 Arastu Sharma1†
Arastu Sharma1† Hargsoon Yoon
Hargsoon Yoon Sung Ung Kang
Sung Ung Kang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 06 August 2020
Sec. Sleep and Circadian Rhythms
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.00844
This article is part of the Research Topic Roles of Sleep Disruption and Circadian Rhythm Alterations on Neurodegeneration and Alzheimer's Disease View all 12 articles
Neurodegenerative disorders have been shown to exhibit substantial interconnectedness with circadian rhythmicity. Alzheimer’s patients exhibit high degradation of the suprachiasmatic nucleus (SCN), the central endogenous circadian timekeeper, and Parkinson’s patients have highly disrupted peripheral clock gene expression. Disrupted sleep patterns are highly evident in patients with neurodegenerative diseases; fragmented sleep has been shown to affect tau-protein accumulation in Alzheimer’s patients, and rapid eye movement (REM) behavioral disorder is observed in a significant amount of Parkinson’s patients. Although numerous studies exist analyzing the mechanisms of neurodegeneration and circadian rhythm function independently, molecular mechanisms establishing specific links between the two must be explored further. Thus, in this review, we explore the possible intersecting molecular mechanisms between circadian rhythm and neurodegeneration, with a particular focus on Parkinson’s disease. We provide evidence for potential influences of E3 ligase and poly adenosine diphosphate (ADP-ribose) polymerase 1 (PARP1) activity on neurodegenerative pathology. The cellular stress and subsequent DNA damage signaling imposed by hyperactivity of these multiple molecular systems in addition to aberrant circadian rhythmicity lead to extensive protein aggregation such as α-synuclein pre-formed fibrils (α-Syn PFFs), suggesting a specific molecular pathway linking circadian rhythmicity, PARP1/E3 ligase activity, and Parkinson’s disease.
Living systems on earth are governed by many natural laws, but circadian rhythms play one of the most important roles in sustaining organisms, acting as the biological timekeepers that perpetuate life from mere seconds to the full Gregorian year. Mammalian circadian rhythms can be observed from the genetic level to the tissue level, and even to the macroscopic level, affecting behavior, biochemical and physiological processes. Genetic and cellular clocks dictate tau (free running period of the organism ∼24 h) and are often entrained by photo-optic cues (Dunlap and Loros, 2017). Light, as the external stimulus, activates intrinsically photosensitive retinal ganglion cells (ipRGCs), which innervate the suprachiasmatic nucleus (SCN) in mammals, entraining the mammal to the 24-h day (Do and Yau, 2010). The SCN assumes the role as the central pacemaker, and through a series of genetic feedback loops and highly coordinated neuronal innervation, endogenous timekeeping activity arises, giving way to the production of circadian rhythm. To garner a more comprehensive understanding of the effects of circadian rhythms on health and neurodegeneration, the underlying fundamental molecular mechanisms and interrelated processes must be explored (Cox and Takahashi, 2019).
A set of core genes constitutes this transcriptional pathway that forms the identity of the endogenous circadian pacemaker. Two heterodimerization transcription factors, CLOCK and BMAL1, activate the production of the period (Per) and cryptochrome (Cry) genes (Yoo et al., 2005; Figure 1). The resulting PER and CRY proteins heterodimerize and are phosphorylated by casein kinase 1 (CK1), allowing for translocation into the nucleus, consequently inhibiting CLOCK and BMAL1 transcriptional activity, preventing the production of PER and CRY, forming the central negative feedback loop that gives rise to endogenous circadian rhythmicity at the most basic cellular level in mammals (Philpott et al., 2020). Mutations in these genes result in altered displays of circadian behavior in affected model organisms (Patke et al., 2020).
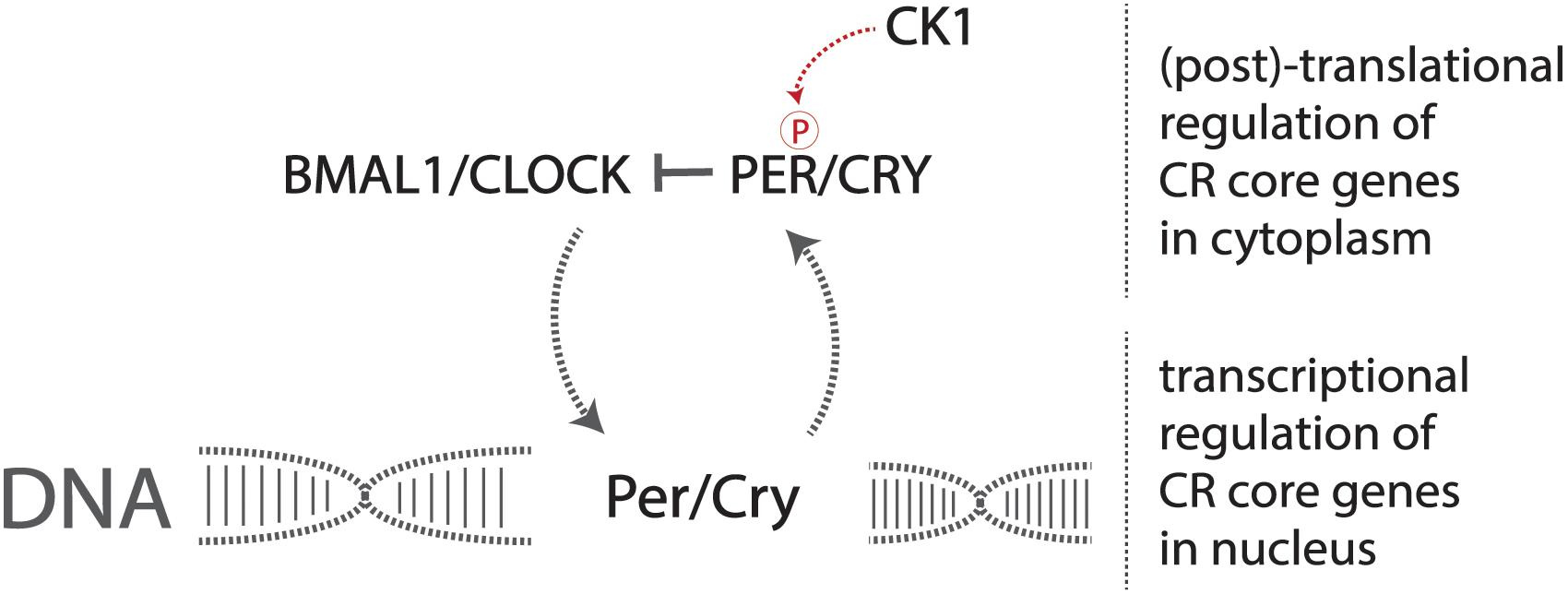
Figure 1. The core circadian molecular machinery gives rise to endogenous timekeeping activity. The CLOCK/BMAL1 heterodimer activates the transcription of Per/Cry genes, and the production and resulting phosphorylation of PER/CRY inhibit the CLOCK/BMAL1 heterodimer, reducing the transcription of Per/Cry forming the negative feedback loop required to maintain circadian rhythmicity at a basic molecular level.
Irregularities in circadian rhythms have been shown to alter sleep–wake cycles, as well as alter metabolic processes that may result in the development of certain diseases or other health-related complications like diabetes, cardiovascular disease, cancer, and neurodegenerative disorders (Wilking et al., 2013; Qian and Scheer, 2016).
More prevalent in old age, neurodegenerative disorders such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) are widely believed to be driven by protein deposition in the form of insoluble aggregates, resulting in loss of physiological functions and pathological dysregulation of neurons (Delenclos et al., 2019; Huseby et al., 2019; Figure 2). Severe degradation of the SCN, as well as negatively altered patterns of circadian expression are prevalent in AD patients, but in PD much more targeted degradation of dopaminergic (DA) striatal neurons occurs (Dauer and Przedborski, 2003). Lower levels of BMAL1 are directly proportional to the gravity of an individual’s development of PD (Cai et al., 2010; Ding et al., 2011). Melatonin treatment was shown to prevent DA neuronal cell degradation, but the mechanism through which the protection of DA neurons occurs is unclear, whether it be oxidative or circadian (Radogna et al., 2010; Wilking et al., 2013).
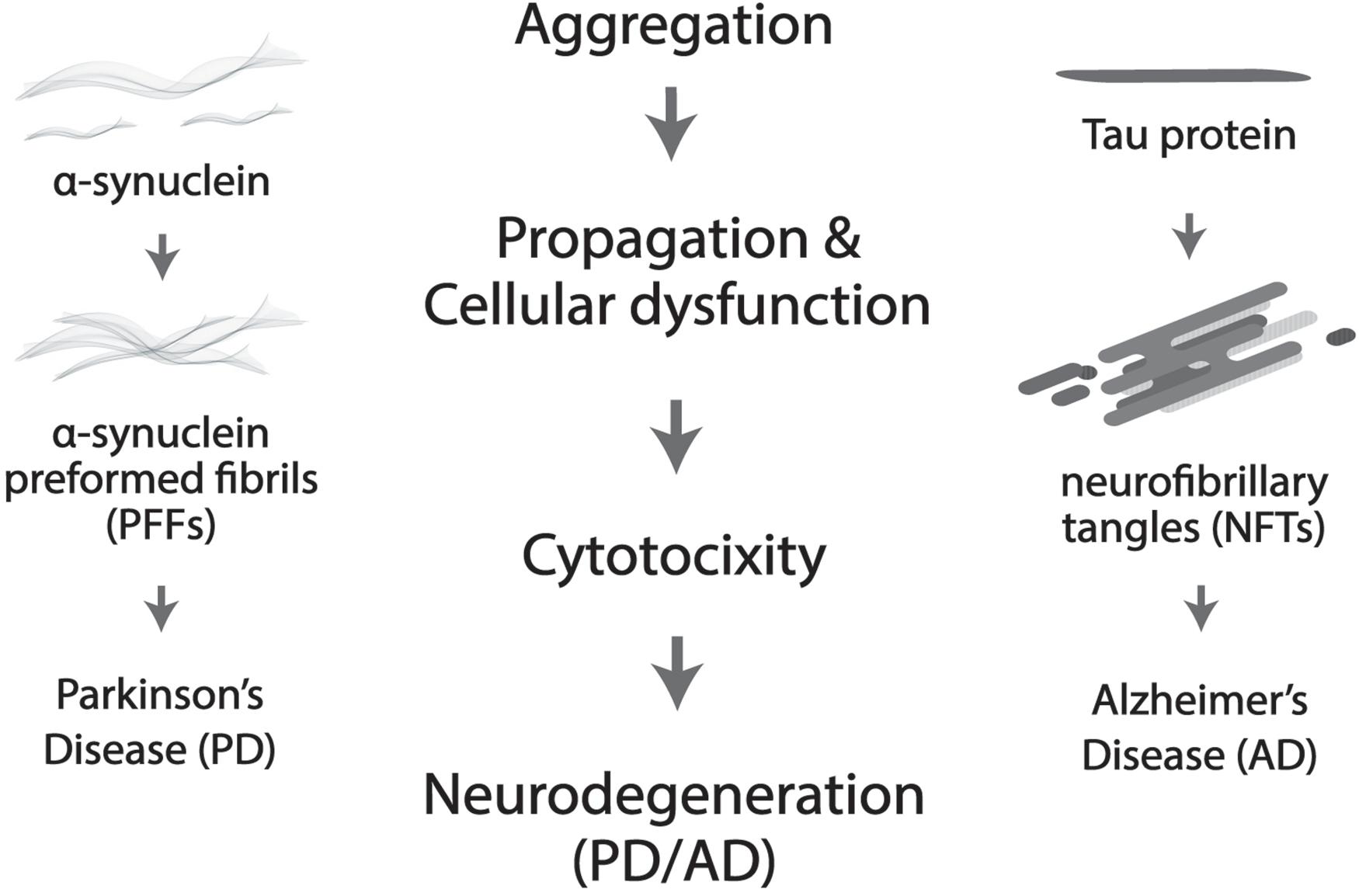
Figure 2. Protein aggregation results in cellular dysfunction and cytotoxicity, which further leads to neurodegeneration. More specifically, neurofibrillary tangles (NTFs) caused by Tau aggregation is the culprit in Alzheimer’s disease (AD) and α-synuclein pre-formed fibrils (α-Syn PFFs) by α-synuclein accumulation in Parkinson’s disease (PD).
Circadian irregularities can also be symptomatic and result in comorbidities, such as cases of individuals with rapid eye movement (REM) behavioral disorder that will ultimately develop PD (Breen et al., 2014). Sleep serves a critical role in the maintenance of circadian rhythms and can strongly affect the buildup of neuronal stress (Breen et al., 2014). In the case of AD, β-amyloid peptide (Aβ) plaques build up in neurons, causing severe systemic stress and eventually leading to the destruction of the cell (Kocahan and Dogan, 2017). Aβ accumulation occurs from natural activity of the cell, and fluctuates with circadian rhythmicity, with higher levels apparent during waking hours and drop-offs during periods of inactivity (Kang et al., 2009).
Alzheimer’s disease models have exhibited disrupted patterns of glymphatic flow (GF) in rodents, and GF has also been shown to be regulated by sleep activity, where slow wave sleep in mice resulted in a 60% increase in interstitial brain fluid and increased GF. This phenomenon is crucial for the removal of excess molecular buildup, particularly Aβ, in the extracellular space of areas with high neuronal activity in the central nervous system (Rasmussen et al., 2018). A vital protein associated with Aβ plaque aggregation and tau protein pathology is Orexin, a compound involved in the sleep–wake pathway that promotes wakefulness. Through analyzing its activity, researchers found that cyclic levels of orexin during normal rhythmic activity were inversely proportional to tau protein aggregation; high Aβ plaque aggregation and tau protein pathology also result in disturbances in non-REM sleep activity, ascertaining the bidirectional influence of circadian rhythmicity and AD pathology (Kilduff et al., 2008; Kang et al., 2009; Liu et al., 2019). Multiple studies found that reduced wakefulness as a result of orexin inhibition reduced levels of Aβ buildup in cells (Davies et al., 2015; Liu et al., 2019). The SCN of affected AD patients was also found to have experienced severe degeneration, and clock gene expression in various regions of the brain was highly asynchronous.
Parkinson’s disease patients exhibit asynchronous peripheral clock gene expression, therefore single nucleotide polymorphisms in various clock genes have been proposed to indicate the risk of PD development in affected individuals (Gu et al., 2015). Patients affected with PD exhibit much more flattened circadian diurnal curves as a result of increased activity during inactive hours and decreased activity during waking hours (Porter et al., 2008; Verbaan et al., 2008). The majority of PD patients suffer from sleep disturbance as one of the primary symptoms, most likely a result of severe degeneration of the raphe nucleus and locus coeruleus (Braak and Del Tredici, 2008). Circadian melatonin rhythmicity is also altered in PD patients, with the curve being flattened over the course of 24 h (Videnovic and Golombek, 2017).
Numerous articles of evidence discovered strongly accentuate the interconnected activity between circadian rhythms, metabolic and molecular pathways, aging, diseases, as well as neurodegenerative disorders. Aberrant normal pathological function in the form of neurodegenerative effects has been discussed, but proper ubiquitination of proteins offers stark neuroprotection against development of neurodegenerative disorders characteristic of protein aggregation. Proteasomal activity has been shown to follow circadian rhythmicity, and distortions in this process may result in increased risks of developing neurodegenerative diseases (Musiek and Holtzman, 2016; Figure 3).
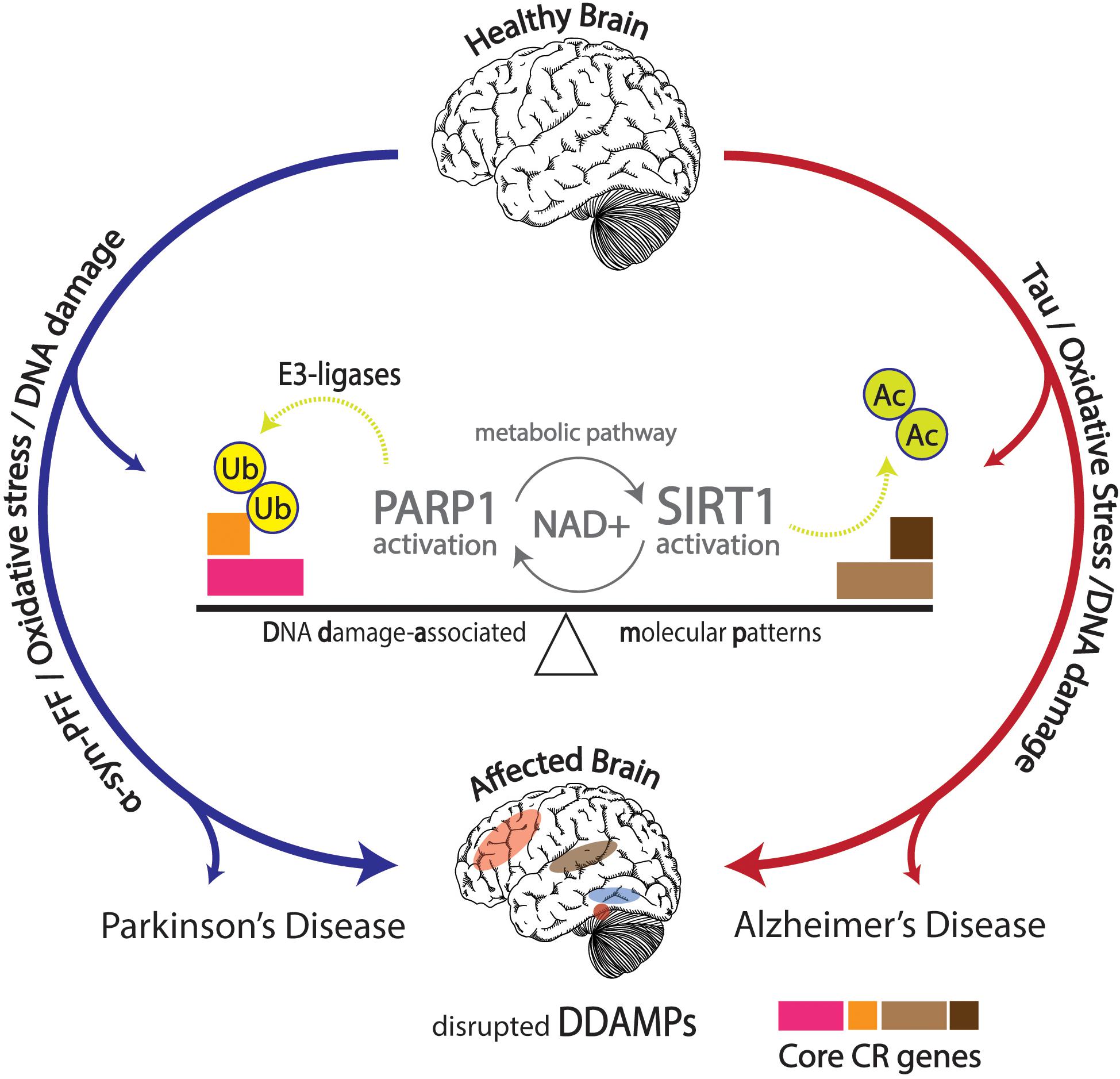
Figure 3. Effects of DNA damage in conjunction with α-synuclein pre-formed fibrils (α-Syn PFFs) and tau pathology shown in the affected brain with disrupted DNA damage-associated molecular patterns (DDAMPs) in Parkinson’s disease (PD) and Alzheimer’s disease (AD), respectively. Disruptions in metabolic pathways lead to altered DNA repair mechanisms, and subsequent poly (ADP-ribose) polymerase 1 (PARP1) and E3 ligase activity, leading to the ubiquitination of core circadian genes in particular, further influencing the course of PD pathology in terms of α-Syn PFFs formation and propagation.
Functional protein families such as kinases, E3 ligases, (de)acetylases and poly adenosine diphosphate (ADP-ribose) polymerases can directly or indirectly affect patterns of circadian behaviors through post-translational modifications of core proteins. Ubiquitination of PER and CRY proteins is a vital step in maintaining the integrity of the cellular circadian clock. The 26S proteasome uses a series of enzymes such as E3 ligases like FBXL3 and FBXL21 to regulate the degradation of PER, CRY, and other proteins, resulting in disinhibition of CLOCK and BMAL1 transcriptional factors, allowing for the molecular circadian clock to reset and begin the negative feedback loop once again (Nandi et al., 2006). Moreover, there are numerous studies that highlight the relationship between other circadian rhythm proteins and E3 ligases (Table 1).

Table 1. Circadian rhythm proteins shown to be associated or dependent on E3 ligase activity.
Furthermore, recent studies have provided strong evidence confirming the link between metabolic processes, neurodegenerative disorders, and circadian dysfunction, through a focus on the E3 ligase pathways, namely mitochondrial ubiquitin ligase 1 pathway (Mul1) and Parkin pathways. Mul1 mutations have been shown to lead to PD symptoms in a similar fashion as Parkin/Pink1 mutations, the usual culprits of the neurodegenerative disorder (Yun et al., 2014). Mul1 and Park1 mutations resulted in shorter lifespans and longer amounts of physical activity during a given 24-h period (Doktor et al., 2019). Mul1 mutants showed a disrupted expression pattern of Per, Tim, and Clock mRNA compared to wild-type flies, and Mul1 mutants exhibited a severe drop-off in ATG5 compared to Park1 mutants and controls (Doktor et al., 2019). ATG5 and other related proteins are critical in autophagy; these proteins form complexes and drive clearance of protein aggregation universally characteristic of neurodegenerative disorders (Doktor et al., 2019).
Competing E3 ligase mechanisms involved with CRY ubiquitination have been shown to affect tau in mice. Missense mutations in the Fbxl21 gene, the gene expressing a protein which targets CRY for ubiquitination, results in shortened circadian periods due to an accelerated degradation of the CRY protein in the nucleus and the cytoplasm, whereas alterations in the competing ligase FBXL3 results in a longer circadian period (Busino et al., 2007; Godinho et al., 2007; Siepka et al., 2007; Yoo et al., 2013). The duality of these two E3 ligases works to preserve the balance of CRY degradation in the cytoplasm and the nucleus (Yoo et al., 2013). An SKP1-Cul1-F-box protein (SCF) E3 ligase complex is formed constituting FBXL3, targeting CRY proteins for proteasomal degradation; FBXL21 has been inferred to exist as a clock-controlled E3 ligase concerning ovine CRY1 degradation (Siepka et al., 2007; Dardente et al., 2008). The existing mutation Overtime (Ovtm) in the FBXL3 protein exists as the functionally antagonistic counterpart to the Psttm mutation in FBXL21 by lengthening the period rather than shortening (Siepka et al., 2007; Yoo et al., 2013). Due to the diminished presence of CRY as a result of the Psttm mutation in the Fbxl21 gene, inhibition of the CLOCK:BMAL1 transcriptionally enhancing complex is further diminished, leading to an increase of RNA expression of target genes. The accelerated degradation of CRY1 is a result of the reduced levels of FBXL21 protein levels. CRY1 was shown to be polyubiquitinated in the presence of the SCFBXL3 complex, but the presence of SCFFBXL21 and SCFPSTTM significantly reduced the ubiquitination of CRY1 by SCFFBXL3, enhancing the antagonistic interactions between FBXL3 and FBXL21. FBXL21 and Psttm have been shown to prevent CRY1 degradation due to FBXL3 as well, further exemplifying the competing characteristics between the two E3 ligases. FBXL21’s specificity for CRY1 degradation may hint to the reduced rate at which it ubiquitinates the CRY1 protein, contrasting from the rather liberal utilization of lysine residues by FBXL3 for CRY1 degradation. The fundamental importance of the family of CRY proteins in regulating circadian period length has been fully emphasized through the analysis of competing E3 ligases FBXL21 and FBXL3 mutations. The “paralogous” nature of FBXL21 and FBXL3 interestingly produced antagonizing behavior in the two E3 ligases, competing for the regulation of CRY1 degradation. The preferential interactions between FBXL21 and CRY may support the finding that FBXL21 is able to protect CRY degradation from SCFFBXL3 activity within the nucleus, accentuating the role of E3 ligases in regulating and dictating circadian period through complex molecular interactions within the nucleus (Yoo et al., 2013; Figure 4).
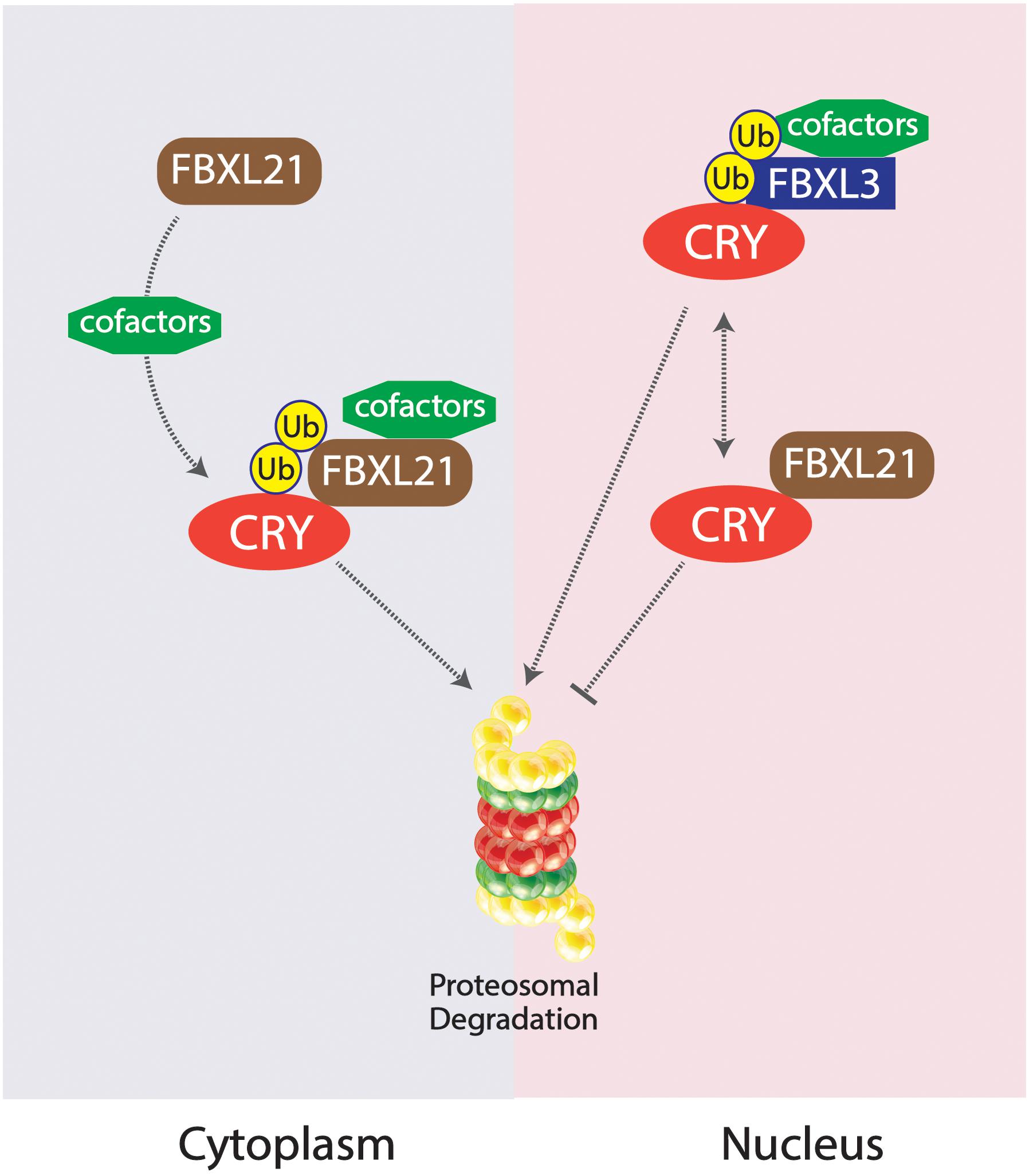
Figure 4. Competing SKP1-Cul1-F-box protein (SCF) E3 ligase complexes SCFFBXL21 and SCFFBXL3 and their effects on CRY degradation in the cytoplasm vs. nucleus. SCFFBXL21 degrades CRY in the cytoplasm but exhibits protective abilities in the nucleus, contrary to the degradative properties of SCFFBXL3 on the CRY protein within the nucleus.
The cyclic influence of core circadian proteins on nicotinamide phosphoribosyltransferase/nicotinamide adenine dinucleotide (NAMPT/NAD) + activity ultimately relates to a vital protein: Poly (ADP-ribose) polymerase 1 (PARP1), a nuclear protein responsible for facilitation of DNA strand repair. It is activated in response to single-strand breaks within DNA, attaching a poly ADP-ribose (PAR) chain for subsequent repair by DNA ligases. After the process nears completion, poly ADP-ribose glycohydrolase (PARGs) degrade the remaining attached PAR chain (Herceg and Wang, 2001). The interwoven nature of the mechanisms between PARP1 and NAD + activity possibly hint to the involvement in the development and course of neurodegenerative disorders (Martire et al., 2015). In addition to PARP1 being a critical component of the DNA repair mechanism, it is also a critical component of parthanatos, the cell death pathway involved in cellularly destructive diseases such as stroke and PD (David et al., 2009). The accumulation of PAR within the cell due to hyperactivity of PARP1 in response to DNA damage in conjunction with the translocation of mitochondrial-associated apoptosis-inducing factor (AIF) into the nucleus results in activation of the parthanatos pathway, leading to extensive DNA fragmentation and cell death (Wang et al., 2011). PARP1 activation occurs following any magnitude of DNA damage, with the amount of activation proportional to the severity of genetic stress. The activity of PARP1 results in the formation of PAR polymer chains which ultimately becomes highly toxic to the cell (Andrabi et al., 2006).
The high dependency of PARP1 on NAD + availability leads to the depletion of the NAD + surplus, leading to subsequent cellular stress. Administration of NAD + to energetically starved cells, or the reinstatement of glycolytic factors within the glycolysis cycle, have been shown to reverse the effects of NAD + depletion, offering neuroprotection and preventing the activation of the parthanatos pathway (Fatokun et al., 2014). Circadian expression patterns of NAMPT, the rate limiting enzyme of the NAD + salvage pathway, results in a circadian rhythm of NAD + abundance within the cell, giving rise to circadian activity of PARP1 (Nakahata et al., 2009; Ramsey et al., 2009). Opposing duality of PARG and PARP1 processes may contribute to rhythmicity, but studies have shown that rhythmicity of PARP1 auto-ADP-ribosylation is maintained in spite of manipulation of PARG activity (Davidovic et al., 2001). Studies have shown that circadian PARP1 activity is independent of central pacemakers and heavily regulated by feeding (Damiola et al., 2000; Stokkan et al., 2001). Moreover, abundance of NAD + has been shown to not sufficiently account for the circadian activity of auto-ADP-ribosylation of PARP1, at least in vitro (Asher et al., 2010). However, unlike PARP1’s endogenous activity, NAD + activity has been shown to be under the influence of the central clock (Ramsey et al., 2009).
Poly (ADP-ribose) polymerase 1 has also been shown to co-immunoprecipitate with CLOCK and BMAL1. PARP1 has the ability to rhythmically affect CLOCK activity, and highly disrupted circadian and rhythmic activity of the CLOCK-BMAL1 dimer is attributed to inhibition of PARP1 activity (Asher et al., 2010). Wild type CLOCK and BMAL1 forms have exhibited circadian rhythmicity when binding to Per2 promoters, whereas PARP1 knockout mice exhibited much higher levels of CLOCK and BMAL1 activity. PARP1 knockout and wild type animals exposed to arrhythmic feeding patterns did not exhibit altered differences in genetic expression, but PARP1 knockout mice exhibited a lower level of mRNA of target genes of CLOCK and BMAL1. Additionally, PARP1 knockout mice exhibited a lengthened Tau compared to wild-type mice, suggesting that direct consequences of PARP1 activity could possibly manifest in the SCN (Asher et al., 2010).
Another highly apparent interaction between E3 ligases, DNA repair and PARP1 activity has been portrayed through the E3 ligase, Iduna. Iduna’s really interesting new gene (RING) finger domain’s zinc binding activity is responsible for its E3 ligase activity, and mutations in the H54A and C60A domains result in absence of ubiquitination activity. The Iduna YRAA mutant disrupts the PAR-binding abilities, and the autoubiquitination abilities of Iduna’s E3 ligase activity have been shown to be directly dependent on PAR concentrations. Two of the primary targets of Iduna have been shown to be PARP1 and PARsylated PARP1. The presence of proteasomal inhibitors, such as MG132, has been shown to inhibit Iduna’s ability to degrade PARP1 and PARsylated PAR, affirming the ubiquitinated PAR-dependent degradation of PARP1 by Iduna. Microirradiation induced DNA damage resulted in an eightfold increase in the amount of apurinic/apyrimidinic sites. The prevention of this increase is possible through overexpression of Iduna. However, Iduna YRAA mutants were unable to suppress the increase of apurinic/apyrimidinic sites due to the lack of PAR binding ability, showing that PAR-binding is crucial for suppression of genetic lesions. Iduna C60A mutants also failed to reduce the increase in DNA lesions, emphasizing that the E3 ligase activity of Iduna is also crucial for DNA maintenance and repair. These complex interactions between DNA damage, PAR-related activity, and E3 ligase activity begin to paint a picture of highly interrelated intricacy, affirming that Iduna acts as a PAR-dependent ubiquitin E3 ligase. Iduna has also been shown to protect the brain from stroke by disruption of the parthanatos mechanism (Kang et al., 2011).
The earlier explored interactions allow for the analysis of the effects of PARP1 activity and subsequent PAR accumulation on the development and course of PD. Accumulation of α-Syn PFFs has been shown to activate PARP1, leading to the generation of PAR aggregates, compounding into large combined aggregates that become toxic for given cells, eventually resulting in cell death via parthanatos (Kam et al., 2018). After administration of α-Syn PFFs in mice, higher PARP1 activity was observed for a period of 2 weeks, whereas administration of PAR inhibitors prevented α-syn PFFs induced cell death. Deletion of PARP1 through CRISPR similarly resulted in prevention of α-syn PFFs induced PARP1 activity, cell death, α-Syn aggregation, as well as cell-to-cell α-Syn PFFs transmission. Nitric oxide (NO) synthesis was also increased after α-syn PFFs administration, whereas NO inhibitors prevented this activation. After administration, increased PAR levels and PARP1 activity were observed, but PAR levels were not increased due to the absence of PARP1 in PARP1 knockout mice, and can be attributed to the fact that PAR aggregation is a result of PARP1 activity. Parthanatos in DA neurons was observed after administration in wild-type mice, but loss of DA neurons was not observed in PARP1 knockout mice and in mice previously injected with ABT-888, a PARP1 inhibitor. PAR has been shown to directly increase the rate of aggregation of α-Syn PFFs in neuronal cells, and 20% of PAR binding to these fibrils was observed in the mouse brain. PAR has also been shown to produce a toxic strain of α-Syn PFFs in the form of more compact and misfolded aggregation. The highly accelerated initial degradation of DA neurons in vivo can be attributed to the presence of PAR α-Syn PFFs rather than the generic species of α-Syn PFFs. PD patients have also exhibited higher levels of PAR in their cerebrospinal fluid as well as in the substantia nigra (Kam et al., 2018). The link between PARP1 and E3 ligase activity, the affected pathology in PD, and underlying circadian processes all ultimately point to a highly interrelated mechanism, implying the possibilities of circadian treatment as a preventative and treatable measure for PD.
The complex web of molecular interactions points to the possibility of much more entangled pathological similarities between PD pathology, DNA damage and repair, and the endogenous regulation of core circadian genetic elements through E3 ligase and PARP1 activity. This specific targeting of PARP1 in respect to PD treatment exhibits mirrored similarity to the mechanism through which SIRT1 is targeted in AD treatment (Wong and Tang, 2016). The interconnectedness between circadian rhythmicity, protein aggregation, and metabolic activity is exemplified by PARP1, suggesting its role as a therapeutic target in treatment for aggressive progression of PD (Figure 3).
Circadian irregularities have been shown to have bi-directional physiological and metabolic implications on general health, as well as influence on and influence from neurodegenerative disorders. E3 ligase and PARP1 activity are essential in regulating both circadian rhythmicity and proper neuronal function. Thus, the dysregulation of these pathways have the capability of causing cellular stress and DNA damage that could further lead to neurodegenerative diseases like AD and PD. DNA damage response (DDR) has been shown to be reliant on the circadian properties of the cell through previously explored mechanisms of PARP1 and E3 ligases. Aberrant circadian qualities may disrupt the DDR or in turn be disrupted by hyperactivity of PARP1 and E3 ligases, ultimately leading to PAR induced accelerated accumulation of neurotoxic strains of α-Syn, further activating PARP1, contributing to the pathogenesis of PD. Regulating proper circadian function may be crucial for preventing the mechanistic onset of molecular complications heralding neurodegenerative distress.
AS, SL, and SK designed and planned this review manuscript. AS, SL, HY, and SK wrote the manuscript. AS, SL, HK, SH, and SK contributed to the literature search, collection, and summary. HY provided amendments to draft versions of the manuscript. All authors contributed to the article and approved the submitted version.
SL was supported by a grant from the Johns Hopkins University, Provost’s Undergraduate Research Award (PURA). HK was supported by Williams Scholar Award and this work was also supported by the NSF 1831962 and 1827847 grants.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Andrabi, S. A., Kim, N. S., Yu, S. W., Wang, H., Koh, D. W., Sasaki, M., et al. (2006). Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. U.S.A. 103, 18308–18313.
Asher, G., Reinke, H., Altmeyer, M., Gutierrez-Arcelus, M., Hottiger, M. O., and Schibler, U. (2010). Poly(ADP-ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell 142, 943–953. doi: 10.1016/j.cell.2010.08.016
Braak, H., and Del Tredici, K. (2008). Invited article: nervous system pathology in sporadic Parkinson disease. Neurology 70, 1916–1925. doi: 10.1212/01.wnl.0000312279.49272.9f
Breen, D. P., Vuono, R., Nawarathna, U., Fisher, K., Shneerson, J. M., Reddy, A. B., et al. (2014). Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol. 71, 589–595.
Busino, L., Bassermann, F., Maiolica, A., Lee, C., Nolan, P. M., Godinho, S. I., et al. (2007). SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science 316, 900–904. doi: 10.1126/science.1141194
Cai, Y., Liu, S., Sothern, R. B., Xu, S., and Chan, P. (2010). Expression of clock genes Per1 and Bmal1 in total leukocytes in health and Parkinson’s disease. Eur. J. Neurol. 17, 550–554. doi: 10.1111/j.1468-1331.2009.02848.x
Chiu, J. C., Vanselow, J. T., Kramer, A., and Edery, I. (2008). The phospho-occupancy of an atypical SLIMB-binding site on PERIOD that is phosphorylated by DOUBLETIME controls the pace of the clock. Genes Dev. 22, 1758–1772. doi: 10.1101/gad.1682708
Cox, K. H., and Takahashi, J. S. (2019). Circadian clock genes and the transcriptional architecture of the clock mechanism. J. Mol. Endocrinol. 63, R93–R102.
Damiola, F., Le Minh, N., Preitner, N., Kornmann, B., Fleury-Olela, F., and Schibler, U. (2000). Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 14, 2950–2961. doi: 10.1101/gad.183500
Dardente, H., Mendoza, J., Fustin, J. M., Challet, E., and Hazlerigg, D. G. (2008). Implication of the F-Box Protein FBXL21 in circadian pacemaker function in mammals. PLoS One 3:e3530. doi: 10.1371/journal.pone.0003530
Dauer, W., and Przedborski, S. (2003). Parkinson’s disease: mechanisms and models. Neuron 39, 889–909.
David, K. K., Andrabi, S. A., Dawson, T. M., and Dawson, V. L. (2009). Parthanatos, a messenger of death. Front. Biosci. 14, 1116–1128. doi: 10.2741/3297
Davidovic, L., Vodenicharov, M., Affar, E. B., and Poirier, G. G. (2001). Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp. Cell. Res. 268, 7–13. doi: 10.1006/excr.2001.5263
Davies, J., Chen, J., Pink, R., Carter, D., Saunders, N., Sotiriadis, G., et al. (2015). Orexin receptors exert a neuroprotective effect in Alzheimer’s disease (AD) via heterodimerization with GPR103. Sci. Rep. 5:12584.
Delenclos, M., Burgess, J. D., Lamprokostopoulou, A., Outeiro, T. F., Vekrellis, K., and Mclean, P. J. (2019). Cellular models of alpha-synuclein toxicity and aggregation. J. Neurochem. 150, 566–576. doi: 10.1111/jnc.14806
Ding, H., Liu, S., Yuan, Y., Lin, Q., Chan, P., and Cai, Y. (2011). Decreased expression of Bmal2 in patients with Parkinson’s disease. Neurosci. Lett. 499, 186–188. doi: 10.1016/j.neulet.2011.05.058
Do, M. T., and Yau, K. W. (2010). Intrinsically photosensitive retinal ganglion cells. Physiol. Rev. 90, 1547–1581.
Doktor, B., Damulewicz, M., and Pyza, E. (2019). Effects of MUL1 and PARKIN on the circadian clock, brain and behaviour in Drosophila Parkinson’s disease models. BMC Neurosci. 20:24. doi: 10.1186/s12868-019-0506-8
Dunlap, J. C., and Loros, J. J. (2017). Making time: conservation of biological clocks from fungi to animals. Microbiol. Spectr. 5:39.
Eide, E. J., Woolf, M. F., Kang, H., Woolf, P., Hurst, W., Camacho, F., et al. (2005). Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Mol. Cell. Biol. 25, 2795–2807.
Fatokun, A. A., Dawson, V. L., and Dawson, T. M. (2014). Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 171, 2000–2016. doi: 10.1111/bph.12416
Godinho, S. I., Maywood, E. S., Shaw, L., Tucci, V., Barnard, A. R., Busino, L., et al. (2007). The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science 316, 897–900. doi: 10.1126/science.1141138
Gossan, N. C., Zhang, F., Guo, B., Jin, D., Yoshitane, H., Yao, A., et al. (2014). The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic Acids Res. 42, 5765–5775. doi: 10.1093/nar/gku225
Gu, Z., Wang, B., Zhang, Y. B., Ding, H., Zhang, Y., Yu, J., et al. (2015). Association of ARNTL and PER1 genes with Parkinson’s disease: a case-control study of Han Chinese. Sci. Rep. 5:15891.
Herceg, Z., and Wang, Z. Q. (2001). Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat. Res. 477, 97–110. doi: 10.1016/s0027-5107(01)00111-7
Hirano, A., Yumimoto, K., Tsunematsu, R., Matsumoto, M., Oyama, M., Kozuka-Hata, H., et al. (2013). FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell 152, 1106–1118. doi: 10.1016/j.cell.2013.01.054
Huseby, C. J., Bundschuh, R., and Kuret, J. (2019). The role of annealing and fragmentation in human tau aggregation dynamics. J. Biol. Chem. 294, 4728–4737. doi: 10.1074/jbc.ra118.006943
Kam, T. I., Mao, X., Park, H., Chou, S. C., Karuppagounder, S. S., Umanah, G. E., et al. (2018). Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science 362:eaat8407. doi: 10.1126/science.aat8407
Kang, H. C., Lee, Y. I., Shin, J. H., Andrabi, S. A., Chi, Z., Gagné, J. P., et al. (2011). Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proc. Natl. Acad. Sci. U.S.A. 108, 14103–14108. doi: 10.1073/pnas.1108799108
Kang, J. E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., et al. (2009). Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. doi: 10.1126/science.1180962
Kilduff, T. S., Lein, E. S., De La Iglesia, H., Sakurai, T., Fu, Y. H., and Shaw, P. (2008). New developments in sleep research: molecular genetics, gene expression, and systems neurobiology. J. Neurosci. 28, 11814–11818. doi: 10.1523/jneurosci.3768-08.2008
Kocahan, S., and Dogan, Z. (2017). Mechanisms of Alzheimer’s disease pathogenesis and prevention: the brain, neural pathology, N-methyl-D-aspartate receptors, tau protein and other risk factors. Clin. Psychopharmacol. Neurosci. 15, 1–8. doi: 10.9758/cpn.2017.15.1.1
Koh, K., Zheng, X., and Sehgal, A. (2006). JETLAG resets the Drosophila circadian clock by promoting light-induced degradation of TIMELESS. Science 312, 1809–1812. doi: 10.1126/science.1124951
Lamaze, A., Lamouroux, A., Vias, C., Hung, H. C., Weber, F., and Rouyer, F. (2011). The E3 ubiquitin ligase CTRIP controls CLOCK levels and PERIOD oscillations in Drosophila. EMBO Rep. 12, 549–557. doi: 10.1038/embor.2011.64
Ling, H. H., Beaule, C., Chiang, C. K., Tian, R., Figeys, D., and Cheng, H. Y. (2014). Time-of-day- and light-dependent expression of ubiquitin protein ligase E3 component N-recognin 4 (UBR4) in the suprachiasmatic nucleus circadian clock. PLoS One 9:e103103. doi: 10.1371/journal.pone.0103103
Liu, J., Zou, X., Gotoh, T., Brown, A. M., Jiang, L., Wisdom, E. L., et al. (2018). Distinct control of PERIOD2 degradation and circadian rhythms by the oncoprotein and ubiquitin ligase MDM2. Sci. Signal. 11:eaau0715. doi: 10.1126/scisignal.aau0715
Liu, Z., Wang, F., Tang, M., Zhao, Y., and Wang, X. (2019). Amyloid beta and tau are involved in sleep disorder in Alzheimer’s disease by orexin A and adenosine A(1) receptor. Int. J. Mol. Med. 43, 435–442.
Martire, S., Mosca, L., and D’erme, M. (2015). PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mech. Ageing Dev. 146-148, 53–64. doi: 10.1016/j.mad.2015.04.001
Musiek, E. S., and Holtzman, D. M. (2016). Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354, 1004–1008. doi: 10.1126/science.aah4968
Naidoo, N., Song, W., Hunter-Ensor, M., and Sehgal, A. (1999). A role for the proteasome in the light response of the timeless clock protein. Science 285, 1737–1741. doi: 10.1126/science.285.5434.1737
Nakahata, Y., Sahar, S., Astarita, G., Kaluzova, M., and Sassone-Corsi, P. (2009). Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 324, 654–657. doi: 10.1126/science.1170803
Nandi, D., Tahiliani, P., Kumar, A., and Chandu, D. (2006). The ubiquitin-proteasome system. J. Biosci. 31, 137–155.
Ohsaki, K., Oishi, K., Kozono, Y., Nakayama, K., Nakayama, K. I., and Ishida, N. (2008). The role of {beta}-TrCP1 and {beta}-TrCP2 in circadian rhythm generation by mediating degradation of clock protein PER2. J. Biochem. 144, 609–618. doi: 10.1093/jb/mvn112
Patke, A., Young, M. W., and Axelrod, S. (2020). Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell. Biol. 21, 67–84. doi: 10.1038/s41580-019-0179-2
Philpott, J. M., Narasimamurthy, R., Ricci, C. G., Freeberg, A. M., Hunt, S. R., Yee, L. E., et al. (2020). Casein kinase 1 dynamics underlie substrate selectivity and the PER2 circadian phosphoswitch. eLife 9:e52343.
Porter, B., Macfarlane, R., and Walker, R. (2008). The frequency and nature of sleep disorders in a community-based population of patients with Parkinson’s disease. Eur. J. Neurol. 15, 50–54.
Qian, J., and Scheer, F. (2016). Circadian system and glucose metabolism: implications for physiology and disease. Trends Endocrinol. Metab. 27, 282–293. doi: 10.1016/j.tem.2016.03.005
Radogna, F., Diederich, M., and Ghibelli, L. (2010). Melatonin: a pleiotropic molecule regulating inflammation. Biochem. Pharmacol. 80, 1844–1852. doi: 10.1016/j.bcp.2010.07.041
Ramsey, K. M., Yoshino, J., Brace, C. S., Abrassart, D., Kobayashi, Y., Marcheva, B., et al. (2009). Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 324, 651–654. doi: 10.1126/science.1171641
Rasmussen, M. K., Mestre, H., and Nedergaard, M. (2018). The glymphatic pathway in neurological disorders. Lancet Neurol. 17, 1016–1024. doi: 10.1016/s1474-4422(18)30318-1
Reischl, S., Vanselow, K., Westermark, P. O., Thierfelder, N., Maier, B., Herzel, H., et al. (2007). Beta-TrCP1-mediated degradation of PERIOD2 is essential for circadian dynamics. J. Biol. Rhythms 22, 375–386. doi: 10.1177/0748730407303926
Sathyanarayanan, S., Zheng, X., Kumar, S., Chen, C. H., Chen, D., Hay, B., et al. (2008). Identification of novel genes involved in light-dependent CRY degradation through a genome-wide RNAi screen. Genes Dev. 22, 1522–1533. doi: 10.1101/gad.1652308
Shirogane, T., Jin, J., Ang, X. L., and Harper, J. W. (2005). SCFbeta-TRCP controls clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. J. Biol. Chem. 280, 26863–26872. doi: 10.1074/jbc.m502862200
Siepka, S. M., Yoo, S. H., Park, J., Song, W., Kumar, V., Hu, Y., et al. (2007). Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell 129, 1011–1023. doi: 10.1016/j.cell.2007.04.030
Stokkan, K. A., Yamazaki, S., Tei, H., Sakaki, Y., and Menaker, M. (2001). Entrainment of the circadian clock in the liver by feeding. Science 291, 490–493. doi: 10.1126/science.291.5503.490
Tong, X., Zhang, D., Guha, A., Arthurs, B., Cazares, V., Gupta, N., et al. (2015). CUL4-DDB1-CDT2 E3 ligase regulates the molecular clock activity by promoting ubiquitination-dependent degradation of the mammalian CRY1. PLoS One 10:e0139725. doi: 10.1371/journal.pone.0139725
Verbaan, D., Van Rooden, S. M., Visser, M., Marinus, J., and Van Hilten, J. J. (2008). Nighttime sleep problems and daytime sleepiness in Parkinson’s disease. Mov. Disord. 23, 35–41. doi: 10.1002/mds.21727
Videnovic, A., and Golombek, D. (2017). Circadian dysregulation in Parkinson’s disease. Neurobiol Sleep Circadian Rhythms 2, 53–58. doi: 10.1016/j.nbscr.2016.11.001
Wang, Y., Kim, N. S., Haince, J. F., Kang, H. C., David, K. K., Andrabi, S. A., et al. (2011). Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 4:ra20. doi: 10.1126/scisignal.2000902
Wilking, M., Ndiaye, M., Mukhtar, H., and Ahmad, N. (2013). Circadian rhythm connections to oxidative stress: implications for human health. Antioxid. Redox Signal. 19, 192–208. doi: 10.1089/ars.2012.4889
Wong, S. Y., and Tang, B. L. (2016). SIRT1 as a therapeutic target for Alzheimer’s disease. Rev. Neurosci. 27, 813–825. doi: 10.1515/revneuro-2016-0023
Yin, L., Joshi, S., Wu, N., Tong, X., and Lazar, M. A. (2010). E3 ligases Arf-bp1 and Pam mediate lithium-stimulated degradation of the circadian heme receptor Rev-erb alpha. Proc. Natl. Acad. Sci. U.S.A. 107, 11614–11619. doi: 10.1073/pnas.1000438107
Yoo, S. H., Ko, C. H., Lowrey, P. L., Buhr, E. D., Song, E. J., Chang, S., et al. (2005). A noncanonical E-box enhancer drives mouse Period2 circadian oscillations in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 2608–2613. doi: 10.1073/pnas.0409763102
Yoo, S. H., Mohawk, J. A., Siepka, S. M., Shan, Y., Huh, S. K., Hong, H. K., et al. (2013). Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell 152, 1091–1105. doi: 10.1016/j.cell.2013.01.055
Keywords: circadian rhythm, DNA damage, PARP1, PAR-dependent E3 ligase, α-synuclein pre-formed fibrils, β-amyloid peptides, Parkinson’s disease, Alzheimer’s disease
Citation: Sharma A, Lee S, Kim H, Yoon H, Ha S and Kang SU (2020) Molecular Crosstalk Between Circadian Rhythmicity and the Development of Neurodegenerative Disorders. Front. Neurosci. 14:844. doi: 10.3389/fnins.2020.00844
Received: 27 April 2020; Accepted: 20 July 2020;
Published: 06 August 2020.
Edited by:
Phyllis Zee, Northwestern University, United StatesReviewed by:
Joanna C. Chiu, University of California, Davis, United StatesCopyright © 2020 Sharma, Lee, Kim, Yoon, Ha and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sung Ung Kang, c2thbmczNEBqaG1pLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.