 Carmen Grijota-Martínez
Carmen Grijota-Martínez Soledad Bárez-López
Soledad Bárez-López David Gómez-Andrés
David Gómez-Andrés Ana Guadaño-Ferraz
Ana Guadaño-Ferraz- 1Department of Endocrine and Nervous System Pathophysiology, Instituto de Investigaciones Biomédicas Alberto Sols, Consejo Superior de Investigaciones Científicas (CSIC), Universidad Autónoma de Madrid (UAM), Madrid, Spain
- 2Center for Biomedical Research on Rare Diseases (Ciberer), Instituto de Salud Carlos III, Madrid, Spain
- 3Department of Cell Biology, Faculty of Biology, Universidad Complutense de Madrid, Madrid, Spain
- 4Translational Health Sciences, Dorothy Hodgkin Building, University of Bristol, Bristol, United Kingdom
- 5Pediatric Neurology, Vall d’Hebron University Hospital and VHIR (Euro-NMD, ERN-RND), Barcelona, Spain
Allan-Herndon-Dudley syndrome is a rare disease caused by inactivating mutations in the SLC16A2 gene, which encodes the monocarboxylate transporter 8 (MCT8), a transmembrane transporter specific for thyroid hormones (T3 and T4). Lack of MCT8 function produces serious neurological disturbances, most likely due to impaired transport of thyroid hormones across brain barriers during development resulting in severe brain hypothyroidism. Patients also suffer from thyrotoxicity in other organs due to the presence of a high concentration of T3 in the serum. An effective therapeutic strategy should restore thyroid hormone serum levels (both T3 and T4) and should address MCT8 transporter deficiency in brain barriers and neural cells, to enable the access of thyroid hormones to target neural cells. Unfortunately, targeted therapeutic options are currently scarce and their effect is limited to an improvement in the thyrotoxic state, with no sign of any neurological improvement. The use of thyroid hormone analogs such as TRIAC, DITPA, or sobetirome, that do not require MCT8 to cross cell membranes and whose controlled thyromimetic activity could potentially restore the normal function of the affected organs, are being explored to improve the cerebral availability of these analogs. Other strategies aiming to restore the transport of THs through MCT8 at the brain barriers and the cellular membranes include gene replacement therapy and the use of pharmacological chaperones. The design of an appropriate therapeutic strategy in combination with an early diagnosis (at prenatal stages), will be key aspects to improve the devastating alterations present in these patients.
Introduction
Thyroid hormones (THs; T4 or thyroxine and T3 or triiodothyronine) are essential for the development and function of the CNS. An insufficient supply of THs during development can hinder essential processes such as neurogenesis, cell migration, synaptogenesis or myelinization. These neurological alterations range from very mild to very severe, and can be even irreversible, depending on the time and duration of the hormonal deficiency.
THs are synthesized in the thyroid gland, mostly as T4, and released by follicular cells to the circulation, reaching the different target tissues. Both genomic and non-genomic actions of THs have been described. The latter are carried out in the cytoplasm or plasma membrane by binding to receptors such as integrin αvβ3 (Davis et al., 2016). On the other hand, in the genomic actions T3 binds to nuclear receptors (TR), regulating the transcription of target genes. The main isoforms of these TR are TRα1, TRα2, TRβ1, and TRβ2, which mediate different actions and whose expression varies throughout ontogeny and different tissues. The amount of the nuclear active hormone T3 is mainly regulated locally in the target tissues by the action of different deiodinase enzymes which activate (converting T4 into T3) or inactivate (deiodinating T4 and T3 into inactive metabolites) THs.
THs use transporter proteins located in cell membranes to reach the inside the cells (Hennemann et al., 2001). Some of the major transporters of THs include the monocarboxylate transporter 8 (MCT8), MCT10 and the solute carrier organic anion transporter 1C1 (OATP1C1), but only MCT8 has been identified as being specific to transport THs. MCT8 is encoded by the SLC16A2 gene (Friesema et al., 2003) and the mutations that cause the inactivation of this transporter result in a rare X-linked disease known as Allan-Herndon-Dudley Syndrome (AHDS or MCT8 deficiency). AHDS (ORPHA:59) is an ultra-rare disorder with an estimated prevalence lower than 1 case per million. Orphanet suggested that 320 patients had been diagnosed within more than 130 families but these numbers are prone to raise given the increasing access to new generation sequencing technologies and the expanding spectrum of severity attributed to the disease (Remerand et al., 2019).
AHDS was described in 1944 (Allan et al., 1944) but its association to mutations in the SLC16A2 gene was established in 2004 (Dumitrescu et al., 2004; Friesema et al., 2004). AHDS patients have a characteristic thyroid profile with elevated plasma T3 levels (peripheral hyperthyroidism), low T4 and normal or slightly elevated TSH with a free T3/T4 ratio greater than 0.75 (Dumitrescu et al., 2004; Friesema et al., 2004; Groeneweg et al., 2019; Remerand et al., 2019).
In addition, the phenotype of AHDS is characterized by severe paraparesis with hypotonia (with poor cephalic control being an early symptom), bradykinesia, spasticity and extrapyramidal manifestations (dystonia or dystonia with choreoathetosis) and moderate to severe intellectual disability with absence of language, associated with low weight and/or low muscle mass and in some cases, epilepsy (Remerand et al., 2019). In recent years, the increasing knowledge about this condition have promoted a growing number of patients that have defined a much broader phenotype, with the appearance of milder forms with motor and cognitive disability of lesser severity (Masnada et al., 2019; Remerand et al., 2019).
These alterations occur as a consequence of two phenomena. On the one hand, the excess of T3 in serum produces peripheral hyperthyroidism with muscle thyrotoxicosis that causes a generalized loss of muscle volume. On the other hand, the absence of MCT8 impairs the effective transport of THs to the CNS, most probably due to the lack of functional MCT8 in brain barriers (Ceballos et al., 2009; Iwayama et al., 2016; Vatine et al., 2017) leading to brain hypothyroidism (Dumitrescu et al., 2006; Trajkovic et al., 2007; López-Espíndola et al., 2014). Indeed, brain tissue necropsies of MCT8-deficient patients have revealed alterations compatible with brain hypothyroidism with anomalies in neuronal differentiation, myelinization and synaptogenesis present already from prenatal stages (López-Espíndola et al., 2014). Consistent with this, MCT8 has been found to be very abundant at the brain barriers in the human fetal brain (Wirth et al., 2009; López-Espíndola et al., 2019; Wilpert et al., 2020).
The rapid diagnosis is a key factor for the patients’ wellbeing. For instance, unlike other similar diseases, there is a marked prevalence of respiratory problems and kyphoscoliosis, so intensive monitoring and an adequate care is important to improve the survival and quality of life of patients. Moreover, due to the fact that brain alterations start during development, an early treatment, including prenatal treatment, takes special importance. Currently, targeted therapies to improve the neurological picture of patients are not available. As MCT8-deficient patients suffer from brain hypothyroidism in combination with peripheral hyperthyroidism, the design of an effective treatment is challenging. The aim of this manuscript is to critically review therapeutic approaches attempted so far both at the clinical level in patients and at the preclinical level in models of the disease to contribute to the development of future solutions for AHDS patients (summarized in Table 1).
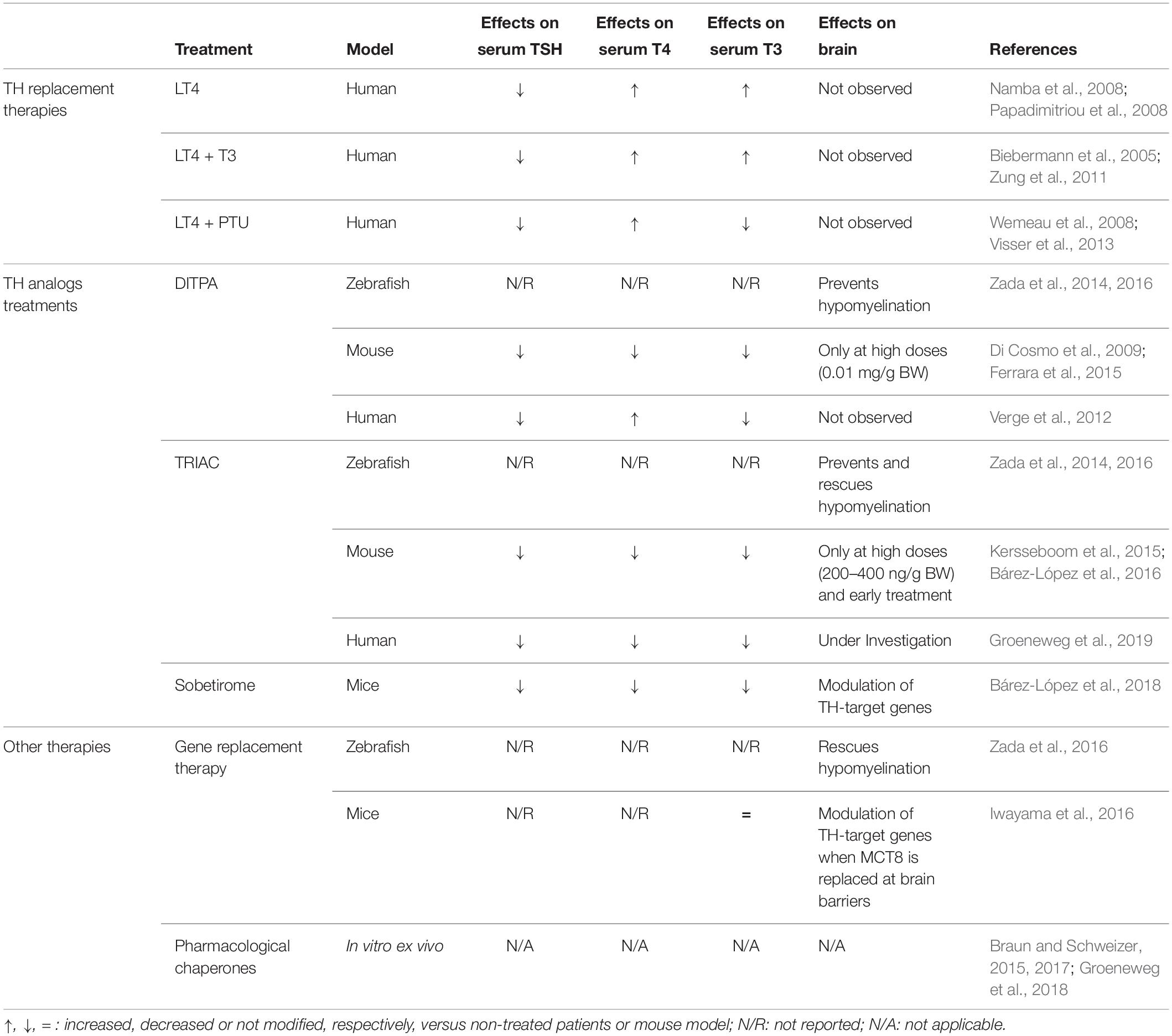
Table 1. Summary of the existing clinical and preclinical therapeutic approaches for AHDS.
Murine Models of the Disease
The development of targeted therapies in rare diseases depends on the availability of good animal models, both to unravel the mechanisms of disease and to carry out preclinical studies to evaluate therapeutic strategies. In the case of MCT8 deficiency, most of the knowledge about the mechanisms underlying the disease as well as the preclinical studies considered so far has arisen from using murine models of the syndrome. However, it has not been straightforward to obtain a suitable model and there is still ongoing work focused on the characterization of different animal models.
Initially, mice deficient of the MCT8 transporter (Mct8KO mice) were proposed as a disease model. Although these mice showed a thyroid profile similar to that of the patients and relatively low levels of T3 in different brain areas (Dumitrescu et al., 2006; Trajkovic et al., 2007), brain histopathology studies did not reveal major defects and only subtle behavioral changes were found (Wirth et al., 2009). It was therefore suggested that these animals present some compensatory mechanism that prevents neurological damage. This compensatory mechanism is due to the complementary action of two proteins: the organic anion transporter OATP1C1 and the deiodinase type 2 (DIO2) enzyme. OATP1C1 transporter is codified by the Slco1c1 gene, it transports mainly T4 and is expressed abundantly in rodent brain barriers, but to a much lesser extent in humans (Roberts et al., 2008; López-Espíndola et al., 2019). The entry of T4 to the brain through this transporter and the subsequent action of DIO2, which converts T4 into T3 in astrocytes and whose expression is increased in these mice, would provide a sufficient amount of the nuclear active hormone T3 in the mouse brain, exerting a compensatory mechanism for the lack of MCT8 (Figure 1). For this reason, two alternative models are currently being used; MCT8 and DIO2 (Mct8/Dio2KO) or MCT8 and OATP1C1 (Mct8/Slc1c1KO) -deficient mice, in which this compensatory mechanism is suppressed. These models present a thyroid profile similar to that of the patients in addition to several neuromotor alterations, offering a more suitable model (Mayerl et al., 2014; Bárez-López et al., 2019a).
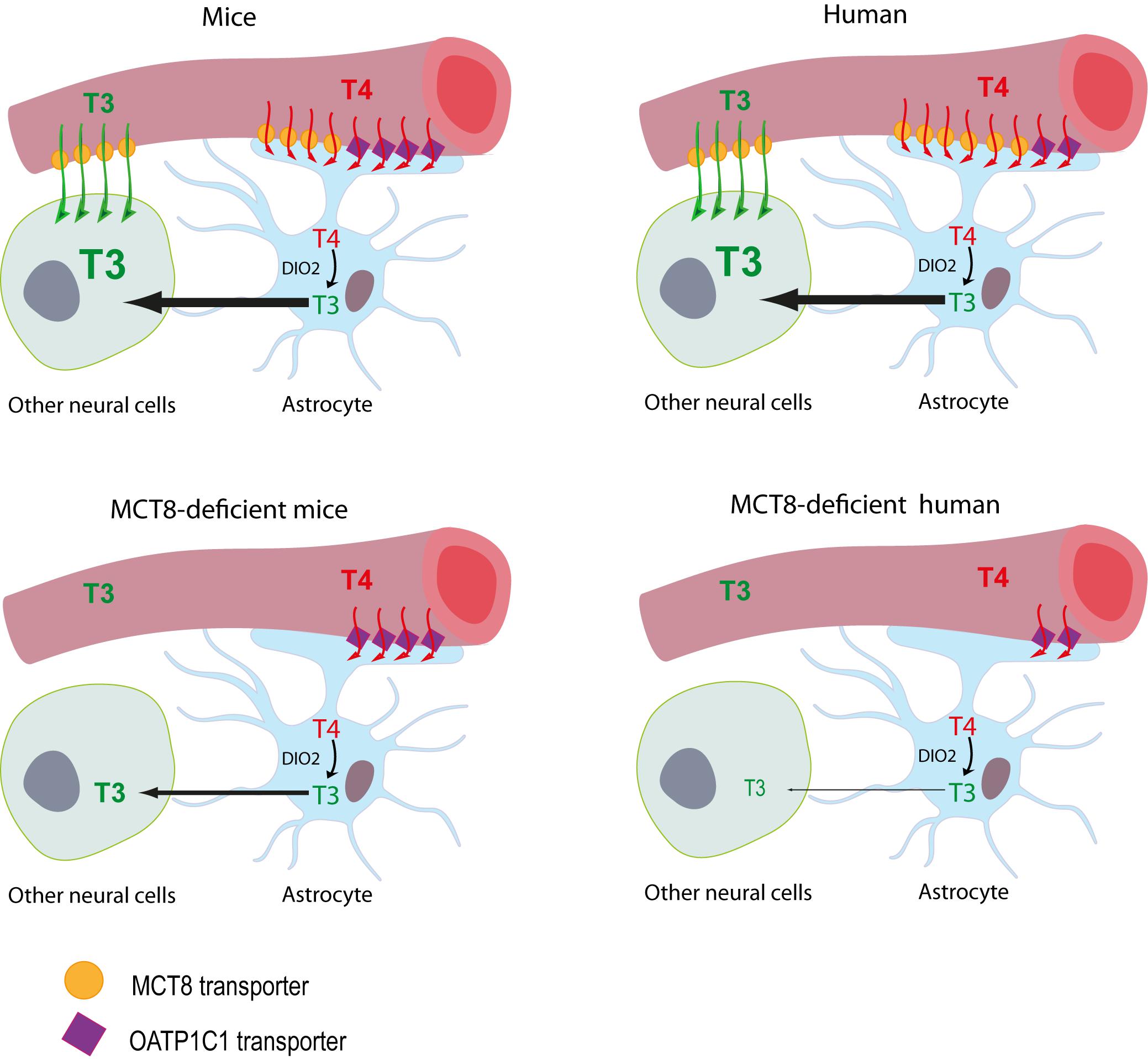
Figure 1. Proposed model illustrating the differences between MCT8-deficient mice and humans in thyroid hormone availability to neural cells. The model for T3 availability to the brain under normal conditions supports that brain T3 can access the target neural cells though two different routes: (1) directly from the circulation, with T3 crossing the BBB mainly via the MCT8 transporter into the extracellular fluid where it directly reaches the target neural cells, or (2) T3 can also be produced locally by DIO2 activity in the astrocytes from T4, which crosses the BBB directly into the astrocytes mainly through MCT8 in humans and through MCT8 and OATP1C1 in mice. In MCT8-deficient mice, the elevated DIO2 activity in the astrocytes converts the T4 available through OATP1C1 into T3, that is subsequently available to target neural cells, compensating for the lack of MCT8. In MCT8-deficient humans, this compensatory mechanism cannot take place as OATP1C1 is hardly present in the human BBB, preventing T4 entry to the brain and its subsequent conversion into T3.
In addition, other alternative models currently available include the use of pluripotent stem cells induced from human patients (Vatine et al., 2017) or the use of other organisms such as zebrafish (Vatine et al., 2013), chicken (Delbaere et al., 2017) or even Xenopus (Mughal et al., 2017). For the first time, this rich cataloge of models is providing the possibility of testing drugs in a fast and accurate way in preclinical stages in AHDS.
Thyroid Hormone Replacement Therapies
As soon as it was found that the syndrome was due to mutations in a transporter of THs, and that it had an endocrine component with altered THs levels, treatments to intervene on the thyroid axis were considered. In Mct8KO mice, the injection of supraphysiological doses of levothyroxine (LT4) was able to reduce the increased expression of TRH mRNA in hypothalamic neurons, indicating that in the absence of MCT8 these neurons can respond to T4 treatment. In contrast, only a high dose of T3 was able to downregulate TSH levels in plasma with a modest reduction of TRH, suggesting an impaired uptake of circulating T3 (Trajkovic et al., 2007).
Based on this, in patients, a LT4 supplement alone or in combination with T3 was initially considered. This treatment suppressed TSH and restored T4; however, it further increased T3 serum levels, worsening the hyperthyroidism in extracerebral tissues, and did not improve the patient’s neurological impairments (Namba et al., 2008; Papadimitriou et al., 2008). After this first approximation, a block-replacement therapy consisting of the administration of propylthiouracil (PTU; an antithyroid drug that also inhibits type 1 deiodinase (DIO1), which transforms T4 into T3) together with LT4, decreased endogenous production of THs and prevented the conversion of LT4 into T3. This therapy normalized TSH levels and significantly decreased serum T3, which was reflected in an improvement in body mass index and a reduction in heart rate. However, none of the patients experienced any improvement at the psychomotor level (Wemeau et al., 2008; Visser et al., 2013).
Treatment With Thyroid Hormone Analogs
The search for T3 analogs that do not require MCT8 to cross the cell membranes is one of the main strategies in the search for an effective therapy. 3,5-diiodo-thyropropionic acid (DITPA) is a T3 analog that binds with similar affinity to the different TR isoforms, although with much lower affinity than T3 (Pennock et al., 1992). Despite this, it has been shown to have inotropic thyromimetic actions but maintaining a low metabolic activity (Pennock et al., 1992; Moreno et al., 2008). In zebrafish, treatment with DITPA was able to prevent hypomyelination in Mct8-deficient embryos but did not fully recovered hypomyelination present in the larvae (Zada et al., 2014, 2016). The administration of this compound to MCT8-deficient mice was able to normalize T3 and TSH while it further decreased T4 levels in serum, ameliorating metabolic parameters related to the hypermetabolism of Mct8KO mice. However, improvements in brain hypothyroidism could only be observed when using high doses of DITPA (Di Cosmo et al., 2009; Ferrara et al., 2015). The administration of DITPA in four patients normalized T3 and TSH and, unlike in mice, increased the levels of T4 to the lower normal range in serum, decreasing also the cardiac rhythm. However, no improvement, or very small, was found in the neurological performance of these patients when they were assessed with the Bayley-III scale (Verge et al., 2012).
Another T3 analog that has received special attention is sobetirome (also called GC-1), a compound that has been used in numerous animal studies (Hartley et al., 2017) as well as in clinical trials for patients with dyslipidemias. Sobetirome binds to TRs with the same affinity for TRβ1 as T3 but 4–10 times less affinity than T3 for TRα1 (Chiellini et al., 1998). In 2018 we published the first study evaluating its action in the absence of MCT8 after intraperitoneal administration to Mct8/Dio2KO mice (Bárez-López et al., 2018). An increase in the content of sobetirome was detected in the brain of the treated animals, demonstrating that it was able to cross the blood-brain barrier of mice in the absence of MCT8. Most importantly, we provided evidence of sobetirome action in brain, regulating the expression of T3 target genes, since it was able to partially recover and even normalize the expression of some genes that are altered in Mct8/Dio2KO animals.
A third and promising alternative is 3,5,3’-triiodothyroacetic acid (TRIAC), a natural metabolite of THs. This compound arises as one of the potential options for the treatment of patients since in vitro studies have shown that it can bind the TR with the same affinity as T3 for the isoform TRα and with greater affinity for TRβ (Takeda et al., 1995; Messier and Langlois, 2000), that it can exert thyromimetic actions, and that can access the inside of the cell in the absence of MCT8 (Horn et al., 2013; Kersseboom et al., 2015). Moreover, treatment with TRIAC in zebrafish was able to prevent and rescue hypomyelination in Mct8-deficient embryos and larvae, respectively (Zada et al., 2014, 2016). The first study evaluating the in vivo action of TRIAC in MCT8 deficiency was carried out in Mct8/Slco1c1KO mice treated with high doses of TRIAC (200-400 ng per gram of weight per day) during early postnatal stages (from postnatal day 1 (P1) to P12). These studies revealed that at high doses TRIAC is able to prevent brain alterations in the absence of MCT8 (Kersseboom et al., 2015). To get a better understanding on the potential of this analog in the clinical practice, we assessed the effects of therapeutic doses of TRIAC at juvenile stages. Mct8KO mice were treated with of 30 ng of TRIAC per gram of weight per day (similar to the dose used in patients) in the drinking water from P21 to P30. This treatment decreased serum levels of both T3 and T4, improving the peripheral hyperthyroidism. However, TRIAC content in the brain did not increase after treatment, and therefore no thyromimetic actions could be observed in this organ. Moreover, we also found that the decrease in T4 plasma levels worsened brain hypothyroidism by further decreasing the brain T3 content and the expression of some target genes (Bárez-López et al., 2016). This highlights the importance of considering therapeutic approaches that do not further decrease serum T4.
Currently, alternative delivery pathways are being explored in order to increase TRIAC content in the brain with the final aim of ameliorating the brain hypothyroidism. In a recent study we administered TRIAC directly into the brain of MCT8-deficient animals through a catheter inserted into the brain lateral ventricle (Bárez-López et al., 2019b). We found that this treatment did not affect T3 or T4 levels in the serum, thus did not aggravate the brain hypothyroidism. Moreover, even though TRIAC content increased in the brain after treatment, this increase was not enough to mediate the expression of T3 target genes. Further studies will be necessary to finally determine if TRIAC serves as a T3 analog mediating brain effects of THs in vivo.
Due to the extensive clinical experience with TRIAC, that has demonstrated its ability to inhibit TSH secretion and therefore decrease the production of endogenous THs, TRIAC has been used in a phase I clinical trial evaluating its effectiveness and safety for treatment of peripheral thyrotoxicosis in MCT8-deficient patients, both pediatric and adult (Groeneweg et al., 2019). In accordance with the results found in the animal model, treatment with 38.3 μg of TRIAC per kg of body weight per day reduced serum T3 levels to the normal range and further decreased serum T4 levels. This study has shown that TRIAC is safe and effective in controlling peripheral thyrotoxicosis improving the altered weight, heart rate and blood pressure. However, it is important to point out that reduced plasma T4 levels could have potential adverse consequences on the brain, as was found in systemically TRIAC-treated Mct8KO mice. Having evaluated the effects of TRIAC on peripheral thyrotoxicosis and showing encouraging effects (a trend toward improvement) in gross motor function when children are treated before 4 year of age, the clinical study is currently in phase II to evaluate the action of this analog on neurological symptoms in pediatric patients (NCT02396459).
Other Therapies for MCT8 Deficiency
In parallel to the use of T3 analogs, other possibilities such as gene replacement therapy and the use of chemical chaperones are also currently being explored. In order to improve brain alterations by gene therapy in mice, MCT8 was administered using adeno-associated virus (AAV) as a vector with the intention of restoring MCT8 expression in the brain. For this purpose, AAV9 (which is transduced in the brain with high efficiency) expressing MCT8 was injected to MCT8-deficient mice at day P1 either intravenously or directly into the brain intracerebroventricularly. The results showed that intravenous administration increased the content of T3 in the brain and recovered the expression of T3 target genes altered in MCT8 deficiency. Although intracerebroventricular administration drove higher expression of MCT8 in the brain than the intravenous delivery, there was no increase in the content of T3 or in the expression of T3 target genes. This could be explained because, unlike the intracerebroventricular route, the intravenous delivery was able to restore MCT8 expression in the choroid plexus demonstrating that for a successful gene replacement therapy, it is essential to restore MCT8 in brain barriers to allow entry of THs into the brain (Iwayama et al., 2016). In zebrafish, the injection of a genetic construct driving Mct8 expression in vascular endothelial cells into Mct8-deficient embryos was able to recover hypomyelination in 10 days post fertilization larvae but not in 3 days post fertilization embryos. This suggests that genetic treatment can be beneficial even after brain damage has occurred and offers a potential treatment for MCT8-deficient patients (Zada et al., 2016).
A different approximation is the use of pharmacological chaperones that allow the conformational stabilization of mutated proteins and assist in their translocation to the plasma membrane. It is believed that this approach would be more effective for those mutations in SLC16A2 that affect the stability of the protein and its intracellular traffic. A recent work using stable overexpression cell models demonstrated that the use of pharmacological chaperones can restore MCT8 in cell membranes and mediate T3 transport in certain types of SLC16A2 mutations (Braun and Schweizer, 2015, 2017). However, similar studies have subsequently been carried out ex vivo on fibroblasts derived from MCT8-deficient patients without functionally restoring MCT8 in cell membranes, and thus demonstrating that the ability of pharmacological chaperones to rescue MCT8 function depends on the cell type used for the assays (Groeneweg et al., 2018).
Conclusion
The complex nature of MCT8 deficiency presents certain difficulties when designing appropriate therapeutic strategies. A suitable therapy should ameliorate the peripheral hyperthyroidism while improving the neurological defects. So far, several therapeutic approaches have been successful in improving the peripheral hyperthyroidism in patients. These include a block-replacement therapy with LT4 in combination with PTU and the T3 analogs DITPA and TRIAC. Treatment with TRIAC further decreases T4 serum levels unlike DITPA and LT4 + PTU treatments. Due to the potential effects that T4 (through local conversion into T3) might be mediating in the brain, an ultimate therapeutic approach should not further decrease but restore serum T4 levels.
The big challenge ahead of us is the improvement of the neurological alterations in patients. To tackle this, several approaches that aim to overcome the limitation of the brain barriers are currently being explored. The use of alternative delivery pathways that can increase the content of T3 analogs in the brain offers the possibility of enhancing the potential action of these analogs in the brain, as long as they are indeed effective in mediating TH-like actions in vivo. The use of the sobetirome, gene replacement therapy and pharmaceutical chaperones offers promising alternative therapies that require further characterization. Moreover, as an early treatment is crucial, the use of T3 analogs that can cross the placental barrier such as TRIAC (Nicolini et al., 1996) or DITPA (Ferrara et al., 2014) or the direct in utero administration of potential therapeutic agents, must be considered.
Due to the great advances in preclinical stages of potential therapies for AHDS, we believe that studies devoted to enhance our understanding about the role of MCT8 in development and the complex changes in the CNS related with this disorder are particularly urgent in order to proceed with an optimal translation of the different therapeutic approaches to patients.
Author Contributions
CG-M, SB-L, DG-A, and AG-F have contributed in the drafting of the article and have revised it critically for important intellectual content and for the final approval of the submitted version. CG-M and SB-L have designed the figures.
Funding
This work was supported by the Spanish Ministry of Economy and Competitiveness, grant number SAF2017-86342-R (MINECO/AEI/FEDER, UE) to AG-F, the Sherman Foundation (Grant Number OTR02211) to AG-F and SB-L, and the BBSRC (Grant Number BB/R016879/1) to SB-L. CG-M is a recipient of a contract from the Center for Biomedical Research on Rare Diseases (CIBERER), Instituto de Salud Carlos III, Madrid. The cost of this publication has been paid in part by FEDER funds.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI). We also thank Javier Pérez García for the artwork.
References
Allan, W., Herndon, C. N., and Dudley, F. C. (1944). Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. Am. J. Ment. Defic. 48, 325–334.
Bárez-López, S., Grijota-Martínez, C., Ausó, E., Fernández-De Frutos, M., Montero-Pedrazuela, A., and Guadaño-Ferraz, A. (2019a). Adult mice lacking Mct8 and Dio2 proteins present alterations in peripheral thyroid hormone levels and severe brain and motor skill impairments. Thyroid 29, 1669–1682. doi: 10.1089/thy.2019.0068
Bárez-López, S., Grijota-Martínez, C., Liao, X. H., Refetoff, S., and Guadaño-Ferraz, A. (2019b). Intracerebroventricular administration of the thyroid hormone analog TRIAC increases its brain content in the absence of MCT8. PLoS One 14:e0226017. doi: 10.1371/journal.pone.0226017
Bárez-López, S., Hartley, M. D., Grijota-Martínez, C., Scanlan, T. S., and Guadaño-Ferraz, A. (2018). Sobetirome and its amide prodrug Sob-AM2 exert thyromimetic actions in Mct8-deficient brain. Thyroid 28, 1211–1220. doi: 10.1089/thy.2018.0008
Bárez-López, S., Obregon, M. J., Martínez-De-Mena, R., Bernal, J., Guadaño-Ferraz, A., and Morte, B. (2016). Effect of triiodothyroacetic acid treatment in Mct8 deficiency: a word of caution. Thyroid 26, 618–626. doi: 10.1089/thy.2015.0388
Biebermann, H., Ambrugger, P., Tarnow, P., Von Moers, A., Schweizer, U., and Grueters, A. (2005). Extended clinical phenotype, endocrine investigations and functional studies of a loss-of-function mutation A150V in the thyroid hormone specific transporter MCT8. Eur. J. Endocrinol. 153, 359–366. doi: 10.1530/eje.1.01980
Braun, D., and Schweizer, U. (2015). Efficient activation of pathogenic DeltaPhe501 mutation in monocarboxylate transporter 8 by chemical and pharmacological chaperones. Endocrinology 156, 4720–4730. doi: 10.1210/en.2015-1393
Braun, D., and Schweizer, U. (2017). The chemical chaperone phenylbutyrate rescues MCT8 mutations associated with milder phenotypes in patients with allan-herndon-dudley syndrome. Endocrinology 158, 678–691. doi: 10.1210/en.2016-1530
Ceballos, A., Belinchón, M. M., Sanchez-Mendoza, E., Grijota-Martínez, C., Dumitrescu, A. M., Refetoff, S., et al. (2009). Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3’-triiodo-L-thyronine. Endocrinology 150, 2491–2496. doi: 10.1210/en.2008-1616
Chiellini, G., Apriletti, J. W., Yoshihara, H. A., Baxter, J. D., Ribeiro, R. C., and Scanlan, T. S. (1998). A high-affinity subtype-selective agonist ligand for the thyroid hormone receptor. Chem. Biol. 5, 299–306. doi: 10.1016/s1074-5521(98)90168-5
Davis, P. J., Goglia, F., and Leonard, J. L. (2016). Nongenomic actions of thyroid hormone. Nat. Rev. Endocrinol. 12, 111–121. doi: 10.1038/nrendo.2015.205
Delbaere, J., Vancamp, P., Van Herck, S. L., Bourgeois, N. M., Green, M. J., Wingate, R. J., et al. (2017). MCT8 deficiency in Purkinje cells disrupts embryonic chicken cerebellar development. J. Endocrinol. 232, 259–272. doi: 10.1530/joe-16-0323
Di Cosmo, C., Liao, X. H., Dumitrescu, A. M., Weiss, R. E., and Refetoff, S. (2009). A thyroid hormone analog with reduced dependence on the monocarboxylate transporter 8 for tissue transport. Endocrinology 150, 4450–4458. doi: 10.1210/en.2009-0209
Dumitrescu, A. M., Liao, X. H., Best, T. B., Brockmann, K., and Refetoff, S. (2004). A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am. J. Hum. Genet. 74, 168–175. doi: 10.1086/380999
Dumitrescu, A. M., Liao, X. H., Weiss, R. E., Millen, K., and Refetoff, S. (2006). Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology 147, 4036–4043. doi: 10.1210/en.2006-0390
Ferrara, A. M., Liao, X. H., Gil-Ibañez, P., Bernal, J., Weiss, R. E., Dumitrescu, A. M., et al. (2014). Placenta passage of the thyroid hormone analog DITPA to male wild-type and Mct8-deficient mice. Endocrinology 155, 4088–4093. doi: 10.1210/en.2014-1085
Ferrara, A. M., Liao, X. H., Ye, H., Weiss, R. E., Dumitrescu, A. M., and Refetoff, S. (2015). The thyroid hormone analog DITPA ameliorates metabolic parameters of male mice with Mct8 deficiency. Endocrinology 156, 3889–3894. doi: 10.1210/en.2015-1234
Friesema, E. C., Ganguly, S., Abdalla, A., Manning Fox, J. E., Halestrap, A. P., and Visser, T. J. (2003). Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J. Biol. Chem. 278, 40128–40135. doi: 10.1074/jbc.m300909200
Friesema, E. C., Grueters, A., Biebermann, H., Krude, H., Von Moers, A., Reeser, M., et al. (2004). Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 364, 1435–1437. doi: 10.1016/s0140-6736(04)17226-7
Groeneweg, S., Peeters, R. P., Moran, C., Stoupa, A., Auriol, F., Tonduti, D., et al. (2019). Effectiveness and safety of the tri-iodothyronine analogue Triac in children and adults with MCT8 deficiency: an international, single-arm, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 7, 695–706. doi: 10.1016/S2213-8587(19)30155-X
Groeneweg, S., Van Den Berge, A., Meima, M. E., Peeters, R. P., Visser, T. J., and Visser, W. E. (2018). Effects of chemical chaperones on thyroid hormone transport by MCT8 mutants in patient-derived fibroblasts. Endocrinology 159, 1290–1302. doi: 10.1210/en.2017-00846
Hartley, M. D., Kirkemo, L. L., Banerji, T., and Scanlan, T. S. (2017). A thyroid hormone-based strategy for correcting the biochemical abnormality in X-linked adrenoleukodystrophy. Endocrinology 158, 1328–1338. doi: 10.1210/en.2016-1842
Hennemann, G., Docter, R., Friesema, E. C., De Jong, M., Krenning, E. P., and Visser, T. J. (2001). Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr. Rev. 22, 451–476. doi: 10.1210/edrv.22.4.0435
Horn, S., Kersseboom, S., Mayerl, S., Muller, J., Groba, C., Trajkovic-Arsic, M., et al. (2013). Tetrac can replace thyroid hormone during brain development in mouse mutants deficient in the thyroid hormone transporter mct8. Endocrinology 154, 968–979. doi: 10.1210/en.2012-1628
Iwayama, H., Liao, X. H., Braun, L., Bárez-López, S., Kaspar, B., Weiss, R. E., et al. (2016). Adeno associated virus 9-based gene therapy delivers a functional monocarboxylate transporter 8, improving thyroid hormone availability to the brain of Mct8-deficient mice. Thyroid 26, 1311–1319. doi: 10.1089/thy.2016.0060
Kersseboom, S., Horn, S., Visser, W. E., Chen, J., Friesema, E. C., Vaurs-Barriere, C., et al. (2015). In vitro and mouse studies support therapeutic utility of triiodothyroacetic acid in MCT8 deficiency. Mol. Endocrinol. 28, 1961–1970. doi: 10.1210/me.2014-1135
López-Espíndola, D., García-Aldea, Á, Gómez-De La Riva, I., Rodríguez-García, A., Salvatore, D., Visser, T., et al. (2019). Thyroid hormones availability in the human fetal brain: novel entry pathways and role of radial glia. Brain Struct. Funct. 224, 2103–2119. doi: 10.1007/s00429-019-01896-8
López-Espíndola, D., Morales-Bastos, C., Grijota-Martínez, C., Liao, X. H., Lev, D., Sugo, E., et al. (2014). Mutations of the thyroid hormone transporter MCT8 cause prenatal brain damage and persistent hypomyelination. J. Clin. Endocrinol. Metab. 99, E2799–E2804. doi: 10.1210/jc.2014-2162
Masnada, S., Groenweg, S., Saletti, V., Chiapparini, L., Castellotti, B., Salsano, E., et al. (2019). Novel mutations in SLC16A2 associated with a less severe phenotype of MCT8 deficiency. Metab Brain Dis. 34, 1565–1575. doi: 10.1007/s11011-019-00464-7
Mayerl, S., Muller, J., Bauer, R., Richert, S., Kassmann, C. M., Darras, V. M., et al. (2014). Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J. Clin. Invest. 124, 1987–1999. doi: 10.1172/jci70324
Messier, N., and Langlois, M. F. (2000). Triac regulation of transcription is T(3) receptor isoform- and response element-specific. Mol. Cell. Endocrinol. 165, 57–66. doi: 10.1016/s0303-7207(00)00266-5
Moreno, M., De Lange, P., Lombardi, A., Silvestri, E., Lanni, A., and Goglia, F. (2008). Metabolic effects of thyroid hormone derivatives. Thyroid 18, 239–253. doi: 10.1089/thy.2007.0248
Mughal, B. B., Leemans, M., Lima, De Souza, E. C., Le Mevel, S., Spirhanzlova, P., et al. (2017). Functional characterization of Xenopus thyroid hormone transporters mct8 and oatp1c1. Endocrinology 158, 2694–2705. doi: 10.1210/en.2017-00108
Namba, N., Etani, Y., Kitaoka, T., Nakamoto, Y., Nakacho, M., Bessho, K., et al. (2008). Clinical phenotype and endocrinological investigations in a patient with a mutation in the MCT8 thyroid hormone transporter. Eur. J. Pediatr. 167, 785–791. doi: 10.1007/s00431-007-0589-6
Nicolini, U., Venegoni, E., Acaia, B., Cortelazzi, D., and Beck-Peccoz, P. (1996). Prenatal treatment of fetal hypothyroidism: is there more than one option? Prenat. Diagn. 16, 443–448. doi: 10.1002/(sici)1097-0223(199605)16:5<443::aid-pd892>3.0.co;2-2
Papadimitriou, A., Dumitrescu, A. M., Papavasiliou, A., Fretzayas, A., Nicolaidou, P., and Refetoff, S. (2008). A novel monocarboxylate transporter 8 gene mutation as a cause of severe neonatal hypotonia and developmental delay. Pediatrics 121, e199–e202. doi: 10.1542/peds.2007-1247
Pennock, G. D., Raya, T. E., Bahl, J. J., Goldman, S., and Morkin, E. (1992). Cardiac effects of 3,5-diiodothyropropionic acid, a thyroid hormone analog with inotropic selectivity. J. Pharmacol. Exp. Ther. 263, 163–169.
Remerand, G., Boespflug-Tanguy, O., Tonduti, D., Touraine, R., Rodriguez, D., Curie, A., et al. (2019). Expanding the phenotypic spectrum of Allan-Herndon-Dudley syndrome in patients with SLC16A2 mutations. Dev. Med. Child Neurol. 61, 1439–1447. doi: 10.1111/dmcn.14332
Roberts, L. M., Woodford, K., Zhou, M., Black, D. S., Haggerty, J. E., Tate, E. H., et al. (2008). Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology 149, 6251–6261. doi: 10.1210/en.2008-0378
Takeda, T., Suzuki, S., Liu, R. T., and Degroot, L. J. (1995). Triiodothyroacetic acid has unique potential for therapy of resistance to thyroid hormone. J. Clin. Endocrinol. Metab. 80, 2033–2040. doi: 10.1210/jc.80.7.2033
Trajkovic, M., Visser, T. J., Mittag, J., Horn, S., Lukas, J., Darras, V. M., et al. (2007). Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J. Clin. Invest. 117, 627–635. doi: 10.1172/jci28253
Vatine, G. D., Al-Ahmad, A., Barriga, B. K., Svendsen, S., Salim, A., Garcia, L., et al. (2017). Modeling psychomotor retardation using iPSCs from MCT8-deficient patients indicates a prominent role for the blood-brain barrier. Cell Stem Cell 20, 831.e5–843.e5.
Vatine, G. D., Zada, D., Lerer-Goldshtein, T., Tovin, A., Malkinson, G., Yaniv, K., et al. (2013). Zebrafish as a model for monocarboxyl transporter 8-deficiency. J. Biol. Chem. 288, 169–180. doi: 10.1074/jbc.M112.413831
Verge, C. F., Konrad, D., Cohen, M., Di Cosmo, C., Dumitrescu, A. M., Marcinkowski, T., et al. (2012). Diiodothyropropionic acid (DITPA) in the treatment of MCT8 deficiency. J. Clin. Endocrinol. Metab. 97, 4515–4523. doi: 10.1210/jc.2012-2556
Visser, W. E., Vrijmoeth, P., Visser, F. E., Arts, W. F., Van Toor, H., and Visser, T. J. (2013). Identification, functional analysis, prevalence and treatment of monocarboxylate transporter 8 (MCT8) mutations in a cohort of adult patients with mental retardation. Clin. Endocrinol. 78, 310–315. doi: 10.1111/cen.12023
Wemeau, J. L., Pigeyre, M., Proust-Lemoine, E., D’herbomez, M., Gottrand, F., Jansen, J., et al. (2008). Beneficial effects of propylthiouracil plus L-thyroxine treatment in a patient with a mutation in MCT8. J. Clin. Endocrinol. Metab. 93, 2084–2088. doi: 10.1210/jc.2007-2719
Wilpert, N. M., Krueger, M., Opitz, R., Sebinger, D., Paisdzior, S., Mages, B., et al. (2020). Spatiotemporal changes of cerebral monocarboxylate transporter 8 expression. Thyroid doi: 10.1089/thy.2019.0544 [Epub ahead of print].
Wirth, E. K., Roth, S., Blechschmidt, C., Holter, S. M., Becker, L., Racz, I., et al. (2009). Neuronal 3’,3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J. Neurosci. 29, 9439–9449. doi: 10.1523/JNEUROSCI.6055-08.2009
Zada, D., Tovin, A., Lerer-Goldshtein, T., and Appelbaum, L. (2016). Pharmacological treatment and BBB-targeted genetic therapy for MCT8-dependent hypomyelination in zebrafish. Dis. Model. Mech. 9, 1339–1348. doi: 10.1242/dmm.027227
Zada, D., Tovin, A., Lerer-Goldshtein, T., Vatine, G. D., and Appelbaum, L. (2014). Altered behavioral performance and live imaging of circuit-specific neural deficiencies in a zebrafish model for psychomotor retardation. PLoS Genet. 10:e1004615. doi: 10.1371/journal.pgen.1004615
Keywords: thyroid hormones, brain, neurodevelopment, MCT8, DITPA, sobetirome, TRIAC
Citation: Grijota-Mart\u00EDnez C, B\u00E1rez-L\u00F3pez S, G\u00F3mez-Andr\u00E9s D and Guada\u00F1o-Ferraz A (2020) MCT8 Deficiency: The Road to Therapies for a Rare Disease. Front. Neurosci. 14:380. doi: 10.3389/fnins.2020.00380
Received: 28 January 2020; Accepted: 27 March 2020;
Published: 28 April 2020.
Edited by:
Riccarda Granata, University of Turin, ItalyReviewed by:
Ulrich E. M. Schweizer, University of Bonn, GermanyLior Appelbaum, Bar-Ilan University, Israel
Copyright © 2020 Grijota-Martínez, Bárez-López, Gómez-Andrés and Guadaño-Ferraz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmen Grijota-Martínez, Y2dyaWpvdGFAaWliLnVhbS5lcw==; Ana Guadaño-Ferraz, YWd1YWRhbm9AaWliLnVhbS5lcw==