 Gautam Wali1,2*†
Gautam Wali1,2*† Kishore Raj Kumar1,2,3,4,5
Kishore Raj Kumar1,2,3,4,5 Erandhi Liyanage1
Erandhi Liyanage1 Ryan L. Davis1,2,4
Ryan L. Davis1,2,4 Alan Mackay-Sim2,6†Carolyn M. Sue1,2,5*†
Alan Mackay-Sim2,6†Carolyn M. Sue1,2,5*†- 1Department of Neurogenetics, Royal North Shore Hospital, Kolling Institute of Medical Research, The University of Sydney, Sydney, NSW, Australia
- 2Faculty of Medicine and Health, Sydney Medical School, The University of Sydney, Sydney, NSW, Australia
- 3Molecular Medicine Laboratory, Department of Neurology, Concord Hospital, Sydney, NSW, Australia
- 4Kinghorn Centre for Clinical Genomics, Garvan Institute of Medical Research, Sydney, NSW, Australia
- 5Department of Neurology, Royal North Shore Hospital, Northern Sydney Local Health District, Sydney, NSW, Australia
- 6Griffith Institute for Drug Discovery, Griffith University, Brisbane, QLD, Australia
Mutations in SPG7 and SPAST are common causes of hereditary spastic paraplegia (HSP). While some SPG7 mutations cause paraplegin deficiency, other SPG7 mutations cause increased paraplegin expression. Mitochondrial function has been studied in models that are paraplegin-deficient (human, mouse, and Drosophila models with large exonic deletions, null mutations, or knockout models) but not in models of mutations that express paraplegin. Here, we evaluated mitochondrial function in olfactory neurosphere-derived cells, derived from patients with a variety of SPG7 mutations that express paraplegin and compared them to cells derived from healthy controls and HSP patients with SPAST mutations, as a disease control. We quantified paraplegin expression and an extensive range of mitochondrial morphology measures (fragmentation, interconnectivity, and mass), mitochondrial function measures (membrane potential, oxidative phosphorylation, and oxidative stress), and cell proliferation. Compared to control cells, SPG7 patient cells had increased paraplegin expression, fragmented mitochondria with low interconnectivity, reduced mitochondrial mass, decreased mitochondrial membrane potential, reduced oxidative phosphorylation, reduced ATP content, increased mitochondrial oxidative stress, and reduced cellular proliferation. Mitochondrial dysfunction was specific to SPG7 patient cells and not present in SPAST patient cells, which displayed mitochondrial functions similar to control cells. The mitochondrial dysfunction observed here in SPG7 patient cells that express paraplegin was similar to the dysfunction reported in cell models without paraplegin expression. The p.A510V mutation was common to all patients and was the likely species associated with increased expression, albeit seemingly non-functional. The lack of a mitochondrial phenotype in SPAST patient cells indicates genotype-specific mechanisms of disease in these HSP patients.
Introduction
Hereditary spastic paraplegia (HSP) is an inherited, progressive neurodegenerative condition causing spasticity and weakness in the lower limbs as a consequence of corticospinal tract degeneration (Hedera et al., 2005; Salinas et al., 2008; Kumar et al., 2015). Mutations in SPG7 and SPAST are the most common causes of HSP. SPG7 encodes for paraplegin, a mitochondrial matrix protease embedded in the inner mitochondrial membrane (Casari et al., 1998). Paraplegin is involved in multiple mitochondrial processes including mitochondrial protein quality surveillance, ribosome assembly, and mitochondrial biogenesis (Quirós et al., 2015). SPG7 mutations can cause loss of paraplegin (Atorino et al., 2003) or increased paraplegin expression (Thal et al., 2015; Pfeffer et al., 2014). The effects of SPG7 mutations leading to a loss of paraplegin expression on mitochondrial function have been characterized. For example, fibroblasts derived from a patient carrying a homozygous 9.5-kb deletion within the SPG7 gene that caused paraplegin deficiency exhibited impaired respiratory chain complex I activity. When the cells were exposed to hydrogen peroxide, to stress mitochondrial function, they were found to have lower ATP synthesis rate and reduced cell viability (Atorino et al., 2003). Consistent with this, a SPG7-null mutant Drosophila model displayed reduced activity of respiratory chain complexes I and II, severely swollen and aberrantly shaped mitochondria in the synaptic terminals of photoreceptors, and neuronal and muscular degeneration (Pareek et al., 2018).
There is limited information on mitochondrial function in patient cells and tissues with SPG7 mutations that do not cause a loss of paraplegin. Post-mortem analysis of a patient with homozygous p.Ala510Val mutations showed an increased expression of paraplegin in neurites of the cerebellar and cerebral cortex (Thal et al., 2015), but mitochondria function was not evaluated. In SPG7 patients carrying compound heterozygous mutations with increased paraplegin expression, the skeletal muscle biopsies showed increased mitochondrial biogenesis, multiple mitochondrial DNA deletions, and cytochrome c oxidase-deficient fibers (Pfeffer et al., 2014). Mitochondrial morphology evaluation of fibroblasts from these patients showed an increased mitochondrial mass and an increased number and length of mitochondrial networks; however, mitochondrial respiratory chain function was not evaluated. Inhibition of the respiratory chain function can lead to excess generation of reactive oxygen species, subsequently leading to cell death mediated by oxidative stress (Keane et al., 2011). This link has been shown for many neurodegenerative diseases (Moreira et al., 2010; Keane et al., 2011). Here, we characterize mitochondria morphology, respiratory chain function, and oxidative stress in paraplegin expressing SPG7 patient cells.
SPAST encodes for spastin, a microtubule-severing protein (Errico et al., 2002; Orlacchio et al., 2004). SPAST patient cells have impaired organelle transport, which includes mitochondria (Abrahamsen et al., 2013; Wali et al., 2020). However, it remains to be determined whether mitochondrial respiratory chain function is affected in SPAST patient cells. A study investigating muscle biopsies from SPAST patients did not show any aberrant activity of complexes I, II, and IV or citrate synthase (McDermott et al., 2003). Histochemical reactions, cytochrome c oxidase, and succinate dehydrogenase were also normal (McDermott et al., 2003). However, a recent study in a single SPAST patient fibroblast cell line showed that the patient cells had less tubular and more fragmented mitochondria, reduced mitochondrial membrane potential, and altered mitochondrial distribution (Dong et al., 2018).
In the present study, we quantified paraplegin expression and a range of mitochondrial morphology measures (fragmentation, interconnectivity, and mass), mitochondrial functions (membrane potential, oxidative phosphorylation, and oxidative stress), and cell proliferation. These measures were performed on olfactory neurosphere-derived stem cells (ONS cells) derived from HSP patients with SPG7 mutations, from healthy controls, and from HSP patients with SPAST mutations.
Materials and Methods
Participants
SPG7 and SPAST HSP patients involved in this study were examined by neurologists CMS and KRK. Details of the patient clinical characteristics and causative gene mutations are listed in Table 1. Control and patient samples used were matched for age and gender. Mean age range of SPG7 patients was 60 years, and those of SPAST patients and healthy controls were 51 and 60 years, respectively. Olfactory neurosphere-derived cells (ONS cells) were prepared from olfactory mucosa biopsies of participants (Féron et al., 1998, 2013) with their informed and written consent.
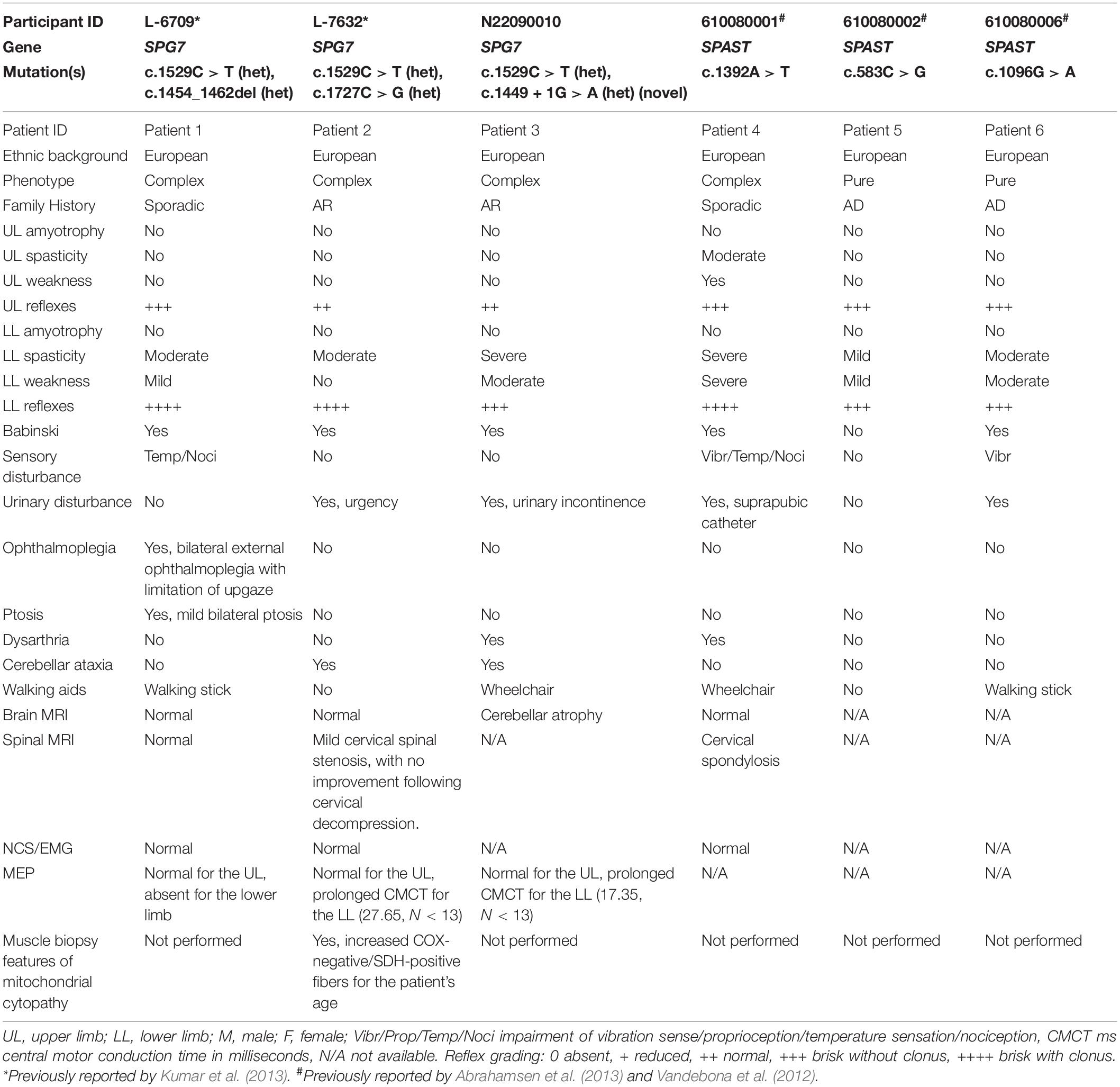
Table 1. Clinical features and mutation details of HSP patients with SPG7 and SPAST mutations.
Targeted Next-Generation Sequencing
All patients except patient 3 have been previously reported (refer to Table 1). Patient 3 was identified using targeted next-generation sequencing, as described elsewhere (Kumar et al., 2013). In brief, patients with an apparent autosomal recessive or sporadic mode of inheritance were screened for nine autosomal recessive HSP-associated genes using a PCR-based library preparation and Roche Junior 454 next-generation sequencing, after exclusion of SPAST mutations. Amplicons that did not achieve 30 reads were resequenced with a conventional Sanger sequencing method. All pathogenic or potentially pathogenic variants were confirmed by Sanger sequencing on an ABI Prism 3130xl Genetic Analyzer platform (Applied Biosystems).
Cell Culture
ONS cells were derived from olfactory mucosa biopsies using our published techniques (Matigian et al., 2010; Féron et al., 2013). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/F12 (Gibco) with 10% fetal bovine serum at 37°C and 5% CO2. Please refer to our previous publication (Féron et al., 2013) for a detailed protocol of generating olfactory neurosphere-derived cells from olfactory mucosal biopsies.
Paraplegin Expression
Protein Isolation
Cells cultured in T-25 flasks were washed three times with Dulbecco’s phosphate-buffered saline (Gibco 14190250) to completely remove culture media. Cells were lysed with cell lysis buffer (Sigma, C3228) supplemented with a protease inhibitor (Thermo Fisher Scientific, 78429). Cells were harvested by scraping and maintained on ice over a rocker for 30 min. Then, the samples were centrifuged at 10,000 g for 10 min at 4°C. The protein samples were then collected and quantified using the BCA pierce TM BCA Protein Assay kit (Thermo Fisher Scientific, 23225).
Western Blot
Total cellular protein (20 μg) was resolved on a 12% Bis-Tris polyacrylamide gel and transferred onto a nitrocellulose membrane. The membrane blot was incubated overnight at 4°C with the primary mouse anti-human SPG7 monoclonal antibody (dilution 1/10,000; OriGene). Following this, the membrane was incubated with goat anti-mouse secondary antibody coupled to horseradish peroxidase (dilution 1/4,000, Sigma-Aldrich). Bands were visualized using Pierce SuperSignal West Femto Chemiluminescent Substrate on a Bio-Rad ChemiDoc XL. Paraplegin expression was normalized to GAPDH (dilution 1/10,000; Abcam) expression levels using ImageJ Version 1.52u software (Schneider et al., 2012).
Western Blot Positive Control
Human HEL293 cells overexpressing paraplegin were used to validate the antibody (OriGene, LC401085). The paraplegin band size was 88 kDa for both the control samples tested and the positive control, showing antibody specificity (Supplementary Figure S1).
Flow Cytometry
Cells were fixed and permeabilized with the BD Cytofix/Cytoperm reagent for 20 min at room temperature. Cells were washed with BD Perm/Wash buffer and then incubated with mouse anti-human SPG7 monoclonal antibody (dilution 1/10,000; OriGene) or the IgG1 isotype control for 1 h. Cells were washed and incubated with Alexa Fluor secondary antibody for 30 min. Cells were washed, and the relative fluorescence intensity was measured for 10,000 events per sample on a BD LSRFortessa, and the data were analyzed using BD FACS Diva software. Mean fluorescence intensity was calculated as the fluorescence intensity of the paraplegin antibody to the intensity of the isotype control antibody.
Mitochondrial Morphology
Cells were treated with 100 nM MitoTracker green for 30 min at 37°C to label active mitochondria (Park et al., 2014). Images of mitochondria-labeled cells were captured using a 63× objective on a confocal microscope. Using ImageJ image analysis software, raw images were binarized, and mitochondrial morphological characteristics of the aspect ratio (AR), indicating mitochondrial length, form factor (FF), and mitochondrial branching, were calculated as described before (Mortiboys et al., 2008). AR is the ratio of the major to minor axis of the mitochondria, and FF is calculated as (perimeter of mitochondria2)/(4π ∗ area of mitochondria) (Mortiboys et al., 2008). Thirty cells were imaged and analyzed per cell line per technical replicate experiment.
Mitochondrial Mass and Mitochondrial Membrane Potential
Mitochondrial mass and membrane potential were assessed by labeling mitochondria with 100 nM MitoTracker green for 30 min (as above) and 50 nM non-quenching TMRM for 30 min at 37°C, respectively. The cells were washed twice with PBS before measuring fluorescence using a BD LSRFortessa™. Fluorescence was measured for 10,000 events per sample, and the data were analyzed using the BD FACS Diva™ software (Gautier et al., 2012). Mitochondrial membrane potential was normalized to MitoTracker green fluorescence, a relative estimate of the mitochondrial mass per cell.
MitoSOX and CM-H2DCFDA
ONS cells (10,000) were plated in each well of a black-walled 96-well plate and cultured for 24 h before being assessed. The cells were stained with 5 μM MitoSOX Red for 15 min (as an indication of mitochondrial oxidative stress) (Teves et al., 2018) or 5 μM CM-H2DCFDA for 15 min (a measure of general cellular oxidative stress) (Wali et al., 2016) at 37°C. The cells were washed twice with PBS before fluorescence was measured using a VICTOR Nivo Multimode Microplate Reader. MitoSOX and CM-H2DCFDA fluorescence levels were normalized to mitochondrial mass, using dual MitoTracker green staining.
ATPlite Assay Measuring ATP Content
ONS cells (10,000 per well) were plated in a white-walled 96-well plate for 24 h before being assessed. The assay was performed as per the manufacturer’s instructions and as described before (Wali et al., 2016). Luminescence was measured using the VICTOR Nivo Multimode Microplate Reader. The ATPlite luminescence values were normalized to mitochondrial mass, using dual MitoTracker green staining.
Oxygen Consumption Rate
A Seahorse XF24-3 Bioanalyzer (Agilent Technologies) was used to measure mitochondrial oxygen consumption as described previously (Koentjoro et al., 2017). On day 1, 70,000 ONS cells per well were plated in a 24-well XF24 V7 cell culture microplate and cultured in DMEM/F12 (Gibco) with 10% fetal bovine serum at 37°C and 5% CO2 for 24 h. On day 2, the DMEM/F12 media was replaced with Seahorse XF Base media supplemented with 2 mM L-glutamine, and the pH was adjusted to 7.35 ± 0.05 (basal media). Cells were incubated in the basal media for 1 h at 37°C in a non-CO2 incubator. The plate was then assembled with a hydrated sensor cartridge and placed in the Seahorse flux analyzer. The oxygen consumption rate (OCR) was measured at baseline and after subsequent stepwise injection of the mitochondrial respiratory chain inhibitors, oligomycin (1 μM), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) (1 μM) and rotenone/antimycin A (0.5 μM). The OCR readings were normalized to cell number measured by CyQUANT assay in a sister plate. Multiple parameters of mitochondrial respiration were calculated based on the response of the cells to the respiratory chain inhibitors: (a) basal respiration, basal OCR - non-mitochondrial respiration rate; (b) ATP production, difference in OCR before and after oligomycin treatment; (c) maximal respiration, OCR measurement after FCCP treatment - OCR measurement after oligomycin treatment; and (d) spare respiratory capacity, maximal respiration - basal respiration.
Cell Proliferation
ONS cells (5,000) were seeded into each well of a 96-well plate. On day 0 (4 h post seeding), day 1 (24 h post seeding), and day 2 (48 h post seeding), the relative cell numbers were assessed using the CyQUANT® NF Cell Proliferation Assay Kit, following the manufacturer’s instructions.
Statistical Analysis
Three technical replicates were performed for all experiments: three control cell lines, three SPG7 patient lines, and three SPAST patient cell lines. One-way analysis of variance with Tukey’s multiple comparisons test was performed to compare control, SPG7, and SPAST sample results. Statistical analysis and graphing were performed with GraphPad (Prism 8).
Results
Clinical Phenotype
All patients with mutations in SPG7 had complex HSP phenotypes (clinical features detailed in Table 1). Complicating features in SPG7 patients included ophthalmoplegia (patient 1), ptosis (patient 1), ataxia (patients 2 and 3), and mitochondrial cytopathy on muscle biopsy (patient 1). In contrast, no SPAST patients (patients 4, 5, and 6) had typical mitochondrial disorder-related clinical signs (such as ocular myopathy, present in a range of mitochondrial myopathy syndromes; Pfeffer and Chinnery, 2013), and only one of three (patient 4) had additional features of dysarthria and upper limb weakness indicative of complicated HSP.
Genetic Studies
Six HSP patients, three with SPG7 mutations and three with SPAST mutations, were recruited to the study. These patients carried a variety of mutations (Table 1). SPG7 patients were patient 1 [c.1454_1462del (het); c.1529C > T (het)], patient 2 [c.1529C > T (het); c.1727C > G (het)], and patient 3 [c.1449 + 1G > A (novel het); c.1529C > T (het)]. SPAST patients were patient 4 (c.1392A > T), patient 5 (c.583C > G), and patient 6 (c.1096G > A). All patients except patient 3 have been reported previously (Abrahamsen et al., 2013; Kumar et al., 2013). SPG7 patient 3 was a 59-year-old man with a severe HSP phenotype complicated by upper limb cerebellar ataxia and cerebellar atrophy on MRI brain (Figures 1A,B). He was found to have the known common mutation NM_003119.3:c.1529C > T (p.Ala510Val) (Brugman et al., 2008; Bonn et al., 2010) and a novel canonical splice variant NM_003119.3:c.1449 + 1G > A, classified as pathogenic by the ACMG criteria (Richards et al., 2015) (PVS1, PM2, and PP3). Both variants were also present in a similarly affected sister (Figure 1C).
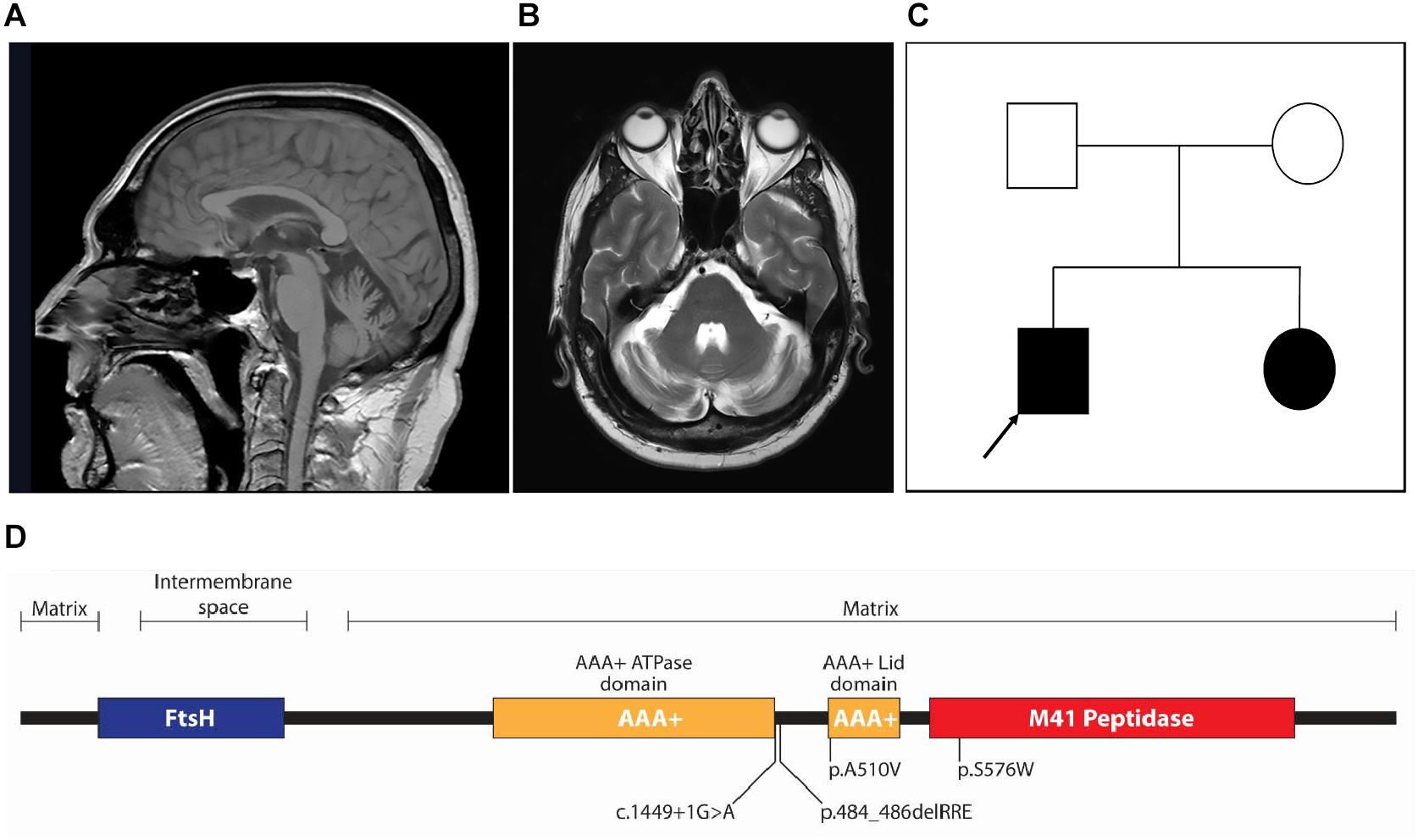
Figure 1. (A) Sagittal T1-weighted fast field echo and (B) transverse T2-weighted MRI images showing mild cerebellar atrophy of the patient (patient 3) carrying novel SPG7 variant. (C) Pedigree of patient 3 (arrow); both the proband and his affected sister carried two heterozygote variants in SPG7: a common mutation [NM_003119.3:c.1529C > T (p.Ala510Val)] and novel canonical splice variant (NM_003119.3:c.1449 + 1G > A). (D) Linear protein domain diagram of paraplegin. The AAA+ domain is shown as two subdomains (ATPase and Lid), separated by a linker region. Three of the four variants in the SPG7 patients in this study locate to the AAA+ ATPase domain and one to the M41 peptidase domain.
Protein Structure
All three SPG7 patients had a common heterozygous variant (c.1529C > T; p.A510V) in addition to different compounding heterozygous variants. All coding variants were predicted to be pathogenic by several prediction algorithms (e.g., MutationTaster, PolyPhen 2, SIFT, and PROVEAN), likely due to high evolutionary conservation of affected amino acids and the considerable physicochemical or likely structural changes imposed. Assessment of possible splicing changes for the non-coding variant using Human Splicing Finder 3.1 provided possible aberrant splicing outcomes (see below).
The pathogenicity of the c.1529C > T variant (p.A510V) common to all SPG7 patients in this study has been extensively studied (Bonn et al., 2010). It affects a highly conserved and completely buried alanine residue at the beginning of the structural AAA+ Lid domain (Figure 1D). Because of the constrained environment around the alanine residue, the introduction of a bulkier valine side chain (89 vs. 117 Da) would be predicted to destabilize the hydrophobic core of the Lid domain. Inspecting the crystal structure of the paraplegin AAA+ ATPase domain (amino acids 305–565; PDB entry 2qz4; Karlberg et al., 2009), of the three possible conformations of the valine side chain, two are predicted to clash with Arg486, and one could potentially be accommodated. This may afford expression of the mutant p.510V protein, although it would likely be dysfunctional for ATPase activity.
The second variant in patient 1 was a deletion variant (c.1454_1462del; p.485_487delRRE) removing three charged amino acids from the highly conserved alpha helix bundle of the AAA+ Lid domain (Figure 1D). The deletion would remove an entire alpha helix turn and three charged residues necessary for structural bonds to other helices in the domain (Karlberg et al., 2009) and is predicted to cause severe structural destabilization.
The second variant in patient 2 was a missense mutation (c.1727C > G; p.S576W) located within the magnesium metal ion coordination and peptidase active site of the M41 peptidase domain (Figure 1D). The substitution of tryptophan for serine is a very large physicochemical change, a mass change of 105 to 204 Da and a hydrophilic serine residue common in active sites vs. tryptophan, which has a bulky hydrophobic aromatic side chain. This would likely cause structural and functional disruption to the peptidase active site. Based on previous mutagenesis studies of this functional site, if the variant protein were expressed, it would likely form dysfunctional complexes (Shanmughapriya et al., 2015). In this situation, peptidase activity could be compensated for by AFG3L2, as demonstrated in yeast (Bonn et al., 2010).
The second variant in patient 3 was a novel canonical donor splice site disrupting mutation (c.1449 + 1G > A) that would impact the polypeptide chain from the end of the AAA+ ATPase domain at the start of the AAA+ domain linker region (Figure 1D). A number of possibilities for the consequent splicing defect include (1) skipping of exon 10 and a frameshift leading to termination after six amino acids; (2) intronic read-through into intron 10 before an alternate donor splice site is encountered 90 nucleotides downstream, resulting in an in-frame inclusion of 30 amino acids that would alter the length of the AAA+ ATPase domain linker; and (3) intronic read-through into intron 10 before encountering a termination codon 96 nucleotides downstream. Transcript analysis would be required to resolve the resulting splicing mechanism, although the variant protein product would almost certainly not be expressed.
Paraplegin Expression
Paraplegin expression in patient and control cells was evaluated by Western blot analysis and by flow cytometry (Figures 2A–D). Western blot analysis showed that SPG7 patient cells had significantly higher expression levels of paraplegin protein, compared to control cells (p < 0.01) (Figures 2A,C). SPAST patient cells had a trend of higher expression levels of paraplegin when compared to control cells, but this difference was not statistically significant (p = 0.06) (Figure 2C). To validate this finding, we evaluated paraplegin expression using flow cytometry. SPG7 patient cells had significantly higher mean fluorescence intensity, compared to control cells (p < 0.01) and SPAST patient cells (p < 0.01) (Figures 2B,D). Both techniques showed that paraplegin expression was significantly increased in SPG7 patient cells compared to control and SPAST patient cells.
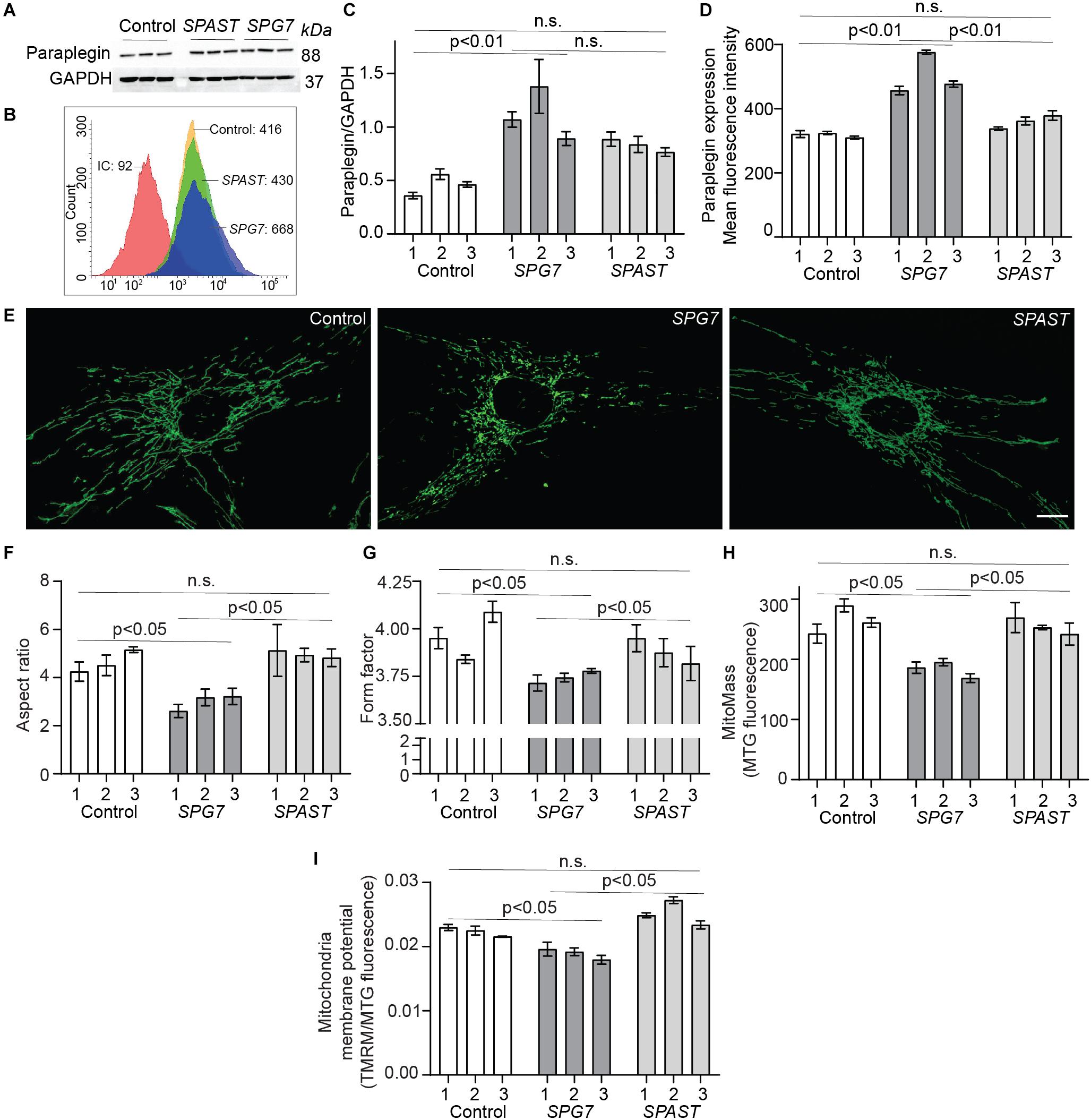
Figure 2. Paraplegin expression, mitochondrial morphology, and membrane potential in control, SPG7, and SPAST patient cells. (A) Western blot analysis of paraplegin and GAPDH expression. (B) Representative flow cytometry histogram of paraplegin fluorescence intensities. IC: Isotype control. (C) Western blot densitometry band quantification showing paraplegin expression normalized to GAPDH expression. (D) Paraplegin mean fluorescence intensity quantified by flow cytometry. (E) Representative images of MitoTracker-labeled mitochondria in cells from control, SPG7, and SPAST patient cells. Scale bar: 10 μm. (F,G) Mitochondria morphological analysis showing aspect ratio (mitochondria length) and form factor (degree of branching). (H) Mitochondrial mass was calculated as the fluorescence intensity of MitoTracker green (MTG) labeled mitochondria. (I) Mitochondrial membrane potential was measured using tetramethylrhodamine methyl ester (TMRM) fluorescence and normalized to mitochondrial mass (MTG fluorescence). Data presented as mean ± SEM. The bars present data from the three technical replicates. One-way analysis of variance with Tukey’s multiple comparisons test was performed to compare control, SPG7, and SPAST groups. n.s., not significant.
Mitochondrial Morphology
Mitochondria in SPG7 patient cells were highly fragmented and short (low AR) with a low degree of branching and interconnectivity (low FF; Figures 2E–G), showing typical signs of a dysfunctional mitochondrial network. Conversely, mitochondria in control cells and SPAST patient cells were longer and interconnected (p < 0.05; Figures 2E–G), indicative of a healthy mitochondrial network.
Consistent with their aberrant mitochondrial morphology, SPG7 patient cells also displayed lower mitochondrial mass (p < 0.05) and lower mitochondrial membrane potential (p < 0.05) (Figures 2H,I).
Oxidative Phosphorylation
To investigate if oxidative phosphorylation was impaired in HSP patient cells, we measured mitochondrial OCR using a Seahorse Bioanalyzer (Figure 3A). Compared to control and SPAST patient cells, SPG7 patient cells had reduced basal respiration (Figure 3B, control vs. SPG7: p < 0.05; SPG7 vs. SPAST: p = 0.09), significantly lower levels of oxygen consumption attributed to ATP production (Figure 3C, control vs. SPG7: p < 0.05; SPG7 vs. SPAST: p < 0.05), reduced maximal respiration (Figure 3D, control vs. SPG7: p < 0.001; SPG7 vs. SPAST: p < 0.01), and reduced spare respiratory capacity (Figure 3E, control vs. SPG7: p < 0.01; SPG7 vs. SPAST: p < 0.05). Consistent with this, SPG7 patient cells had lower total ATP content compared to both control and SPAST patient cells (p < 0.05) (Figure 3F). SPAST patient cells were not significantly different from control cells on any of these measures (Figures 3B–F).
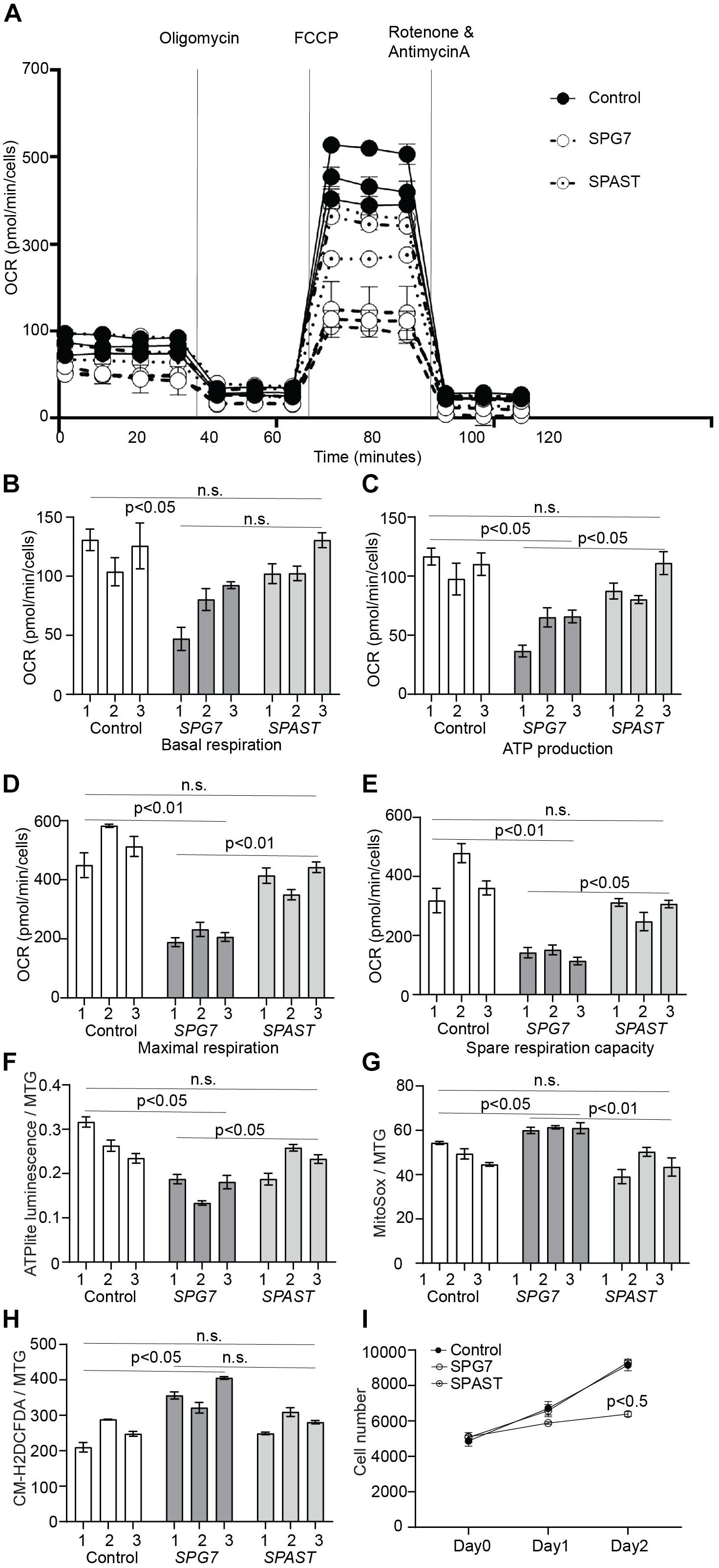
Figure 3. Oxidative respiration and oxidative stress in control, SPG7, and SPAST patient cells. (A) Representative graph of mitochondrial respiration determined by measuring OCR (mean ± SE) using the Seahorse-based assay. (B–E) Comparison of different aspects of mitochondrial respiration (F) ATP content in the cells was measured using the ATPlite assay kit and normalized to mitochondrial mass [MitoTracker green (MTG) fluorescence]. (G) Mitochondrial oxidative stress was measured using MitoSOX (mitochondrial superoxide indicator) fluorescence and normalized to mitochondrial mass (MTG fluorescence). (H) General oxidative stress was measured using the CM-H2DCFDA (general oxidative stress indicator) assay kit and normalized to mitochondrial mass (MTG fluorescence). (I) Cellular proliferation was measured for on day 0 (4 h after cell seeding), day 1, and day 2 using CyQUANT assay. Data presented as mean ± SEM. The bars present data from three technical replicates. One-way analysis of variance with Tukey’s multiple comparisons test was performed to compare control, SPG7, and SPAST groups. n.s., not significant.
Oxidative Stress
MitoSOX fluorescence, a mitochondria-specific superoxide indicator, was significantly higher in SPG7 patient cells compared to controls (p < 0.05) and SPAST patient cells (p < 0.01) (Figure 3G). CM-H2DCFDA fluorescence, a general reactive oxygen species indicator, was significantly higher in SPG7 patient cells compared to control cells (p < 0.05) and SPAST patient cells (trend, p = 0.08) (Figure 3H).
Cell Proliferation
MELAS and MERRF mitochondrial disease patient cells have increased oxidative stress and reduced cellular proliferation (James et al., 1996). To test if SPG7 patient cells were similarly affected, we evaluated cellular proliferation in SPG7, SPAST, and control cells. SPG7 patient cells had reduced cellular proliferation compared to both control and SPAST patient cells (p < 0.05) (Figure 3I).
Discussion
We show here that SPG7 patient ONS cells with compound heterozygous mutations have increased paraplegin expression accompanied by multiple mitochondrial dysfunctions, including aberrant mitochondrial morphology (reduced length and interconnectivity), reduced mitochondrial mass, reduced mitochondrial membrane potential, impaired oxidative phosphorylation, reduced ATP production and content, and increased oxidative stress. SPG7 patient cells also showed reduced cellular proliferation. These cell function deficits were observed in all three SPG7 cell lines carrying different compound heterozygous mutations including a novel SPG7 mutation (NM_003119.3:c.1449 + 1G > A). These mitochondrial dysfunctions have previously been associated with cell and animal models lacking paraplegin (Atorino et al., 2003; Ferreirinha et al., 2004; Pareek et al., 2018). SPAST patient cells had a trend of lower respiratory chain function, but this was not statistically significant, indicating less severe respiratory chain dysfunction compared to SPG7 patient cells.
In addition to spasticity, SPG7 HSP patients generally have additional clinical features (refer to Table 1). The SPG7 patients, but not SPAST patients, had clinical features typically associated with mitochondrial diseases, such as ophthalmoplegia, ptosis, ataxia, and mitochondrial cytopathy on muscle biopsy. This is in accordance with the considerable mitochondrial dysfunction in SPG7 patient cells. These more extensive deficits in mitochondrial function may underlie the impaired cell proliferation in SPG7 patient cells, not seen in SPAST patient cells.
SPG7, but not SPAST, patient cells have increased levels of mitochondrial oxidative stress and reduced oxidative phosphorylation. Consistently, dysfunctional mitochondria are often accompanied by increased production of reactive oxygen species and associated with other neurodegenerative disorders (Leadsham Jane et al., 2013; Angelova and Abramov, 2018). Paraplegin maintains mitochondrial protein quality by degrading subunits of the respiratory chain that are damaged or not assembled (Gerdes et al., 2012) and by acting as an ATP-dependent matrix-AAA protease located in the inner mitochondrial membrane that harbors the respiratory chain complexes (Quirós et al., 2015). All SPG7 patients showed increased expression of paraplegin, despite having mutant alleles that were unlikely to be expressed. This is consistent with yeast complementation assays showing that when the AAA+ ATPase region was deleted, paraplegin was expressed but protease function was impaired (Bonn et al., 2010).
The SPG7 patient cells in this study were compound heterozygous, of which the p.Ala510Val mutation was common to all. This variant has a carrier rate in the general population of 0.5–1% (Bonn et al., 2010; Pfeffer et al., 2014), and its association to HSP pathogenicity has been explored extensively (Karlberg et al., 2009; Bonn et al., 2010; Roxburgh et al., 2013; Pfeffer et al., 2014). In particular, an apparent increased expression of paraplegin has been observed in SPG7 patient fibroblast cells (Pfeffer et al., 2014) and in neurites of the cerebellar and cerebral cortex on post-mortem in a patient with homozygous p.Ala510Val mutations (Thal et al., 2015). It is likely that the commonly expressed p.Ala510Val variant leads to the increased levels of paraplegin seen in our patient samples.
We have previously shown that SPAST patients have reduced levels of stabilized microtubules, leading to reduced organelle transport, thereby making them more sensitive to oxidative stress (Abrahamsen et al., 2013). Microtubule-dependent mitochondrial movement is necessary to maintain healthy mitochondrial structure and bioenergetics (Bartolák-Suki et al., 2017). We show here that mitochondrial function was not affected by SPAST mutations as severely as in SPG7 patient cells. However, a trend toward lower respiratory chain function was observed.
We establish here that SPG7 patient cells with increased paraplegin expression have mitochondrial dysfunction similar to patient cells that do not express paraplegin (Atorino et al., 2003). The severity of mitochondrial dysfunction in SPG7 patient cells compared to SPAST patient cells suggests that the cellular mechanisms responsible for the diagnosis of HSP do not necessarily converge on mitochondrial function. Our results highlight a need for understanding the distinct biology of patient cells with different mutations. This understanding is helpful for drug screening and developing drug treatments. A common drug treatment that can rectify the reduced respiratory chain function may be useful for all SPG7 patients.
Data Availability Statement
All datasets presented in this study are included in the article.
Ethics Statement
This study was reviewed and approved by the Human Research Ethics Committee of the Northern Sydney Local Health District, New South Wales Government, Australia (ethics committee reference number: RESP/15/314). The patients/participants provided written informed consent to participate in this study.
Author Contributions
GW, AM-S, and CS conceived and designed the experiments and provided funding for the research. GW, EL, and KK performed the experiments. RD performed the protein structure and function analysis. GW performed the statistical analysis. GW, AM-S, RD, KK, and CS wrote the main manuscript text. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants to CS, AM-S, and GW from the Spastic Paraplegia Foundation Incorporated and Hereditary Spastic Paraplegia Research Foundation Incorporated. CS was supported by an NHMRC Practitioner Fellowship (App1136800). KK was supported by the Paul Ainsworth Family Foundation and the Michael J. Fox Foundation, Aligning Science Across Parkinson’s (ASAP) initiative.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We express our thanks to the patients who consented to contribute to this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2020.00820/full#supplementary-material
FIGURE S1 | The paraplegin antibody was validated using a positive control, i.e., human HEK293 cells over-expressing paraplegin protein. The paraplegin band size was 88 kDa for both the control samples tested and the positive control, showing antibody specificity.
References
Abrahamsen, G., Fan, Y., Matigian, N., Wali, G., Bellette, B., Sutharsan, R., et al. (2013). A patient-derived stem cell model of hereditary spastic paraplegia with SPAST mutations. Dis. Models Mech. 6, 489–502. doi: 10.1242/dmm.010884
Angelova, P. R., and Abramov, A. Y. (2018). Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. 592, 692–702. doi: 10.1002/1873-3468.12964
Atorino, L., Silvestri, L., Koppen, M., Cassina, L., Ballabio, A., Marconi, R., et al. (2003). Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J. Cell Biol. 163, 777–787. doi: 10.1083/jcb.200304112
Bartolák-Suki, E., Imsirovic, J., Nishibori, Y., Krishnan, R., and Suki, B. (2017). Regulation of mitochondrial structure and dynamics by the cytoskeleton and mechanical factors. Int. J. Mol. Sci. 18:1812. doi: 10.3390/ijms18081812
Bonn, F., Pantakani, K., Shoukier, M., Langer, T., and Mannan, A. U. (2010). Functional evaluation of paraplegin mutations by a yeast complementation assay. Hum. Mutat. 31, 617–621.
Brugman, F., Scheffer, H., Wokke, J. H. J., Nillesen, W. M., de Visser, M., Aronica, E., et al. (2008). Paraplegin mutations in sporadic adult-onset upper motor neuron syndromes. Neurology 71, 1500–1505. doi: 10.1212/01.wnl.0000319700.11606.21
Casari, G., De Fusco, M., Ciarmatori, S., Zeviani, M., Mora, M., Fernandez, P., et al. (1998). Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 93, 973–983. doi: 10.1016/s0092-8674(00)81203-9
Dong, E.-L., Wang, C., Wu, S., Lu, Y.-Q., Lin, X.-H., Su, H.-Z., et al. (2018). Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol. Neurodegener. 13:36.
Errico, A., Ballabio, A., and Rugarli, E. (2002). Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum. Mol. Genet. 11, 153–163. doi: 10.1093/hmg/11.2.153
Féron, F., Perry, C., Girard, S. D., and Mackay-Sim, A. (2013). “Isolation of adult stem cells from the human olfactory mucosa,” in Neural Progenitor Cells: Methods and Protocols, eds A. B. Reynolds and P. L. Deleyrolle (Totowa, NJ: Humana Press), 107–114. doi: 10.1007/978-1-62703-574-3_10
Féron, F., Perry, C., McGrath, J. J., and Mackay-Sim, A. (1998). NEw techniques for biopsy and culture of human olfactory epithelial neurons. Arch. Otolaryngol. Head Neck Surg. 124, 861–866. doi: 10.1001/archotol.124.8.861
Ferreirinha, F., Quattrini, A., Pirozzi, M., Valsecchi, V., Dina, G., Broccoli, V., et al. (2004). Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J. Clin. Investig. 113, 231–242. doi: 10.1172/jci200420138
Gautier, C. A., Giaime, E., Caballero, E., Núñez, L., Song, Z., Chan, D., et al. (2012). Regulation of mitochondrial permeability transition pore by PINK1. Mol. Neurodegener. 7, 1–15.
Gerdes, F., Tatsuta, T., and Langer, T. (2012). Mitochondrial AAA proteases — Towards a molecular understanding of membrane-bound proteolytic machines. Biochim. Biophys. Acta Mol. Cell Res. 1823, 49–55. doi: 10.1016/j.bbamcr.2011.09.015
Hedera, P., Eldevik, O. P., Maly, P., Rainier, S., and Fink, J. K. (2005). Spinal cord magnetic resonance imaging in autosomal dominant hereditary spastic paraplegia. Neuroradiology 47, 730–734. doi: 10.1007/s00234-005-1415-3
James, A. M., Wei, Y.-H., Pang, C.-Y., and Murphy, M. P. (1996). Altered mitochondrial function in fibroblasts containing MELAS or MERRF mitochondrial DNA mutations. Biochem. J. 318, 401–407. doi: 10.1042/bj3180401
Karlberg, T., van den Berg, S., Hammarström, M., Sagemark, J., Johansson, I., Holmberg-Schiavone, L., et al. (2009). Crystal structure of the ATPase domain of the human AAA+ protein paraplegin/SPG7. PLoS One 4:e6975. doi: 10.1371/journal.pone.0006975
Keane, P. C., Kurzawa, M., Blain, P. G., and Morris, C. M. (2011). Mitochondrial dysfunction in Parkinson’s disease. Parkinson Dis. 2011:716871.
Koentjoro, B., Park, J.-S., and Sue, C. M. (2017). Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 7:44373.
Kumar, K. R., Blair, N. F., Vandebona, H., Liang, C., Ng, K., Sharpe, D. M., et al. (2013). Targeted next generation sequencing in SPAST-negative hereditary spastic paraplegia. J. Neurol. 260, 2516–2522. doi: 10.1007/s00415-013-7008-x
Kumar, K. R., Blair, N. F., and Sue, C. M. (2015). An update on the hereditary spastic paraplegias: new genes and new disease models. Mov. Disord. Clin. Pract. 2, 213–223. doi: 10.1002/mdc3.12184
Leadsham Jane, E., Sanders, G., Giannaki, S., Bastow Emma, L., Hutton, R., Naeimi Wesley, R., et al. (2013). Loss of cytochrome c oxidase promotes RAS-dependent ROS production from the ER resident NADPH Oxidase, Yno1p, in yeast. Cell Metab. 18, 279–286. doi: 10.1016/j.cmet.2013.07.005
Matigian, N., Abrahamsen, G., Sutharsan, R., Cook, A. L., Vitale, A. M., Nouwens, A., et al. (2010). Disease-specific, neurosphere-derived cells as models for brain disorders. Dis. Models Mech. 3, 785–798. doi: 10.1242/dmm.005447
McDermott, C. J., Taylor, R. W., Hayes, C., Johnson, M., Bushby, K. M. D., Turnbull, D. M., et al. (2003). Investigation of mitochondrial function in hereditary spastic paraparesis. NeuroReport 14, 485–488. doi: 10.1097/00001756-200303030-00038
Moreira, P. I., Carvalho, C., Zhu, X., Smith, M. A., and Perry, G. (2010). Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta Mol. Basis Dis. 1802, 2–10. doi: 10.1016/j.bbadis.2009.10.006
Mortiboys, H., Thomas, K., Koopman, W. J. H., Klaffke, S., Abou-Sleiman, P., Olpin, S., et al. (2008). Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol. 64, 555–565. doi: 10.1002/ana.21492
Orlacchio, A., Kawarai, T., Totaro, A., Errico, A., St George-Hyslop, P. H., Rugarli, E. I., et al. (2004). Hereditary spastic paraplegia: clinical genetic Study of 15 families. Arch. Neurol. 61, 849–855. doi: 10.1001/archneur.61.6.849
Pareek, G., Thomas, R. E., and Pallanck, L. J. (2018). Loss of the Drosophila m-AAA mitochondrial protease paraplegin results in mitochondrial dysfunction, shortened lifespan, and neuronal and muscular degeneration. Cell Death Dis. 9:304.
Park, J.-S., Koentjoro, B., Veivers, D., Mackay-Sim, A., and Sue, C. M. (2014). Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 23, 2802–2815. doi: 10.1093/hmg/ddt623
Pfeffer, G., and Chinnery, P. F. (2013). Diagnosis and treatment of mitochondrial myopathies. Ann. Med. 45, 4–16. doi: 10.3109/07853890.2011.605389
Pfeffer, G., Gorman, G. S., Griffin, H., Kurzawa-Akanbi, M., Blakely, E. L., Wilson, I., et al. (2014). Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 137, 1323–1336. doi: 10.1093/brain/awu060
Quirós, P. M., Langer, T., and López-Otín, C. (2015). New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 16, 345–359. doi: 10.1038/nrm3984
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Roxburgh, R. H., Marquis-Nicholson, R., Ashton, F., George, A. M., Lea, R. A., Eccles, D., et al. (2013). The p.Ala510Val mutation in the SPG7 (paraplegin) gene is the most common mutation causing adult onset neurogenetic disease in patients of British ancestry. J. Neurol. 260, 1286–1294. doi: 10.1007/s00415-012-6792-z
Salinas, S., Proukakis, C., Crosby, A., and Warner, T. (2008). Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 7, 1127–1138. doi: 10.1016/s1474-4422(08)70258-8
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671. doi: 10.1038/nmeth.2089
Shanmughapriya, S., Rajan, S., Hoffman Nicholas, E., Higgins Andrew, M., Tomar, D., Nemani, N., et al. (2015). SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Mol. Cell 60, 47–62. doi: 10.1016/j.molcel.2015.08.009
Teves, J. M. Y., Bhargava, V., Kirwan, K. R., Corenblum, M. J., Justiniano, R., Wondrak, G. T., et al. (2018). Parkinson’s disease skin fibroblasts display signature alterations in growth, redox homeostasis, mitochondrial function, and autophagy. Front. Neurosci. 11:737. doi: 10.3389/fnins.2017.00737
Thal, D. R., Züchner, S., Gierer, S., Schulte, C., Schöls, L., Schüle, R., et al. (2015). Abnormal paraplegin expression in swollen neurites, τ- and α-synuclein pathology in a case of hereditary spastic paraplegia SPG7 with an Ala510Val mutation. Int. J. Mol. Sci. 16, 25050–25066. doi: 10.3390/ijms161025050
Vandebona, H., Kerr, N., Liang, C., and Sue, C. (2012). SPAST mutations in Australian patients with hereditary spastic paraplegia. Intern. Med. J. 42, 1342–1347.
Wali, G., Liyanage, E., Blair, N. F., Sutharsan, R., Park, J.-S., Mackay-Sim, A., et al. (2020). Oxidative stress-induced axon fragmentation is a consequence of reduced axonal transport in hereditary spastic paraplegia SPAST patient neurons. Front. Neurosci. 14:401. doi: 10.3389/fnins.2020.00401
Keywords: hereditary spastic paraplegia, SPG7, SPAST, spastin, paraplegin, mitochondria, oxidative phosphorylation
Citation: Wali G, Kumar KR, Liyanage E, Davis RL, Mackay-Sim A and Sue CM (2020) Mitochondrial Function in Hereditary Spastic Paraplegia: Deficits in SPG7 but Not SPAST Patient-Derived Stem Cells. Front. Neurosci. 14:820. doi: 10.3389/fnins.2020.00820
Received: 15 April 2020; Accepted: 14 July 2020;
Published: 20 August 2020.
Edited by:
Vincenzo La Bella, University of Palermo, ItalyReviewed by:
Stefan Hauser, Helmholtz-Gemeinschaft Deutscher Forschungszentren (HZ), GermanyAntonio Orlacchio, Santa Lucia Foundation (IRCCS), Italy
Copyright © 2020 Wali, Kumar, Liyanage, Davis, Mackay-Sim and Sue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gautam Wali, Z2F1dGFtLndhbGlAc3lkbmV5LmVkdS5hdQ==; Carolyn M. Sue, Y2Fyb2x5bi5zdWVAc3lkbmV5LmVkdS5hdQ==
†These authors share senior authorship