 Yilan Xu
Yilan Xu Manna Zhao
Manna Zhao Yuying Han
Yuying Han Heng Zhang
Heng Zhang- Neurodegeneration and Neuroregeneration Laboratory, Department of Basic Medicine, School of Medicine, Shaoxing University, Shaoxing, China
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized clinically by severe cognitive deficits and pathologically by amyloid plaques, neuronal loss, and neurofibrillary tangles. Abnormal amyloid β-protein (Aβ) deposition in the brain is often thought of as a major initiating factor in AD neuropathology. However, gamma-aminobutyric acid (GABA) inhibitory interneurons are resistant to Aβ deposition, and Aβ decreases synaptic glutamatergic transmission to decrease neural network activity. Furthermore, there is now evidence suggesting that neural network activity is aberrantly increased in AD patients and animal models due to functional deficits in and decreased activity of GABA inhibitory interneurons, contributing to cognitive deficits. Here we describe the roles played by excitatory neurons and GABA inhibitory interneurons in Aβ-induced cognitive deficits and how altered GABA interneurons regulate AD neuropathology. We also comprehensively review recent studies on how GABA interneurons and GABA receptors can be exploited for therapeutic benefit. GABA interneurons are an emerging therapeutic target in AD, with further clinical trials urgently warranted.
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disease. AD is characterized clinically by severe cognitive deficits and pathologically by amyloid plaques, neuronal loss, and neurofibrillary tangles (Selkoe, 1999; Selkoe and Hardy, 2016; Wyss-Coray, 2016; Polanco et al., 2018). AD patients often have learning deficits, memory loss, language difficulties, confusion in place and time, and emotional and other behavioral changes (Selkoe and Hardy, 2016; Polanco et al., 2018). These changes are not only a burden to the individual but also to families, communities, and healthcare systems (Alzheimer’s Association, 2019). However, the molecular mechanisms underlying the cognitive deficits seen in AD are unclear, and there remains no effective treatment to slow or halt progression of AD.
Many factors are known to contribute to AD pathophysiology (Mucke et al., 2000; Mucke, 2009; Mucke and Selkoe, 2012), with abnormal aggregation of amyloid β-protein (Aβ) in the brain a likely dominant initiating factor (Huang and Mucke, 2012; Barage and Sonawane, 2015; Musiek and Holtzman, 2015; Wiseman et al., 2015; Donohue et al., 2017). Aβ is formed from cleavage of amyloid precursor protein (APP) by beta (β)-(BACE1) and gamma (γ)-secretase, and Aβ exists in different forms including monomers, oligomers, fibrils, and senile plaques (Whitehouse et al., 1982; Tanzi and Bertram, 2005; Canter et al., 2016; Riek and Eisenberg, 2016). Excessive Aβ can affect the expression of synapse-related proteins, decrease dendritic spine density, inhibit excitatory synaptic transmission, affect synaptic plasticity, and cause cognitive impairments both in AD patients (Donohue et al., 2017; Evered et al., 2019; Wen et al., 2019) and AD mouse models (Selkoe, 2019; Zott et al., 2019) including Tg2576, 5xFAD, 3xTg-AD, APP/PS1, hAPP-J20, and hAPPJ9/FYN mice (Verret et al., 2012; Flanigan et al., 2014; Palop and Mucke, 2016; Shu et al., 2016; Cattaud et al., 2018; Lerdkrai et al., 2018). A novel AppNL–F/NL–F knock-in AD model mouse shows age-dependent accumulation of Aβ, neuroinflammation, and neurodegeneration similar to AD patients (Wang et al., 2017c; Shah et al., 2018). Recent studies performed in this model indicate that, in the early stages of AD, synaptic dysfunction originates in the lateral entorhinal cortex (LEC) and then spreads to other brain areas including the hippocampus in combination with synaptic hyperexcitation, severely disrupting excitatory–inhibitory inputs and resulting in synaptic imbalance and dysfunctional synaptic homeostasis (Petrache et al., 2019; Shi et al., 2019). Dysfunctional synaptic homeostasis induces a decrease in long-term potentiation (LTP), and it has been shown that high Aβ concentrations inhibit LTP, which is essential for the normal development of cognitive functions such as learning and memory, whilst enhancing long-term depression (LTD) (Wang et al., 2004; Shankar et al., 2007; Li G. et al., 2009; Tackenberg and Brandt, 2009; Mucke and Selkoe, 2012). LTP inhibition and LTD enhancement disrupt normal neural network activity and may induce cognitive impairment in AD (Palop and Mucke, 2010). Network activities supporting cognition, including activation and deactivation deficits, abnormal oscillatory rhythmic activity, and network hypersynchrony, are altered in AD (Sperling et al., 2009), schizophrenia (Seidman et al., 2014), epilepsy (Fahoum et al., 2013; Oser et al., 2014), and other neurological and psychiatric diseases (Anticevic et al., 2012). It has been shown that network hypersynchrony and altered oscillatory rhythmic activity may lead to cognitive abnormalities in AD (Palop and Mucke, 2016). During normal brain activity, excitatory and inhibitory neurons are co-activated and maintain normal network activities (Iaccarino et al., 2016). The balance between neuronal excitation and inhibition is disrupted in neurodegenerative and psychiatric disorders including epilepsy, schizophrenia, Parkinson’s disease (PD), and AD (Shetty and Turner, 2000; Marin, 2012; Southwell et al., 2014; Tyson and Anderson, 2014). γ-aminobutyric acid (GABA) inhibitory interneurons (GABAergic neurons) regulate excitatory neurons in different brain regions through GABA release. Imbalanced GABAergic and glutamatergic transmission has been shown to impair adult neurogenesis in animal models of AD (Sun et al., 2009). Thus, abnormally increased neural network activity in AD may be due to excitatory-inhibitory imbalance.
A recent clinical imaging study combining positron emission tomography with a novel analytical framework showed that distant Aβ induces regional metabolic vulnerability, and the interaction between local Aβ and a vulnerable environment drives the clinical progression of dementia (Pascoal et al., 2019). Similar results were obtained in transgenic Aβ rats that do not form neurofibrillary tangles, suggesting a novel mechanism of cognitive deterioration (Pascoal et al., 2019). Therefore, the causal link between Aβ and AD remains controversial, and further work is still required to fully establish how Aβ participates in the definition, etiology, and diagnosis of AD. The genetic risk factors for AD may act through both Aβ-dependent and Aβ−independent mechanisms to influence disease onset and progression (Morris et al., 2014, 2018; van der Kant et al., 2020). Despite this, the presence of extracellular Aβ deposition as neuritic plaques and intracellular accumulation of hyperphosphorylated Tau (pTau) as neurofibrillary tangles remain the primary neuropathological diagnostic criteria for AD (Long and Holtzman, 2019). Moreover, plasma Aβ is a reliable biomarker for AD in the clinical setting, along with other biomarkers (Wang et al., 2019). Aβ still plays an important role in AD pathology, and here we explore both Aβ−dependent and Aβ−independent mechanisms of neurocognitive decline and how they might be exploited for clinical benefit.
Studies on Excitatory Neurons in AD
Initial studies on the pathological changes and cognitive impairment induced by Aβ focused on Aβ’s effect on excitatory neurons. In excitatory neurons, Aβ can reduce synaptic transmission by activating the glutamate N-methyl-D-aspartate receptor (NMDAR) (Liu et al., 2004; Kullmann and Lamsa, 2007; Sinnen et al., 2016; Wang and Reddy, 2017). Aβ inhibits excitatory synaptic transmission, reducing neuronal network excitability (Wang et al., 2004; Shankar et al., 2007; Li S. et al., 2009; Tackenberg and Brandt, 2009; Mucke and Selkoe, 2012). However, many studies have shown that Aβ can also induce aberrant neural network activity (Verret et al., 2012; Palop and Mucke, 2016). Aβ leads to rapid excitatory amino acid transporter (EAATs) mislocalization and internalization and alters the clearance of glutamate, resulting in excitatory neurotoxicity during AD development (Scimemi et al., 2013). Synaptic damage can be significantly inhibited by treatment with the vitamin E derivative, Trolox (Scimemi et al., 2013). An intrasynaptic vesicle APP domain also promoted the release of the excitatory neurotransmitter glutamate (Yao et al., 2019). Recent studies in APP/PS1 mice have shown that, in the early stages of AD, microRNA-34a expression increases before Aβ production and cognitive impairment (Xu et al., 2018). MicroRNA-34a caused synaptic damage and cognitive impairment by inhibiting NMDAR and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor expression (Xu et al., 2018). Moreover, increased microRNA-34a expression inhibited synaptogenesis by suppressing the expression of two synaptic proteins, synaptotagmin I and syntaxin 1A, in AD patients (Agostini et al., 2011; Sarkar et al., 2016). microRNA-34a knockdown in vitro significantly increased NMDA and AMPA receptor expression, thereby inhibiting synaptic damage and cognitive deficits (Agostini et al., 2011; Sarkar et al., 2016).
AD pathogenesis is also associated with significant cholinergic and glutamatergic neurotransmitter system dysfunction, including changes in the levels of these neurotransmitters and neuronal network dysfunction in AD patients (Rossor et al., 1981; Dickson and Murray, 2015; Ferreira-Vieira et al., 2016; Kamkwalala and Newhouse, 2017; Mesulam et al., 2019). Furthermore, the basal forebrain cholinergic system (BFCS) undergoes severe atrophy in AD patients (Grothe et al., 2012) as well as in AD animals (Salehi et al., 2006; Burke et al., 2013; Zhao et al., 2017). Based on these studies, US Food and Drug Administration (FDA)-approved drugs for AD can be divided into two categories: the acetylcholine inhibitors including tacrine, donepezil, galanthamine, and rivastigmine, which enhance residual cholinergic activity to treat mild AD, and memantine, an NMDAR antagonist (Santos et al., 2016; Michalska et al., 2017; Rojas-Gutierrez et al., 2017; Behrens et al., 2018; Canevelli et al., 2018; Ettcheto et al., 2018; Khoury et al., 2018; Sahoo et al., 2018). However, these therapeutic drugs do not fundamentally delay disease progression and are required at large doses that induce adverse reactions; they are also too expensive for many patients (Alzheimer’s Association, 2019). Therefore, there is a clinical imperative to develop different treatment schemes for different stages of AD development (Graham et al., 2017). Regulating the expression of NMDA and AMPA receptors using NMDAR antagonists to inhibit excitatory neurons and EAAT expression may improve synaptic damage and cognitive deficits, but there is clearly a need for further studies on regulating excitatory neurons and the cholinergic system to identify new therapeutic modalities.
The Role of γ-Aminobutyric Acid Inhibitory Interneurons in AD Pathogenesis
Studies have demonstrated that GABA is an important inhibitory neurotransmitter that balances neural excitability and inhibition and is ubiquitously expressed in the brains of mammals including humans (Isaacson and Scanziani, 2011; Bown and Shelp, 2016; Chen et al., 2017; Tang, 2019). GABA’s inhibitory function is mainly mediated by three different GABA receptors: GABAA, GABAB, and GABAC (Li et al., 2016). GABA inhibitory activities can be divided into two main types: phasic inhibition and tonic inhibition. In phasic inhibition, during the action potential, the membrane depolarizes and GABA is released from presynaptic vesicles to rapidly increase GABA concentrations in the synaptic cleft. GABA activates GABA receptors in the postsynaptic membrane, decreasing postsynaptic neuron excitability (Farrant and Nusser, 2005). Phasic inhibition is induced by the transient or phasic activation of GABA receptors by GABA from presynaptic vesicles (Farrant and Nusser, 2005; Li et al., 2016). The postsynaptic γ2 subunits of GABAA receptors have been shown to be the primary mediators of phasic inhibition (Schweizer et al., 2003; Farrant and Nusser, 2005). Extrasynaptic GABAA receptors containing the pi (π) subunit mediate tonic inhibition in most brain regions, and the alpha (α)-5 and delta (δ) subunits are the major GABAA receptors mediating inhibition in the hippocampus (Glykys et al., 2008). Hippocampal neurons receive inhibitory charges from tonic inhibition that account for ~75% of the total inhibitory charge received by hippocampal neurons (Mody and Pearce, 2004). In this way, GABA released from the synaptic cleft can activate extrasynaptic GABA receptors to persistently inhibit neurons (Li et al., 2016). Furthermore, α5 GABAA receptor-mediated tonic inhibitory conductance in hippocampal pyramidal neurons may regulate memory and neuroexcitatory disorders (Caraiscos et al., 2004). Moreover, α5 GABAA receptor activity predominates over synaptic inhibition in modifying the strength of both synaptic plasticity in vitro and certain forms of memory in vivo under specific conditions (Martin et al., 2010). Tonic inhibition could be deemed a continuous “brake on the system,” regulating excitation due to long-lasting hyperpolarization (Schipper et al., 2016). Therefore, tonic currents play an important role in mediating neuronal excitability, network oscillations (Mann and Mody, 2010), synaptic plasticity, neurogenesis (Ge et al., 2006; Duveau et al., 2011), neuronal development (Holter et al., 2010), information processing, and cognition (Martin et al., 2010).
Cortical microcircuits containing the hippocampus are critical for normal cognitive functions in the mammalian brain (Booker and Vida, 2018). Microcircuits comprise two major neuronal classes: excitatory principal cells and inhibitory interneurons, which release the neurotransmitters glutamate and GABA, respectively (Maccaferri and Lacaille, 2003; Booker and Vida, 2018; Huang and Paul, 2019). GABA inhibitory interneurons account for 10–20% of cortical neurons (Xu et al., 2010; Meyer et al., 2011; Moore and Wehr, 2013; Hu et al., 2014). They can regulate the activity of excitatory neurons to maintain normal neural circuitry and neural network activities (Verret et al., 2012; Palop and Mucke, 2016). According to differential neuronal molecular expression, cortical interneurons are divided into at least five types including parvalbumin (PV) neurons, neuropeptide somatostatin (SST/SOM) neurons, neuropeptide Y (NPY) neurons, vasoactive intestinal peptide (VIP) neurons, and cholecystokinin (CCK) neurons (DeFelipe et al., 2013). Of these, SST neurons account for 20–30% and PV inhibitory interneurons 40–50% of GABA inhibitory interneurons (Wonders and Anderson, 2006). Wamsley et al. classified cortical interneurons into four types that do not overlap according to their expressed molecular markers, namely PV, SST, VIP, and RELN (Reelin, non-SST) neurons (Wamsley and Fishell, 2017). Tremblinglay et al. divided cortical interneurons into PV, SST, and ionotropic serotonin receptor 5HT3α (5HT3αR) neurons, which were further divided into VIP and non-VIP neurons (Tremblay et al., 2016). Rudy et al. found that PV, SST, and 5HT3αR interneurons account for nearly 100% of neocortical GABAergic neurons, representing 40, 30, and 30%, respectively (Rudy et al., 2011). Neurogliaform cells, also called spiderweb cells, belong to VIP-negative/SST-negative 5HT3αR neurons (Rudy et al., 2011). In rat and human cerebral cortex, neurogliaform cells are unique in that they elicit slow, long-lasting, inhibitory postsynaptic potentials (IPSPs) on pyramidal cells and other interneurons through the combined activation of slow GABAA and GABAB receptors (Tamas et al., 2003; Olah et al., 2007). Neurogliaform cells produce hyperpolarizing responses in a large fraction of nearby neurons via axonal varicosities containing synaptic vesicles without requiring synapses to produce inhibitory responses, suggesting that the cells influence target neurons by volume release of GABA (Olah et al., 2009). Moreover, the inhibitory effect of neurogliaform cells is dependent on GABAAδ receptors, which preferentially localize to neurogliaform cells of all the cortical interneurons (Olah et al., 2009).
Early studies showed that GABA inhibitory interneurons are not vulnerable to Aβ attack (Rossor et al., 1982; Pike and Cotman, 1993). Therefore, research efforts have mostly focused on the effect of Aβ on excitatory neurons or excitatory synaptic transmission (Wang et al., 2004; Shankar et al., 2007; Li G. et al., 2009; Tackenberg and Brandt, 2009; Mucke and Selkoe, 2012). Recent studies, however, have shown that AD patients suffer from memory and cognitive impairment that is obviously different to normal age-related decline and partly due to hippocampal neuron over-activity caused by GABA inhibitory interneuron dysfunction (Huang and Mucke, 2012; Govindpani et al., 2017; Selkoe, 2019; Villette and Dutar, 2017; Table 1). These neural circuits are overactive in different transgenic AD mouse models including hAPP-J20, Tg2576, 5xFAD, 3xTg-AD, and APP/PS1 mice (Verret et al., 2012; Flanigan et al., 2014; Palop and Mucke, 2016; Shu et al., 2016; Cattaud et al., 2018; Lerdkrai et al., 2018; Table 2). The brain wave activity recorded by electroencephalography (EEG) in APP transgenic mice contains epileptiform discharges, suggesting that neural network excitability and neuronal discharge synchronization are enhanced in AD (Verret et al., 2012; Palop and Mucke, 2016; Lerdkrai et al., 2018). Since neural network activity is regulated by both excitatory and inhibitory neurons, these seemingly contradictory results suggest that Aβ not only affects the function of excitatory neurons but also inhibitory neurons, breaking down the balance between neuronal excitability and inhibition. Therefore, the role played by GABA inhibitory interneurons in AD development is attracting increasing attention.
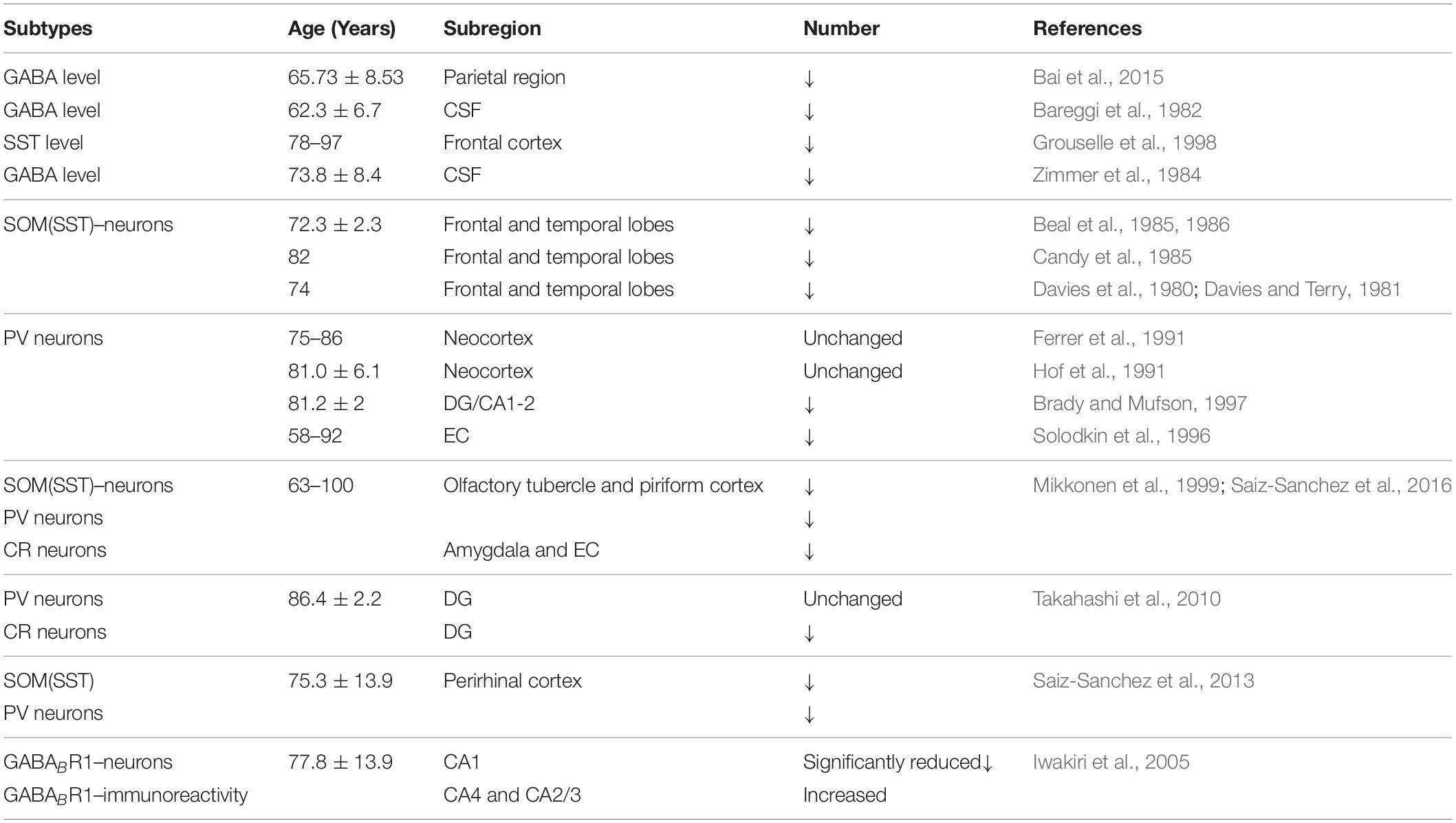
Table 1. Alterations in GABA levels and GABAergic interneurons in AD patients.
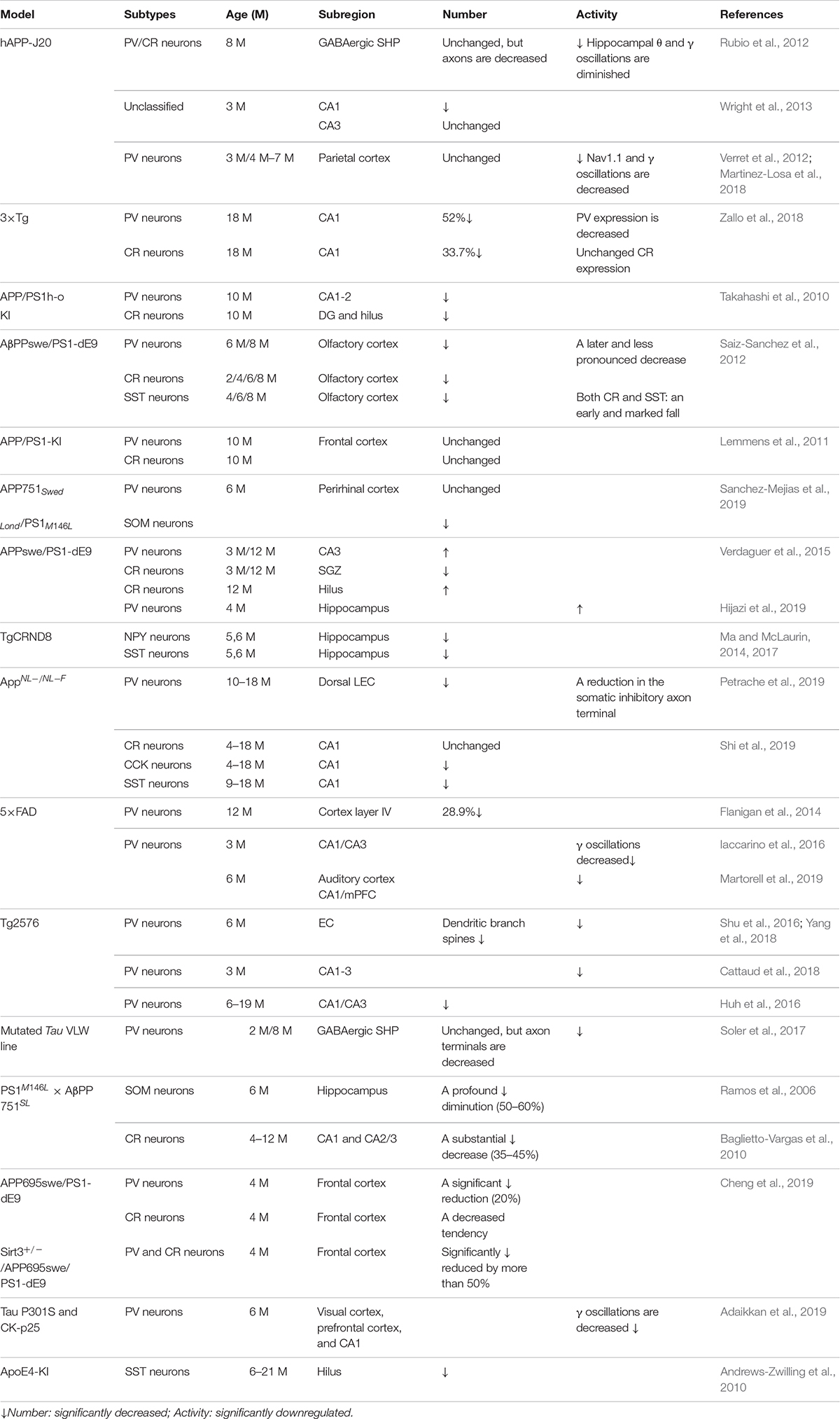
Table 2. Alterations in specific subtypes of GABA interneuron in typical AD mouse models.
Further studies have indicated that abnormal Aβ production and aggregation interfere with GABA inhibitory interneuron function, aberrantly activating hippocampal neurons and promoting cognitive impairment in AD mice (Verret et al., 2012). There is abnormal hippocampal overactivity in patients with moderate AD and young people carrying the apolipoprotein E (APOE4) gene (Filippini et al., 2009; Yassa et al., 2010; Bakker et al., 2012). Imbalanced excitatory and inhibitory neurons in the hippocampus may be important in the cognitive impairment seen in aging and AD patients (Palop and Mucke, 2016; Li et al., 2017; Selkoe, 2019). Aβ can reduce the number and activity of GABA inhibitory interneurons, resulting in abnormal synaptic transmission and aberrant neural network activity, ultimately causing cognitive impairment in both AD patients and mice (Verret et al., 2012; Palop and Mucke, 2016; Walsh et al., 2017; Frere and Slutsky, 2018). Moreover, the altered patterns of synchronous activity might be caused by the loss of GABAergic septohippocampal pathway (SHP) axons in AD patients, which might also modulate hippocampal network activities (Rubio et al., 2012; Soler et al., 2017). Therefore, targeting GABA inhibitory interneurons to rescue cognitive impairment might be a valuable therapeutic option, perhaps by increasing the number or activity of GABA inhibitory interneurons or the number of GABA inhibitory interneuron synapses or regulating GABA neurotransmitters (Figure 1 and Table 3).
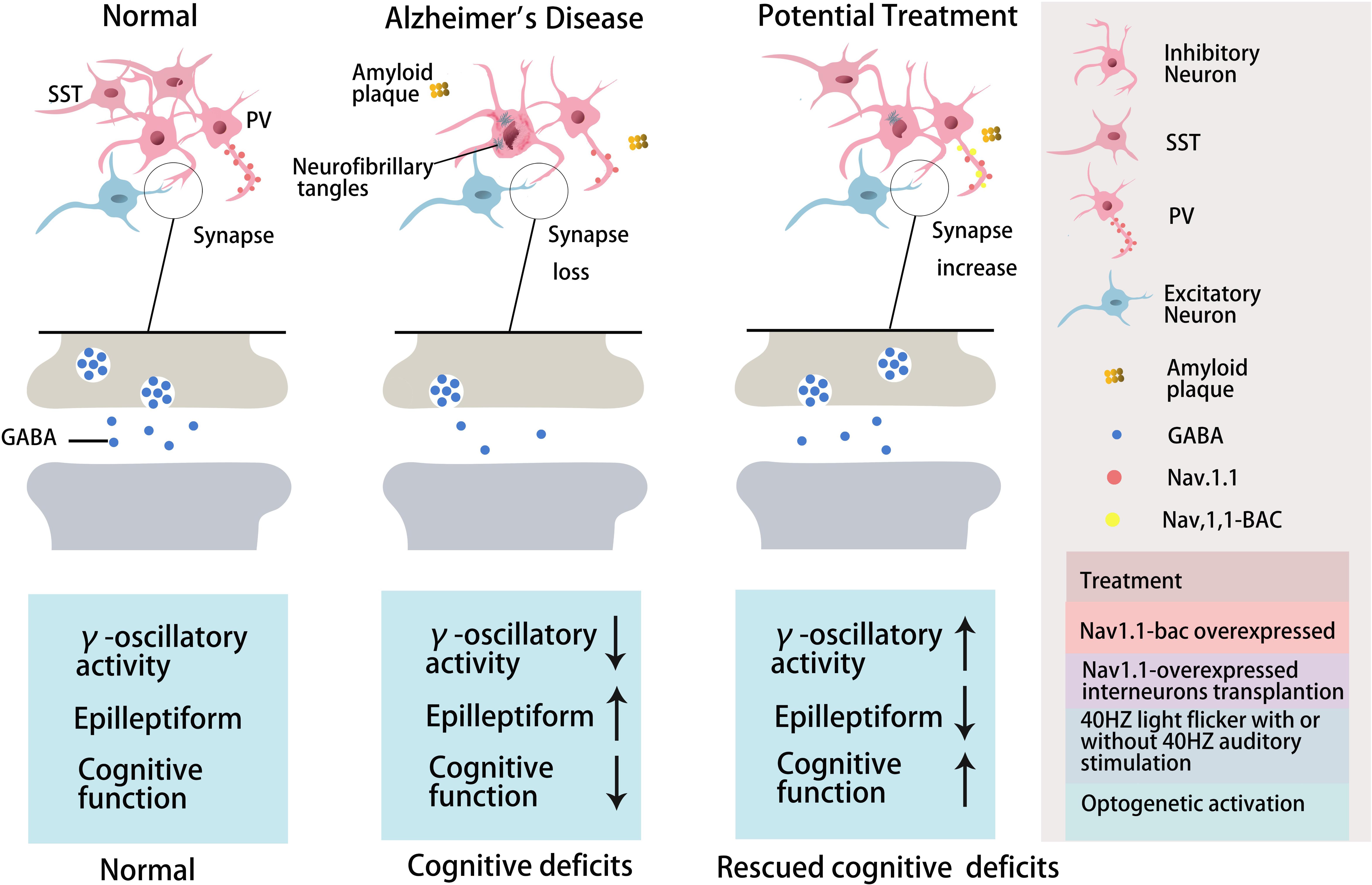
Figure 1. The role of GABA inhibitory interneurons, especially PV neurons and SST neurons, during AD progression and as potential treatment targets. Abnormal increased network activity in AD pathogenesis may be due to GABA inhibitory interneuron loss, synapse loss, or GABA inhibitory interneuron dysfunction, eventually leading to the development of the disease. GABA inhibitory interneurons are a potential target for AD treatment by increasing neuron number, enhancing neuronal activity, inhibiting synapses loss, or promoting GABA release. PV inhibitory interneurons in AD are dysfunctional with decreased γ-oscillatory activity, Nav1.1 expression, and GABA release (Verret et al., 2012). Improving Nav1.1 expression (Verret et al., 2012), enhancing γ-oscillatory activity in PV inhibitory interneurons by 40 Hz light flickering with/without 40 Hz auditory stimulation (Iaccarino et al., 2016; Adaikkan et al., 2019; Martorell et al., 2019), or transplanting Nav1.1-overexpressing interneurons (Verret et al., 2012; Martinez-Losa et al., 2018) could inhibit epileptiform phenomena and rescue cognitive deficits. Optogentic activation of PV and SST neurons rescued network oscillations (Chung et al., 2020; Park et al., 2020).
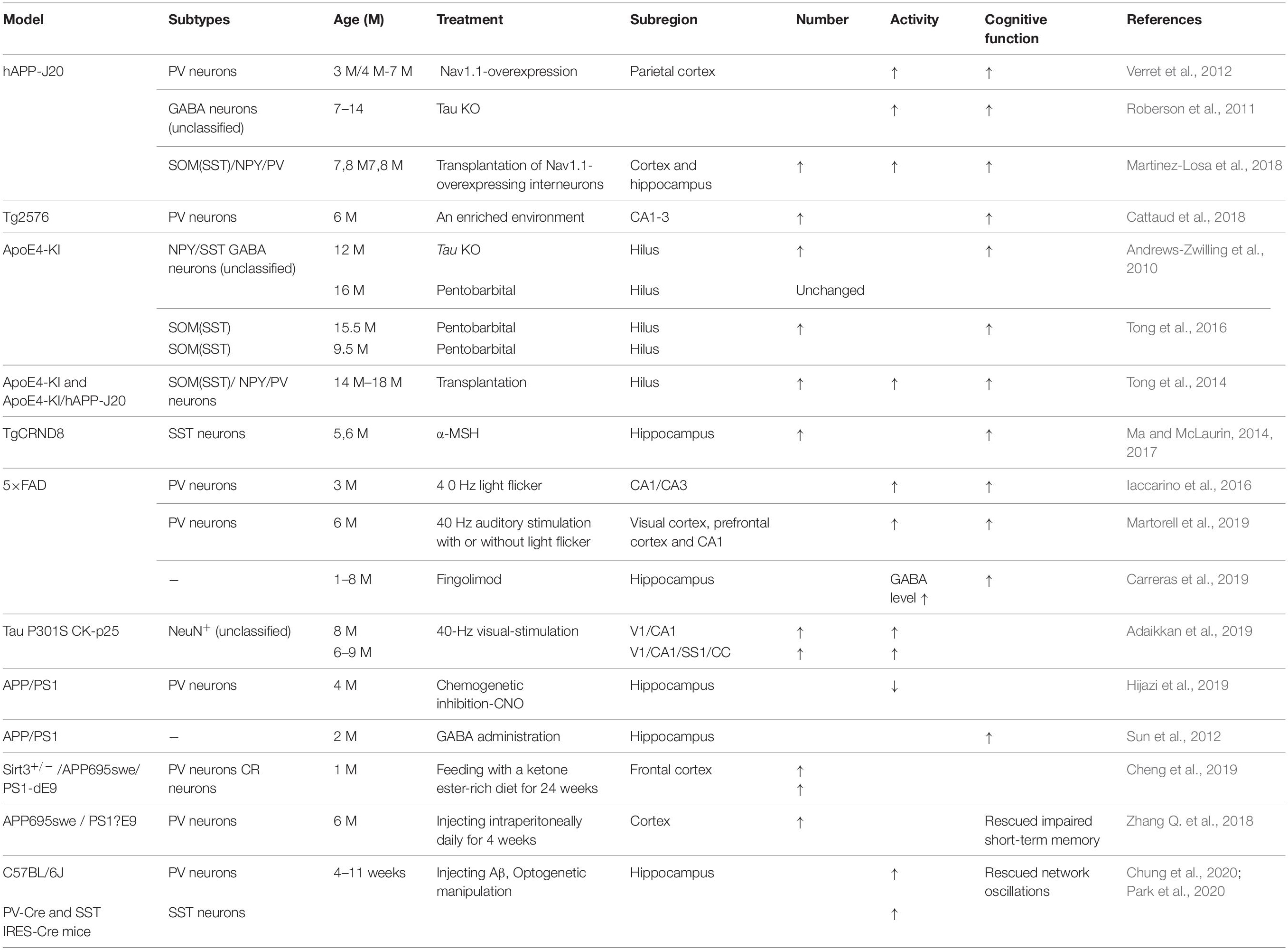
Table 3. GABA inhibitory interneurons as treatment targets in AD.
Targeting Increased Numbers of GABA Inhibitory Interneurons
Previous studies have shown that neuronal loss is common in AD (Wright et al., 2013; Figures 1, 2 and Tables 1, 2). Excessive Aβ may downregulate GABA inhibitory interneuron activity, cause GABA inhibitory interneuron loss, and thereby induce functional deficiencies in GABA inhibitory interneurons (Verret et al., 2012; Palop and Mucke, 2016; Lerdkrai et al., 2018). A recent study showed that pTau accumulates in the hippocampal subgranular cell zone and hilus in AD patients. Most of these pTau-positive cells have been identified as GABAergic interneurons by co-labeling with glutamate decarboxylase 67 (GAD67) in PV and SST neurons, further suggesting GABA inhibitory interneuron function is disturbed in AD (Zheng et al., 2020). Compared with controls, GABA inhibitory interneurons and SST neurons in the brains of AD patients are abnormally reduced, along with the inhibitory neurotransmitters (Bareggi et al., 1982; Zimmer et al., 1984; Grouselle et al., 1998; Bai et al., 2015; Shetty and Bates, 2016; Table 1). There are significantly fewer SST neurons in the frontal and temporal lobes of AD patients as measured by high-performance liquid chromatography analysis or immunohistochemical staining (Davies et al., 1980; Davies and Terry, 1981; Beal et al., 1985, 1986; Candy et al., 1985). PV neurons in the temporal neocortex have been shown to be relatively resistant to degeneration in AD, with only one in seven patients with AD having significantly reduced PV neuron numbers (Ferrer et al., 1991; Hof et al., 1991). However, PV neurons are decreased in dentate gyrus (DG)-CA4 and CA1–CA2 but not in CA3 of the hippocampus in AD patients (Brady and Mufson, 1997). Furthermore, there is a clear decrease in PV neurons in parts of the entorhinal cortex in AD patients (Solodkin et al., 1996). Other studies have shown that SST neurons, calretinin (CR) neurons, and PV neurons are decreased in the olfactory tubercle, piriform cortex, amygdala, and EC of AD patients (Mikkonen et al., 1999; Saiz-Sanchez et al., 2016). Takahashi et al. observed a dramatic reduction in the mean densities of CR neurons in the hippocampal DG in AD patients compared to controls by immunofluorescent detection. However, compared to controls, there were no differences in PV neuron densities in the same region (Takahashi et al., 2010).
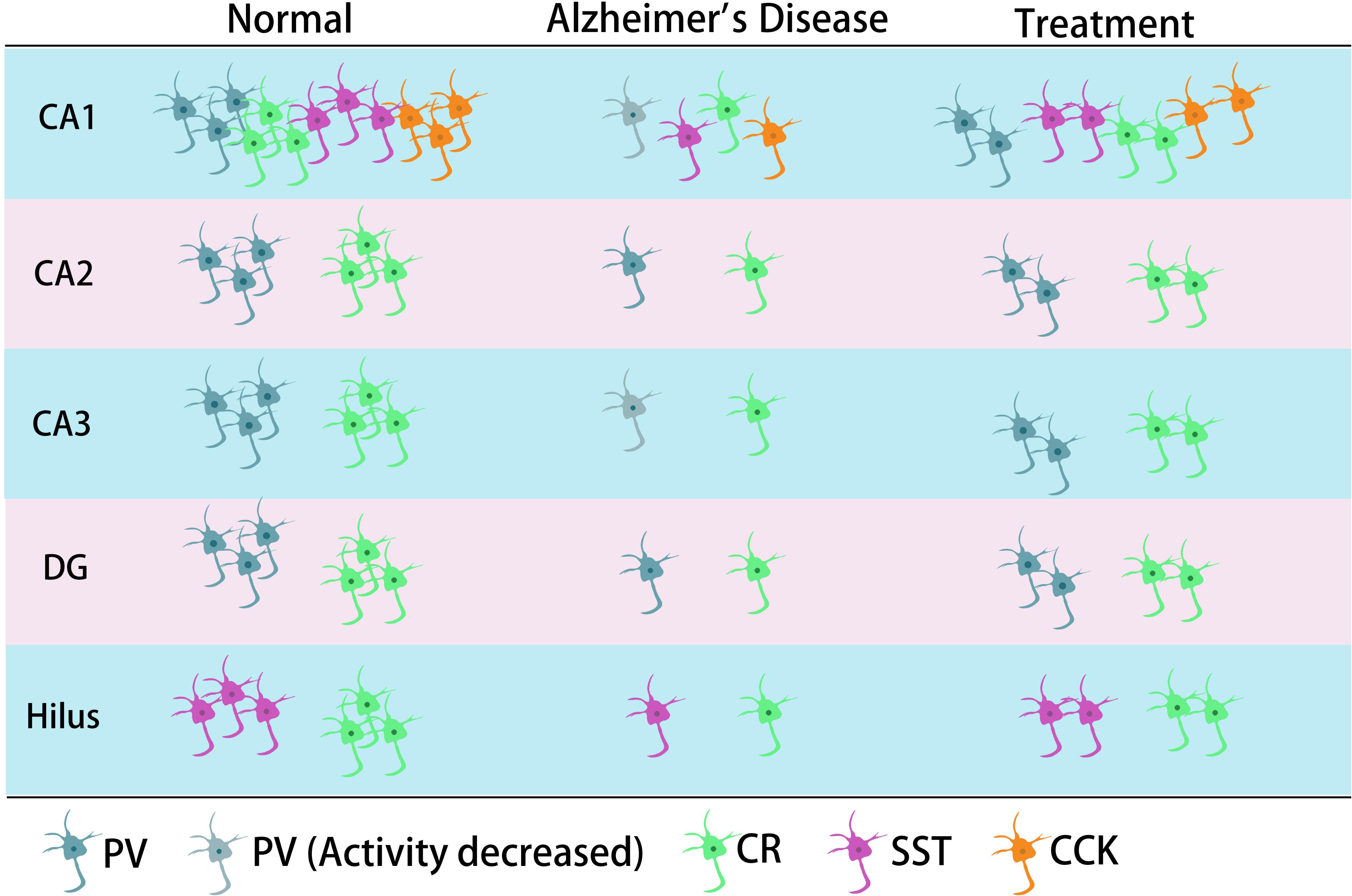
Figure 2. The main typical GABA inhibitory interneurons in the hippocampus involved in AD. In the CA1 region of hippocampus, it has been shown that all four types of GABA inhibitory interneuron change in number during AD. CR neurons decreased in different regions of hippocampus. The neural activity of PV neurons decreased in CA1 or/and CA3 in their corresponding studies. SST cells might also decrease in the hilus.
Later studies indicated that the numbers of GABA inhibitory interneurons in the hippocampus were significantly decreased in transgenic mice such as TgCRND8, Tg2576, 5xFAD, and TauPS2APP (Krantic et al., 2012; Loreth et al., 2012; Verret et al., 2012; Flanigan et al., 2014; Huh et al., 2016; Palop and Mucke, 2016; Shu et al., 2016; Cattaud et al., 2018; Lerdkrai et al., 2018; Carreras et al., 2019; Table 2). Loss of PV and CR interneurons in the hippocampal CA1 of aged 3xTg-AD mice might be in part due to the global network excitability defects associated with AD (Zallo et al., 2018). In APP/PS1 mice, there was a loss of PV neurons (40–50%) in CA1-2 and a loss of CR neurons (37–52%) in the DG and hilus (Brady and Mufson, 1997; Takahashi et al., 2010), as well as a loss of SST neurons (50–60%) in the hippocampus (Ramos et al., 2006). Another immunohistochemical study in this model showed a substantial decrease (35–45%) in CR-positive interneurons in CA1 and CA2/3 hippocampal subfields in very young mice (4 months) compared to age-matched controls (Baglietto-Vargas et al., 2010). At 4 months, Cheng et al. found that there was a significant reduction (20%) in PV neurons in the frontal cortex and also some decreases in CR neurons in APP/PS1 mice. SIRT3 haploinsufficiency (Sirt3+/–AppPs1) aggravated the loss of both PV and CR neurons by >50% compared to control mice and Sirt3 haploinsufficiency alone mice (Cheng et al., 2019). Feeding with a ketone ester-rich diet increased SIRT3 expression and prevented PV and CR neuron degeneration in Sirt3+/–AppPs1 AD mice, indicating that the aggravated GABAergic neuronal loss seen in these mice may be caused by reductions in SIRT3 that could be rescued by increased SIRT3 expression (Cheng et al., 2019). Further results obtained from APP/PS1 AD mice showed that there was also a decrease in PV, SST, and CR neurons in the olfactory cortex (Saiz-Sanchez et al., 2012). Moreover, CR neurons decreased during the early stages of the pathology, while PV neurons decreased at the later stages of the disease in the olfactory bulbs of APP/PS1 mice (Saiz-Sanchez et al., 2013; Zallo et al., 2018). However, there have been contradictory analyses of these neurons in AD mice, with other studies revealing no changes in either the hippocampus or the entorhinal cortex (Lemmens et al., 2011; Verdaguer et al., 2015). A recent study showed that SOM (SST)-positive neurons were significantly less frequent in the perirhinal cortex of 6-month-old APP/PS1 mice and in AD patients (91% in Braak V, VI cases) (Sanchez-Mejias et al., 2019). PV interneurons in the perirhinal cortex of APP/PS1 mice were unaffected, but a pronounced and significant loss (69%) has been detected in the corresponding area in AD patients (Sanchez-Mejias et al., 2019). Therefore, further studies are needed to ascertain the changes in the numbers of these neurons in AD models and their clinicopathological correlation with human disease (Table 2).
The synaptic imbalance seen in AppNL–F/NL–F AD mice results from a reduction in the number of PV interneurons and a reduction in the somatic inhibitory axon terminals in the lateral entorhinal cortex (Petrache et al., 2019). Another study indicated that CCK and SST interneurons decrease in an age-dependent manner in AppNL–F/NL–F AD mice (Shi et al., 2019). A novel mechanism of memory loss in AD suggests that the synaptic terminals of pyramidal neurons in EC layer II directly innervate CA1 PV neurons and are selectively degenerated in AD mice (Yang et al., 2018). Synaptic loss between the pyramidal and PV neurons disturbs the excitatory and inhibitory balance in the CA1 circuit and ultimately impairs learning and memory (Yang et al., 2018). APOE4 is an AD susceptibility factor and high-risk gene that can significantly lower the age of onset of AD (Genin et al., 2011; Hu et al., 2015; Liu et al., 2015; Najm et al., 2019); about 60–75% of AD patients are APOE4 gene carriers (Genin et al., 2011; Hu et al., 2015; Liu et al., 2015; Najm et al., 2019). There are GABA and SST decreases in APOE4 carriers, resulting in higher brain activity during rest and in the face of work requiring memory (Filippini et al., 2009; Dennis et al., 2010). In APOE knock-in mice, the hilar GABAergic interneurons, especially SST neurons, significantly decline with age (Andrews-Zwilling et al., 2010). Enhancing GABA signaling in APOE4 knock-in mice prevented age-dependent GABAergic interneuron decline and learning and memory deficits during middle adulthood (Tong et al., 2016). The APPE693Δ (Osaka) mutation is associated with familial AD, reduces the number of GABA inhibitory interneurons, and inhibits their functional activity, thereby causing a series of downstream AD symptoms (Umeda et al., 2017). The studies presented above suggest that the number of GABA inhibitory interneurons is significantly reduced during the process of cognitive deficits induced by excessive Aβ and may be accompanied by reduced neural activity. Thus, increasing numbers of GABA inhibitory interneurons might be potential therapeutic option in AD.
It has been reported that the treatment of transgenic CRND8 mice with alpha-melanocyte stimulating hormone (MSH) can prevent cognitive deficits by inhibiting the loss of hippocampal SST inhibitory interneurons (Ma and McLaurin, 2014, 2017). Enriched environment rescues PV cell numbers and improves cognitive deficits (Cattaud et al., 2018). Citalopram, a selective serotonin reuptake inhibitor (SSRI), can reduce Aβ formation in vitro and reduce Aβ plaques in APP/PS1 mice (Dhami et al., 2013). Furthermore, citalopram administration decreases the quantity of newly generated Aβ in young healthy humans (Sheline et al., 2014). Clinical studies indicate that citalopram has therapeutic effects in AD patients, including rescuing non-cognitive neuropsychiatric behaviors such as depression, anxiety, irritability, and apathy (Zhang H. et al., 2018), although the benefits on agitation and cognition remain controversial (Siddique et al., 2009; Porsteinsson et al., 2014). Results from socially isolated rats suggested that citalopram improves learning and memory by promoting synaptic plasticity and hippocampal neurogenesis (Gong et al., 2017). Furthermore, chronic citalopram administration in APP/PS1 mice rescued impaired short-term memory and ameliorated non-cognitive behavioral deficits such as sociability dysfunction, depressive-like behaviors, and repetition-like behaviors. After treatment with citalopram, PV neurons increased in the cortex but not in the hippocampus of APP/PS1 mice (Zhang Q. et al., 2018). The improvements in behavior might be due to an increase in PV neurons, since PV neurons are essential for social interactions, the progression of depression (Zhang Q. et al., 2018), and PV neurons in the prefrontal cortex can affect short-term memory in mice (Murray et al., 2015; Shang et al., 2015; Kim et al., 2016). Cortical PV neurons are indispensable for short-term memory and social interactions via cortical circuit plasticity (Murray et al., 2015). PV neurons were found to be decreased in the cortex of 6-month-old APP/PS1 mice, and citalopram treatment increased the numbers of cortical PV neurons, thus rescuing behavioral performance (Zhang Q. et al., 2018).
Transplantation of GABA progenitor neurons into the hippocampus also improved the learning and memory function of Aβ-overexpressing mice (Tong et al., 2014). Recent studies have shown that medial ganglionic eminence (MGE) progenitor cells derived from day 13.5 (E13.5) mouse embryos and transplanted into the DG hilum of 14-month-old AD model mice had the same electrophysiological characteristics as endogenous normal GABA inhibitory interneurons after 90 days (Tong et al., 2014; Shetty and Bates, 2016). These neurons regulated neuronal activity in the DG region, significantly inhibiting memory impairment in AD mice and reducing anxiety-related behaviors to normal levels (Tong et al., 2014; Shetty and Bates, 2016). APOE4-overexpressing mice showed GABA inhibitory interneuron loss and cognitive impairment, while APOE4 knockout significantly increased the number of GABA inhibitory interneurons and rescued cognitive deficits (Knoferle et al., 2014). In addition, cells derived from human embryonic stem cells (hESCs) or human pluripotent stem cells (hPSCs) can be differentiated into MGE progenitor cells after induction and then transplanted into the hippocampus of AD mice (Maroof et al., 2013; Nicholas et al., 2013). In these experiments, transplanted MGE progenitor cells differentiated into GABA inhibitory interneurons with normal function and significantly improved neuronal circuit dysfunction, thereby improving the cognitive ability of AD mice (Maroof et al., 2013; Nicholas et al., 2013; Southwell et al., 2014; Shetty and Bates, 2016). Therefore, GABA inhibitory interneurons are dysfunctional during AD development, and transplantation of GABA progenitor cells may improve cognition in AD mice (Table 3). In the early stages of AD, it might be possible to improve the cognitive ability of AD patients by increasing the number of normal GABA inhibitory interneurons by transplantation.
Targets to Improve GABA Inhibitory Interneuron Activity
There are now several studies suggesting that abnormal neuronal activity is associated with the increase in Aβ seen in AD patients (Donohue et al., 2017; Evered et al., 2019; Wen et al., 2019). Synaptic inhibition and abnormally active neural network activity may coexist in both AD mice and AD patients (Selkoe, 2019), although further studies are needed to confirm this. Clusters of hyperactive neurons have been detected near amyloid plaques, and the hyperactivity is presumably due to a relative decrease in synaptic inhibition (Busche et al., 2008). Previous studies have shown that glutamate decarboxylase 65 (GAD65) protein levels are significantly reduced in AD patients and that, in these patients, the GABAergic system is severely affected (Schwab et al., 2013). GAD65 deficits may contribute to AD pathogenesis through a loss of GABAergic inhibitory activity (Schwab et al., 2013). In hAPP-J20 mice, without inhibiting the action potential, the frequency of spontaneous inhibitory postsynaptic currents (sIPSCs) significantly decreases, as recorded in the granular cells in the hippocampal DG and pyramidal neurons in cortical layers II/III (Verret et al., 2012; Palop and Mucke, 2016), suggesting that Aβ overexpression affects the activity of GABA inhibitory interneurons (Roberson et al., 2011). Subsequent studies have shown that these GABA inhibitory interneurons are dysfunctional, and GABA release is impaired in the hippocampus of hAPP-J20 AD mice, leading to the dysfunction of neural circuits and cognitive impairment during AD development (Verret et al., 2012; Villette and Dutar, 2017). A relative decrease in synaptic inhibition contributed to neuronal hyperactivity near amyloid plaques in the cerebral cortex of APP23 × PS45 mice (Busche et al., 2008), indicating a reduction in activity of GABA inhibitory interneurons. Oriens lacunosum-moleculare (O-LM) interneurons are one type of SST-positive interneuron in the hippocampus. In APP/PS1 mice, progressive axonal loss and increased turnover of dendritic spines suggested defective O-LM interneuron connectivity, resulting in O-LM interneuron dysfunction associated with memory deficits (Schmid et al., 2016). Similarly, significant GABA inhibitory interneuron functional impairment and GABA release inhibition were found in the hippocampi of APOE4 transgenic AD mice (Li S. et al., 2009). Therefore, GABA inhibitory interneurons not only decrease in number but also in activity during excessive Aβ-induced cognitive deficits, resulting in GABA inhibitory interneuron functional impairment. However, further studies are necessary to clarify the mechanisms of functional impairment mediated by GABA inhibitory interneurons.
With respect to the molecular mechanism underpinning GABA inhibitory interneuron functional impairment, it has been shown that the amplitude of the action potential induced in PV inhibitory interneurons in the cortex of hAPP-J20 mice is smaller than that of control mice (Verret et al., 2012). This further confirms that Aβ overexpression affects GABA inhibitory interneuron activity. Recent studies have shown that Aβ may inhibit PV inhibitory interneuron activity by interacting with ErbB4, subsequently causing pathological abnormalities and cognitive impairment (Zhang et al., 2017). Further studies have shown that APP can regulate GABA inhibitory interneuron function by changing L-type Ca2+ channels. The number of L-type Ca2+ channels and Ca2+ current in GABA inhibitory interneurons were obviously increased after knocking out APP by enhancing the function and plasticity of GABA inhibitory interneurons, a phenomenon reversed by re-expressing APP (Yang et al., 2009). Aβ may also affect GABA inhibitory interneuron function by regulating other ion channels. For example, the deleterious voltage-gated sodium channel Nav1.1 mutation impairs telencephalic inhibitory neurons and results in Dravet Syndrome, an intractable form of childhood epilepsy (Sun et al., 2016). Nav1.1 expression is decreased in PV inhibitory interneurons of hAPP-J20 AD mice, affecting their intrinsic excitability and inhibiting γ oscillations, thereby triggering the synchronization of excitatory pyramidal neurons and increasing abnormal network activity with characteristics similar to epilepsy (Verret et al., 2012). The γ oscillation is crucial for maintaining normal learning and memory (Verret et al., 2012; Lundqvist et al., 2016; Struber et al., 2017; Martinez-Losa et al., 2018). Upregulation of Nav1.1 in PV inhibitory interneurons enhanced PV inhibitory interneuron activity, significantly increased PV inhibitory interneuron-dependent γ oscillation, inhibited the synchronization of neural network activity, and improved the cognitive ability of AD mice (Verret et al., 2012). A further study indicated that Nav1.1-overexpressing interneuron transplants derived from the embryonic MGE enhanced PV inhibitory interneuron-dependent γ oscillatory activity, reduced network hypersynchrony, and improved cognitive functions in hAPP-J20 AD mice (Martinez-Losa et al., 2018). Since γ oscillations play a vital role in cognitive activity and neural network regulation (Sohal et al., 2009; Buzsaki and Wang, 2012; Xenos et al., 2018), many studies have shown that Aβ deposition in the brain can be reduced by enhancing γ oscillations in PV inhibitory interneurons, which may improve cognition in AD mice (Verret et al., 2012; Iaccarino et al., 2016). A non-invasive light flicker stimulus obviously enhanced PV inhibitory interneuron activity, increased γ oscillations, reduced Aβ plaque deposition and pTau levels, and ultimately improved the cognition of AD mice (Iaccarino et al., 2016; Adaikkan et al., 2019). 40 Hz visual stimulation entrained γ oscillations in the visual cortex (V1), CA1, and pre-frontal cortex, reduced neuronal and synaptic loss, modified synaptic signaling and synaptic plasticity-related proteins, and rescued spatial learning and memory in both Tau P301S and CK-p25 mice (Adaikkan et al., 2019). The Aβ levels in the cortex and hippocampus as well as the number of plaques in 5xFAD mice were significantly reduced after continuous treatment with a 40 Hz auditory stimulation, thereby inhibiting the cognitive deficits (Martorell et al., 2019). Combined 40 Hz auditory stimulation with 40 Hz light flicker further induced γ oscillations in the hippocampal CA1 and auditory cortex, reducing amyloid levels and improving memory in AD models (Martorell et al., 2019). Hippocampal in vivo multi-electrode recordings revealed that optogenetic activation of channelrhodopsin-2 (ChR2)-expressing SST and PV interneurons in Aβ-injected mice selectively restored Aβ induced reduction of the peak power of theta and gamma oscillations, respectively, and resynchronized CA1 pyramidal cell spikes (Chung et al., 2020). Similar studies by Park et al. (2020) revealed that Aβ-induced impairments of gamma oscillogenesis and oscillation-induced timing-dependent LTP were fully restored by optogenetic activation of PV and SST interneurons, respectively. Hence, these results suggest that SST neurons and PV neurons are potential therapeutic targets for restoring hippocampal network oscillations in early AD (Table 3 and Figures 1, 2).
It has been shown that Tau reduction prevents Aβ-induced defects in axonal transport (Vossel et al., 2010, 2015). Tau knockout in hAPP-J20 mice can inhibit epileptic-like EEG activity and improve learning and memory (Roberson et al., 2007, 2011), perhaps due to the increased activity of GABA inhibitory interneurons (Roberson et al., 2011). Expressing mutant Tau in the entorhinal cortex also induces excitatory neuron loss, grid cell dysfunction, and spatial memory deficits (Fu et al., 2017). Further studies have shown that Tau accumulates predominantly in excitatory neurons rather than inhibitory neurons, not only early in the entorhinal cortex but also in areas later affected by AD (Fu H. et al., 2019). These studies suggest that both Aβ and Tau can affect excitatory neurons and GABA inhibitory interneurons during AD development, probably via different mechanisms, but ultimately disturbing the balance between neuronal excitation and inhibition (Lei et al., 2016; Fu Y. et al., 2019). High levels of Tau phosphorylation induce excessive metabolism of excitatory glutamatergic neurons and GABA inhibitory interneurons, disrupting the excitation-inhibition balance and resulting in abnormal neural network activity in TauP301L mice (Nilsen et al., 2013).
Recent studies demonstrated that ErbB4 protein knockdown in PV inhibitory interneurons significantly increased LTP and improved memory ability in hAPP-J20 mice without changing Aβ plaque deposition (Zhang et al., 2017). One plausible mechanism for this is that ErbB4 ablation increases PV inhibitory interneuron activity (Zhang et al., 2017; Zhang H. et al., 2018), although this requires further confirmation. By regulating GAD67 expression in GABA inhibitory interneurons, half-quantity expression of GAD67 significantly reduced Aβ deposition and improved cognitive performance in 5xFAD mice due to elevated GABA inhibitory interneuron activity (Wang et al., 2017a). Moreover, nicotine enhanced synaptic plasticity in the CA3–CA1 synapses, and GABA receptor antagonists inhibited this enhancement, suggesting that GABAergic interneurons are required for nicotine-treated adult anti-NGF mice (AD11), a comprehensive animal model of AD (Rosato-Siri et al., 2006). However, recent studies demonstrated that at the early stage of AD (16 weeks), Aβ induces hyperexcitability of hippocampal PV interneurons and contributes to neural network dysfunction and memory impairment in APP/PS1 mice (Hijazi et al., 2019). Suppressing PV interneuron hyperexcitability restored PV interneuron properties to wild-type levels, thereby reducing inhibitory inputs into pyramidal cells and rescuing memory deficits (Hijazi et al., 2019). One possible reason for the discrepancy may be due to the different ages and models of mice used in different studies. While further studies are needed to clarify this complex issue, these results suggest that targeting GABAergic interneurons might be a potential therapeutic target for AD patients.
Targeting GABA Neurotransmitters
Both GABA and glutamate levels are significantly decreased in the temporal cortex of AD patients, suggesting deficient synaptic function and neuronal transmission in AD (Gueli and Taibi, 2013; Li et al., 2016). Using magnetic resonance spectroscopy (1H-MRS), a significant decrease in GABA levels was detected in the parietal region of AD patients (Bai et al., 2015). Further studies showed that the decreased GABA neurotransmitter levels in the CSF were associated with age and AD (Bareggi et al., 1982; Zimmer et al., 1984; Grouselle et al., 1998; Li et al., 2016). SST expression was reduced by 50% in AD, which was related to the formation of Aβ oligomers (Saiz-Sanchez et al., 2020). SST might inhibit Aβ metabolism by upregulating neprilysin activity, with elevated neprilysin activity reducing the accumulation of both soluble and fibrillar Aβ in APP transgenic mice (Saito et al., 2005). Recent studies revealed that SST interfered with Aβ fibrillization and promoted the formation of Aβ assemblies characterized by a 50–60 kDa core, and these findings may signify a new role for SST in AD (Wang et al., 2017b; Solarski et al., 2018). A liquid chromatography-mass spectrometry study suggested that GABA levels are significantly reduced in cultured APP/PS1 mouse hippocampal neurons and the CSF collected from the hippocampal regions in 0-, 2-, 6-, and 8-month-old APP/PS1 mice compared to age-matched controls (Sun et al., 2012). Direct administration of GABA into APP/PS1 AD mice at 2 months but not 6 or 8 months improved cognitive function, suggesting that GABA administration during early life may have potential as a treatment for AD (Sun et al., 2012). 5xFAD transgenic AD mice treated with fingolimod had restored hippocampal GABA, decreased brain Aβ levels, and inhibited activation of microglia and astrocytes, ultimately improving memory (Carreras et al., 2019). Therefore, it is possible to improve the cognitive impairment seen in AD model mice and AD patients by regulating the levels and release of GABA from GABAergic neurons during AD pathogenesis (Calvo-Flores et al., 2018; Table 4).
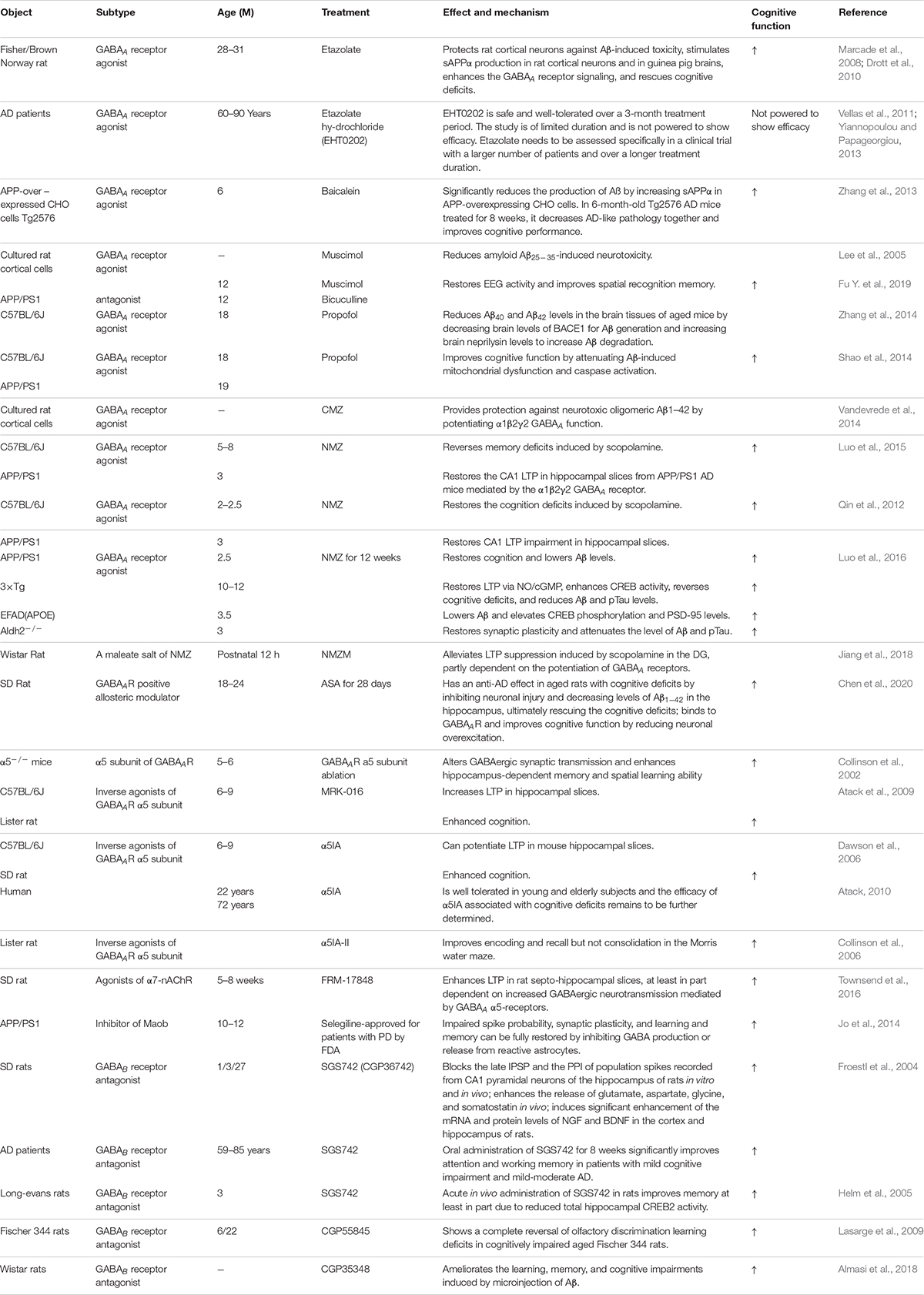
Table 4. GABA receptors as treatment targets in AD.
Previous in vitro and in vivo studies have indicated that Aβ suppresses synaptic inhibition via downregulation of GABAA receptors (Gutierres et al., 2014; Revilla et al., 2014; Bearer et al., 2015). Therefore, upregulation of GABAA receptors may be a useful AD treatment. GABAA receptor agonists have been tested in vitro and in vivo and shown to have a positive effect on AD pathological changes. In the foraging/homing task, etazolate, a selective GABAA receptor modulator, significantly improved the cognitive deficits in aged rats at 28–31 months (Drott et al., 2010). A further study indicated that etazolate can protect cortical neurons against Aβ-induced toxicity due to etazolate stimulating sAPPα production in rat cortical neurons and in guinea pig brains, enhancing GABAA receptor signaling and rescuing the cognitive deficits (Marcade et al., 2008). In a later clinical Phase IIA study, EHT0202 (etazolate hydrochloride) was shown to be safe and generally well tolerated in a 3 month, randomized, placebo-controlled, double-blind study in AD patients (Vellas et al., 2011; Yiannopoulou and Papageorgiou, 2013). Similar results were obtained using another GABAA receptor modulator baicalein. Zhang et al. found that activating GABAA receptors with baicalein significantly reduced Aβ production by increasing sAPPα in wild-type APP-overexpressing CHO cells and also had an effect in 6-month-old Tg2576 AD mice treated for 8 weeks, decreasing AD-like pathology and improving cognitive performance (Zhang et al., 2013). Chronic stimulation of GABAA receptors with their agonist muscimol reduced amyloid Aβ25–35-induced neurotoxicity in cultured rat cortical cells, and the effect was completely reversed by the GABAA receptor antagonist bicuculine (Lee et al., 2005). A further study showed that AD mice exhibit increased spontaneous EEG delta (2–4 Hz) and decreased spontaneous EEG alpha (8–12 Hz) activity in the hippocampus and decreased Y-maze EEG theta (4–8 Hz) activity in the pre-frontal cortex. Low-dose muscimol and the GABAA receptor antagonist bicuculine reduced EEG delta activity and increased EEG theta activity in the pre-frontal cortex and improved spatial recognition memory during Y-maze testing in APP/PS1 mice (Fu H. et al., 2019). Another GABAA receptor agonist, propofol, is a commonly used anesthetic that has been demonstrated to reduce Aβ40 and Aβ42 levels in the brain tissues of aged (18 month) mice by decreasing brain levels of BACE1 to decrease Aβ generation and increasing brain neprilysin levels to increase Aβ degradation (Zhang et al., 2014). Moreover, propofol treatment improved cognitive function by attenuating Aβ-induced mitochondrial dysfunction and caspase activation in both aged (18 month) wide-type mice and 19 month APP/PS1 mice (Shao et al., 2014).
The clinical sedative and hypnotic chlomethiazole (CMZ) is neuroprotective and has anti-inflammatory properties in animal models due to GABAA-potentiating actions in vitro and sedative activity in vivo (Marshall et al., 2000; Luo et al., 2015). CMZ protects against neurotoxic oligomeric Aβ1–42 in primary neurons (Vandevrede et al., 2014). A CMZ analogue, 4-methyl-5-(2-(nitrooxy)ethyl) thiazol-3-ium chloride (NMZ), retains the GABAA potentiating actions of CMZ in vitro and sedative activity in vivo. NMZ, but not CMZ, restored the LTP in hippocampal slices from APP/PS1 AD mice and also restored memory consolidation in these mice (Qin et al., 2012; Luo et al., 2015). These effects of NMZ can be antagonized by the GABAA receptor antagonist bicuculline, an allosteric inhibitor of GABAA receptor channel opening, indicating that NMZ’s neuroprotective effect is mediated by the GABAA receptor (Qin et al., 2012; Luo et al., 2015). A further study revealed that the effect of NMZ is mediated by the α1β2γ2 GABAA receptor without having direct actions on the ion current gated by this receptor (Luo et al., 2015). However, the mechanisms of action of NMZ as an anti-AD agent remains unclear. Previous studies have indicated that the NO/sGC/cGMP/CREB pathway is vital for cognition, learning and memory, and neuroprotective effects (Thatcher et al., 2004). NMZ is one of a series of NO-releasing hybrid agents that can be used as potential therapeutic compound to treat AD. A recent study showed that NMZ is a multifunctional drug that does not directly target Aβ and Tau pathology. NMZ successfully attenuated the hallmarks of AD pathology and rescued cognitive deficits in different mouse models of AD including three familial AD models (APP/PS1, 3xTg, APOE4) and a novel model of sporadic Aldh2–/– AD mice in different ways (Luo et al., 2016). In APP/PS1 mice, NMZ restored cognition and reduced Aβ levels. In 3xTg mice, NMZ restored LTP via NO/cGMP, enhanced CREB activity, reversed cognitive deficits, and reduced Aβ and pTau levels. In APOE4 transgenic AD mice, NMZ lowered Aβ and elevated CREB phosphorylation and PSD-95. In Aldh2–/– mice, NMZ restored synaptic plasticity and attenuated Aβ and pTau levels (Luo et al., 2016). These multifunctional properties of NMZ make it a potential drug to treat the mixed pathology seen in AD patients. NMZM is a maleate salt of NMZ with higher efficacy due to its improved solubility and absorption. A recent study showed that NMZM may improve learning and memory by alleviating LTP suppression induced by scopolamine in rat DGs. Moreover, the protective effects of NMZM against scopolamine-induced depression of LTP was partly dependent on the potentiation of GABAA receptors (Jiang et al., 2018).
Alpha-asarone (ASA) is an essential oil isolated from the traditional Chinese medicinal herb Acorus gramineus that has been used to treat respiratory diseases and neural disorders for centuries in traditional Chinese and Indian herbal medicine (Huang et al., 2013; Rajput et al., 2014; Chen et al., 2020). ASA has positive neuroprotective effects and improves cognition in rodent models (Gu et al., 2010; Kumar et al., 2012; Mao et al., 2015). Chen et al. found that ASA had an anti-AD effect in aged rats with cognitive deficits by inhibiting neuronal injury and decreasing Aβ1–42 levels in the hippocampus, ultimately rescuing the cognitive deficits (Chen et al., 2020). ASA also had neuroprotective effects on primary hippocampal neurons impaired by glutamate. Further computer modeling and whole-cell patch-clamp recording studies suggested that ASA, as a GABAAR-positive allosteric modulator, can bind to GABAAR and improve cognitive function by reducing neuronal overexcitation (Chen et al., 2020). Therefore, these studies suggest that treatment with GABAA receptor agonists or positive allosteric regulators could be a potential strategy for improving cognitive function in the elderly and in AD patients.
GABAA receptors are ligand-gated pentameric structures around a central chloride channel assembled from different combinations of 19 subunits including α1–6, β1–3,γ1–3, ρ1–3, δ, θ, ε, and π (Olsen and Sieghart, 2008; Sigel and Steinmann, 2012; Han et al., 2019). Different subunits may have specific functions. For example, the postsynaptic γ2 subunits mainly mediate phasic inhibition (Schweizer et al., 2003; Farrant and Nusser, 2005), while extrasynaptic GABAA receptors contain π subunits that mediate tonic inhibition in most brain regions, with the α5 and δ subunits the major GABAA receptors for mediating inhibition in the hippocampus (Glykys et al., 2008). α5-containing GABAA receptors play an important role in cognitive processes by controlling a component of synaptic transmission in the hippocampus (Crestani et al., 2002). Ablation or reduction of the a5 subunit of GABAA receptors alters GABAergic synaptic transmission and enhances hippocampus-dependent memory and spatial learning in mice (Collinson et al., 2002). Many studies have shown that inverse agonists of the GABAA receptor α5 subunit negatively regulate receptor activity by binding to the α5 subunit to improve learning and memory. MRK-016 (Atack et al., 2009), α5IA (Dawson et al., 2006), and α5IA-II (Collinson et al., 2006) have shown positive effects in improving cognition in animal models (Gabriella and Giovanna, 2010). In a preclinical and clinical study, α5IA was well tolerated in young and elderly subjects, and the efficacy of α5IA with respect to cognitive deficits requires further clarification (Atack, 2010). 3-iodo-8-(pyridin-4-ylmethoxy)pyrazolo[5,1-c][1,2,4]benzotriazine 5-oxide 1 selectively, safely, and significantly improved mouse memory processes in strict chemical relationships with α5IA, α5IA-II, and MRK-016 (Guerrini et al., 2009). Several other compounds with pyrazolo[5,1-c][1,2,4]benzotriazine cores acting at GABAA receptors have been confirmed to selectively display anti-amnesic and pro-cognitive activities in vitro and in vivo (Guerrini et al., 2013). α7-nicotinic acetylcholine receptor (α7-nAChR) agonists have entered clinical trials as pro-cognitive agents for treating schizophrenia and AD (Stoiljkovic et al., 2015; Townsend et al., 2016). FRM-17848, an α7-nAChR agonist, enhanced LTP in rat septo-hippocampal slices, at least in part dependent on increased GABAergic neurotransmission mediated by GABAA α5-receptors (Townsend et al., 2016). Therefore, targeting α5-containing GABAA receptors is an attractive strategy for treating disorders associated with cognitive defects such as AD.
A recent study found that astrocytes are also involved in tonic inhibition (Wu et al., 2014). In the DG of 5xFAD AD mice (6–8 months) and AD patients, compared with normal controls, the GABA content in astrocytes was significantly increased and the GABA release mediated by GABA transporter GAT3/4 obviously enhanced the tonic inhibition of hippocampal DG cells (Wu et al., 2014). Moreover, suppressing tonic inhibition either with SNAP-5114 to block astrocytic GABA release or with L-655,708 to block α5 GABAA receptors rescued the LTP and memory deficits in 5xFAD AD mice (Wu et al., 2014). Consistent with this study, Jo et al. found that in APP/PS1 AD mice, activated astrocytes can induce excessive tonic gliotransmitter GABA secretion through B-type monoamine oxidase-B (Maob) and promote GABA release through the bestrophin 1 channel in DG (Jo et al., 2014). The released GABA decreased the spike probability of granule cells by acting on presynaptic GABA receptors (Jo et al., 2014). More importantly, astrocytic GABA and Maob were significantly upregulated in AD patients (Jo et al., 2014). However, the impaired spike probability, synaptic plasticity, and learning and memory could be fully restored by inhibiting GABA production or release from reactive astrocytes with selegiline, a selective and irreversible Maob inhibitor, which has been FDA-approved for patients with PD (Jo et al., 2014). Therefore, selective inhibition of astrocytic GABA synthesis, release, or GABAA receptors may be a potential effective therapeutic strategy for treating memory impairment in AD.
GABAB receptors are located pre-synaptically, post-synaptically, and on extrasynaptic membranes in the hippocampus (Cryan and Kaupmann, 2005). Synaptic transmission in the brain is tightly regulated by GABAB receptors in two ways: presynaptic GABAB autoreceptors inhibit the release of a variety of neurotransmitters, and postsynaptic GABAB receptors generate inhibitory K+ currents that hyperpolarize the membrane and inhibit neuronal activity (Cryan and Kaupmann, 2005; Dinamarca et al., 2019). Extrasynaptic GABAB receptors can probably be activated by ‘spill-over’ of GABA from neighboring synapses (Cryan and Kaupmann, 2005). The presynaptic GABAB receptors decline in response to neural activity (Guetg et al., 2010; Terunuma et al., 2010; Orts-Del’Immagine and Pugh, 2018) and in AD (Chu et al., 1987; Iwakiri et al., 2005). In AD patients, the numbers of GABAB receptor R1 protein (GABABR1)-positive neurons were found to be significantly reduced in the CA1 field of the hippocampus, and GABABR1 immunoreactivity but not neuron number was increased in the CA4 and CA3/2 fields. The changes in hippocampal GABABR1 may reflect the balance between excitatory and inhibitory neurotransmitter systems and result in dysfunction of the hippocampal circuitry in AD (Iwakiri et al., 2005). Recent studies have indicated that secreted Aβ precursor proteins such as sAPPα can act as GABABR1α ligands to modulate synaptic transmission, thereby providing a potential target for the development of GABABR signaling–specific therapeutics in AD (Rice et al., 2019). Previous reports have suggested that astrocytes are abnormally activated in AD mouse models and human AD patients (Jo et al., 2014; Wu et al., 2014). Reactive astrocytes can induce excessive tonic gliotransmitter GABA secretion, and the released GABA binds to neuronal GABAB receptors at extrasynaptic sites, inhibiting synaptic release in APP/PS1 AD mice (Jo et al., 2014). Thus, suppressing GABAB receptor function may rescue synaptic release and improve the cognitive deficits seen in AD. The first GABAB receptor antagonist in clinical trials, SGS742 (CGP36742), displays pronounced cognition-enhancing effects in mice, young and old rats, and in Rhesus monkeys according to several different cognitive and learning-related behavioral tests (Getova and Bowery, 2001; Froestl et al., 2004; Helm et al., 2005). A further study has shown that SGS742 blocks the late IPSP and the paired-pulse inhibition (PPI) of population spikes recorded from rat CA1 pyramidal neurons in vitro and in vivo. SGS742 significantly enhanced the release of glutamate, aspartate, glycine, and somatostatin in vivo. Moreover, SGS742 induced significant enhancements in mRNA and protein levels of nerve growth factor (NGF) and brain-derived natriuretic factor (BDNF) in the cortex and hippocampus of rats (Froestl et al., 2004). Acute in vivo administration of SGS742 in rats improved memory, at least in part due to reduced total hippocampal CREB2 activity (Helm et al., 2005). Oral administration of SGS742 for 8 weeks in a Phase II clinical trial significantly improved attention and working memory in patients with mild cognitive impairment and mild-moderate AD (Froestl et al., 2004; Cryan and Kaupmann, 2005). Another GABAB receptor antagonist, CGP55845, showed a complete reversal of olfactory discrimination learning deficits in cognitively impaired aged Fischer 344 rats (Lasarge et al., 2009). Further results revealed that the intra-hippocampal microinjection of the GABAB receptor antagonist, CGP35348, ameliorated the learning, memory, and cognitive impairments induced by microinjection of Aβ in rats, suggesting that GABAB receptor antagonism may be a therapeutic agent against the progression of acute Aβ toxicity-induced memory impairment (Almasi et al., 2018). Therefore, it is possible to improve the cognitive impairment seen in AD model mice and AD patients by regulating the level and release of GABA from GABAergic neurons and astrocytes during AD pathogenesis (Figure 3). However, the mechanisms underpinning GABA content and release remain unclear, with more research needed in this area.
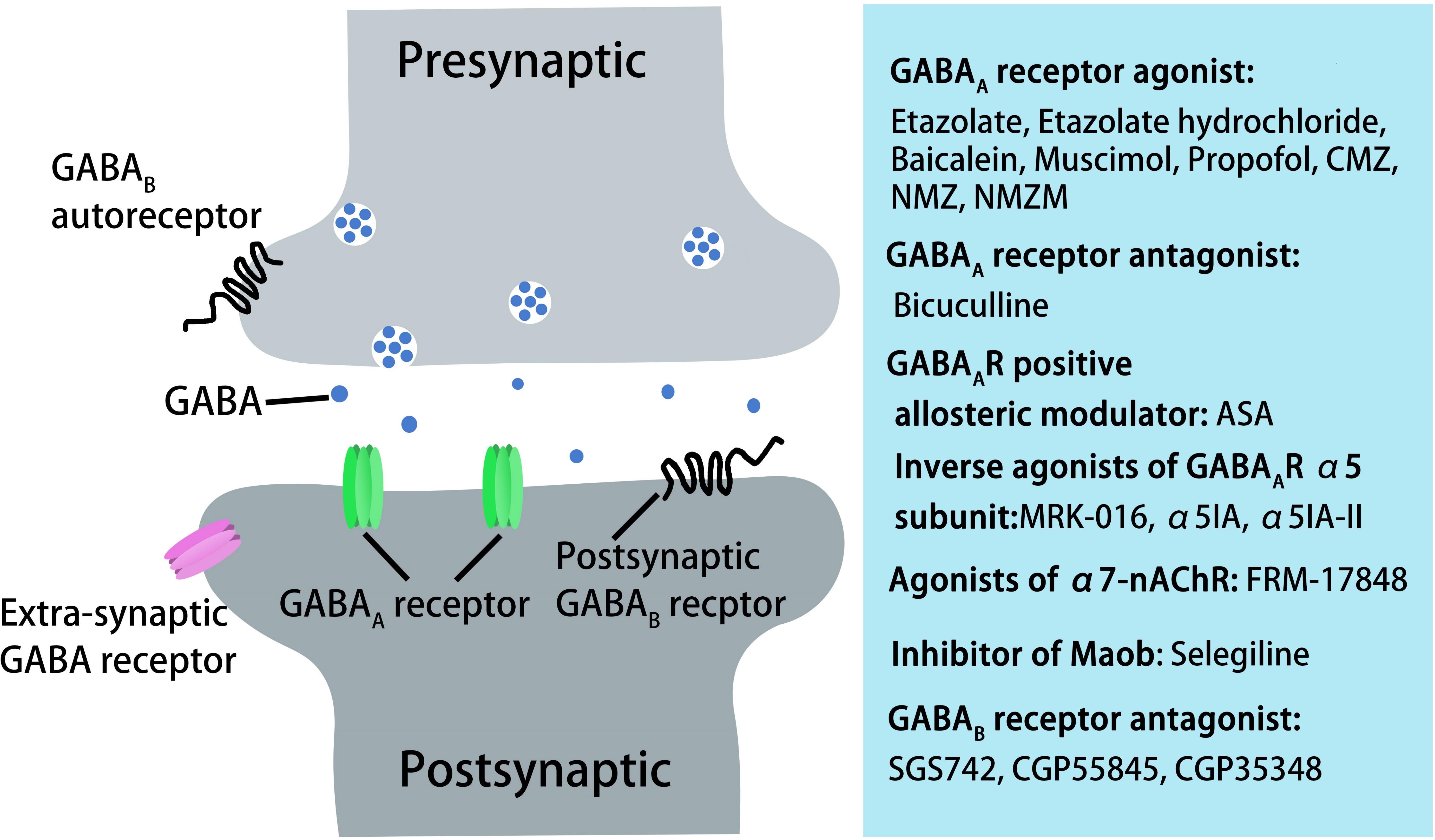
Figure 3. AD treatment through pharmacological manipulation of GABAergic transmission via GABAA and GABAB receptors. GABAA receptor agonists or antagonist, GABAA receptor-positive allosteric modulators, inverse agonists of GABAA receptor α5 subunit, agonists of α7-nAChR, inhibitors of Maob, or GABAB receptor antagonists can rescue cognitive deficits in aged rats, AD mice, or in AD patients.
Conclusion and Future Perspectives
During AD development, Aβ leads to the loss and dysfunction of GABA inhibitory interneurons, with abnormal activity resulting in structural and functional impairment of nerve circuits and ultimately the cognitive deficits seen in AD patients and mice. Targeting GABA inhibitory interneurons by transplantation or functional enhancement may rescue the cognitive impairment seen in AD animals. Further studies of AD animal models transplanted with GABA inhibitory interneurons to stimulate GABA release or GABA receptor expression are needed. It will also be necessary to clarify the molecular mechanisms underpinning Aβ-induced dysfunction of GABA inhibitory interneurons, the aberrant activity of neuronal networks, and the cognitive deficits seen in AD. Nevertheless, GABA inhibitory interneurons represent a promising therapeutic target for the treatment of AD.
Author Contributions
HZ and YX conceived and designed the study. YX, MZ, and YH summarized the related literature. HZ and MZ conceived the images. MZ drew the images. HZ, YX, and MZ wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 31800891 to HZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge editorial assistance from Nextgenediting (www.nextgenediting.com).
References
Adaikkan, C., Middleton, S. J., Marco, A., Pao, P. C., Mathys, H., Kim, D. N., et al. (2019). Gamma entrainment binds higher-order brain regions and offers neuroprotection. Neuron 102, 929–943.e8. doi: 10.1016/j.neuron.2019.04.011
Agostini, M., Tucci, P., Killick, R., Candi, E., Sayan, B. S., Rivetti di Val Cervo, P., et al. (2011). Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proc. Natl. Acad. Sci. U.S.A. 108, 21093–21098. doi: 10.1073/pnas.1112061109
Almasi, A., Zarei, M., Raoufi, S., Sarihi, A., Salehi, I., Komaki, A., et al. (2018). Influence of hippocampal GABAB receptor inhibition on memory in rats with acute beta-amyloid toxicity. Metab. Brain Dis. 33, 1859–1867. doi: 10.1007/s11011-018-0292-5
Alzheimer’s Association (2019). Alzheimer’s disease facts and figures. Alzheimers Dement. 15, 321–387. doi: 10.1016/j.jalz.2019.01.010
Andrews-Zwilling, Y., Bien-Ly, N., Xu, Q., Li, G., Bernardo, A., Yoon, S. Y., et al. (2010). Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. 30, 13707–13717. doi: 10.1523/jneurosci.4040-10.2010
Anticevic, A., Cole, M. W., Murray, J. D., Corlett, P. R., Wang, X. J., and Krystal, J. H. (2012). The role of default network deactivation in cognition and disease. Trends Cogn. Sci. 16, 584–592. doi: 10.1016/j.tics.2012.10.008
Atack, J. R. (2010). Preclinical and clinical pharmacology of the GABAA receptor alpha5 subtype-selective inverse agonist alpha5IA. Pharmacol. Ther. 125, 11–26. doi: 10.1016/j.pharmthera.2009.09.001
Atack, J. R., Maubach, K. A., Wafford, K. A., O’Connor, D., Rodrigues, A. D., Evans, D. C., et al. (2009). In vitro and in vivo properties of 3-tert-butyl-7-(5-methylisoxazol-3-yl)-2-(1-methyl-1H-1,2,4-triazol-5-ylmethoxy)- pyrazolo[1,5-d]-[1,2,4]triazine (MRK-016), a GABAA receptor alpha5 subtype-selective inverse agonist. J. Pharmacol. Exp. Ther. 331, 470–484. doi: 10.1124/jpet.109.157636
Baglietto-Vargas, D., Moreno-Gonzalez, I., Sanchez-Varo, R., Jimenez, S., Trujillo-Estrada, L., Sanchez-Mejias, E., et al. (2010). Calretinin interneurons are early targets of extracellular amyloid-beta pathology in PS1/AbetaPP Alzheimer mice hippocampus. J. Alzheimers Dis. 21, 119–132. doi: 10.3233/jad-2010-100066
Bai, X., Edden, R. A., Gao, F., Wang, G., Wu, L., Zhao, B., et al. (2015). Decreased gamma-aminobutyric acid levels in the parietal region of patients with Alzheimer’s disease. J. Magn. Reson. Imaging 41, 1326–1331. doi: 10.1002/jmri.24665
Bakker, A., Krauss, G. L., Albert, M. S., Speck, C. L., Jones, L. R., Stark, C. E., et al. (2012). Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74, 467–474. doi: 10.1016/j.neuron.2012.03.023
Barage, S. H., and Sonawane, K. D. (2015). Amyloid cascade hypothesis: pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 52, 1–18. doi: 10.1016/j.npep.2015.06.008
Bareggi, S. R., Franceschi, M., Bonini, L., Zecca, L., and Smirne, S. (1982). Decreased CSF concentrations of homovanillic acid and gamma-aminobutyric acid in Alzheimer’s disease. Age- or disease-related modifications? Arch. Neurol. 39, 709–712. doi: 10.1001/archneur.1982.00510230035010
Beal, M. F., Mazurek, M. F., Svendsen, C. N., Bird, E. D., and Martin, J. B. (1986). Widespread reduction of somatostatin-like immunoreactivity in the cerebral cortex in Alzheimer’s disease. Ann. Neurol. 20, 489–495. doi: 10.1002/ana.410200408
Beal, M. F., Mazurek, M. F., Tran, V. T., Chattha, G., Bird, E. D., and Martin, J. B. (1985). Reduced numbers of somatostatin receptors in the cerebral cortex in Alzheimer’s disease. Science 229, 289–291. doi: 10.1126/science.2861661
Bearer, C. F., Wellmann, K. A., Tang, N., He, M., and Mooney, S. M. (2015). Choline ameliorates deficits in balance caused by acute neonatal ethanol exposure. Cerebellum 14, 413–420. doi: 10.1007/s12311-015-0691-7
Behrens, S., Rattinger, G. B., Schwartz, S., Matyi, J., Sanders, C., DeBerard, M. S., et al. (2018). Use of FDA approved medications for Alzheimer’s disease in mild dementia is associated with reduced informal costs of care. Int. Psychogeriatr. 30, 1499–1507. doi: 10.1017/S104161021800011X
Booker, S. A., and Vida, I. (2018). Morphological diversity and connectivity of hippocampal interneurons. Cell Tissue Res. 373, 619–641. doi: 10.1007/s00441-018-2882-2
Bown, A. W., and Shelp, B. J. (2016). Plant GABA: not just a metabolite. Trends Plant Sci. 21, 811–813. doi: 10.1016/j.tplants.2016.08.001
Brady, D. R., and Mufson, E. J. (1997). Parvalbumin-immunoreactive neurons in the hippocampal formation of Alzheimer’s diseased brain. Neuroscience 80, 1113–1125. doi: 10.1016/s0306-4522(97)00068-7
Burke, R. M., Norman, T. A., Haydar, T. F., Slack, B. E., Leeman, S. E., Blusztajn, J. K., et al. (2013). BMP9 ameliorates amyloidosis and the cholinergic defect in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 110, 19567–19572. doi: 10.1073/pnas.1319297110
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Wiederhold, K. H., Haass, C., et al. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689. doi: 10.1126/science.1162844
Buzsaki, G., and Wang, X. J. (2012). Mechanisms of gamma oscillations. Annu. Rev. Neurosci. 35, 203–225. doi: 10.1146/annurev-neuro-062111-150444
Calvo-Flores, G. B., Vinnakota, C., Govindpani, K., Waldvogel, H. J., Faull, R. L. M., and Kwakowsky, A. (2018). The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 146, 649–669. doi: 10.1111/jnc.14345
Candy, J. M., Gascoigne, A. D., Biggins, J. A., Smith, A. I., Perry, R. H., Perry, E. K., et al. (1985). Somatostatin immunoreactivity in cortical and some subcortical regions in Alzheimer’s disease. J. Neurol. Sci. 71, 315–323. doi: 10.1016/0022-510x(85)90070-x
Canevelli, M., Bruno, G., Vico, C., Zaccaria, V., Lacorte, E., Iavicoli, I., et al. (2018). Socioeconomic disparities in clinical trials on Alzheimer’s disease: a systematic review. Eur. J. Neurol. 25, e26–e43. doi: 10.1111/ene.13587
Canter, R. G., Penney, J., and Tsai, L. H. (2016). The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 539, 187–196. doi: 10.1038/nature20412
Caraiscos, V. B., Elliott, E. M., You-Ten, K. E., Cheng, V. Y., Belelli, D., Newell, J. G., et al. (2004). Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. U.S.A. 101, 3662–3667. doi: 10.1073/pnas.0307231101
Carreras, I., Aytan, N., Choi, J. K., Tognoni, C. M., Kowall, N. W., Jenkins, B. G., et al. (2019). Dual dose-dependent effects of fingolimod in a mouse model of Alzheimer’s disease. Sci. Rep. 9:10972.
Cattaud, V., Bezzina, C., Rey, C. C., Lejards, C., Dahan, L., and Verret, L. (2018). Early disruption of parvalbumin expression and perineuronal nets in the hippocampus of the Tg2576 mouse model of Alzheimer’s disease can be rescued by enriched environment. Neurobiol. Aging 72, 147–158. doi: 10.1016/j.neurobiolaging.2018.08.024
Chen, C., Sigurdsson, H. P., Pepes, S. E., Auer, D. P., Morris, P. G., Morgan, P. S., et al. (2017). Activation induced changes in GABA: functional MRS at 7T with MEGA-sLASER. Neuroimage 156, 207–213. doi: 10.1016/j.neuroimage.2017.05.044
Chen, Y., Gao, X., Liu, Q., Zeng, L., Zhang, K., Mu, K., et al. (2020). Alpha-asarone improves cognitive function of aged rats by alleviating neuronal excitotoxicity via GABAA receptors. Neuropharmacology 162:107843. doi: 10.1016/j.neuropharm.2019.107843
Cheng, A., Wang, J., Ghena, N., Zhao, Q., Perone, I., King, M. T., et al. (2019). SIRT3 haploinsufficiency aggravates loss of GABAergic interneurons and neuronal network hyperexcitability in an Alzheimer’s disease model. J. Neurosci. 40, 694–709. doi: 10.1523/JNEUROSCI.1446-19.2019
Chu, D. C., Penney, J. B., and Young, A. B. (1987). Cortical GABAB and GABAA receptors in Alzheimer’s disease: a quantitative autoradiographic study. Neurology 37, 1454–1459. doi: 10.1212/wnl.37.9.1454
Chung, H., Park, K., Jang, H. J., Kohl, M. M., and Kwag, J. (2020). Dissociation of somatostatin and parvalbumin interneurons circuit dysfunctions underlying hippocampal theta and gamma oscillations impaired by amyloid beta oligomers in vivo. Brain Struct. Funct. 225, 935–954. doi: 10.1007/s00429-020-02044-3
Collinson, N., Atack, J. R., Laughton, P., Dawson, G. R., and Stephens, D. N. (2006). An inverse agonist selective for alpha5 subunit-containing GABAA receptors improves encoding and recall but not consolidation in the Morris water maze. Psychopharmacology 188, 619–628. doi: 10.1007/s00213-006-0361-z
Collinson, N., Kuenzi, F. M., Jarolimek, W., Maubach, K. A., Cothliff, R., Sur, C., et al. (2002). Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J. Neurosci. 22, 5572–5580. doi: 10.1523/jneurosci.22-13-05572.2002
Crestani, F., Keist, R., Fritschy, J. M., Benke, D., Vogt, K., Prut, L., et al. (2002). Trace fear conditioning involves hippocampal alpha5 GABA(A) receptors. Proc. Natl. Acad. Sci. U.S.A. 99, 8980–8985. doi: 10.1073/pnas.142288699
Cryan, J. F., and Kaupmann, K. (2005). Don’t worry ‘B’ happy!: a role for GABA(B) receptors in anxiety and depression. Trends Pharmacol. Sci. 26, 36–43. doi: 10.1016/j.tips.2004.11.004
Davies, P., Katzman, R., and Terry, R. D. (1980). Reduced somatostatin-like immunoreactivity in cerebral cortex from cases of Alzheimer disease and Alzheimer senile dementa. Nature 288, 279–280. doi: 10.1038/288279a0
Davies, P., and Terry, R. D. (1981). Cortical somatostatin-like immunoreactivity in cases of Alzheimer’s disease and senile dementia of the Alzheimer type. Neurobiol. Aging 2, 9–14. doi: 10.1016/0197-4580(81)90053-1
Dawson, G. R., Maubach, K. A., Collinson, N., Cobain, M., Everitt, B. J., MacLeod, A. M., et al. (2006). An inverse agonist selective for alpha5 subunit-containing GABAA receptors enhances cognition. J. Pharmacol. Exp. Ther. 316, 1335–1345. doi: 10.1124/jpet.105.092320
DeFelipe, J., Lopez-Cruz, P. L., Benavides-Piccione, R., Bielza, C., Larranaga, P., Anderson, S., et al. (2013). New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat. Rev. Neurosci. 14, 202–216.
Dennis, N. A., Browndyke, J. N., Stokes, J., Need, A., Burke, J. R., Welsh-Bohmer, K. A., et al. (2010). Temporal lobe functional activity and connectivity in young adult APOE varepsilon4 carriers. Alzheimers Dement. 6, 303–311. doi: 10.1016/j.jalz.2009.07.003
Dhami, K. S., Churchward, M. A., Baker, G. B., and Todd, K. G. (2013). Fluoxetine and citalopram decrease microglial release of glutamate and D-serine to promote cortical neuronal viability following ischemic insult. Mol. Cell. Neurosci. 56, 365–374. doi: 10.1016/j.mcn.2013.07.006
Dickson, D. W., and Murray, M. E. (2015). Intraneuronal amyloid-beta accumulation in basal forebrain cholinergic neurons: a marker of vulnerability, yet inversely related to neurodegeneration. Brain 138(Pt 6), 1444–1445. doi: 10.1093/brain/awv097
Dinamarca, M. C., Raveh, A., Schneider, A., Fritzius, T., Fruh, S., Rem, P. D., et al. (2019). Complex formation of APP with GABAB receptors links axonal trafficking to amyloidogenic processing. Nat. Commun. 10:1331.
Donohue, M. C., Sperling, R. A., Petersen, R., Sun, C. K., Weiner, M. W., Aisen, P. S., et al. (2017). Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA 317, 2305–2316. doi: 10.1001/jama.2017.6669
Drott, J., Desire, L., Drouin, D., Pando, M., and Haun, F. (2010). Etazolate improves performance in a foraging and homing task in aged rats. Eur. J. Pharmacol. 634, 95–100. doi: 10.1016/j.ejphar.2010.02.036
Duveau, V., Laustela, S., Barth, L., Gianolini, F., Vogt, K. E., Keist, R., et al. (2011). Spatiotemporal specificity of GABAA receptor-mediated regulation of adult hippocampal neurogenesis. Eur. J. Neurosci. 34, 362–373. doi: 10.1111/j.1460-9568.2011.07782.x
Ettcheto, M., Sanchez-Lopez, E., Gomez-Minguez, Y., Cabrera, H., Busquets, O., Beas-Zarate, C., et al. (2018). Peripheral and central effects of memantine in a mixed preclinical mice model of obesity and familial Alzheimer’s disease. Mol. Neurobiol. 57, 2887–2888. doi: 10.1007/s12035-020-01918-1
Evered, L., Silbert, B., Scott, D. A., and Eckenhoff, R. G. (2019). Recommendations for a new perioperative cognitive impairment nomenclature. Alzheimers Dement. 15, 1115–1116. doi: 10.1016/j.jalz.2019.05.005
Fahoum, F., Zelmann, R., Tyvaert, L., Dubeau, F., and Gotman, J. (2013). Epileptic discharges affect the default mode network–FMRI and intracerebral EEG evidence. PLoS One 8:e68038. doi: 10.1371/journal.pone.0068038
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat. Rev. 6, 215–229. doi: 10.1038/nrn1625
Ferreira-Vieira, T. H., Guimaraes, I. M., Silva, F. R., and Ribeiro, F. M. (2016). Alzheimer’s disease: targeting the cholinergic system. Curr. Neuropharmacol. 14, 101–115.
Ferrer, I., Soriano, E., Tunon, T., Fonseca, M., and Guionnet, N. (1991). Parvalbumin immunoreactive neurons in normal human temporal neocortex and in patients with Alzheimer’s disease. J. Neurol. Sci. 106, 135–141. doi: 10.1016/0022-510x(91)90250-b
Filippini, N., MacIntosh, B. J., Hough, M. G., Goodwin, G. M., Frisoni, G. B., Smith, S. M., et al. (2009). Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc. Natl. Acad. Sci. U.S.A. 106, 7209–7214. doi: 10.1073/pnas.0811879106
Flanigan, T. J., Xue, Y., Kishan Rao, S., Dhanushkodi, A., and McDonald, M. P. (2014). Abnormal vibrissa-related behavior and loss of barrel field inhibitory neurons in 5xFAD transgenics. Genes Brain Behav. 13, 488–500. doi: 10.1111/gbb.12133
Frere, S., and Slutsky, I. (2018). Alzheimer’s disease: from firing instability to homeostasis network collapse. Neuron 97, 32–58. doi: 10.1016/j.neuron.2017.11.028
Froestl, W., Gallagher, M., Jenkins, H., Madrid, A., Melcher, T., Teichman, S., et al. (2004). SGS742: the first GABA(B) receptor antagonist in clinical trials. Biochem. Pharmacol. 68, 1479–1487. doi: 10.1016/j.bcp.2004.07.030
Fu, H., Possenti, A., Freer, R., Nakano, Y., Hernandez Villegas, N. C., Tang, M., et al. (2019). A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat. Neurosci. 22, 47–56. doi: 10.1038/s41593-018-0298-7
Fu, H., Rodriguez, G. A., Herman, M., Emrani, S., Nahmani, E., Barrett, G., et al. (2017). Tau pathology induces excitatory neuron loss, grid cell dysfunction, and spatial memory deficits reminiscent of early Alzheimer’s disease. Neuron 93, 533–541.e5. doi: 10.1016/j.neuron.2016.12.023
Fu, Y., Li, L., Wang, Y., Chu, G., Kong, X., and Wang, J. (2019). Role of GABAA receptors in EEG activity and spatial recognition memory in aged APP and PS1 double transgenic mice. Neurochem. Int. 131;104542. doi: 10.1016/j.neuint.2019.104542
Gabriella, G., and Giovanna, C. (2010). gamma-Aminobutyric acid type A (GABA(A)) receptor subtype inverse agonists as therapeutic agents in cognition. Methods Enzymol. 485, 197–211. doi: 10.1016/b978-0-12-381296-4.00011-7
Ge, S., Goh, E. L., Sailor, K. A., Kitabatake, Y., Ming, G. L., and Song, H. (2006). GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439, 589–593. doi: 10.1038/nature04404
Genin, E., Hannequin, D., Wallon, D., Sleegers, K., Hiltunen, M., Combarros, O., et al. (2011). APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16, 903–907. doi: 10.1038/mp.2011.52
Getova, D. P., and Bowery, N. G. (2001). Effects of high-affinity GABAB receptor antagonists on active and passive avoidance responding in rodents with gamma-hydroxybutyrolactone-induced absence syndrome. Psychopharmacology 157, 89–95. doi: 10.1007/s002130100766
Glykys, J., Mann, E. O., and Mody, I. (2008). Which GABA(A) receptor subunits are necessary for tonic inhibition in the hippocampus? J. Neurosci. 28, 1421–1426. doi: 10.1523/JNEUROSCI.4751-07.2008
Gong, W. G., Wang, Y. J., Zhou, H., Li, X. L., Bai, F., Ren, Q. G., et al. (2017). Citalopram ameliorates synaptic plasticity deficits in different cognition-associated brain regions induced by social isolation in middle-aged rats. Mol. Neurobiol. 54, 1927–1938. doi: 10.1007/s12035-016-9781-x
Govindpani, K., Calvo-Flores, G. B., Vinnakota, C., Waldvogel, H. J., Faull, R. L., and Kwakowsky, A. (2017). Towards a better understanding of GABAergic remodeling in Alzheimer’s disease. Int. J. Mol. Sci. 18:1813. doi: 10.3390/ijms18081813
Graham, W. V., Bonito-Oliva, A., and Sakmar, T. P. (2017). Update on Alzheimer’s disease therapy and prevention strategies. Annu. Rev. Med. 68, 413–430.
Grothe, M., Heinsen, H., and Teipel, S. J. (2012). Atrophy of the cholinergic Basal forebrain over the adult age range and in early stages of Alzheimer’s disease. Biol. Psychiatry 71, 805–813. doi: 10.1016/j.biopsych.2011.06.019
Grouselle, D., Winsky-Sommerer, R., David, J. P., Delacourte, A., Dournaud, P., and Epelbaum, J. (1998). Loss of somatostatin-like immunoreactivity in the frontal cortex of Alzheimer patients carrying the apolipoprotein epsilon 4 allele. Neurosci. Lett. 255, 21–24. doi: 10.1016/s0304-3940(98)00698-3
Gu, Q., Du, H., Ma, C., Fotis, H., Wu, B., Huang, C., et al. (2010). Effects of alpha-asarone on the glutamate transporter EAAC1 in Xenopus oocytes. Planta Med. 76, 595–598. doi: 10.1055/s-0029-1240613
Gueli, M. C., and Taibi, G. (2013). Alzheimer’s disease: amino acid levels and brain metabolic status. Neurol. Sci. 34, 1575–1579. doi: 10.1007/s10072-013-1289-9
Guerrini, G., Ciciani, G., Cambi, G., Bruni, F., Selleri, S., Guarino, C., et al. (2009). Synthesis, in vivo evaluation, and molecular modeling studies of new pyrazolo[5,1-c][1,2,4]benzotriazine 5-oxide derivatives. Identification of a bifunctional hydrogen bond area related to the inverse agonism. J. Med. Chem. 52, 4668–4682. doi: 10.1021/jm801599a
Guerrini, G., Ciciani, G., Costanzo, A., Daniele, S., Martini, C., Ghelardini, C., et al. (2013). Synthesis of novel cognition enhancers with pyrazolo[5,1-c][1,2,4]benzotriazine core acting at gamma-aminobutyric acid type A (GABA(A)) receptor. Bioorg. Med. Chem. 21, 2186–2198. doi: 10.1016/j.bmc.2013.02.027
Guetg, N., Abdel Aziz, S., Holbro, N., Turecek, R., Rose, T., Seddik, R., et al. (2010). NMDA receptor-dependent GABAB receptor internalization via CaMKII phosphorylation of serine 867 in GABAB1. Proc. Natl. Acad. Sci. U.S.A. 107, 13924–13929. doi: 10.1073/pnas.1000909107
Gutierres, J. M., Carvalho, F. B., Schetinger, M. R., Marisco, P., Agostinho, P., Rodrigues, M., et al. (2014). Anthocyanins restore behavioral and biochemical changes caused by streptozotocin-induced sporadic dementia of Alzheimer’s type. Life Sci. 96, 7–17. doi: 10.1016/j.lfs.2013.11.014
Han, W., Li, J., Pelkey, K. A., Pandey, S., Chen, X., Wang, Y. X., et al. (2019). Shisa7 is a GABAA receptor auxiliary subunit controlling benzodiazepine actions. Science 366, 246–250. doi: 10.1126/science.aax5719
Helm, K. A., Haberman, R. P., Dean, S. L., Hoyt, E. C., Melcher, T., Lund, P. K., et al. (2005). GABAB receptor antagonist SGS742 improves spatial memory and reduces protein binding to the cAMP response element (CRE) in the hippocampus. Neuropharmacology 48, 956–964. doi: 10.1016/j.neuropharm.2005.01.019
Hijazi, S., Heistek, T. S., Scheltens, P., Neumann, U., Shimshek, D. R., Mansvelder, H. D., et al. (2019). Early restoration of parvalbumin interneuron activity prevents memory loss and network hyperexcitability in a mouse model of Alzheimer’s disease. Mol. Psychiatry doi: 10.1038/s41380-019-0483-4 [Epub ahead of print].
Hof, P. R., Cox, K., Young, W. G., Celio, M. R., Rogers, J., and Morrison, J. H. (1991). Parvalbumin-immunoreactive neurons in the neocortex are resistant to degeneration in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 50, 451–462. doi: 10.1097/00005072-199107000-00006
Holter, N. I., Zylla, M. M., Zuber, N., Bruehl, C., and Draguhn, A. (2010). Tonic GABAergic control of mouse dentate granule cells during postnatal development. Eur. J. Neurosci. 32, 1300–1309. doi: 10.1111/j.1460-9568.2010.07331.x
Hu, H., Gan, J., and Jonas, P. (2014). Interneurons. Fast-spiking, parvalbumin(+) GABAergic interneurons: from cellular design to microcircuit function. Science 345:1255263. doi: 10.1126/science.1255263
Hu, J., Liu, C. C., Chen, X. F., Zhang, Y. W., Xu, H., and Bu, G. (2015). Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol. Neurodegener. 10:6.
Huang, C., Li, W. G., Zhang, X. B., Wang, L., Xu, T. L., Wu, D., et al. (2013). alpha-asarone from Acorus gramineus alleviates epilepsy by modulating A-type GABA receptors. Neuropharmacology 65, 1–11. doi: 10.1016/j.neuropharm.2012.09.001
Huang, Y., and Mucke, L. (2012). Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222. doi: 10.1016/j.cell.2012.02.040
Huang, Z. J., and Paul, A. (2019). The diversity of GABAergic neurons and neural communication elements. Nat. Rev. Neurosci. 20, 563–572. doi: 10.1038/s41583-019-0195-4
Huh, S., Baek, S. J., Lee, K. H., Whitcomb, D. J., Jo, J., Choi, S. M., et al. (2016). The reemergence of long-term potentiation in aged Alzheimer’s disease mouse model. Sci. Rep. 6:29152.
Iaccarino, H. F., Singer, A. C., Martorell, A. J., Rudenko, A., Gao, F., Gillingham, T. Z., et al. (2016). Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540, 230–235. doi: 10.1038/nature20587
Isaacson, J. S., and Scanziani, M. (2011). How inhibition shapes cortical activity. Neuron 72, 231–243. doi: 10.1016/j.neuron.2011.09.027
Iwakiri, M., Mizukami, K., Ikonomovic, M. D., Ishikawa, M., Hidaka, S., Abrahamson, E. E., et al. (2005). Changes in hippocampal GABABR1 subunit expression in Alzheimer’s patients: association with Braak staging. Acta Neuropathol. 109, 467–474. doi: 10.1007/s00401-005-0985-9
Jiang, X. M., Wang, W. P., Liu, Z. H., Yin, H. J., Ma, H., Feng, N., et al. (2018). 2-(4-methyl-thiazol-5-yl) ethyl nitrate maleate-potentiated GABAA receptor response in hippocampal neurons. CNS Neurosci. Ther. 24, 1231–1240. doi: 10.1111/cns.13033
Jo, S., Yarishkin, O., Hwang, Y. J., Chun, Y. E., Park, M., Woo, D. H., et al. (2014). GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 20, 886–896. doi: 10.1038/nm.3639
Kamkwalala, A. R., and Newhouse, P. A. (2017). Beyond acetylcholinesterase inhibitors: novel cholinergic treatments for Alzheimer’s disease. Curr. Alzheimer Res. 14, 377–392. doi: 10.2174/1567205013666160930112625
Khoury, R., Rajamanickam, J., and Grossberg, G. T. (2018). An update on the safety of current therapies for Alzheimer’s disease: focus on rivastigmine. Ther. Adv. Drug Saf. 9, 171–178. doi: 10.1177/2042098617750555
Kim, H., Ahrlund-Richter, S., Wang, X., Deisseroth, K., and Carlen, M. (2016). Prefrontal parvalbumin neurons in control of attention. Cell 164, 208–218. doi: 10.1016/j.cell.2015.11.038
Knoferle, J., Yoon, S. Y., Walker, D., Leung, L., Gillespie, A. K., Tong, L. M., et al. (2014). Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J. Neurosci. 34, 14069–14078. doi: 10.1523/JNEUROSCI.2281-14.2014
Krantic, S., Isorce, N., Mechawar, N., Davoli, M. A., Vignault, E., Albuquerque, M., et al. (2012). Hippocampal GABAergic neurons are susceptible to amyloid-beta toxicity in vitro and are decreased in number in the Alzheimer’s disease TgCRND8 mouse model. J. Alzheimers Dis. 29, 293–308. doi: 10.3233/jad-2011-110830
Kullmann, D. M., and Lamsa, K. P. (2007). Long-term synaptic plasticity in hippocampal interneurons. Nat. Rev. Neurosci. 8, 687–699. doi: 10.1038/nrn2207
Kumar, H., Kim, B. W., Song, S. Y., Kim, J. S., Kim, I. S., Kwon, Y. S., et al. (2012). Cognitive enhancing effects of alpha asarone in amnesic mice by influencing cholinergic and antioxidant defense mechanisms. Biosci. Biotechnol. Biochem. 76, 1518–1522. doi: 10.1271/bbb.120247
Lasarge, C. L., Banuelos, C., Mayse, J. D., and Bizon, J. L. (2009). Blockade of GABA(B) receptors completely reverses age-related learning impairment. Neuroscience 164, 941–947. doi: 10.1016/j.neuroscience.2009.08.055
Lee, B. Y., Ban, J. Y., and Seong, Y. H. (2005). Chronic stimulation of GABAA receptor with muscimol reduces amyloid beta protein (25-35)-induced neurotoxicity in cultured rat cortical cells. Neurosci. Res. 52, 347–356. doi: 10.1016/j.neures.2005.04.008
Lei, M., Xu, H., Li, Z., Wang, Z., O’Malley, T. T., Zhang, D., et al. (2016). Soluble Abeta oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol. Dis. 85, 111–121. doi: 10.1016/j.nbd.2015.10.019
Lemmens, M. A., Sierksma, A. S., Rutten, B. P., Dennissen, F., Steinbusch, H. W., Lucassen, P. J., et al. (2011). Age-related changes of neuron numbers in the frontal cortex of a transgenic mouse model of Alzheimer’s disease. Brain Struct. Funct. 216, 227–237. doi: 10.1007/s00429-011-0305-1
Lerdkrai, C., Asavapanumas, N., Brawek, B., Kovalchuk, Y., Mojtahedi, N., Olmedillas Del Moral, M., et al. (2018). Intracellular Ca(2+) stores control in vivo neuronal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 115, E1279–E1288. doi: 10.1073/pnas.1714409115
Li, G., Bien-Ly, N., Andrews-Zwilling, Y., Xu, Q., Bernardo, A., Ring, K., et al. (2009). GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell 5, 634–645. doi: 10.1016/j.stem.2009.10.015
Li, S., Hong, S., Shepardson, N. E., Walsh, D. M., Shankar, G. M., and Selkoe, D. (2009). Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801. doi: 10.1016/j.neuron.2009.05.012
Li, Y., Chen, Z., Gao, Y., Pan, G., Zheng, H., Zhang, Y., et al. (2017). Synaptic adhesion molecule Pcdh-gammaC5 mediates synaptic dysfunction in Alzheimer’s disease. J. Neurosci. 37, 9259–9268. doi: 10.1523/JNEUROSCI.1051-17.2017
Li, Y., Sun, H., Chen, Z., Xu, H., Bu, G., and Zheng, H. (2016). Implications of GABAergic neurotransmission in Alzheimer’s disease. Front. Aging Neurosci. 8:31. doi: 10.3389/fnagi.2016.00031
Liu, D. S., Pan, X. D., Zhang, J., Shen, H., Collins, N. C., Cole, A. M., et al. (2015). APOE4 enhances age-dependent decline in cognitive function by down-regulating an NMDA receptor pathway in EFAD-Tg mice. Mol. Neurodegener. 10:7.
Liu, L., Wong, T. P., Pozza, M. F., Lingenhoehl, K., Wang, Y., Sheng, M., et al. (2004). Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304, 1021–1024. doi: 10.1126/science.1096615
Long, J. M., and Holtzman, D. M. (2019). Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179, 312–339. doi: 10.1016/j.cell.2019.09.001
Loreth, D., Ozmen, L., Revel, F. G., Knoflach, F., Wetzel, P., Frotscher, M., et al. (2012). Selective degeneration of septal and hippocampal GABAergic neurons in a mouse model of amyloidosis and tauopathy. Neurobiol. Dis. 47, 1–12. doi: 10.1016/j.nbd.2012.03.011
Lundqvist, M., Rose, J., Herman, P., Brincat, S. L., Buschman, T. J., and Miller, E. K. (2016). Gamma and beta bursts underlie working memory. Neuron 90, 152–164. doi: 10.1016/j.neuron.2016.02.028
Luo, J., Lee, S. H., VandeVrede, L., Qin, Z., Ben Aissa, M., Larson, J., et al. (2016). A multifunctional therapeutic approach to disease modification in multiple familial mouse models and a novel sporadic model of Alzheimer’s disease. Mol. Neurodegener. 11:35.
Luo, J., Lee, S. H., VandeVrede, L., Qin, Z., Piyankarage, S., Tavassoli, E., et al. (2015). Re-engineering a neuroprotective, clinical drug as a procognitive agent with high in vivo potency and with GABAA potentiating activity for use in dementia. BMC Neurosci. 16:67. doi: 10.1186/s12868-015-0208-9
Ma, K., and McLaurin, J. (2014). alpha-Melanocyte stimulating hormone prevents GABAergic neuronal loss and improves cognitive function in Alzheimer’s disease. J. Neurosci. 34, 6736–6745. doi: 10.1523/JNEUROSCI.5075-13.2014
Ma, K., and McLaurin, J. (2017). alpha-Melanocyte stimulating hormone as a potential therapy for Alzheimer‘s disease. Curr. Alzheimer Res. 14, 18–29. doi: 10.2174/1567205013666160819130641
Maccaferri, G., and Lacaille, J. C. (2003). Interneuron diversity series: hippocampal interneuron classifications–making things as simple as possible, not simpler. Trends Neurosci. 26, 564–571. doi: 10.1016/j.tins.2003.08.002
Mann, E. O., and Mody, I. (2010). Control of hippocampal gamma oscillation frequency by tonic inhibition and excitation of interneurons. Nat. Neurosci. 13, 205–212. doi: 10.1038/nn.2464
Mao, J., Huang, S., Liu, S., Feng, X. L., Yu, M., Liu, J., et al. (2015). A herbal medicine for Alzheimer’s disease and its active constituents promote neural progenitor proliferation. Aging Cell 14, 784–796. doi: 10.1111/acel.12356
Marcade, M., Bourdin, J., Loiseau, N., Peillon, H., Rayer, A., Drouin, D., et al. (2008). Etazolate, a neuroprotective drug linking GABA(A) receptor pharmacology to amyloid precursor protein processing. J. Neurochem. 106, 392–404. doi: 10.1111/j.1471-4159.2008.05396.x
Marin, O. (2012). Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 13, 107–120. doi: 10.1038/nrn3155
Maroof, A. M., Keros, S., Tyson, J. A., Ying, S. W., Ganat, Y. M., Merkle, F. T., et al. (2013). Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12, 559–572. doi: 10.1016/j.stem.2013.04.008
Marshall, J. W., Cross, A. J., Jackson, D. M., Green, A. R., Baker, H. F., and Ridley, R. M. (2000). Clomethiazole protects against hemineglect in a primate model of stroke. Brain Res. Bull. 52, 21–29. doi: 10.1016/s0361-9230(99)00275-0
Martin, L. J., Zurek, A. A., MacDonald, J. F., Roder, J. C., Jackson, M. F., and Orser, B. A. (2010). Alpha5GABAA receptor activity sets the threshold for long-term potentiation and constrains hippocampus-dependent memory. J. Neurosci. 30, 5269–5282. doi: 10.1523/JNEUROSCI.4209-09.2010
Martinez-Losa, M., Tracy, T. E., Ma, K., Verret, L., Clemente-Perez, A., Khan, A. S., et al. (2018). Nav1.1-Overexpressing interneuron transplants restore brain rhythms and cognition in a mouse model of Alzheimer’s disease. Neuron 98, 75–89.e5. doi: 10.1016/j.neuron.2018.02.029
Martorell, A. J., Paulson, A. L., Suk, H. J., Abdurrob, F., Drummond, G. T., Guan, W., et al. (2019). Multi-sensory gamma stimulation ameliorates Alzheimer’s-associated pathology and improves cognition. Cell 177, 256–271.e22. doi: 10.1016/j.cell.2019.02.014
Mesulam, M. M., Lalehzari, N., Rahmani, F., Ohm, D., Shahidehpour, R., Kim, G., et al. (2019). Cortical cholinergic denervation in primary progressive aphasia with Alzheimer pathology. Neurology 92, e1580–e1588. doi: 10.1212/WNL.0000000000007247
Meyer, H. S., Schwarz, D., Wimmer, V. C., Schmitt, A. C., Kerr, J. N., Sakmann, B., et al. (2011). Inhibitory interneurons in a cortical column form hot zones of inhibition in layers 2 and 5A. Proc. Natl. Acad. Sci. U.S.A. 108, 16807–16812. doi: 10.1073/pnas.1113648108
Michalska, P., Buendia, I., Del Barrio, L., and Leon, R. (2017). Novel multitarget hybrid compounds for the treatment of Alzheimer’s disease. Curr. Top. Med. Chem. 17, 1027–1043. doi: 10.2174/1568026616666160927154116
Mikkonen, M., Alafuzoff, I., Tapiola, T., Soininen, H., and Miettinen, R. (1999). Subfield- and layer-specific changes in parvalbumin, calretinin and calbindin-D28K immunoreactivity in the entorhinal cortex in Alzheimer’s disease. Neuroscience 92, 515–532. doi: 10.1016/s0306-4522(99)00047-0
Mody, I., and Pearce, R. A. (2004). Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. 27, 569–575. doi: 10.1016/j.tins.2004.07.002
Moore, A. K., and Wehr, M. (2013). Parvalbumin-expressing inhibitory interneurons in auditory cortex are well-tuned for frequency. J. Neurosci. 33, 13713–13723. doi: 10.1523/JNEUROSCI.0663-13.2013
Morris, G. P., Clark, I. A., and Vissel, B. (2014). Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2:135. doi: 10.1186/preaccept-1342777270140958
Morris, G. P., Clark, I. A., and Vissel, B. (2018). Questions concerning the role of amyloid-beta in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 136, 663–689. doi: 10.1007/s00401-018-1918-8
Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., et al. (2000). High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058. doi: 10.1523/jneurosci.20-11-04050.2000
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2:a006338. doi: 10.1101/cshperspect.a006338
Murray, A. J., Woloszynowska-Fraser, M. U., Ansel-Bollepalli, L., Cole, K. L., Foggetti, A., Crouch, B., et al. (2015). Parvalbumin-positive interneurons of the prefrontal cortex support working memory and cognitive flexibility. Sci. Rep. 5:16778. doi: 10.1038/srep16778
Musiek, E. S., and Holtzman, D. M. (2015). Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat. Neurosci. 18, 800–806. doi: 10.1038/nn.4018
Najm, R., Jones, E. A., and Huang, Y. (2019). Apolipoprotein E4, inhibitory network dysfunction, and Alzheimer’s disease. Mol. Neurodegener. 14:24.
Nicholas, C. R., Chen, J., Tang, Y., Southwell, D. G., Chalmers, N., Vogt, D., et al. (2013). Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 12, 573–586. doi: 10.1016/j.stem.2013.04.005
Nilsen, L. H., Rae, C., Ittner, L. M., Gotz, J., and Sonnewald, U. (2013). Glutamate metabolism is impaired in transgenic mice with tau hyperphosphorylation. J. Cereb. Blood Flow Metab. 33, 684–691. doi: 10.1038/jcbfm.2012.212
Olah, S., Fule, M., Komlosi, G., Varga, C., Baldi, R., Barzo, P., et al. (2009). Regulation of cortical microcircuits by unitary GABA-mediated volume transmission. Nature 461, 1278–1281. doi: 10.1038/nature08503
Olah, S., Komlosi, G., Szabadics, J., Varga, C., Toth, E., Barzo, P., et al. (2007). Output of neurogliaform cells to various neuron types in the human and rat cerebral cortex. Front. Neural Circuits 1:4. doi: 10.3389/neuro.04.004.2007
Olsen, R. W., and Sieghart, W. (2008). International union of pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev. 60, 243–260. doi: 10.1124/pr.108.00505
Orts-Del’Immagine, A., and Pugh, J. R. (2018). Activity-dependent plasticity of presynaptic GABAB receptors at parallel fiber synapses. Synapse 72:e22027. doi: 10.1002/syn.22027
Oser, N., Hubacher, M., Specht, K., Datta, A. N., Weber, P., and Penner, I. K. (2014). Default mode network alterations during language task performance in children with benign epilepsy with centrotemporal spikes (BECTS). Epilepsy Behav. 33, 12–17. doi: 10.1016/j.yebeh.2014.01.008
Palop, J. J., and Mucke, L. (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818. doi: 10.1038/nn.2583
Palop, J. J., and Mucke, L. (2016). Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. 17, 777–792. doi: 10.1038/nrn.2016.141
Park, K., Lee, J., Jang, H. J., Richards, B. A., Kohl, M. M., and Kwag, J. (2020). Optogenetic activation of parvalbumin and somatostatin interneurons selectively restores theta-nested gamma oscillations and oscillation-induced spike timing-dependent long-term potentiation impaired by amyloid beta oligomers. BMC Biol. 18:7. doi: 10.1186/s12915-019-0732-7
Pascoal, T. A., Mathotaarachchi, S., Kang, M. S., Mohaddes, S., Shin, M., Park, A. Y., et al. (2019). Abeta-induced vulnerability propagates via the brain’s default mode network. Nat. Commun. 10:2353. doi: 10.1038/s41467-019-10217-w
Petrache, A. L., Rajulawalla, A., Shi, A., Wetzel, A., Saito, T., Saido, T. C., et al. (2019). Aberrant excitatory-inhibitory synaptic mechanisms in entorhinal cortex microcircuits during the pathogenesis of Alzheimer’s disease. Cereb. Cortex 29, 1834–1850. doi: 10.1093/cercor/bhz016
Pike, C. J., and Cotman, C. W. (1993). Cultured GABA-immunoreactive neurons are resistant to toxicity induced by beta-amyloid. Neuroscience 56, 269–274. doi: 10.1016/0306-4522(93)90331-9
Polanco, J. C., Li, C., Bodea, L. G., Martinez-Marmol, R., Meunier, F. A., and Gotz, J. (2018). Amyloid-beta and tau complexity - towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 14, 22–39. doi: 10.1038/nrneurol.2017.162
Porsteinsson, A. P., Drye, L. T., Pollock, B. G., Devanand, D. P., Frangakis, C., Ismail, Z., et al. (2014). Effect of citalopram on agitation in Alzheimer disease: the CitAD randomized clinical trial. JAMA 311, 682–691. doi: 10.1001/jama.2014.93
Qin, Z., Luo, J., VandeVrede, L., Tavassoli, E., Fa, M., Teich, A. F., et al. (2012). Design and synthesis of neuroprotective methylthiazoles and modification as NO-chimeras for neurodegenerative therapy. J. Med. Chem. 55, 6784–6801. doi: 10.1021/jm300353r
Rajput, S. B., Tonge, M. B., and Karuppayil, S. M. (2014). An overview on traditional uses and pharmacological profile of Acorus calamus Linn. (Sweet flag) and other Acorus species. Phytomedicine 21, 268–276. doi: 10.1016/j.phymed.2013.09.020
Ramos, B., Baglietto-Vargas, D., del Rio, J. C., Moreno-Gonzalez, I., Santa-Maria, C., Jimenez, S., et al. (2006). Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenic model of Alzheimer’s disease. Neurobiol. Aging 27, 1658–1672. doi: 10.1016/j.neurobiolaging.2005.09.022
Revilla, S., Sunol, C., Garcia-Mesa, Y., Gimenez-Llort, L., Sanfeliu, C., and Cristofol, R. (2014). Physical exercise improves synaptic dysfunction and recovers the loss of survival factors in 3xTg-AD mouse brain. Neuropharmacology 81, 55–63. doi: 10.1016/j.neuropharm.2014.01.037
Rice, H. C., de Malmazet, D., Schreurs, A., Frere, S., Van Molle, I., Volkov, A. N., et al. (2019). Secreted amyloid-beta precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science 363:eaao4827. doi: 10.1126/science.aao4827
Riek, R., and Eisenberg, D. S. (2016). The activities of amyloids from a structural perspective. Nature 539, 227–235. doi: 10.1038/nature20416
Roberson, E. D., Halabisky, B., Yoo, J. W., Yao, J., Chin, J., Yan, F., et al. (2011). Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 31, 700–711. doi: 10.1523/jneurosci.4152-10.2011
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., et al. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. doi: 10.1126/science.1141736
Rojas-Gutierrez, E., Munoz-Arenas, G., Trevino, S., Espinosa, B., Chavez, R., Rojas, K., et al. (2017). Alzheimer’s disease and metabolic syndrome: a link from oxidative stress and inflammation to neurodegeneration. Synapse 71:e21990. doi: 10.1002/syn.21990
Rosato-Siri, M., Cattaneo, A., and Cherubini, E. (2006). Nicotine-induced enhancement of synaptic plasticity at CA3-CA1 synapses requires GABAergic interneurons in adult anti-NGF mice. J. Physiol. 576(Pt 2), 361–377. doi: 10.1113/jphysiol.2006.114587
Rossor, M. N., Garrett, N. J., Johnson, A. L., Mountjoy, C. Q., Roth, M., and Iversen, L. L. (1982). A post-mortem study of the cholinergic and GABA systems in senile dementia. Brain 105(Pt 2), 313–330. doi: 10.1093/brain/105.2.313
Rossor, M. N., Iversen, L. L., Johnson, A. J., Mountjoy, C. Q., and Roth, M. (1981). Cholinergic deficit in frontal cerebral cortex in Alzheimer’s disease is age dependent. Lancet 2:1422. doi: 10.1016/s0140-6736(81)92836-1
Rubio, S. E., Vega-Flores, G., Martinez, A., Bosch, C., Perez-Mediavilla, A., del Rio, J., et al. (2012). Accelerated aging of the GABAergic septohippocampal pathway and decreased hippocampal rhythms in a mouse model of Alzheimer’s disease. FASEB J. 26, 4458–4467. doi: 10.1096/fj.12-208413
Rudy, B., Fishell, G., Lee, S., and Hjerling-Leffler, J. (2011). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 71, 45–61. doi: 10.1002/dneu.20853
Sahoo, A. K., Dandapat, J., Dash, U. C., and Kanhar, S. (2018). Features and outcomes of drugs for combination therapy as multi-targets strategy to combat Alzheimer’s disease. J. Ethnopharmacol. 215, 42–73. doi: 10.1016/j.jep.2017.12.015
Saito, T., Iwata, N., Tsubuki, S., Takaki, Y., Takano, J., Huang, S. M., et al. (2005). Somatostatin regulates brain amyloid beta peptide Abeta42 through modulation of proteolytic degradation. Nat. Med. 11, 434–439. doi: 10.1038/nm1206
Saiz-Sanchez, D., De La Rosa-Prieto, C., Ubeda-Banon, I., and Martinez-Marcos, A. (2013). Interneurons and beta-amyloid in the olfactory bulb, anterior olfactory nucleus and olfactory tubercle in APPxPS1 transgenic mice model of Alzheimer’s disease. Anat. Rec. 296, 1413–1423. doi: 10.1002/ar.22750
Saiz-Sanchez, D., Flores-Cuadrado, A., Ubeda-Banon, I., de la Rosa-Prieto, C., and Martinez-Marcos, A. (2016). Interneurons in the human olfactory system in Alzheimer’s disease. Exp. Neurol. 276, 13–21. doi: 10.1016/j.expneurol.2015.11.009
Saiz-Sanchez, D., Ubeda-Banon, I., De la Rosa-Prieto, C., and Martinez-Marcos, A. (2012). Differential expression of interneuron populations and correlation with amyloid-beta deposition in the olfactory cortex of an AbetaPP/PS1 transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 31, 113–129. doi: 10.3233/jad-2012-111889
Saiz-Sanchez, D., Ubeda-Banon, I., Flores-Cuadrado, A., Gonzalez-Rodriguez, M., Villar-Conde, S., Astillero-Lopez, V., et al. (2020). Somatostatin, olfaction, and neurodegeneration. Front. Neurosci. 14:96. doi: 10.3389/fnins.2020.00096
Salehi, A., Delcroix, J. D., Belichenko, P. V., Zhan, K., Wu, C., Valletta, J. S., et al. (2006). Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51, 29–42. doi: 10.1016/j.neuron.2006.05.022
Sanchez-Mejias, E., Nunez-Diaz, C., Sanchez-Varo, R., Gomez-Arboledas, A., Garcia-Leon, J. A., Fernandez-Valenzuela, J. J., et al. (2019). Distinct disease-sensitive GABAergic neurons in the perirhinal cortex of Alzheimer’s mice and patients. Brain Pathol. 30, 345–363. doi: 10.1111/bpa.12785
Santos, M. A., Chand, K., and Chaves, S. (2016). Recent progress in repositioning Alzheimer’s disease drugs based on a multitarget strategy. Future Med. Chem. 8, 2113–2142. doi: 10.4155/fmc-2016-0103
Sarkar, S., Jun, S., Rellick, S., Quintana, D. D., Cavendish, J. Z., and Simpkins, J. W. (2016). Expression of microRNA-34a in Alzheimer’s disease brain targets genes linked to synaptic plasticity, energy metabolism, and resting state network activity. Brain Res. 1646, 139–151. doi: 10.1016/j.brainres.2016.05.026
Schipper, S., Aalbers, M. W., Rijkers, K., Swijsen, A., Rigo, J. M., Hoogland, G., et al. (2016). Tonic GABAA receptors as potential target for the treatment of temporal lobe epilepsy. Mol. Neurobiol. 53, 5252–5265. doi: 10.1007/s12035-015-9423-8
Schmid, L. C., Mittag, M., Poll, S., Steffen, J., Wagner, J., Geis, H. R., et al. (2016). Dysfunction of somatostatin-positive interneurons associated with memory deficits in an Alzheimer’s disease model. Neuron 92, 114–125. doi: 10.1016/j.neuron.2016.08.034
Schwab, C., Yu, S., Wong, W., McGeer, E. G., and McGeer, P. L. (2013). GAD65, GAD67, and GABAT immunostaining in human brain and apparent GAD65 loss in Alzheimer’s disease. J. Alzheimers Dis. 33, 1073–1088. doi: 10.3233/jad-2012-121330
Schweizer, C., Balsiger, S., Bluethmann, H., Mansuy, I. M., Fritschy, J. M., Mohler, H., et al. (2003). The gamma 2 subunit of GABA(A) receptors is required for maintenance of receptors at mature synapses. Mol. Cell. Neurosci. 24, 442–450. doi: 10.1016/s1044-7431(03)00202-1
Scimemi, A., Meabon, J. S., Woltjer, R. L., Sullivan, J. M., Diamond, J. S., and Cook, D. G. (2013). Amyloid-beta1-42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J. Neurosci. 33, 5312–5318. doi: 10.1523/JNEUROSCI.5274-12.2013
Seidman, L. J., Rosso, I. M., Thermenos, H. W., Makris, N., Juelich, R., Gabrieli, J. D., et al. (2014). Medial temporal lobe default mode functioning and hippocampal structure as vulnerability indicators for schizophrenia: a MRI study of non-psychotic adolescent first-degree relatives. Schizophr. Res. 159, 426–434. doi: 10.1016/j.schres.2014.09.011
Selkoe, D. J. (1999). Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 399(Suppl. 6738), A23–A31.
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608.
Shah, D., Latif-Hernandez, A., De Strooper, B., Saito, T., Saido, T., Verhoye, M., et al. (2018). Spatial reversal learning defect coincides with hypersynchronous telencephalic BOLD functional connectivity in APP(NL-F/NL-F) knock-in mice. Sci. Rep. 8:6264.
Shang, C., Liu, Z., Chen, Z., Shi, Y., Wang, Q., Liu, S., et al. (2015). BRAIN CIRCUITS. A parvalbumin-positive excitatory visual pathway to trigger fear responses in mice. Science 348, 1472–1477. doi: 10.1126/science.aaa8694
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. doi: 10.1523/jneurosci.4970-06.2007
Shao, H., Zhang, Y., Dong, Y., Yu, B., Xia, W., and Xie, Z. (2014). Chronic treatment with anesthetic propofol improves cognitive function and attenuates caspase activation in both aged and Alzheimer’s disease transgenic mice. J. Alzheimers Dis. 41, 499–513. doi: 10.3233/JAD-132792
Sheline, Y. I., West, T., Yarasheski, K., Swarm, R., Jasielec, M. S., Fisher, J. R., et al. (2014). An antidepressant decreases CSF Abeta production in healthy individuals and in transgenic AD mice. Sci. Transl. Med. 6:236re4. doi: 10.1126/scitranslmed.3008169
Shetty, A. K., and Bates, A. (2016). Potential of GABA-ergic cell therapy for schizophrenia, neuropathic pain, and Alzheimer’s and Parkinson’s diseases. Brain Res. 1638(Pt A), 74–87. doi: 10.1016/j.brainres.2015.09.019
Shetty, A. K., and Turner, D. A. (2000). Fetal hippocampal grafts containing CA3 cells restore host hippocampal glutamate decarboxylase-positive interneuron numbers in a rat model of temporal lobe epilepsy. J. Neurosci. 20, 8788–8801. doi: 10.1523/jneurosci.20-23-08788.2000
Shi, A., Petrache, A. L., Shi, J., and Ali, A. B. (2019). Preserved calretinin interneurons in an app model of Alzheimer’s disease disrupt hippocampal inhibition via upregulated P2Y1 purinoreceptors. Cereb. Cortex 30, 1272–1290. doi: 10.1093/cercor/bhz165
Shu, S., Zhu, H., Tang, N., Chen, W., Li, X., Li, H., et al. (2016). Selective degeneration of entorhinal-CA1 synapses in Alzheimer’s disease via activation of DAPK1. J. Neurosci. 36, 10843–10852. doi: 10.1523/JNEUROSCI.2258-16.2016
Siddique, H., Hynan, L. S., and Weiner, M. F. (2009). Effect of a serotonin reuptake inhibitor on irritability, apathy, and psychotic symptoms in patients with Alzheimer’s disease. J. Clin. Psychiatry 70, 915–918. doi: 10.4088/JCP.08m04828
Sigel, E., and Steinmann, M. E. (2012). Structure, function, and modulation of GABA(A) receptors. J. Biol. Chem. 287, 40224–40231. doi: 10.1074/jbc.R112.386664
Sinnen, B. L., Bowen, A. B., Gibson, E. S., and Kennedy, M. J. (2016). Local and use-dependent effects of beta-amyloid oligomers on NMDA receptor function revealed by optical quantal analysis. J. Neurosci. 36, 11532–11543. doi: 10.1523/JNEUROSCI.1603-16.2016
Sohal, V. S., Zhang, F., Yizhar, O., and Deisseroth, K. (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702. doi: 10.1038/nature07991
Solarski, M., Wang, H., Wille, H., and Schmitt-Ulms, G. (2018). Somatostatin in Alzheimer’s disease: a new role for an old player. Prion 12, 1–8. doi: 10.1080/19336896.2017.1405207
Soler, H., Dorca-Arevalo, J., Gonzalez, M., Rubio, S. E., Avila, J., Soriano, E., et al. (2017). The GABAergic septohippocampal connection is impaired in a mouse model of tauopathy. Neurobiol. Aging 49, 40–51. doi: 10.1016/j.neurobiolaging.2016.09.006
Solodkin, A., Veldhuizen, S. D., and Van Hoesen, G. W. (1996). Contingent vulnerability of entorhinal parvalbumin-containing neurons in Alzheimer’s disease. J. Neurosci. 16, 3311–3321. doi: 10.1523/jneurosci.16-10-03311.1996
Southwell, D. G., Nicholas, C. R., Basbaum, A. I., Stryker, M. P., Kriegstein, A. R., Rubenstein, J. L., et al. (2014). Interneurons from embryonic development to cell-based therapy. Science 344:1240622. doi: 10.1126/science.1240622
Sperling, R. A., Laviolette, P. S., O’Keefe, K., O’Brien, J., Rentz, D. M., Pihlajamaki, M., et al. (2009). Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63, 178–188. doi: 10.1016/j.neuron.2009.07.003
Stoiljkovic, M., Leventhal, L., Chen, A., Chen, T., Driscoll, R., Flood, D., et al. (2015). Concentration-response relationship of the alpha7 nicotinic acetylcholine receptor agonist FRM-17874 across multiple in vitro and in vivo assays. Biochem. Pharmacol. 97, 576–589. doi: 10.1016/j.bcp.2015.07.006
Struber, M., Sauer, J. F., Jonas, P., and Bartos, M. (2017). Distance-dependent inhibition facilitates focality of gamma oscillations in the dentate gyrus. Nat. Commun. 8:758.
Sun, B., Halabisky, B., Zhou, Y., Palop, J. J., Yu, G., Mucke, L., et al. (2009). Imbalance between GABAergic and glutamatergic transmission impairs adult neurogenesis in an animal model of Alzheimer’s disease. Cell Stem Cell 5, 624–633. doi: 10.1016/j.stem.2009.10.003
Sun, X., Meng, X., Zhang, J., Li, Y., Wang, L., Qin, X., et al. (2012). GABA attenuates amyloid toxicity by downregulating its endocytosis and improves cognitive impairment. J. Alzheimers Dis. 31, 635–649. doi: 10.3233/jad-2012-120535
Sun, Y., Pasca, S. P., Portmann, T., Goold, C., Worringer, K. A., Guan, W., et al. (2016). A deleterious Nav1.1 mutation selectively impairs telencephalic inhibitory neurons derived from Dravet Syndrome patients. eLife 5:e13073. doi: 10.7554/eLife.13073
Tackenberg, C., and Brandt, R. (2009). Divergent pathways mediate spine alterations and cell death induced by amyloid-beta, wild-type tau, and R406W tau. J. Neurosci. 29, 14439–14450. doi: 10.1523/jneurosci.3590-09.2009
Takahashi, H., Brasnjevic, I., Rutten, B. P., Van Der Kolk, N., Perl, D. P., Bouras, C., et al. (2010). Hippocampal interneuron loss in an APP/PS1 double mutant mouse and in Alzheimer’s disease. Brain Struct. Funct. 214, 145–160. doi: 10.1007/s00429-010-0242-4
Tamas, G., Lorincz, A., Simon, A., and Szabadics, J. (2003). Identified sources and targets of slow inhibition in the neocortex. Science 299, 1902–1905. doi: 10.1126/science.1082053
Tang, B. L. (2019). Amyloid precursor protein (APP) and GABAergic neurotransmission. Cells 8:E550. doi: 10.3390/cells8060550
Tanzi, R. E., and Bertram, L. (2005). Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. doi: 10.1016/j.cell.2005.02.008
Terunuma, M., Vargas, K. J., Wilkins, M. E., Ramirez, O. A., Jaureguiberry-Bravo, M., Pangalos, M. N., et al. (2010). Prolonged activation of NMDA receptors promotes dephosphorylation and alters postendocytic sorting of GABAB receptors. Proc. Natl. Acad. Sci. U.S.A. 107, 13918–13923. doi: 10.1073/pnas.1000853107
Thatcher, G. R., Bennett, B. M., Dringenberg, H. C., and Reynolds, J. N. (2004). Novel nitrates as NO mimetics directed at Alzheimer’s disease. J. Alzheimers Dis. 6(Suppl. 6), S75–S84. doi: 10.3233/jad-2004-6s614
Tong, L. M., Djukic, B., Arnold, C., Gillespie, A. K., Yoon, S. Y., Wang, M. M., et al. (2014). Inhibitory interneuron progenitor transplantation restores normal learning and memory in ApoE4 knock-in mice without or with Abeta accumulation. J. Neurosci. 34, 9506–9515. doi: 10.1523/jneurosci.0693-14.2014
Tong, L. M., Yoon, S. Y., Andrews-Zwilling, Y., Yang, A., Lin, V., Lei, H., et al. (2016). Enhancing GABA signaling during middle adulthood prevents age-dependent GABAergic interneuron decline and learning and memory deficits in ApoE4 mice. J. Neurosci. 36, 2316–2322. doi: 10.1523/JNEUROSCI.3815-15.2016
Townsend, M., Whyment, A., Walczak, J. S., Jeggo, R., van den Top, M., Flood, D. G., et al. (2016). alpha7-nAChR agonist enhances neural plasticity in the hippocampus via a GABAergic circuit. J. Neurophysiol. 116, 2663–2675. doi: 10.1152/jn.00243.2016
Tremblay, R., Lee, S., and Rudy, B. (2016). GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91, 260–292. doi: 10.1016/j.neuron.2016.06.033
Tyson, J. A., and Anderson, S. A. (2014). GABAergic interneuron transplants to study development and treat disease. Trends Neurosci. 37, 169–177. doi: 10.1016/j.tins.2014.01.003
Umeda, T., Kimura, T., Yoshida, K., Takao, K., Fujita, Y., Matsuyama, S., et al. (2017). Mutation-induced loss of APP function causes GABAergic depletion in recessive familial Alzheimer’s disease: analysis of Osaka mutation-knockin mice. Acta Neuropathol. Commun. 5:59.
van der Kant, R., Goldstein, L. S. B., and Ossenkoppele, R. (2020). Amyloid-beta-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 21, 21–35. doi: 10.1038/s41583-019-0240-3
Vandevrede, L., Tavassoli, E., Luo, J., Qin, Z., Yue, L., Pepperberg, D. R., et al. (2014). Novel analogues of chlormethiazole are neuroprotective in four cellular models of neurodegeneration by a mechanism with variable dependence on GABA(A) receptor potentiation. Br. J. Pharmacol. 171, 389–402. doi: 10.1111/bph.12454
Vellas, B., Sol, O., Snyder, P. J., Ousset, P. J., Haddad, R., Maurin, M., et al. (2011). EHT0202 in Alzheimer’s disease: a 3-month, randomized, placebo-controlled, double-blind study. Curr. Alzheimer Res. 8, 203–212. doi: 10.2174/156720511795256053
Verdaguer, E., Brox, S., Petrov, D., Olloquequi, J., Romero, R., de Lemos, M. L., et al. (2015). Vulnerability of calbindin, calretinin and parvalbumin in a transgenic/knock-in APPswe/PS1dE9 mouse model of Alzheimer disease together with disruption of hippocampal neurogenesis. Exp. Gerontol. 69, 176–188. doi: 10.1016/j.exger.2015.06.013
Verret, L., Mann, E. O., Hang, G. B., Barth, A. M., Cobos, I., Ho, K., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. doi: 10.1016/j.cell.2012.02.046
Villette, V., and Dutar, P. (2017). GABAergic microcircuits in Alzheimer’s disease models. Curr. Alzheimer Res. 14, 30–39. doi: 10.2174/1567205013666160819125757
Vossel, K. A., Xu, J. C., Fomenko, V., Miyamoto, T., Suberbielle, E., Knox, J. A., et al. (2015). Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J. Cell Biol. 209, 419–433. doi: 10.1083/jcb.201407065
Vossel, K. A., Zhang, K., Brodbeck, J., Daub, A. C., Sharma, P., Finkbeiner, S., et al. (2010). Tau reduction prevents Abeta-induced defects in axonal transport. Science 330:198. doi: 10.1126/science.1194653
Walsh, C., Drinkenburg, W. H., and Ahnaou, A. (2017). Neurophysiological assessment of neural network plasticity and connectivity: progress towards early functional biomarkers for disease interception therapies in Alzheimer’s disease. Neurosci. Biobehav. Rev. 73, 340–358. doi: 10.1016/j.neubiorev.2016.12.020
Wamsley, B., and Fishell, G. (2017). Genetic and activity-dependent mechanisms underlying interneuron diversity. Nat. Rev. Neurosci. 18, 299–309. doi: 10.1038/nrn.2017.30
Wang, H., Muiznieks, L. D., Ghosh, P., Williams, D., Solarski, M., Fang, A., et al. (2017a). Somatostatin binds to the human amyloid beta peptide and favors the formation of distinct oligomers. eLife 6:e28401. doi: 10.7554/eLife.28401
Wang, H., Williams, D., Griffin, J., Saito, T., Saido, T. C., Fraser, P. E., et al. (2017b). Time-course global proteome analyses reveal an inverse correlation between Abeta burden and immunoglobulin M levels in the APPNL-F mouse model of Alzheimer disease. PLoS One 12:e0182844. doi: 10.1371/journal.pone.0182844
Wang, Q., Walsh, D. M., Rowan, M. J., Selkoe, D. J., and Anwyl, R. (2004). Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J. Neurosci. 24, 3370–3378. doi: 10.1523/jneurosci.1633-03.2004
Wang, R., and Reddy, P. H. (2017). Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimers Dis. 57, 1041–1048. doi: 10.3233/JAD-160763
Wang, X., Sun, Y., Li, T., Cai, Y., and Han, Y. (2019). Amyloid-beta as a blood biomarker for Alzheimer’s disease: a review of recent literature. J. Alzheimers Dis. 73, 819–832. doi: 10.3233/JAD-190714
Wang, Y., Wu, Z., Bai, Y. T., Wu, G. Y., and Chen, G. (2017c). Gad67 haploinsufficiency reduces amyloid pathology and rescues olfactory memory deficits in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 12:73.
Wen, Q., Mustafi, S. M., Li, J., Risacher, S. L., Tallman, E., Brown, S. A., et al. (2019). White matter alterations in early-stage Alzheimer’s disease: a tract-specific study. Alzheimers Dement. 11, 576–587. doi: 10.1016/j.dadm.2019.06.003
Whitehouse, P. J., Price, D. L., Struble, R. G., Clark, A. W., Coyle, J. T., and Delon, M. R. (1982). Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science 215, 1237–1239. doi: 10.1126/science.7058341
Wiseman, F. K., Al-Janabi, T., Hardy, J., Karmiloff-Smith, A., Nizetic, D., Tybulewicz, V. L., et al. (2015). A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 16, 564–574. doi: 10.1038/nrn3983
Wonders, C. P., and Anderson, S. A. (2006). The origin and specification of cortical interneurons. Nat. Rev. Neurosci. 7, 687–696. doi: 10.1038/nrn1954
Wright, A. L., Zinn, R., Hohensinn, B., Konen, L. M., Beynon, S. B., Tan, R. P., et al. (2013). Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS One 8:e59586. doi: 10.1371/journal.pone.0059586
Wu, Z., Guo, Z., Gearing, M., and Chen, G. (2014). Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat. Commun. 5:4159. doi: 10.1038/ncomms5159
Wyss-Coray, T. (2016). Ageing, neurodegeneration and brain rejuvenation. Nature 539, 180–186. doi: 10.1038/nature20411
Xenos, D., Kamceva, M., Tomasi, S., Cardin, J. A., Schwartz, M. L., and Vaccarino, F. M. (2018). Loss of TrkB Signaling in parvalbumin-expressing basket cells results in network activity disruption and abnormal behavior. Cereb. Cortex 28, 3399–3413. doi: 10.1093/cercor/bhx173
Xu, X., Roby, K. D., and Callaway, E. M. (2010). Immunochemical characterization of inhibitory mouse cortical neurons: three chemically distinct classes of inhibitory cells. J. Comp. Neurol. 518, 389–404. doi: 10.1002/cne.22229
Xu, Y., Li, X., Wang, X., Yao, J., and Zhuang, S. (2018). miR-34a deficiency in APP/PS1 mice promotes cognitive function by increasing synaptic plasticity via AMPA and NMDA receptors. Neurosci. Lett. 670, 94–104. doi: 10.1016/j.neulet.2018.01.045
Yang, L., Wang, Z., Wang, B., Justice, N. J., and Zheng, H. (2009). Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J. Neurosci. 29, 15660–15668. doi: 10.1523/jneurosci.4104-09.2009
Yang, X., Yao, C., Tian, T., Li, X., Yan, H., Wu, J., et al. (2018). A novel mechanism of memory loss in Alzheimer’s disease mice via the degeneration of entorhinal-CA1 synapses. Mol. Psychiatry 23, 199–210. doi: 10.1038/mp.2016.151
Yao, W., Tambini, M. D., Liu, X., and D’Adamio, L. (2019). Tuning of glutamate, but not GABA, release by an intrasynaptic vesicle APP domain whose function can be modulated by beta- or alpha-secretase cleavage. J. Neurosci. 39, 6992–7005. doi: 10.1523/JNEUROSCI.0207-19.2019
Yassa, M. A., Stark, S. M., Bakker, A., Albert, M. S., Gallagher, M., and Stark, C. E. (2010). High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic mild cognitive impairment. Neuroimage 51, 1242–1252. doi: 10.1016/j.neuroimage.2010.03.040
Yiannopoulou, K. G., and Papageorgiou, S. G. (2013). Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 6, 19–33. doi: 10.1177/1756285612461679
Zallo, F., Gardenal, E., Verkhratsky, A., and Rodriguez, J. J. (2018). Loss of calretinin and parvalbumin positive interneurones in the hippocampal CA1 of aged Alzheimer’s disease mice. Neurosci. Lett. 681, 19–25. doi: 10.1016/j.neulet.2018.05.027
Zhang, H., He, X., Mei, Y., and Ling, Q. (2018). Ablation of ErbB4 in parvalbumin-positive interneurons inhibits adult hippocampal neurogenesis through down-regulating BDNF/TrkB expression. J. Comp. Neurol. 526, 2482–2492. doi: 10.1002/cne.24506
Zhang, H., Zhang, L., Zhou, D., He, X., Wang, D., Pan, H., et al. (2017). Ablating ErbB4 in PV neurons attenuates synaptic and cognitive deficits in an animal model of Alzheimer’s disease. Neurobiol. Dis. 106, 171–180. doi: 10.1016/j.nbd.2017.07.001
Zhang, Q., Yang, C., Liu, T., Liu, L., Li, F., Cai, Y., et al. (2018). Citalopram restores short-term memory deficit and non-cognitive behaviors in APP/PS1 mice while halting the advance of Alzheimer’s disease-like pathology. Neuropharmacology 131, 475–486. doi: 10.1016/j.neuropharm.2017.12.021
Zhang, S. Q., Obregon, D., Ehrhart, J., Deng, J., Tian, J., Hou, H., et al. (2013). Baicalein reduces beta-amyloid and promotes nonamyloidogenic amyloid precursor protein processing in an Alzheimer’s disease transgenic mouse model. J. Neurosci. Res. 91, 1239–1246. doi: 10.1002/jnr.23244
Zhang, Y., Shao, H., Dong, Y., Swain, C. A., Yu, B., Xia, W., et al. (2014). Chronic treatment with anesthetic propofol attenuates beta-amyloid protein levels in brain tissues of aged mice. Transl. Neurodegener. 3:8. doi: 10.1186/2047-9158-3-8
Zhao, C., Zhang, H., Li, H., Lv, C., Liu, X., Li, Z., et al. (2017). Geniposide ameliorates cognitive deficits by attenuating the cholinergic defect and amyloidosis in middle-aged Alzheimer model mice. Neuropharmacology 116, 18–29. doi: 10.1016/j.neuropharm.2016.12.002
Zheng, J., Li, H. L., Tian, N., Liu, F., Wang, L., Yin, Y., et al. (2020). Interneuron accumulation of phosphorylated tau impairs adult hippocampal neurogenesis by suppressing GABAergic transmission. Cell Stem Cell 26, 331–345.e6. doi: 10.1016/j.stem.2019.12.015
Zimmer, R., Teelken, A. W., Trieling, W. B., Weber, W., Weihmayr, T., and Lauter, H. (1984). Gamma-aminobutyric acid and homovanillic acid concentration in the CSF of patients with senile dementia of Alzheimer’s type. Arch. Neurol. 41, 602–604. doi: 10.1001/archneur.1984.04210080010005
Keywords: GABA inhibitory interneurons, cognitive deficits, Alzheimer’s disease, amyloid β-protein, PV inhibitory interneurons
Citation: Xu Y, Zhao M, Han Y and Zhang H (2020) GABAergic Inhibitory Interneuron Deficits in Alzheimer’s Disease: Implications for Treatment. Front. Neurosci. 14:660. doi: 10.3389/fnins.2020.00660
Received: 21 February 2020; Accepted: 28 May 2020;
Published: 30 June 2020.
Edited by:
Francesco Fazio, Albert Einstein College of Medicine, United StatesReviewed by:
Maxime Assous, Rutgers University, Newark, United StatesJohn J. Hablitz, University of Alabama at Birmingham, United States
Daniel Saiz Sanchez, University of Castilla-La Mancha, Spain
Copyright © 2020 Xu, Zhao, Han and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heng Zhang, emhhbmdoZW5nQHVzeC5lZHUuY24=
†These authors have contributed equally to this work