
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Neurosci., 12 May 2020
Sec. Neuroenergetics and Brain Health
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.00453
This article is part of the Research TopicCutting-Edge Approaches for CNS Protection and Repair: Focus on Vascular and Degenerative DisordersView all 83 articles
 Koji Tanaka1
Koji Tanaka1 Shoji Matsumoto2
Shoji Matsumoto2 Takeshi Yamada3
Takeshi Yamada3 Ryo Yamasaki1Makoto Suzuki4
Ryo Yamasaki1Makoto Suzuki4 Mizuho A. Kido5
Mizuho A. Kido5 Jun-Ichi Kira1*
Jun-Ichi Kira1*Background and Purpose: In the acute phase of ischemia-reperfusion, hypoperfusion associated with ischemia and reperfusion in microvascular regions and disruption of the blood–brain barrier (BBB) contribute to post-ischemic brain injury. We aimed to clarify whether brain injury following transient middle cerebral artery occlusion (tMCAO) is ameliorated in Transient receptor potential vanilloid 4 knockout (Trpv4–/–) mice.
Methods: tMCAO was induced in wild-type (WT) and Trpv4–/– mice aged 8–10 weeks. Ischemia-induced lesion volume was evaluated by 2,3,5-triphenyltetrazolium chloride staining at 24 h post-tMCAO. Tissue water content and Evans blue leakage in the ipsilateral hemisphere and a neurological score were evaluated at 48 h post-tMCAO. Transmission electron microscopy (TEM) was performed to assess the morphological changes in microvasculature in the ischemic lesions at 6 h post-tMCAO.
Results: Compared with WT mice, Trpv4–/– mice showed reduced ischemia-induced lesion volume and reduced water content and Evans blue leakage in the ipsilateral hemisphere alongside milder neurological symptoms. The loss of zonula occludens-1 and occludin proteins in the ipsilateral hemisphere was attenuated in Trpv4–/– mice. TEM revealed that parenchymal microvessels in the ischemic lesion were compressed and narrowed by the swollen endfeet of astrocytes in WT mice, but these effects were markedly ameliorated in Trpv4–/– mice.
Conclusion: The present results demonstrate that TRPV4 contributes to post-ischemic brain injury. The preserved microcirculation and BBB function shortly after reperfusion are the key neuroprotective roles of TRPV4 inhibition, which represents a promising target for the treatment of acute ischemic stroke.
Acute ischemic stroke secondary to large artery occlusion is a common and devastating condition that results in death or long-term disability in a high proportion of patients. Recent advances in revascularization therapies, including mechanical thrombectomy, have enabled rapid and effective recanalization for large artery occlusions in patients with acute ischemic stroke (Goyal et al., 2016). However, despite the high rates of early recanalization, some patients still experience poor outcomes. In these cases, lack of reperfusion due to blockade of the microcirculation leading to hypoperfusion associated with ischemia and reperfusion and disruption of the blood–brain barrier (BBB) may contribute to post-ischemic brain injury (Molina and Alvarez-Sabín, 2009).
Transient receptor potential vanilloid 4 (TRPV4), a member of the TRP vanilloid subfamily, is a non-selective cation channel permeable to Ca2+, Na+, and Mg2+ ions (Ramsey et al., 2006). TRPV4 was first described as a cellular osmotic sensor that detects extracellular hypo-osmolarity, and was subsequently shown to be activated by multiple stimuli. TRPV4 is widely expressed in the central nervous system (Nilius and Voets, 2013), and its localization on astrocyte endfeet plays an essential role in the regulation of vascular tone in brain microvessels (Dunn et al., 2013; Filosa et al., 2013). Enrichment of TRPV4 and aquaporins at the endfeet of astrocytes ensures that even small changes in conditions, such as extracellular ion concentrations, result in astroglial swelling, volume regulation, and reorganization of downstream signaling pathways (Ryskamp et al., 2015; Iuso and Križaj, 2016). Pharmacological blockade of TRPV4 results in reduction of brain ischemia-induced lesion volume through inhibition of matrix metalloprotease (MMP) activation, thereby providing protection against BBB disruption following transient focal cerebral ischemia (Li et al., 2013; Jie et al., 2015a, b, 2016). Moreover, TRPV4 contributes to Ca2+ influx in both astrocytes and neurons and to extracellular glutamate accumulation during peri-infarct depolarizations (Rakers et al., 2017). These findings indicate that TRPV4 has a large influence on ischemia-induced brain injury and indicate that inhibition of TRPV4 has a neuroprotective role in acute ischemic stroke. However, a recent report demonstrated exacerbation of ischemia-induced brain edema formation in Trpv4 knockout (Trpv4–/–) mice (Pivonkova et al., 2018). Specifically, Trpv4–/– mice showed larger T2-hyperintensity lesions following transcranial permanent middle cerebral artery occlusion (pMCAO) compared with wild-type (WT) mice. Moreover, the report described a minimal involvement of TRPV4 in astrocyte volume regulation in vivo. Thus, the mechanism for the neuroprotective effects of TRPV4 in acute ischemic stroke remains inconclusive. Given this context, we aimed to investigate the differences in post-ischemic brain injury between Trpv4–/– and WT mice, and to clarify the mechanism for any identified differences using a transient MCAO (tMCAO) model in the C57BL/6N mouse strain.
The animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and all procedures were approved by the Animal Ethics Committee of Kyushu University (Approval Nos. A28-08-4, A30-138-2, 26-60, and 1-48).
Trpv4–/– mice were transferred from Jichi Medical University (Suzuki et al., 2003). C57BL/6N mice were purchased from Kyudo (Fukuoka, Japan). The Trpv4–/– mouse strain had been backcrossed with the C57BL/6N strain for >10 generations. Inbred adult male mice at 8–10 weeks of age (median [interquartile range] weight: 23.3 [21.7–24.9] g for WT mice and 24.1 [20.8–26.2] g for Trpv4–/– mice) were maintained in a temperature-controlled (22–25°C) specific pathogen-free animal facility on a 12-h/12-h light/dark cycle with free access to food and water.
tMCAO was performed using a previously described method with modifications (Engel et al., 2011). Mice were anesthetized with 1.5–2.0% isoflurane (Escain; Mylan Seiyaku, Tokyo, Japan) in 30% oxygen-mixed gas under spontaneous respiration. Focal cerebral ischemia was induced by a silicone-coated 6-0 monofilament (#602356; Doccol, Redlands, CA, United States) inserted through a small incision in the right common carotid artery and then advanced through the internal carotid artery to occlude the origin of the MCA. The filament was withdrawn to allow reperfusion of the ischemic hemisphere. Regional cerebral blood flow in the right MCA territory was monitored by laser Doppler flowmetry (Omegaflo FLO-C1; Omegawave, Tokyo, Japan) to confirm vessel occlusion, shown by a rapid and dramatic decrease in brain perfusion (mean reduction: 70–80%) that remained stable until reperfusion. Body temperature was maintained at 37°C based on the rectal temperature. In sham-operated animals, the same procedure was performed except for insertion of the intraluminal filament. The optimal time of tMCAO was determined by a preliminarily experiment in which tMCAO was induced in WT mice for 15, 30, or 45 min. The lesion size at 24 h post-tMCAO became larger and more stable while the mortality rate increased with longer tMCAO induction (≈0% mortality for 15 min, ≈10% for 30 min, ≈20% for 45 min tMCAO). Based on the lesion size and mortality rate, 30 min tMCAO was used in this study.
Transient focal cerebral ischemia was induced in 12 Trpv4–/– and 12 WT mice. At 24 h post-tMCAO, mice were transcardially perfused with cold saline. The brain was removed immediately and cut into serial 1-mm-thick coronal sections. The sections were incubated in a 2% solution of TTC (Sigma-Aldrich Japan, Tokyo, Japan) for 15 min at 37°C. Images were captured with a Nikon D7100 digital camera (Nikon, Tokyo, Japan). Unstained areas were measured in TTC-stained sections using ImageJ software, version 1.46r (National Institutes of Health, Bethesda, MD, United States) to calculate the infarct volume.
Transient focal cerebral ischemia was induced in 12 Trpv4–/– and 12 WT mice. At 48 h post-tMCAO, mice were decapitated under anesthesia and the olfactory bulb, cerebellum, and brainstem were removed from each dissected brain. The ipsilateral and contralateral hemispheres were separated and their wet weights immediately recorded. The dry weights were determined after drying the tissues to a constant weight at 100°C for 48 h. Tissue water content was calculated as % H2O = (1 − dry weight/wet weight) × 100%.
Transient focal cerebral ischemia was induced in 12 Trpv4–/– and 12 WT mice. At 46 h post-tMCAO, 2% EB dye (Sigma-Aldrich Japan) in normal saline was injected intraperitoneally at a dose of 4 ml/kg body weight. Subsequently, the mice were perfused with cold saline at 48 h post-tMCAO. The ipsilateral hemisphere was collected and incubated with 500 μl formamide (Sigma-Aldrich Japan) at room temperature for 24 h to extract EB. The concentration of dye in the extract was calculated by reference to a standard curve of EB in formamide quantified at 620 nm by spectrofluorophotometry (PerkinElmer Ensight; PerkinElmer, Waltham, MA, United States).
Neurological scores were evaluated at 24 and 48 h post-tMCAO in the Trpv4–/– and WT mice subjected to evaluation of tissue water content and EB leakage (n = 24 per group) using a scoring system modified from a neurological scoring system for sensorimotor deficits (Garcia et al., 1995). The highest possible score of 18 indicated no neurological deficit and the lowest score of 3 indicated the most severe impairment.
Transient focal cerebral ischemia was induced or a sham operation performed in 9 Trpv4–/– and 9 WT mice. At 48 h post-tMCAO, mice were transcardially perfused with cold saline and a 1-mm coronal slice of the ipsilateral hemisphere at 4 mm posterior to the olfactory bulb was collected. The brain slice was homogenized in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, 1% aprotinin, 50 mM NaF, 0.1 mM Na3VO4, 1% Triton-X100) containing anti-phosphatase PhoSTOP (Roche, Mannheim, Germany) and then centrifuged at 10,000 × g for 10 min at 4°C. The protein sample was separated by 4–15% SDS-PAGE and transferred to a polyvinylidene fluoride membrane. The membrane was incubated with anti-zonula occludens (ZO)-1 (61-7300; Zymed Laboratories, San Francisco, CA, United States; 1:1,000 dilution), anti-occludin (ab167161; Abcam, Cambridge, MA, United States; 1:2,000 dilution), or anti-β-actin (A5441; Sigma-Aldrich Japan; 1:20,000 dilution) primary antibodies at 4°C overnight. After washing in Tris-buffered saline with 0.5% Tween-20, the membrane was incubated with a horseradish peroxidase-labeled secondary antibody (Abcam). Antibody-bound protein bands were visualized by enhanced chemiluminescence (ECL prime; Amersham Biosciences, Piscataway, NJ, United States). The protein levels were quantitatively analyzed using a ChemiDoc Touch Imaging System (Bio-Rad, Hercules, CA, United States) and Image Lab software version 5.2.1 (Bio-Rad). The amount of occludin was defined as the sum of the amounts of its dimeric (120 kDa) and monomeric (64 kDa) forms (Yanai et al., 2017). The protein levels of ZO-1 and occludin were normalized by those of β-actin. One mouse without surgery was used as an external control.
Five Trpv4–/– and five WT mice without surgery were evaluated for their baseline gene expression levels of Trpv4, Aqp4, and Kcnj10. The mice were transcardially perfused with cold saline and a 1-mm-thick coronal slice of the hemisphere at 4 mm posterior to the olfactory bulb was collected. Total RNA was extracted using an RNeasy Mini Kit (Qiagen, Valencia, CA, United States). The A260/A280 ratio was measured using a spectrophotometer (Nanodrop 2000; Thermo Fisher Scientific, Wilmington, DE, United States) to determine the purity and concentration of the RNA. Complementary DNA was synthesized from the total RNA using ReverTra Ace reverse transcriptase (Toyobo, Osaka, Japan). Subsequently, RT-qPCR analysis was performed in a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, United States) using TaqMan Fast Universal PCR master mixture and TaqMan probes (Applied Biosystems) for Trpv4 (Hs01099348_m1), Aqp4 (Hs00242342_m1), Kcnj10 (Hs01922935_s1), and Gapdh (Sp03785097_s1) under standard reaction conditions recommended by the manufacturer. The mRNA levels were normalized by those of Gapdh.
Transient focal cerebral ischemia was induced in one Trpv4–/– and one WT mouse. At 6 h post-tMCAO (Kurisu et al., 2016), mice were transcardially perfused with 4% paraformaldehyde with 3.4% sucrose. Brain slices including the right striatum were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer at 4°C overnight, post-fixed with 1% OsO4 in 0.1 M cacodylate buffer on ice for 2 h, dehydrated in a graded ethanol series and propylene oxide, and embedded in epoxy resin. Ultrathin sections were cut at 80-nm thickness with an EM UC7 ultramicrotome (Leica Microsystems, Wetzlar, Germany) and stained with 2% uranyl acetate followed by a mixture of lead nitrate-lead acetate-lead citrate. The sections were observed with a Tecnai 20 electron microscope (FEI Co., Eindhoven, Netherlands) operated at an accelerating voltage of 200 kV and micrographs were recorded at a magnification of 5,000× for at least 10 microvasculatures.
Statistical analyses were performed using JMP version 9.0 software (SAS Institute, Cary, NC, United States). Data are expressed as the median and interquartile range. Statistically significant differences were evaluated by the Wilcoxon rank-sum test or the Kruskal–Wallis test with post hoc comparison by Dunn’s multiple comparison test. Values of p < 0.05 were considered to indicate statistical significance.
To assess the ischemia-induced lesion volume, TTC staining was performed at 24 h post-tMCAO. The ischemia-induced lesion volume was significantly reduced in Trpv4–/– mice compared with WT mice (p = 0.002; Figures 1A,B). Because brain edema progressed to the maximum level on the second and third days, the tissue water content in the ipsilateral hemisphere was measured at 48 h post-tMCAO. The increase in tissue water in the ipsilateral hemisphere was significantly decreased in Trpv4–/– mice compared with WT mice (p = 0.004; Figure 1C). To assess the BBB function, EB leakage in the ipsilateral hemisphere was measured at 48 h post-tMCAO. The extent of EB leakage was significantly smaller in Trpv4–/– mice compared with WT mice (p = 0.002; Figure 1D). To assess the sensorimotor performance, the neurological score was evaluated at 24 and 48 h post-tMCAO in the two types of mice. Performance was impaired in both Trpv4–/– and WT mice after tMCAO with no significant difference between the groups at 24 h (12 [11–13] vs. 12 [10–12], p = 0.307; Figure 1E). However, Trpv4–/– mice showed significantly milder symptoms than WT mice at 48 h post-tMCAO (12 [10–13] vs. 10 [9–11], p = 0.009).
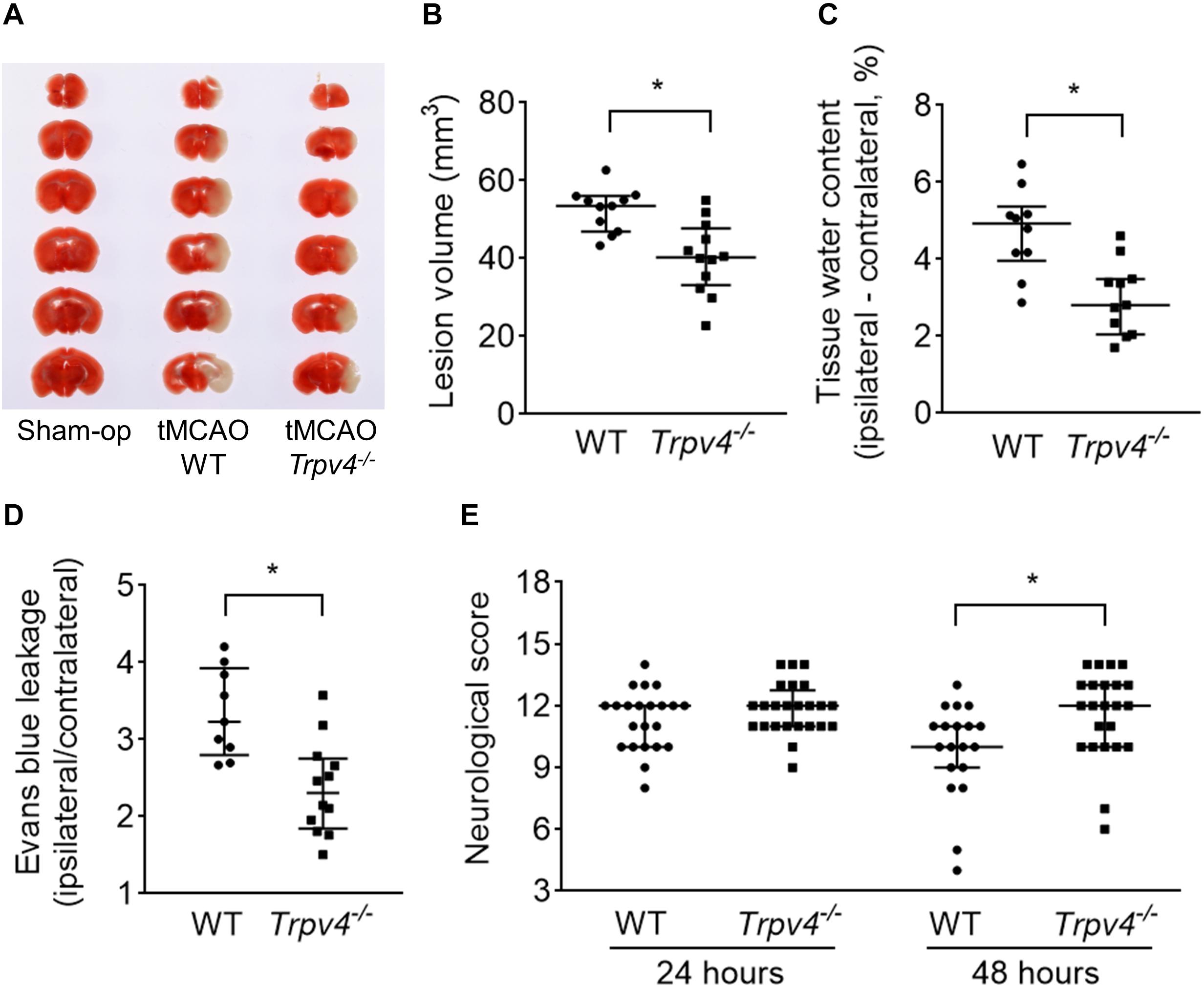
Figure 1. Effects of Trpv4–/– on neurological and pathophysiological findings. (A,B) Evaluation of ischemia-induced lesion volumes at 24 h post-tMCAO (one WT mouse died within 24 h; therefore, n = 11 for WT mice and n = 12 for Trpv4–/– mice). (C) Evaluation of tissue water content by subtraction of the contralateral hemisphere from the ipsilateral hemisphere at 48 h post-tMCAO (two WT mice and one Trpv4–/– mice died within 48 h; therefore, n = 10 for WT mice and n = 11 for Trpv4–/– mice). (D) Evans blue leakage presented as the ratio of leakage in the ipsilateral hemisphere to that in the contralateral hemisphere at 48 h post-tMCAO (three WT mice died within 48 h; therefore, n = 9 for WT mice and n = 12 for Trpv4–/– mice). (E) Evaluation of neurological scores at 24 and 48 h post-tMCAO (n = 22 for WT mice and n = 24 for Trpv4–/– mice at 24 h post-tMCAO; n = 19 for WT mice and n = 23 for Trpv4–/– mice at 48 h post-tMCAO). *p < 0.05.
To assess protein levels in tight junctions of the BBB, western blotting was performed at 48 h post-tMCAO (Figures 2A,B). The occludin and ZO-1 protein levels in the ipsilateral hemisphere were significantly lower in WT mice after tMCAO than after sham operation (p = 0.044 and p = 0.019, respectively), while these changes were not statistically significant in Trpv4–/– mice (p = 0.400 and p = 0.535, respectively).
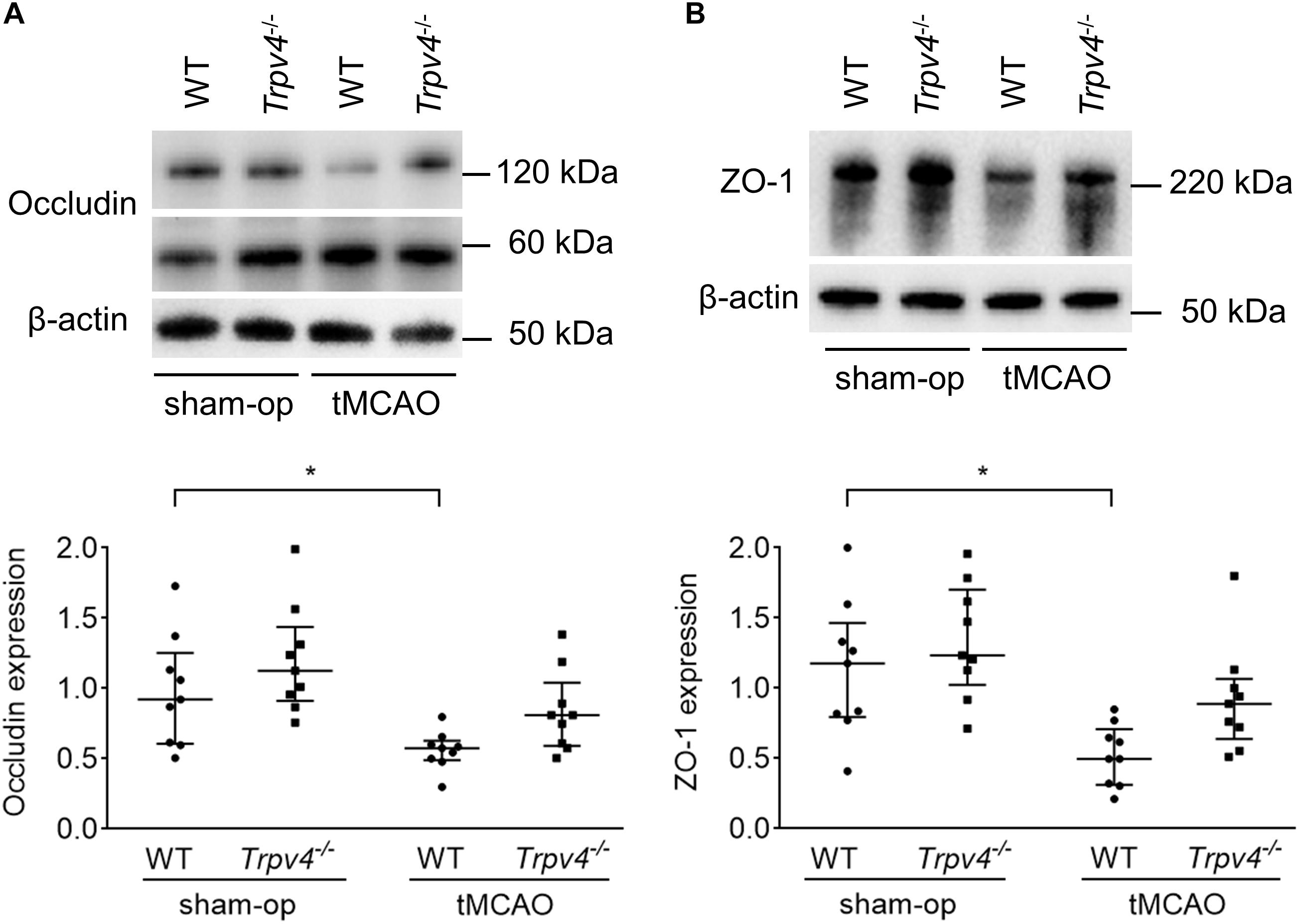
Figure 2. Effects of Trpv4–/– on changes in tight junction proteins. (A,B) Western blot analysis of occludin (A) and ZO-1 (B) protein levels in the ipsilateral hemisphere at 48 h after tMCAO or sham operation in WT and Trpv4–/– mice. Each group contained nine mice. Protein levels were corrected by β-actin. *p < 0.05.
The expression levels of Trpv4 and genes associated with astrocyte volume regulation (Aqp4 and Kcnj10) were evaluated by RT-qPCR. The data revealed significantly decreased expression of Trpv4 in the Trpv4–/– mouse brain compared with the WT mouse brain (p = 0.009), while Aqp4 and Kcnj10 showed no differences (p = 0.347 and p = 0.465, respectively; Figure 3).
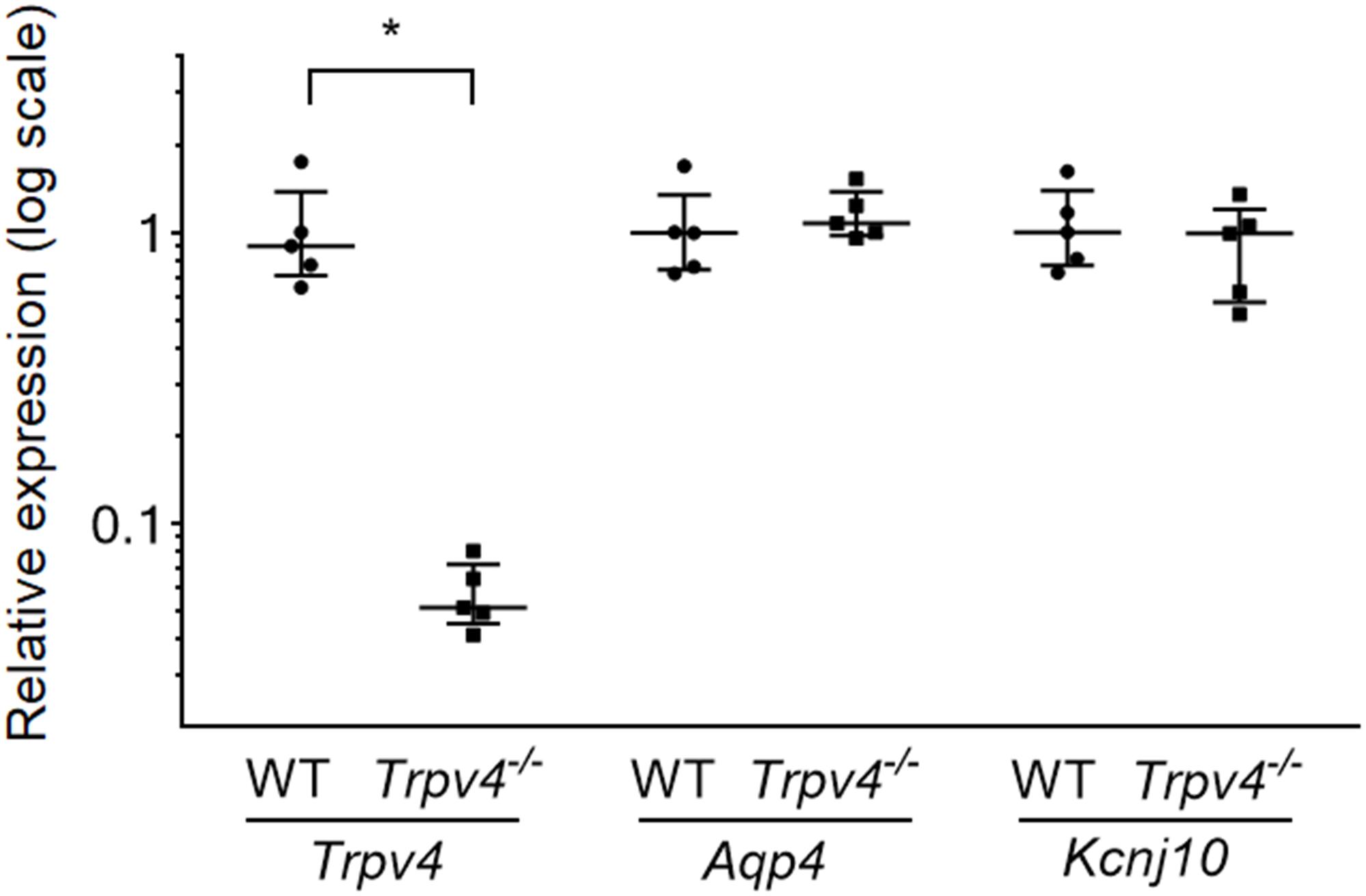
Figure 3. Expression levels of Trpv4, Aqp4, and Kcnj10 in WT and Trpv4–/– mouse brains. RT-qPCR analyses were performed to measure the mRNA levels of Trpv4, Aqp4, and Kcnj10 in the brains of WT and Trpv4–/– mice. Expression levels were corrected by Gapdh. Each group contained five mice. *p < 0.05.
Given the highly polarized localization of TRPV4 in the astrocyte endfeet surrounding brain capillaries, microvascular morphological changes in the ischemic lesion were analyzed by TEM at 6 h post-tMCAO. As shown in Figure 4, there was evidence of widespread endfeet swelling of pericapillary astrocytes in most of the microvasculature in the ischemic lesion. The cytoplasm was expanded, and mitochondria in the endfeet were enlarged and degenerated with swollen cristae. The residual lumen of microvessels was narrowed through compression by the extensive swollen endfeet of perivascular astrocytes in WT mice, but this was markedly suppressed in Trpv4–/– mice.
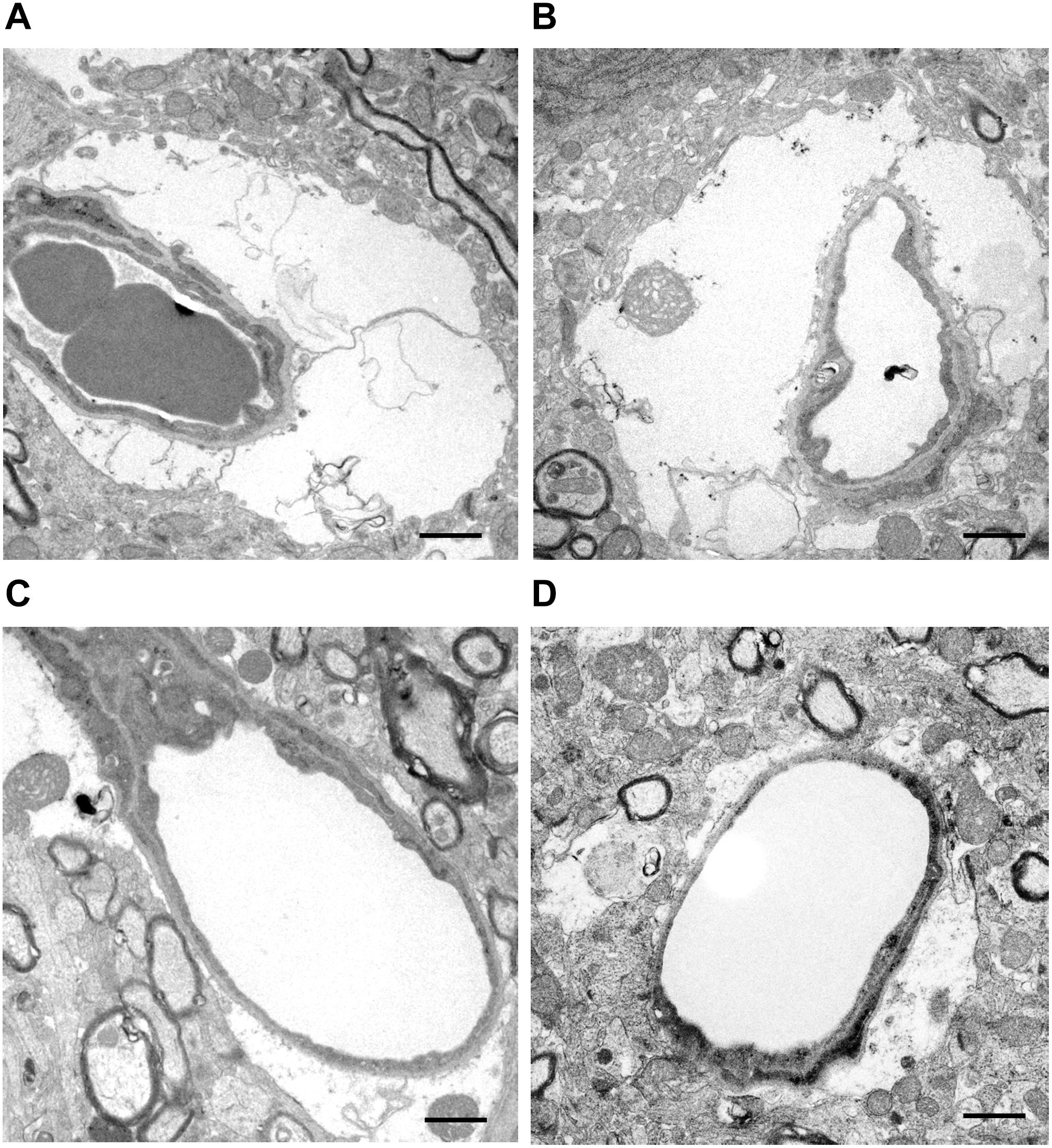
Figure 4. Representative images of microvascular morphological changes evaluated by TEM. (A,B) Microvessels are compressed and narrowed by swollen astrocyte endfeet in WT mice. (C,D) By contrast, the astrocyte endfeet swelling is milder and the residual vascular lumen is well-preserved in Trpv4–/– mice. One WT mouse and one Trpv4–/– mouse were assessed. Scale bars = 1 μm.
This study demonstrated that Trpv4–/– mice showed reduced brain ischemia-induced lesion size and preserved BBB compared with WT mice in the acute phase. Astrocyte endfeet swelling and microvascular narrowing shortly after reperfusion were markedly suppressed in Trpv4–/– mice.
Previous research using ischemia-reperfusion animal models demonstrated decreased capillary blood flow with narrowed capillary diameters (Hauck et al., 2004). This microvessel narrowing was caused by swelling of perivascular astrocyte endfeet shortly after ischemia-reperfusion (Ito et al., 2011; Kurisu et al., 2016). Furthermore, the astrocyte endfeet swelling was markedly ameliorated by Aqp4 knockout (Manley et al., 2000) or infusion of cold saline or mannitol (Ito et al., 2014; Kurisu et al., 2016), leading to reduced infarct volumes. Shimbo et al. (2017) demonstrated that infusion of hemoglobin-encapsulating liposomes, which were smaller than red blood cells and could pass through narrowed blood vessels, reduced the infarction size. These reports indicate that microvascular narrowing may contribute to hypoperfusion associated with ischemia and reperfusion. The mechanism for astrocyte endfeet swelling in acute ischemia was assumed to involve upregulated expression and polarized localization of AQP4 at astrocyte endfeet shortly after reperfusion (Steiner et al., 2012; Kurisu et al., 2016). The present study found similar mRNA expression patterns of Aqp4 and Kcnj10 between WT and Trpv4–/– mouse brains, in accordance with a previous study (Pivonkova et al., 2018). Blockade of TRPV4 altered hypothermia-induced surface localization of AQP4, despite there being no change in total protein expression levels (Salman et al., 2017). Thus, TRPV4 deficiency may reduce astrocyte endfeet swelling by inhibiting the AQP4 surge or AQP4 surface localization at astrocyte endfeet after ischemia-reperfusion.
This study demonstrated preserved BBB function shortly after reperfusion in Trpv4–/– mice, which was similar to that observed after pharmacological blockade of TRPV4 (Jie et al., 2015b). TRPV4-mediated Ca2+ influx led to activation of MMPs in lung tissue (Villalta et al., 2014), with subsequent disruption of tight junctions among alveolar epithelial cells (Dagenais et al., 2018). Therefore, TRPV4 deficiency may maintain the BBB via MMP inhibition to preserve tight junction proteins. A TRPV4 antagonist, GSK2798745, is currently being administered to patients in a phase II clinical trial for treatment of congestive heart failure-induced pulmonary edema (NTC02497937) (Stewart et al., 2020). EB leakage might have occurred because of BBB disruption and also because of ischemia-induced opening of connexin hemichannels in neurovascular unit constituent cells, making them permeable to EB dye (Orellana et al., 2013); however, TRPV4 has potential as a therapeutic target for post-ischemic brain injury from the perspective of BBB protection.
An important concern against clinical application of TRPV4 inhibition is that it may lead to increased lesion volumes (Pivonkova et al., 2018). However, this was inconsistent with the findings of the present study. The difference between the two studies may be because of the different model used (pMCAO vs. tMCAO). Although recanalization is considered the most important event, development of collateral circulation is another major determinant for severity of ischemic stroke with large artery occlusions. Dilation of leptomeningeal arteries occurs as early as 6 h after pMCAO via increased phosphorylation of endothelial nitric oxide synthase (eNOS) in response to shear stress (Iwasawa et al., 2018). Meanwhile, Trpv4–/– mice exhibited loss of shear stress-induced vasodilatation of the carotid artery (Hartmannsgruber et al., 2007), and endothelial downregulation of TRPV4 was shown to contribute to impairment of eNOS-mediated relaxation of the mesenteric artery (Boudaka et al., 2019). These findings suggest that TRPV4 deficiency may exacerbate ischemic brain injury through impairment of acute collateral development in cases of recanalization failure, such as the pMCAO model.
The neuroprotective effects of TRPV4 inhibition may not be solely derived from astrocytic TRPV4 as suggested in a previous report (Pivonkova et al., 2018), because TRPV4 is also expressed in endothelial cells and neutrophils. TRPV4 deficiency prevented neutrophil responses to proinflammatory stimuli, including formation of reactive oxygen species (ROS), adhesion, chemotaxis to break the alveolar–capillary barrier, and development of acute lung injury (Yin et al., 2016). Neutrophils play an important role in acute stroke pathology, wherein reperfusion provides neutrophils with access to the vulnerable brain (Stowe et al., 2009). TRPV4 deficiency or blockade may suppress neutrophil activation to break the BBB. Moreover, free radical damage was shown to play a key role in ischemia-reperfusion injury (Sun et al., 2018). Activation of TRPV4 enhanced oxidative stress in hippocampal neurons (Hong et al., 2016), and TRPV4 was simultaneously activated by ROS (Badr et al., 2016; Suresh et al., 2018). These observations indicate that TRPV4 and free radicals form a positive feedback loop that is involved in neurotoxicity. Thus, blockade of TRPV4 may have other neuroprotective effects in terms of ROS-mediated brain injury.
This study has several limitations. First, the mouse groups were not randomized for surgery and other tests because of the study design, which may have caused some selection biases. Second, in the TEM analysis, we could not perform a statistical evaluation because of a shortage in the number of detected microvessels in our data. Therefore, we qualitatively described the main observed findings in terms of microvascular morphological changes. Third, the neuroprotective effects in Trpv4–/– mice were only assessed in the acute phase of transient focal cerebral ischemia because most of the mice showed continuous body weight loss that exceeded 20% by 48 h post-tMCAO and thus met the humane endpoint for laboratory animals (Science Council of Japan, 2006). Therefore, the effects of Tprv4 deficiency on chronic ischemia and delayed ischemic neurological deficits remain to be established. Activation of TRPV4 via 4α-phorbol 12,13-didecanoate was reported to contribute to functional recovery through angiogenesis and neurogenesis at 5 days post-tMCAO (Chen et al., 2018). Further investigations are needed to confirm whether long-term inhibition of TRPV4 in the subacute to convalescent phases plays a neuroprotective role. Finally, we only examined the expression of two genes that regulate astrocyte volume, Aqp4 and Kcnj10 (Iuso and Križaj, 2016); therefore, further unbiased gene expression studies using microarray methodology are necessary to reveal any gene expression changes that contribute to ischemia-resistance due to knockout of Trpv4.
TRPV4 contributes to post-ischemic brain injury following transient focal cerebral ischemia. The preserved BBB function and microcirculation shortly after reperfusion are the key neuroprotective roles of TRPV4 inhibition, which represents a promising target for the treatment of acute ischemic stroke.
The datasets generated for this study are available on request to the corresponding author.
The animal study was reviewed and approved by Animal Ethics Committee of Kyushu University.
KT, SM, and TY made substantial contributions to the concept and design of the study and analysis and interpretation of data. KT acquired data and drafted the manuscript. SM, TY, MS, MK, RY, and J-IK revised the manuscript critically for important intellectual content. KT approved the version to be published.
This study was supported by a Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (grant number 16K10727) and a Grant-in-Aid for Research Activity Start-up (grant number 19K21303).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We appreciate the technical assistance from The Research Support Center, Research Center for Human Disease Modeling, Kyushu University. We thank Mr. Ryo Ugawa, Laboratory for Technical Support, Medical Institute of Bioregulation, Kyushu University for electron microscopy. We also thank Alison Sherwin, Ph.D., from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Badr, H., Kozai, D., Sakaguchi, R., Numata, T., and Mori, Y. (2016). Different contribution of redox-sensitive transient receptor potential channels to acetaminophen-induced death of human hepatoma cell line. Front. Pharmacol. 7:19. doi: 10.3389/fphar.2016.00019
Boudaka, A., Al-Suleimani, M., Al-Lawati, I., Baomar, H., Al-Siyabi, S., and Zadjali, F. (2019). Downregulation of endothelial transient receptor potential vanilloid type 4 channel underlines impaired endothelial nitric oxide-mediated relaxation in the mesenteric arteries of hypertensive rats. Physiol. Res. 68, 219–231. doi: 10.33549/physiolres.933952
Chen, C. K., Hsu, P. Y., Wang, T. M., Miao, Z. F., Lin, R. T., and Juo, S. H. (2018). TRPV4 activation contributes functional recovery from ischemic stroke via angiogenesis and neurogenesis. Mol. Neurobiol. 55, 4127–4135. doi: 10.1007/s12035-017-0625-0
Dagenais, A., Desjardins, J., Shabbir, W., Roy, A., Filion, D., Sauvé, R., et al. (2018). Loss of barrier integrity in alveolar epithelial cells downregulates ENaC expression and activity via Ca2+ and TRPV4 activation. Pflugers. Arch. 470, 1615–1631. doi: 10.1007/s00424-018-2182-4
Dunn, K. M., Hill-Eubanks, D. C., Liedtke, W. B., and Nelson, M. T. (2013). TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc. Natl. Acad. Sci. U.S.A. 110, 6157–6162. doi: 10.1073/pnas.1216514110
Engel, O., Kolodziej, S., Dirnagl, U., and Prinz, V. (2011). Modeling stroke in mice - middle cerebral artery occlusion with the filament model. J. Vis. Exp. 47:2423. doi: 10.3791/2423
Filosa, J. A., Yao, X., and Rath, G. (2013). TRPV4 and the regulation of vascular tone. J. Cardiovasc. Pharmachol. 61, 113–119. doi: 10.1097/FJC.0b013e318279ba42
Garcia, J. H., Wagner, S., Liu, K. F., and Hu, X. J. (1995). Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 26, 627–634. doi: 10.1161/01.str.26.4.627
Goyal, M., Menon, B. K., van Zwam, W. H., Dippel, D. W., Mitchell, P. J., Demchuk, A. M., et al. (2016). Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet 387, 1723–1731. doi: 10.1016/S0140-6736(16)00163-X
Hartmannsgruber, V., Heyken, W. T., Kacik, M., Kaistha, A., Grgic, I., Harteneck, C., et al. (2007). Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS One 2:e827. doi: 10.1371/journal.pone.0000827
Hauck, E. F., Apostel, S., Hoffmann, J. F., Heimann, A., and Kempski, O. (2004). Capillary flow and diameter changes during reperfusion after global cerebral ischemia studied by intravital video microscopy. J. Cereb. Blood Flow Metab. 24, 383–391. doi: 10.1097/00004647-200404000-00003
Hong, Z., Tian, Y., Yuan, Y., Qi, M., Li, Y., Du, Y., et al. (2016). Enhanced oxidative stress is responsible for TRPV4-induced neurotoxicity. Front. Cell Neurosci. 10:232. doi: 10.3389/fncel.2016.00232
Ito, U., Hakamata, Y., Kawakami, E., and Oyanagi, K. (2011). Temporary cerebral ischemia results in swollen astrocytic end-feet that compress microvessels and lead to delayed focal cortical infarction. J. Cereb. Blood Flow Metab. 31, 328–338. doi: 10.1038/jcbfm.2010.97
Ito, U., Hakamata, Y., Watabe, K., and Oyanagi, K. (2014). Mannitol infusion immediately after reperfusion suppresses the development of focal cortical infarction after temporary cerebral ischemia in gerbils. Neuropathology 34, 360–369. doi: 10.1111/neup.12113
Iuso, A., and Križaj, D. (2016). TRPV4-AQP4 interactions ‘turbocharge’ astroglial sensitivity to small osmotic gradients. Channels 10, 172–174. doi: 10.1080/19336950.2016.1140956
Iwasawa, E., Ishibashi, S., Suzuki, M., Li, F., Ichijo, M., Miki, K., et al. (2018). Sphingosine-1-phosphate receptor 1 activation enhances leptomeningeal collateral development and improves outcome after stroke in mice. J. Stroke Cerebrovasc. Dis. 27, 1237–1251. doi: 10.1016/j.jstrokecerebrovasdis.2017.11.040
Jie, P., Lu, Z., Hong, Z., Li, L., Zhou, L., Li, Y., et al. (2016). Activation of transient receptor potential vanilloid 4 is involved in neuronal injury in middle cerebral artery occlusion in mice. Mol. Neurobiol. 53, 8–17. doi: 10.1007/s12035-014-8992-2
Jie, P., Hong, Z., Tian, Y., Li, Y., Lin, L., Zhou, L., et al. (2015a). Activation of transient receptor potential vanilloid 4 induces apoptosis in hippocampus through downregulating PI3K/Akt and upregulating p38 MAPK signaling pathways. Cell Death Dis. 6:e1775. doi: 10.1038/cddis.2015.146
Jie, P., Tian, Y., Hong, Z., Li, L., Zhou, L., Chen, L., et al. (2015b). Blockage of transient receptor potential vanilloid 4 inhibits brain edema in middle cerebral artery occlusion mice. Front. Cell Neurosci. 9:141. doi: 10.3389/fncel.2015.00141
Kurisu, K., Abumiya, T., Nakamura, H., Shimbo, D., Shichinohe, H., Nakayama, N., et al. (2016). Transarterial regional brain hypothermia inhibits acute aquaporin-4 surge and sequential microvascular events in ischemia/reperfusion injury. Neurosurgery 79, 125–134. doi: 10.1227/NEU.0000000000001088
Li, L., Qu, W., Zhou, L., Lu, Z., Jie, P., Chen, L., et al. (2013). Activation of transient receptor potential vanilloid 4 increases NMDA-activated current in hippocampal pyramidal neurons. Front. Cell Neurosci. 7:17. doi: 10.3389/fncel.2013.00017
Manley, G. T., Fujimura, M., Ma, T., Noshita, N., Filiz, F., Bollen, A. W., et al. (2000). Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 6, 159–163. doi: 10.1038/72256
Molina, C. A., and Alvarez-Sabín, J. (2009). Recanalization and reperfusion therapies for acute ischemic stroke. Cerebrovasc. Dis. 27(Suppl. 1), 162–167. doi: 10.1159/000200455
Nilius, B., and Voets, T. (2013). The puzzle of TRPV4 channelopathies. EMBO Rep. 14, 152–163. doi: 10.1038/embor.2012.219
Orellana, J. A., Martinez, A. D., and Retamal, M. A. (2013). Gap junction channels and hemichannels in the CNS: regulation by signaling molecules. Neuropharmacology 75, 567–582. doi: 10.1016/j.neuropharm.2013.02.020
Pivonkova, H., Hermanova, Z., Kirdajova, D., Awadova, T., Malinsky, J., Valihrach, L., et al. (2018). The contribution of TRPV4 channels to astrocyte volume regulation and brain edema formation. Neuroscience 394, 127–143. doi: 10.1016/j.neuroscience.2018.10.028
Rakers, C., Schmid, M., and Petzold, G. C. (2017). TRPV4 channels contribute to calcium transients in astrocytes and neurons during peri-infarct depolarizations in a stroke model. Glia 65, 1550–1561. doi: 10.1002/glia.23183
Ramsey, I. S., Delling, M., and Clapham, D. E. (2006). An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647. doi: 10.1146/annurev.physiol.68.040204.100431
Ryskamp, D. A., Iuso, A., and Križaj, D. (2015). TRPV4 links inflammatory signaling and neuroglial swelling. Channels 9, 70–72. doi: 10.1080/19336950.2015.1017998
Salman, M. M., Kitchen, P., Woodroofe, M. N., Brown, J. E., Bill, R. M., Conner, A. C., et al. (2017). Hypothermia increases aquaporin 4 (AQP4) plasma membrane abundance in human primary cortical astrocytes via a calcium/transient receptor potential vanilloid 4 (TRPV4)- and calmodulin-mediated mechanism. Eur. J. Neurosci. 46, 2542–2547. doi: 10.1111/ejn.13723
Science Council of Japan (2006). Guidelines for Proper Conduct of Animal Experiments. Available online at: http://www.scj.go.jp/ja/info/kohyo/pdf/kohyo-20-k16-2e.pdf (accessed October 23, 2019).
Shimbo, D., Abumiya, T., Kurisu, K., Osanai, T., Shichinohe, H., Nakayama, N., et al. (2017). Superior microvascular perfusion of infused liposome-encapsulated hemoglobin prior to reductions in infarctions after transient focal cerebral ischemia. J. Stroke Cerebrovasc. Dis. 26, 2994–3003. doi: 10.1016/j.jstrokecerebrovasdis.2017.07.026
Steiner, E., Enzmann, G. U., Lin, S., Ghavampour, S., Hannocks, M. J., Zuber, B., et al. (2012). Loss of astrocyte polarization upon transient focal brain ischemia as a possible mechanism to counteract early edema formation. Glia 60, 1646–1659. doi: 10.1002/glia.22383
Stewart, G. M., Johnson, B. D., Sprecher, D. L., Reddy, Y. N. V., Obokata, M., Goldsmith, S., et al. (2020). Targeting pulmonary capillary permeability to reduce lung congestion in heart failure: a randomized, controlled pilot trial. Eur. J. Heart Fail. doi: 10.1002/ejhf.1809 [Epub ahead of print].
Stowe, A. M., Adair-Kirk, T. L., Gonzales, E. R., Perez, R. S., Shah, A. R., Park, T. S., et al. (2009). Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol. Dis. 35, 82–90. doi: 10.1016/j.nbd.2009.04.006
Sun, M. S., Jin, H., Sun, X., Huang, S., Zhang, F. L., Guo, Z. N., et al. (2018). Free radical damage in ischemia-reperfusion injury: an obstacle in acute ischemic stroke after revascularization therapy. Oxid. Med. Cell Longev. 2018:3804979. doi: 10.1155/2018/3804979
Suresh, K., Servinsky, L., Jiang, H., Bigham, Z., Yun, X., Kliment, C., et al. (2018). Reactive oxygen species induced Ca2+ influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 314, L893–L907. doi: 10.1152/ajplung.00430.2017
Suzuki, M., Mizuno, A., Kodaira, K., and Imai, M. (2003). Impaired pressure sensation in mice lacking TRPV4. J. Biol. Chem. 278, 22664–22668. doi: 10.1074/jbc.M302561200
Villalta, P. C., Rocic, P., and Townsley, M. I. (2014). Role of MMP2 and MMP9 in TRPV4-induced lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 307, L652–L659. doi: 10.1152/ajplung.00113.2014
Yanai, S., Toyohara, J., Ishiwata, K., Ito, H., and Endo, S. (2017). Long-term cilostazol administration ameliorates memory decline in senescence-accelerated mouse prone 8 (SAMP8) through a dual effect on cAMP and blood-brain barrier. Neuropharmacology 116, 247–259. doi: 10.1016/j.neuropharm.2016.12.006
Keywords: ischemic stroke, transient receptor potential vanilloid 4, knockout mice, brain edema, blood–brain barrier
Citation: Tanaka K, Matsumoto S, Yamada T, Yamasaki R, Suzuki M, Kido MA and Kira J-I (2020) Reduced Post-ischemic Brain Injury in Transient Receptor Potential Vanilloid 4 Knockout Mice. Front. Neurosci. 14:453. doi: 10.3389/fnins.2020.00453
Received: 23 November 2019; Accepted: 14 April 2020;
Published: 12 May 2020.
Edited by:
Mathias Gelderblom, University Medical Center Hamburg-Eppendorf, GermanyReviewed by:
Juan C. Saez, Pontifical Catholic University of Chile, ChileCopyright © 2020 Tanaka, Matsumoto, Yamada, Yamasaki, Suzuki, Kido and Kira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun-Ichi Kira, a2lyYUBuZXVyby5tZWQua3l1c2h1LXUuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.