 Ying Zhang1,2†
Ying Zhang1,2† Xinyang Xie1,2,3†Jiangnan Hu4Kazi Sabrina Afreen5
Xinyang Xie1,2,3†Jiangnan Hu4Kazi Sabrina Afreen5 Chun-Li Zhang6Qichuan Zhuge1,2*
Chun-Li Zhang6Qichuan Zhuge1,2* Jianjing Yang1,2*
Jianjing Yang1,2*- 1Zhejiang Provincial Key Laboratory of Aging and Neurological Disorder Research, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China
- 2Department of Neurosurgery, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China
- 3International Department of The Affiliated High School of South China Normal University (HFI), Guangzhou, China
- 4Department of Pharmaceutical Sciences, University of North Texas Health Science Center, Fort Worth, TX, United States
- 5Department of Microbiology & Immunology, Rosalind Franklin University of Medicine and Science, North Chicago, IL, United States
- 6Department of Molecular Biology, UT Southwestern Medical Center, Dallas, TX, United States
A reliable disease model is critical to the study of specific disease mechanisms as well as for the discovery and development of new drugs. Despite providing crucial insights into the mechanisms of neurodegenerative diseases, translation of this information to develop therapeutics in clinical trials have been unsuccessful. Reprogramming technology to convert adult somatic cells to induced Pluripotent Stem Cells (iPSCs) or directly reprogramming adult somatic cells to induced Neurons (iN), has allowed for the creation of better models to understand the molecular mechanisms and design of new drugs. In recent times, iPSC technology has been commonly used for modeling neurodegenerative diseases and drug discovery. However, several technological challenges have limited the application of iN. As evidence suggests, iN for the modeling of neurodegenerative disorders is advantageous compared to those derived from iPSCs. In this review, we will compare iPSCs and iN models for neurodegenerative diseases and their potential applications in the future.
Introduction
Neurodegenerative diseases comprised of a group of complicated disorders of the central nervous system among the aged population. To design effective treatment strategies to cure these diseases, scientists are in desperate need of convenient and reliable disease models. Previous neurodegenerative disease models based on genetic manipulations include transgene integration or gene knockout systems. These systems can only be utilized partially to understand disease mechanisms, pathology, and progression (Hargus et al., 2014; Heilker et al., 2014; Imaizumi and Okano, 2014; Zhao et al., 2014). These current models cannot be used as accurate models for neurodegenerative diseases especially due to specific limitations. First, although the fibroblasts or disease-associated mutation transformed cell lines of patients have enabled detailed mechanistic studies to be carried out, the biology of cell lines does not resemble the biology of primary neurons (Hargus et al., 2014). Thus it is often unclear whether the mechanisms studied are directly comparable to patients’ pathology. Second, animal models-such as dogs, flies, monkeys, and especially rodents (Zhao et al., 2014), is another method of studying neurodegenerative diseases (Gitler et al., 2017). However, these models often cannot accurately recapitulate human disease and animal models of the sporadic forms of neurodegenerative diseases due to species-specific differences. In addition, it is difficult to manipulate affected cell types in neurodegenerative disorders in vitro. Due to these limitations, a number of preclinical trials that aimed to identify drugs have failed to successfully translate into therapeutics in clinical settings (Kraljevic et al., 2004; Ledford, 2011; Ke et al., 2016). In summary, it is important to develop accurate and predictive disease models as they are essential to providing key insights to understanding disease mechanisms and the development of drugs to cure neurodegenerative diseases.
Innovations in cellular reprogramming technology have provided us with a promising tool to solve this problem. Takahashi and Yamanaka (2006) established a unique method of reprogramming somatic cells to iPSCs, which can be differentiated into cell types of all the three germ layers including non-proliferating neurons. The neurons derived from iPSCs would have the same genetic information as the individual patient and can be differentiated from iPSCs. This technology has been utilized by other investigators for neurodegenerative disease modeling (Table 1; Wan et al., 2015; Haston and Finkbeiner, 2016; Liu and Deng, 2016; Csobonyeiova et al., 2017). Moreover, in recent years, the discovery of direct reprogramming technology has enabled the reprogramming of somatic cells to neurons, bypassing the iPSC stage (Vierbuchen et al., 2010; Ambasudhan et al., 2011; Li et al., 2015, 2017; Karow et al., 2018; Tanabe et al., 2018; Xiao et al., 2018). With the advancement of these technologies, scientists have been able to create highly efficient and lineage-specific neurons through the reprogramming of somatic cells (Marro et al., 2011; Xu Z. et al., 2015; Black et al., 2016; Mall et al., 2017). Altogether, these technologies can be used for modeling neurodegenerative diseases (Shi et al., 2017; Sun et al., 2017; Han et al., 2018; Farkhondeh et al., 2019).
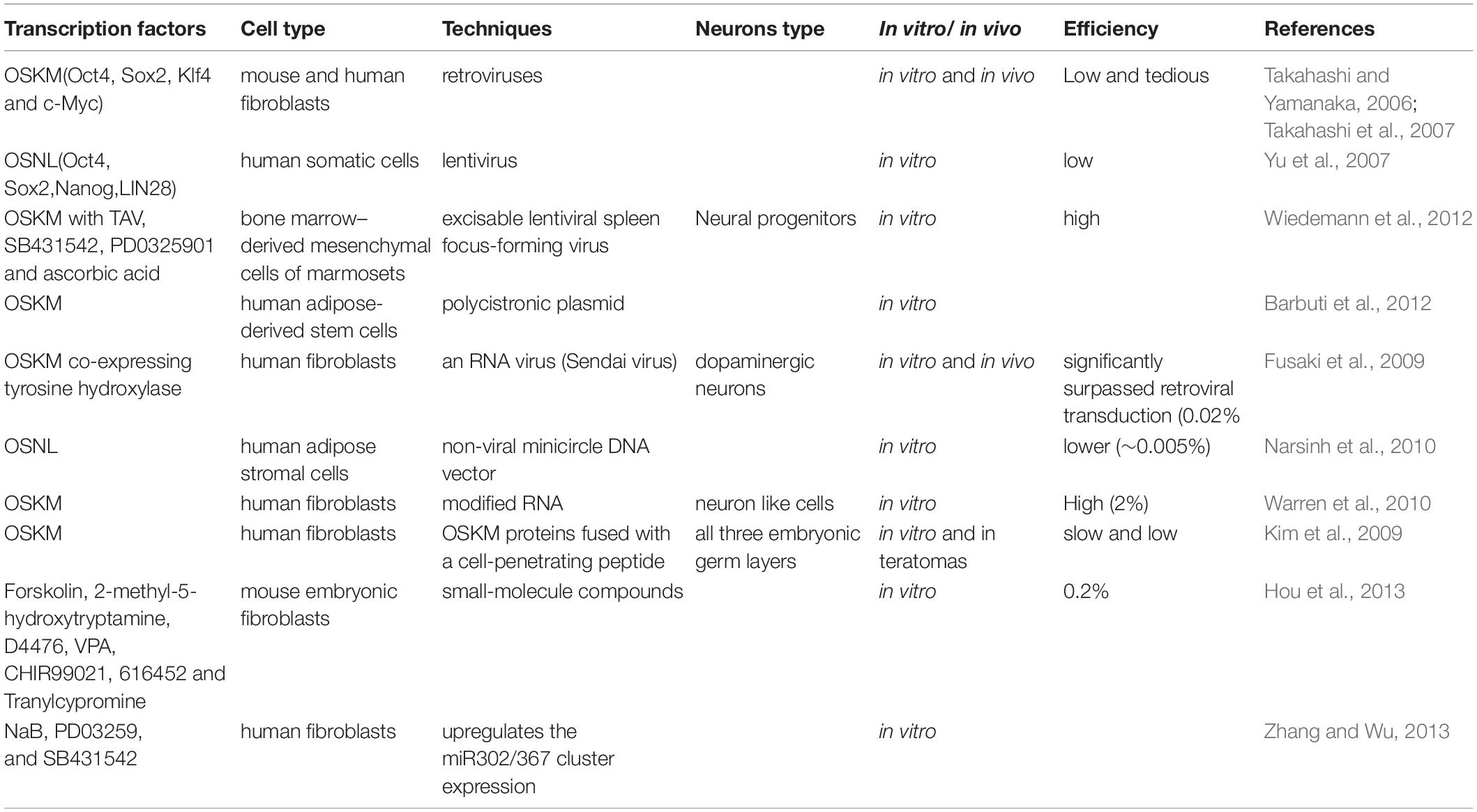
Table 1. Neurons derived from iPSC technology.
Even though, the mechanisms of iPSCs or iN reprogramming are still unclear (Xu J. et al., 2015; Omole and Fakoya, 2018), there are some obvious differences between iPSCs-derived neurons and iN. Among them, recent studies have indicated that the application of iN for aging-related neurodegenerative diseases would be a better choice, as it does not reset aging information (Mertens et al., 2015, 2018; Tang et al., 2017; Bohnke et al., 2018; Traxler et al., 2019). In this review, we summarize recent studies involving iPSCs and in neurodegenerative disease modeling and its advantages and limitations.
Reprogramming Somatic Cells to Neuron Cells
iPSC Technology
In 2006, a phenomenal study conducted in Yamanaka lab demonstrated that viral vectors carrying a combination of pluripotent transcription factors, including Oct4, Sox2, Klf4, and c-Myc (OSKM), were sufficient to effectively reprogram mouse fibroblasts cells to iPSCs (Takahashi and Yamanaka, 2006; Okano and Yamanaka, 2014). In 2007, their laboratory also demonstrated that OSKM could reprogram human fibroblasts to iPSCs by the retroviral system (Takahashi et al., 2007). The generated iPSCs had the potential to be differentiated into all three germ layers of cell type with the unlimited ability of self-renewal. Besides OSKM, the combination of other transcription factors, including Oct4, Sox2, Nanog, and LIN28, has also been demonstrated to be able to convert human somatic cells into iPSCs with a lentiviral system (Yu et al., 2007). In addition, this technology has been successfully used for translating into other somatic cell types, such as neural stem cells (Eminli et al., 2008; Kim et al., 2008), stomach and liver cells (Aoi et al., 2008), mature β lymphocytes (Hanna et al., 2008), melanocytes (Utikal et al., 2009), adipose stem cells (Sun et al., 2009), and keratinocytes (Maherali et al., 2008). iPSC technology provides a platform that can be used as a model system for neurodegenerative diseases to design new therapeutics. However, the current iPSC technology still has some limitations, including low efficiency and a long reprogramming process, which are primarily due to the existence of several roadblocks (Ebrahimi, 2015; Haridhasapavalan et al., 2020). Another problem is that iPSCs may cause cancerous tumor formation due to an undifferentiated pluripotent stem cell after transplantation (Choi and Hong, 2017). In recent years, researchers put tremendous efforts into refining and optimizing approaches to improve reprogramming efficiency and safety (O’Malley et al., 2009; Sommer and Mostoslavsky, 2010, 2013; Gonzalez et al., 2011; Morris and Daley, 2013; Omole and Fakoya, 2018; Borgohain et al., 2019; Haridhasapavalan et al., 2019). Maherali et al. (2008) created a doxycycline-inducible lentiviral system including OSKM, which had a higher frequency of converting primary fibroblasts into iPSCs. This system could even reprogram keratinocytes into iPSCs within 10 days (Maherali et al., 2008). In addition, using lentivirus or retrovirus to deliver OSKM may cause insertional mutagenesis when integrating gene sequences in the genomic DNA of the cells. To improve technical safety, other delivery methods, including non-viral or non-integrating viral vectors, have been attempted, such as protein transduction, the transfection of modified mRNA transcripts, small molecules, sendai virus, and episomal vectors (Sommer and Mostoslavsky, 2010, 2013; Gonzalez et al., 2011; Morris and Daley, 2013; Omole and Fakoya, 2018; Borgohain et al., 2019; Haridhasapavalan et al., 2019). However, compared to the traditional viral gene delivery method, these alternative methods had poorer outcomes.
iN Technology
After the establishment of iPSC reprogramming technology, researchers are continuously seeking effective ways to improve the reprogramming condition. The main challenge is to rapidly and efficiently change cell fate by reprogramming using minimal transcription factors. In 2010, Vierbuchen and his group succeeded in directly reprogramming mouse fibroblasts to functional neurons by overexpression of three transcription factors, including Ascl1, Brn2, and Myt1l (Vierbuchen et al., 2010). Subsequently, several studies showed some other transcription factors (Ngn2, Ascl1, and Dlx2) also could convert mouse postnatal astrocytes into both GABAergic and cholinergic neurons (Berninger et al., 2007; Heinrich et al., 2011; Xiao et al., 2018; Huang et al., 2019; Wazan et al., 2019). Moreover, only one transcription factor NGN2, when supplemented with chemicals including dorsomorphin and forskolin, could directly reprogram human fibroblasts (MRC5) to neurons (Liu et al., 2013) with high efficiency. The neurons generated are functional and mostly cholinergic neurons (Liu et al., 2013). Only epigenetic chemicals without transcription factors have been demonstrated to directly reprogram human and mouse fibroblasts into functional neuron cells (Hu et al., 2015; Li et al., 2015, 2017; Smith et al., 2016; Qin et al., 2017). Other studies have shown that some defined tissue-specific transcription factors (TFs), such as Sox2, Zfp521 (a single zinc-finger TF), and Ptfa1, directly reprogram human fibroblasts into a neural stem cell (Maucksch et al., 2013; Shahbazi et al., 2016; Xiao et al., 2018). In addition, in Rubio et al. (2016) used the CRISPR/Cas9 platform to inactivate two neurological disorder genes, TSC2 and KCNQ2 and subsequently combined with a multicistronic lentivirus expressing the Ascl1, Lmx1a, and Nurr1 genes to directly convert fibroblasts into neuropathological-resistant neuronal cells. Although several other cell types can also be reprogrammed into neurons, like hepatocytes and pericytes cells (Marro et al., 2011; Karow et al., 2012), fibroblasts are still the most popular original cell type for reprogramming. Together these findings supported that iN can be directly derived from different cell types by certain combinations of transcription factors (Table 2). This technology for the generation of iN from other cell types could be useful for the development of neurological disease models (Ruggieri et al., 2014; Gascon et al., 2017; Gao et al., 2019; Pereira et al., 2019).
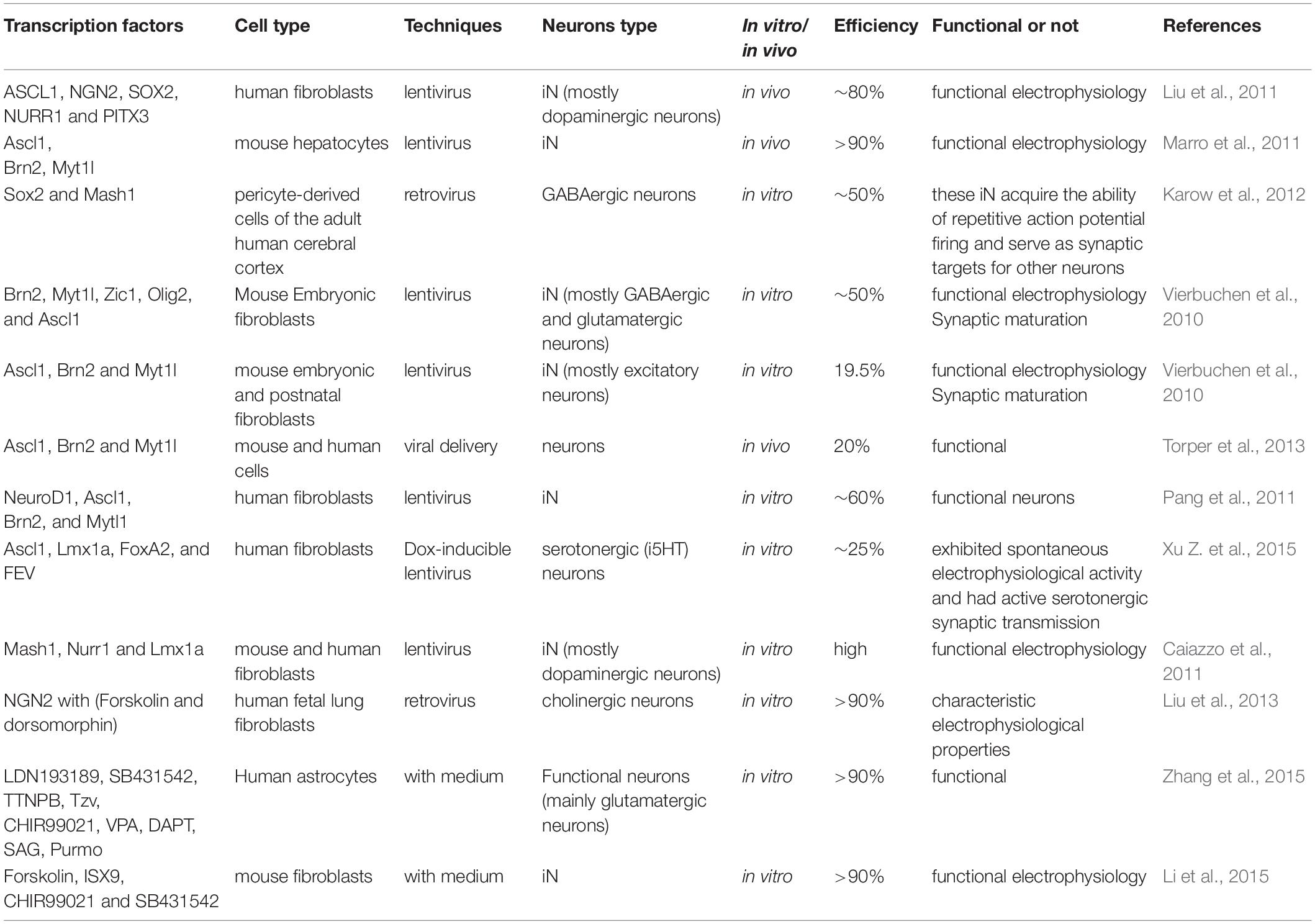
Table 2. Neurons derived from direct reprogramming technology.
Direct Reprogramming to Generate Specific Neuronal Subtypes
During the early stages, just after the discovery of direct reprogramming technology, investigators paid more attention to the efficacy of reprogramming and whether the neurons generated are physiologically functional. Subsequently, investigators tried to control the reprogramming process to convert somatic cells to specific neuronal subtypes. Reprogramming somatic cells into defined neuronal subtypes is a crucial step for the application of iN reprogramming technology into clinical trials. In recent years, technical improvements in this field have made substantial progress, which would dramatically increase the applications of iN technology.
Dopaminergic Neurons
Parkinson’s disease is a neurodegenerative disorder with progressive loss of dopaminergic neurons in the midbrain (Alexander, 2004). Thus, using reprogramming technology for the generation of defined dopaminergic neurons could be an interesting approach for the treatment of Parkinson’s disease. According to neuronal system development, several transcription factors play a critical role in the generation and specification of dopaminergic neurons, including Otx2, FoxA1/2 Lmx1a/b, Ascl1, Ngn2, Pitx3, and Nurr1 (Nr4a2) (Arenas et al., 2015). Several studies have reported the successful reprogramming of fibroblasts or astrocytes into induced dopaminergic (iDA) neurons. Among them, the minimal combination is Ascl1, Nurr1, and Lmx1a (Kim et al., 2011; Pfisterer et al., 2011; Torper et al., 2013; Caiazzo et al., 2015). The iDA neurons that are generated are functional, can produce dopamine, and have firing of action potentials and functional D2 auto receptors (Caiazzo et al., 2011). Moreover, transplantation of these functional iDA neurons could improve the behavior deficit caused by the loss of endogenous DA neurons (Dell’Anno et al., 2014). De Gregorio et al. (2018) found that, when combined with transcription factors ASCL1 and NURR1, miR-34b/c could double the yield of transdifferentiated fibroblasts into dopaminergic neurons. The iDA neurons that are generated synthesize dopamine and showed spontaneous electrical activity and are reversibly blocked by tetrodotoxin, which is consistent with the electrophysiological properties featured by brain dopaminergic neurons (De Gregorio et al., 2018).
Spinal Motor Neurons
Genetic disorders like Amyotrophic Lateral Sclerosis (ALS) result in the loss of motor neurons (Robberecht and Philips, 2013). Regeneration of new motor neurons is important for potential therapy and disease models for ALS. Studies on mouse models have demonstrated that reprogramming of mouse embryonic fibroblasts into induced motor neurons (iMN) could be achieved by combined overexpression of common transcription factors [Ascl1, Neurog2, Myt1l, and Brn2 (Pou3f2)] with some specific TFs (Lhx3, Isl1, and Hb9) for spinal cord motor neurons (Lee et al., 2009; Son et al., 2011; Tang et al., 2017; Zhang et al., 2017). These iMNs could survive after being transplanted into the spinal cord and are capable of forming a neuromuscular junction with myotube cells in vitro (Son et al., 2011). To optimize the reprogramming condition, four TFs (Neurog2, Sox11, LHX3, and Isl1), when supplemented with forskolin, dorsomorphin, and FGF2 could directly reprogram human fibroblasts into motor neurons, which are HB9 and ChAT-positive, have action potentials and can form a neuromuscular junction with extremely high efficiency (>80%) (Liu et al., 2016).
GABAergic Neurons (Interneurons)
The GABAergic neurons are inhibitory interneurons located in the cortex, which play crucial roles in regulating the excitation and inhibition of nervous system activation (Tremblay et al., 2016). The loss or malfunction of GABAergic neurons would also result in neurological diseases, such as epilepsies, cognitive disorders, autism, schizophrenia, and intellectual disabilities (Woo and Lu, 2006; Brooks-Kayal, 2010; Marin, 2012). Colasante et al. (2015) demonstrated the use of five TFs (Foxg1, Ascl1, Sox2, Dlx5, and Lhx6) for reprogramming human and mouse fibroblasts into induced GABA (iGABA) interneurons. The generated iGABA interneurons could survive and mature after being transplanted into the hippocampus (Colasante et al., 2015). The new iGABA interneurons can form functional synapses, and release GABA (Colasante et al., 2015). Importantly, the transplanted iGABA interneurons can integrate into host circuitry and play inhibitory functions (Colasante et al., 2015). A great part of the GABAergic neurons also showed Parvalbumin (PV) protein and gene expression. Soon after, another research group obtained induced PV (iPV) neurons by Ascl1 from mouse fibroblasts (Shi et al., 2016). These reports showed that the controlled reprogramming process by some specific regional TFs would lead to lineage reprogramming of neuronal subtypes (Masserdotti et al., 2016).
iPSCs Application for Neurodegenerative Diseases Modeling and Drug Discovery
Neurodegenerative diseases including Alzheimer’s Disease (AD), Parkinson’s Disease (PD) and Amyotrophic Lateral Sclerosis (ALS) are aging-related disorders in which several genetic mutations have been identified before the onset of the diseases. However, even with a clearer understanding of the mechanisms of neurodegenerative diseases, the progression of designing therapy is going slow (Finkbeiner, 2010; Mason et al., 2014; Wyss-Coray, 2016). Based on these genetic mutations, different animal models have been established to study the underlying disease mechanisms and explore the potential drugs for treatment. Unfortunately, due to the variations among different species and the irreproducibility of human disease pathology, current animal models cannot ideally model neurodegenerative diseases as the data generated from these models cannot be successfully translated into clinical applications (Jucker, 2010; Imaizumi and Okano, 2014; Mitsumoto et al., 2014). In this scenario, iPSC technology brought new hope for neurodegenerative disease modeling and drug discovery in vitro. Nowadays, iPSC technology has been widely applied for disease modeling, mechanism study, and the screening of drugs for neurodegenerative diseases (Figure 1).
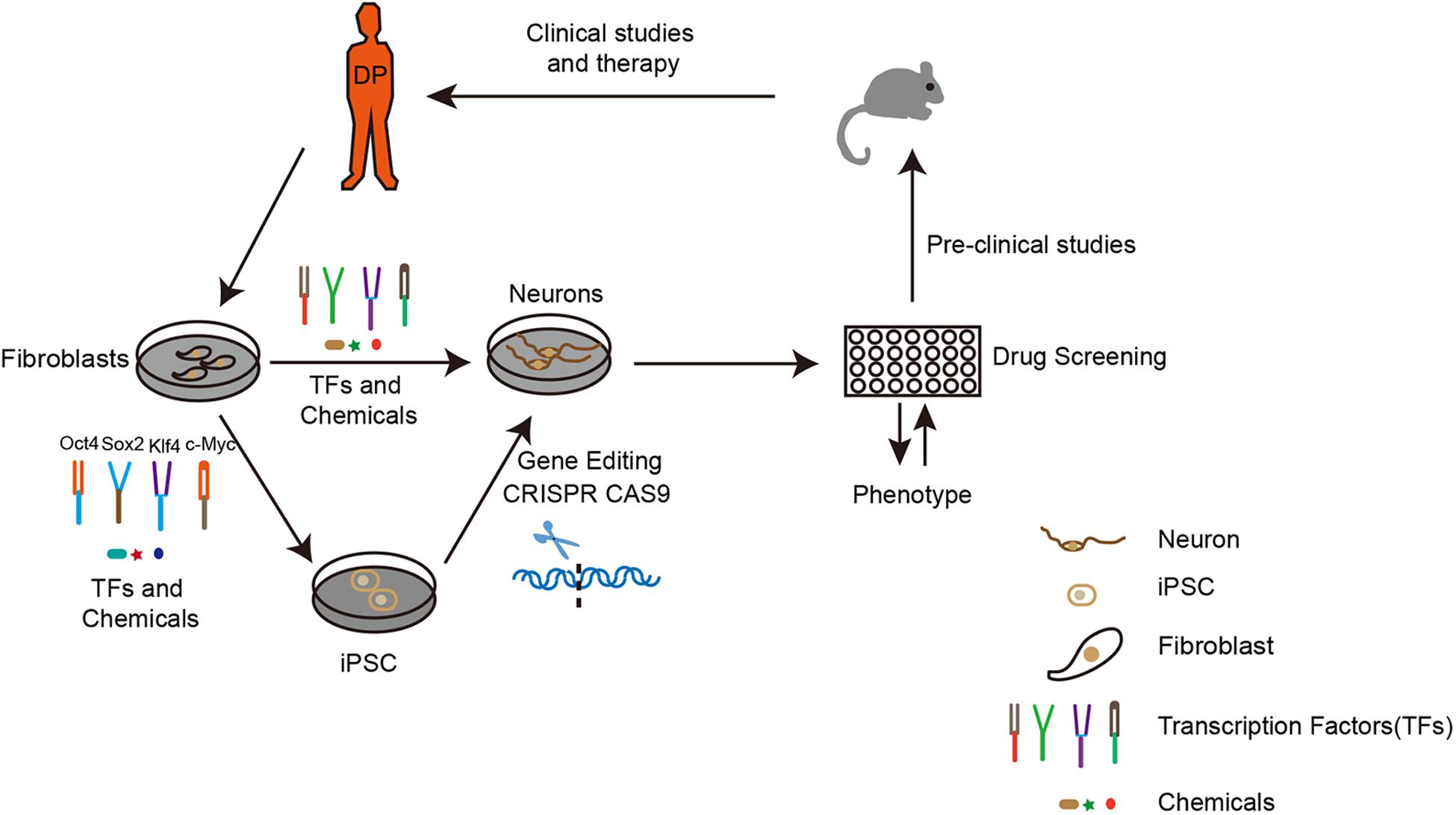
Figure 1. The route to apply iPSCs and iN technology for neurodegenerative disease modeling and drug discovery. To establish an in vitro disease model, the first step is to obtain fibroblasts from a diseased person. Upon overexpression of certain transcription factors, fibroblasts can be directly or indirectly reprogrammed to neurons. Depending on research purposes, the fibroblasts from original iPSCs can be modified by gene editing (CRISPR CAS9). The neurons generated would be applied for drug screening according to the disease phenotype. Subsequently, the best candidates could then be used for pre-clinical studies on drug toxicity, bio-availability, pharmacology, and metabolism in animals. Finally, the potential drug would be used for clinical studies and therapy. DP: disease person.
Alzheimer’s Disease (AD)
Alzheimer’s Disease is the most common chronic progressive neurodegenerative disease. In recent decades, researchers have focused on the study of the pathogenesis of AD. Several genetic mutations have been identified in genes namely, APP, presenilin 1/2 (PS1/2), and APOE, to cause Familial Alzheimer’s disease (Armstrong, 2013; Karch et al., 2014; Shen, 2014; Moreno et al., 2018; Wang et al., 2018). However, the mechanisms of the neurons and synapse damage in AD remain unclear. The new iPSC technology from AD patients can provide sufficient quantity or quality neurons for the discovery of potential therapeutics (Byrne, 2014; Tcw, 2019).
In recent years, researchers have been successful in reprogramming fibroblasts carrying with different genetic mutations to iPSCs. Yagi et al. (2011) pioneered the use of iPSC technology to establish an in vitro model for AD, which was derived from iPSCs with PS1/2 mutation. The expression of Aβ42 was dramatically increased in neurons derived from iPSCs (Yagi et al., 2011). In addition to this, the APP gene mutation has also been investigated by Israel et al. (2012). They demonstrated that the levels of Aβ42 and tau are significantly increased in neurons derived from iPSCs (Israel et al., 2012; Ochalek et al., 2017). Subsequently, studies conducted by several groups of investigators used iPSCs to produce neurons derived to model AD where the properties of pathogenic Aβ42 and tau were reserved (Shi et al., 2012; Duan et al., 2014; Muratore et al., 2014; Sproul et al., 2014; Chang et al., 2015; Moore et al., 2015; Rowland et al., 2018; Tcw, 2019). In summary, the novel iPSCs in vitro model can be utilized as an excellent tool to study AD.
Parkinson’s Disease (PD)
The loss of dopaminergic neurons in the neuropathology of PD, which causes motor problems, including bradykinesia, resting tremor, rigidity, flexed posture, “freezing,” and lose of postural reflexes (Seibler et al., 2011; Postuma et al., 2015). Similar to AD, the deficit of reliable in vitro models has limited the progression of drug discovery for PD. Several groups obtained iPSCs from patient somatic cells with different genetic mutations including LRRK2, SNCA, PARK2, or PINK1, which are related to familial PD, and the DA neurons derived from iPSCs has been used to investigate the molecular mechanisms (Ke et al., 2019). The dopaminergic neurons derived from LRRK2 iPSCs have some important PD features, including (α-Syn aggregates, overexpression of oxidative stress genes, lower number of neurites, and caspase-3 activation (Nguyen et al., 2011; Sanchez-Danes et al., 2012). Importantly, after correction of LRRK2 mutation in iPSCs, they can rescue the pathogenic phenotypes of neurite shortening and mitochondrial DNA damage (Reinhardt et al., 2013; Sanders et al., 2014). iPSCs derived from PD patients, who received triplication of the SNCA gene, have also been shown to have PD pathogenic neuron properties (Oliveira et al., 2015). PARK2 gene mutation has been shown to play a critical role in neuron morphology by iPSCs-derived neuron model (Imaizumi et al., 2012; Ren et al., 2015). In addition, (Seibler et al., 2011) also reported that iPSCs reprogrammed from PD patients’ fibroblasts with PINK1 mutations can generate DA neurons. The new DA neurons showed properties of upregulation of PGC-1α, which can be reversed after overexpression of wild-type PINK1 in new DA neurons. Together, all these studies demonstrated that iPSCs is a better in vitro model for PD with genetic mutations.
Amyotrophic Lateral Sclerosis (ALS)
Induced Pluripotent Stem Cells technology also has been widely applied for ALS. The pathology of ALS includes the progressive loss of motor neurons in the brain and spinal cord. Several genes have been identified to be associated with ALS, such as SOD1, C9orf2, and TDP-43 (Rosen et al., 1993; Sreedharan et al., 2008; DeJesus-Hernandez et al., 2011). Among them, the SOD1 gene mutation is the most studied genetic alteration in ALS. Compare to wild type SOD1, motor neurons (MN) derived from SOD1 mutated patients’ iPSCs showed the features of decreased survival rate, smaller soma size, and shorter neurite (Chen et al., 2014; Kiskinis et al., 2014). In addition, MN derived from SOD1 mutated iPSCs showed impaired mitochondrial function and increased oxidative stress (Chen et al., 2014). Importantly, the correction of the SOD1 mutation could rescue these phenotypes in iPSCs (Chen et al., 2014; Kiskinis et al., 2014). iPSC-derived motor neurons retaining the patients’ full genetic information, therefore, scientists established a large number of in vitro cellular models for sporadic ALS. The sufficient utility of sporadic ALS models is useful for elucidating the pathological characteristics of specific cases and identifying novel candidate drugs (Fujimori et al., 2018). On the other hand, many investigators have studied the phenotypes of MN derived from C9orf72 mutant iPSCs (Donnelly et al., 2013; Sareen et al., 2013; Devlin et al., 2015; Dafinca et al., 2016). Abnormalities of electrophysiology, calcium homeostasis, ER stress, and mitochondrial membrane potential have been identified in MN from iPSCs carrying C9orf72 mutation (Devlin et al., 2015; Dafinca et al., 2016). In addition, the C9orf72 mutant has been demonstrated to cause oxidative and neurotoxicity in MN from iPSCs (Donnelly et al., 2013; Sareen et al., 2013; Birger et al., 2019).
iN for Neurodegenerative Disease Modeling and Drug Discovery
With a specific combination of reprogramming factors, somatic cells can be directly converted into neurons bypassing the iPSC stage. Along with the advancement of direct reprogramming technology, the new generation of iN has also been applied for modeling neurogenerative diseases and drug discovery (Figure 1). Liu et al. (2016) have used direct reprogramming technology through using a combination of TFs and small molecules and efficiently reprogrammed ALS patients’ fibroblasts to motor neurons with FUS gene mutation. The new iMN from ALS patients was unable to form neuromuscular junctions with muscle cells. Moreover, after the chemical screening, they found the chemical kenpaullone can rescue the disease phenotype. Recently, (Chang et al., 2018) utilized the mesoporous silica nanoparticles (MSNs) as a non-viral delivery system for the transduction of the three key factors to achieve the conversion of mouse fibroblasts (MFs) into functional dopaminergic neuron-like cells. These recent studies are the beginning of developments that will enable us to apply iN for neurodegenerative disease modeling and drug discovery. Before applying this technique for large scale drug screening, the problems associated with efficiency and the homogeneity of direct reprogramming needs to be further improved.
Comparison of iPSCs-Derived Neurons to iN
In contrast to the application of iPSC technology, the application of the iN approach is new and emerging in the field of neurodegenerative diseases. Like any technique, iN technology has some obvious advantages and disadvantages (Table 3). Because direct reprogramming does not involve the iPSC stage and the differentiation step, which saves a lot of time, iPSC technology may take several months, depending on the protocol. In addition, the technical challenges of iN are less compared to iPSCs culture technology. The most important difference between iPSCs and iN is epigenetic reset.

Table 3. The different features between iN and iPSCs-derived neurons.
As we know, a healthy and diseased person not only differs in genomic levels but also has different epigenetics. Epigenetic information is crucial for disease onset, especially for aging-related diseases. A recent study conducted by Tang et al. (2017) found iPSCs derived motor neurons did not show age-related differences, while iN, in contrast, age-equivalent induced motor neurons showed nuclear envelope defects. Mertens et al. (2015) provided interesting evidence for iN as it can reserve aging signatures of the original patient, which is not observed in iPSCs. Furthermore, they have also found downregulation of RanBP1 in aged fibroblasts and iN derived from aged fibroblasts, and when RanBP1 was knocked down, the transcriptional markers shifted from young to aged (Mertens et al., 2015). Therefore, iN is a more reliable model for neurodegenerative diseases and drug discovery, which could model natural disease progression, especially age-related information. On the other hand, iPSCs can maintain self-renewal but not iN, which is required for maintenance and stock. Due to the unlimited self-renewal of iPSCs, the neurons derived from iPSCs can be unlimited. Thus, without an iPSC stage, investigators might need to acquire a larger quantity of original cells from a patient to obtain enough iN. In addition, identification of the right combination of transcription factors, the inclusion of chemical compounds (small molecules), and the efficiency of reprogramming are also very important. However, to realize the application of iN in neurodegenerative diseases, the underlying mechanisms of direct reprogramming need to be further addressed.
Conclusion and Perspective
In conclusion, this review has discussed recent iPSCs and iN technology and their application for neurodegenerative disease modeling. Compared them to traditional disease models, both iPSCs and iN are more accurate models for studying diseases and drug discovery. For iPSCs and iN disease models, there are still some challenges that need to be further investigated to optimize reprogramming conditions, especially the efficiency of direct reprogramming and lineage-specific reprogramming. For the modeling of neurodegenerative diseases, iN could be a better model for disease and the development of drugs, without epigenetic reset. In the coming years, we expect there to be extensive improvements in reprogramming technology for the application of iPSCs and iN for disease modeling and drug discovery.
Author Contributions
YZ, XX, and JY performed the literature research and wrote the manuscript. C-LZ and QZ proposed the framework. JH and KA critically revised the manuscript. QZ and JY provided the funding support. All authors read and approved the manuscript.
Funding
This study was partly supported by the National Natural Science Foundation of China (No. 81771262), the Zhejiang Health Science and Technology Project (2016RCA022), Zhejiang Key Research and Development Project (2017C03027), and Zhejiang Provincial Natural Science Foundation of China (LQ20H090005).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ABM, Ascl1, Brn2, and Myt1l; AD, Alzheimer’s Disease; ALS, Amyotrophic Lateral Sclerosis; ANL, Ascl1, Nurr1, and Lmx1a; APOE, apolipoprotein E; APP, amyloid beta precursor protein; ASCL1, achaete-scute family BHLH transcription factor 1; Brn2, POU domain, class 3, transcription factor 2; Dlx2, distal-less homeobox 2; Dlx5, distal-less homeobox 5; FGF2, fibroblasts growth factor 2; FoxA1/2, forkhead box A1/2; Foxg1, forkhead box G1; Hb9, motor neurons and pancreas homeobox 1; iDA, induced dopaminergic; iMN, induced motor neurons; iN, induced neurons; iPSCs, induced pluripotent stem cells; Isl1, ISL LIM homeobox 1; Klf4, Kruppel-like factor 4; Lhx3, LIM homeobox protein 3; Lhx6, LIM homeobox protein 6; Lmx1a/b, LIM homeobox transcription factor 1 a/b; LRRK2, leucine rich repeat kinase 2; Myt1l, myelin transcription factor 1 like; Ngn2, neurogenin 2; Nurr1, (Nr4a2), nuclear receptor subfamily 4, group A, member 2; Oct4, octamer-binding transcription factor 4; OHDA, hydroxydopamine; OSKM, Oct4, Sox2, Klf4, and c-Myc, otx2, orthodenticle homeobox 2; PARK2, parkin RBR E3 ubiquitin protein ligase; PD, Parkinson’s Disease; PGC-1 α, PPARG coactivator 1 alpha; PINK1, PTEN induced putative kinase 1; Pitx3, paired-like homeodomain transcription factor 3; PS1/2, presenilin 1/2; PV, Parvalbumin; RanBP1, RAN binding protein 1; SNCA, synuclein alpha; SOD1, superoxide dismutase 1; Sox11, SRY (sex determining region Y)-box 11; Sox2, SRY (sex determining region Y)-box 2; TDP-43, TAR DNA binding protein; TFs, transcription factors; Zfp521, Zinc Finger Protein 521.
References
Alexander, G. E. (2004). Biology of Parkinson’s disease: pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialog. Clin. Neurosci. 6, 259–280.
Ambasudhan, R., Talantova, M., Coleman, R., Yuan, X., Zhu, S., Lipton, S. A., et al. (2011). Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 9, 113–118. doi: 10.1016/j.stem.2011.07.002
Aoi, T., Yae, K., Nakagawa, M., Ichisaka, T., Okita, K., Takahashi, K., et al. (2008). Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science 321, 699–702. doi: 10.1126/science.1154884
Arenas, E., Denham, M., and Villaescusa, J. C. (2015). How to make a midbrain dopaminergic neuron. Development 142, 1918–1936. doi: 10.1242/dev.097394
Armstrong, R. (2013). Review article What causes alzheimer’s disease? Folia Neuropathol. 3, 169–188. doi: 10.5114/fn.2013.37702
Barbuti, A., Qu, X., Liu, T., Song, K., Li, X., and Ge, D. (2012). Induced pluripotent stem cells generated from human adipose-derived stem cells using a non-viral polycistronic plasmid in feeder-free conditions. PLoS One 7:e48161. doi: 10.1371/journal.pone.0048161
Berninger, B., Costa, M. R., Koch, U., Schroeder, T., Sutor, B., Grothe, B., et al. (2007). Functional properties of neurons derived from in vitro reprogrammed postnatal astroglia. J. Neurosci. 27, 8654–8664. doi: 10.1523/JNEUROSCI.1615-07.2007
Birger, A., Ben-Dor, I., Ottolenghi, M., Turetsky, T., Gil, Y., Sweetat, S., et al. (2019). Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. eBio Med. 50, 274–289. doi: 10.1016/j.ebiom.2019.11.026
Black, J. B., Adler, A. F., Wang, H. G., D’Ippolito, A. M., Hutchinson, H. A., Reddy, T. E., et al. (2016). Targeted epigenetic remodeling of endogenous loci by CRISPR/Cas9-based transcriptional activators directly converts fibroblasts to neuronal cells. Cell Stem Cell 19, 406–414. doi: 10.1016/j.stem.2016.07.001
Bohnke, L., Traxler, L., Herdy, J. R., and Mertens, J. (2018). Human neurons to model aging: a dish best served old. Drug Discov. Today Dis. Models 27, 43–49. doi: 10.1016/j.ddmod.2019.01.001
Borgohain, M. P., Haridhasapavalan, K. K., Dey, C., Adhikari, P., and Thummer, R. P. (2019). An insight into DNA-free reprogramming approaches to generate integration-free induced pluripotent stem cells for prospective biomedical applications. Stem Cell Rev. Rep. 15, 286–313. doi: 10.1007/s12015-018-9861-6
Brooks-Kayal, A. (2010). Epilepsy and autism spectrum disorders: are there common developmental mechanisms? Brain Dev. 32, 731–738. doi: 10.1016/j.braindev.2010.04.010
Byrne, J. A. (2014). Developing neural stem cell-based treatments for neurodegenerative diseases. Stem Cell Res. Ther. 5:72. doi: 10.1186/scrt461
Caiazzo, M., Dell’Anno, M. T., Dvoretskova, E., Lazarevic, D., Taverna, S., Leo, D., et al. (2011). Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476, 224–227. doi: 10.1038/nature10284
Caiazzo, M., Giannelli, S., Valente, P., Lignani, G., Carissimo, A., Sessa, A., et al. (2015). Direct conversion of fibroblasts into functional astrocytes by defined transcription factors. Stem Cell Rep. 4, 25–36. doi: 10.1016/j.stemcr.2014.12.002
Chang, C. Y., Chen, S. M., Lu, H. E., Lai, S. M., Lai, P. S., Shen, P. W., et al. (2015). N-butylidenephthalide attenuates Alzheimer’s disease-like cytopathy in Down syndrome induced pluripotent stem cell-derived neurons. Sci. Rep. 5:8744. doi: 10.1038/srep08744
Chang, J. H., Tsai, P. H., Wang, K. Y., Wei, Y. T., Chiou, S. H., and Mou, C. Y. (2018). Generation of functional dopaminergic neurons from reprogramming fibroblasts by nonviral-based mesoporous silica nanoparticles. Sci. Rep. 8:11. doi: 10.1038/s41598-017-18324-8
Chen, H., Qian, K., Du, Z., Cao, J., Petersen, A., Liu, H., et al. (2014). Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 14, 796–809. doi: 10.1016/j.stem.2014.02.004
Choi, K. A., and Hong, S. (2017). Induced neural stem cells as a means of treatment in Huntington’s disease. Expert Opin. Biol. Ther. 17, 1333–1343. doi: 10.1080/14712598.2017.1365133
Colasante, G., Lignani, G., Rubio, A., Medrihan, L., Yekhlef, L., Sessa, A., et al. (2015). Rapid conversion of fibroblasts into functional forebrain GABAergic interneurons by direct genetic reprogramming. Cell Stem Cell 17, 719–734. doi: 10.1016/j.stem.2015.09.002
Csobonyeiova, M., Polak, S., Nicodemou, A., and Danisovic, L. (2017). Induced pluripotent stem cells in modeling and cell-based therapy of amyotrophic lateral sclerosis. J. Physiol. Pharmacol. 68, 649–657.
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34, 2063–2078. doi: 10.1002/stem.2388
De Gregorio, R., Pulcrano, S., De Sanctis, C., Volpicelli, F., Guatteo, E., von Oerthel, L., et al. (2018). miR-34b/c regulates Wnt1 and enhances mesencephalic dopaminergic neuron differentiation. Stem Cell Rep. 10, 1237–1250. doi: 10.1016/j.stemcr.2018.02.006
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Dell’Anno, M. T., Caiazzo, M., Leo, D., Dvoretskova, E., Medrihan, L., Colasante, G., et al. (2014). Remote control of induced dopaminergic neurons in parkinsonian rats. J. Clin. Invest. 124, 3215–3229. doi: 10.1172/JCI74664
Devlin, A. C., Burr, K., Borooah, S., Foster, J. D., Cleary, E. M., Geti, I., et al. (2015). Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 6:5999. doi: 10.1038/ncomms6999
Donnelly, C. J., Zhang, P.-W., Pham, J. T., Haeusler, A. R., Mistry, N. A., Vidensky, S., et al. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428. doi: 10.1016/j.neuron.2013.10.015
Duan, L., Bhattacharyya, B. J., Belmadani, A., Pan, L., Miller, R. J., and Kessler, J. A. (2014). Stem cell derived basal forebrain cholinergic neurons from Alzheimer’s disease patients are more susceptible to cell death. Mol. Neurodegener. 9:3. doi: 10.1186/1750-1326-9-3
Ebrahimi, B. (2015). Reprogramming barriers and enhancers: strategies to enhance the efficiency and kinetics of induced pluripotency. Cell Regen. 4:10. doi: 10.1186/s13619-015-0024-9
Eminli, S., Utikal, J., Arnold, K., Jaenisch, R., and Hochedlinger, K. (2008). Reprogramming of neural progenitor cells into induced pluripotent stem cells in the absence of exogenous Sox2 expression. Stem Cells 26, 2467–2474. doi: 10.1634/stemcells.2008-0317
Farkhondeh, A., Li, R., Gorshkov, K., Chen, K. G., Might, M., Rodems, S., et al. (2019). Induced pluripotent stem cells for neural drug discovery. Drug Discov. Today 24, 992–999. doi: 10.1016/j.drudis.2019.01.007
Finkbeiner, S. (2010). Bridging the Valley of death of therapeutics for neurodegeneration. Nat. Med. 16, 1227–1232. doi: 10.1038/nm.2222
Fujimori, K., Ishikawa, M., Otomo, A., Atsuta, N., Nakamura, R., Akiyama, T., et al. (2018). Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 24, 1579–1589. doi: 10.1038/s41591-018-0140-5
Fusaki, N., Ban, H., Nishiyama, A., Saeki, K., and Hasegawa, M. (2009). Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn Acad. Ser. B 85, 348–362. doi: 10.2183/pjab.85.348
Gao, L., Huang, S., Zhang, H., Hua, W., Xin, S., Cheng, L., et al. (2019). Suppression of glioblastoma by a drug cocktail reprogramming tumor cells into neuronal like cells. Sci. Rep. 9:3462. doi: 10.1038/s41598-019-39852-5
Gascon, S., Masserdotti, G., Russo, G. L., and Gotz, M. (2017). Direct neuronal reprogramming: achievements, hurdles, and new roads to success. Cell Stem Cell 21, 18–34. doi: 10.1016/j.stem.2017.06.011
Gitler, A. D., Dhillon, P., and Shorter, J. (2017). Neurodegenerative disease: models, mechanisms, and a new hope. Dis. Models Mech. 10, 499–502. doi: 10.1242/dmm.030205
Gonzalez, F., Boue, S., and Belmonte, J. C. I. (2011). Methods for making induced pluripotent stem cells: reprogramming a la carte. Nat. Rev. Genet. 12, 231–242. doi: 10.1038/nrg2937
Han, C., Chaineau, M., Chen, C. X., Beitel, L. K., and Durcan, T. M. (2018). Open science meets stem cells: a new drug discovery approach for neurodegenerative disorders. Front. Neurosci. 12:47. doi: 10.3389/fnins.2018.00047
Hanna, J., Markoulaki, S., Schorderet, P., Carey, B. W., Beard, C., Wernig, M., et al. (2008). Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell 133, 250–264. doi: 10.1016/j.cell.2008.03.028
Hargus, G., Ehrlich, M., Hallmann, A. L., and Kuhlmann, T. (2014). Human stem cell models of neurodegeneration: a novel approach to study mechanisms of disease development. Acta Neuropathol. 127, 151–173. doi: 10.1007/s00401-013-1222-6
Haridhasapavalan, K. K., Borgohain, M. P., Dey, C., Saha, B., Narayan, G., Kumar, S., et al. (2019). An insight into non-integrative gene delivery approaches to generate transgene-free induced pluripotent stem cells. Gene 686, 146–159. doi: 10.1016/j.gene.2018.11.069
Haridhasapavalan, K. K., Raina, K., Dey, C., Adhikari, P., and Thummer, R. P. (2020). An insight into reprogramming barriers to iPSC generation. Stem Cell Rev. Rep. 16, 56–81. doi: 10.1007/s12015-019-09931-1
Haston, K. M., and Finkbeiner, S. (2016). Clinical trials in a dish: the potential of pluripotent stem cells to develop therapies for neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 56, 489–510. doi: 10.1146/annurev-pharmtox-010715-103548
Heilker, R., Traub, S., Reinhardt, P., Scholer, H. R., and Sterneckert, J. (2014). iPS cell derived neuronal cells for drug discovery. Trends Pharmacol. Sci. 35, 510–519. doi: 10.1016/j.tips.2014.07.003
Heinrich, C., Gascon, S., Masserdotti, G., Lepier, A., Sanchez, R., Simon-Ebert, T., et al. (2011). Generation of subtype-specific neurons from postnatal astroglia of the mouse cerebral cortex. Nat. Protoc. 6, 214–228. doi: 10.1038/nprot.2010.188
Hou, P., Li, Y., Zhang, X., Liu, C., Guan, J., Li, H., et al. (2013). Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 341, 651–654. doi: 10.1126/science.1239278
Hu, W., Qiu, B., Guan, W., Wang, Q., Wang, M., Li, W., et al. (2015). Direct conversion of normal and Alzheimer’s disease human fibroblasts into neuronal cells by small molecules. Cell Stem Cell 17, 204–212. doi: 10.1016/j.stem.2015.07.006
Huang, L., Wang, J., Huang, S., Siaw-Debrah, F., Nyanzu, M., and Zhuge, Q. (2019). Polyacrylic acid-coated nanoparticles loaded with recombinant tissue plasminogen activator for the treatment of mice with ischemic stroke. Biochem. Biophys. Res. Commun. 516, 565–570. doi: 10.1016/j.bbrc.2019.06.079
Imaizumi, Y., Okada, Y., Akamatsu, W., Koike, M., Kuzumaki, N., Hayakawa, H., et al. (2012). Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 5:35. doi: 10.1186/1756-6606-5-35
Imaizumi, Y., and Okano, H. (2014). Modeling human neurological disorders with induced pluripotent stem cells. J. Neurochem. 129, 388–399. doi: 10.1111/jnc.12625
Israel, M. A., Yuan, S. H., Bardy, C., Reyna, S. M., Mu, Y., Herrera, C., et al. (2012). Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220. doi: 10.1038/nature10821
Jucker, M. (2010). The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat. Med. 16, 1210–1214. doi: 10.1038/nm.2224
Karch, C. M., Cruchaga, C., and Goate, A. M. (2014). Alzheimer’s disease genetics: from the bench to the clinic. Neuron 83, 11–26. doi: 10.1016/j.neuron.2014.05.041
Karow, M., Camp, J. G., Falk, S., Gerber, T., Pataskar, A., Gac-Santel, M., et al. (2018). Direct pericyte-to-neuron reprogramming via unfolding of a neural stem cell-like program. Nat. Neurosci. 21, 932–940. doi: 10.1038/s41593-018-0168-3
Karow, M., Sánchez, R., Schichor, C., Masserdotti, G., Ortega, F., Heinrich, C., et al. (2012). Reprogramming of pericyte-derived cells of the adult human brain into induced neuronal cells. Cell Stem Cell 11, 471–476. doi: 10.1016/j.stem.2012.07.007
Ke, M., Chong, C.-M., and Su, H. (2019). Using induced pluripotent stem cells for modeling Parkinson’s disease. World J. Stem Cells 11, 634–649. doi: 10.4252/wjsc.v11.i9.634
Ke, Z., Zhang, X., Cao, Z., Ding, Y., Li, N., Cao, L., et al. (2016). Drug discovery of neurodegenerative disease through network pharmacology approach in herbs. Biomed. Pharmacother. 78, 272–279. doi: 10.1016/j.biopha.2016.01.021
Kim, D., Kim, C. H., Moon, J. I., Chung, Y. G., Chang, M. Y., Han, B. S., et al. (2009). Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 4, 472–476. doi: 10.1016/j.stem.2009.05.005
Kim, J., Su, S. C., Wang, H., Cheng, A. W., Cassady, J. P., Lodato, M. A., et al. (2011). Functional integration of dopaminergic neurons directly converted from mouse fibroblasts. Cell Stem Cell 9, 413–419. doi: 10.1016/j.stem.2011.09.011
Kim, J. B., Zaehres, H., Wu, G., Gentile, L., Ko, K., Sebastiano, V., et al. (2008). Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature 454, 646–650. doi: 10.1038/nature07061
Kiskinis, E., Sandoe, J., Williams, L. A., Boulting, G. L., Moccia, R., Wainger, B. J., et al. (2014). Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795. doi: 10.1016/j.stem.2014.03.004
Kraljevic, S., Stambrook, P. J., and Pavelic, K. (2004). Accelerating drug discovery. EMBO Rep. 5, 837–842. doi: 10.1038/sj.embor.7400236
Ledford, H. (2011). Translational research: 4 ways to fix the clinical trial. Nature 477, 526–528. doi: 10.1038/477526a
Lee, S., Lee, B., Lee, J. W., and Lee, S. K. (2009). Retinoid signaling and neurogenin2 function are coupled for the specification of spinal motor neurons through a chromatin modifier CBP. Neuron 62, 641–654. doi: 10.1016/j.neuron.2009.04.025
Li, X., Liu, D., Ma, Y., Du, X., Jing, J., Wang, L., et al. (2017). Direct reprogramming of fibroblasts via a chemically induced XEN-like State. Cell Stem Cell 21, 264–273.e267. doi: 10.1016/j.stem.2017.05.019
Li, X., Zuo, X., Jing, J., Ma, Y., Wang, J., Liu, D., et al. (2015). Small-molecule-driven direct reprogramming of mouse fibroblasts into functional neurons. Cell Stem Cell 17, 195–203. doi: 10.1016/j.stem.2015.06.003
Liu, M. L., Zang, T., and Zhang, C. L. (2016). Direct lineage reprogramming reveals disease-specific phenotypes of motor neurons from human ALS patients. Cell Rep. 14, 115–128. doi: 10.1016/j.celrep.2015.12.018
Liu, M.-L., Zang, T., Zou, Y., Chang, J. C., Gibson, J. R., Huber, K. M., et al. (2013). Small molecules enable neurogenin 2 to efficiently convert human fibroblasts into cholinergic neurons. Nat. Commun. 4:2183. doi: 10.1038/ncomms3183
Liu, X., Li, F., Stubblefield, E. A., Blanchard, B., Richards, T. L., Larson, G. A., et al. (2011). Direct reprogramming of human fibroblasts into dopaminergic neuron-like cells. Cell Res 22, 321–332. doi: 10.1038/cr.2011.181
Liu, Y., and Deng, W. (2016). Reverse engineering human neurodegenerative disease using pluripotent stem cell technology. Brain Res. 1638, 30–41. doi: 10.1016/j.brainres.2015.09.023
Maherali, N., Ahfeldt, T., Rigamonti, A., Utikal, J., Cowan, C., and Hochedlinger, K. (2008). A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell 3, 340–345. doi: 10.1016/j.stem.2008.08.003
Mall, M., Kareta, M. S., Chanda, S., Ahlenius, H., Perotti, N., Zhou, B., et al. (2017). Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature 544, 245–249. doi: 10.1038/nature21722
Marin, O. (2012). Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 13, 107–120. doi: 10.1038/nrn3155
Marro, S., Pang, Z. P., Yang, N., Tsai, M. C., Qu, K., Chang, H. Y., et al. (2011). Direct lineage conversion of terminally differentiated hepatocytes to functional neurons. Cell Stem Cell 9, 374–382. doi: 10.1016/j.stem.2011.09.002
Mason, A. R., Ziemann, A., and Finkbeiner, S. (2014). Targeting the low-hanging fruit of neurodegeneration. Neurology 83, 1470–1473. doi: 10.1212/WNL.0000000000000894
Masserdotti, G., Gascon, S., and Gotz, M. (2016). Direct neuronal reprogramming: learning from and for development. Development 143, 2494–2510. doi: 10.1242/dev.092163
Maucksch, C., Jones, K. S., and Connor, B. (2013). Concise review: the involvement of SOX2 in direct reprogramming of induced neural stem/precursor cells. Stem Cells Transl. Med. 2, 579–583. doi: 10.5966/sctm.2012-0179
Mertens, J., Paquola, A. C. M., Ku, M., Hatch, E., Bohnke, L., Ladjevardi, S., et al. (2015). Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17, 705–718. doi: 10.1016/j.stem.2015.09.001
Mertens, J., Reid, D., Lau, S., Kim, Y., and Gage, F. H. (2018). Aging in a dish: iPSC-derived and directly induced neurons for studying brain aging and age-related neurodegenerative diseases. Annu. Rev. Genet. 52, 271–293. doi: 10.1146/annurev-genet-120417-031534
Mitsumoto, H., Brooks, B. R., and Silani, V. (2014). Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol. 13, 1127–1138. doi: 10.1016/s1474-4422(14)70129-2
Moore, S., Evans, L. D., Andersson, T., Portelius, E., Smith, J., Dias, T. B., et al. (2015). APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 11, 689–696. doi: 10.1016/j.celrep.2015.03.068
Moreno, C. L., Della Guardia, L., Shnyder, V., Ortiz-Virumbrales, M., Kruglikov, I., Zhang, B., et al. (2018). iPSC-derived familial Alzheimer’s PSEN2 (N141I) cholinergic neurons exhibit mutation-dependent molecular pathology corrected by insulin signaling. Mol. Neurodegener. 13:33. doi: 10.1186/s13024-018-0265-5
Morris, S. A., and Daley, G. Q. (2013). A blueprint for engineering cell fate: current technologies to reprogram cell identity. Cell Res. 23, 33–48. doi: 10.1038/cr.2013.1
Muratore, C. R., Rice, H. C., Srikanth, P., Callahan, D. G., Shin, T., Benjamin, L. N., et al. (2014). The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 23, 3523–3536. doi: 10.1093/hmg/ddu064
Narsinh, K. H., Jia, F., Robbins, R. C., Kay, M. A., Longaker, M. T., and Wu, J. C. (2010). Generation of adult human induced pluripotent stem cells using nonviral minicircle DNA vectors. Nat. Protoc. 6, 78–88. doi: 10.1038/nprot.2010.173
Nguyen, H. N., Byers, B., Cord, B., Shcheglovitov, A., Byrne, J., Gujar, P., et al. (2011). LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280. doi: 10.1016/j.stem.2011.01.013
Ochalek, A., Mihalik, B., Avci, H. X., Chandrasekaran, A., Teglasi, A., Bock, I., et al. (2017). Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res. Ther. 9:90. doi: 10.1186/s13195-017-0317-z
Okano, H., and Yamanaka, S. (2014). iPS cell technologies: significance and applications to CNS regeneration and disease. Mol. Brain 7:22. doi: 10.1186/1756-6606-7-22
Oliveira, L. M., Falomir-Lockhart, L. J., Botelho, M. G., Lin, K. H., Wales, P., Koch, J. C., et al. (2015). Elevated alpha-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis. 6:e1994. doi: 10.1038/cddis.2015.318
O’Malley, J., Woltjen, K., and Kaji, K. (2009). New strategies to generate induced pluripotent stem cells. Curr. Opin. Biotechnol. 20, 516–521. doi: 10.1016/j.copbio.2009.09.005
Omole, A. E., and Fakoya, A. O. J. (2018). Ten years of progress and promise of induced pluripotent stem cells: historical origins, characteristics, mechanisms, limitations, and potential applications. PeerJ 6:e4370. doi: 10.7717/peerj.4370
Pang, Z. P., Yang, N., Vierbuchen, T., Ostermeier, A., Fuentes, D. R., Yang, T. Q., et al. (2011). Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223. doi: 10.1038/nature10202
Pereira, M., Birtele, M., and Rylander Ottosson, D. (2019). Direct reprogramming into interneurons: potential for brain repair. Cell. Mol. Life Sci. 76, 3953–3967. doi: 10.1007/s00018-019-03193-3193
Pfisterer, U., Kirkeby, A., Torper, O., Wood, J., Nelander, J., Dufour, A., et al. (2011). Direct conversion of human fibroblasts to dopaminergic neurons. Proc. Natl. Acad. Sci. U.S.A. 108, 10343–10348. doi: 10.1073/pnas.1105135108
Postuma, R. B., Berg, D., Stern, M., Poewe, W., Olanow, C. W., Oertel, W., et al. (2015). MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 30, 1591–1601. doi: 10.1002/mds.26424
Qin, H., Zhao, A., and Fu, X. (2017). Small molecules for reprogramming and transdifferentiation. Cell. Mol. Life Sci. 74, 3553–3575. doi: 10.1007/s00018-017-2586-x
Reinhardt, P., Schmid, B., Burbulla, L. F., Schondorf, D. C., Wagner, L., Glatza, M., et al. (2013). Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12, 354–367. doi: 10.1016/j.stem.2013.01.008
Ren, Y., Jiang, H., Hu, Z., Fan, K., Wang, J., Janoschka, S., et al. (2015). Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cells 33, 68–78. doi: 10.1002/stem.1854
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi: 10.1038/nrn3430
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rowland, H. A., Hooper, N. M., and Kellett, K. A. B. (2018). Modelling sporadic Alzheimer’s disease using induced pluripotent stem cells. Neurochem. Res. 43, 2179–2198. doi: 10.1007/s11064-018-2663-z
Rubio, A., Luoni, M., Giannelli, S. G., Radice, I., Iannielli, A., Cancellieri, C., et al. (2016). Rapid and efficient CRISPR/Cas9 gene inactivation in human neurons during human pluripotent stem cell differentiation and direct reprogramming. Sci. Rep. 6:37540. doi: 10.1038/srep37540
Ruggieri, M., Riboldi, G., Brajkovic, S., Bucchia, M., Bresolin, N., Comi, G. P., et al. (2014). Induced neural stem cells: methods of reprogramming and potential therapeutic applications. Prog. Neurobiol. 114, 15–24. doi: 10.1016/j.pneurobio.2013.11.001
Sanchez-Danes, A., Richaud-Patin, Y., Carballo-Carbajal, I., Jimenez-Delgado, S., Caig, C., Mora, S., et al. (2012). Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 4, 380–395. doi: 10.1002/emmm.201200215
Sanders, L. H., Laganiere, J., Cooper, O., Mak, S. K., Vu, B. J., Huang, Y. A., et al. (2014). LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol. Dis. 62, 381–386. doi: 10.1016/j.nbd.2013.10.013
Sareen, D., O’Rourke, J. G., Meera, P., Muhammad, A. K., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5:208ra149. doi: 10.1126/scitranslmed.3007529
Seibler, P., Graziotto, J., Jeong, H., Simunovic, F., Klein, C., and Krainc, D. (2011). Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 31, 5970–5976. doi: 10.1523/JNEUROSCI.4441-10.2011
Shahbazi, E., Moradi, S., Nemati, S., Satarian, L., Basiri, M., Gourabi, H., et al. (2016). Conversion of human fibroblasts to stably self-renewing neural stem cells with a single zinc-finger transcription factor. Stem Cell Rep. 6, 539–551. doi: 10.1016/j.stemcr.2016.02.013
Shen, J. (2014). Function and dysfunction of presenilin. Neurodegener. Dis. 13, 61–63. doi: 10.1159/000354971
Shi, Y., Inoue, H., Wu, J. C., and Yamanaka, S. (2017). Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov. 16, 115–130. doi: 10.1038/nrd.2016.245
Shi, Y., Kirwan, P., Smith, J., MacLean, G., Orkin, S. H., and Livesey, F. J. (2012). A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci. Transl. Med. 4:124ra129. doi: 10.1126/scitranslmed.3003771
Shi, Z., Zhang, J., Chen, S., Li, Y., Lei, X., Qiao, H., et al. (2016). Conversion of fibroblasts to parvalbumin neurons by one transcription factor, Ascl1, and the chemical compound forskolin. J. Biol. Chem. 291, 13560–13570. doi: 10.1074/jbc.M115.709808
Smith, D. K., Yang, J., Liu, M. L., and Zhang, C. L. (2016). Small molecules modulate chromatin accessibility to promote NEUROG2-mediated fibroblast-to-neuron reprogramming. Stem Cell Rep. 7, 955–969. doi: 10.1016/j.stemcr.2016.09.013
Sommer, C. A., and Mostoslavsky, G. (2010). Experimental approaches for the generation of induced pluripotent stem cells. Stem Cell Res. Ther. 1:26. doi: 10.1186/Scrt26
Sommer, C. A., and Mostoslavsky, G. (2013). The evolving field of induced pluripotency: recent progress and future challenges. J. Cell. Physiol. 228, 267–275. doi: 10.1002/jcp.24155
Son, E. Y., Ichida, J. K., Wainger, B. J., Toma, J. S., Rafuse, V. F., Woolf, C. J., et al. (2011). Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 9, 205–218. doi: 10.1016/j.stem.2011.07.014
Sproul, A. A., Jacob, S., Pre, D., Kim, S. H., Nestor, M. W., Navarro-Sobrino, M., et al. (2014). Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS One 9:e84547. doi: 10.1371/journal.pone.0084547
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Sun, N., Panetta, N. J., Gupta, D. M., Wilson, K. D., Lee, A., Jia, F., et al. (2009). Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc. Natl. Acad. Sci. U.S.A. 106, 15720–15725. doi: 10.1073/pnas.0908450106
Sun, W., Zheng, W., and Simeonov, A. (2017). Drug discovery and development for rare genetic disorders. Am. J. Med. Genet. Part A 173, 2307–2322. doi: 10.1002/ajmg.a.38326
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Tanabe, K., Ang, C. E., Chanda, S., Olmos, V. H., Haag, D., Levinson, D. F., et al. (2018). Transdifferentiation of human adult peripheral blood T cells into neurons. Proc. Natl. Acad. Sci. U.S.A. 115, 6470–6475. doi: 10.1073/pnas.1720273115
Tang, Y., Liu, M. L., Zang, T., and Zhang, C. L. (2017). Direct reprogramming rather than iPSC-Based reprogramming maintains aging hallmarks in human motor neurons. Front. Mol. Neurosci. 10:359. doi: 10.3389/fnmol.2017.00359
Tcw, J. (2019). Human iPSC application in Alzheimer’s disease and Tau-related neurodegenerative diseases. Neurosci. Lett. 699, 31–40. doi: 10.1016/j.neulet.2019.01.043
Torper, O., Pfisterer, U., Wolf, D. A., Pereira, M., Lau, S., Jakobsson, J., et al. (2013). Generation of induced neurons via direct conversion in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, 7038–7043. doi: 10.1073/pnas.1303829110
Traxler, L., Edenhofer, F., and Mertens, J. (2019). Next-generation disease modeling with direct conversion: a new path to old neurons. FEBS Lett. 593, 3316–3337. doi: 10.1002/1873-3468.13678
Tremblay, R., Lee, S., and Rudy, B. (2016). GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91, 260–292. doi: 10.1016/j.neuron.2016.06.033
Utikal, J., Maherali, N., Kulalert, W., and Hochedlinger, K. (2009). Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J. Cell Sci. 122, 3502–3510. doi: 10.1242/jcs.054783
Vierbuchen, T., Ostermeier, A., Pang, Z. P., Kokubu, Y., Sudhof, T. C., and Wernig, M. (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041. doi: 10.1038/nature08797
Wan, W., Cao, L., Kalionis, B., Xia, S., and Tai, X. (2015). Applications of induced pluripotent stem cells in studying the neurodegenerative diseases. Stem Cells Int. 2015:382530. doi: 10.1155/2015/382530
Wang, C., Najm, R., Xu, Q., Jeong, D.-E., Walker, D., Balestra, M. E., et al. (2018). Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 24, 647–657. doi: 10.1038/s41591-018-0004-z
Warren, L., Manos, P. D., Ahfeldt, T., Loh, Y. H., Li, H., Lau, F., et al. (2010). Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 618–630. doi: 10.1016/j.stem.2010.08.012
Wazan, L. E., Urrutia-Cabrera, D., and Wong, R. C.-B. (2019). Using transcription factors for direct reprogramming of neurons in vitro. World J. Stem Cells 11, 431–444. doi: 10.4252/wjsc.v11.i7.431
Wiedemann, A., Hemmer, K., Bernemann, I., Göhring, G., Pogozhykh, O., Figueiredo, C., et al. (2012). Induced pluripotent stem cells generated from adult bone marrow-derived cells of the nonhuman primate (Callithrix jacchus) using a novel quad-cistronic and excisable lentiviral vector. Cell. Reprogramm. 14, 485–496. doi: 10.1089/cell.2012.0036
Woo, N. H., and Lu, B. (2006). Regulation of cortical interneurons by neurotrophins: from development to cognitive disorders. Neuroscientist 12, 43–56. doi: 10.1177/1073858405284360
Wyss-Coray, T. (2016). Ageing, neurodegeneration and brain rejuvenation. Nature 539, 180–186. doi: 10.1038/nature20411
Xiao, D., Liu, X., Zhang, M., Zou, M., Deng, Q., Sun, D., et al. (2018). Direct reprogramming of fibroblasts into neural stem cells by single non-neural progenitor transcription factor Ptf1a. Nat. Commun. 9:2865. doi: 10.1038/s41467-018-05209-1
Xu, J., Du, Y., and Deng, H. (2015). Direct lineage reprogramming: strategies, mechanisms, and applications. Cell Stem Cell 16, 119–134. doi: 10.1016/j.stem.2015.01.013
Xu, Z., Jiang, H., Zhong, P., Yan, Z., Chen, S., and Feng, J. (2015). Direct conversion of human fibroblasts to induced serotonergic neurons. Mol. Psychiatry 21, 62–70. doi: 10.1038/mp.2015.101
Yagi, T., Ito, D., Okada, Y., Akamatsu, W., Nihei, Y., Yoshizaki, T., et al. (2011). Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 20, 4530–4539. doi: 10.1093/hmg/ddr394
Yu, J., Vodyanik, M. A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920. doi: 10.1126/science.1151526
Zhang, L., Yin, J. C., Yeh, H., Ma, N. X., Lee, G., Chen, X. A., et al. (2015). Small molecules efficiently reprogram human astroglial cells into functional neurons. Cell Stem Cell 17, 735–747. doi: 10.1016/j.stem.2015.09.012
Zhang, Q. J., Li, J. J., Lin, X., Lu, Y. Q., Guo, X. X., Dong, E. L., et al. (2017). Modeling the phenotype of spinal muscular atrophy by the direct conversion of human fibroblasts to motor neurons. Oncotarget 8, 10945–10953. doi: 10.18632/oncotarget.14641
Zhang, Z., and Wu, W.-S. (2013). Sodium butyrate promotes generation of human induced pluripotent stem cells through induction of the miR302/367 cluster. Stem Cells Dev. 22, 2268–2277. doi: 10.1089/scd.2012.0650
Keywords: iN, iPSCs, disease modeling, drug screening, neurodegenerative disease
Citation: Zhang Y, Xie X, Hu J, Afreen KS, Zhang C-L, Zhuge Q and Yang J (2020) Prospects of Directly Reprogrammed Adult Human Neurons for Neurodegenerative Disease Modeling and Drug Discovery: iN vs. iPSCs Models. Front. Neurosci. 14:546484. doi: 10.3389/fnins.2020.546484
Received: 28 March 2020; Accepted: 12 October 2020;
Published: 19 November 2020.
Edited by:
Kuangyu Shi, University of Bern, SwitzerlandReviewed by:
Anandhan Annadurai, University of Arizona, United StatesRajkumar P. Thummer, Indian Institute of Technology Guwahati, India
Copyright © 2020 Zhang, Xie, Hu, Afreen, Zhang, Zhuge and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qichuan Zhuge, cWMuemh1Z2VAd211LmVkdS5jbg==; Jianjing Yang, eWFuZ2ppYW5qaW5nMkAxNjMuY29t
†These authors have contributed equally to this work