 Jiayan Jin2,5
Jiayan Jin2,5 Yuan Gao
Yuan Gao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 26 June 2020
Sec. Neurodegeneration
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.00595
Yes-associated protein (YAP) is a key effector downstream of the Hippo signaling pathway and plays an important role in the development of the physiology and pathology of the central nervous system (CNS), especially regulating cell proliferation, differentiation, migration, and apoptosis. However, the roles and underlying mechanisms of YAP in CNS diseases are still puzzling. Here, this review will systematically and comprehensively summarize the biological feature, pathological role, and underlying mechanisms of YAP in normal and pathologic CNS, which aims to provide insights into the potential molecular targets and new therapeutic strategies for CNS diseases.
The Hippo signaling pathway was first discovered in the Drosophila wts gene mutation, causing the growth phenotype of the eyes and wings (Justice et al., 1995). However, the components of the signaling pathway are highly conserved from Drosophila to mammals (Zhao et al., 2010a), including mammalian STE20-like protein kinase 1/2 (Mst1/2), Salvador family WW domain-containing protein 1 (SAV1), large tumor suppressor substance 1/2 (LATs1/2), MOB kinase activator (MOB1), Yes-associated protein (YAP) and its paralog, and TAZ and TEA domain-containing sequence-specific transcription factors (TEAD1–4; Chan et al., 2005; Lei et al., 2008; Wang et al., 2019). The Hippo signaling pathway is relatively highly conserved in mammals and is mainly composed of a series of core kinases, including MST1/2, SAV1, LATs1/2, and MOB1A/B. When receiving upstream signal stimulation, phosphorylation-activated MST1/2 in turn leads to phosphorylation activation of other core kinases, SAV1, LATs1/2, and MOB1. The interaction between MST1/2 and SAV1 enhances the activity of MST1/2 kinase, which in turn promotes the phosphorylation and activation of LATs1/2. The activation of the Hippo signaling pathway then causes YAP/TAZ phosphorylation to remain in the cytoplasm, eventually leading to its chelation and proteasome-mediated degradation. Conversely, when the Hippo signaling pathway is inactivated, dephosphorylated YAP/TAZ enters the nucleus from the cytoplasm, promoting its downstream gene expression that mediates cell proliferation and migration. Because YAP lacks a DNA-binding domain, YAP needs to combine with other transcription factors to form a transcriptional regulatory complex, such as TEAD, RUNT-related transcription factor 1/2 (RUNX1/2), and T-box transcription factor 5 (TBX5), especially TEAD (Zhao et al., 2007, 2010a; Figure 1).
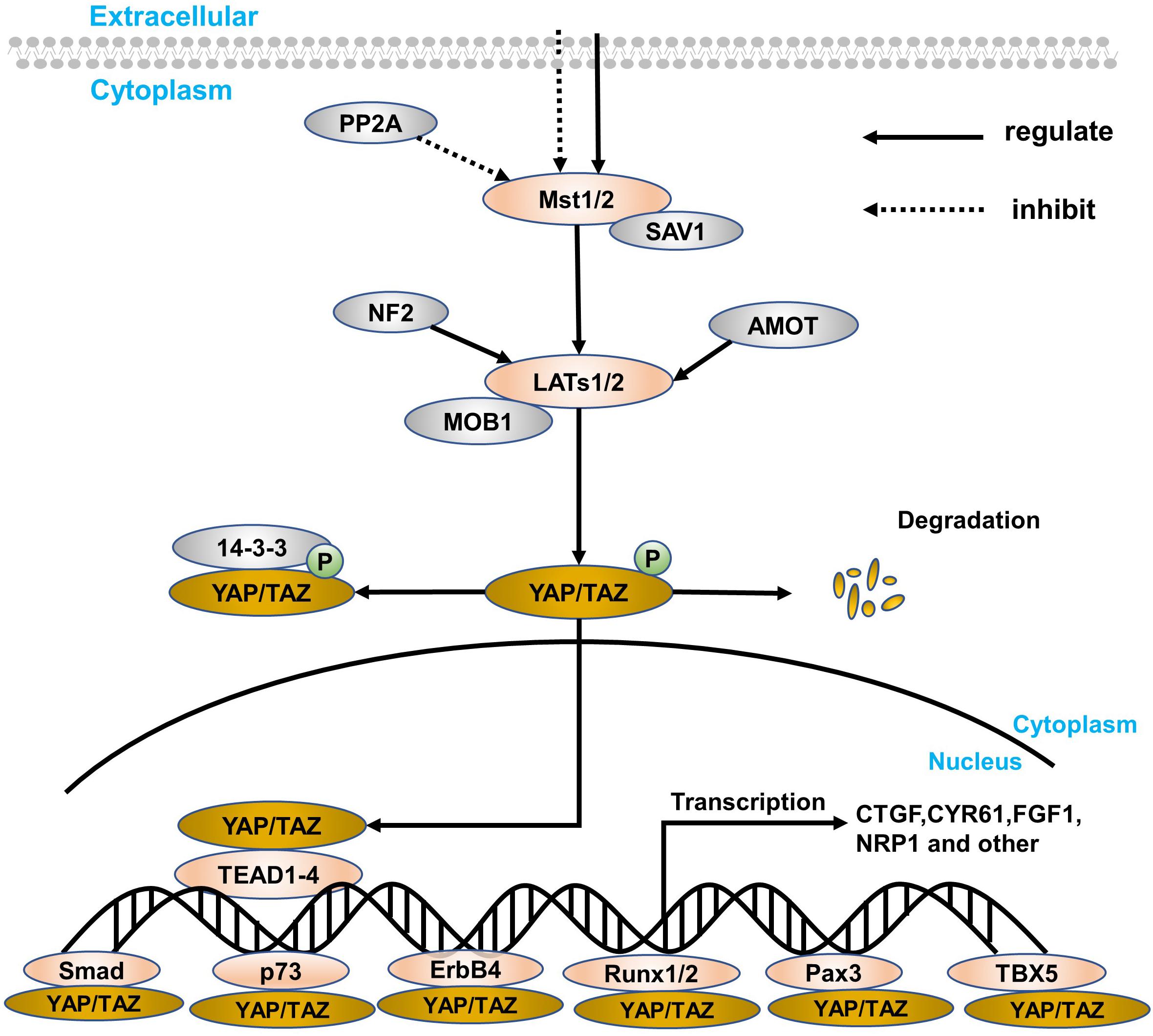
Figure 1. A simple overview of the Hippo signaling pathway and YAP. The core components of the Hippo signaling pathway are Mst1/2, LATs1/2, YAP/TAZ, and target genes, which are closely connected, especially YAP. The retention method for YAP is in the nucleus and cytoplasm. In the cytoplasm, YAP/TAZ can blind to some specific proteins (such as the 14-3-3 protein) through direct LATs1/2 phosphorylation and indirect Mst1/2 activation. At the same time, SAV1 and MOB1 are defined as regulatory proteins of Mst1/2 and LATs1/2, respectively. Besides, PP2A can inhibit the Mst1/2–SAV1 complex, while both NF2 and AMOT can regulate the LATs1/2–MOB1 complex. In the nucleus transferred from the cytoplasm, YAP can express a series of target genes through interacting with transcription factors, like TEAD family, Smad, p73, ErbB4, Runx1/2, Pax, and TBX5. Finally, when suffering from certain physiological and pathological damage, YAP will degrade.
Yes-associated protein is the main downstream effector of the Hippo signaling pathway in mammals. As a transcriptional activator, YAP has no DNA-binding domain and cannot directly bind to DNA. When the signaling pathway is inhibited, it is necessary to interact with some transcription factors to mediate transcription factors of multiple genes (Mo et al., 2014), like CTGF, CYP61, NRP1, and FGF1. However, LATs1/2 phosphorylates YAP, and proteins in the cytoplasm, like 14-3-3 protein and AMOT, are then ubiquitinated and degraded (Figure 1). What’s more amazing is that the position of YAP can regulate the volume of the organ and the regeneration of tissues to maintain cell proliferation and apoptosis and participate in the inhibition of cell contact (Zhao et al., 2007, 2010a). Thus, research regarding the Hippo signaling pathway has been paid more and more attention, and YAP is considered to be the most important downstream effector in the current research (Vassilev et al., 2001; Zhao et al., 2008b, 2010a).
As we all know, YAP can regulate the volume of organ and regeneration of injured tissues, maintain the balance of cell proliferation and apoptosis via the downstream gene expression degree, and participate in cell contact inhibition regulation. In this section, we set forth that YAP keeps a balance between apoptosis and proliferation in neural progenitor cell (NPCs) to reach a steady state, responds to neuroinflammation, ensures the smooth signal transmission via downstream pathway or genes, and interacts with endothelial cells with different approaches during angiogenesis (Table 1).
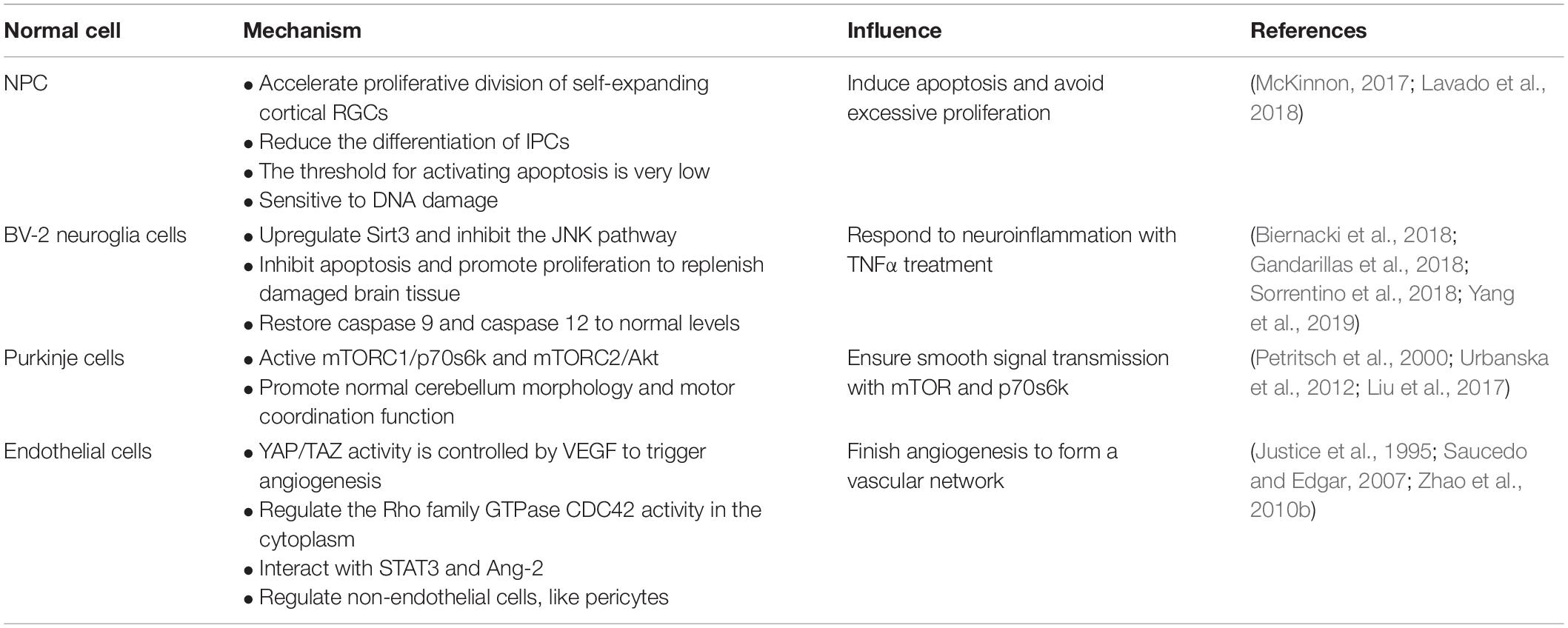
Table 1. Summary of the role of YAP in normal cells of CNS.
NPCs are a kind of precursor cells differentiated from neural stem cells (NSCs) and specifically differentiated into neurons (Lee et al., 2016). Their proliferation, differentiation, and migration are closely related to embryonic development and the number and precise distribution of neurons after birth. The normal size, composition, and function of the nervous system depend on the precise self-renewal and specific differentiation of NPCs. Studying the mechanism of neural precursor cell differentiation and proliferation can also provide a new scientific basis for neurodegenerative diseases and nerve damage. Recent studies have found that the Hippo/YAP signaling pathway plays an important role in the proliferation, differentiation, and apoptosis of NPCs (Lee et al., 2016; Lavado et al., 2018).
In the developing neural tube of vertebrates (chicken embryos), the ependymal NPCs express YAP. Overexpression of YAP or transcription-activated TEAD in the neural tube can cause a decrease in neuronal differentiation and a significant increase in the number of NPCs. YAP deficiency causes cell death and promotes early neuronal differentiation. These effects are mainly related to the upregulation of cyclin D1 expression induced by YAP/TEADs. Moreover, conditional knockout of LATs1/2 results in a decrease in phosphorylated YAP and an increase in the level of YAP/TAZ, which triggers the shift of most YAP/TAZ proteins from the cytoplasm to the nucleus. The strong nuclear localization of YAP/TAZ enhances global transcription activity and upregulates many genes, thereby boosting the capacity of biosynthesis and proliferation (Lavado et al., 2018), which is consistent with the known Hippo kinase cascade. Nevertheless, in NPCs, LATs1/2 deletion accelerates proliferative division of self-expanding cortical radial glial cells (RGCs) (Kriegstein and Alvarez-Buylla, 2009) and reduces the differentiation of intermediate progenitor cells (IPCs) (Lavado et al., 2018), which are sufficient to induce apoptosis dependent on TEAD-guided transcription and ultimately exacerbate DNA replication pressure and DNA damage and worsen brain morphology (Figure 2). More interestingly, the mode of action of YAP is distinct from the normal context of the upregulation of antiapoptotic genes, probably because NPCs are particularly sensitive to DNA damage and their threshold for activating apoptosis is very low (McKinnon, 2017). Consequently, the YAP signaling pathway plays an important role in cell proliferation and differentiation. Its overexpression or overactivation can promote both cell proliferation and cell apoptosis, which helps maintain the homeostasis.
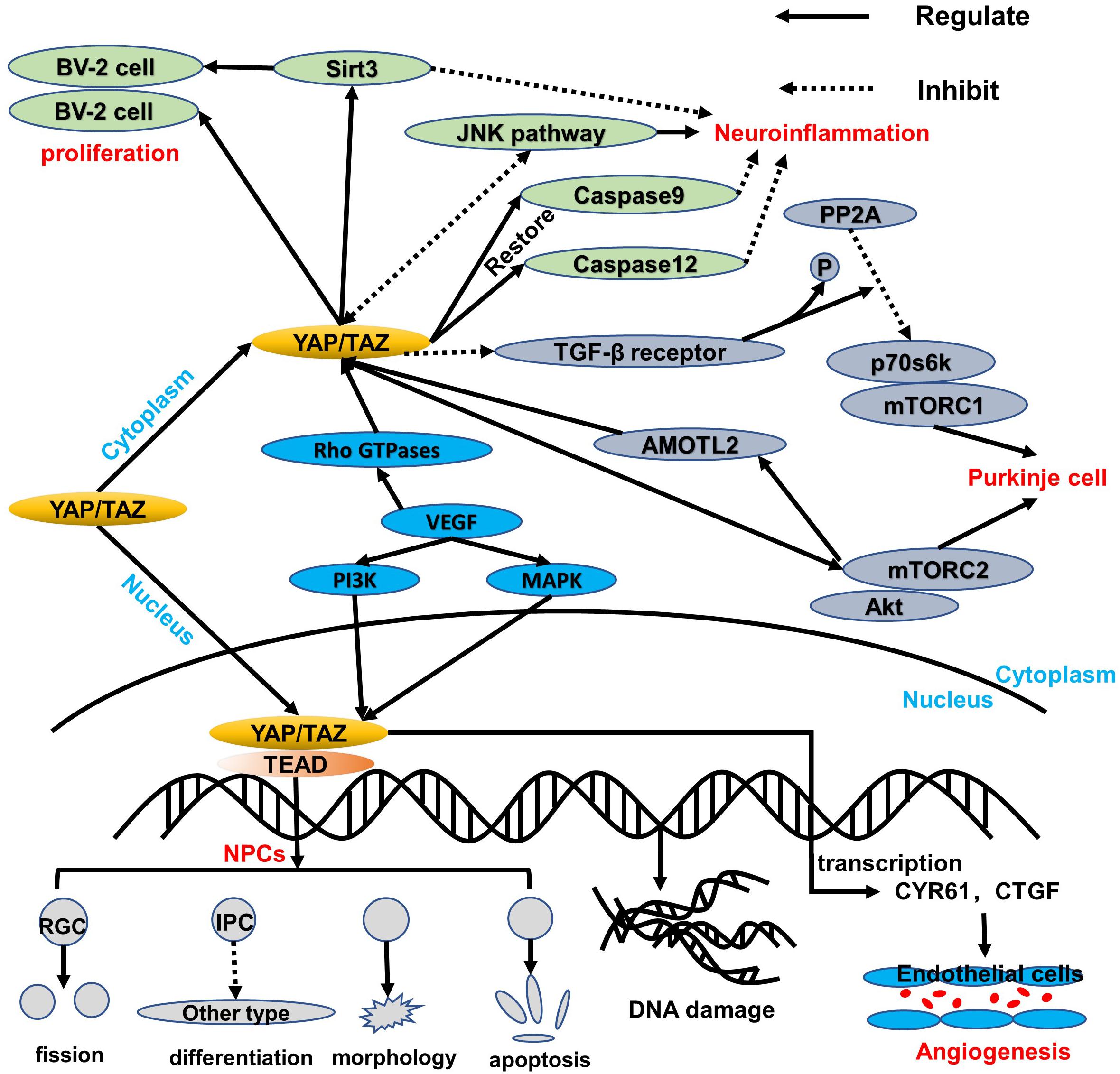
Figure 2. An overview of the regulation of YAP/TAZ in healthy tissue. YAP/TAZ regulates the healthy cells of the central nervous system. In NPCs, YAP/TAZ shifts to the nucleus to interact with the TEAD family, thereby leading to a balance of proliferation and apoptosis via blocking IPC differentiation, damaging morphology, accelerating the RGC fission, and inducing DNA replication stress and damage. Additionally, YAP/TAZ in the cytoplasm can upregulate Sirt3 and inhibit the JNK pathway, which both are beneficial to protect BV-2 cell proliferation and respond to neuroinflammation. Moreover, caspase 9 and caspase 12 can be restored to reduce mitochondrial damage and endoplasmic reticulum stress. Like the BV-2 cells in the cytoplasm, the p70s6k-and-mTORC1 complex is regulated via downregulation of PP2A and TGF-β receptor, while the Akt-and-mTORC2 complex can interact with YAP/TAZ through AMOTL2. More significantly, VEGF induces YAP/TAZ via Rho GTPases and subsequently triggers YAP/TAZ target genes to protect the mitosis and survival of endothelial cells during angiogenesis through PI3K and MAPK regulation.
Under the condition of TNFα-induced neuroinflammation, the viability of BV-2 cells decreased while the apoptosis index increased. At the same time, YAP expression is significantly reduced, which indicates that YAP may aggravate BV-2 cell death in neuroinflammation (Yang et al., 2019; Figure 2). Studies have found that overexpression of YAP induced by adenovirus transfection can reverse the viability of BV-2 cells, thereby reducing the apoptosis index of TNFα-treated BV-2 cells. The molecular mechanism study found that YAP overexpression is closely related to Sirt3 upregulation (Cheignon et al., 2018). Sirt3 has been validated as the biomarker of the viability of BV-2 neuroglia cells (Cui et al., 2018) and is required for YAP-induced BV-2 cell protection (Li et al., 2018; Sorrentino et al., 2018; Yang et al., 2019). Increased Sirt3 expression can reduce ER stress response, reduce mitochondrial damage, and block the JNK pro-apoptotic pathway, and the knockout of Sirt3 eliminates the protective effect of YAP overexpression on TNFα-treated BV-2 cells, indicating that YAP overexpression can protect BV-2 cells from TNFα-mediated cells. In addition to Sirt3, the JNK pathway, which is found as the classical downstream pathway of neuroinflammation response (Yang et al., 2019), frequently triggers oxidative stress, endoplasmic reticulum (ER) stress, irradiation, infection, and interaction of the pro-apoptosis genes. Thus, Sirt3 upregulation and JNK pathway inhibition via YAP activation protect BV-2 cells from excessive apoptosis and relieve severe inflammation symptoms in the neuroinflammation microenvironment. Interestingly, reactivation of the JNK pathway activity significantly attenuates the beneficial effects exerted by YAP (Yang et al., 2019), whereas high YAP expression inhibits JNK pathway activity, which may form dynamic circulation stability in vivo. Apart from those mentioned above, YAP can also inhibit apoptosis and promote proliferation to replenish damaged brain tissue in BV-2 cells of the neuroinflammation microenvironment (Biernacki et al., 2018; Figure 2). Meanwhile, caspase 9 and caspase 12, respectively restored to normal levels via the upregulation of the YAP level, which can attenuate the alternations of mitochondrial damage and ER stress (Gandarillas et al., 2018; Figure 2). Consequently, YAP of BV-2 cells can exert corresponding defenses to cope with the neuroinflammation microenvironment in physiological conditions.
Purkinje cells are an important part of the signal transmission (Fang et al., 2018), and dendritic dysfunction can cause the related signal transmission to be blocked. The current study found that the YAP signaling pathway can maintain the normal structure of the Purkinje cell dendrites, thereby promoting normal cerebellum morphology and motor coordination function (Liu et al., 2017). YAP participates in the regulation of transforming growth factor-β (TGF-β)/Smad signaling (Ferrigno et al., 2002), mainly interacts with Smad7, and inhibits TGF-β receptor activity. Accordingly, the protein phosphatase 2A (PP2A) is regulated via TGF-β dephosphorylation and downregulation of p70s6k (Petritsch et al., 2000), which can also be phosphorylated by mTORC1 (Figure 2). Thus, the complex of mTORC1 and p70s6k, which is defined as one of the significant regulatory regulators of dendritic arbor development (Moser et al., 1997), is required for dendritic arbor development (Urbanska et al., 2012), including the width and number of dendritic branching point (Rojek et al., 2019). In addition to the above, the downstream effectors of mTORC2, which are essential to the growth of dendritic trees, are related to Akt. Even more surprising is that mTORC2 kinase phosphorylates AMOTL2, which is part of the Hippo pathway and promotes YAP signaling (Artinian et al., 2015). Of note, mTORC1/p70s6k and mTORC2/Akt are not separated from each other; in contrast, they jointly control the dendritic arbor morphology of the cerebellum, which is mainly that mTORC1/p70s6k drives mTORC2/Akt to work (Urbanska et al., 2012) and mTORC2/Akt can also influence mTORC1/p70s6k, whereas the detailed interaction between mTORC1/p70s6k and mTORC2/Akt needs to be further explored.
The formation of vascular networks in embryos requires angiogenesis, including sprouting angiogenesis and intussusception angiogenesis (Azad et al., 2019). Of note, the YAP expression is dynamic and widespread throughout the whole development of blood vessels. In the early stages of embryos, YAP/TAZ is predominantly located in the nucleus, whereas it is located in the cytoplasm mostly at the later period. During angiogenesis development, YAP/TAZ activity is controlled by vascular endothelial growth factor (VEGF). vascular endothelial growth factor is involved in angiogenesis, microvascular permeability, and mitosis and survival of endothelial cells (Azad et al., 2019), which may be positively correlated with PI3K and MAPK activities. vascular endothelial growth factor receptor activated by blinding to the ligand can trigger PI3K and MAPK signaling (Figure 2), which have been identified as the major inducible genes of YAP/TAZ activation. Subsequently, a large number of transcription factors, such as CYR61 and CTGF, can modulate the shape and behavior of endothelial cells associated with YAP/TAZ (Azad et al., 2019). The absence of YAP/TAZ causes major defects in retinal vascular development, including a 21% reduction in radial dilation, a 26% decrease in capillary density, and a 55% decrease in branch frequency. During vascular regression, YAP/TAZ is activated and promotes actin polymerization and CTGF induction (Brigstock, 2002; Jiang et al., 2004; Chaqour and Goppelt-Struebe, 2006; Nagasawa-Masuda and Terai, 2017), which indicates that YAP/TAZ may relieve or even restore vascular degeneration via upregulating the transcription level. Intriguingly, YAP/TAZ in the cytoplasm triggers proliferation and migration of vascular endothelial cells via regulating the Rho family GTPase CDC42 activity (Sakabe et al., 2017; Figure 2). Thus, YAP/TAZ in the endothelium is required for vascular growth, branching, and regularity of the vascular network.
Also, YAP interacts with interleukin 6 (IL-6)-driven signal trans-activator transcription 3 (STAT3; He et al., 2018) and enhances the expression of angiopoietin 2 (Ang-2), both of which trigger blood vessels generated, especially in the retina. In addition to the above, YAP/TAZ can also regulate non-endothelial cells during angiogenesis development (Azad et al., 2019), such as pericytes, edema, diabetic retinopathy, and embryonic lethality due to overdilation and blood loss of blood vessels (Bergers and Song, 2005).
After in-depth research, Hippo-YAP, which is defined as the core target of certain central nervous system diseases (Liu et al., 2017; Mueller et al., 2018; Rojek et al., 2019), usually transfers from the cytoplasm that binds to TEAD to the nucleus. The binding method is that the end of N YAP interacts with the spherical structure of the C-terminal of TEAD (four α-helices around β-sheets; Chen et al., 2010; Tian et al., 2010). However, in certain central nervous system diseases, the cytoplasmic localization of YAP is widely distributed through interaction with certain specific proteins (14-3-3 protein). Thus, in various central nervous system diseases, the location of YAP and interaction with protein are ultimately different. Tumor proliferation is considered to be a prominent feature in glioma, and at the same time, Hippo-YAP expression is upregulated. Of note, YAP is related to the mutant p53, which enhances the proliferation and transcriptional activity of the tumor. The correlation between p53 and YAP can be mediated via the control of the WASP interaction protein (WIP; Zhao et al., 2007; Rivas et al., 2018), which is also regulated by the PI3K–Akt pathway. Additionally, neurofibromatosis 2 (NF2) inhibits YAP nuclear translocation and triggers the deletion of transcription factors, thereby increasing invasiveness and affecting the SATA5A–SOCS2–SATA3 pathway (Sen et al., 2012; Lee et al., 2016). In subarachnoid hemorrhage (SAH), YAP usually upregulates the expressions of Nrg1β1, ErbB4, Mst1, and PIK3CB and inhibits the cytoplasmic retention of NF-κB and the activity and degradation of MMP-9 tight junction proteins (Yan et al., 2017; Qu et al., 2018). In Huntington’s disease (HD), a mutant Huntington gene is observed, which manifests a repeat of the CAG sequence (Mueller et al., 2018). Due to the activation of Mst1 in HD, the phosphorylation of YAP increases, which induces mutations in the Huntington gene via excitotoxicity and calcium signal disturbances, mitochondrial dynamics changes, transcriptional interference, cytoskeletal disruption, and improper protein processing (Morrison, 2009; Mueller et al., 2018). Besides, in neurodegenerative diseases such as HD and Alzheimer’s disease (AD), ballooning cell death (BCD) and transcriptional repression-induced atypical cell death (TRIAD) both occur with the interaction between YAP and p73 (Pardo et al., 2006). In AD, the amyloid-β (Aβ) peptide aggregation has been proven to be a prominent feature of the pathological process. More significantly, the disease is positively correlated with the YAP/p73/Bax-mediated apoptosis and intracellular Aβ-induced neuron necrosis (Cosentino et al., 2009; Tanaka et al., 2020; Table 2).
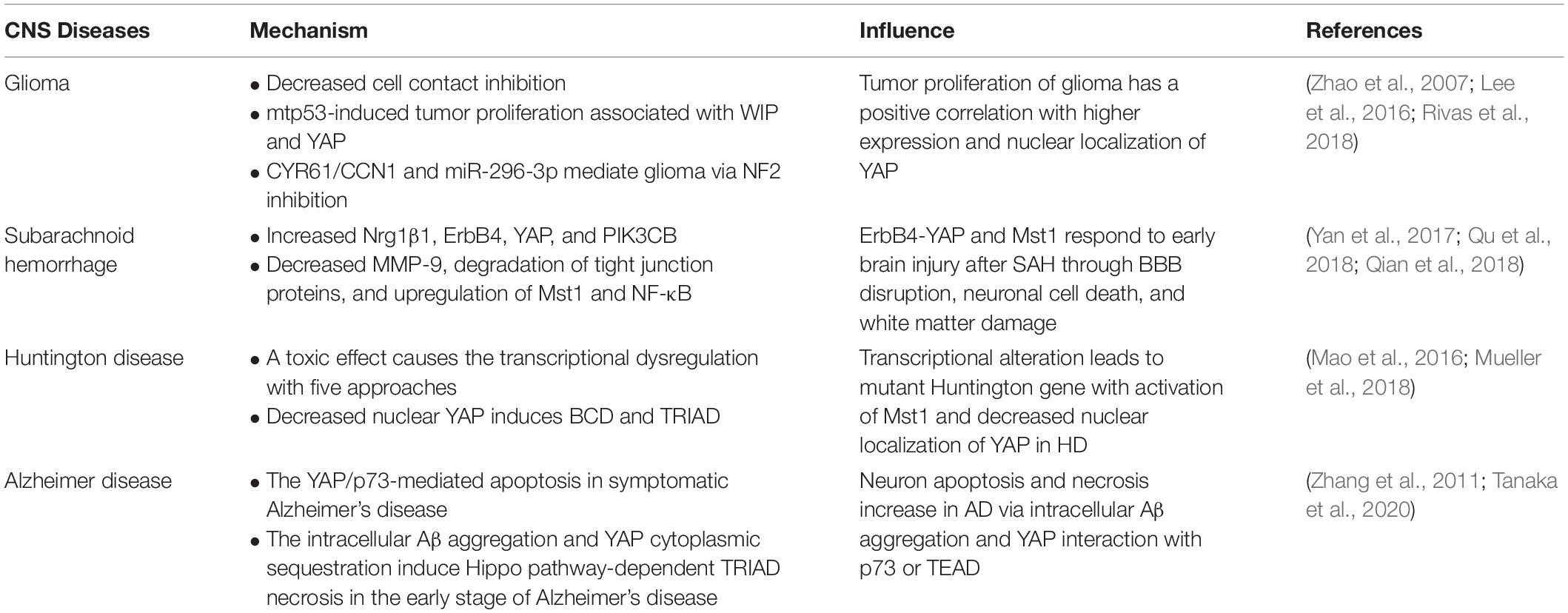
Table 2. Summary of the regulation of YAP in central nervous system diseases.
Yes-associated protein is a proline-rich phosphoprotein with two subtypes of splicing, namely, YAP1 and YAP2, mainly synthesized by the YAP1 gene. Due to this gene sequence, the subcellular localization of YAP1 is similar to YAP and has basic characteristics with YAP. In addition to the above characteristic, previous studies have revealed that increased nuclear localization of YAP can be observed in different types of cancer tissues (Zhao et al., 2007). More interestingly, YAP/TAZ activation can induce epithelial–mesenchymal transition (EMT), cell proliferation, and anti-apoptotic programs through binding to TEAD, thereby promoting the occurrence and development of tumors (Lei et al., 2008; Nishio et al., 2013; Piccolo et al., 2014). Besides, it is believed that the activation of YAP transport into the nucleus is closely related to the formation of gliomas. What’s more, the expression of Hippo signaling pathway molecules LATs2 and Mst2 and Merlin can cause cytoplasmic translocation of YAP, while phosphorylation of LATs2 significantly enhances the effect of YAP translocation (Zhao et al., 2007). Of note, effective regulation of YAP intercellular transport between cytoplasm and nucleus is essential for normal cellular development, controlling cell proliferation, organ size, and cancer development (Zeng and Hong, 2008; Harvey et al., 2013).
As a transcriptional coactivator, YAP is usually associated with oncogenes, and in many types of human cancers, elevated or overactivated expression of YAP has been observed (Bleeker et al., 2012). Glioma derives from glial cells in the brain, which can keep neurons in proper position and perform good functions. It is the most common type of tumor with about 30% occurrence ratio in central nervous system tumors and 80% occurrence ratio in malignant brain tumors (Lau et al., 2008; Goodenberger and Jenkins, 2012). Zhao et al. (2007) revealed that YAP overexpression caused the cells to fail to exit the cell cycle at confluence and to uninterruptedly grow to a higher density. At the same time, the dominant-positive YAP leads to cell transformation and escapes the inhibition of cell junctions that fundamentally destroys the limits of splitting and proliferation, which makes it difficult to control cell division and proliferation that triggers excessive cell growth and uncontrolled tumor progression (Liu and Wang, 2015) and even augments their capacity to invade host tissues and metastasize (Ribatti, 2017). However, YAP is inhibited by Merlin, which has been implicated in mediating cell contact inhibition (Okada et al., 2005; Zhao et al., 2007). In Salvador (Sav)-knockout cancer cell lines, inhibition of YAP function can restore contact inhibition in human cancer cell lines. Indeed, the loss of cell contact inhibition almost can be restored through blocking endogenous YAP function. Therefore, through the verification of YAP’s upregulation and downregulation, it can be concluded that the division and proliferation of glial cells are positively correlated with the expression of YAP.
On the one hand, YAP can enhance the tumor proliferative transcriptional activity of mutant p53 (mtp53) protein (Di Agostino et al., 2016) through the ARF-NDM2-P53 INK4a-RB pathway, which has been defined as a significant approach of promoting oncogenic capacity. In the regulation approach between YAP and mtp53 (Rivas et al., 2018; Figure 3), the transcription of YAP/TAZ can be influenced via actin, although it is not completely dependent on actin. More obviously, WIP also mediates the YAP transcription level. Thus, WIP exerts powerful control on YAP/TAZ in glioma. Additionally, the Akt, which is considered to be a major pro-tumor factor, can regulate WIP and is then mediated by PI3K depending on phosphoinositide-dependent kinase-1 (PDK1) and mammalian target of rapamycin complex 2 (mTORC2), respectively (Yang et al., 2004; Figure 3). So Akt plays an essential role in the brand-new WIP-YAP/TAZ-mtp53 pathway (Rivas et al., 2018). However, the precise interaction mechanism between WIP and Akt is still confusing. Intriguingly, both RAC and PAK also regulate WIP besides Akt (Rivas et al., 2018).
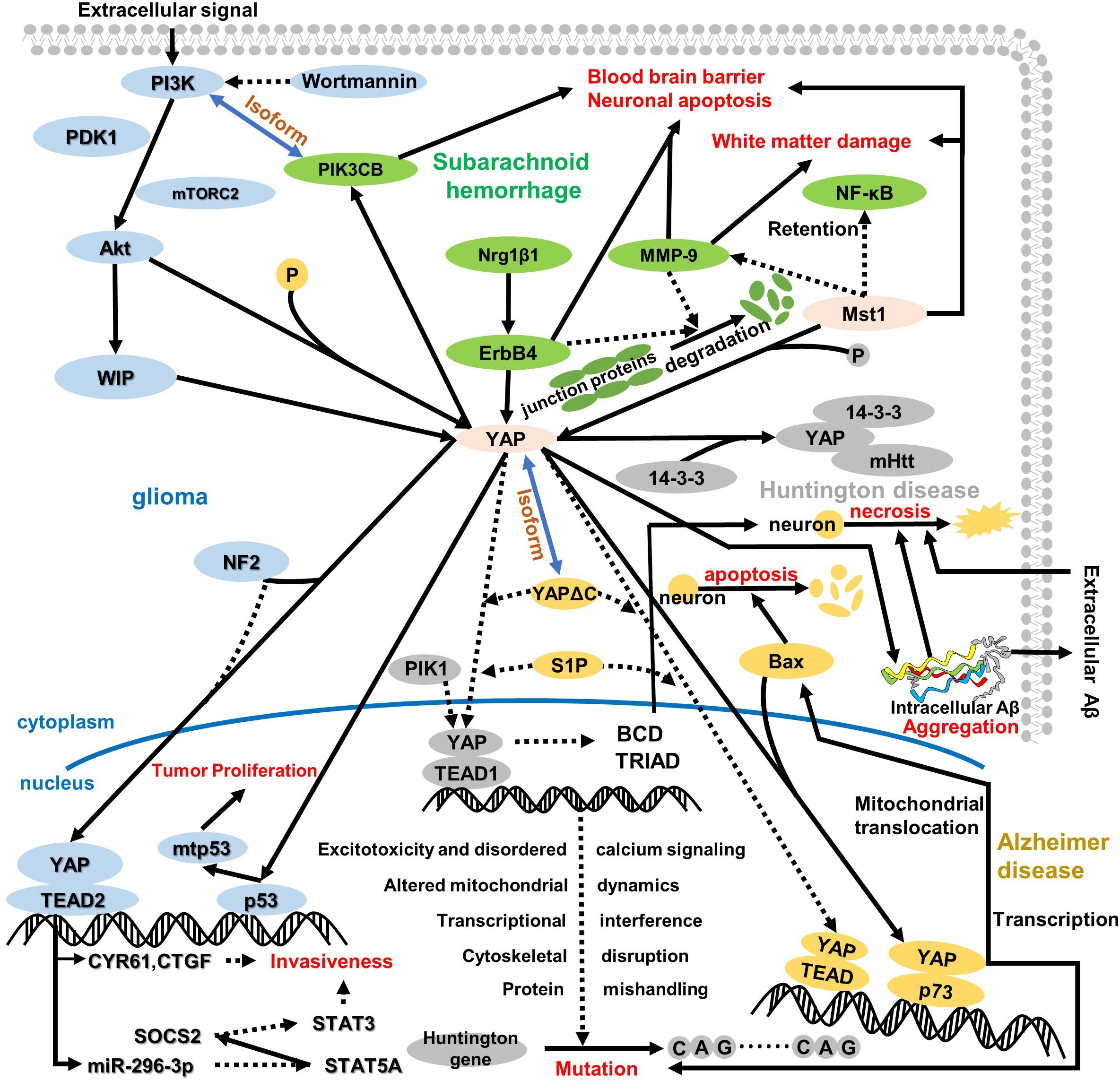
Figure 3. An overview of the role of YAP in the cytoplasm and nucleus regarding central nervous system disease. YAP plays a pivotal role in the whole pathologic process, which depends on its location and pathway. In glioma, YAP is regulated by WIP to induce p53 mutation in the nucleus, further resulting in tumor proliferation and attenuating contact cell inhibition. Of note, WIP expression is regulated via the P13K/Akt pathway, which is assisted by some regulatory proteins, like mTORC2 and PDK1. Meanwhile, NF2 suppresses the interaction between YAP and TEAD2, which triggers transcription target gene CYR61 and CTGF expressions and mediates miR-296-3p regulation. Both CYR61/CTGF and miR-296-3p can inhibit glioma invasiveness, whereas the invasiveness for miR-296-3p is fulfilled by decreased SATA3 and SATA5A and increased SOCS2. After subarachnoid hemorrhage, ErbB4 and Mst1 could respond to the BBB disruption, neuronal apoptosis, and white matter damage. One pathway regarding ErbB4 is upregulation of Nrg1β1, ErbB4, and PIK3CB; another pathway on Mst1 is the degradation of tight junction protein and inhibition of MMP-9 and NF-κB. Intriguingly, the isoform relationship between PI3K and PIK3CB connects glioma and subarachnoid hemorrhage. Additionally, HD is closely related to AD as a neurodegenerative disease. In HD, both decreased interaction between YAP and TEAD1 and increased YAP phosphorylation induce Huntington gene mutation. In the nucleus, the downregulation of YAP/TEAD1 stimulates CAG sequence repetition via excitotoxicity and disordered calcium signaling, altered mitochondrial dynamics, transcriptional interference, cytoskeletal disruption, and protein mishandling. Moreover, the decreased nuclear location of YAP inhibits BCD and TRIAD in cells, which stimulates neuro-necrosis. More interestingly, the mutant Huntington gene interacting with the YAP 14-3-3 complex aggravates the original pathologic symptom. In AD, YAP remarkably reduces interaction with TEAD and augments the combination with p73 that triggers Bax expression to aggravate neuron apoptosis, and YAP/p73 also aggravates the mutation in HD. Moreover, the cytoplasm retention of YAP and TRIAD/BCD mediates intracellular Aβ aggregation further, which results in neuron cell necrosis different from neuron apoptosis. Incidentally, intracellular Aβ aggregation transports outside to accumulate extracellular Aβ aggregation during the late period in AD, and extracellular Aβ aggregation can also result in neuro-necrosis with non-cell-autonomous approaches.
On the other hand, the tumor suppressor NF2, a core molecule governing cell survival, motility, and invasiveness, attenuates the proliferation of glioma cells in the central nervous system (Lau et al., 2008). Of note, NF2 can induce the phosphorylation of YAP, inhibit YAP nuclear translocation, and also bind to many transmembrane receptors and intercellular proteins (Stamenkovic and Yu, 2010), like CD44, which usually promotes phosphorylation/inactivation of NF2. Meanwhile, the deletion or silencing of NF2 causes increased invasiveness of glioblastoma multiforme cells in a YAP- and TEAD2-dependent manner. When the NF2 is knocked out, YAP shifts to the nuclear rather than cytoplasm retention as the normal pathway; subsequently, the YAP target genes are highly expressed (Lee et al., 2016), such as neuronal growth regulator 1 (NEGR1), cysteine-rich angiogenic inducer 61 (CYR61/CCN1), and connective tissue growth factor (CTGF/CCN2). Among them, CYR61/CCN1 and CTGF/CCN2 have been demonstrated to be involved in the invasiveness of glioma cells, which is consistent with the research result that knockdown or silencing of CYR61/CCN1 and CTGF/CCN2 hampers the invasive phenotype (Lee et al., 2016; Figure 3). Additionally, miRNA has also been defined to control the invasiveness of glioma, especially miR-296-3p associated with increased phosphorylated STAT3 level and decreased expressions of STAT5A and SOCS2 (suppressor of cytokine signaling 2; Sen et al., 2012; Figure 3). Indeed, the regulatory functions of CYR61/CCN1, CTGF/CCN2, and miRNA are not separate; reversely, they are correlated with each other. Thus, NF2 controls the invasiveness of glioma through YAP-dependent expression of CYR61/CCN1 and miR-296-3p.
In addition to the information mentioned above, YAP also enhances the invasion and migration of glioma cells by regulating the expression and alterations of Twist and N-cadherin which could promote the transition from epithelial cells to mesenchymal cells (Goodenberger and Jenkins, 2012; Ouyang et al., 2020).
SAH is a fatal disease with high mortality and poor prognosis (Connolly et al., 2012). It is identified as a subtype of hemorrhage stroke caused by the rupture of damaged blood vessels at the bottom of the brain or on the surface of the brain (van Lieshout et al., 2018). Some diseases usually are a positive correlation with SAHs, such as aneurysm and cerebrovascular malformation. More significantly, after SAH, both blood–brain barrier (BBB) disruption and neuronal apoptosis contribute to the pathogenesis of early brain injury (Yan et al., 2017). Meanwhile, after SAH, the expression levels of ErbB4 (EGFR family member v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 4) were notably increased, which has been demonstrated as the pivotal mechanism of the regulation of neurite outgrowth, axonal guidance, and synaptic signaling (Shamir et al., 2012). In addition to ErbB4, Nrg1β1, YAP, and PIK3CB are all involved in early brain injury regulation within 72 h after SAH. It is worth noting that a large number of studies have indicated that ErbB4 and its downstream YAP/PIK3CB signaling pathway exert a neuroprotective effect in early brain injury after SAH. Additionally, acute white matter injury is also a crucial pathological process in early brain injury (Kummer et al., 2015; Wu et al., 2017), which is associated with the regulation of Mst1, NF-κB, MMP-9, and tight junction proteins.
After SAH, the trophic factor Nrg1β1 with an epidermal growth factor (EGF)-like domain, begins to increase as early as 3 h and peaks at about 72 h, whereas endogenous ErbB4 augments at 3 h and keeps stable subsequently (Yan et al., 2017). High-dosage Nrg1β1 treatment significantly augments neurobehavioral performance, alleviates brain swelling, and attenuates brain water content, all of which contribute to BBB permeability and integrity within 24 and 72 h after SAH. The ErbB4 expression is augmented via Nrg1β1 administration; simultaneously, both YAP and PIK3CB exert the same regulatory tendency. Activation of ErbB4 via Nrg1β1 treatment alleviates the neurobehavioral deficit and attenuates the brain water content and albumin extravasation within 24 and 72 h after SAH (Qian et al., 2018). Of note, increased expressions of YAP and its downstream PIK3CB collectively protect the positive effect of ErbB4 activation about the BBB after SAH. Indeed, BBB disruption causes brain edema and brain swelling, which in turn lead to adverse outcomes, like neuronal death and neurological deficits. Thus, decreased YAP expression induces brain injury after SAH via BBB disruption. In addition to the BBB, Nrg1β1-ErbB4-YAP-PIK3CB can alleviate neurologic dysfunction and reduce neuronal cell apoptosis, which is consistent with the research result of ErbB4-knockout neuronal cell death after SAH, and the specific mechanism of action is almost similar to the mechanism mentioned above. Besides the fundamental function of the Nrg1β1-ErbB4-YAP-PIK3CB pathway (Figure 3), ErbB4 can also preserve the endothelial barrier function and attenuate the expression of inflammatory genes (Xu et al., 2005; Lok et al., 2012), which deserve to proceed with in-depth exploration.
Compared with the regulation of ErbB4 and YAP in BBB destruction and neuronal cell death, Mst1 can mediate the NF-κB/MMP-9 pathway and tight junction protein to restore white matter damage in early brain injury after SAH (Figure 3). Mst1, the upstream factor of the YAP, has been demonstrated to exert fundamental function in many central nervous system diseases, like SAH, white matter damage, and BBB disruption (Zheng et al., 2010; Lee et al., 2013; Tavares et al., 2013; Yang et al., 2016). Some experimental studies have shown that Mst1 gene knockout can improve neurobehavior and affect the expression of p65NF-κB and MMP-9, as well as tight junction proteins, such as ZO-1, occludin, and claudin-5 (Qu et al., 2018). However, the comprehensive pathway response to early brain injury is that the increased phosphorylation level of Mst1, associated negatively with NF-κB regulation in the nucleus, is negatively correlated with MMP-9 activity, which contributes to the SAH-mediated white matter injury and BBB repair via alleviating the degradation of tight junction proteins from 12 to 72 h after SAH (Pan et al., 2017). Thus, Mst1 upregulation aggravates the BBB disruption, neuronal cell death, and neurological deficits via promoting the NF-κB/MMP-9 pathway and attenuating tight junction proteins.
Overall, during SAH, the BBB, white matter damage, neuronal apoptosis, and neurobehavioral defects play a role in early brain damage. In view of physiological homeostasis, the regulation of Nrg1β1, ErbB4, YAP, and PIK3CB all increased, while the regulation of Mst1, NF-κB, MMP-9, and tight junction protein increased early brain damage. During SAH, the variation of Hippo-YAP and Mst1 reached consistency, but the effect was completely opposite. More interestingly, the start times of Nrg1β1 and Mst1 are 3 and 12 h respectively, which is worth pondering.
HD is an autosomal dominant hereditary neurodegenerative disorder characterized by progressive dyskinesia, progressive cognitive decline, and mental disorders, usually within 15–20 years of diagnosis. The major manifestation of dyskinesia is chorea associated with degeneration or hereditary defects of GABAergic interneurons in the neostriatum and striatum atrophy (Lasker et al., 1987). Meanwhile, the main clinical manifestations of cognitive impairment are the decline of memory, attention, and language function (Paulsen et al., 2013), and mental disorders are principally emotional outbursts, depression, and anxiety. More significantly, the disease is almost caused by a mutation in the Huntington gene of chromosome 4, subsequently producing mutant proteins, which continuously accumulate in the central nervous system and finally intervene in the normal function of the nerve cell (Dayalu and Albin, 2015). Previous studies have indicated that the extended CAG repeat sequence in the Huntington gene, averaging 17–20 CAG repeats (Kremer et al., 1994), leads to an over-long polyglutamine extension near the N-terminus of the protein. Interestingly, the occurrence and development of HD are due to the CAG gene being repeated at least 40 times, which has been proven (Mueller et al., 2018). Therefore, mutations in the Huntington gene on chromosome 4 can cause CAG duplication, which can easily lead to HD.
Many research results have demonstrated that both activation of Mst1 and the decline of nuclear YAP expression lead to transcriptional dysregulation with the occurrence of HD (Mueller et al., 2018). Thus, the Mst1 activation and the YAP phosphorylation suppress the normal transcriptional function and induce the mutation of the Huntington gene, both of which contribute to the occurrence of HD. Due to cytoplasm retention of YAP, YAP frequently binds to the chaperone 14-3-3 regarded as an abundant protein in the brain before degradation (Morrison, 2009). Correspondingly, decreased YAP nuclear localization may translate to alterations in gene expression via reducing YAP/TEAD interactions. During HD, transcriptional dysregulation is a pivotal pathogenic mechanism (Cha, 2007; Glajch and Sadri-Vakili, 2015), just like the mutant Huntington gene. Of note, the cortex and striatum have been observed to have increased phosphorylated Mst1 and YAP levels, the combination of YAP and TEAD in the nucleus is decreased when transcriptional dysregulation occurs, and the alteration of Mst1 phosphorylation and YAP nuclear localization is almost basically consistent via LATs1/2-mediated activation (Mueller et al., 2018). In exactly the above alteration, the transcription disorder induces the mutation of the Huntington gene, especially CAG sequence repetition (Figure 3). More interestingly, with the disruption of the YAP/TEAD interaction, the transcriptional dysregulation is accompanied by a toxic effect during Huntington gene mutation, mainly the following five core mechanisms:
(1) Transcriptional interference. The mutant Huntington gene enters the nucleus and directly interferes with gene transcription, thereby affecting neurotransmitter receptors and ion channels (Hodges et al., 2006), as well as BDNF (Zuccato et al., 2001).
(2) Cytoskeletal disruption. The mutant Huntington gene affects the microtubule system, leading to failure of vesicle transport (Caviston and Holzbaur, 2009).
(3) Protein mishandling. The mutant Huntington gene may overwhelm the neuron’s ability to tag and clear degraded and misfolded proteins via the ubiquitin–proteasome (Wang et al., 2008) and autophagy–lysosome (Wong and Holzbaur, 2014) pathways.
(4) Altered mitochondrial dynamics. The mutant Huntington gene alters many mitochondria-associated messenger RNA levels (Shirendeb et al., 2011); reduces transcription of PGC-α, which itself is a pivotal transcriptional regulator to activate a series of important roles in mitochondrial structure and function (Weydt et al., 2006); and leads to a decline in ATP levels and oxidative stress (Mochel et al., 2012), which all cause brain metabolic defects and increased cerebrospinal fluid lactate levels (Koroshetz et al., 1997).
(5) Excitotoxicity and disordered calcium signaling. The mutant Huntington gene directly sensitizes NMDA receptors via the NR2B subunit, which results in excitotoxicity (Zeron et al., 2001). Meanwhile, NMDA results in excessive calcium influx by interacting with type 1 inositol triphosphate receptors, which all exert huge pressure on neurons and ultimately result in cell abnormity and apoptosis (Zeron et al., 2002).
Given the above mechanism regarding the mutant Huntington gene, a slightly integrated pathway can be summarized in which both Mst1 activation and YAP phosphorylation attenuate the combination of YAP and TEAD via reducing the nuclear translocation of YAP, which all induce transcriptional interference, cytoskeletal disruption, protein mishandling, altered mitochondrial dynamics, and excitotoxicity and disordered calcium signaling to cause the Huntington gene mutation, like the CAG repeat that easily occurs in HD.
In addition to the above mechanism, decreased nuclear YAP induces TRIAD (Pardo et al., 2006), which is similar to mutant-Huntington-mediated neuronal death termed BCD rather than the p73/YAP-induced apoptosis. Intriguingly, both YAP and YAPdeltaC can recover BCD and TRIAD via binding to TEAD1 that is validated as a target transcription in BCD (Mao et al., 2016). BCD has proven to be a type of necrotic cell death, similar to TRIAD, which expands the ER and destroys asymmetric cell bodies. Therefore, YAP nuclear translocation attenuates BCD in mutant Huntington by necessarily binding to TEAD1. Additionally, PIK1 has been certified to switch the partner of YAP from TEAD1 to p73, which forms the balance of TEAD/YAP-dependent necrosis and p73/YAP-dependent apoptosis (Mao et al., 2016; Figure 3). However, SIP can promote the combination of TEAD1 and YAP. Unfortunately, this theory has not yet been applied to HD maturely, but interventions for TRIAD and BCD may lead to the discovery of new therapeutic targets for HD.
Alzheimer’s disease is a common neurodegenerative disease in the central nervous system with progressive cognitive disorder and neurobehavioral disruption. Its pathological features are amyloid plaques, neurofibrillary tangles, and neuronal loss (Ikonomovic et al., 2008). Indeed, the deposition of Aβ peptide in the brain has been demonstrated as a pathological hallmark of AD, and it has also been validated to result in neurotoxicity and cell apoptosis in vivo and in vitro (Cancino et al., 2008; Lauren et al., 2009). Intriguingly, multiple types of previous experimental studies indicated that neuronal cell apoptosis was implicated in the pathogenesis of AD (Yuan and Yankner, 2000; Gupta et al., 2006). In addition to the Aβ peptide deposition, the pathogenesis of AD is closely associated with tau protein neurofibrillary tangles (Xia et al., 2017), neuroinflammatory mechanism theory (Kamat et al., 2016; Trovato Salinaro et al., 2018), and neurovascular damage theory and oxidative stress (Zhao and Zhao, 2013). In the following section, the relationship between pathological hallmarks of AD and YAP could be emphatically elaborated.
During symptomatic AD, YAP is implicated in the deposition of the Aβ peptide via an apoptotic medium, which differs from the necrosis in AD. To be precise, apoptosis plays a vital role in the body’s physiological survival, such as cell renewal and homeostasis, but it can also lead to diseases such as AD (Gupta et al., 2006; Cancino et al., 2008; Zhao et al., 2008c). As a key factor in inhibiting the apoptosis pathway, the nuclear YAP expression level of neurons during AD decreased significantly, and the cytoplasmic retention of YAP was severely phosphorylated. The retention of YAP in the cytoplasm usually reduces the interaction with TEAD1. If YAP is transferred from the cytoplasm to the nucleus, it can be combined with p73 to promote apoptosis and necrosis (Basu et al., 2003; Strano et al., 2005; Hoshino et al., 2006; Levy et al., 2008), rather than promoting cell proliferation, differentiation, and survival through TEAD interaction (Saucedo and Edgar, 2007; Zhao et al., 2008a, 2010b). Besides binding to p73, YAP maintains the protein stabilization to augment p73-mediated apoptosis. After the interaction between YAP and p73 in the nucleus, the complex stimulates pro-apoptotic gene Bax mitochondrial translocation rather than direct transcription (Zhang et al., 2011). Interestingly, the gene Bax is positively correlated with p73-induced mitochondrial translocation and neuron apoptosis (Figure 3).
However, in the early stage of AD, including preclinical AD, mild cognitive impairment, or ultra-early stage of AD, YAP deprivation mediates Hippo pathway-dependent necrosis via intracellular Aβ, which is similar to that induced by YAP cytoplasmic retention in mutant Huntington gene (Mao et al., 2016) and differs from the apoptosis belonging to type II cell death. Of note, during AD, numbers of intracellular Aβ and TEAD-YAP transcription are closely related to ER ballooning and the Hippo pathway-dependent TRIAD. Before ER ballooning, intracellular Aβs were increased, and TEAD-YAP transcriptional activity was reduced. Thus, intracellular Aβ aggregation is inversely correlated in the transcription of TEAD and YAP (Figure 3). In addition to the above in AD, the Hippo pathway-dependent TRIAD necrosis has been validated as a pivotal role in AD pathology, and the necrosis ultimately depends on the correlation of intracellular Aβ and YAP (Tanaka et al., 2020). Therefore, the approaches between YAP and necrosis could be elucidated as follows (Strano et al., 2005; Levy et al., 2008):
(1) Due to intracellular Aβ-mediated necrosis, vast quantities of cerebral neurons are remarkably decreased through the YAP retention during a cell-autonomous degradative process.
(2) Secondary cell damage is implicated in necrotic neurons via release of alarmins to expand bystander neuron degeneration during a non-cell-autonomous process.
(3) Extracellular Aβ, which is aggregated via the medium of intracellular Aβ, aggravates degeneration by another non-cell-autonomous approach.
Therefore, the Hippo pathway-dependent TRIAD necrosis and intracellular Aβ aggregation are induced by YAP deprivation, occurring most actively in the early stages of AD before extracellular Aβ aggregation.
More intriguingly, both S1P and YAPdeltaC have been validated as a candidate therapeutic strategy in human AD. Slices of results have revealed that S1P and YAPdeltaC reduced the extracellular Aβ burden and increased nuclear YAP/YAPdeltaC, which decreases ER instability, necrosis, and cognitive disorder. Thus, the repression of TEAD-YAP/YAPdeltaC-induced transcription could be rescued via S1P and YAPdeltaC under YAP sequestration into intracellular Aβ aggregates, which was not affected by the treatments. Additionally, due to Akt activation, the wortmannin, an inhibitor of PI3K upstream of Akt, could remarkably abolish Akt-induced protection from apoptosis Aβ25–35-treated cells (Figure 3).
Collectively, in AD, neuron cell death occurs throughout the whole degradative process, including apoptosis and necrosis, both of which are correlated with YAP and intracellular Aβ. During the apoptosis of AD, nuclear YAP binds to p73 which induces cell apoptosis to express the transcription gene Bax, which has been certified as a pivotal pro-apoptotic target (Mao et al., 2016). Additionally, in the early stage of AD, both intracellular Aβ aggregation and YAP sequestration are positively correlated with the Hippo pathway-dependent TRIAD neuron necrosis through about three expansive approaches (Tanaka et al., 2020). With the in-depth exploration, slices of experimental results have revealed that S1P, YAPdeltaC, and wortmannin may suppress Aβ aggregation and cell death via increasing YAP/YAPdeltaC nuclear translocation.
Collectively, YAP plays a pivotal role in the healthy and pathologic central nervous system. The role of YAP in the central nervous system is diverse due to its location and pathway with other proteins or genes, which are all based on cell proliferation, apoptosis, cell contact inhibition, and organ volume control. Thus, portions of increased YAP expression can reduce cell death, thereby ameliorating the original pathologic symptoms, like neuroinflammation, whereas parts of decreased expression aggravate adverse elements. In a healthy central nervous system, we unravel some correlation between healthy cells and YAP, such as NPC, BV-2 neuroglia cells, Purkinje cells, and endothelial cells. The specific function includes a balance between proliferation and apoptosis, the response to neuroinflammation with Sirt3 and JNK pathways, the smooth signal transmission with mTORC1/p70s6k and mTORC2/Akt regulation, and the formation of vascular networks with CTGF and CYR61. In addition to the healthy system, glioma, SAH, HD, and AD are all elucidated as part of CNS diseases. In glioma, the YAP expression is upregulated and implicated in the mutant p53, which enhances the tumor proliferative transcriptional activity. Besides, YAP nuclear translocation is inhibited through NF2 to aggravate tumor invasiveness related to miR-296-3p and CTGF/CYR61. In SAH, the YAP is positively correlated with the regulation of Nrg1β1, ErbB4, Mst1, and PIK3CB, whereas inversely with NF-κB cytoplasmic retention, MMP-9 activity, and degradation of tight junction proteins. Additionally, Huntington gene mutation, observed in HD, is induced via increased YAP phosphorylation and TRIAD/BCD-mediated cell death. Ultimately, the Aβ peptide aggregation has been validated as a remarkable characteristic of the AD pathology process, which is closely associated with the intracellular Aβ-induced neuron necrosis and YAP/p73-mediated apoptosis.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This study was supported by the Shanghai Key Lab of Forensic Medicine (No. KF1805), Zhejiang Provincial Natural Science Foundation of China (No. LQ19H090010), and Wenzhou Medical University Research Development Fund (No. QTJ17021).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Artinian, N., Cloninger, C., Holmes, B., Benavides-Serrato, A., Bashir, T., and Gera, J. (2015). Phosphorylation of the hippo pathway component AMOTL2 by the mTORC2 kinase promotes YAP signaling, resulting in enhanced glioblastoma growth and invasiveness. J. Biol. Chem. 290, 19387–19401. doi: 10.1074/jbc.m115.656587
Azad, T., Ghahremani, M., and Yang, X. (2019). The role of YAP and TAZ in angiogenesis and vascular mimicry. Cells 8:E407.
Basu, S., Totty, N. F., Irwin, M. S., Sudol, M., and Downward, J. (2003). Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 11, 11–23. doi: 10.1016/s1097-2765(02)00776-1
Bergers, G., and Song, S. (2005). The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 7, 452–464. doi: 10.1215/s1152851705000232
Biernacki, M., Ambrozewicz, E., Gegotek, A., Toczek, M., Bielawska, K., and Skrzydlewska, E. (2018). Redox system and phospholipid metabolism in the kidney of hypertensive rats after FAAH inhibitor URB597 administration. Redox Biol. 15, 41–50. doi: 10.1016/j.redox.2017.11.022
Bleeker, F. E., Molenaar, R. J., and Leenstra, S. (2012). Recent advances in the molecular understanding of glioblastoma. J. Neurooncol. 108, 11–27. doi: 10.1007/s11060-011-0793-0
Brigstock, D. R. (2002). Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis 5, 153–165.
Cancino, G. I., Toledo, E. M., Leal, N. R., Hernandez, D. E., Yevenes, L. F., Inestrosa, N. C., et al. (2008). STI571 prevents apoptosis, tau phosphorylation and behavioural impairments induced by Alzheimer’s beta-amyloid deposits. Brain 131(Pt 9), 2425–2442. doi: 10.1093/brain/awn125
Caviston, J. P., and Holzbaur, E. L. (2009). Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 19, 147–155. doi: 10.1016/j.tcb.2009.01.005
Cha, J. H. (2007). Transcriptional signatures in Huntington’s disease. Prog. Neurobiol. 83, 228–248. doi: 10.1016/j.pneurobio.2007.03.004
Chan, E. H., Nousiainen, M., Chalamalasetty, R. B., Schafer, A., Nigg, E. A., and Sillje, H. H. (2005). The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 24, 2076–2086. doi: 10.1038/sj.onc.1208445
Chaqour, B., and Goppelt-Struebe, M. (2006). Mechanical regulation of the Cyr61/CCN1 and CTGF/CCN2 proteins. FEBS J. 273, 3639–3649. doi: 10.1111/j.1742-4658.2006.05360.x
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464.
Chen, L., Chan, S. W., Zhang, X., Walsh, M., Lim, C. J., Hong, W., et al. (2010). Structural basis of YAP recognition by TEAD4 in the hippo pathway. Genes Dev. 24, 290–300. doi: 10.1101/gad.1865310
Connolly, E. S. Jr., Rabinstein, A. A., Carhuapoma, J. R., Derdeyn, C. P., Dion, J., Higashida, R. T., et al. (2012). Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 43, 1711–1737. doi: 10.1161/str.0b013e3182587839
Cosentino, M., Colombo, C., Mauri, M., Ferrari, M., Corbetta, S., Marino, F., et al. (2009). Expression of apoptosis-related proteins and of mRNA for dopaminergic receptors in peripheral blood mononuclear cells from patients with Alzheimer disease. Alzheimer Dis. Assoc. Disord. 23, 88–90. doi: 10.1097/wad.0b013e318184807d
Cui, Z., Ni, N. C., Wu, J., Du, G. Q., He, S., Yau, T. M., et al. (2018). Polypyrrole-chitosan conductive biomaterial synchronizes cardiomyocyte contraction and improves myocardial electrical impulse propagation. Theranostics 8, 2752–2764. doi: 10.7150/thno.22599
Dayalu, P., and Albin, R. L. (2015). Huntington disease: pathogenesis and treatment. Neurol. Clin. 33, 101–114.
Di Agostino, S., Sorrentino, G., Ingallina, E., Valenti, F., Ferraiuolo, M., Bicciato, S., et al. (2016). YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 17, 188–201. doi: 10.15252/embr.201540488
Fang, H., Wang, Z. H., Bu, Y. J., Yuan, Z. J., Wang, G. Q., Guo, Y., et al. (2018). Repeated inhalation of sevoflurane inhibits the information transmission of Purkinje cells and delays motor development via the GABAA receptor epsilon subunit in neonatal mice. Mol. Med. Rep. 17, 1083–1092.
Ferrigno, O., Lallemand, F., Verrecchia, F., L’Hoste, S., Camonis, J., Atfi, A., et al. (2002). Yes-associated protein (YAP65) interacts with Smad7 and potentiates its inhibitory activity against TGF-beta/Smad signaling. Oncogene 21, 4879–4884. doi: 10.1038/sj.onc.1205623
Gandarillas, A., Molinuevo, R., and Sanz-Gomez, N. (2018). Mammalian endoreplication emerges to reveal a potential developmental timer. Cell Death Differ. 25, 471–476. doi: 10.1038/s41418-017-0040-0
Glajch, K. E., and Sadri-Vakili, G. (2015). Epigenetic mechanisms involved in Huntington’s disease pathogenesis. J. Huntingtons Dis. 4, 1–15. doi: 10.3233/jhd-140134
Goodenberger, M. L., and Jenkins, R. B. (2012). Genetics of adult glioma. Cancer Genet. 205, 613–621. doi: 10.1016/j.cancergen.2012.10.009
Gupta, V. B., Hegde, M. L., and Rao, K. S. (2006). Role of protein conformational dynamics and DNA integrity in relevance to neuronal cell death in neurodegeneration. Curr. Alzheimer Res. 3, 297–309. doi: 10.2174/156720506778249452
Harvey, K. F., Zhang, X., and Thomas, D. M. (2013). The hippo pathway, and human cancer. Nat. Rev. Cancer 13, 246–257.
He, J., Bao, Q., Zhang, Y., Liu, M., Lv, H., Liu, Y., et al. (2018). Yes-associated protein promotes angiogenesis via signal transducer and activator of transcription 3 in endothelial cells. Circ. Res. 122, 591–605. doi: 10.1161/circresaha.117.311950
Hodges, A., Strand, A. D., Aragaki, A. K., Kuhn, A., Sengstag, T., Hughes, G., et al. (2006). Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 15, 965–977. doi: 10.1093/hmg/ddl013
Hoshino, M., Qi, M. L., Yoshimura, N., Miyashita, T., Tagawa, K., Wada, Y., et al. (2006). Transcriptional repression induces a slowly progressive atypical neuronal death associated with changes of YAP isoforms and p73. J. Cell Biol. 172, 589–604. doi: 10.1083/jcb.200509132
Ikonomovic, M. D., Klunk, W. E., Abrahamson, E. E., Mathis, C. A., Price, J. C., Tsopelas, N. D., et al. (2008). Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 131(Pt 6), 1630–1645. doi: 10.1093/brain/awn016
Jiang, W. G., Watkins, G., Fodstad, O., Douglas-Jones, A., Mokbel, K., and Mansel, R. E. (2004). Differential expression of the CCN family members Cyr61, CTGF and Nov in human breast cancer. Endocr. Relat. Cancer 11, 781–791. doi: 10.1677/erc.1.00825
Justice, R. W., Zilian, O., Woods, D. F., Noll, M., and Bryant, P. J. (1995). The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 9, 534–546. doi: 10.1101/gad.9.5.534
Kamat, P. K., Kalani, A., Rai, S., Swarnkar, S., Tota, S., Nath, C., et al. (2016). Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of Alzheimer’s disease: understanding the therapeutics strategies. Mol. Neurobiol. 53, 648–661. doi: 10.1007/s12035-014-9053-6
Koroshetz, W. J., Jenkins, B. G., Rosen, B. R., and Beal, M. F. (1997). Energy metabolism defects in Huntington’s disease and effects of coenzyme Q10. Ann. Neurol. 41, 160–165. doi: 10.1002/ana.410410206
Kremer, B., Goldberg, P., Andrew, S. E., Theilmann, J., Telenius, H., Zeisler, J., et al. (1994). A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 330, 1401–1406. doi: 10.1056/nejm199405193302001
Kriegstein, A., and Alvarez-Buylla, A. (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184. doi: 10.1146/annurev.neuro.051508.135600
Kummer, T. T., Magnoni, S., MacDonald, C. L., Dikranian, K., Milner, E., and Sorrell, J. (2015). Experimental subarachnoid haemorrhage results in multifocal axonal injury. Brain 138(Pt 9), 2608–2618. doi: 10.1093/brain/awv180
Lasker, A. G., Zee, D. S., Hain, T. C., Folstein, S. E., and Singer, H. S. (1987). Saccades in Huntington’s disease: initiation defects and distractibility. Neurology 37, 364–370.
Lau, Y. K., Murray, L. B., Houshmandi, S. S., Xu, Y., Gutmann, D. H., and Yu, Q. (2008). Merlin is a potent inhibitor of glioma growth. Cancer Res. 68, 5733–5742. doi: 10.1158/0008-5472.can-08-0190
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132. doi: 10.1038/nature07761
Lavado, A., Park, J. Y., Pare, J., Finkelstein, D., Pan, H., Xu, B., et al. (2018). The hippo pathway prevents YAP/TAZ-driven hypertranscription and controls neural progenitor number. Dev. Cell 47, 576–591.e8. doi: 10.1016/j.devcel.2018.09.021
Lee, H., Hwang, S. J., Kim, H. R., Shin, C. H., Choi, K. H., Joung, J. G., et al. (2016). Neurofibromatosis 2 (NF2) controls the invasiveness of glioblastoma through YAP-dependent expression of CYR61/CCN1 and miR-296-3p. Biochim. Biophys. Acta 1859, 599–611. doi: 10.1016/j.bbagrm.2016.02.010
Lee, J. K., Shin, J. H., Hwang, S. G., Gwag, B. J., McKee, A. C., Lee, J., et al. (2013). MST1 functions as a key modulator of neurodegeneration in a mouse model of ALS. Proc. Natl. Acad. Sci. U.S.A. 110, 12066–12071. doi: 10.1073/pnas.1300894110
Lei, Q. Y., Zhang, H., Zhao, B., Zha, Z. Y., Bai, F., Pei, X. H., et al. (2008). TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell. Biol. 28, 2426–2436. doi: 10.1128/mcb.01874-07
Levy, D., Adamovich, Y., Reuven, N., and Shaul, Y. (2008). Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 29, 350–361. doi: 10.1016/j.molcel.2007.12.022
Li, R., Xin, T., Li, D., Wang, C., Zhu, H., and Zhou, H. (2018). Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: the role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 18, 229–243. doi: 10.1016/j.redox.2018.07.011
Liu, P., Calvisi, D. F., Kiss, A., Cigliano, A., Schaff, Z., Che, L., et al. (2017). Central role of mTORC1 downstream of YAP/TAZ in hepatoblastoma development. Oncotarget 8, 73433–73447. doi: 10.18632/oncotarget.20622
Liu, Y. C., and Wang, Y. Z. (2015). Role of Yes-associated protein 1 in gliomas: pathologic and therapeutic aspects. Tumour Biol. 36, 2223–2227. doi: 10.1007/s13277-015-3297-2
Lok, J., Zhao, S., Leung, W., Seo, J. H., Navaratna, D., Wang, X., et al. (2012). Neuregulin-1 effects on endothelial and blood-brain-barrier permeability after experimental injury. Transl. Stroke Res. 3(Suppl. 1), S119–S124.
Mao, Y., Chen, X., Xu, M., Fujita, K., Motoki, K., Sasabe, T., et al. (2016). Targeting TEAD/YAP-transcription-dependent necrosis, TRIAD, ameliorates Huntington’s disease pathology. Hum. Mol. Genet. 25, 4749–4770.
McKinnon, P. J. (2017). Genome integrity and disease prevention in the nervous system. Genes Dev. 31, 1180–1194. doi: 10.1101/gad.301325.117
Mo, J. S., Park, H. W., and Guan, K. L. (2014). The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 15, 642–656. doi: 10.15252/embr.201438638
Mochel, F., Durant, B., Meng, X., O’Callaghan, J., Yu, H., Brouillet, E., et al. (2012). Early alterations of brain cellular energy homeostasis in Huntington disease models. J. Biol. Chem. 287, 1361–1370. doi: 10.1074/jbc.m111.309849
Morrison, D. K. (2009). The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol. 19, 16–23. doi: 10.1016/j.tcb.2008.10.003
Moser, B. A., Dennis, P. B., Pullen, N., Pearson, R. B., Williamson, N. A., Wettenhall, R. E., et al. (1997). Dual requirement for a newly identified phosphorylation site in p70s6k. Mol. Cell. Biol. 17, 5648–5655. doi: 10.1128/mcb.17.9.5648
Mueller, K. A., Glajch, K. E., Huizenga, M. N., Wilson, R. A., Granucci, E. J., Dios, A. M., et al. (2018). Hippo signaling pathway dysregulation in human Huntington’s disease brain and neuronal stem cells. Sci. Rep. 8:11355.
Nagasawa-Masuda, A., and Terai, K. (2017). Yap/Taz transcriptional activity is essential for vascular regression via Ctgf expression and actin polymerization. PLoS One 12:e0174633. doi: 10.1371/journal.pone.0174633
Nishio, M., Otsubo, K., Maehama, T., Mimori, K., and Suzuki, A. (2013). Capturing the mammalian Hippo: elucidating its role in cancer. Cancer Sci. 104, 1271–1277. doi: 10.1111/cas.12227
Okada, T., Lopez-Lago, M., and Giancotti, F. G. (2005). Merlin/NF-2 mediates contact inhibition of growth by suppressing recruitment of Rac to the plasma membrane. J. Cell Biol. 171, 361–371. doi: 10.1083/jcb.200503165
Ouyang, T., Meng, W., Li, M., Hong, T., and Zhang, N. (2020). Recent advances of the Hippo/YAP signaling pathway in brain development and glioma. Cell. Mol. Neurobiol. 40, 495–510. doi: 10.1007/s10571-019-00762-9
Pan, P., Zhang, X., Li, Q., Zhao, H., Qu, J., Zhang, J. H., et al. (2017). Cyclosporine A alleviated matrix metalloproteinase 9 associated blood-brain barrier disruption after subarachnoid hemorrhage in mice. Neurosci. Lett. 649, 7–13. doi: 10.1016/j.neulet.2017.03.050
Pardo, R., Colin, E., Regulier, E., Aebischer, P., Deglon, N., Humbert, S., et al. (2006). Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J. Neurosci. 26, 1635–1645. doi: 10.1523/jneurosci.3706-05.2006
Paulsen, J. S., Smith, M. M., Long, J. D., and investigators, P. H. (2013). Coordinators of the Huntington study G. Cognitive decline in prodromal Huntington disease: implications for clinical trials. J. Neurol. Neurosurg. Psychiatry 84, 1233–1239. doi: 10.1136/jnnp-2013-305114
Petritsch, C., Beug, H., Balmain, A., and Oft, M. (2000). TGF-beta inhibits p70 S6 kinase via protein phosphatase 2A to induce G(1) arrest. Genes Dev. 14, 3093–3101. doi: 10.1101/gad.854200
Piccolo, S., Dupont, S., and Cordenonsi, M. (2014). The biology of YAP/TAZ: hippo signaling and beyond. Physiol. Rev. 94, 1287–1312. doi: 10.1152/physrev.00005.2014
Qian, H., Dou, Z., Ruan, W., He, P., Zhang, J. H., and Yan, F. (2018). ErbB4 preserves blood-brain barrier integrity via the YAP/PIK3CB pathway after subarachnoid hemorrhage in rats. Front. Neurosci. 12:492. doi: 10.3389/fnins.2018.00492
Qu, J., Zhao, H., Li, Q., Pan, P., Ma, K., Liu, X., et al. (2018). MST1 suppression reduces early brain injury by inhibiting the NF-kappaB/MMP-9 pathway after subarachnoid hemorrhage in mice. Behav. Neurol. 2018:6470957.
Ribatti, D. (2017). A revisited concept: contact inhibition of growth. From cell biology to malignancy. Exp. Cell Res. 359, 17–19. doi: 10.1016/j.yexcr.2017.06.012
Rivas, S., Anton, I. M., and Wandosell, F. (2018). WIP-YAP/TAZ as a new pro-oncogenic pathway in glioma. Cancers 10:191. doi: 10.3390/cancers10060191
Rojek, K. O., Krzemien, J., Dolezyczek, H., Boguszewski, P. M., Kaczmarek, L., Konopka, W., et al. (2019). Amot and Yap1 regulate neuronal dendritic tree complexity and locomotor coordination in mice. PLoS Biol. 17:e3000253. doi: 10.1371/journal.pbio.3000253
Sakabe, M., Fan, J., Odaka, Y., Liu, N., Hassan, A., Duan, X., et al. (2017). YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc. Natl. Acad. Sci. U.S.A. 114, 10918–10923. doi: 10.1073/pnas.1704030114
Saucedo, L. J., and Edgar, B. A. (2007). Filling out the Hippo pathway. Nat. Rev. Mol. Cell Biol. 8, 613–621. doi: 10.1038/nrm2221
Sen, B., Peng, S., Woods, D. M., Wistuba, I., Bell, D., El-Naggar, A. K., et al. (2012). STAT5A-mediated SOCS2 expression regulates Jak2 and STAT3 activity following c-Src inhibition in head and neck squamous carcinoma. Clin. Cancer Res. 18, 127–139. doi: 10.1158/1078-0432.ccr-11-1889
Shamir, A., Kwon, O. B., Karavanova, I., Vullhorst, D., Leiva-Salcedo, E., Janssen, M. J., et al. (2012). The importance of the NRG-1/ErbB4 pathway for synaptic plasticity and behaviors associated with psychiatric disorders. J. Neurosci. 32, 2988–2997. doi: 10.1523/jneurosci.1899-11.2012
Shirendeb, U., Reddy, A. P., Manczak, M., Calkins, M. J., Mao, P., Tagle, D. A., et al. (2011). Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum. Mol. Genet. 20, 1438–1455. doi: 10.1093/hmg/ddr024
Sorrentino, G., Mantovani, F., and Del Sal, G. (2018). The stiff RhoAd from mevalonate to mutant p53. Cell Death Differ. 25, 645–647. doi: 10.1038/s41418-018-0091-x
Stamenkovic, I., and Yu, Q. (2010). Merlin, a “magic” linker between extracellular cues and intracellular signaling pathways that regulate cell motility, proliferation, and survival. Curr. Protein Pept. Sci. 11, 471–484. doi: 10.2174/138920310791824011
Strano, S., Monti, O., Pediconi, N., Baccarini, A., Fontemaggi, G., Lapi, E., et al. (2005). The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Mol. Cell 18, 447–459. doi: 10.1016/j.molcel.2005.04.008
Tanaka, H., Homma, H., Fujita, K., Kondo, K., Yamada, S., Jin, X., et al. (2020). YAP-dependent necrosis occurs in early stages of Alzheimer’s disease and regulates mouse model pathology. Nat. Commun. 11:507.
Tavares, I. A., Touma, D., Lynham, S., Troakes, C., Schober, M., Causevic, M., et al. (2013). Prostate-derived sterile 20-like kinases (PSKs/TAOKs) phosphorylate tau protein and are activated in tangle-bearing neurons in Alzheimer disease. J. Biol. Chem. 288, 15418–15429. doi: 10.1074/jbc.m112.448183
Tian, W., Yu, J., Tomchick, D. R., Pan, D., and Luo, X. (2010). Structural and functional analysis of the YAP-binding domain of human TEAD2. Proc. Natl. Acad. Sci. U.S.A. 107, 7293–7298. doi: 10.1073/pnas.1000293107
Trovato Salinaro, A., Pennisi, M., Di Paola, R., Scuto, M., Crupi, R., Cambria, M. T., et al. (2018). Neuroinflammation and neurohormesis in the pathogenesis of Alzheimer’s disease and Alzheimer-linked pathologies: modulation by nutritional mushrooms. Immun. Ageing 15:8.
Urbanska, M., Gozdz, A., Swiech, L. J., and Jaworski, J. (2012). Mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) control the dendritic arbor morphology of hippocampal neurons. J. Biol. Chem. 287, 30240–30256. doi: 10.1074/jbc.m112.374405
van Lieshout, J. H., Dibue-Adjei, M., Cornelius, J. F., Slotty, P. J., Schneider, T., Restin, T., et al. (2018). An introduction to the pathophysiology of aneurysmal subarachnoid hemorrhage. Neurosurg. Rev. 41, 917–930.
Vassilev, A., Kaneko, K. J., Shu, H., Zhao, Y., and DePamphilis, M. L. (2001). TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 15, 1229–1241. doi: 10.1101/gad.888601
Wang, J., Wang, C. E., Orr, A., Tydlacka, S., Li, S. H., and Li, X. J. (2008). Impaired ubiquitin-proteasome system activity in the synapses of Huntington’s disease mice. J. Cell Biol. 180, 1177–1189. doi: 10.1083/jcb.200709080
Wang, Z., Lu, W., Zhang, Y., Zou, F., Jin, Z., and Zhao, T. (2019). The Hippo pathway and viral infections. Front. Microbiol. 10:3033. doi: 10.3389/fmicb.2019.03033
Weydt, P., Pineda, V. V., Torrence, A. E., Libby, R. T., Satterfield, T. F., Lazarowski, E. R., et al. (2006). Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 4, 349–362. doi: 10.1016/j.cmet.2006.10.004
Wong, Y. C., and Holzbaur, E. L. (2014). The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 34, 1293–1305. doi: 10.1523/jneurosci.1870-13.2014
Wu, Y., Peng, J., Pang, J., Sun, X., and Jiang, Y. (2017). Potential mechanisms of white matter injury in the acute phase of experimental subarachnoid haemorrhage. Brain 140:e36. doi: 10.1093/brain/awx084
Xia, C., Makaretz, S. J., Caso, C., McGinnis, S., Gomperts, S. N., Sepulcre, J., et al. (2017). Association of in vivo [18F]AV-1451 tau PET imaging results with cortical atrophy and symptoms in typical and atypical Alzheimer disease. JAMA Neurol. 74, 427–436.
Xu, Z., Ford, G. D., Croslan, D. R., Jiang, J., Gates, A., Allen, R., et al. (2005). Neuroprotection by neuregulin-1 following focal stroke is associated with the attenuation of ischemia-induced pro-inflammatory and stress gene expression. Neurobiol. Dis. 19, 461–470. doi: 10.1016/j.nbd.2005.01.027
Yan, F., Tan, X., Wan, W., Dixon, B. J., Fan, R., Enkhjargal, B., et al. (2017). ErbB4 protects against neuronal apoptosis via activation of YAP/PIK3CB signaling pathway in a rat model of subarachnoid hemorrhage. Exp. Neurol. 297, 92–100. doi: 10.1016/j.expneurol.2017.07.014
Yang, L., Dan, H. C., Sun, M., Liu, Q., Sun, X. M., Feldman, R. I., et al. (2004). Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 64, 4394–4399. doi: 10.1158/0008-5472.can-04-0343
Yang, Y., Gong, Z., and Wang, Z. (2019). Yes-associated protein reduces neuroinflammation through upregulation of Sirt3 and inhibition of JNK signaling pathway. J. Recept. Signal Transduct. Res. 39, 479–487. doi: 10.1080/10799893.2019.1705339
Yang, Y., Wang, L., Wu, Y., Su, D., Wang, N., Wang, J., et al. (2016). Tanshinol suppresses inflammatory factors in a rat model of vascular dementia and protects LPS-treated neurons via the MST1-FOXO3 signaling pathway. Brain Res. 1646, 304–314. doi: 10.1016/j.brainres.2016.06.017
Zeng, Q., and Hong, W. (2008). The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell 13, 188–192. doi: 10.1016/j.ccr.2008.02.011
Zeron, M. M., Chen, N., Moshaver, A., Lee, A. T., Wellington, C. L., Hayden, M. R., et al. (2001). Mutant huntingtin enhances excitotoxic cell death. Mol. Cell. Neurosci. 17, 41–53. doi: 10.1006/mcne.2000.0909
Zeron, M. M., Hansson, O., Chen, N., Wellington, C. L., Leavitt, B. R., Brundin, P., et al. (2002). Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron 33, 849–860. doi: 10.1016/s0896-6273(02)00615-3
Zhang, H., Wu, S., and Xing, D. (2011). YAP accelerates Abeta(25-35)-induced apoptosis through upregulation of Bax expression by interaction with p73. Apoptosis 16, 808–821. doi: 10.1007/s10495-011-0608-y
Zhao, B., Lei, Q. Y., and Guan, K. L. (2008a). The Hippo-YAP pathway: new connections between regulation of organ size and cancer. Curr. Opin. Cell Biol. 20, 638–646. doi: 10.1016/j.ceb.2008.10.001
Zhao, B., Ye, X., Yu, J., Li, L., Li, W., Li, S., et al. (2008b). TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971. doi: 10.1101/gad.1664408
Zhao, L., Qian, Z. M., Zhang, C., Wing, H. Y., Du, F., and Ya, K. (2008c). Amyloid beta-peptide 31-35-induced neuronal apoptosis is mediated by caspase-dependent pathways via cAMP-dependent protein kinase A activation. Aging Cell 7, 47–57. doi: 10.1111/j.1474-9726.2007.00352.x
Zhao, B., Li, L., Lei, Q., and Guan, K. L. (2010a). The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 24, 862–874. doi: 10.1101/gad.1909210
Zhao, B., Li, L., Tumaneng, K., Wang, C. Y., and Guan, K. L. (2010b). A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 24, 72–85. doi: 10.1101/gad.1843810
Zhao, B., Wei, X., Li, W., Udan, R. S., Yang, Q., Kim, J., et al. (2007). Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761. doi: 10.1101/gad.1602907
Zhao, Y., and Zhao, B. (2013). Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013:316523.
Zheng, X., Xu, C., Di Lorenzo, A., Kleaveland, B., Zou, Z., Seiler, C., et al. (2010). CCM3 signaling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. J. Clin. Invest. 120, 2795–2804. doi: 10.1172/jci39679
Keywords: central nervous system diseases, Hippo signaling pathway, YAP, effect, mechanism
Citation: Jin J, Zhao X, Fu H and Gao Y (2020) The Effects of YAP and Its Related Mechanisms in Central Nervous System Diseases. Front. Neurosci. 14:595. doi: 10.3389/fnins.2020.00595
Received: 25 February 2020; Accepted: 15 May 2020;
Published: 26 June 2020.
Edited by:
Mohammad Badruzzaman Khan, Augusta University, United StatesReviewed by:
Hyun Woo Park, Yonsei University, South KoreaCopyright © 2020 Jin, Zhao, Fu and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan Gao, d3lkZ2FveXVhbkAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.