 Nicholas G. Norwitz1*
Nicholas G. Norwitz1* Henry Querfurth2
Henry Querfurth2- 1Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, United Kingdom
- 2Department of Neurology, Tufts Medical Center, Boston, MA, United States
The mechanistic target of rapamycin protein complex, mTORC1, has received attention in recent years for its role in aging and neurodegenerative diseases, such as Alzheimer’s disease. Numerous excellent reviews have been written on the pathways and drug targeting of this keystone regulator of metabolism. However, none have specifically highlighted several important nuances of mTOR regulation as relates to neurodegeneration. Herein, we focus on six such nuances/open questions: (1) “Antagonistic pleiotropy” – Should we weigh the beneficial anabolic functions of mTORC1 against its harmful inhibition of autophagy? (2) “Early/late-stage specificity” – Does the relative importance of these neuroprotective/neurotoxic actions change as a disease progresses? (3) “Regional specificity” – Does mTOR signaling respond differently to the same interventions in different brain regions? (4) “Disease specificity” – Could the same intervention to inhibit mTORC1 help in one disease and cause harm in another disease? (5) “Personalized therapy” – Might genetically-informed personalized therapies that inhibit particular nodes in the mTORC1 regulatory network be more effective than generalized therapies? (6) “Lifestyle interventions” – Could specific diets, micronutrients, or exercise alter mTORC1 signaling to prevent or improve the progression neurodegenerative diseases? This manuscript is devoted to discussing recent research findings that offer insights into these gaps in the literature, with the aim of inspiring further inquiry.
Introduction
Neurodegenerative diseases are an accelerating pandemic. The burden of Alzheimer disease (AD) alone is staggering and climbing at a precipitous rate. 5.8 million Americans over the age of 65 suffer from AD, a number that is expected to triple to 13.8 million by 2050 (Alzheimer’s Association, 2020). AD is not alone in its ascent. Parkinson’s disease (PD), the second most common form of neurodegeneration, is increasing in prevalence at a similarly alarming rate (Rocca, 2018). As there are currently no effective long-term treatments for these diseases, new therapies are desperately needed. One potential molecular target of such therapies is the mechanistic target of rapamycin complex 1 (mTORC1), a nutrient sensor and metabolic regulator heavily implicated in the process of aging (Sharp and Strong, 2010; Papadopoli et al., 2019; Heras-Sandoval et al., 2020).
While this manuscript will be primarily devoted to discussing and gaps in the literature surrounding mTORC1, a succinct overview of mTOR signaling and regulation is warranted as a preface to this discussion and is depicted in Figure 1 [For a more comprehensive overview, Heras-Sandoval et al. (2020) recently published an excellent review on mTOR signaling, regulation, and drug-targeting]. mTORC1 is composed of the proteins mTOR kinase and its regulator protein, Raptor, as well as mLST8, PRAS40, and Deptor. Its primary function is to sense intracellular nutrient status and extracellular trophic factors [including, but not exclusive to insulin, shown in Figure 1 as an example], integrate these signals, and ultimately regulate the balance between cells’ anabolic and catabolic processes. Specifically, mTORC1 is a positive regulator of protein synthesis and negative regulator of autophagy.
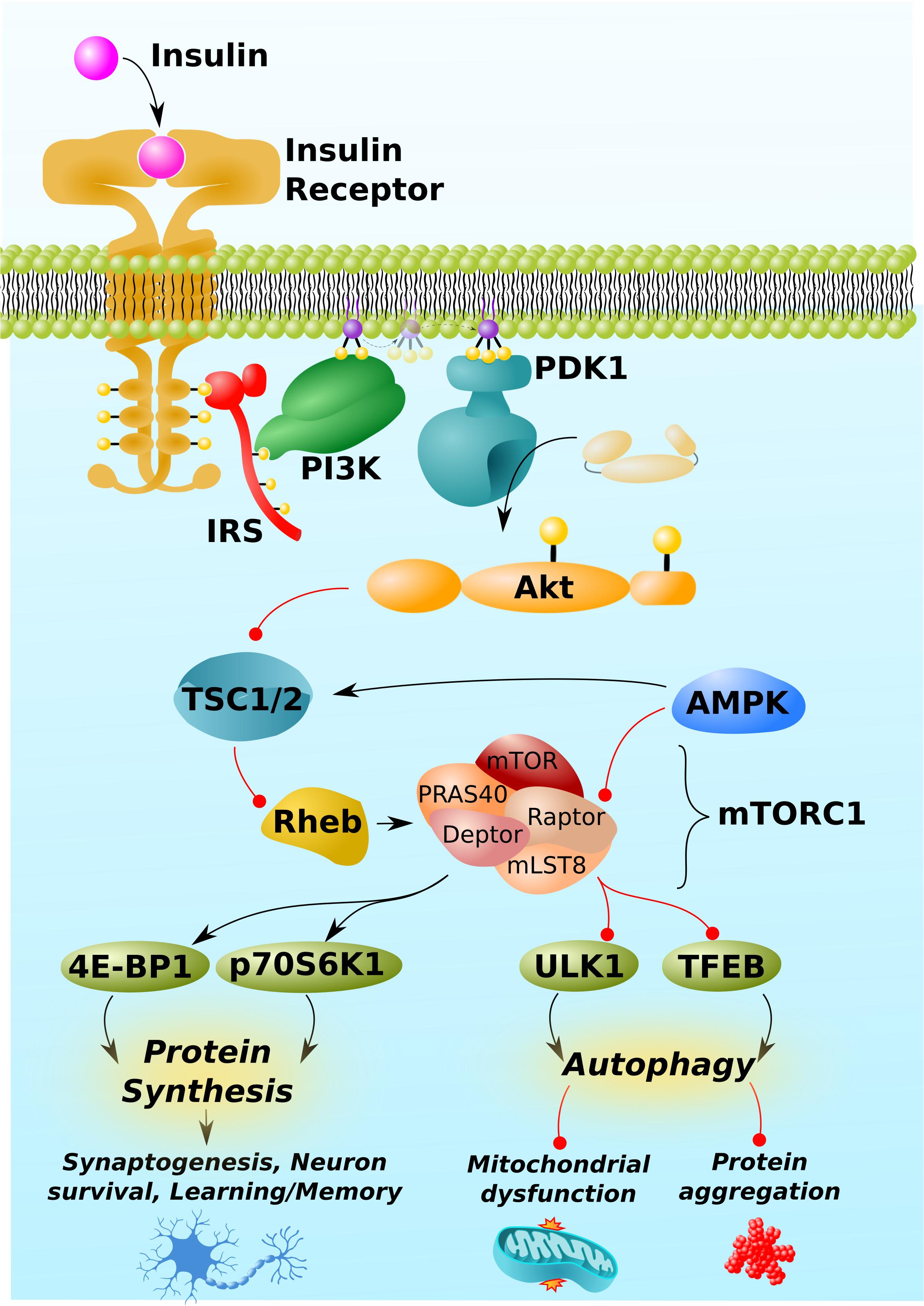
Figure 1. mTORC1 pathway and regulation. mTORC1 is activated by insulin. Insulin/Akt signaling inhibits TSC1/2, thereby permitting the activation of the GTP-binding protein, Rheb. Rheb is the proximal activator of mTORC1. AMPK inhibits mTORC1 activity through indirect and direct mechanisms, phosphorylating TSC1/2 and the Raptor regulatory component of mTORC1. (Other trophic factors and pathways beyond insulin/Akt and AMPK, not shown for simplicity, also regulate mTORC1). mTORC1 downstream targets include proteins involved the mRNA translation, 4E-BP1 and p70S6K1, and those involved in autophagy, such as the initiator of autophagy, ULK1, and the master regulator of lysosomal biogenesis, TFEB. By regulating the activity of these and other proteins, mTORC1 promotes protein synthesis, which is required for synaptogenesis, learning, and memory, but can also impair autophagy, leading to mitochondrial dysfunction and neurotoxic protein aggregation (Aβ, phospho-tau, α-synuclein, etc.). Black arrows and red lines respectively represent positive and negative regulation.
mTORC1 itself is regulated positively by insulin-signaling and negatively by AMPK. Insulin/Akt signaling inhibits the protein complex, TSC1/2, which itself prevents the conversion of the mTORC1 activator, Rheb, into its active GTP-bound form (Inoki et al., 2002; Hers et al., 2011). Insulin/Akt signaling turns off TSC1/2, thereby activating Rheb and mTORC1. By contrast, AMP-activated protein kinase (AMPK) activates TSC1/2 (Inoki et al., 2003) and directly inhibits mTORC1 by phosphorylating Raptor (Gwinn et al., 2008). In brief, the respective growth and preservation functions of insulin and AMPK align with their respective stimulatory and inhibitory regulations of mTORC1.
mTORC1 promotes protein synthesis by phosphorylating and activating the downstream targets, 4E-BP1 and p70S6K1, which directly promote the initiation and elongation phases of translation (Graber et al., 2013). Critically, mTORC1-mediated anabolic signaling promotes the development of neuronal synapses (Dwyer and Duman, 2013, in part, by responding to established neuronal growth factors like BDNF; Takei et al., 2004) and inhibits apoptosis (Chen et al., 2010; Chong et al., 2013). Through these two mechanisms, mTORC1 activity has the potential to promote learning and memory and protect against neurodegeneration. Correspondingly, excessive inhibition of mTORC1 can impair learning and memory and permit neuronal death (Blundell et al., 2008; Belelovsky et al., 2009; Gafford et al., 2011; Jobim et al., 2012; Graber et al., 2013).
Despite these potentially positive functions of mTORC1 signaling in the brain, far more attention has been paid to its negative regulation of autophagy, an intracellular recycling process essential to maintaining neuronal integrity and protecting against neurodegenerative diseases (Oddo, 2012; Sarkar, 2013; Heras-Sandoval et al., 2020). mTORC1 inhibits autophagy at multiple levels, including the inhibitory phosphorylation of ULK1 and transcription factor EB (TFEB), which respectively initiate autophagy and promote the lysosomal biogenesis required to break down the contents of autophagosomes (Kim et al., 2011; Napolitano et al., 2018).
Importantly, multiple independent human post-mortem studies confirm levels of phosphorylated mTOR and its downstream targets are elevated in the AD brain as compared to those of controls (An et al., 2003; Li et al., 2004, 2005; Griffin et al., 2005). Dysregulated autophagy is also a hallmark of multiple neurodegenerative conditions (Fujikake et al., 2018), which is not surprising because autophagy is required to prevent the accumulation of toxic intracellular protein aggregates that contribute to neurodegenerative diseases, such as amyloid-β (Aβ) (Nixon, 2007; Nilsson et al., 2013; Yang et al., 2014), phospho-tau (Hamano et al., 2008; Kruger et al., 2012; Wang and Mandelkow, 2012), α-synuclein (Webb et al., 2003; Lee et al., 2004; Xilouri et al., 2016), and mutant huntingtin (Martin et al., 2015). Autophagy is also required to recycle mitochondria and prevent mitochondrial dysfunction (Chakravorty et al., 2019; Li et al., 2019a), another hallmark of neurodegenerative diseases, and one which can further lead to the pathologies of oxidative stress and inflammation (Lopez-Armada et al., 2013; Norwitz et al., 2019a, b). Given these data and the clinical burden of neurodegenerative disease, it’s reasonable that translational research generally focuses on the inhibition of mTOR (and promotion of autophagy), rather than on its activation.
Antagonistic Pleiotropy
“Antagonistic pleiotropy” is a term typically used to refer to an evolutionary tradeoff between fitness in an organism’s early life at the expense of health later in life (Schmeisser and Parker, 2019). An example of antagonistic pleiotropy is the ApoE4 allele, the leading genetic risk factor of AD (Yamazaki et al., 2019). This allele sensitizes the immune system and protected ancestral humans from infections that compromised reproductive fitness and cognition (Vasunilashorn et al., 2011; Trumble et al., 2017). Further relevant to modern contexts, ApoE4 is associated with accelerated neurodevelopment (Wright et al., 2003) and improved memory during youth (Mondadori et al., 2007).
Another example of possible antagonistic pleiotropy in neurodegenerative disease is that of adiponectin (APN), a hormone secreted by adipose tissue. APN has broad beneficial functions on metabolism, including stimulating neurogenesis, and is generally thought to be neuroprotective (Zhang et al., 2011, 2016). On the other hand, APN can induce astrocyte mediated neuroinflammation (Wan et al., 2014), oxidative stress (Fujimoto et al., 2010), and plasma levels of APN are correlated with the severity of cognitive decline and Aβ accumulation (Wennberg et al., 2016). [Waragai et al. (2020) provide a comprehensive overview of antagonistic pleiotropy with regards to APN].
Hashimoto et al. (2018) have even proposed that amyloidogenic proteins, including Aβ in AD and α-synuclein in PD, might exhibit antagonistic pleiotropy. They hypothesize that the heterogeneity of amyloidogenic aggregates reflects the heterogeneity of metabolic stressors to which the human brain is exposed, and that specific amyloidogenic aggregates may serve to “precondition” the brain against future toxic exposures (Hashimoto et al., 2018). In effect, Aβ and α-synuclein could serve, in youth, as adaptive hormetic stressors. [As an aside, the Aβ/α-synuclein antagonistic pleiotropy hypothesis is intertwined with the “evolvability hypothesis” of amyloidogenic proteins, which is beyond the scope of this piece and reviewed by Hashimoto et al. (2018)].
The moral of these examples – ApoE4, APN, and Aβ/α-synuclein – is that the trade of better health and cognition during youth, at the expense of cognition during non-reproductive years, was evolutionarily judicious. Furthermore, each these examples would not have been specifically mentioned if they did not plausibly involve mTORC1. With respect to ApoE4, mTORC1 activates pathways that promote synaptogenesis and neuronal development, which would benefit cognition during youth at the expense of decreased autophagy and increased risk of accumulating mitochondrial damage and neurotoxic protein aggregates over time, as in the case of ApoE4 (Wright et al., 2003; Mondadori et al., 2007). Indeed, the ApoE4 genotype is associated with elevated mTOR signaling (Li et al., 2019b). APN has been shown to induce oxidative stress in an mTORC1-dependent manner by modulating both insulin and AMPK signaling (Fujimoto et al., 2010; Figure 1). And, of course, mTORC1 activity is assumed to be culpable for dysfunctional autophagy and accumulation of neurotoxic protein aggregates in neurodegenerative diseases, as noted in the introduction. Thus, mTORC1 may be a keystone player of antagonistic pleiotropy in neurodegenerative disease.
Consideration of antagonistic pleiotropy is important for evaluating the preventative value of inhibiting mTORC1 prior to the development of symptoms. No doubt, it’s important to prevent the development of the pathologies underlying neurodegenerative diseases, which are established decades before symptoms develop (Braak et al., 2003; Dickson et al., 2010; Hoglund et al., 2017). But when and by how much? During mid-life, should one strive for mTORC1 inhibition, or value activating mTORC1 in a cyclic manner in order to build neural networks and increase her/his cognitive reserve, thus protecting against cognitive decline later in life? These are open questions.
Early/Late-Stage Specificity
Although inhibiting mTORC1 to increase autophagy (and therefore clear damaged mitochondria and protein aggregates) may seem like a prudent intervention for neurodegenerative diseases, that may not be universally true. What if a disease progresses past a threshold beyond which the pathology is too well-established to be meaningfully improved by an upregulation in autophagy? For instance, the mTORC1 inhibitor, rapamycin, does not reverse pathology or benefit cognition in late-stage AD models (Majumder et al., 2011). More importantly, because mTORC1 can inhibit apoptosis by activating p70S6K, which itself inhibits the pro-apoptotic protein BAD (Harada et al., 2001; Castedo et al., 2002), what if inhibiting mTORC1 beyond this hypothetical threshold increases apoptotic cell death?
Evidence consistent with this hypothesis is provided by multiple independent cell and rodent models of PD. These models of established late-stage disease suggest that increasing, rather that decreasing, mTOR activity could be beneficial under certain circumstances. In MPP+-treated SH-SY5Y cells, activation of mTOR with cannabidiol led to protection against MPP+-induced cell death (Gugliandolo et al., 2020). In genetic and pharmacologic mouse models, upregulation of mTOR signaling (through PTEN ablation) is likewise associated with less cell death and improved symptomology (Domanskyi et al., 2011). A limitation of these early PD studies is that they do not involve α-synuclein accumulation, which may better recapitulate the human form of the disease and relative importance of autophagy therein. Nevertheless, given the knowledge that mTORC1 can inhibit apoptosis, and distinct possibility that there may be a point past which activation of autophagy is insufficient to improve disease course (Majumder et al., 2011), it’s worth questioning whether mTORC1 inhibition could actually be harmful in late-stage neurodegenerative disease.
Regional Specificity
In addition to considering the temporal dimension (early/late-stage disease), it’s important to consider the spatial dimension. As the brain is partitioned into networks, nuclei, and cell types, a given intervention may impact one region differently than another. For example, Ramalingam et al. (2019) discovered that rotenone injections (used to generate murine models of PD) oppositely impact mTORC1 activity in different regions of mouse brains, increasing activity in the midbrain and decreasing activity in the striatum. Lifestyle interventions (more on this below), such as exercise, may also alter mTOR activity in a region-specific manner. In mice, wheel running regulates mTORC1 signaling most strongly in the nucleus accumbens and hippocampus, as compared to other brain regions (Lloyd et al., 2017). This is notable because atrophy of nucleus accumbens and hippocampus is most strongly associated with AD (Nie et al., 2017).
The data are nascent but sufficient to issue caution. What if a PD patient suffering from substantia nigra atrophy were treated with an mTORC1 inhibitor based on a rationale from data collected from hippocampal pathology? What if a frontotemporal dementia (FTD) patient suffering from primarily temporal lobe pathologies was treated with an mTORC1-targeting drug based on frontal lobe data? As there is limited evidence to support that mTORC1 responds consistently to a wide range of interventions across brain regions, and some evidence to the contrary, it’s responsible to not overgeneralize and assume globalized impact on the brain. More research needs to be conducted on the region-specific impacts of different mTORC1-directed interventions.
Disease Specificity
While many neurodegenerative diseases share several key core pathologies, including mitochondrial dysfunction, protein aggregation, oxidative stress, and inflammation, it’s also important to consider disease-specific aspects of neurometabolim that could interact with mTORC1. For example, Zhuang et al. (2020) recently discovered that TFEB activity (which stimulates lysosomal biogenesis and promotes autophagy) is increased in a 6-OHDA-treated SH-5YSY model of PD, as well as in dopaminergic neurons, and that TFEB activity is calcium/calcineurin-dependent. This is important because PD is characterized by loss of substantia nigra pars compacta neurons, which exhibit a unique form of calcium pacemaking activity not seen in most other neurons. This suggests that regulation of autophagy may be different in the brain region most affected by PD as compared to brain regions impacted in other diseases.
Another example is Amyotrophic Lateral Sclerosis (ALS), which can be caused by loss-of-function mutations in the UBQLN2/4 genes. While the products of these genes, ubiquilin proteins, are known best as components of the ubiquitin-proteasome system, they are also important in autophagy. Specifically, ubiquilins are required to maintain the vacuolar H+-ATPase function that acidifies lysosomes (Senturk et al., 2019). In a scenario in which mTORC1 were inhibited to induce autophagy in ALS, induction of autophagy and lysosomal biogenesis may be increased (Figure 1), but if lysosomes are not sufficiently acidic to destroy the contents of the autophagosome, the contents could accumulate and exacerbate cellular stress. Therefore, inhibiting mTORC1 to upregulate autophagy could impair autophagic flux, leading to a back-up of components, and be harmful in such genetic cases of ALS.
Personalized Therapy
There is a need for informed, disease-specific interventions. In this section, we provide three hypothetical examples of personalized interventions involving mTORC1. These will include glutamatergic antagonism for Huntington’s disease (HD) (Abd-Elrahman and Ferguson, 2019), metformin treatment for multiple sclerosis (MS) (Sanadgol et al., 2019), and SMCR8-centered therapy for ALS and FTD (Lan et al., 2019).
Glutamate hyperactivity plays a prominent role in HD (Andre et al., 2010) and can activate mTORC1 via the mGluR5-PDK1-Akt-mTORC1 pathway (Abd-Elrahman and Ferguson, 2019). Correspondingly, Abd-Elrahman and Ferguson (2019) recently demonstrated, in a mouse model of HD, that antagonism of the mGluR5 metabotropic glutamate receptor can correct overactive mTORC1 signaling and, consequently, increase autophagic clearance of mutant huntingtin protein. The authors of this paper also point out that huntingtin aggregates sequester the transcription factor, CREB, leading to a down regulation of neuroprotective BDNF. They show that mGluR5 inhibitors not only clear pathological aggregates, but also increase BDNF expression (Abd-Elrahman and Ferguson, 2019). Therefore, mGluR5 antagonism, by inhibiting hyperactive mTORC1, could simultaneously promote the clearance of pathological huntingtin aggregates and increase neurotrophic factor signaling.
MS is characterized by demyelination of nerve cell axons. As oligodendrocytes are responsible for building myelin sheaths within the central nervous system, a goal of MS treatments is to boost oligodendrocyte renewal and remyelination. In a cuprizone-challenge mouse model of MS, Sanadgol et al. (2019) recently reported that the diabetes drug, metformin, did precisely that: it increased oligodendrocyte renewal and remyelination. These beneficial effects were mediated by a direct stimulatory interaction between metformin and AMPK, and subsequent inhibition of mTORC1 (Sanadgol et al., 2019; Figure 1). Thus, metformin is a candidate for an mTORC1-targeting therapy for MS.
Mutations in the C9orf72 gene are the leading cause of inherited ALS and FTD. Only recently was it discovered that another protein, SMCR8, complexes with the C9orf72 protein to form a heterodimer that negatively regulates mTORC1 activity (Lan et al., 2019). Furthermore, a SMCR8-deficient mouse model recapitulates the C9orf72-deficient phenotype, leads to a decrease in C9orf72 protein, and is associated with upregulation of mTORC1 activity and decreased autophagy (Lan et al., 2019). Future treatments for genetic causes of ALS and FTD might consider SMCR8 therapy or other interventions that target the SMCR8-mTORC1-autophagy axis.
These examples were chosen because HD, MS, ALS, and FTD are lesser studied than AD and PD. However, the same personalization principle applies to all conditions in which mTORC1 plays a role. In PD, for example, levodopa-induced dyskinesia is thought to be induced by D1-receptor-mediated phosphorylation of mTORC1, a hypothesis supported by the fact that genetic variability in mTOR pathway components is associated with PD dyskinesia (Zhu et al., 2019). The development of useful future interventions for neurodegenerative disorders would benefit from a deeper consideration of the interactions between mTORC1 signaling and disease/patient-specific mechanisms.
Lifestyle Interventions
Two reasons most neurodegenerative diseases are refractory to treatment are that interventions may be initiated too late in the disease process and/or are too specific. These limitations are a function of the pharmacologic approach to neurodegenerative disease in which symptomatic patients, who have usually been afflicted by the underlying disease for years to decades, are prescribed drugs not available for prevention during the preclinical stage. Certainly, drugs have their place. But to quell the neurodegenerative disease pandemic will require universally accessible preventative measures based on safe lifestyle interventions, including diet and exercise. Evidence suggests such interventions could operate, in part, through mTORC1-mediated mechanisms.
Turmeric is the best-studied nutraceutical for neurodegenerative diseases. In a genetic mouse model of AD, turmeric’s active component, curcumin, inhibited mTORC1 to increase autophagy and prevent Aβ accumulation (Wang et al., 2014a). Correspondingly, curcumin-induced inhibition of mTORC1 protected against memory impairments in this model (Wang et al., 2014a). A more specific dietary example would be the mineral manganese in HD. As manganese deficiency might contribute to the pathogenesis of HD by affecting the insulin/Akt/mTORC1 pathway, correcting a simple micronutrient deficiency could be protective in some cases of HD (Bryan and Bowman, 2017). A third example is that of PPARs, a family of transcription factors that can inhibit mTORC1 and promote autophagy to protect against neurodegenerative disease (San et al., 2015; Heras-Sandoval et al., 2020). Many nutrients and their derivates activate PPARs, including oleoylethanolamide derived from oleic acid in olive oil (Rodriguez de Fonseca et al., 2001; Fu et al., 2005) and the monoterpenes carvacrol and thymol found in mint family plants (basil, mint, rosemary, sage) (Hotta et al., 2010; Rigano et al., 2017). Curcumin, manganese, and dietary PPAR activators are just three examples of nutraceuticals from different classes that, when combined in a well-formulated diet and with other dietary mTOR regulators (Wang et al., 2014b; Rigano et al., 2017), could have a meaningful impact on cognitive longevity.
In addition to nutraceuticals and micronutrients, shifts in macronutrient intake can also impact mTORC1 activity. The most evident examples are intermittent fasting and high-fat, low-carbohydrate ketogenic diets, which can modulate mTORC1 activity through at least three mechanisms. First, fasting and ketogenic diets diminish insulin-mediated mTORC1 activation. Second, they activate AMPK (by altering the AMP/ATP ratio and causing glycogen depletion) to inhibit mTORC1 and induce autophagy (Alirezaei et al., 2010; Miller et al., 2018). Third, fasting and ketogenic diets share the common feature of stimulating hepatic production of the ketone body, β-hydroxybutyrate, which itself is a signaling molecule that regulates mTORC1 (Li et al., 2017; Newman and Verdin, 2017; Norwitz et al., 2019a). Interestingly, it has recently been demonstrated that both short-term ketogenic diets and acute administration of exogenous β-hydroxybutyrate improve a marker of brain aging called “brain network stability,” in contrast to standard Western diets and sugar which decrease network stability (Mujica-Parodi et al., 2020). Long-term prospective studies will need to be conducted to determine whether fasting and ketogenic diets are truly neuroprotective in humans. Nevertheless, these mechanisms and data coincide with the growing popularity of intermittent fasting and ketogenic diets as prevention or treatment strategies for neurodegenerative conditions (Roberts et al., 2017; Zhang et al., 2017; Shin et al., 2018; Sohn, 2018; Taylor et al., 2018, 2019; Broom et al., 2019; Norwitz et al., 2019a; Wlodarek, 2019; Mujica-Parodi et al., 2020; Soto-Mota et al., 2020).
Exercise is another lifestyle intervention that benefits brain health. Prospective cohort and randomized controlled studies have found that exercise reduces the risk of developing dementia by as much as 38% (Larson et al., 2006) and improves cognitive function in those already living with AD (Groot et al., 2016; Jia et al., 2019). Kou et al. (2019) recently published a compelling review arguing that the benefits of exercise on cognitive function and AD may be mediated by mTORC1 regulation. Even a cursory consideration of this hypothesis suggests it has merit. Exercise alters nutrient flux, trophic factor signaling, and can activate AMPK. Exercise can also correct overactive mTORC1 signaling to increase autophagy by correcting dysfunctional microRNA expression in a mouse model of AD (Kou et al., 2017; Chen et al., 2019). These particular studies focus on microRNA-34a, but there is reason to believe that exercise can influence mTORC1, autophagy, and cognitive aging by regulating a wide network of microRNAs (Kou et al., 2019). In another rodent model of AD, treadmill exercise decreased phospho-mTOR levels [Ser-2,448, Akt target residue (Nave et al., 1999)], increased autophagy, and completely rescued cognitive function on the Morris water maze test (Kang and Cho, 2015).
Dietary micronutrients, fasting and ketogenic diets, and exercise are but a few illustrative examples of lifestyle interventions that may interact with mTORC1 to modulate the course of neurodegenerative diseases. Additional therapies include probiotics to modulate the gut-brain axis, which has been heavily implicated in the development of neurodegenerative diseases (Sampson et al., 2016; Sochocka et al., 2019), and heat therapy to induce chaperone heat shock proteins [whose expression is at least partially mediated by mTORC1 (Sun et al., 2011)] that could promote the proper folding of amyloidogenic proteins (Singh et al., 2006, 2010; Laukkanen et al., 2017). At the present time, clinical studies examining the impact of lifestyle interventions on mTORC1 signaling for cognitive decline are few (Halikas and Gibas, 2018; Kou et al., 2019) and more research needs to be conducted in this area to inform holistic and universally available best practices for the treatment and prevention of neurodegenerative disease.
Conclusion
While references to the most pressing open questions are scattered throughout the abundant literature on mTOR and neurodegenerative disease, herein, we have consolidated these gaps in the literature (Figure 2). How do we balance the beneficial effects of mTORC1 against its negative effects? How does this balance shift with disease progression or brain region? How can we use knowledge of biochemical pathways, specific to diseases and even individual cases, to inform personalized therapy? And what universally available lifestyle interventions might help in the prevention of neurodegeneration? Consideration of these mTOR mysteries will inform future research.

Figure 2. mTORC1 mysteries. Six nuances regarding mTORC1 in neurodegenerative disease. The questions and examples below each topic are illustrative, not comprehensive, of the literature covered in this review. Disease abbreviations: ALS, Amyotrophic Lateral Sclerosis; FTD, frontotemporal dementia; HD, Huntington’s disease; MS, multiple sclerosis; PD, Parkinson’s disease.
Author Contributions
Both authors contributed to this work.
Funding
This publication was funded by the NIA NIH AGO44871-01 to HQ, institutional funds, and through an award from the Bennett Foundation. The Keasbey Memorial Foundation funds NN’s research at Oxford.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
A β, amyloid- β; AD, Alzheimer’s disease; Akt, protein kinase B; ALS, Amyotrophic Lateral Sclerosis; AMPK, AMP-activated protein kinase; APN, adiponectin; BAD, Bcl2 associated agonist of cell death; CREB, cAMP response element-binding protein; C9orf72, chromosome 9 open reading frame 72; Deptor, domain-containing mTOR-interacting protein; FTD, frontotemporal dementia; HD, Huntington’s disease; IRS, insulin receptor substrate; mGluR5, metabotropic glutamate receptor type 5; mLST8, mammalian lethal with SEC13 protein 8; MPP+, 1-methyl-4-phenylpyridinium; MS, multiple sclerosis; mTOR, mechanistic target of rapamycin; PD, Parkinson’s disease; PDK-1, phosphoinositide-dependent kinase-1; PI3K, phosphoinositide 3-kinase; PPAR, peroxisome proliferator-activated receptor; PRAS40, proline-rich Akt substrate of 40 kDa; PTEN, phosphatase and tensin homolog; p70S6K1, p70 ribosomal S6 protein kinase 1; Raptor, regulatory-associated protein of mTOR; Rheb, Ras homolog enriched in brain protein; SMCR8, Smith-Magenis syndrome chromosome region 8; TFEB, transcription factor EB; TSC1/2, Tuberous sclerosis protein-complex; UBQLN2/4, ubiquilin genes; ULK1, Unc-51-like kinase 1; 4E-BP1, 4E-binding protein-1; 6-OHDA, 6-hydroxydopamine.
References
Abd-Elrahman, K. S., and Ferguson, S. S. G. (2019). Modulation of mTOR and CREB pathways following mGluR5 blockade contribute to improved Huntington’s pathology in zQ175 mice. Mol. Brain 12:35.
Alirezaei, M., Kemball, C. C., Flynn, C. T., Wood, M. R., Whitton, J. L., and Kiosses, W. B. (2010). Short-term fasting induces profound neuronal autophagy. Autophagy 6, 702–710. doi: 10.4161/auto.6.6.12376
Alzheimer’s Association (2020). Alzheimer’s Disease Facts and Figures. Alzheimers Dement. Chicago, IL: Alzheimer’s Association.
An, W. L., Cowburn, R. F., Li, L., Braak, H., Alafuzoff, I., Iqbal, K., et al. (2003). Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol. 163, 591–607. doi: 10.1016/s0002-9440(10)63687-5
Andre, V. M., Cepeda, C., and Levine, M. S. (2010). Dopamine and glutamate in Huntington’s disease: a balancing act. CNS Neurosci. Ther. 16, 163–178. doi: 10.1111/j.1755-5949.2010.00134.x
Belelovsky, K., Kaphzan, H., Elkobi, A., and Rosenblum, K. (2009). Biphasic activation of the mTOR pathway in the gustatory cortex is correlated with and necessary for taste learning. J. Neurosci. 29, 7424–7431. doi: 10.1523/jneurosci.3809-08.2009
Blundell, J., Kouser, M., and Powell, C. M. (2008). Systemic inhibition of mammalian target of rapamycin inhibits fear memory reconsolidation. Neurobiol. Learn. Mem. 90, 28–35. doi: 10.1016/j.nlm.2007.12.004
Braak, H., Del Tredici, K., Rub, U., de Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/s0197-4580(02)00065-9
Broom, G. M., Shaw, I. C., and Rucklidge, J. J. (2019). The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition 60, 118–121. doi: 10.1016/j.nut.2018.10.003
Bryan, M. R., and Bowman, A. B. (2017). Manganese and the insulin-IGF signaling network in huntington’s disease and other neurodegenerative disorders. Adv. Neurobiol. 18, 113–142. doi: 10.1007/978-3-319-60189-2_6
Castedo, M., Ferri, K. F., and Kroemer, G. (2002). Mammalian target of rapamycin (mTOR): pro- and anti-apoptotic. Cell Death Differ. 9, 99–100. doi: 10.1038/sj.cdd.4400978
Chakravorty, A., Jetto, C. T., and Manjithaya, R. (2019). Dysfunctional mitochondria and mitophagy as drivers of Alzheimer’s disease pathogenesis. Front. Aging Neurosci. 11:311.
Chen, L., Xu, B., Liu, L., Luo, Y., Yin, J., Zhou, H., et al. (2010). Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab. Invest. 90, 762–773. doi: 10.1038/labinvest.2010.36
Chen, P., Chen, F., Lei, J., Li, Q., and Zhou, B. (2019). Activation of the miR-34a-mediated SIRT1/mTOR signaling pathway by urolithin a attenuates d-galactose-induced brain aging in mice. Neurotherapeutics 16, 1269–1282. doi: 10.1007/s13311-019-00753-0
Chong, Z. Z., Yao, Q., and Li, H. H. (2013). The rationale of targeting mammalian target of rapamycin for ischemic stroke. Cell Signal 25, 1598–1607. doi: 10.1016/j.cellsig.2013.03.017
Dickson, D. W., Uchikado, H., Fujishiro, H., and Tsuboi, Y. (2010). Evidence in favor of braak staging of Parkinson’s disease. Mov. Disord. 25(Suppl. 1), S78–S82.
Domanskyi, A., Geissler, C., Vinnikov, I. A., Alter, H., Schober, A., Vogt, M. A., et al. (2011). Pten ablation in adult dopaminergic neurons is neuroprotective in Parkinson’s disease models. FASEB J. 25, 2898–2910. doi: 10.1096/fj.11-181958
Dwyer, J. M., and Duman, R. S. (2013). Activation of mammalian target of rapamycin and synaptogenesis: role in the actions of rapid-acting antidepressants. Biol. Psychiatry 73, 1189–1198. doi: 10.1016/j.biopsych.2012.11.011
Fu, J., Oveisi, F., Gaetani, S., Lin, E., and Piomelli, D. (2005). Oleoylethanolamide, an endogenous PPAR-alpha agonist, lowers body weight and hyperlipidemia in obese rats. Neuropharmacology 48, 1147–1153. doi: 10.1016/j.neuropharm.2005.02.013
Fujikake, N., Shin, M., and Shimizu, S. (2018). Association between autophagy and neurodegenerative diseases. Front. Neurosci. 12:255.
Fujimoto, A., Akifusa, S., Kamio, N., Hirofuji, T., Nonaka, K., and Yamashita, Y. (2010). Involvement of mTOR in globular adiponectin-induced generation of reactive oxygen species. Free Radic Res. 44, 128–134. doi: 10.3109/10715760903348328
Gafford, G. M., Parsons, R. G., and Helmstetter, F. J. (2011). Consolidation and reconsolidation of contextual fear memory requires mammalian target of rapamycin-dependent translation in the dorsal hippocampus. Neuroscience 182, 98–104. doi: 10.1016/j.neuroscience.2011.03.023
Graber, T. E., McCamphill, P. K., and Sossin, W. S. (2013). A recollection of mTOR signaling in learning and memory. Learn. Mem. 20, 518–530. doi: 10.1101/lm.027664.112
Griffin, R. J., Moloney, A., Kelliher, M., Johnston, J. A., Ravid, R., Dockery, P., et al. (2005). Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem. 93, 105–117. doi: 10.1111/j.1471-4159.2004.02949.x
Groot, C., Hooghiemstra, A. M., Raijmakers, P. G., van Berckel, B. N., Scheltens, P., Scherder, E. J., et al. (2016). The effect of physical activity on cognitive function in patients with dementia: a meta-analysis of randomized control trials. Ageing Res. Rev. 25, 13–23. doi: 10.1016/j.arr.2015.11.005
Gugliandolo, A., Pollastro, F., Bramanti, P., and Mazzon, E. (2020). Cannabidiol exerts protective effects in an in vitro model of Parkinson’s disease activating AKT/mTOR pathway. Fitoterapia 143:104553. doi: 10.1016/j.fitote.2020.104553
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. doi: 10.1016/j.molcel.2008.03.003
Halikas, A., and Gibas, K. J. (2018). AMPK induced memory improvements in the diabetic population: a case study. Diabetes Metab. Syndr. 12, 1141–1146. doi: 10.1016/j.dsx.2018.04.033
Hamano, T., Gendron, T. F., Causevic, E., Yen, S. H., Lin, W. L., Isidoro, C., et al. (2008). Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur. J. Neurosci. 27, 1119–1130. doi: 10.1111/j.1460-9568.2008.06084.x
Harada, H., Andersen, J. S., Mann, M., Terada, N., and Korsmeyer, S. J. (2001). p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. U.S.A. 98, 9666–9670. doi: 10.1073/pnas.171301998
Hashimoto, M., Ho, G., Sugama, S., Takamatsu, Y., Shimizu, Y., Takenouchi, T., et al. (2018). Evolvability of amyloidogenic proteins in human brain. J. Alzheimers Dis. 62, 73–83. doi: 10.3233/jad-170894
Heras-Sandoval, D., Perez-Rojas, J. M., and Pedraza-Chaverri, J. (2020). Novel compounds for the modulation of mTOR and autophagy to treat neurodegenerative diseases. Cell Signal 65:109442. doi: 10.1016/j.cellsig.2019.109442
Hers, I., Vincent, E. E., and Tavare, J. M. (2011). Akt signalling in health and disease. Cell Signal 23, 1515–1527. doi: 10.1016/j.cellsig.2011.05.004
Hoglund, K., Kern, S., Zettergren, A., Borjesson-Hansson, A., Zetterberg, H., Skoog, I., et al. (2017). Preclinical amyloid pathology biomarker positivity: effects on tau pathology and neurodegeneration. Transl. Psychiatry 7:e995. doi: 10.1038/tp.2016.252
Hotta, M., Nakata, R., Katsukawa, M., Hori, K., Takahashi, S., and Inoue, H. (2010). Carvacrol, a component of thyme oil, activates PPARalpha and gamma and suppresses COX-2 expression. J. Lipid Res. 51, 132–139. doi: 10.1194/jlr.m900255-jlr200
Inoki, K., Li, Y., Zhu, T., Wu, J., and Guan, K. L. (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657. doi: 10.1038/ncb839
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi: 10.1016/s0092-8674(03)00929-2
Jia, R. X., Liang, J. H., Xu, Y., and Wang, Y. Q. (2019). Effects of physical activity and exercise on the cognitive function of patients with Alzheimer disease: a meta-analysis. BMC Geriatr. 19:181.
Jobim, P. F., Pedroso, T. R., Christoff, R. R., Werenicz, A., Maurmann, N., Reolon, G. K., et al. (2012). Inhibition of mTOR by rapamycin in the amygdala or hippocampus impairs formation and reconsolidation of inhibitory avoidance memory. Neurobiol. Learn Mem. 97, 105–112. doi: 10.1016/j.nlm.2011.10.002
Kang, E. B., and Cho, J. Y. (2015). Effect of treadmill exercise on PI3K/AKT/mTOR, autophagy, and Tau hyperphosphorylation in the cerebral cortex of NSE/htau23 transgenic mice. J. Exerc. Nutr. Biochem. 19, 199–209. doi: 10.5717/jenb.2015.15090806
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kou, X., Chen, D., and Chen, N. (2019). Physical activity alleviates cognitive dysfunction of Alzheimer’s disease through regulating the mtor signaling pathway. Int. J. Mol. Sci. 20:1591. doi: 10.3390/ijms20071591
Kou, X., Li, J., Liu, X., Chang, J., Zhao, Q., Jia, S., et al. (2017). Swimming attenuates d-galactose-induced brain aging via suppressing miR-34a-mediated autophagy impairment and abnormal mitochondrial dynamics. J. Appl. Physiol. (1985) 122, 1462–1469. doi: 10.1152/japplphysiol.00018.2017
Kruger, U., Wang, Y., Kumar, S., and Mandelkow, E. M. (2012). Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol. Aging 33, 2291–2305. doi: 10.1016/j.neurobiolaging.2011.11.009
Lan, Y., Sullivan, P. M., and Hu, F. (2019). SMCR8 negatively regulates AKT and MTORC1 signaling to modulate lysosome biogenesis and tissue homeostasis. Autophagy 15, 871–885. doi: 10.1080/15548627.2019.1569914
Larson, E. B., Wang, L., Bowen, J. D., McCormick, W. C., Teri, L., Crane, P., et al. (2006). Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann. Intern. Med. 144, 73–81.
Laukkanen, T., Kunutsor, S., Kauhanen, J., and Laukkanen, J. A. (2017). Sauna bathing is inversely associated with dementia and Alzheimer’s disease in middle-aged Finnish men. Age Ageing 46, 245–249. doi: 10.1093/ageing/afw212
Lee, H. J., Khoshaghideh, F., Patel, S., and Lee, S. J. (2004). Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci. 24, 1888–1896. doi: 10.1523/jneurosci.3809-03.2004
Li, H., Ham, A., Ma, T. C., Kuo, S. H., Kanter, E., Kim, D., et al. (2019a). Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 15, 113–130. doi: 10.1080/15548627.2018.1509818
Li, W., Su, D., Zhai, Q., Chi, H., She, X., Gao, X., et al. (2019b). Proteomes analysis reveals the involvement of autophagy in AD-like neuropathology induced by noise exposure and ApoE4. Environ. Res. 176:108537. doi: 10.1016/j.envres.2019.108537
Li, X., Alafuzoff, I., Soininen, H., Winblad, B., and Pei, J. J. (2005). Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 272, 4211–4220. doi: 10.1111/j.1742-4658.2005.04833.x
Li, X., An, W. L., Alafuzoff, I., Soininen, H., Winblad, B., and Pei, J. J. (2004). Phosphorylated eukaryotic translation factor 4E is elevated in Alzheimer brain. Neuroreport 15, 2237–2240. doi: 10.1097/00001756-200410050-00019
Li, Z., Li, X., Lin, S., Chen, Y., Ma, S., Fu, Y., et al. (2017). Nicotinic acid receptor GPR109A exerts anti-inflammatory effects through inhibiting the Akt/mTOR signaling pathway in MIN6 pancreatic beta cells. Ann. Clin. Lab. Sci. 47, 729–737.
Lloyd, B. A., Hake, H. S., Ishiwata, T., Farmer, C. E., Loetz, E. C., Fleshner, M., et al. (2017). Exercise increases mTOR signaling in brain regions involved in cognition and emotional behavior. Behav. Brain Res. 323, 56–67. doi: 10.1016/j.bbr.2017.01.033
Lopez-Armada, M. J., Riveiro-Naveira, R. R., Vaamonde-Garcia, C., and Valcarcel-Ares, M. N. (2013). Mitochondrial dysfunction and the inflammatory response. Mitochondrion 13, 106–118. doi: 10.1016/j.mito.2013.01.003
Majumder, S., Richardson, A., Strong, R., and Oddo, S. (2011). Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 6:e25416. doi: 10.1371/journal.pone.0025416
Martin, D. D., Ladha, S., Ehrnhoefer, D. E., and Hayden, M. R. (2015). Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 38, 26–35. doi: 10.1016/j.tins.2014.09.003
Miller, V. J., Villamena, F. A., and Volek, J. S. (2018). Nutritional ketosis and mitohormesis: potential implications for mitochondrial function and human health. J. Nutr. Metab. 2018:5157645.
Mondadori, C. R., de Quervain, D. J., Buchmann, A., Mustovic, H., Wollmer, M. A., Schmidt, C. F., et al. (2007). Better memory and neural efficiency in young apolipoprotein E epsilon4 carriers. Cereb. Cortex 17, 1934–1947. doi: 10.1093/cercor/bhl103
Mujica-Parodi, L. R., Amgalan, A., Sultan, S. F., Antal, B., Sun, X., Skiena, S., et al. (2020). Diet modulates brain network stability, a biomarker for brain aging, in young adults. Proc. Natl. Acad. Sci. U.S.A. 117, 6170–6177. doi: 10.1073/pnas.1913042117
Napolitano, G., Esposito, A., Choi, H., Matarese, M., Benedetti, V., Di Malta, C., et al. (2018). mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun. 9:3312.
Nave, B. T., Ouwens, M., Withers, D. J., Alessi, D. R., and Shepherd, P. R. (1999). Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem. J. 344(Pt 2), 427–431. doi: 10.1042/bj3440427
Newman, J. C., and Verdin, E. (2017). Beta-hydroxybutyrate: a signaling metabolite. Annu. Rev. Nutr. 37, 51–76. doi: 10.1146/annurev-nutr-071816-064916
Nie, X., Sun, Y., Wan, S., Zhao, H., Liu, R., Li, X., et al. (2017). Subregional structural alterations in hippocampus and nucleus accumbens correlate with the clinical impairment in patients with Alzheimer’s disease clinical spectrum: parallel combining volume and vertex-based approach. Front. Neurol. 8:399.
Nilsson, P., Loganathan, K., Sekiguchi, M., Matsuba, Y., Hui, K., Tsubuki, S., et al. (2013). Abeta secretion and plaque formation depend on autophagy. Cell Rep. 5, 61–69. doi: 10.1016/j.celrep.2013.08.042
Nixon, R. A. (2007). Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 120, 4081–4091. doi: 10.1242/jcs.019265
Norwitz, N. G., Hu, M. T., and Clarke, K. (2019a). The Mechanisms by which the ketone body d-beta-hydroxybutyrate may improve the multiple cellular pathologies of Parkinson’s disease. Front. Nutr. 6:63.
Norwitz, N. G., Mota, A. S., Norwitz, S. G., and Clarke, K. (2019b). Multi-loop model of alzheimer disease: an integrated perspective on the Wnt/GSK3beta, alpha-synuclein, and Type 3 diabetes hypotheses. Front. Aging Neurosci. 11:184.
Oddo, S. (2012). The role of mTOR signaling in Alzheimer disease. Front. Biosci. (Schl. Ed.) 4, 941–952. doi: 10.2741/s310
Papadopoli, D., Boulay, K., Kazak, L., Pollak, M., Mallette, F., Topisirovic, I., et al. (2019). mTOR as a central regulator of lifespan and aging. F1000Res 8:F1000FacultyRev–998.
Ramalingam, M., Huh, Y. J., and Lee, Y. I. (2019). The impairments of alpha-synuclein and mechanistic target of rapamycin in rotenone-induced SH-SY5Y cells and mice model of Parkinson’s disease. Front. Neurosci. 13:1028.
Rigano, D., Sirignano, C., and Taglialatela-Scafati, O. (2017). The potential of natural products for targeting PPARalpha. Acta Pharm. Sin. B 7, 427–438.
Roberts, M. N., Wallace, M. A., Tomilov, A. A., Zhou, Z., Marcotte, G. R., Tran, D., et al. (2017). A ketogenic diet extends longevity and healthspan in adult mice. Cell Metab 26, 539–546.e535.
Rocca, W. A. (2018). The burden of Parkinson’s disease: a worldwide perspective. Lancet Neurol. 17, 928–929. doi: 10.1016/s1474-4422(18)30355-7
Rodriguez de Fonseca, F., Navarro, M., Gomez, R., Escuredo, L., Nava, F., et al. (2001). An anorexic lipid mediator regulated by feeding. Nature 414, 209–212. doi: 10.1038/35102582
Sampson, T. R., Debelius, J. W., Thron, T., Janssen, S., Shastri, G. G., Ilhan, Z. E., et al. (2016). Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 16, 1469–1480.e1412.
San, Y. Z., Liu, Y., Zhang, Y., Shi, P. P., and Zhu, Y. L. (2015). Peroxisome proliferator-activated receptor-gamma agonist inhibits the mammalian target of rapamycin signaling pathway and has a protective effect in a rat model of status epilepticus. Mol. Med. Rep. 12, 1877–1883. doi: 10.3892/mmr.2015.3641
Sanadgol, N., Barati, M., Houshmand, F., Hassani, S., Clarner, T., Shahlaei, M., et al. (2019). Metformin accelerates myelin recovery and ameliorates behavioral deficits in the animal model of multiple sclerosis via adjustment of AMPK/Nrf2/mTOR signaling and maintenance of endogenous oligodendrogenesis during brain self-repairing period. Pharmacol. Rep. 72, 641–658. doi: 10.1007/s43440-019-00019-8
Sarkar, S. (2013). Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 41, 1103–1130. doi: 10.1042/bst20130134
Schmeisser, K., and Parker, J. A. (2019). Pleiotropic effects of mTOR and autophagy during development and aging. Front. Cell Dev. Biol. 7:192.
Senturk, M., Lin, G., Zuo, Z., Mao, D., Watson, E., Mikos, A. G., et al. (2019). Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat. Cell Biol. 21, 384–396. doi: 10.1038/s41556-019-0281-x
Sharp, Z. D., and Strong, R. (2010). The role of mTOR signaling in controlling mammalian life span: what a fungicide teaches us about longevity. J. Gerontol. A Biol. Sci. Med. Sci. 65, 580–589. doi: 10.1093/gerona/glp212
Shin, B. K., Kang, S., Kim, D. S., and Park, S. (2018). Intermittent fasting protects against the deterioration of cognitive function, energy metabolism and dyslipidemia in Alzheimer’s disease-induced estrogen deficient rats. Exp. Biol. Med. (Maywood) 243, 334–343. doi: 10.1177/1535370217751610
Singh, R., Kolvraa, S., Bross, P., Christensen, K., Bathum, L., Gregersen, N., et al. (2010). Anti-inflammatory heat shock protein 70 genes are positively associated with human survival. Curr. Pharm. Des. 16, 796–801. doi: 10.2174/138161210790883499
Singh, R., Kolvraa, S., Bross, P., Christensen, K., Gregersen, N., Tan, Q., et al. (2006). Heat-shock protein 70 genes and human longevity: a view from Denmark. Ann. N. Y. Acad. Sci. 1067, 301–308. doi: 10.1196/annals.1354.040
Sochocka, M., Donskow-Lysoniewska, K., Diniz, B. S., Kurpas, D., Brzozowska, E., and Leszek, J. (2019). The Gut microbiome alterations and inflammation-driven pathogenesis of Alzheimer’s disease-a critical review. Mol. Neurobiol. 56, 1841–1851. doi: 10.1007/s12035-018-1188-4
Sohn, E. (2018). How the evidence stacks up for preventing Alzheimer’s disease. Nature 559, S18–S20.
Soto-Mota, A., Norwitz, N. G., and Clarke, K. (2020). Why a d-beta-hydroxybutyrate monoester? Biochem. Soc. Trans. 48, 51–59. doi: 10.1042/bst20190240
Sun, J., Conn, C. S., Han, Y., Yeung, V., and Qian, S. B. (2011). PI3K-mTORC1 attenuates stress response by inhibiting cap-independent Hsp70 translation. J. Biol. Chem. 286, 6791–6800. doi: 10.1074/jbc.m110.172882
Takei, N., Inamura, N., Kawamura, M., Namba, H., Hara, K., Yonezawa, K., et al. (2004). Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J. Neurosci. 24, 9760–9769. doi: 10.1523/jneurosci.1427-04.2004
Taylor, M. K., Sullivan, D. K., Mahnken, J. D., Burns, J. M., and Swerdlow, R. H. (2018). Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement (N. Y.) 4, 28–36. doi: 10.1016/j.trci.2017.11.002
Taylor, M. K., Swerdlow, R. H., Burns, J. M., and Sullivan, D. K. (2019). An experimental ketogenic diet for Alzheimer disease was nutritionally dense and rich in vegetables and avocado. Curr. Dev. Nutr. 3:nzz003.
Trumble, B. C., Stieglitz, J., Blackwell, A. D., Allayee, H., Beheim, B., Finch, C. E., et al. (2017). Apolipoprotein E4 is associated with improved cognitive function in Amazonian forager-horticulturalists with a high parasite burden. FASEB J. 31, 1508–1515. doi: 10.1096/fj.201601084r
Vasunilashorn, S., Finch, C. E., Crimmins, E. M., Vikman, S. A., Stieglitz, J., Gurven, M., et al. (2011). Inflammatory gene variants in the Tsimane, an indigenous Bolivian population with a high infectious load. Biodemogr. Soc. Biol. 57, 33–52. doi: 10.1080/19485565.2011.564475
Wan, Z., Mah, D., Simtchouk, S., Klegeris, A., and Little, J. P. (2014). Globular adiponectin induces a pro-inflammatory response in human astrocytic cells. Biochem. Biophys. Res. Commun. 446, 37–42. doi: 10.1016/j.bbrc.2014.02.077
Wang, C., Zhang, X., Teng, Z., Zhang, T., and Li, Y. (2014a). Downregulation of PI3K/Akt/mTOR signaling pathway in curcumin-induced autophagy in APP/PS1 double transgenic mice. Eur. J. Pharmacol. 740, 312–320. doi: 10.1016/j.ejphar.2014.06.051
Wang, L., Waltenberger, B., Pferschy-Wenzig, E. M., Blunder, M., Liu, X., Malainer, C., et al. (2014b). Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARgamma): a review. Biochem. Pharmacol. 92, 73–89.
Wang, Y., and Mandelkow, E. (2012). Degradation of tau protein by autophagy and proteasomal pathways. Biochem. Soc. Trans. 40, 644–652. doi: 10.1042/bst20120071
Waragai, M., Ho, G., Takamatsu, Y., Wada, R., Sugama, S., Takenouchi, T., et al. (2020). Adiponectin Paradox in Alzheimer’s disease; relevance to amyloidogenic evolvability? Front. Endocrinol. (Lausanne) 11:108.
Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N., and Rubinsztein, D. C. (2003). Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013. doi: 10.1074/jbc.m300227200
Wennberg, A. M., Gustafson, D., Hagen, C. E., Roberts, R. O., Knopman, D., Jack, C., et al. (2016). Serum adiponectin levels, neuroimaging, and cognition in the mayo clinic study of aging. J. Alzheimers Dis. 53, 573–581. doi: 10.3233/jad-151201
Wlodarek, D. (2019). Role of ketogenic diets in neurodegenerative diseases (Alzheimer’s disease and Parkinson’s disease). Nutrients 11:169. doi: 10.3390/nu11010169
Wright, R. O., Hu, H., Silverman, E. K., Tsaih, S. W., Schwartz, J., Bellinger, D., et al. (2003). Apolipoprotein E genotype predicts 24-month bayley scales infant development score. Pediatr Res. 54, 819–825. doi: 10.1203/01.pdr.0000090927.53818.de
Xilouri, M., Brekk, O. R., and Stefanis, L. (2016). Autophagy and alpha-synuclein: relevance to parkinson’s disease and related synucleopathies. Mov. Disord. 31, 178–192. doi: 10.1002/mds.26477
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C., and Bu, G. (2019). Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518. doi: 10.1038/s41582-019-0228-7
Yang, Y., Chen, S., Zhang, J., Li, C., Sun, Y., Zhang, L., et al. (2014). Stimulation of autophagy prevents amyloid-beta peptide-induced neuritic degeneration in PC12 cells. J. Alzheimers Dis. 40, 929–939. doi: 10.3233/jad-132270
Zhang, D., Guo, M., Zhang, W., and Lu, X. Y. (2011). Adiponectin stimulates proliferation of adult hippocampal neural stem/progenitor cells through activation of p38 mitogen-activated protein kinase (p38MAPK)/glycogen synthase kinase 3beta (GSK-3beta)/beta-catenin signaling cascade. J. Biol. Chem. 286, 44913–44920. doi: 10.1074/jbc.m111.310052
Zhang, D., Wang, X., and Lu, X. Y. (2016). Adiponectin exerts neurotrophic effects on dendritic arborization, spinogenesis, and neurogenesis of the dentate gyrus of male mice. Endocrinology 157, 2853–2869. doi: 10.1210/en.2015-2078
Zhang, J., Zhan, Z., Li, X., Xing, A., Jiang, C., Chen, Y., et al. (2017). Intermittent fasting protects against Alzheimer’s disease possible through restoring aquaporin-4 polarity. Front. Mol. Neurosci. 10:395.
Zhu, Z., Yang, C., Iyaswamy, A., Krishnamoorthi, S., Sreenivasmurthy, S. G., Liu, J., et al. (2019). Balancing mTOR signaling and autophagy in the treatment of Parkinson’s disease. Int. J. Mol. Sci. 20:728.
Keywords: Alzheimer’s disease, antagonistic pleiotropy, autophagy, insulin/Akt, mTOR, Parkinson’s disease
Citation: Norwitz NG and Querfurth H (2020) mTOR Mysteries: Nuances and Questions About the Mechanistic Target of Rapamycin in Neurodegeneration. Front. Neurosci. 14:775. doi: 10.3389/fnins.2020.00775
Received: 25 May 2020; Accepted: 01 July 2020;
Published: 29 July 2020.
Edited by:
Heather M. Wilkins, University of Kansas Medical Center Research Institute, United StatesReviewed by:
Antonella Tramutola, Sapienza University of Rome, ItalyRussell H. Swerdlow, University of Kansas, United States
Copyright © 2020 Norwitz and Querfurth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas G. Norwitz, bmljaG9sYXMubm9yd2l0ekBkcGFnLm94LmFjLnVr