolic changes that can distinguish the placebo from the olexosime group but also to reveal metabolic pathways specifically altered after treatment with olesoxime and riluzole in combination in comparison to riluzole therapy alone, supporting the value of blood metabolomic profiles as biomarkers for evaluating the individual response to drug treatments and their side effects (Blasco et al., 2018).
Another layer of complexity to the understanding of complex interactions between the genome and the environment is represented by epigenetic modifications, including DNA methylation, histone post-translational modifications, ATP-dependent chromatin remodeling and RNA-dependent gene silencing (Jirtle, 2009; Dolinar et al., 2018; Douglas, 2018; Ebbert et al., 2018; Bennett et al., 2019; Calió et al., 2020; Klingl et al., 2020; Wang et al., 2020). Several lines of evidence associate epigenome modifications to ALS development, with alterations in DNA methylation and DNA-(cytosine-5)- methyltransferase (DNMT) enzyme activity, as well as alterations to the balance between histone acetylation and deacetylation observed in blood and post-mortem neural tissue from patients with ALS and in different experimental models (Paez-Colasante et al., 2015; Dolinar et al., 2018; Bennett et al., 2019). Of note, variations in epigenetic marks and modifier enzymes, and alterations in the methylation status of some ALS-related genes promoters were also determined, including hypomethylation of OPTN, hypermethylation of C9orf72 expansion CpG islands in the blood of FTD/ALS patients, whereas mutant SOD1, FUS and TDP43 contribute to global epigenome alteration by inducing alterations in histone post-translational modifications and DNA methylation (Masala et al., 2018). While several high-density microarrays or sequencing-based epigenomic technologies are available, particular attention should be paid to EpiSwitchTM, a high-resolution platform, recently developed by Oxford BioDynamics, for analyzing structural-functional epigenetic changes in genomic architecture associated with pathological phenotypes called “chromosome conformation signatures.” Using this innovative technological platform, Salter and colleagues performed a comparative interrogation of the genomic architecture from healthy and ALS-patient blood samples revealing unique chromosomal conformation signatures with the ability to discern between diseased subjects and healthy controls, predict faster versus slower progressing patients at baseline and stratify responsive and non-responsive patients, representing a crucial step toward personalized medicine in ALS (Poesen, 2018; Salter et al., 2018).
MicroRNAs (miRNAs), small non-coding molecules of about 20–22 nucleotides, represent an additional layer of epigenetic regulation that, thanks to their capability to be highly stable in human body fluids, are considered promising biomarkers for neurodegenerative diseases, including ALS (Ricci et al., 2018; Sharma and Lu, 2018). Over the last few years, several whole-genome miRNA profiling studies have identified a panel of a dozen miRNAs that can distinguish ALS from controls with high accuracy in blood cells, serum and CSF, and may be altered in pre-symptomatic ALS mutation carriers even years before the estimated disease onset, representing potentially useful biomarkers of early-stage ALS in coming years (Figueroa-Romero et al., 2016; Rizzuti et al., 2018; Joilin et al., 2019). Despite the heterogeneous nature of ALS may prevent a significant correlation of miRNA levels with clinical disease parameters, down-regulation of two miRNAs, miR-1234-3p and miR-1825, not only is specific for ALS, at least when compared with cohorts of Alzheimer’s and Huntington’s disease, but also significantly correlated with disease characteristics like age of onset, disease severity and duration (Freischmidt et al., 2015; Takahashi et al., 2015). In particular, while the downregulation of miR-1825 is a general early feature in both FALS and SALS, miR-1234-3p is significantly downregulated only in SALS patients. Of note, a large proportion of SALS patients showed miRNA signatures resembling those of FALS patients and mutation carriers, suggesting alteration of common pathways and a high contribution of genetic factors also in SALS (Freischmidt et al., 2015; Takahashi et al., 2015). Other examples include down-regulation of miR-206, a specific modulator of skeletal muscle growth involved in nerve regeneration after injury, which accelerates disease progression in ALS mice, whereas up-regulation of miR-208B and miR-499 is found in the skeletal muscles of patients with slower disease progression, suggesting the potential utility of these microRNAs as promising candidate biomarkers and targets for this motor neuron disease (Toivonen et al., 2014; Ma et al., 2015; Di Pietro et al., 2017; De Luna et al., 2020).
As detailed in the previous paragraphs, omics technologies have been used to identify and/or provide functional supporting information for deciphering important players and pathways involved in ALS pathogenesis and identifying a panel of candidate therapeutic targets and biomarkers that will assist in the rapid diagnosis and prognosis assessment of the disease, and in the stratification of patients into different subgroups for specific targeted therapies. However, if considered individually, these technologies are insufficient to clarify the intricate disease mechanisms implicated in ALS. Taking a holistic molecular approach, based on the integration of multiple types of omics data with existing biological knowledge, has the potential role in improving the knowledge of the molecular basis underlying complex and heterogeneous diseases, establishing different molecular subtypes and patient stratification, thus providing a rational foundation for designing new studies to identify novel targets and clinical trials (Figure 1; Mitropoulos et al., 2018; Yu and Zeng, 2018; Mirza et al., 2019; Nguyen and Wang, 2020). Numerous studies have demonstrated the utility of whole- and multi-omics strategies for deciphering the molecular landscape of neurodegenerative diseases, including ALS, providing a feasible opportunity to develop an efficient and effective personalized diagnostics and patient-guided therapies (Bu et al., 2016; Santiago et al., 2017; Castrillo et al., 2018; Hampel et al., 2018a,b; Mitropoulos et al., 2018; Olivier et al., 2019; Vijayakumar et al., 2019; Lam et al., 2020).
An interesting example of applying integrated omics approaches to define an individual’s molecular profile useful for the development and application of personalized medicine in ALS, is represented by recent studies carried out by our research groups. As previously described, transcriptional profiling of post-mortem motor cortex samples from SALS patients has allowed to differentiate two distinct patient subgroups characterized by different deregulated genes and pathways (Aronica et al., 2015; Morello and Cavallaro, 2015; Morello et al., 2017a,b,c). To investigate whether these transcriptional alterations may be related to genomic DNA alterations, and thus represent potential markers for a molecular-based stratification of SALS patients, we integrated gene expression profiling with the analysis of genomic structural aberrations occurring in the motor cortex of the same set of SALS samples (Morello et al., 2019). This comprehensive molecular characterization at the genomic and transcriptomic levels revealed subtype-specific genomic alterations positively correlating with transcriptional signature profiles, further confirming the existence of molecular and functional heterogeneity in SALS and suggesting that genomic and transcriptomic events complement each other in driving disease pathogenesis (Morello et al., 2019). Beyond refining ALS molecular architecture, our results also pinpointed candidate driver genes potentially useful as therapeutic targets and biomarkers for genomic-based patient stratification and individualized treatment (Morello and Cavallaro, 2015; Morello et al., 2017c, 2019; Maugeri et al., 2019). Among these, numerous genes involved in histamine receptors, metabolism, transport, secretion and signal transduction, were differentially expressed in the motor cortex as well as in the spinal cord of two molecular-based subgroups of SALS patients and, of note, some of these genes are located within genomic regions disrupted by DNA copy number occurring in SALS patients (Apolloni et al., 2019). By integrating our data with the known pathogenic variants of ALS-related gene reported in the ALSOD database, we identified a good number of coding variants in these genes, supporting the hypothesis that histamine-related genes might represent candidate biomarkers and targets for patient-oriented ALS care (Apolloni et al., 2019). In this regard, pharmacological modulation of the histamine-related pathway has already proved broad efficacy in ameliorating ALS features, improving motor performance and survival in ALS mice and increasing motor neurons survival in vivo and in vitro ALS models (Apolloni et al., 2017, 2019).
In the past decade, advanced omics technologies have fostered our understanding of the complex molecular architecture of ALS, contributing in part to explain its clinical heterogeneity, and providing a basis for a molecular taxonomy that may radically change our medical approach to ALS. The identification of relevant classifiers and subgroup-specific diagnostic, prognostic and predictive biomarkers is in fact urgently needed for accelerating the development of effective and personalized treatment approaches in ALS. In this review, we discuss the most significant contributions of omics approaches in unraveling the biological complexity of ALS, highlight how holistic systems biology approaches and multi-omics data integration are ideal to provide a comprehensive characterization of patient-specific molecular signatures that could potentially guide therapeutic decisions. We strongly believe that the future research in ALS, as well as in other neurodegenerative diseases, calls a multidisciplinary holistic approach, integrating multi-layer omics data with multimodal neuroimaging and clinical data. This approach will provide a clear understanding of disease prognosis and progression and accelerate development of innovative, effective and personalized strategies for ALS.
GM wrote the manuscript. SS, VD’A, and FC participated in revising the manuscript. SC conceived, directed, and supervised the project. All authors contributed to the article and approved the submitted version.
This work was supported by the Italian Ministry of Education, Universities and Research through grant CTN01_00177_817708 and the international Ph.D. programs in Neuroscience of the University of Catania.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge Cristina Calì, Alfia Corsino, Maria Patrizia D’Angelo, and Francesco Marino for their administrative and technical support.
ALS, amyotrophic lateral sclerosis; MNs, motor neurons; FDA, Food and Drug Administration; UMN, upper motor neuron; LMN, lower motor neuron; FTD, frontotemporal dementia; FALS, familial ALS; SALS, sporadic ALS; SNPs, single nucleotide polymorphisms; SOD1, Superoxide dismutase 1 [Cu-Zn]; C9orf72, chromosome 9 open reading frame 72; FUS, Fused in Sarcoma RNA binding protein; TDP-43/TARDBP, TAR DNA binding protein; GWAS, genome-wide association studies; WGS, whole-genome sequencing; WES, whole-exome sequencing; KIF5A, kinesin family member 5A; ATXN2, ataxin 2; SPAST, spastin; FIG4, FIG4 phosphoinositide 5-phosphatase; SETX, senataxin; DCTN1, dynactin subunit 1; MATR3, matrin 3; CHCHD10, coiled-coil-helix-coiled-coil-helix domain containing 10; SQSTM1, sequestosome 1; VAPB, VAMP associated protein B and C; HNRNPA1, heterogeneous nuclear ribonucleoprotein A1; VCP, valosin containing protein; OPTN, optineurin; EPHA4, Ephrin type-A receptor 4; KIFAP3, Kinesin Associated Protein 3; UNC13A, Unc-13 Homolog A; CNVs, copy-number variations; SMN, survival motor neuron; PMA, progressive muscular atrophy; mtDNA, mitochondrial DNA; EPHA3, Ephrin type-A receptor 3; iPSC, induced pluripotent stem cells; LCM, laser capture microdissection; MS, Mass Spectrometry; CSF, cerebrospinal fluid; NF-L, neurofilament light chain; pNFH, phosphorylated neurofilament heavy chain; IL-10, interleukin 10; IL-6, interleukin 6; IL-2, interleukin 2; IL-15, interleukin 15; IL-8, interleukin 8; GM-CSF, Granulocyte-Macrophage Colony-Stimulating Factor; MIP-1 α, Macrophage Inflammatory Proteins 1-alpha; wrCRP, wide-range C-reactive protein; HMGB, High Mobility Group Box 1; GPNMB, glycoprotein NMB; UCHL1, ubiquitin C-terminal hydrolase L1; bFGF, basic fibroblast growth factor; VGF, Nerve Growth Factor Inducible; PPI, protein-protein interaction; LDL, low-density lipoprotein; HDL, high- density lipoprotein; DNMT, DNA-(cytosine-5)-methyltransferase; miRNA, MicroRNA.
Abrahams, S., Newton, J., Niven, E., Foley, J., and Bak, T. H. (2014). Screening for cognition and behaviour changes in ALS. Amyotroph. Lateral Scler. Frontotemporal. Degener. 15, 9–14. doi: 10.3109/21678421.2013.805784
Achi, E. Y., and Rudnicki, S. A. (2012). ALS and frontotemporal dysfunction: a review. Neurol. Res. Int. 2012:806306. doi: 10.1155/2012/806306
Al-Chalabi, A., Van Den Berg, L. H., and Veldink, J. (2017). Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat. Rev. Neurol. 13, 96–104. doi: 10.1038/nrneurol.2016.182
Amador-Ortiz, C., Lin, W. L., Ahmed, Z., Personett, D., Davies, P., Duara, R., et al. (2007). TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 61, 435–445. doi: 10.1002/ana.21154
Andrés-Benito, P., Moreno, J., Domínguez, R., Aso, E., Povedano, M., and Ferrer, I. (2017). Inflammatory gene expression in whole peripheral blood at early stages of sporadic amyotrophic lateral sclerosis. Front. Neurol. 8:546. doi: 10.3389/fneur.2017.00546
Apolloni, S., Amadio, S., Fabbrizio, P., Morello, G., Spampinato, A. G., Latagliata, E. C., et al. (2019). Histaminergic transmission slows progression of amyotrophic lateral sclerosis. J. Chachexia Sarcopenia Muscle. 10, 872–893. doi: 10.1002/jcsm.12422
Apolloni, S., Fabbrizio, P., Amadio, S., Napoli, G., Verdile, V., Morello, G., et al. (2017). Histamine regulates the inflammatory profile of SOD1-G93A microglia and the histaminergic system is dysregulated in amyotrophic lateral sclerosis. Front. Immunol. 8:1689. doi: 10.3389/fimmu.2017.01689
Aronica, E., Baas, F., Iyer, A., ten Asbroek, A. L. M. A., Morello, G., and Cavallaro, S. (2015). Molecular classification of amyotrophic lateral sclerosis by unsupervised clustering of gene expression in motor cortex. Neurobiol. Dis. 74, 359–376. doi: 10.1016/j.nbd.2014.12.002
Artemiadis, A. K., Peppas, C., Giannopoulos, S., Zouvelou, V., and Triantafyllou, N. (2016). Case of young-onset sporadic amyotrophic lateral sclerosis. J. Clin. Neuromuscul. Dis. 17, 220–222. doi: 10.1097/CND.0000000000000107
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558. doi: 10.1038/s41582-018-0047-2
Balendra, R., Moens, T. G., and Isaacs, A. M. (2017). Specific biomarkers for C9orf72 FTD / ALS could expedite the journey towards effective therapies. EMBO Mol. Med. 9, 853–855. doi: 10.15252/emmm.201707848
Baloh, R. H. (2011). TDP-43: the relationship between protein aggregation and neurodegeneration in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. FEBS J. 278, 3539–3549. doi: 10.1111/j.1742-4658.2011.08256.x
Barker, H. V., Niblock, M., Lee, Y. B., Shaw, C. E., and Gallo, J. M. (2017). RNA misprocessing in C9orf72-linked neurodegeneration. Front. Cell. Neurosci. 11:195. doi: 10.3389/fncel.2017.00195
Barschke, P., Oeckl, P., Steinacker, P., Ludolph, A., and Otto, M. (2017). Proteomic studies in the discovery of cerebrospinal fluid biomarkers for amyotrophic lateral sclerosis. Expert Rev. Proteomics 14, 769–777. doi: 10.1080/14789450.2017.1365602
Bean, D. M., Al-Chalabi, A., Dobson, R. J. B., and Iacoangeli, A. (2020). A knowledge-based machine learning approach to gene prioritisation in amyotrophic lateral sclerosis. Genes (Basel) 11:668. doi: 10.3390/genes11060668
Benatar, M., Wuu, J., Andersen, P. M., Lombardi, V., and Malaspina, A. (2018). Neurofilament light: a candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann. Neurol. 84, 130–139. doi: 10.1002/ana.25276
Bennett, S. A., Tanaz, R., Cobos, S. N., and Torrente, M. P. (2019). Epigenetics in amyotrophic lateral sclerosis: a role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 204, 19–30. doi: 10.1016/j.trsl.2018.10.002
Benninger, F., Glat, M. J., Offen, D., and Steiner, I. (2016). Glial fibrillary acidic protein as a marker of astrocytic activation in the cerebrospinal fluid of patients with amyotrophic lateral sclerosis. J. Clin. Neurosci. 26, 75–78. doi: 10.1016/j.jocn.2015.10.008
Beuche, W., Yushchenko, M., Mäder, M., Maliszewska, M., Felgenhauer, K., and Weber, F. (2000). Matrix metalloproteinase-9 is elevated in serum of patients with amyotrophic lateral sclerosis. Neuroreport 11, 3419–3422. doi: 10.1097/00001756-200011090-00003
Bhandari, R., Kuhad, A., and Kuhad, A. (2018). Edaravone: a new hope for deadly amyotrophic lateral sclerosis. Drugs Today 54, 349–360. doi: 10.1358/dot.2018.54.6.2828189
Bilic, E., Bilic, E., Rudan, I., Kusec, V., Zurak, N., Delimar, D., et al. (2006). Comparison of the growth hormone, IGF-1 and insulin in cerebrospinal fluid and serum between patients with motor neuron disease and healthy controls. Eur. J. Neurol. 13, 1340–1345. doi: 10.1111/j.1468-1331.2006.01503.x
Blasco, H., Patin, F., Descat, A., Garçon, G., Corcia, P., Gelé, P., et al. (2018). A pharmaco-metabolomics approach in a clinical trial of ALS: identification of predictive markers of progression. PLoS One 13:e0198116. doi: 10.1371/journal.pone.0198116
Blasco, H., Patin, F., Madji Hounoum, B., Gordon, P. H., Vourc’h, P., Andres, C. R., et al. (2016). Metabolomics in amyotrophic lateral sclerosis: how far can it take us? Eur. J. Neurol. 23, 447–454. doi: 10.1111/ene.12956
Blauw, H. M., Al-Chalabi, A., Andersen, P. M., van Vught, P. W. J., Diekstra, F. P., van Es, M. A., et al. (2010). A large genome scan for rare CNVs in amyotrophic lateral sclerosis. Hum. Mol. Genet. 19, 4091–4099. doi: 10.1093/hmg/ddq323
Blauw, H. M., Barnes, C. P., Van Vught, P. W. J., Van Rheenen, W., Verheul, M., Cuppen, E., et al. (2012). SMN1 gene duplications are associated with sporadic ALS. Neurology 78, 776–780. doi: 10.1212/WNL.0b013e318249f697
Blauw, H. M., Veldink, J. H., van Es, M. A., van Vught, P. W., Saris, C. G., van der Zwaag, B., et al. (2008). Copy-number variation in sporadic amyotrophic lateral sclerosis: a genome-wide screen. Lancet Neurol. 7, 319–326. doi: 10.1016/S1474-4422(08)70048-6
Bohl, D., Pochet, R., Mitrecic, D., and Nicaise, C. (2016). Modelling and treating amyotrophic lateral sclerosis through induced- pluripotent stem cells technology. Curr. Stem Cell Res. Ther. 11, 301–312. doi: 10.2174/1574888x10666150528144303
Borghero, G., Pugliatti, M., Marrosu, F., Marrosu, M. G., Murru, M. R., Floris, G., et al. (2015). ATXN2 is a modifier of phenotype in ALS patients of Sardinian ancestry. Neurobiol. Aging 36, 2906.e1–2906.e5. doi: 10.1016/j.neurobiolaging.2015.06.013
Bourinaris, T., and Houlden, H. (2018). C9orf72 and its relevance in parkinsonism and movement disorders: a comprehensive review of the literature. Mov. Disord. Clin. Pract. 5, 575–585. doi: 10.1002/mdc3.12677
Bowser, R., Cudkowicz, M., and Kaddurah-Daouk, R. (2006). Biomarkers for amyotrophic lateral sclerosis. Expert Rev. Mol. Diagn. 6, 387–398. doi: 10.1586/14737159.6.3.387
Brenner, D., Yilmaz, R., Müller, K., Grehl, T., Petri, S., Meyer, T., et al. (2018). Hot-spot KIF5A mutations cause familial ALS. Brain 141, 688–697. doi: 10.1093/brain/awx370
Brettschneider, J., Petzold, A., Süßmuth, S. D., Landwehrmeyer, G. B., Ludolph, A. C., Kassubek, J., et al. (2006). Neurofilament heavy-chain NfHSMI35 in cerebrospinal fluid supports the differential diagnosis of Parkinsonian syndromes. Mov. Disord. 21, 2224–2227. doi: 10.1002/mds.21124
Brown, R. H., and Al-Chalabi, A. (2017). Amyotrophic lateral sclerosis. N. Engl. J. Med. 377, 162–172. doi: 10.1056/NEJMra1603471
Bu, L. L., Yang, K., Xiong, W. X., Liu, F. T., Anderson, B., Wang, Y., et al. (2016). Toward precision medicine in Parkinson’s disease. Ann. Transl. Med. 4:26. doi: 10.3978/j.issn.2305-5839.2016.01.21
Butchbach, M. E. R. (2016). Copy number variations in the survival motor neuron genes: implications for spinal muscular atrophy and other neurodegenerative diseases. Front. Mol. Biosci. 3:7. doi: 10.3389/fmolb.2016.00007
Cady, J., Allred, P., Bali, T., Pestronk, A., Goate, A., Miller, T. M., et al. (2015). Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol. 77, 100–113. doi: 10.1002/ana.24306
Calió, M. L., Henriques, E., Siena, A., Bertoncini, C. R. A., Gil-Mohapel, J., and Rosenstock, T. R. (2020). Mitochondrial dysfunction, neurogenesis, and epigenetics: putative implications for amyotrophic lateral sclerosis neurodegeneration and treatment. Front. Neurosci. 14:679. doi: 10.3389/fnins.2020.00679
Cappella, M., Ciotti, C., Cohen-Tannoudji, M., and Biferi, M. G. (2019). Gene therapy for ALS-A perspective. Int. J. Mol. Sci. 20:4388. doi: 10.3390/ijms20184388
Castrillo, J. I., Lista, S., Hampel, H., and Ritchie, C. W. (2018). Systems biology methods for Alzheimer’s disease research toward molecular signatures, subtypes, and stages and precision medicine: application in cohort studies and trials. Methods Mol. Biol. 1750, 31–66. doi: 10.1007/978-1-4939-7704-8_3
Centeno, E. G. Z., Cimarosti, H., and Bithell, A. (2018). 2D versus 3D human induced pluripotent stem cell-derived cultures for neurodegenerative disease modelling. Mol. Neurodegener. 13:27. doi: 10.1186/s13024-018-0258-4
Chang, C. Y., Ting, H. C., Liu, C. A., Su, H. L., Chiou, T. W., Lin, S. Z., et al. (2020). Induced pluripotent stem cell (iPSC)-based neurodegenerative disease models for phenotype recapitulation and drug screening. Molecules 25:2000. doi: 10.3390/molecules25082000
Chew, J., Gendron, T. F., Prudencio, M., Sasaguri, H., Zhang, Y. J., Castanedes-Casey, M., et al. (2015). C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348, 1151–1154. doi: 10.1126/science.aaa9344
Chiò, A., Calvo, A., Moglia, C., Canosa, A., Brunetti, M., Barberis, M., et al. (2015). ATXN2 polyQ intermediate repeats are a modifier of ALS survival. Neurology 84, 251–258. doi: 10.1212/WNL.0000000000001159
Chiò, A., Logroscino, G., Traynor, B. J., Collins, J., Simeone, J. C., Goldstein, L. A., et al. (2013). Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41, 118–130. doi: 10.1159/000351153
Chiò, A., Mazzini, L., and Mora, G. (2020). Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology 167:107986. doi: 10.1016/j.neuropharm.2020.107986
Chiò, A., Moglia, C., Canosa, A., Manera, U., Vasta, R., Brunetti, M., et al. (2019). Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology 93, E984–E994. doi: 10.1212/WNL.0000000000008063
Chipika, R. H., Finegan, E., Li Hi Shing, S., Hardiman, O., and Bede, P. (2019). Tracking a fast-moving disease: longitudinal markers, monitoring, and clinical trial endpoints in ALS. Front. Neurol. 10:229. doi: 10.3389/fneur.2019.00229
Cirulli, E. T., Lasseigne, B. N., Petrovski, S., Sapp, P. C., Dion, P. A., Leblond, C. S., et al. (2015). Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441. doi: 10.1126/science.aaa3650
Coatti, G. C., Beccari, M. S., Olávio, T. R., Mitne-Neto, M., Okamoto, O. K., and Zatz, M. (2015). Stem cells for amyotrophic lateral sclerosis modeling and therapy: myth or fact? Cytom. Part A 87, 197–211. doi: 10.1002/cyto.a.22630
Collins, M. A. (2016). Identification of Amyotrophic Lateral Sclerosis Disease Mechanisms by Cerebrospinal Fluid Proteomic Profiling. Doctoral Dissertation, University of Pittsburgh, Pittsburgh, PA.
Collins, M. A., An, J., Hood, B. L., Conrads, T. P., and Bowser, R. P. (2015). Label-free LC-MS/MS proteomic analysis of cerebrospinal fluid identifies protein/pathway alterations and candidate biomarkers for amyotrophic lateral sclerosis. J. Proteome Res. 14, 4486–4501. doi: 10.1021/acs.jproteome.5b00804
Cook, C., and Petrucelli, L. (2019). Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron 101, 1057–1069. doi: 10.1016/j.neuron.2019.02.032
Cooper-Knock, J., Kirby, J., Ferraiuolo, L., Heath, P. R., Rattray, M., and Shaw, P. J. (2012). Gene expression profiling in human neurodegenerative disease. Nat. Rev. Neurol. 8, 518–530. doi: 10.1038/nrneurol.2012.156
Csobonyeiova, M., Polak, S., Nicodemou, A., and Danisovic, L. (2017). Induced pluripotent stem cells in modeling and cell-based therapy of amyotrophic lateral sclerosis. J. Physiol. Pharmacol. 68, 649–657.
Dangond, F., Hwang, D., Camelo, S., Pasinelli, P., Frosch, M. P., Stephanopoulos, G., et al. (2004). Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol. Genomics 16, 229–239. doi: 10.1152/physiolgenomics.00087.2001
Daoud, H., Belzil, V., Desjarlais, A., Camu, W., Dion, P. A., and Rouleau, G. A. (2010). Analysis of the UNC13A gene as a risk factor for sporadic amyotrophic lateral sclerosis. Arch. Neurol. 67, 516–517. doi: 10.1001/archneurol.2010.46
Dash, R. P., Babu, R. J., and Srinivas, N. R. (2018). Two decades-long journey from riluzole to edaravone: revisiting the clinical pharmacokinetics of the only two amyotrophic lateral sclerosis therapeutics. Clin. Pharmacokinet. 57, 1385–1398. doi: 10.1007/s40262-018-0655-4
De Aguilar, J. L. G. (2019). Lipid biomarkers for amyotrophic lateral sclerosis. Front. Neurol. 10:284. doi: 10.3389/fneur.2019.00284
De Luna, N., Turon-Sans, J., Cortes-Vicente, E., Carrasco-Rozas, A., Illán-Gala, I., Dols-Icardo, O., et al. (2020). Downregulation of miR-335-5P in amyotrophic lateral sclerosis can contribute to neuronal mitochondrial dysfunction and apoptosis. Sci. Rep. 10, 1–12. doi: 10.1038/s41598-020-61246-1
de Oliveira, G. P., Alves, C. J., and Chadi, G. (2013). Early gene expression changes in spinal cord from SOD1G93A amyotrophic lateral sclerosis animal model. Front. Cell. Neurosci. 6:216. doi: 10.3389/fncel.2013.00216
De Schaepdryver, M., Goossens, J., De Meyer, S., Jeromin, A., Masrori, P., Brix, B., et al. (2019). Serum neurofilament heavy chains as early marker of motor neuron degeneration. Ann. Clin. Transl. Neurol. 6, 1971–1979. doi: 10.1002/acn3.50890
De Schaepdryver, M., Jeromin, A., Gille, B., Claeys, K. G., Herbst, V., Brix, B., et al. (2018). Comparison of elevated phosphorylated neurofilament heavy chains in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 89, 367–373. doi: 10.1136/jnnp-2017-316605
Di Pietro, L., Baranzini, M., Berardinelli, M. G., Lattanzi, W., Monforte, M., Tasca, G., et al. (2017). Potential therapeutic targets for ALS: MIR206, MIR208b and MIR499 are modulated during disease progression in the skeletal muscle of patients. Sci. Rep. 7, 1–11. doi: 10.1038/s41598-017-10161-z
Dickson, D. W., Baker, M. C., Jackson, J. L., Dejesus-Hernandez, M., Finch, N. C. A., Tian, S., et al. (2019). Extensive transcriptomic study emphasizes importance of vesicular transport in C9orf72 expansion carriers. Acta Neuropathol. Commun. 7:150. doi: 10.1186/s40478-019-0797-0
Diekstra, F. P., van Vught, P. W. J., van Rheenen, W., Koppers, M., Pasterkamp, R. J., van Es, M. A., et al. (2012). UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol. Aging 33, 630.e3–8. doi: 10.1016/j.neurobiolaging.2011.10.029
Dolinar, A., Ravnik-Glavač, M., and Glavač, D. (2018). Epigenetic mechanisms in amyotrophic lateral sclerosis: a short review. Mech. Ageing Dev. 174, 103–110. doi: 10.1016/j.mad.2018.03.005
Dorst, J., Kühnlein, P., Hendrich, C., Kassubek, J., Sperfeld, A. D., and Ludolph, A. C. (2011). Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J. Neurol. 258, 613–617. doi: 10.1007/s00415-010-5805-z
Douglas, A. G. L. (2018). Non-coding RNA in C9orf72-related amyotrophic lateral sclerosis and frontotemporal dementia: a perfect storm of dysfunction. Non Coding RNA Res. 3, 178–187. doi: 10.1016/j.ncrna.2018.09.001
Duque, T., Gromicho, M., Pronto-Laborinho, A. C., and de Carvalho, M. (2020). Transforming growth factor-β plasma levels and its role in amyotrophic lateral sclerosis. Med. Hypotheses 139:109632. doi: 10.1016/j.mehy.2020.109632
Ebbert, M. T. W., Lank, R. J., and Belzil, V. V. (2018). An epigenetic spin to ALS and FTD. Adv. Neurobiol. 20, 1–29. doi: 10.1007/978-3-319-89689-2_1
Ekegren, T., Hanrieder, J., and Bergquist, J. (2008). Clinical perspectives of high-resolution mass spectrometry-based proteomics in neuroscience: exemplified in amyotrophic lateral sclerosis biomarker discovery research. J. Mass Spectrom. 43, 559–571. doi: 10.1002/jms.1409
Fang, T., Jozsa, F., and Al-Chalabi, A. (2017). Nonmotor symptoms in amyotrophic lateral sclerosis: a systematic review. Int. Rev. Neurobiol. 134, 1409–1441. doi: 10.1016/bs.irn.2017.04.009
Farrawell, N. E., Lambert-Smith, I. A., Warraich, S. T., Blair, I. P., Saunders, D. N., Hatters, D. M., et al. (2015). Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci. Rep. 5:13416. doi: 10.1038/srep13416
Ferrari, R., Kapogiannis, D., Huey, E. D., and Momeni, P. (2011). FTD and ALS: a tale of two diseases. Curr. Alzheimer Res. 8, 273–294. doi: 10.2174/156720511795563700
Figueroa-Romero, C., Hur, J., Lunn, J. S., Paez-Colasante, X., Bender, D. E., Yung, R., et al. (2016). Expression of microRNAs in human post-mortem amyotrophic lateral sclerosis spinal cords provides insight into disease mechanisms. Mol. Cell Neurosci. 71, 34–45. doi: 10.1016/j.mcn.2015.12.008
Floeter, M. K., and Gendron, T. F. (2018). Biomarkers for amyotrophic lateral sclerosis and frontotemporal dementia associated with hexanucleotide expansion mutations in C9orf72. Front. Neurol. 9:1063. doi: 10.3389/fneur.2018.01063
Foxe, D., Elan, E., Burrell, J. R., Felicity, F. V., Devenney, E., Kwok, J. B., et al. (2018). Intrafamilial phenotypic variability in the C9orf72 gene expansion: 2 case studies. Front. Psychol. 9:1615. doi: 10.3389/fpsyg.2018.01615
Freischmidt, A., Müller, K., Zondler, L., Weydt, P., Mayer, B., von Arnim, C. A. F., et al. (2015). Serum microRNAs in sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 36, 2660.e15–20. doi: 10.1016/j.neurobiolaging.2015.06.003
Fujimori, K., Ishikawa, M., Otomo, A., Atsuta, N., Nakamura, R., Akiyama, T., et al. (2018). Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 24, 1579–1589. doi: 10.1038/s41591-018-0140-5
Furukawa, T., Matsui, N., Fujita, K., Nodera, H., Shimizu, F., Miyamoto, K., et al. (2015). CSF cytokine profile distinguishes multifocal motor neuropathy from progressive muscular atrophy. Neurol. Neuroimmunol. NeuroInflammation 2:e138. doi: 10.1212/NXI.0000000000000138
Gaastra, B., Shatunov, A., Pulit, S., Jones, A. R., Sproviero, W., Gillett, A., et al. (2016). Rare genetic variation in UNC13A may modify survival in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 17, 593–599. doi: 10.1080/21678421.2016.1213852
Gaiottino, J., Norgren, N., Dobson, R., Topping, J., Nissim, A., Malaspina, A., et al. (2013). Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS One 8:e0075091. doi: 10.1371/journal.pone.0075091
Gall, L., Anakor, E., Connolly, O., Vijayakumar, U. G., Duddy, W. J., and Duguez, S. (2020). Molecular and cellular mechanisms affected in ALS. J. Pers. Med. 10, E101. doi: 10.3390/JPM10030101
Gendron, T. F., Chew, J., Stankowski, J. N., Hayes, L. R., Zhang, Y. J., Prudencio, M., et al. (2017). Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci. Transl. Med. 9:eaai7866. doi: 10.1126/scitranslmed.aai7866
Germeys, C., Vandoorne, T., Bercier, V., and Van Den Bosch, L. (2019). Existing and emerging metabolomic tools for ALS research. Genes (Basel) 10:1011. doi: 10.3390/genes10121011
Geyer, F. C., Decker, T., and Reis-Filho, J. S. (2009). Genomweite Expressionsprofile als klinische Entscheidungshilfe: bereit für die Praxis? Pathologe 30, 141–146. doi: 10.1007/s00292-008-1104-1
Ghaffari, L. T., Starr, A., Nelson, A. T., and Sattler, R. (2018). Representing diversity in the dish: using patient-derived in vitro models to recreate the heterogeneity of neurological disease. Front. Neurosci. 12:56. doi: 10.3389/fnins.2018.00056
Gross, S., Chen, Q., Sandhu, D., Konrad, C., Roychoudhury, D., Schwartz, B. I., et al. (2018). Identification of a distinct metabolomic subtype of sporadic ALS Patients. bioRxiv[Preprint] doi: 10.1101/416396
Guo, J., Yang, X., Gao, L., and Zang, D. (2017). Evaluating the levels of CSF and serum factors in ALS. Brain Behav. 7:e00637. doi: 10.1002/brb3.637
Guo, W., Fumagalli, L., Prior, R., and van den Bosch, L. (2017). Current advances and limitations in modeling ALS/FTD in a dish using induced pluripotent stem cells. Front. Neurosci. 11:671. doi: 10.3389/fnins.2017.00671
Halpern, M., Brennand, K. J., and Gregory, J. (2019). Examining the relationship between astrocyte dysfunction and neurodegeneration in ALS using hiPSCs. Neurobiol. Dis. 132:104562. doi: 10.1016/j.nbd.2019.104562
Hampel, H., Toschi, N., Babiloni, C., Baldacci, F., Black, K. L., Bokde, A. L. W., et al. (2018a). Revolution of Alzheimer precision neurology. Passageway of systems biology and neurophysiology. J. Alzheimers Dis. 64, S47–S105. doi: 10.3233/JAD-179932
Hampel, H., Vergallo, A., Aguilar, L. F., Benda, N., Broich, K., Cuello, A. C., et al. (2018b). Precision pharmacology for Alzheimer’s disease. Pharmacol. Res. 130, 331–365. doi: 10.1016/j.phrs.2018.02.014
Harms, M. B., and Baloh, R. H. (2013). Clinical neurogenetics: amyotrophic lateral sclerosis. Neurol. Clin. 31, 929–950. doi: 10.1016/j.ncl.2013.05.003
Hawrot, J., Imhof, S., and Wainger, B. J. (2020). Modeling cell-autonomous motor neuron phenotypes in ALS using iPSCs. Neurobiol. Dis. 134:104680. doi: 10.1016/j.nbd.2019.104680
He, J., Mangelsdorf, M., Fan, D., Bartlett, P., and Brown, M. A. (2015). Amyotrophic lateral sclerosis genetic studies: from genome-wide association mapping to genome sequencing. Neuroscientist 21, 599–615. doi: 10.1177/1073858414555404
Heath, P. R., Kirby, J., and Shaw, P. J. (2013). Investigating cell death mechanisms in amyotrophic lateral sclerosis using transcriptomics. Front. Cell. Neurosci. 7:259. doi: 10.3389/fncel.2013.00259
Hedges, E. C., Mehler, V. J., and Nishimura, A. L. (2016). The use of stem cells to model amyotrophic lateral sclerosis and frontotemporal dementia: from basic research to regenerative medicine. Stem Cells Int. 2016:9279516. doi: 10.1155/2016/9279516
Hedl, T. J., Gil, R. S., Cheng, F., Rayner, S. L., Davidson, J. M., De Luca, A., et al. (2019). Proteomics approaches for biomarker and drug target discovery in ALS and FTD. Front. Neurosci. 13:548. doi: 10.3389/fnins.2019.00548
Hergesheimer, R., Lanznaster, D., Vourc’h, P., Andres, C., Bakkouche, S., Beltran, S., et al. (2020). Advances in disease-modifying pharmacotherapies for the treatment of amyotrophic lateral sclerosis. Expert Opin. Pharmacother. 21, 1103–1110. doi: 10.1080/14656566.2020.1746270
Hosback, S., Hardiman, O., Nolan, C. M., Doyle, M. A. C., Gorman, G., Lynch, C., et al. (2007). Circulating insulin-like growth factors and related binding proteins are selectively altered in amyotrophic lateral sclerosis and multiple sclerosis. Growth Horm. IGF Res. 17, 472–479. doi: 10.1016/j.ghir.2007.06.002
Hutten, S., and Dormann, D. (2019). RAN translation down. Nat. Neurosci. 22, 1379–1380. doi: 10.1038/s41593-019-0482-4
Jääskeläinen, O., Solje, E., Hall, A., Katisko, K., Korhonen, V., Tiainen, M., et al. (2019). Low serum high-density lipoprotein cholesterol levels associate with the C9orf72 repeat expansion in frontotemporal lobar degeneration patients. J. Alzheimers Dis. 72, 127–137. doi: 10.3233/JAD-190132
Jacobsson, J., Jonsson, P. A., Andersen, P. M., Forsgren, L., and Marklund, S. L. (2001). Superoxide dismutase in CSF from amyotrophic lateral sclerosis patients with and without CuZn-superoxide dismutase mutations. Brain 124, 1461–1466. doi: 10.1093/brain/124.7.1461
Jaiswal, M. K. (2019). Riluzole and edaravone: a tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 39, 733–748. doi: 10.1002/med.21528
Jeon, G. S., Shim, Y. M., Lee, D. Y., Kim, J. S., Kang, M. J., Ahn, S. H., et al. (2019). Pathological modification of TDP-43 in amyotrophic lateral sclerosis with SOD1 Mutations. Mol. Neurobiol. 56, 2007–2021. doi: 10.1007/s12035-018-1218-2
Jiang, Y. M., Yamamoto, M., Kobayashi, Y., Yoshihara, T., Liang, Y., Terao, S., et al. (2005). Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 57, 236–251. doi: 10.1002/ana.20379
Jirtle, R. L. (2009). Epigenome: the program for human health and disease. Epigenomics 1, 13–16. doi: 10.2217/epi.09.16
Johansson, A., Larsson, A., Nygren, I., Blennow, K., and Askmark, H. (2003). Increased serum and cerebrospinal fluid FGF-2 levels in amyotrophic lateral sclerosis. Neuroreport 14, 1867–1869. doi: 10.1097/00001756-200310060-00022
Joilin, G., Leigh, P. N., Newbury, S. F., and Hafezparast, M. (2019). An overview of microRNAs as biomarkers of ALS. Front. Neurol. 10:186. doi: 10.3389/fneur.2019.00186
Jones, A. R., Troakes, C., King, A., Sahni, V., De Jong, S., Bossers, K., et al. (2015). Stratified gene expression analysis identifies major amyotrophic lateral sclerosis genes. Neurobiol. Aging 36, 2006.e1–9. doi: 10.1016/j.neurobiolaging.2015.02.017
Kasai, T., Kojima, Y., Ohmichi, T., Tatebe, H., Tsuji, Y., Noto, Y. I., et al. (2019). Combined use of CSF NfL and CSF TDP-43 improves diagnostic performance in ALS. Ann. Clin. Transl. Neurol. 6, 2489–2502. doi: 10.1002/acn3.50943
Keeney, P. M., and Bennett, J. P. (2010). ALS spinal neurons show varied and reduced mtDNA gene copy numbers and increased mtDNA gene deletions. Mol. Neurodegener. 5:21. doi: 10.1186/1750-1326-5-21
Kirby, J., Al Sultan, A., Waller, R., and Heath, P. (2016). The genetics of amyotrophic lateral sclerosis: current insights. Degener. Neurol. Neuromuscul. Dis. 6:49. doi: 10.2147/dnnd.s84956
Kirby, J., Halligan, E., Baptista, M. J., Allen, S., Heath, P. R., Holden, H., et al. (2005). Mutant SOD1 alters the motor neuronal transcriptome: implications for familial ALS. Brain 128, 1686–1706. doi: 10.1093/brain/awh503
Klingl, Y. E., Pakravan, D., and Van Den Bosch, L. (2020). Opportunities for histone deacetylase inhibition in amyotrophic lateral sclerosis. Br. J. Pharmacol. doi: 10.1111/bph.15217 [Epub ahead of print].
Krokidis, M. G. (2020). Transcriptomics and metabolomics in amyotrophic lateral sclerosis. Adv. Exp. Med. Biol. 1195, 205–212. doi: 10.1007/978-3-030-32633-3_29
Krokidis, M. G., and Vlamos, P. (2018). Transcriptomics in amyotrophic lateral sclerosis. Front. Biosci. 10:103–121. doi: 10.2741/e811
Krüger, T., Lautenschläger, J., Grosskreutz, J., and Rhode, H. (2013). Proteome analysis of body fluids for amyotrophic lateral sclerosis biomarker discovery. Proteomics Clin. Appl. 7, 123–135. doi: 10.1002/prca.201200067
Kumar, A., Ghosh, D., and Singh, R. L. (2013). Amyotrophic lateral sclerosis and metabolomics: clinical implication and therapeutic approach. J. Biomarkers 2013, 1–15. doi: 10.1155/2013/538765
La Cognata, V., Gentile, G., Aronica, E., and Cavallaro, S. (2020). Splicing players are differently expressed in sporadic amyotrophic lateral sclerosis molecular clusters and brain regions. Cells 9:159. doi: 10.3390/cells9010159
Lacomblez, L., Doppler, V., Beucler, I., Costes, G., Salachas, F., Raisonnier, A., et al. (2002). APOE: a potential marker of disease progression in ALS. Neurology 58, 1112–1114. doi: 10.1212/WNL.58.7.1112
Laffita-Mesa, J. M., Rodríguez Pupo, J. M., Moreno Sera, R., Vázquez Mojena, Y., Kourí, V., Laguna-Salvia, L., et al. (2013). De novo mutations in Ataxin-2 Gene and ALS Risk. PLoS One 8:e70560. doi: 10.1371/journal.pone.0070560
Lam, S., Bayraktar, A., Zhang, C., Turkez, H., Nielsen, J., Boren, J., et al. (2020). A systems biology approach for studying neurodegenerative diseases. Drug Discov. Today 25, 1146–1159. doi: 10.1016/j.drudis.2020.05.010
Landers, J. E., Melki, J., Meininger, V., Glass, J. D., Van Den Berg, L. H., Van Es, M. A., et al. (2009). Reduced expression of the Kinesin-Associated Protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 106, 9004–9009. doi: 10.1073/pnas.0812937106
Lanznaster, D., de Assis, D. R., Corcia, P., Pradat, P.-F., and Blasco, H. (2018). Metabolomics biomarkers: a strategy toward therapeutics improvement in ALS. Front. Neurol. 9:1126. doi: 10.3389/fneur.2018.01126
Lanznaster, D., Veyrat-Durebex, C., Vourc’h, P., Andres, C. R., Blasco, H., and Corcia, P. (2020). Metabolomics: a tool to understand the impact of genetic mutations in amyotrophic lateral sclerosis. Genes (Basel) 11:537. doi: 10.3390/genes11050537
Lederer, C. W., Torrisi, A., Pantelidou, M., Santama, N., and Cavallaro, S. (2007). Pathways and genes differentially expressed in the motor cortex of patients with sporadic amyotrophic lateral sclerosis. BMC Genomics 8:26. doi: 10.1186/1471-2164-8-26
Lee, J. H., Liu, J. W., Lin, S. Z., Harn, H. J., and Chiou, T. W. (2018). Advances in patient-specific induced pluripotent stem cells shed light on drug discovery for amyotrophic lateral sclerosis. Cell Transplant. 27, 1301–1312. doi: 10.1177/0963689718785154
Lehmer, C., Oeckl, P., Weishaupt, J. H., Volk, A. E., Diehl-Schmid, J., Schroeter, M. L., et al. (2017). Poly-GP in cerebrospinal fluid links C9orf72-associated dipeptide repeat expression to the asymptomatic phase of ALS/FTD. EMBO Mol. Med. 9, 859–868. doi: 10.15252/emmm.201607486
Li, Y. J., Pericak-Vance, M. A., Haines, J. L., Siddique, N., McKenna-Yasek, D., Hung, W. Y., et al. (2004). Apolipoprotein E is associated with age at onset of amyotrophic lateral sclerosis. Neurogenetics 5, 209–213. doi: 10.1007/s10048-004-0193-0
Little, J., Barakat-Haddad, C., Martino, R., Pringsheim, T., Tremlett, H., McKay, K. A., et al. (2017). Genetic variation associated with the occurrence and progression of neurological disorders. Neurotoxicology 61, 243–264. doi: 10.1016/j.neuro.2016.09.018
Liu, W., Venugopal, S., Majid, S., Ahn, I. S., Diamante, G., Hong, J., et al. (2020). Single-cell RNA-seq analysis of the brainstem of mutant SOD1 mice reveals perturbed cell types and pathways of amyotrophic lateral sclerosis. Neurobiol. Dis. 141:104877. doi: 10.1016/j.nbd.2020.104877
Lorenzl, S., Albers, D. S., Narr, S., Chirichigno, J., and Beal, M. F. (2002). Expression of MMP-2, MMP-9, and MMP-1 and their endogenous counterregulators TIMP-1 and TIMP-2 in postmortem brain tissue of Parkinson’s disease. Exp. Neurol. 178, 13–20. doi: 10.1006/exnr.2002.8019
Lu, C.-H., Allen, K., Oei, F., Leoni, E., Kuhle, J., Tree, T., et al. (2016). Systemic inflammatory response and neuromuscular involvement in amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 3:e244. doi: 10.1212/nxi.0000000000000244
Lu, C. H., Macdonald-Wallis, C., Gray, E., Pearce, N., Petzold, A., Norgren, N., et al. (2015). Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology 84, 2247–2257. doi: 10.1212/WNL.0000000000001642
Ma, G., Wang, Y., Li, Y., Cui, L., Zhao, Y., Zhao, B., et al. (2015). MiR-206, a key modulator of skeletal muscle development and disease. Int. J. Biol. Sci. 11, 345–352. doi: 10.7150/ijbs.10921
Mackenzie, I. R. A., Bigio, E. H., Ince, P. G., Geser, F., Neumann, M., Cairns, N. J., et al. (2007). Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 61, 427–434. doi: 10.1002/ana.21147
Mackenzie, I. R. A., and Rademakers, R. (2008). The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 21, 693–700. doi: 10.1097/WCO.0b013e3283168d1d
Majumder, V., Gregory, J. M., Barria, M. A., Green, A., and Pal, S. (2018). TDP-43 as a potential biomarker for amyotrophic lateral sclerosis: a systematic review and meta-analysis. BMC Neurol. 18:90. doi: 10.1186/s12883-018-1091-7
Malaspina, A., and de Belleroche, J. (2004). Spinal cord molecular profiling provides a better understanding of amyotrophic lateral sclerosis pathogenesis. Brain Res. Brain Res. Rev. 45, 213–229. doi: 10.1016/j.brainresrev.2004.04.002
Malaspina, A., Kaushik, N., and De Belleroche, J. (2008). Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J. Neurochem. 77, 132–145. doi: 10.1046/j.1471-4159.2001.00231.x
Malik, R., and Wiedau, M. (2020). Therapeutic approaches targeting protein aggregation in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 13:98. doi: 10.3389/fnmol.2020.00098
Maniatis, S., Äijö, T., Vickovic, S., Braine, C., Kang, K., Mollbrink, A., et al. (2019). Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science 364, 89–93. doi: 10.1126/science.aav9776
Mao, Y., Kuo, S.-W., Chen, L., Heckman, C. J., and Jiang, M. C. (2017). The essential and downstream common proteins of amyotrophic lateral sclerosis: a protein-protein interaction network analysis. PLoS One 12:e0172246. doi: 10.1371/journal.pone.0172246
Maria D’erchia, A., Gallo, A., Manzari, C., Raho, S., Horner, D. S., Chiara, M., et al. (2017). Massive transcriptome sequencing of human spinal cord tissues provides new insights into motor neuron degeneration in ALS. Sci. Rep. 7:10046. doi: 10.1038/s41598-017-10488-7
Masala, A., Sanna, S., Esposito, S., Rassu, M., Galioto, M., Zinellu, A., et al. (2018). Epigenetic changes associated with the expression of Amyotrophic Lateral Sclerosis (ALS) causing genes. Neuroscience 390, 1–11. doi: 10.1016/j.neuroscience.2018.08.009
Maugeri, G., D’Amico, A. G., Rasà, D. M., Federico, C., Saccone, S., Morello, G., et al. (2019). Molecular mechanisms involved in the protective effect of pituitary adenylate cyclase-activating polypeptide in an in vitro model of amyotrophic lateral sclerosis. J. Cell. Physiol. 234, 5203–5214. doi: 10.1002/jcp.27328
Maurel, C., Dangoumau, A., Marouillat, S., Brulard, C., Chami, A., Hergesheimer, R., et al. (2018). Causative genes in amyotrophic lateral sclerosis and protein degradation pathways: a link to neurodegeneration. Mol. Neurobiol. 55, 6480–6499. doi: 10.1007/s12035-017-0856-0
Mawrin, C., Kirches, E., Krause, G., Wiedemann, F. R., Vorwerk, C. K., Bogerts, B., et al. (2004). Single-cell analysis of mtDNA deletion levels in sporadic amyotrophic lateral sclerosis. Neuroreport 15, 939–943. doi: 10.1097/00001756-200404290-00002
McCann, E. P., Williams, K. L., Fifita, J. A., Tarr, I. S., O’Connor, J., Rowe, D. B., et al. (2017). The genotype-phenotype landscape of familial amyotrophic lateral sclerosis in Australia. Clin. Genet. 92, 259–266. doi: 10.1111/cge.12973
Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., and Akkari, P. A. (2019). ALS genetics, mechanisms, and therapeutics: where are we now? Front. Neurosci. 13:1310. doi: 10.3389/fnins.2019.01310
Mirza, B., Wang, W., Wang, J., Choi, H., Chung, N. C., and Ping, P. (2019). Machine learning and integrative analysis of biomedical big data. Genes (Basel) 10:87. doi: 10.3390/genes10020087
Mitchell, R. M., Freeman, W. M., Randazzo, W. T., Stephens, H. E., Beard, J. L., Simmons, Z., et al. (2009). A CSF biomarker panel for identification of patients with amyotrophic lateral sclerosis. Neurology 72, 14–19. doi: 10.1212/01.wnl.0000333251.36681.a5
Mitropoulos, K., Katsila, T., Patrinos, G. P., and Pampalakis, G. (2018). Multi-omics for biomarker discovery and target validation in biofluids for amyotrophic lateral sclerosis diagnosis. OMICS 22, 52–64. doi: 10.1089/omi.2017.0183
Moreau, C., Devos, D., Brunaud-Danel, V., Defebvre, L., Perez, T., Destée, A., et al. (2006). Paradoxical response of VEGF expression to hypoxia in CSF of patients with ALS. J. Neurol. Neurosurg. Psychiatry 77, 255–257. doi: 10.1136/jnnp.2005.070904
Morello, G., and Cavallaro, S. (2015). Transcriptional analysis reveals distinct subtypes in amyotrophic lateral sclerosis: implications for personalized therapy. Future Med. Chem. 7, 1335–1359. doi: 10.4155/fmc.15.60
Morello, G., Conforti, F. L., Parenti, R., D’Agata, V., and Cavallaro, S. (2015). Selection of potential pharmacological targets in ALS based on whole-genome expression profiling. Curr. Med. Chem. 22, 2004–2021. doi: 10.2174/0929867322666150408112135
Morello, G., Guarnaccia, M., Spampinato, A. G., La Cognata, V., D’Agata, V., and Cavallaro, S. (2018a). Copy number variations in amyotrophic lateral sclerosis: piecing the mosaic tiles together through a systems biology approach. Mol. Neurobiol. 55, 1299–1322. doi: 10.1007/s12035-017-0393-x
Morello, G., Guarnaccia, M., Spampinato, A. G., Salomone, S., D’Agata, V., Conforti, F. L., et al. (2019). Integrative multi-omic analysis identifies new drivers and pathways in molecularly distinct subtypes of ALS. Sci. Rep. 9:9968. doi: 10.1038/s41598-019-46355-w
Morello, G., Spampinato, A. G., and Cavallaro, S. (2017a). Molecular taxonomy of sporadic amyotrophic lateral sclerosis using disease-associated genes. Front. Neurol. 8:152. doi: 10.3389/fneur.2017.00152
Morello, G., Spampinato, A. G., and Cavallaro, S. (2017b). Neuroinflammation and ALS: transcriptomic insights into molecular disease mechanisms and therapeutic targets. Mediat. Inflamm. 2017:7070469. doi: 10.1155/2017/7070469
Morello, G., Spampinato, A. G., Conforti, F. L., and Cavallaro, S. (2018b). Taxonomy meets neurology, the case of amyotrophic lateral sclerosis. Front. Neurosci. 12:673. doi: 10.3389/fnins.2018.00673
Morello, G., Spampinato, A. G., Conforti, F. L., D’Agata, V., and Cavallaro, S. (2017c). Selection and prioritization of candidate drug targets for amyotrophic lateral sclerosis through a meta-analysis approach. J. Mol. Neurosci. 61, 563–580. doi: 10.1007/s12031-017-0898-9
Moulard, B., Sefiani, A., Laamri, A., Malafosse, A., and Camu, W. (1996). Apolipoprotein E genotyping in sporadic amyotrophic lateral sclerosis, evidence for a major influence on the clinical presentation and prognosis. J. Neurol. Sci. 139, 34–37. doi: 10.1016/0022-510X(96)00085-8
Myszczynska, M., and Ferraiuolo, L. (2016). New in vitro models to study amyotrophic lateral sclerosis. Brain Pathol. 26, 258–265. doi: 10.1111/bpa.12353
Namboori, S. C., Thomas, P., Ames, R., Garrett, L. O., Willis, C. R. G., Stanton, L. W., et al. (2019). Single cell transcriptomics identifies master regulators of dysfunctional pathways in SOD1 ALS motor neurons. bioRxiv[Preprint] doi: 10.1101/593129
Naruse, H., Ishiura, H., Mitsui, J., Takahashi, Y., Matsukawa, T., Tanaka, M., et al. (2019). Burden of rare variants in causative genes for amyotrophic lateral sclerosis (ALS) accelerates age at onset of ALS. J. Neurol. Neurosurg. Psychiatry 90, 537–542. doi: 10.1136/jnnp-2018-318568
Nardo, G., Iennaco, R., Fusi, N., Heath, P. R., Marino, M., Trolese, M. C., et al. (2013). Transcriptomic indices of fast and slow disease progression in two mouse models of amyotrophic lateral sclerosis. Brain 136, 3305–3332. doi: 10.1093/brain/awt250
Navone, F., Genevini, P., and Borgese, N. (2015). Autophagy and neurodegeneration: insights from a cultured cell model of ALS. Cells 4, 354–386. doi: 10.3390/cells4030354
Nguyen, D. K. H., Thombre, R., and Wang, J. (2019). Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci. Lett. 697, 34–48. doi: 10.1016/j.neulet.2018.04.006
Nguyen, N. D., and Wang, D. (2020). Multiview learning for understanding functional multiomics. PLoS Comput. Biol. 16:e1007677. doi: 10.1371/journal.pcbi.1007677
Nicolas, A., Kenna, K., Renton, A. E., Ticozzi, N., Faghri, F., Chia, R., et al. (2018). Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 97, 1268–1283.e6. doi: 10.1016/j.neuron.2018.02.027
Niebroj-Dobosz, I., Janik, P., Sokołowska, B., and Kwiecinski, H. (2010). Matrix metalloproteinases and their tissue inhibitors in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 17, 226–231. doi: 10.1111/j.1468-1331.2009.02775.x
Oeckl, P., Weydt, P., Thal, D. R., Weishaupt, J. H., Ludolph, A. C., and Otto, M. (2020). Proteomics in cerebrospinal fluid and spinal cord suggests UCHL1, MAP2 and GPNMB as biomarkers and underpins importance of transcriptional pathways in amyotrophic lateral sclerosis. Acta Neuropathol. 139, 119–134. doi: 10.1007/s00401-019-02093-x
Olivier, M., Asmis, R., Hawkins, G. A., Howard, T. D., and Cox, L. A. (2019). The need for multi-omics biomarker signatures in precision medicine. Int. J. Mol. Sci. 20:4781. doi: 10.3390/ijms20194781
Online, R., Hedl, T., Gil, R. S., Cheng, F., Rayner, S. L., Davidson, J., et al. (2019). Proteomics approaches for biomarker and drug target discovery in ALS and FTD publication details. Front Neurosci. 13:548.
Paez-Colasante, X., Figueroa-Romero, C., Sakowski, S. A., Goutman, S. A., and Feldman, E. L. (2015). Amyotrophic lateral sclerosis: mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurol. 11, 266–279. doi: 10.1038/nrneurol.2015.57
Parakh, S., and Atkin, J. D. (2016). Protein folding alterations in amyotrophic lateral sclerosis. Brain Res. 1648, 633–649. doi: 10.1016/j.brainres.2016.04.010
Pasinelli, P., and Brown, R. H. (2006). Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7, 710–723. doi: 10.1038/nrn1971
Pasinetti, G. M., Ungar, L. H., Lange, D. J., Yemul, S., Deng, H., Yuan, X., et al. (2006). Identification of potential CSF biomarkers in ALS. Neurology 66, 1218–1222. doi: 10.1212/01.wnl.0000203129.82104.07
Perlson, E., Maday, S., Fu, M. M., Moughamian, A. J., and Holzbaur, E. L. F. (2010). Retrograde axonal transport: pathways to cell death? Trends Neurosci. 33, 335–344. doi: 10.1016/j.tins.2010.03.006
Poesen, K. (2018). The chromosomal conformation signature: a new kid on the block in ALS biomarker research? EBioMedicine 33, 6–7. doi: 10.1016/j.ebiom.2018.07.003
Prell, T., Lautenschläger, J., and Grosskreutz, J. (2013). Calcium-dependent protein folding in amyotrophic lateral sclerosis. Cell Calcium 54, 132–143. doi: 10.1016/j.ceca.2013.05.007
Prudencio, M., Belzil, V. V., Batra, R., Ross, C. A., Gendron, T. F., Pregent, L. J., et al. (2015). Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 18, 1175–1182. doi: 10.1038/nn.4065
Rahman, M. R., Islam, T., Huq, F., Quinn, J. M. W., and Moni, M. A. (2019). Identification of molecular signatures and pathways common to blood cells and brain tissue of amyotrophic lateral sclerosis patients. Inform. Med. Unlock. 16:100193. doi: 10.1016/j.imu.2019.100193
Raman, R., Allen, S. P., Goodall, E. F., Kramer, S., Ponger, L.-L., Heath, P. R., et al. (2015). Gene expression signatures in motor neurone disease fibroblasts reveal dysregulation of metabolism, hypoxia-response and RNA processing functions. Neuropathol. Appl. Neurobiol. 41, 201–226. doi: 10.1111/nan.12147
Ramanan, V. K., and Saykin, A. J. (2013). Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. Am. J. Neurodegener. Dis. 2, 145–175.
Ranganathan, S., Williams, E., Ganchev, P., Gopalakrishnan, V., Lacomis, D., Urbinelli, L., et al. (2005). Proteomic profiling of cerebrospinal fluid identifies biomarkers for amyotrophic lateral sclerosis. J. Neurochem. 95, 1461–1471. doi: 10.1111/j.1471-4159.2005.03478.x
Rao, V. S., Srinivas, K., Sujini, G. N., and Kumar, G. N. S. (2014). Protein-protein interaction detection: methods and analysis. Int. J. Proteomics 2014:147648. doi: 10.1155/2014/147648
Rattray, N. J. W., and Daouk, R. K. (2017). Pharmacometabolomics and precision medicine special issue editorial. Metabolomics 13, 1–4. doi: 10.1007/s11306-017-1191-1
Recabarren-Leiva, D., and Alarcón, M. (2018). New insights into the gene expression associated to amyotrophic lateral sclerosis. Life Sci. 193, 110–123. doi: 10.1016/j.lfs.2017.12.016
Renton, A. E., Chiò, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23. doi: 10.1038/nn.3584
Ricci, C., Marzocchi, C., and Battistini, S. (2018). MicroRNAs as biomarkers in amyotrophic lateral sclerosis. Cells 7:219. doi: 10.3390/cells7110219
Rizzuti, M., Filosa, G., Melzi, V., Calandriello, L., Dioni, L., Bollati, V., et al. (2018). MicroRNA expression analysis identifies a subset of downregulated miRNAs in ALS motor neuron progenitors. Sci. Rep. 8, 1–12. doi: 10.1038/s41598-018-28366-1
Robberecht, W., and Eykens, C. (2015). The genetic basis of amyotrophic lateral sclerosis: recent breakthroughs. Adv. Genomics Genet. 5:327. doi: 10.2147/agg.s57397
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rosengren, L. E., Karlsson, J.-E., Karlsson, J.-O., Persson, L. I., and Wikkelsø, C. (2002). Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. J. Neurochem. 67, 2013–2018. doi: 10.1046/j.1471-4159.1996.67052013.x
Rué, L., Timmers, M., Lenaerts, A., Smolders, S., Poppe, L., de Boer, A., et al. (2019). Reducing EphA4 before disease onset does not affect survival in a mouse model of Amyotrophic Lateral Sclerosis. Sci. Rep. 9, 1–9. doi: 10.1038/s41598-019-50615-0
Ruegsegger, C., and Saxena, S. (2016). Proteostasis impairment in ALS. Brain Res. 1648, 571–579. doi: 10.1016/j.brainres.2016.03.032
Ryan, M., Heverin, M., Doherty, M. A., Davis, N., Corr, E. M., Vajda, A., et al. (2018). Determining the incidence of familiality in ALS. Neurol. Genet. 4:e239. doi: 10.1212/NXG.0000000000000239
Ryberg, H., and Bowser, R. (2008). Protein biomarkers for amyotrophic lateral sclerosis. Expert Rev. Proteomics 5, 249–262. doi: 10.1586/14789450.5.2.249
Salter, M., Corfield, E., Ramadass, A., Grand, F., Green, J., Westra, J., et al. (2018). Initial identification of a blood-based chromosome conformation signature for aiding in the diagnosis of amyotrophic lateral sclerosis. EBioMedicine 33, 169–184. doi: 10.1016/j.ebiom.2018.06.015
Sances, S., Bruijn, L. I., Chandran, S., Eggan, K., Ho, R., Klim, J. R., et al. (2016). Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat. Neurosci. 19, 542–553. doi: 10.1038/nn.4273
Sangare, M., Dicko, I., Guinto, C. O., Sissoko, A., Dembele, K., Coulibaly, Y., et al. (2016). Does the survival motor neuron copy number variation play a role in the onset and severity of sporadic amyotrophic lateral sclerosis in Malians? eNeurologicalSci 3, 17–20. doi: 10.1016/j.ensci.2015.12.001
Santiago, J. A., Bottero, V., and Potashkin, J. A. (2017). Dissecting the molecular mechanisms of neurodegenerative diseases through network biology. Front. Aging Neurosci. 9:166. doi: 10.3389/fnagi.2017.00166
Saris, C. G. J., Groen, E. J. N., Koekkoek, J. A. F., Veldink, J. H., and Van Den Berg, L. H. (2013). Meta-analysis of gene expression profiling in amyotrophic lateral sclerosis: a comparison between transgenic mouse models and human patients. Amyotroph. Lateral Scler. Front. Degener. 14, 177–189. doi: 10.3109/21678421.2012.729842
Scoles, D. R., and Pulst, S. M. (2018). Oligonucleotide therapeutics in neurodegenerative diseases. RNA Biol. 15, 707–714. doi: 10.1080/15476286.2018.1454812
Selvaraj, B. T., Livesey, M. R., and Chandran, S. (2017). Modeling the C9ORF72 repeat expansion mutation using human induced pluripotent stem cells. Brain Pathol. 27, 518–524. doi: 10.1111/bpa.12520
Sharma, S., and Lu, H. C. (2018). microRNAs in neurodegeneration: current findings and potential impacts. J. Alzheimers Dis. Park. 9, 166. doi: 10.4172/2161-0460.1000420
Shi, N., Kawano, Y., Tateishi, T., Kikuchi, H., Osoegawa, M., Ohyagi, Y., et al. (2007). Increased IL-13-producing T cells in ALS: positive correlations with disease severity and progression rate. J. Neuroimmunol. 182, 232–235. doi: 10.1016/j.jneuroim.2006.10.001
Shurte, L. (2016). Determining Protein-Protein Interactions of ALS-Associated SOD1. Brows. all Theses Diss. Available online at: https://corescholar.libraries.wright.edu/etd_all/2043 (accessed June 3, 2020).
Snider, J., Kotlyar, M., Saraon, P., Yao, Z., Jurisica, I., and Stagljar, I. (2015). Fundamentals of protein interaction network mapping. Mol. Syst. Biol. 11:848. doi: 10.15252/msb.20156351
Sproviero, W., Shatunov, A., Stahl, D., Shoai, M., van Rheenen, W., Jones, A. R., et al. (2017). ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol. Aging 51, 178.e1–178.e9. doi: 10.1016/j.neurobiolaging.2016.11.010
Su, X. W., Broach, J. R., Connor, J. R., Gerhard, G. S., and Simmons, Z. (2014). Genetic heterogeneity of amyotrophic lateral sclerosis: implications for clinical practice and research. Muscle and Nerve 49, 786–803. doi: 10.1002/mus.24198
Süssmuth, S. D., Tumani, H., Ecker, D., and Ludolph, A. C. (2003). Amyotrophic lateral sclerosis: disease stage related changes of tau protein and S100 beta in cerebrospinal fluid and creatine kinase in serum. Neurosci. Lett. 353, 57–60. doi: 10.1016/j.neulet.2003.09.018
Takahashi, I., Hama, Y., Matsushima, M., Hirotani, M., Kano, T., Hohzen, H., et al. (2015). Identification of plasma microRNAs as a biomarker of sporadic Amyotrophic Lateral Sclerosis. Mol. Brain 8:67. doi: 10.1186/s13041-015-0161-7
Tam, O. H., Rozhkov, N. V., Shaw, R., Ravits, J., Dubnau, J., Gale, M., et al. (2019). Postmortem cortex samples identify distinct molecular subtypes of ALS: retrotransposon activation, oxidative stress, and activated glia. CellReports 29, 1164–1177.e5. doi: 10.1016/j.celrep.2019.09.066
Tanaka, F., Sone, J., Atsuta, N., Nakamura, R., Doi, H., Koyano, S., et al. (2013). Personal genome analysis in amyotrophic lateral sclerosis. Brain Nerve 65, 257–265.
Tanaka, M., Kikuchi, H., Ishizu, T., Minohara, M., Osoegawa, M., Motomura, K., et al. (2006). Intrathecal upregulation of granulocyte colony stimulating factor and its neuroprotective actions on motor neurons in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 65, 816–825. doi: 10.1097/01.jnen.0000232025.84238.e1
Tang, A. Y. (2014). RNA-binding proteins associated molecular mechanisms of motor neuron degeneration pathogenesis. Mol. Biotechnol. 56, 779–786. doi: 10.1007/s12033-014-9785-6
Tateishi, T., Yamasaki, R., Tanaka, M., Matsushita, T., Kikuchi, H., Isobe, N., et al. (2010). CSF chemokine alterations related to the clinical course of amyotrophic lateral sclerosis. J. Neuroimmunol. 222, 76–81. doi: 10.1016/j.jneuroim.2010.03.004
Taylor, J. P., Brown, R. H., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Thompson, A. G., Gray, E., Thézénas, M. L., Charles, P. D., Evetts, S., Hu, M. T., et al. (2018). Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann. Neurol. 83, 258–268. doi: 10.1002/ana.25143
Toivonen, J. M., Manzano, R., Oliván, S., Zaragoza, P., García-Redondo, A., and Osta, R. (2014). MicroRNA-206: a potential circulating biomarker candidate for amyotrophic lateral sclerosis. PLoS One 9:e89065. doi: 10.1371/journal.pone.0089065
Tsuboi, Y., Kakimoto, K., Akatsu, H., Daikuhara, Y., and Yamada, T. (2002). Hepatocyte growth factor in cerebrospinal fluid in neurologic disease. Acta Neurol. Scand. 106, 99–103. doi: 10.1034/j.1600-0404.2002.01125.x
Umoh, M. E., Dammer, E. B., Dai, J., Duong, D. M., Lah, J. J., Levey, A. I., et al. (2018). A proteomic network approach across the ALS – FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol. Med. 10, 48–62. doi: 10.15252/emmm.201708202
Uyan, Ö, Ömür, Ö, Ağım, Z. S., Özoğuz, A., Li, H., Parman, Y., et al. (2013). Genome-wide copy number variation in sporadic amyotrophic lateral sclerosis in the Turkish population: deletion of EPHA3 is a possible protective factor. PLoS One 8:e72381. doi: 10.1371/journal.pone.0072381
Vadgama, N., Pittman, A., Simpson, M., Nirmalananthan, N., Murray, R., Yoshikawa, T., et al. (2019). De novo single-nucleotide and copy number variation in discordant monozygotic twins reveals disease-related genes. Eur. J. Hum. Genet. 27, 1121–1133. doi: 10.1038/s41431-019-0376-7
van Blitterswijk, M., Mullen, B., Heckman, M. G., Baker, M. C., DeJesus-Hernandez, M., Brown, P. H., et al. (2014a). Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol. Aging 35, 2421.e13–7. doi: 10.1016/j.neurobiolaging.2014.04.016
van Blitterswijk, M., Mullen, B., Wojtas, A., Heckman, M. G., Diehl, N. N., Baker, M. C., et al. (2014b). Genetic modifiers in carriers of repeat expansions in the C9ORF72 gene. Mol. Neurodegener. 9:38. doi: 10.1186/1750-1326-9-38
Van Den Heuvel, D. M. A., Harschnitz, O., Van Den Berg, L. H., and Pasterkamp, R. J. (2014). Taking a risk: a therapeutic focus on ataxin-2 in amyotrophic lateral sclerosis? Trends Mol. Med. 20, 25–35. doi: 10.1016/j.molmed.2013.09.001
van der Spek, R. A. A., van Rheenen, W., Pulit, S. L., Kenna, K. P., van den Berg, L. H., and Veldink, J. H. (2019). The project MinE databrowser: bringing large-scale whole-genome sequencing in ALS to researchers and the public. Amyotroph. Lateral Scler. Front. Degener. 20, 432–440. doi: 10.1080/21678421.2019.1606244
Van Hoecke, A., Schoonaert, L., Lemmens, R., Timmers, M., Staats, K. A., Laird, A. S., et al. (2012). EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat. Med. 18, 1418–1422. doi: 10.1038/nm.2901
Van Rheenen, W., Pulit, S. L., Dekker, A. M., Al Khleifat, A., Brands, W. J., Iacoangeli, A., et al. (2018). Project MinE: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur. J. Hum. Genet. 26, 1537–1546. doi: 10.1038/s41431-018-0177-4
Van Rheenen, W., Shatunov, A., Dekker, A. M., McLaughlin, R. L., Diekstra, F. P., Pulit, S. L., et al. (2016). Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048. doi: 10.1038/ng.3622
Vella, D., Zoppis, I., Mauri, G., Mauri, P., and Di Silvestre, D. (2017). From protein-protein interactions to protein co-expression networks: a new perspective to evaluate large-scale proteomic data. Eurasip J. Bioinforma. Syst. Biol. 2017, 1–16. doi: 10.1186/s13637-017-0059-z
Verde, F., Steinacker, P., Weishaupt, J. H., Kassubek, J., Oeckl, P., Halbgebauer, S., et al. (2019). Neurofilament light chain in serum for the diagnosis of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 90, 157–164. doi: 10.1136/jnnp-2018-318704
Vijayakumar, U. G., Milla, V., Stafford, M. Y. C., Bjourson, A. J., Duddy, W., and Duguez, S. M. R. (2019). A systematic review of suggested molecular strata, biomarkers and their tissue sources in ALS. Front. Neurol. 10:400. doi: 10.3389/fneur.2019.00400
Volonté, C., Morello, G., Spampinato, A. G., Amadio, S., Apolloni, S., D’Agata, V., et al. (2020). Omics-based exploration and functional validation of neurotrophic factors and histamine as therapeutic targets in ALS. Ageing Res. Rev. 62, 101121. doi: 10.1016/j.arr.2020.101121
von Neuhoff, N., Oumeraci, T., Wolf, T., Kollewe, K., Bewerunge, P., Neumann, B., et al. (2012). Monitoring CSF proteome alterations in amyotrophic lateral sclerosis: obstacles and perspectives in translating a novel marker panel to the clinic. PLoS One 7:e44401. doi: 10.1371/journal.pone.0044401
Wain, L. V., Pedroso, I., Landers, J. E., Breen, G., Shaw, C. E., Leigh, P. N., et al. (2009). The role of copy number variation in susceptibility to amyotrophic lateral sclerosis: genome-wide association study and comparison with published loci. PLoS One 4:e8175. doi: 10.1371/journal.pone.0008175
Wang, T., Zhang, J., and Xu, Y. (2020). Epigenetic basis of lead-induced neurological disorders. Int. J. Environ. Res. Public Health 17, 1–23. doi: 10.3390/ijerph17134878
Wang, X. S., Simmons, Z., Liu, W., Boyer, P., and Connor, J. (2006). Differential expression of genes in amyotrophic lateral sclerosis revealed by profiling the post mortem cortex. Amyotroph. Lateral Scler. 7, 201–216. doi: 10.1080/17482960600947689
Webster, C. P., Smith, E. F., Shaw, P. J., and De Vos, K. J. (2017). Protein homeostasis in amyotrophic lateral sclerosis: therapeutic opportunities? Front. Mol. Neurosci. 10:123. doi: 10.3389/fnmol.2017.00123
Weskamp, K., and Barmada, S. J. (2018). TDP43 and RNA instability in amyotrophic lateral sclerosis. Brain Res. 1693, 67–74. doi: 10.1016/j.brainres.2018.01.015
Wuolikainen, A., Andersen, P. M., Moritz, T., Marklund, S. L., and Antti, H. (2012). ALS patients with mutations in the SOD1 gene have an unique metabolomic profile in the cerebrospinal fluid compared with ALS patients without mutations. Mol. Genet. Metab. 105, 472–478. doi: 10.1016/j.ymgme.2011.11.201
Yang, B., Jiang, H., Wang, F., Li, S., Wu, C., Bao, J., et al. (2019). UNC13A variant rs12608932 is associated with increased risk of amyotrophic lateral sclerosis and reduced patient survival: a meta-analysis. Neurol. Sci. 40, 2293–2302. doi: 10.1007/s10072-019-03951-y
Yerbury, J. J., Farrawell, N. E., and McAlary, L. (2020). Proteome homeostasis dysfunction: a unifying principle in ALS pathogenesis. Trends Neurosci. 43, 274–284. doi: 10.1016/j.tins.2020.03.002
Yu, X. T., and Zeng, T. (2018). Integrative analysis of omics big data. Methods Mol. Biol. 1754, 109–135. doi: 10.1007/978-1-4939-7717-8_7
Zeng, P., and Zhou, X. (2019). Causal effects of blood lipids on amyotrophic lateral sclerosis: a Mendelian randomization study. Hum. Mol. Genet. 28, 688–697. doi: 10.1093/hmg/ddy384
Zetterberg, H., Jacobsson, J., Rosengren, L., Blennow, K., and Andersen, P. M. (2007). Cerebrospinal fluid neurofilament light levels in amyotrophic lateral sclerosis: impact of SOD1 genotype. Eur. J. Neurol. 14, 1329–1333. doi: 10.1111/j.1468-1331.2007.01972.x
Zhao, Z., Lange, D. J., Ho, L., Bonini, S., Shao, B., Salton, S. R., et al. (2008). Vgf is a novel biomarker associated with muscle weakness in amyotrophic lateral sclerosis (ALS), with a potential role in disease pathogenesis. Int. J. Med. Sci. 5, 92–99. doi: 10.7150/ijms.5.92
Zhu, S., Wuolikainen, A., Wu, J., Öhman, A., Wingsle, G., Moritz, T., et al. (2019). Targeted multiple reaction monitoring analysis of CSF identifies UCHL1 and GPNMB as candidate biomarkers for ALS. J. Mol. Neurosci. 69, 643–657. doi: 10.1007/s12031-019-01411-y
Ziff, O. J., and Patani, R. (2019). Harnessing cellular aging in human stem cell models of amyotrophic lateral sclerosis. Aging Cell 18:e12862. doi: 10.1111/acel.12862
Zubiri, I., Lombardi, V., Bremang, M., Mitra, V., Nardo, G., Adiutori, R., et al. (2018). Tissue-enhanced plasma proteomic analysis for disease stratification in amyotrophic lateral sclerosis. Mol. Neurodegener. 13:60. doi: 10.1186/s13024-018-0292-2
Zucchi, E., Ticozzi, N., and Mandrioli, J. (2019). Psychiatric symptoms in amyotrophic lateral sclerosis: beyond a motor neuron disorder. Front. Neurosci. 13:175. doi: 10.3389/fnins.2019.00175
Keywords: amyotrophic lateral sclerosis, ALS-FTD, personalized medicine, molecular taxonomy, multi-omics, systems biology
Citation: Morello G, Salomone S, D’Agata V, Conforti FL and Cavallaro S (2020) From Multi-Omics Approaches to Precision Medicine in Amyotrophic Lateral Sclerosis. Front. Neurosci. 14:577755. doi: 10.3389/fnins.2020.577755
Received: 29 June 2020; Accepted: 13 October 2020;
Published: 30 October 2020.
Edited by:
Ruth Luthi-Carter, University of Leicester, United KingdomReviewed by:
Janine Kirby, The University of Sheffield, United KingdomCopyright © 2020 Morello, Salomone, D’Agata, Conforti and Cavallaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastiano Cavallaro, c2ViYXN0aWFuby5jYXZhbGxhcm9AY25yLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.