 Luis A. De Jesús-Olmo*
Luis A. De Jesús-Olmo* Norma Rodríguez
Norma Rodríguez Marcelo FranciaJonathan Alemán-RiosCarlos J. Pacheco-AgostoJoselyn Ortega-TorresRichard NievesNicolás Fuenzalida-Uribe
Marcelo FranciaJonathan Alemán-RiosCarlos J. Pacheco-AgostoJoselyn Ortega-TorresRichard NievesNicolás Fuenzalida-Uribe Alfredo Ghezzi
Alfredo Ghezzi José L. Agosto*
José L. Agosto*- Department of Biology, University of Puerto Rico, Rio Piedras, Puerto Rico
Recent studies have identified the Drosophila brain circuits involved in the sleep/wake switch and have pointed to the modulation of neuronal excitability as one of the underlying mechanisms triggering sleep need. In this study we aimed to explore the link between the homeostatic regulation of neuronal excitability and sleep behavior in the circadian circuit. For this purpose, we selected Pumilio (Pum), whose main function is to repress protein translation and has been linked to modulation of neuronal excitability during chronic patterns of altered neuronal activity. Here we explore the effects of Pum on sleep homeostasis in Drosophila melanogaster, which shares most of the major features of mammalian sleep homeostasis. Our evidence indicates that Pum is necessary for sleep rebound and that its effect is more pronounced during chronic sleep deprivation (84 h) than acute deprivation (12 h). Knockdown of pum, results in a reduction of sleep rebound during acute sleep deprivation and the complete abolishment of sleep rebound during chronic sleep deprivation. Based on these findings, we propose that Pum is a critical regulator of sleep homeostasis through neural adaptations triggered during sleep deprivation.
Introduction
It is well established, even by our own experience, that the urge to sleep increases as a function of time awake. This urge, or sleep drive, triggers a prolonged compensatory sleep after the organism is sleep deprived (Daan et al., 1984; Allada et al., 2017). This compensatory sleep, which is also called sleep rebound, is a key indicator of the homeostatic regulation of sleep (Vyazovskiy et al., 2009). In this process, deviations from a reference level of sleep are compensated, i.e., lack of sleep fosters compensatory increase in the intensity and duration of sleep, whereas excessive sleep counteracts the sleep need (Tobler and Achermann, 2007). More than a century of sleep research has made important progress in understanding the function of sleep and its regulatory circuitry, but the molecular basis of sleep homeostasis remains elusive (Sehgal et al., 2007; Cirelli and Tononi, 2008; Siegel, 2008; Donlea, 2017). Understanding the molecular mechanisms involved in the regulation of sleep homeostasis is key for the overall understanding the regulation of both the sleep circuit and the sleep function. To achieve that level of understanding, we need to study the link between molecular markers, sleep brain circuits and homeostatic sleep behavior.
The fruit fly Drosophila melanogaster is an ideal model to study the molecular markers impacting sleep behavior. Sleep rebound is a stable phenotype in flies which shares most major features of mammalian sleep homeostasis (Huber et al., 2004). Drosophila shows easily measurable and recognizable sleep patterns linked to reduced brain activity (Nitz et al., 2002; Van Swinderen et al., 2004), limited sensory responsiveness during sleep and display a robust homeostatic sleep rebound (Hendricks et al., 2000; Shaw et al., 2000) as occurs in mammals. In addition, it has been demonstrated that humans and fruit flies have a common sleep control mechanism involving GABA receptors in brain neurons linked to the circadian clock (Parisky et al., 2008; Chung et al., 2009). Moreover, fly genetics has been used as a tool to validate human sleep biomarkers affected by sleep deprivation (Thimgan et al., 2013). Hence, we circumscribed our study of the relationship between homeostatic markers and sleep behavior to the fly model.
Recent studies have shown that two structures of Drosophila’s brain central complex, the Ellipsoid Body (EB) and the fan body (FB), induce sleep when artificially activated, and produce insomnia, when inhibited (Donlea et al., 2011; Liu et al., 2016). Other studies have shown that neuronal microcircuits in the mushroom body (MB) drives rebound recovery after sleep deprivation (Sitaraman et al., 2015). Follow up studies have produced important progress by identifying dopamine as the neuromodulator responsible for the homeostatic switch operation between sleep/wake, which is mediated by potassium currents (Pimentel et al., 2016). Homeostatic sleep seems to be controlled by the dorsal FB neurons, which are electrically active during wake and electrically silent during rest (Pimentel et al., 2016). These studies point to the regulation of neuronal excitability as an important effector of the sleep regulation. Nevertheless, the underlying molecular framework that connects neuronal excitability with sleep behavior is a relatively unexplored area of research.
Several genes have been identified to regulate normal sleep, but only a few genes have been linked to the molecular regulation of homeostatic sleep compensation after sleep deprivation. A mutation in the Shaker (Sh) gene, which encodes a voltage dependent potassium channel involved in membrane repolarization, increases neuronal excitability and reduces normal sleep (Cirelli et al., 2005), but fails to alter sleep rebound. Interestingly, the Shaker activator sleepless (sss), which encodes for a brain-enriched glycosyl-phosphatidylinositol-anchored protein, impairs sleep rebound (Koh et al., 2008), perhaps by a mechanism independent of Shaker. The gene crossveinless (cv-c), which codes for a Rho-GTPase-activating protein, is necessary for dorsal FB neurons to transduce the excitability produced by sleep pressure into homeostatic sleep (Donlea et al., 2014). Knocking down the Cullin 3 (Cul3) ubiquitin ligase gene and its putative adaptor insomniac (inc), reduces sleep rebound after sleep deprivation (Pfeiffenberger and Allada, 2012). Mutants of fragile X mental retardation gene (Fmr1), a translational inhibitor that causes the most common form of inherited mental retardation in humans, have also been reported to reduce sleep rebound (Bushey et al., 2009). In addition, it was reported that interfering with the expression of the genes sandman (sand) and Sh in the dorsal FB neurons, increased or decreased sleep respectively as part of the sleep/wake switch (Pimentel et al., 2016). The regulatory picture presented by these genes and the other neuromodulators and proteins known to affect homeostatic sleep compensation seems far from complete, although together, they also point to neuronal excitability as a key component of sleep homeostatic regulation.
Unregulated neuronal excitability may lead to a potentially disruptive positive feedback. To cope with this, neurons have evolved compensatory (homeostatic) mechanisms to reduce excitability. The mechanisms by which neurons stabilize firing activity have been collectively termed “homeostatic plasticity” (Marder and Prinz, 2003; Turrigiano and Nelson, 2004; Davis, 2006; Turrigiano, 2008, 2012; Pozo and Goda, 2010). Therefore, it is plausible that wake promoting neurons, after prolonged times of wakefulness, would utilize one of the homeostatic plasticity mechanisms to regulate neuronal excitability. In this study, we begin to explore the relationship between neuronal homeostasis mechanisms and sleep regulation by testing the role of the neuronal homeostasis gene pumilio (pum) on the regulation of compensatory sleep.
The protein encoded by pum is characterized by a highly conserved RNA-binding domain, which acts as a post-transcriptional repressor of mRNA targets. Binding occurs through an RNA consensus sequence in the 3’-UTR of target transcripts—the Pumilio Response Element (PRE), 5′-UGUANAUA-3′, that is related to the Nanos Response Element (NRE) (Wang et al., 2018). While it was originally described in Drosophila for its critical role in embryonic development, Pum has an important role in the development of the nervous system. Pum is known for controlling the elaboration of dendritic branches (Ye et al., 2004), and is also required for proper adaptive responses and memory storage (Dubnau et al., 2003). Evidence of its regulatory role in neuronal excitability (Schweers et al., 2002) and homeostatic processes include Pum’s repression of translation of the Drosophila voltage-gated sodium channel (paralytic) in an activity dependent manner (Mee et al., 2004; Muraro et al., 2008). Pum-mediated repression of the voltage gated sodium channel plays a pivotal role in the regulation of neuronal homeostasis, given the central role of the sodium channel in the regulation of membrane excitability (Weston and Baines, 2007). Furthermore, Pum was found to be necessary for the homeostatic compensation of increased neuronal activity, or what is known as homeostatic synaptic depression (Fiore et al., 2014). In addition, Pum has been found to influence synaptic bouton size/number, synaptic growth and function by regulating expression of eukaryotic initiation factor 4E (eIF4E), which is the limiting factor for the initiation of the CAP dependent translation in Eukaryotes (Menon et al., 2004; Vessey et al., 2006; Cao et al., 2009). Pum was our first choice to study neuronal homeostasis effects on compensatory sleep because microarray experiments show that pum is expressed in Pdf-expressing neurons, which are key circadian cells known to promote wakefulness in Drosophila (Parisky et al., 2008; Kula-Eversole et al., 2010). With over 1000 potential targets and many others indirect targets through its eIF4E regulatory role, based on the cumulative evidence, Pum could be considered a master regulator of neuronal homeostatic processes (Menon et al., 2004; Gerber et al., 2006; Chen et al., 2008).
Studies exploring the mechanisms of neuronal homeostasis often involve long-term manipulations of neural activity, spanning from 48 h to the entire life span (Turrigiano et al., 1998; Turrigiano, 2012; Davis, 2013). Moreover, studies linking pum with neuronal homeostasis primarily use genetic manipulations that alter neuronal activity throughout the lifetime of the organisms (Mee et al., 2004; Weston and Baines, 2007; Muraro et al., 2008). Thus, in this study we decided to explore the role of pum in the regulation of compensatory sleep induced by chronic (long-term) sleep deprivation as well as acute sleep deprivation (SD).
Results
Knockdown of pumilio Abolishes Sleep Rebound After Chronic Mechanical Sleep Deprivation
We knocked down the expression of pum using a transgenic fly containing a pum RNA interference construct (pumRNAi) under control of the upstream activating sequence (UAS) of the yeast transcription factor Gal4. To activate the UAS-pumRNAi we used a second transgenic construct that expressed Gal4 under control of the timeless (tim) gene promoter (tim-Gal4). When both transgenes are present in the same fly (tim-Gal4/UAS-pumRNAi), the pumRNAi construct is expressed constitutively in tim expressing neurons. We selected the tim-Gal4 driver because it is a strong and broadly expressed promoter targeting circadian cells found in several brain structures including the wake promoting, Pdf-expressing ventral lateral neurons and glia (Kaneko and Hall, 2000).
In our first set of experiments, we subjected the parental controls (tim-Gal4/+), the pumRNAi (UAS-pumRNAi/tim-Gal4) experimental flies and their “sibling” control flies (UAS-pumRNAi/+), which carry the pumRNAi construct by itself and has a closer genetic similarity to the experimental flies than parental controls, to a chronic (84 h) mechanical SD protocol (see section “Materials and Methods”). The results from the chronic SD showed a strong effectiveness of the sleep deprivation method during the first 12 h (Figure 1A). As time progressed, we noticed a gradual increase in the amount of sleep in all the sleep deprived genotypes during sustained mechanical deprivation (Figures 1A–C), which we first though could be an adaptation to the SD method. However, this increase in sleep through time did not seem to affect the sleep rebound, as both parental and “sibling” control flies were able to produce a normal sleep rebound pattern that initiated at the 84th hour—immediately after the SD protocol was terminated (Figures 1A,B). If we look more closely at the recovery period in Figure 1C (which could be better viewed in Figure 2D that has the x axis expanded), we can see that sleep-deprived flies with silenced pum are more active than non-deprived flies. This difference becomes more obvious in the sleep recovery plot (Figure 1E), where sleep levels are normalized against the non-deprived flies, and it is evident that flies with silenced pum begin the sleep recovery period close to zero percent but slowly decreased with time. This tendency continued decreasing for the next 48–72 h until it stabilized (Supplementary Figure S2). To determine if this lack of sleep rebound was related to adaptation to the SD method leading to insufficient sleep deprivation, we quantified the sleep loss of all experimental groups (Figure 1D). This quantification of cumulative sleep loss during the 84-h deprivation period showed a significant difference between the pumRNAi/tim-Gal4 flies and the tim-Gal4/+ parental control flies, but no difference between the pumRNAi/tim-Gal4 flies and the UAS-pumRNAi/+ “sibling” controls (Figure 1D). The fact that this difference was not significant between the genetically closer “sibling” control and experimental pumRNAi flies, suggests the difference in deprivation effectiveness between parental controls and pumRNAi flies could be due to an adaptation to the SD method influenced by the genetic background. We used this sleep lost value to normalize the sleep recovery calculation (see section “Materials and Methods”) (Figure 1E). The results for sleep recovery show a normal recovery pattern, as indicated by the increase in cumulative sleep recovered during the first hours after SD. This result is indicative of compensatory sleep present in both parental and “sibling” controls after sleep deprivation (Figure 1E). However, pumRNAi flies showed a negative sleep recovery, which indicates pumRNAi flies were even more active than non-deprived flies after 84 h. of continuous deprivation (Figure 1E). This loss of homeostatic regulation in the recovery of pumRNAi flies was maintained up to 96 h post-deprivation with no mortality in any of the groups after 108 h post deprivation (see Supplementary Figure S3).
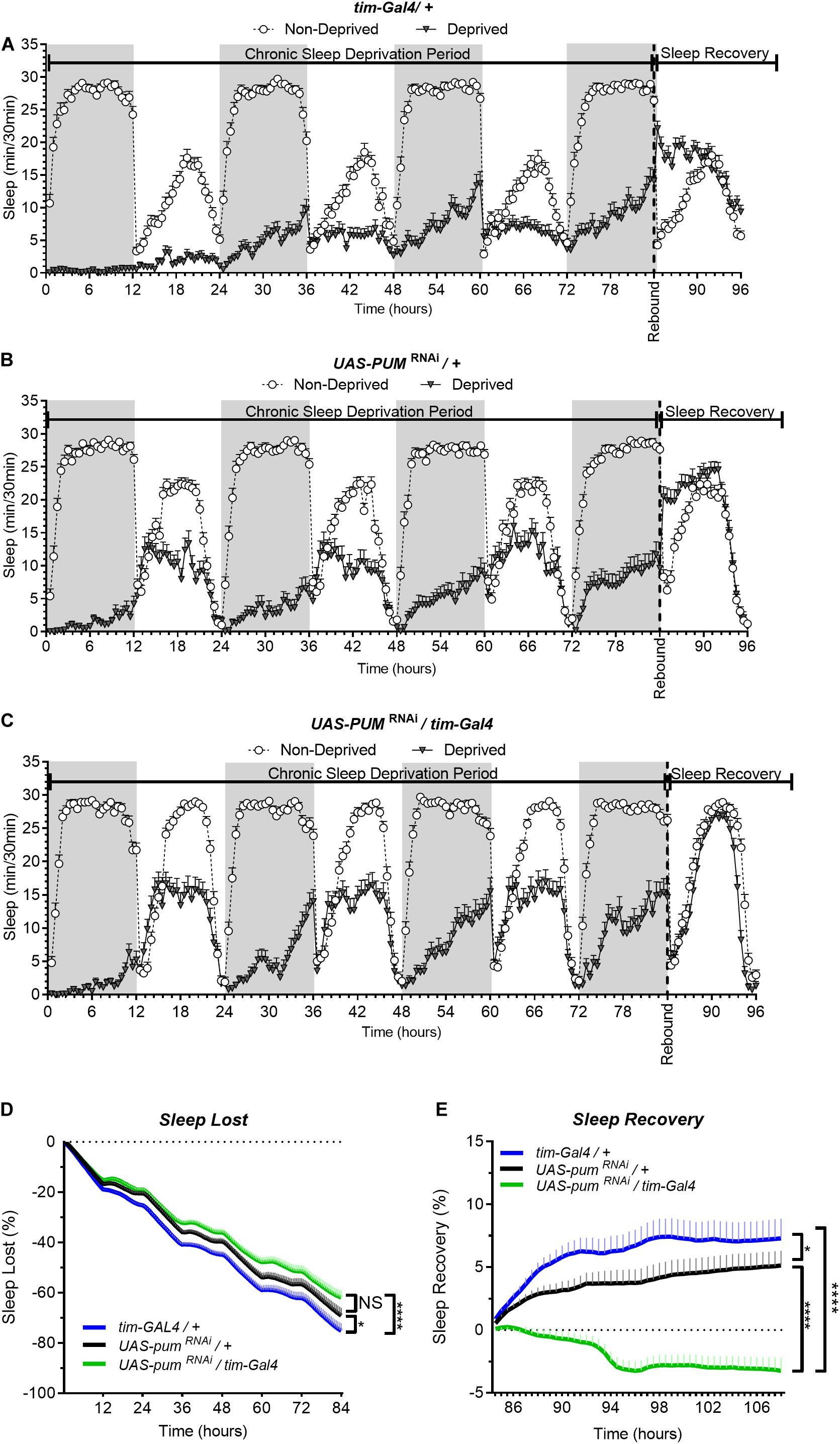
Figure 1. Knockdown of pum eliminates sleep recovery after chronic mechanical sleep deprivation. Sleep comparison of UAS-pumRNAi/tim-Gal4 (experimental) vs. tim-Gal4/+ (parental) flies and UAS-pumRNAi/+ (“sibling” controls) during chronic SD. The X axis indicates time after start of sleep deprivation. (A–C) Depiction of sleep activity during the sleep deprivation and sleep rebound period for all genotypes. (D) Cumulative sleep lost during deprivation expressed as a percentage of total sleep in non-deprived flies of the same genotype. Two-way ANOVA using “genotype” as a factor and “time” as a repeated measure showed significant differences in genotypes [F(2,132) = 11.24 P < 0.0001], time [F(167,22044) = 1033, P < 0.0001] and interaction [F(334,22044) = 3.066, P < 0.0001]. Post hoc analysis using Dunnett’s multiple comparisons test showed significant differences between UAS-pumRNAi/tim-Gal4 vs. tim-Gal4/+ flies (P < 0.0001). (E) Percent sleep recovery after SD. Two-Way ANOVA with repeat measures indicated significant differences in genotypes [F(2,132) = 18.58, P < 0.0001] and interaction [F(94,6204) = 13.73 P < 0.0001]. Post hoc analysis using Sidak’s multiple comparisons test comparing both control genotypes against experimental flies, revealed significant differences (P < 0.0001) between UAS-pumRNAi/tim-Gal4 vs. tim-Gal4/+ flies and UAS-pumRNAi/+ throughout the recovery period. The data shown represents two experiments with the following sample sizes (N): tim-Gal4/+ Non-Deprived (N = 56) and Deprived (N = 53); UAS-pumRNAi/+ Non-Deprived (N = 60) and Deprived (N = 35); UAS-pumRNAi/+ Non-Deprived (N = 63) and Deprived (N = 39). Because the calculations of sleep lost and sleep recovery involve both the Non-Deprived and Deprived groups (see section “Materials and Methods”), the N for panels (D,E) is equal to the N of the Deprived group. SD. Data points and error bars represent means ± SEM. Stars indicate significance level (*p < 0.05; ****p < 0.0001).
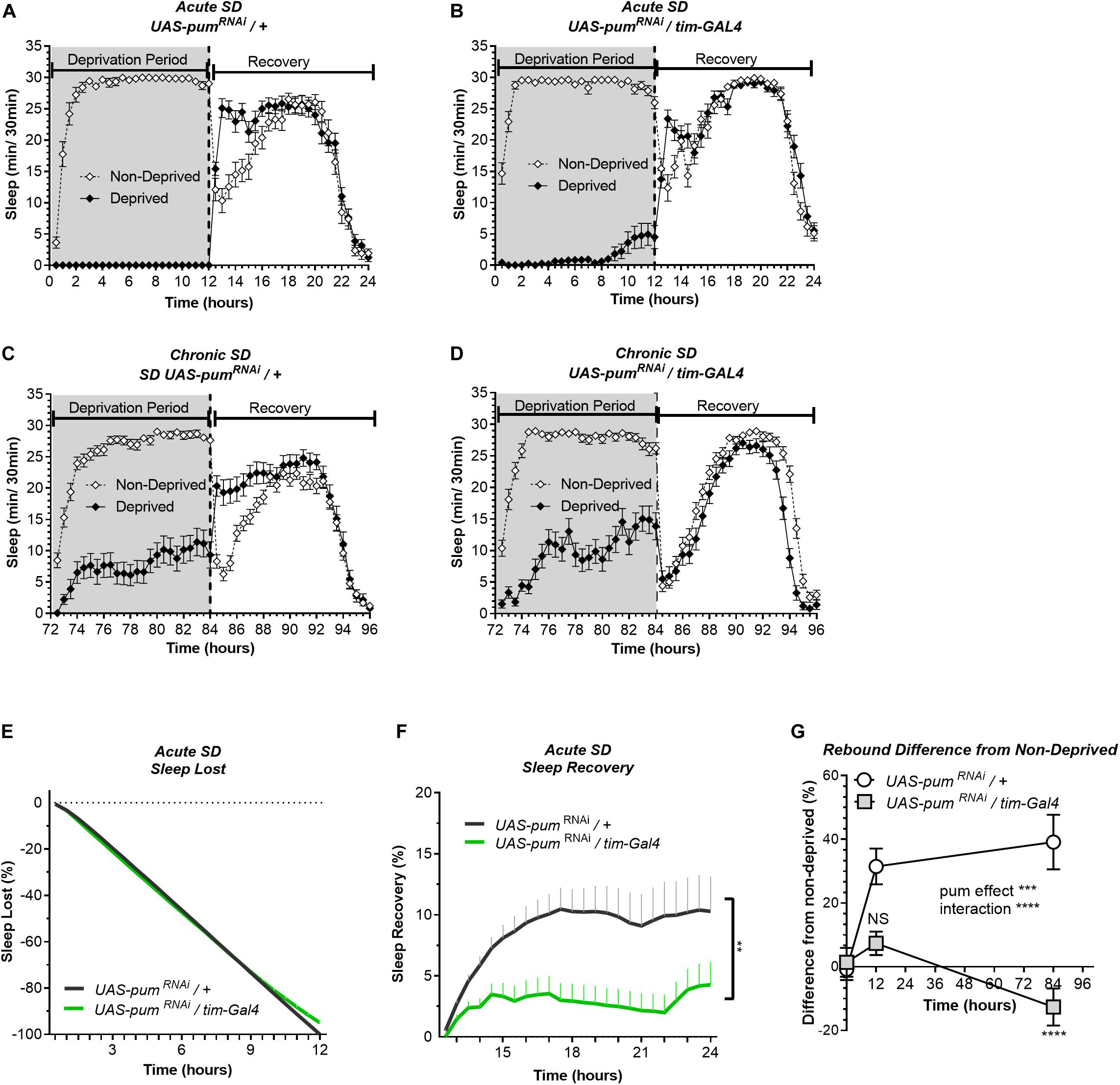
Figure 2. Knockdown of pum differentially reduces sleep recovery in acute vs. chronic SD. Sleep comparison of UAS-pumRNAi/tim-Gal4 (experimental) vs. UAS-pumRNAi/+ (“sibling” controls) during acute (12 h) mechanical SD. The X axis indicates time after sleep deprivation. (A,B) Depiction of sleep activity during the sleep deprivation and sleep rebound period for both genotypes during acute SD. (C,D) Depiction of the sleep activity during sleep deprivation and sleep rebound period for both genotypes during hours 72–96 of chronic mechanical SD included for ease of comparison. The y-axis shows the number of minutes that flies slept in intervals of 30 min. (E) Cumulative sleep lost during deprivation expressed as a percentage of total sleep in non-deprived flies of the same genotype. Two-way ANOVA, using “genotype” as a factor and “time” as a repeated measure, did not showed significant differences between the genotypes (P = 0.8664). (F) Percent sleep recovery after SD. Two-Way ANOVA with repeat measures indicated significant difference in genotypes [F(1,58) = 7.114, P < 0.0099] and interaction [F(23,1334) = 3.054, P < 0.0001]. (G) Percent difference in rebound between deprived and non-deprived flies after acute and chronic sleep deprivation protocols of UAS-pumRNAi/+ and UAS-pumRNAi/tim-Gal4 flies. Two-way ANOVA with repeated measures showed a significant difference in genotype [F(1,91) = 13.72, P = 0.0004] and time vs. genotype interaction [F(2,106) = 13.97, P < 0.0001]. Post hoc analysis using Tukey’s multiple comparisons test revealed significant differences between UAS-pumRNAi/+ and UAS-pumRNAi/tim-Gal4 at 84 h of deprivation (P < 0.0001) no difference was observed at 12 h (acute SD) (P = 0.0735). The data shown represents one experiment with the following sample sizes (N): UAS-pumRNAi/+ Non-Deprived (N = 31) and Deprived (N = 32); UAS-pumRNAi/tim-Gal4 Non-Deprived (N = 31) and Deprived (N = 28). Because the calculations of sleep lost and sleep recovery involve both the Non-Deprived and Deprived groups (see section “Materials and Methods”), the N for panels (E,F) is equal to the N of the Deprived group. Data points and error bars represent means ± SEM. Stars indicate significance level (**p < 0.01; ***p < 0.001; ****p < 0.0001).
In our experiments, the UAS-pumRNAi/+ control lines are “siblings” of the UAS-pumRNAi/tim-Gal4 flies. Meanwhile the tim-Gal4/+ controls were generated directly by crossing the parental tim-Gal4 line with a non-transgenic wild type (CS), which can introduce differences in genetic background. Thus, our conclusions are based mostly on the results from “sibling” controls because they have a greater genetic similarity, which results in a more similar baseline sleep pattern than parental controls (Figures 1A–C). Hence, for the following acute SD experiments, parental controls were not used.
Pumilio Regulates Sleep Rebound Differentially Between Acute and Chronic Mechanical Sleep Deprivation
The results from the 12 h acute SD showed sleep lost effectivity close to 100% for both pumRNAi and “sibling” controls (Figures 2A,B). During the deprivation period (0–12 h), the cumulative sleep loss in deprived flies did not show a significant difference between the two genotypes (Figure 2E). Once again, controls showed an effective sleep rebound (Figure 2A), while pumRNAi flies showed a reduction in sleep rebound (Figure 2B). However, this time the rebound was not completely abolished as we observed during chronic SD (Figure 2B vs. Figure 2D). We included the chronic deprivation rebound period as a point of comparison between acute vs. chronic (Figures 2C,D). The results from the acute SD sleep recovery resembled the results from chronic SD with a normal rebound in “sibling” controls and reduced sleep recovery in pumRNAi flies. Nevertheless, the sleep recovery of pumRNAi flies was not negative as we observed during chronic SD (Figure 2F). When acute vs. chronic SD results are compared (Figure 2G), we see significant differences, not only between the genotypes, but also within pumRNAi flies exposed to acute vs. chronic SD, while the rebound difference of the “sibling” control between acute vs. chronic SD remains constant. These results suggest that pum differentially regulates acute vs. chronic SD. This interpretation is in fact reinforced by our molecular experiments contrasting gene expression changes between acute and chronic SD as reported in the Supplementary Figure S5.
So far, our findings link the duration of sleep deprivation to increased pum regulation, which is consistent with our expectations. Since we observed greater homeostatic changes during chronic SD, we continued throughout the study using only chronic SD to measure pum’s regulatory effects in compensatory sleep. The difference in sleep rebound between pumRNAi vs. parental flies does not seem to be related to non-specific effects of the genetic background affecting baseline sleep because daytime baseline sleep of pumRNAi flies is higher than both parental and “sibling” controls (Supplementary Figure S2). If baseline sleep would have been a contributing factor for the recovery results, we should have expected a higher sleep rebound, not lower. The fact that we obtained a lower rebound indicates a Pum knockdown effect rather that genetic differences influencing baseline sleep are the culprit of our results.
Pumilio Mutants Show Reduced Sleep Rebound
To confirm the effects of pum knockdown in sleep homeostasis independently of transgenic flies, we tested mutant fly lines to further validate our results. First, we selected the classical loss of function allele pum13 (also known as pum680). Pum13 is a dominant negative allele that bears a single amino acid substitution, which not only knocks down Pum function but also interferes with normal Pum function in heterozygotes (Wharton et al., 1998). Thus, in addition to the semi-lethal pum13 homozygous mutants, we used pum13/TM3 heterozygotes in our experiments.
The sleep deprivation produced similar sleep lost amounts in each of the lines tested. Figures 3A–C,D). Nonetheless, the sleep recovery showed a significant difference between both wild type (+/+) and pum13/+flies compared to pum13/pum13 flies (Figure 3E). By the end of the recovery period, the differences between pum13/+ and the knockout pum13/pum13 were still maintained. Moreover, pum13/pum13 escaper flies completely abolished rebound to chronic sleep deprivation for the first 12 h of the recovery period (Figure 3E). This suggests that differential pum levels between the heterozygote and the pum13 homozygote, have correlative regulatory effects in sleep rebound.
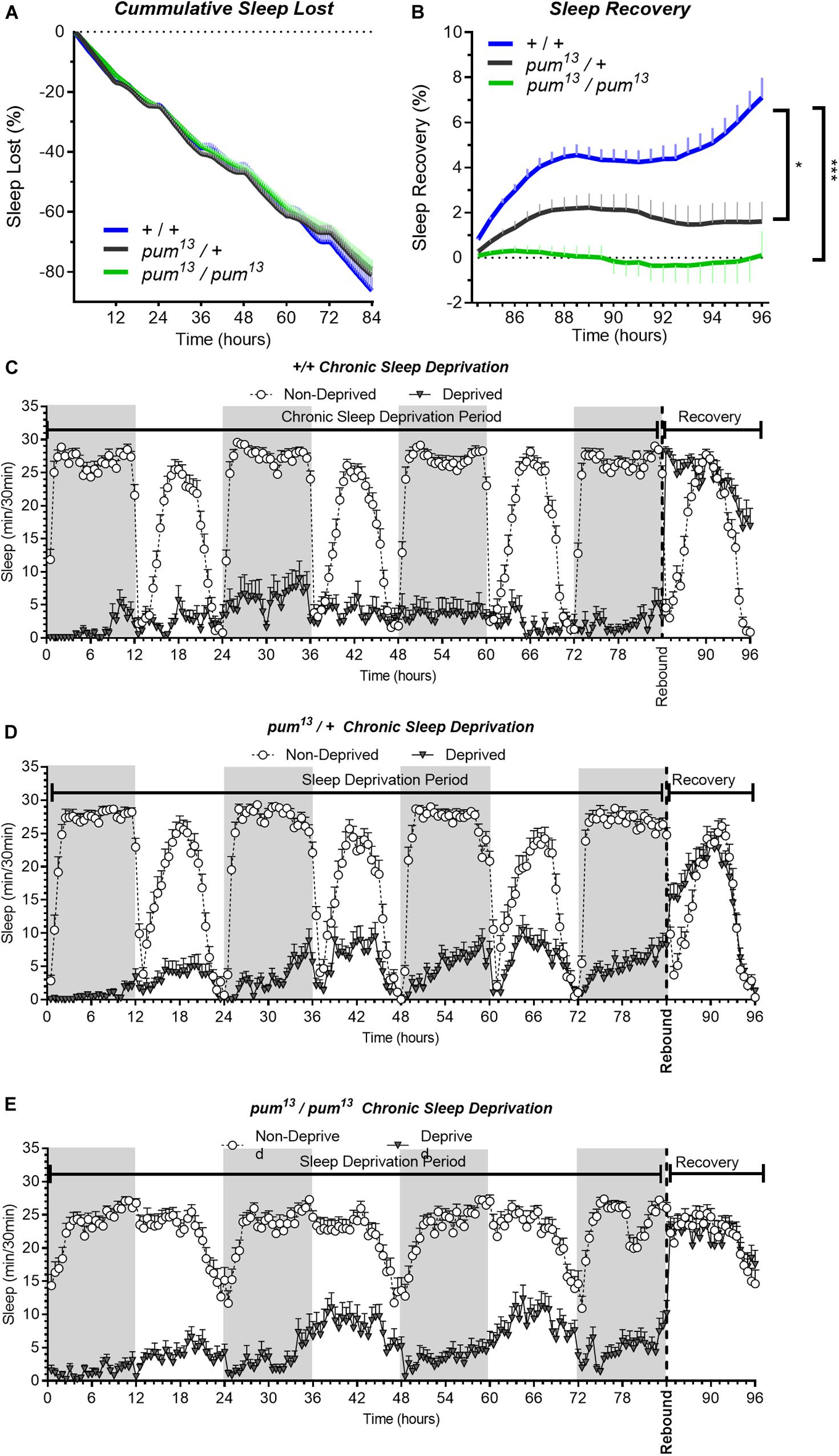
Figure 3. pum13 mutant show reduced sleep rebound after chronic SD. Sleep comparison of wild type, heterozygous and homozygous flies for the pum13 allele. (A–C) Depiction of sleep activity during the sleep deprivation and sleep rebound period for all genotypes. The X axis indicates time after the start of the sleep deprivation protocol. The y-axis shows the number of minutes that flies slept in intervals of 30 min. (D) Cumulative sleep lost during deprivation expressed as a percentage of total sleep in non-deprived flies of the same genotype. Two-way ANOVA, using “genotype” as a factor and “time” as a repeated measure, did not show significant differences between the genotypes [F(2,63) = 0.3635, P = 0.6967]. (E) Percent sleep recovery after SD. Two-Way ANOVA with repeat measures indicated significant difference in genotypes [F(2,63) = 11.29, P < 0.0001] and interaction [F(46,1449) = 5.667, P < 0.0001]. Post hoc analysis using Uncorrected Fisher’s LSD comparisons test comparing all genotypes against pum13/pum13 flies revealed significant differences with pum13/+ flies (P = 0.0319) and with pum13/pum13 (P < 0.0001). The comparison between pum13/+ and pum13/pum13 show no difference (P = 0.0728). The data shown represents one experiment with the following sample sizes (N): (1) Canton-S (+/+), Non-Deprived (N = 30) and Deprived (N = 17); pum13/+, Non-Deprived (N = 28) and Deprived (N = 28); pum13/pum13, Non-Deprived (N = 30) and Deprived (N = 22). Because the calculations of sleep lost and sleep recovery involve both the Non-Deprived and Deprived groups (see methods), the N for panels (A,B) is equal to the N of the Deprived group. Data points and error bars represent means ± SEM. Stars indicate significance level (*p < 0.05; ***p < 0.001).
Additionally, we used the p-element insertion pum allele, Milord-1, to confirm the mutant results with another independent line. This line was generated by single transposon mutagenesis inserted in the pum transcriptional unit (Dubnau et al., 2003). We compared this line with controls obtained from a wild type stock Canton S flies. As expected, Milord-1 flies showed a significant sleep rebound reduction (Figure 4D). Although there was a significant sleep lost difference between the genotypes at the end of the deprivation period (Figure 4C), the ANOVA table results did not show a significant difference between the genotypes for the whole deprivation period. In addition, the sleep recovery calculation normalizes by the sleep lost, therefore, any sleep lost differences affecting the results have already been considered.
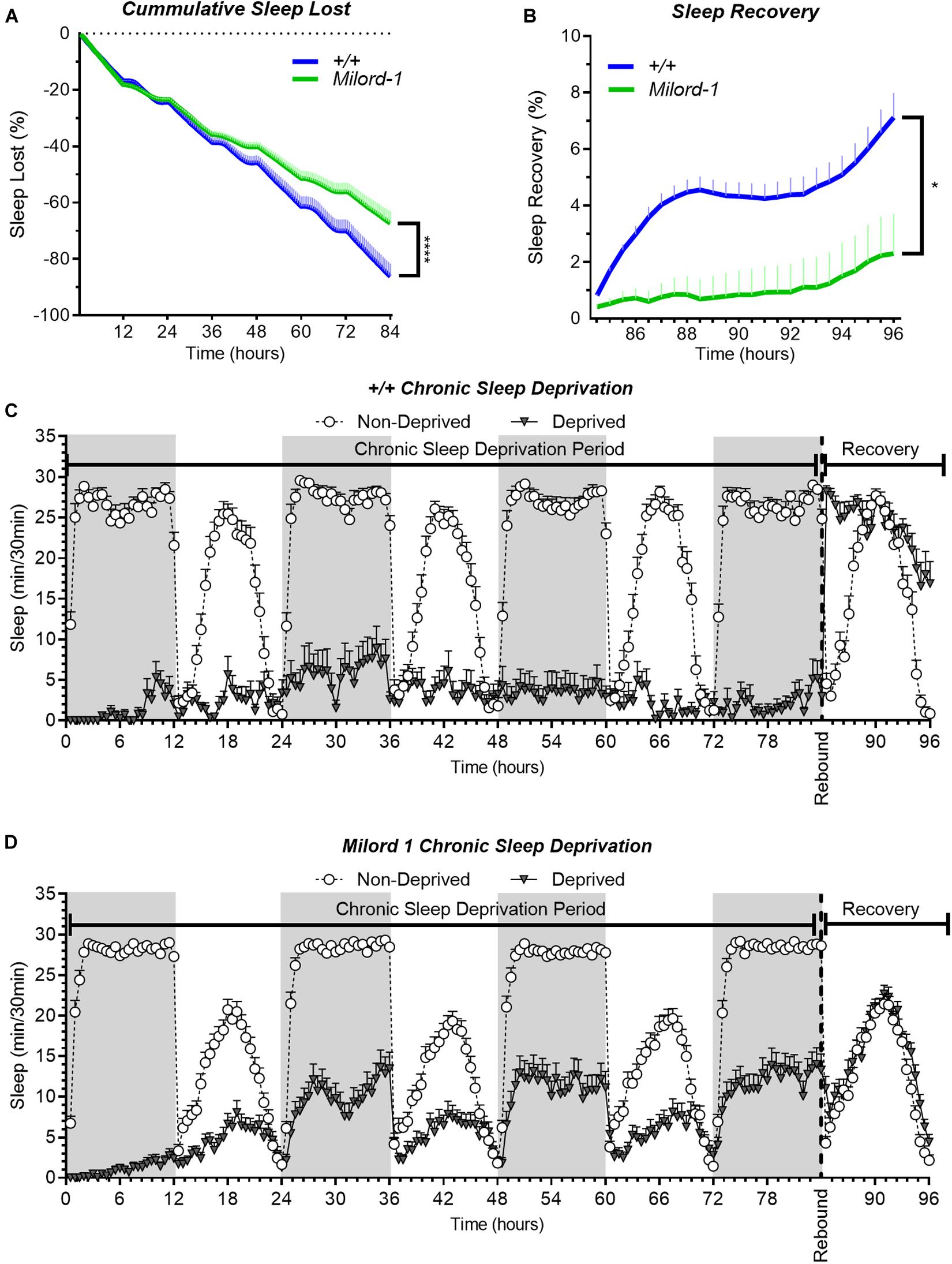
Figure 4. The Milord-1 fly line shows reduced sleep rebound after chronic SD. Sleep comparison of wild type and Milord-1 flies. (A,B) Depiction of sleep activity during the sleep deprivation and sleep rebound period for all genotypes. The X axis indicates time after the start of the sleep deprivation protocol. The y-axis shows the number of minutes that flies slept in intervals of 30 min. (C) Cumulative sleep lost during deprivation expressed as a percentage of total sleep in non-deprived flies of the same genotype. Two-way ANOVA using “genotype” as a factor and “time” as a repeated measure showed no significant differences between the genotypes [F(1,58) = 3.712, P = 0.0589]. (D) Percent sleep recovery after SD. Two-Way ANOVA with repeat measures indicated significant difference in genotypes [F(1,58) = 5.193, P = 0.0264] and interaction [F(23,1334) = 1.695 P < 0.0213]. The data shown represents two experiments with the following sample sizes (N): Canton-S (+/+) Non-Deprived (N = 30) and Deprived (N = 17); Milord-1 Non-Deprived (N = 62) and Deprived (N = 45). Because the calculations of sleep lost and sleep recovery involve both the Non-Deprived and Deprived groups (see methods), the N for panels (A,B) is equal to the N of the Deprived group. Data points and error bars represent means ± SEM. Stars indicate significance level (*p < 0.05; ****p < 0.0001).
Pumilio Knockdown in Tim Neurons Increases the Number of Pdf Puncta
In order to directly observe the effects of pum knockdown in the Drosophila brain, we selected the small ventral Lateral Neurons (sLNv) which are an important part of the circadian wake promoting system in Drosophila (Parisky et al., 2008). This subset of tim positive neurons (Seluzicki et al., 2014) are characterized by the secretion of the neuropeptide Pigment Dispensing Factor (Pdf). We wanted to evaluate the effect of pum knockdown on the morphology of these neurons firstly because it has been reported that flies lacking pum exhibit abnormalities in dendrite morphology (Ye et al., 2004) and secondly because Pdf secretion have been shown to increase during sleep deprivation in Drosophila neurons (Bushey et al., 2011). Therefore, we should expect to see an overall increase in Pdf immunofluorescent signal as a result of the sleep deprivation method in sleep deprived flies.
For illustration purposes, we selected 4 representative brain lobes from each of the 4 experimental groups in the study: non-deprived “sibling” controls (Figure 5A), deprived “sibling” controls (Figure 5B), non-deprived pumRNAi (Figure 5C), and deprived pumRNAi flies (Figure 5D) and adjusted all the images to the same gain. We did not observe morphological abnormalities in any of the groups. The effect of pum knockdown in pumRNAi flies could be clearly observed in both the fluorescence intensity and the number of Pdf puncta by comparing non-deprived “sibling” controls vs. non-deprived pumRNAi flies (Figure 5A vs. Figure 5C). Quantification from anti-Pdf immunofluorescence throughout the sLNv arbor in both the genetic “sibling” control and pumRNAi flies, based on counting Pdf-reactive (Pdf+) puncta, showed that pumRNAi non-deprived flies had a significant increase in Pdf puncta when compared with non-deprived controls (Figure 5E). Nevertheless, we did not see a significant increase in deprived pumRNAi flies vs. deprived “sibling” controls (Figure 5D vs. Figure 5B) perhaps due to some ceiling effect when the increase observed in pumRNAi flies (Figure 5A vs. Figure 5C) is combined with the increase seen during deprivation (Figure 5A vs. Figure 5B). These results indicates that pum knockdown has a direct effect circadian Pdf wake promoting neurons, without affecting their morphology.
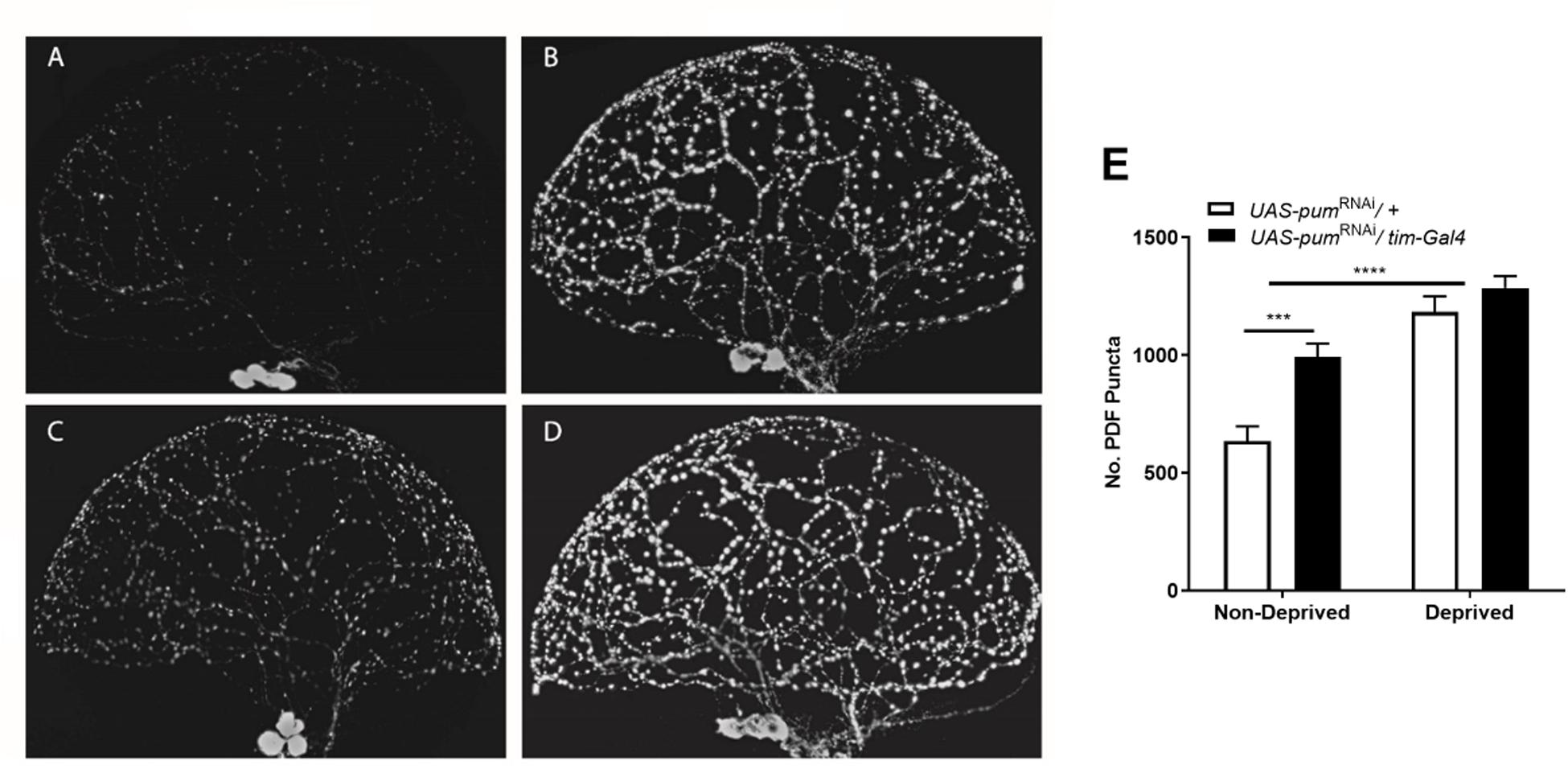
Figure 5. Knockdown of pum flies showed increased Pdf neurons puncta and pdf expression after chronic sleep deprivation. (A–D) Representative confocal images of anti-pdf immunofluorescence of Drosophila brain optic lobes showing Pdf neurons terminals. All panels were adjusted to the same gain intensity. (A) Representative immunofluorescence for UAS-pumRNAi/+ non-deprived “sibling” controls (n = 10). (B) Representative immunofluorescence for UAS-pumRNAi/+ deprived “sibling” controls (n = 12). (C) Representative immunofluorescence for UAS-pumRNAi/tim-Gal4 non-deprived experimental flies (n = 12). (D) Representative immunofluorescence for UAS-pumRNAi/tim-Gal4 deprived experimental flies (n = 9). (E) Quantification of Pdf neurons puncta from anti-Pdf immunofluorescence labeled Drosophila brain lobes. A Two-way ANOVA shows a significant interaction [p = 0.0438, F(1,39) = 4.339] and significant differences between the factors deprivation [p = 0.0006, F(1,39) = 14.04] and genotype [p < 0.0001, F(1,39) = 47.55]. Post hoc using Tukey’s multiple comparisons test showed differences within genotypes for deprived vs. non-deprived flies. Stars indicate significance level (***p < 0.001; ****p < 0.0001).
Discussion
Through the behavioral data of transgenic RNAi knockdowns and mutant behavioral analysis, our results indicate that pum is necessary for the compensatory sleep behavior displayed after sleep deprivation in Drosophila. The pum-dependent regulation of sleep compensation increases as sleep needs increases as demonstrated by the sleep rebound differences between acute vs. chronic SD, together with the differential gene expression patterns. Compensatory sleep rebound after a 12-h sleep deprivation protocol (acute SD) was slightly reduced by knockdown of pum in tim neurons, but completely abolished after 84-h of sleep deprivation (chronic SD).
Interestingly, we also observed that pumRNAi flies have increased day-time sleep in non-deprived conditions (Figure 1 and Supplementary Figure S2A), suggesting that other sleep behaviors are also regulated by pum. This effect of pum could perhaps be explained by the increased expression levels of Gad1 and Shal in pumRNAi non-deprived flies, as both genes are associated with a depression in overall neural activity (Supplementary Figure S5A). Additionally, the role of pum on regulating baseline sleep seems to be disconnected from its role in regulating sleep rebound. For instance, the daytime baseline sleep, in pumRNAi flies is about two times the baseline of both control flies (Supplementary Figure S2A), but the same flies showed no rebound sleep after SD, suggesting that the homeostatic sleep rebound is independently regulated from baseline sleep. This interpretation is supported by reports from other groups. Shaw et al. (2002) previously reported that cycle (cyc01) mutants showed an exaggerated response to sleep deprivation, which was 3 times as high as baseline sleep. In addition, Seidner et al. (2015), found evidence suggesting that baseline sleep and homeostatic sleep are regulated by distinct neural circuits, which has been also independently corroborated by other studies (Liu et al., 2016). As reported by Seidner et al. (2015), thermo-activation of selected cholinergic neurons activates the sleep homeostat to promote rebound, whereas thermo-activation of octopaminergic neurons suppresses the sleep homeostat and produce “negative rebound.” In this context, a potential explanation for our finding that flies with pum silenced in the tim-Gal4 circuit exhibited a “negative rebound” after chronic sleep deprivation is that the processes that promote and suppress sleep homeostasis are recruited by our deprivation protocol. In the presence of Pum the homeostatic promoting process dominates, whereas when Pum is reduced, the homeostatic suppressing process dominates.
It is important to note that the tim-Gal4 driver used in these experiments has broad expression pattern, which is not limited to circadian neurons, and includes non-circadian neurons as well as glial cells. Interestingly, recent studies have pointed to a possible role of glia in the homeostatic control of sleep (Krzeptowski et al., 2018). Thus, in our view, including both clock neurons and glia in our analysis of sleep homeostasis is a reasonable choice to use as a primary probe. Restricting our manipulation to just neurons (as with elav-Gal4) or to only specific clock neurons may have precluded a positive result. Furthermore, tim-Gal4 expression is very strong in adults compared to other drivers. In fact, tim expression during development is highest in the adult brain and lowest during the developing embryo and larva. In contrast, elav-Gal4 has very high expression levels during development, but relatively low adult expression. While our choice certainly leaves the precise anatomical location of Pum’s effects in sleep rebound undefined, it has narrowed down the possibilities for future research.
As an RNA-binding protein regulating the translation of many target genes, Pum regulates many biological processes. Among these processes some could be associated with the results presented in this study. These include the regulation of neuronal excitability, glial function, Epidermal Growth Factor Receptor (EGFR) signaling, inflammatory pathways, the cell cycle gene network, and neural development. With regards to neuronal excitability, a number of studies have shown that neural plastic changes that increase the excitability of specific sleep-promoting neurons are fundamental for encoding sleep pressure (Donlea et al., 2014; Sitaraman et al., 2015; Liu et al., 2016; Guo et al., 2018; Ni et al., 2019). In contrast to sleep-promoting neurons, wake-promoting neurons exhibit decreased neuronal excitability after deprivation and this change is also associated with the generation of sleep pressure (Sitaraman et al., 2015). Since Pum is known to decrease neuronal excitability under a wide range of experimental conditions (Schweers et al., 2002; Mee et al., 2004; Driscoll et al., 2013; Lin and Baines, 2015), a potential mechanism is that Pum contributes to sleep homeostasis in the following manner: (1) participating in the generation of sleep pressure by decreasing the excitability of wake-promoting neurons during deprivation and (2) participating in the dissipation of sleep pressure after sleep deprivation by decreasing the excitability of sleep promoting neurons during rebound. In addition, a network of cell cycle genes has recently been implicated in sleep homeostasis through a post-mitotic function, presumably by regulating the excitability of the Pars Lateralis (PL) neurons by the CDK1 kinase (Afonso et al., 2015). Interestingly, a regulator of the CDK1 kinase, CycB, is a known target of Pum. Aside from the lack of experimental evidence directly testing these hypotheses, other limitations of these interpretations are that the main Gal4 line used in this study, tim-Gal4, has little or no expression in some of the main sleep homeostasis centers (Ellipsoid Body, dorsal Fan Shape body, Mushroom Body and Pars Lateralis) (Kaneko and Hall, 2000; Rogulja and Young, 2012), and that the role of circadian neurons on sleep homeostasis is just beginning to be elucidated (Guo et al., 2018; Lamaze et al., 2018; Ni et al., 2019).
Another site of convergence between biological processes affected by Pum and sleep homeostasis is the glial cells. For instance, Pum directly interacts with various components of the Notch signaling pathway in mammals, and it has been shown that glial Notch signaling in Drosophila negatively regulates sleep homeostasis (Seugnet et al., 2011). Importantly, Pum is expressed in Drosophila glial cells (Ng et al., 2016) and tim-Gal4 expression is very strong in these cells (Kaneko and Hall, 2000). The role of glial cells on sleep homeostasis is slowly becoming clearer and has been shown in both flies and mammals (Halassa et al., 2009; Chen et al., 2015; Vanderheyden et al., 2018, 2019; Frank, 2019). A key role of mammalian Pum in reactive astrogliosis after brain injury has also been described, which further strengthens the idea that this gene plays an important role in glial cell function (Kanemaru et al., 2013).
Other possible Pum functions that could have played a role in our results include EGFR and the regulation of inflammatory pathways. EGFR is a direct target of Pum in Drosophila and has been implicated in sleep regulation in flies, worms, zebrafish and humans (Foltenyi et al., 2007; Kim et al., 2012; Lee et al., 2019; Konietzka et al., 2020). These studies showed that EGFR signaling promotes sleep in all organisms tested but its role in sleep homeostasis was only examined in zebrafish. Although it is clear that Pum suppresses EGFR signaling in Drosophila, the sleep homeostasis effects observed in this study are unlikely due to EGFR regulation since Foltenyi et al. (2007) tested various circadian drivers, including the tim-Gal4 driver, and did not observe any effect of EGFR signaling manipulations with these Gal4 lines. Regarding the contribution of Pum’s inflammatory pathways regulation to our results, a genome-wide identification of mRNAs carrying Pum binding sites, found that 27% of Pum-regulated genes were immunity genes including various antimicrobial peptides and cytokines (Gerber et al., 2006). Cytokines are increased in chronically sleep deprived patients and a number of them have been shown to be sleep-regulatory substances (SRSs) mediating sleep homeostasis (Krueger, 2008; Allada et al., 2017; Nguyen et al., 2019; Tan et al., 2019). Gila cells and plasmatocytes are a major source of cytokines; therefore, these potential Pum effects cannot be ruled out.
The anatomical loci for Pum effects on sleep homeostasis remains unclear but based on the rapidly growing literature on the cellular and molecular bases of sleep homeostasis, we can speculate potential cellular groups. Accumulating evidence indicates that although there are many wake and sleep promoting cellular groups in the brain, only a small fraction participates in the homeostatic control of sleep. For instance, Seidner et al. (2015) screened 374 randomly selected Gal4 drivers from the Janelia Farm library. Interestingly, they found that while 10% were robust wake-promoting drivers, only 11% of those, which represent 1% of the total, exhibited a sleep homeostatic response. Within the expression pattern of the tim-Gal4 driver used in this study, the only cellular groups that have been directly linked to sleep homeostasis regulation are a subset of the dorsal neurons (the DN1p), the lateral posterior neurons (LPNs) and glial cells (Kaneko and Hall, 2000; Seugnet et al., 2011; Chen et al., 2015; Guo et al., 2018; Hsu and Sehgal, 2018; Lamaze et al., 2018; Vanderheyden et al., 2018, 2019; Ni et al., 2019). Although core clock neurons such as the Pdf cells and the lateral neurons dorsal group (LNd) have been shown to be wake-promoting, their role on sleep homeostasis has not been directly described. Pivotal neurons in the control of sleep homeostasis such as those in the ellipsoid bodies (EB) and fan-shape body (FB) are not included in the tim-Gal4 circuit. However, these regions are surrounded by tim-positive glial cells (Kaneko and Hall, 2000). A brain region that has undetectable Tim protein levels but has strong expression in the tim-Gal4 driver is the sub-esophageal ganglion (SEG). This region is relevant in this context since the set of wake-promoting neurons with the strongest influence on sleep homeosis described, are cholinergic neurons that reside on this region (Seidner et al., 2015).
Although our chronic sleep deprivation protocol is very effective during the first 12 h of deprivation, we consistently observed a gradual sleep increase in all transgenic and mutant lines tested. The degree of this adaptation varies greatly among the different lines and is not consistently associated with pum knockdown. Comparisons between UAS-pumRNAi/+ and UAS-pumRNAi/tim-Gal4 line (Figure 1), or pum13/+ and pum13/pum13 (Figure 4), did not show significant differences in sleep lost. The only comparison showing a significant difference in sleep lost during the deprivation period is the wild type Canton S flies vs. Milord-1 allele. This is in great contrast with the sleep rebound phase, in which all comparisons consistently show a reduction on sleep rebound. Even though there is not an association with pum manipulation, we believe that this adaptation is a sleep homeostatic response since we have consistently observed in wild type flies that individuals that are able to sleep during the deprivation period, show reduced sleep rebound compared to individuals more effectively deprived (data not shown).
Our finding that pum knockdown had a more dramatic effect after chronic SD compared to acute SD indicates that pum’s control of translation becomes progressively more important with deprivation time. Moreover, this suggests that sleep homeostasis involves a gradual recruitment of different processes to achieve its homeostatic goal depending on the chronicity of the sleep depriving insult. Consistent with this idea, Liu et al. (2016) pointed out that the short half-life’s (minutes) of the sleep-regulatory substances (SRSs) thought to underlie sleep pressure, was inconsistent with the time course for dissipating sleep pressure (hours or even days). Furthermore, they showed that neural plastic changes in specific circuits within the ellipsoid body are necessary and sufficient for generating sleep pressure. Initial studies of chronic SD in other species have also pointed to a potential difference in the regulatory mechanisms between acute vs. chronic SD. Rats exposed to chronic SD do not seem to regain the sleep lost even after a full 3-day recovery period, whereas in acute deprivation, most of the sleep was regained (Kim et al., 2007). Critics attributed these differences, between acute and chronic SD, to the increase in sleep pressure, which force micro-sleep episodes or EEG artifacts during chronic SD (Leemburg et al., 2010). A more recent study showed that chronically sleep deprived animals no longer express the compensatory increases that characterize sleep homeostasis in daily sleep time and sleep intensity (Kim et al., 2013). The authors of the study suggested that this decoupling of sleepiness from sleep time/intensity imply that there is one sleep regulation system mediating sleepiness (homeostatic), and another regulatory system for sleep time/intensity (allostatic) (Kim et al., 2013). Whether the lack of sleep compensation observed during chronic SD is a real mechanistic phenomenon or an artifact of the deprivation method remained controversial. In our study, we wanted to test if the behavioral differences reported by the literature, between acute and chronic SD, were regulated by the same mechanism under the pum gene. Our results point to the presence of a differential homeostatic response between acute vs. chronic SD in pum knockdowns, which suggests that pum participation in sleep homeostatic regulation is proportional to sleep need. Our data also suggests that pum regulation of sleep rebound is done through differential gene activation between acute and chronic SD. This difference seems to be aligned with fast action ion channel genes for acute SD and translation related and/or genes in which we expect to require more time to become active for chronic SD.
Another consideration is that the decrease in sleep rebound observed during pum knockdown is associated an increase in fly activity, which is related to increased neuronal excitability (Tabuchi et al., 2015). One possible explanation for these results is Pum known regulation of sodium currents (Ina) and excitability in Drosophila motor neurons through translational repression and binding with para-RNA (Baines, 2003; Driscoll et al., 2013). Reducing pum expression means there could be more sodium channels available and consequently, more neurons excited. Those excited neurons would have a diminished homeostatic mechanism to couple with the increase in excitability, resulting in prolonged wakefulness even after sleep deprivation stimulus was discontinued. Additional evidence in the literature supports the notion of a direct correlation between ion channels activity and wakefulness. Parisky et al. (2008), expressed the EKO potassium channel to hyperpolarize Ventral Lateral neurons (LNv) to reduce their excitability. In addition, they knocked down the Shaw potassium channel gene or expressed a dominant-negative Na+/K+-ATPase α subunit in the Pdf LNv neurons in order to increase neuronal excitability. The results showed that suppressed LNvs increased sleep whereas hyperactive LNvs increased wake. Furthermore, studies in rats have shown increases in cortical neurons firing with an increase in time awake (Vyazovskiy et al., 2009). Moreover, Donlea et al. (2014) found that the crossveinless (cv-c) mutants show decreased electrical activity in sleep promoting dorsal fan neurons. Additionally, the same study found that sleep pressure increases electrical excitability of sleep promoting neurons and this mechanism was blunted in cv-c mutants. This favors the alternative that pum regulates sleep homeostasis through the regulation of neuronal excitability.
Gene expression studies of pum mRNA levels in heads of pumRNAi flies and their respective “sibling” controls indicated that the RNA interference leads to a 50% reduction in non-deprived flies. Nevertheless, when we examined pum mRNA levels in sleep-deprived animals, we found a dramatic increase in pumRNAi flies and their respective “sibling” controls. Although this is a very counterintuitive result, we speculate that this is probably the result of a sleep-deprivation-induced increase in pum levels in cells outside the tim-Gal4 circuit. Furthermore, this finding is consistent with the idea that the “negative rebound” observed in pumRNAi flies, but not in pum13 and Milord-1 hypomorphs, is due to pum actions in cells outside the tim-Gal4 circuit.
Finally, our analysis of gene expression presented in the Supplementary Material indicates that pum knockdown in tim cells induces a series of global changes in gene expression that may contribute to the lack of homeostatic sleep response. However, this analysis suffers from major limitations. Because the qRT-PCR analysis uses RNA extracted from whole heads, which include many more cells than covered by the driver line used to knockdown pum, it is difficult to interpret the results. Small changes in expression in tim cells will likely be lost in noise, whilst larger changes may persist. Furthermore, we do not know whether the changes in mRNA are happening within tim cells or somewhere else. Thus, we cannot infer a direct role of pum in any of the changes in mRNA observed. Nonetheless, one striking observation is that, in general, the expression levels of most genes tested remains relatively stable upon acute and chronic sleep deprivation (<2-fold change). However, when pum is knocked down specifically in tim cells, global expression of the same genes is dramatically altered upon deprivation (4 to 16-fold change). This suggests that Pum is necessary, either directly or indirectly, in keeping these genes in check after deprivation. Another important observation is that in most cases the genes affected by Pum knockdown after acute deprivation are not the same as those affected after chronic deprivation. This suggests that the two responses are controlled, at least partially, by distinct mechanisms.
In summary, our results indicate that Pum is necessary for a normal sleep rebound after sleep deprivation. In addition, we showed that this effect is more pronounced during chronic sleep deprivation (84 h) than acute deprivation (12 h). These behavioral changes were associated with accompanying differential changes in the expression of genes involved in synaptic translation and the regulation of neuronal excitability. Based on these findings, we propose that Pum is an important regulator of sleep homeostasis through neural adaptations triggered during sleep deprivation and induces rebound sleep. Further studies characterizing additional Pum targets warrant exciting findings about the molecular control of sleep. Moreover, future studies identifying the specific circuits where Pum is required for sleep regulation could provide a better picture of the mechanistic relationship between sleep function and molecular sleep regulation.
Materials and Methods
Fly Stocks
Drosophila stocks were raised on standard Drosophila medium in a 12/12 h light/dark cycle. The following stocks were used in this study: The UAS-pumRNAi (stock #26725: y[1] v[1]; P{y[+t7.7] v[+t1.8]=TRiP.JF02267}attP2) fly line was obtained from Bloomington Stock Center; The tim-Gal4 transgenic line: yw; cyo/tim-Gal4 was obtained from Dr. Leslie Griffith’s and Dr. Michael Rosbash’s labs at Brandeis University. These two lines were crossed to obtain both UAS-pumRNAi/tim-Gal4 experimental flies and the “sibling” control flies UAS-pumRNAi/+. The Milord-1 P{lacZ}pummilord–1 was obtained from Dr. Josh Dubnau. The mutant pum13 (pum680) and Canton S wild type flies were also obtained from Bloomington Stock Center and crossed to obtain both pum13/ + and pum13/pum13 flies used in Figure 5.
Sleep Assays
Sleep assays used 1–2 days old female flies. The individuals were collected, separated by phenotype and placed into controlled temperature for 6–7 days under 12 h:12 h light dark cycles for entrainment. The individuals were then anesthetized with CO2 and placed in individual tubes containing fly food (5% sucrose, 2% agar). Tubes were then placed in Drosophila Activity Monitors (DAM) within an environmentally controlled incubator (26°C, 80% humidity, light intensity of 800 lux) and connected to the monitoring system (TriKinetics, Waltham, MA, United States) under 12 h:12 h light dark cycles. After 6 days of baseline recordings, after changing the fly food to avoid dryness and microbial growth, the different groups of flies were sleep deprived with the methods described below. The genetic controls (“siblings”) were handled and tested side by side to the experimental flies. Flies with less than 80% deprivation within the first 12 h were excluded from the analysis. Number of individuals tested and number of experiment replications depicted are stated in figure legends. A cumulative sleep lost plot was produced by calculating the cumulative sleep difference between the deprivation period and the baseline period before sleep deprivation. The individual sleep recovery (rebound) was calculated by dividing the cumulative amount of sleep regained by the total amount of sleep lost during deprivation.
Mechanical Sleep Deprivation
Mechanical deprivation was performed using a commercially available Drosophila sleep deprivation apparatus (Trikinetics Inc., VMP Vortexer Mounting Plate). The apparatus was controlled by the Trikinetics software, shaking the monitors for 30 s on alternate settings of 4, 5, and 8 min to create an apparently random shaking pattern. The same pattern was used for all experiments. The flies were placed in the Drosophila Activity Monitors to be monitored for 6 days for baseline sleep. After the 6th day, flies were subjected to mechanical SD. Both chronic and acute deprivation protocols were identical in terms of stimulus intensity and pattern; the only difference was the duration of the deprivation period. For chronic sleep deprivation, the SD protocol was active for the first 84 h starting at the beginning of the first dark period (Figure 1), while for acute sleep deprivation, the SD protocol lasted only 12 h, which encompassed the entirety of the dark period preceding the sleep recovery period. For the acute SD experiment, the same set up was used but for only 12 h of the deprivation night. Although this protocol results in partial sleep deprivation, rather than total deprivation, it induces significant sleep lost, normally around 80%, and allows the flies to survive through the chronic sleep deprivation period. Due to the long SD time of 84 h and the baseline period, we perform a fly food change the day before SD to avoid microbial growth and food dryness. This change is coordinated with the morning cycle and performed simultaneously for all experimental groups.
Immunofluorescence
Flies were frozen in dry ice immediately after sleep deprivation. Brains were dissected and fixed with 4% formaldehyde in 100 mm phosphate buffer (PBS) for 30 min at room temperature. The brains were then washed and rinsed four times in PBS with 0.3% Triton X-100 (PB-T) for 10 min to remove the formaldehyde. Brains were then were blocked in 5% normal goat serum in PB-T for 1 h and incubated with primary antibody (rabbit anti-RFP 1:1000; Invitrogen), at 4°C overnight in a humid chamber. The next day, the brains were washed four times for 10 min each time in PB-T and incubated with secondary antibody at 1:500 for 2 h at RT. The secondary antibody was washed four times for 10 min each time in PB-T. To remove the PB-T, the brains were washed two times for 5 min with PBS and mounted in 80% Prolong Anti-fading Agent. Images were taken either on a Zeiss Pascal LSM or a Zeiss LSM 510 Meta confocal microscope at 40X magnification. The same gain was used for all images after calibrating the gain with the group of brains of the pumRNAi deprived flies, which showed the highest level of fluorescence of all experimental groups. After acquisition, images were processed employing Imaris (Redicon) software program.
Statistical Methods
All statistical comparisons for significance between control and experimental groups was calculated using a significance cut off p < 0.05. All statistical analyses were performed using GraphPad Prism 8 software. Statistical analyses performed are included in the figure legends.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
JA, NR, and LD designed the study. JA-R, NR, CP-A, JO-T, RN, MF, and LD performed the experiments and data analysis. JA, AG, NF-U, and LD wrote and reviewed the manuscript.
Funding
This work has been partially supported by the NIH-RISE Grant # 5R25GM061151-12, the NSF REU-CRIB Program Grant 1156810, the NIGMS COBRE Grant # 2P20GM103642-06 (Subaward # 14625747), and the NIH NeuroID Grant 5R25NS080687-10.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Tugrul Giray, Dr. Adrienel Vazquez, Dr. Adrian Ovalos, Dr. Manuel Giannioni, Hidequel Rodriguez, Bismark Madera, Gabriel Diaz, Maria del Mar Reyes, Franshesca Rivera, Alejandro Medina, Lizangelis Cueto, Melina Torres, Carlos Billini, Rosa Alers, Wilfredo Soto, Rubielis Serrano, Keila Velazquez, Marcelo Francia, Oto Mendez, Norelis Diaz, and the students from the genetics lab at UPR-RP biol3350 for their support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2020.00319/full#supplementary-material
References
Afonso, D. J., Machado, D. R., and Koh, K. (2015). Control of sleep by a network of cell cycle genes. Fly 9, 165–172. doi: 10.1080/19336934.2016.1153776
Allada, R., Cirelli, C., and Sehgal, A. (2017). Molecular mechanisms of sleep homeostasis in flies and mammals. Cold Spring Harb. Perspect. Biol. 9:a027730. doi: 10.1101/cshperspect.a027730
Baines, R. A. (2003). Postsynaptic protein kinase A reduces neuronal excitability in response to increased synaptic excitation in the Drosophila CNS. J. Neurosci. 23, 8664–8672. doi: 10.1523/jneurosci.23-25-08664.2003
Bushey, D., Tononi, G., and Cirelli, C. (2009). The Drosophila fragile X mental retardation gene regulates sleep need. J. Neurosci. 29, 1948–1961. doi: 10.1523/jneurosci.4830-08.2009
Bushey, D., Tononi, G., and Cirelli, C. (2011). Sleep and synaptic homeostasis: structural evidence in Drosophila. Science 332, 1576–1581. doi: 10.1126/science.1202839
Cao, Q., Padmanabhan, K., and Richter, J. D. (2009). Pumilio 2 controls translation by competing with eIF4E for 7-methyl guanosine cap recognition. RNA 16, 221–227. doi: 10.1261/rna.1884610
Chen, G., Li, W., Zhang, Q. S., Regulski, M., Sinha, N., Barditch, J., et al. (2008). Identification of synaptic targets of Drosophila pumilio. PLoS Comput. Biol. 4:e1000026. doi: 10.1371/journal.pcbi.1000026
Chen, W. F., Maguire, S., Sowcik, M., Luo, W., Koh, K., and Sehgal, A. (2015). A neuron-glia interaction involving GABA transaminase contributes to sleep loss in sleepless mutants. Mol. Psychiatry 20, 240–251. doi: 10.1038/mp.2014.11
Chung, B. Y., Kilman, V. L., Russel, K. J., Pitman, J. L., and Allada, R. (2009). The GABAA receptor RDL acts in peptidergic PDF neurons to promote sleep in Drosophila. Curr. Biol. 19, 386–390. doi: 10.1016/j.cub.2009.01.040
Cirelli, C., Bushey, D., Hill, S., Huber, R., Kreber, R., Ganetzky, B., et al. (2005). Reduced sleep in Drosophila shaker mutants. Nature 434, 1087–1092. doi: 10.1038/nature03486
Cirelli, C., and Tononi, G. (2008). Is sleep essential? PLoS Biol. 6:e216. doi: 10.1371/journal.pbio.0060216
Daan, S., Beersma, D. G., and Borbély, A. A. (1984). Timing of human sleep: recovery process gated by a circadian pacemaker. Am. J. Physiol. 246(2 Pt 2), R161–R183.
Davis, G. W. (2006). Homeostatic control of neural activity: from phenomenology to molecular design. Annu. Rev. Neurosci. 29, 307–323. doi: 10.1146/annurev.neuro.28.061604.135751
Davis, G. W. (2013). Homeostatic signaling and the stabilization of neural function. Neuron 80, 718–728. doi: 10.1016/j.neuron.2013.09.044
Donlea, J. M. (2017). Neuronal and molecular mechanisms of sleep homeostasis. Curr. Opin. Insect Sci. 24, 51–57. doi: 10.1016/j.cois.2017.09.008
Donlea, J. M., Pimentel, D., and Miesenböck, G. (2014). Neuronal machinery of sleep homeostasis in Drosophila. Neuron 81, 860–872. doi: 10.1016/j.neuron.2013.12.013
Donlea, J. M., Thimgan, M. S., Suzuki, Y., Gottschalk, L., and Shaw, P. J. (2011). Inducing sleep by remote control facilitates memory consolidation in Drosophila. Science 332, 1571–1576. doi: 10.1126/science.1202249
Driscoll, H. E., Muraro, N. I., He, M., and Baines, R. A. (2013). Pumilio-2 regulates translation of Nav1.6 to mediate homeostasis of membrane excitability. J. Neurosci. 33, 9644–9654. doi: 10.1523/jneurosci.0921-13.2013
Dubnau, J., Chiang, A. S., Grady, L., Barditch, J., Gossweiler, S., McNeil, J., et al. (2003). The staufen/pumilio pathway is involved in Drosophila long-term memory. Curr. Biol. 13, 286–296. doi: 10.1016/s0960-9822(03)00064-2
Fiore, R., Rajman, M., Schwale, C., Bicker, S., Antoniou, A., Bruehl, C., et al. (2014). MiR-134-dependent regulation of Pumilio-2 is necessary for homeostatic synaptic depression. EMBO J. 33, 2231–2246. doi: 10.15252/embj.201487921
Foltenyi, K., Greenspan, R. J., and Newport, J. W. (2007). Activation of EGFR and ERK by rhomboid signaling regulates the consolidation and maintenance of sleep in Drosophila. Nat. Neurosci. 10, 1160–1167. doi: 10.1038/nn1957
Frank, M. G. (2019). The role of glia in sleep regulation and function. Handb. Exp. Pharmacol. 253, 83–96. doi: 10.1007/164_2017_87
Gerber, A. P., Luschnig, S., Krasnow, M. A., Brown, P. O., and Herschlag, D. (2006). Genome-wide identification of mRNAs associated with the translational regulator PUMILIO in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 103, 4487–4492. doi: 10.1073/pnas.0509260103
Guo, F., Holla, M., Díaz, M. M., and Rosbash, M. A. (2018). Circadian output circuit controls sleep-wake arousal in Drosophila. Neuron 100, 624.e4–635.e4.
Halassa, M. M., Florian, C., Fellin, T., Munoz, J. R., Lee, S. Y., Abel, T., et al. (2009). Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 61, 213–219. doi: 10.1016/j.neuron.2008.11.024
Hendricks, J. C., Sehgal, A., and Pack, A. I. (2000). The need for a simple animal model to understand sleep. Prog. Neurobiol. 61, 339–351. doi: 10.1016/s0301-0082(99)00048-9
Hsu, C. T., and Sehgal, A. (2018). Circadian and sleep circuits ring together. Neuron 100, 514–516. doi: 10.1016/j.neuron.2018.10.030
Huber, R., Hill, S. L., Holladay, C., Biesiadecki, M., Tononi, G., and Cirelli, C. (2004). Sleep homeostasis in Drosophila melanogaster. Sleep 27, 628–639. doi: 10.1093/sleep/27.4.628
Kaneko, M., and Hall, J. C. (2000). Neuroanatomy of cells expressing clock genes in Drosophila: transgenic manipulation of the period and timeless genes to mark the perikadya of circadian pacemaker neurons and their projections. J. Comp. Neurol. 422, 66–94. doi: 10.1002/(sici)1096-9861(20000619)422:1<66::aid-cne5>3.0.co;2-2
Kanemaru, K., Kubota, J., Sekiya, H., Hirose, K., Okubo, Y., and Iino, M. (2013). Calcium-dependent N-cadherin up-regulation mediates reactive astrogliosis and neuroprotection after brain injury. Proc. Natl. Acad. Sci. U.S.A. 110, 11612–11617. doi: 10.1073/pnas.1300378110
Kim, S. Y., Kim, J. Y., Malik, S., Son, W., Kwon, K. S., and Kim, C. (2012). Negative regulation of EGFR/MAPK pathway by Pumilio in Drosophila melanogaster. PLoS One 7:e34016. doi: 10.1371/journal.pone.0034016
Kim, Y., Chen, L., McCarley, R. W., and Strecker, R. E. (2013). Sleep allostasis in chronic sleep restriction: the role of the norepinephrine system. Brain Res. 1531, 9–16. doi: 10.1016/j.brainres.2013.07.048
Kim, Y., Laposky, A. D., Bergmann, B. M., and Turek, F. W. (2007). Repeated sleep restriction in rats leads to homeostatic and allostatic responses during recovery sleep. Proc. Natl. Acad. Sci. U.S.A. 104, 10697–10702. doi: 10.1073/pnas.0610351104
Koh, K., Joiner, W. J., Wu, M. N., Yue, Z., Smith, C. J., and Sehgal, A. (2008). Identification of SLEEPLESS, a sleep-promoting factor. Science 321, 372–376. doi: 10.1126/science.1155942
Konietzka, J., Fritz, M., Spiri, S., McWhirter, R., Leha, A., Palumbos, S., et al. (2020). Epidermal growth factor signaling promotes sleep through a combined series and parallel neural circuit. Curr. Biol. 30, 1–16.
Krueger, J. M. (2008). The role of cytokines in sleep regulation. Curr. Pharm. Des 14, 3408–3416. doi: 10.2174/138161208786549281
Krzeptowski, W., Walkowicz, L., Płonczyńska, A., and Górska-Andrzejak, J. (2018). Different levels of expression of the clock protein PER and the glial marker REPO in ensheathing and astrocyte-like glia of the distal medulla of drosophila optic lobe. Front. Physiol. 9:361. doi: 10.3389/fphys.2018.00361
Kula-Eversole, E., Nagoshi, E., Shang, Y., Rodriguez, J., Allada, R., and Rosbash, M. (2010). Surprising gene expression patterns within and between PDF-containing circadian neurons in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 107, 13497–13502. doi: 10.1073/pnas.1002081107
Lamaze, A., Krätschmer, P., Chen, K. F., Lowe, S., and Jepson, J. E. C. (2018). A wake-promoting circadian output circuit in Drosophila. Curr. Biol. 28, 3098–3105.
Lee, D. A., Liu, J., Hong, Y., Lane, J. M., Hill, A. J., Hou, S. L., et al. (2019). Evolutionarily conserved regulation of sleep by epidermal growth factor receptor signaling. Sci. Adv. 5:eaax4249. doi: 10.1126/sciadv.aax4249
Leemburg, S., Vyazovskiy, V. V., Olcese, U., Bassetti, C. L., Tononi, G., and Cirelli, C. (2010). Sleep homeostasis in the rat is preserved during chronic sleep restriction. Proc. Natl. Acad. Sci. U.S.A. 107, 15939–15944. doi: 10.1073/pnas.1002570107
Lin, W. H., and Baines, R. A. (2015). Regulation of membrane excitability: a convergence on voltage-gated sodium conductance. Mol. Neurobiol. 51, 57–67. doi: 10.1007/s12035-014-8674-0
Liu, S., Liu, Q., Tabuchi, M., and Wu, M. N. (2016). Sleep drive is encoded by neural plastic changes in a dedicated circuit. Cell 165, 1347–1360. doi: 10.1016/j.cell.2016.04.013
Marder, E., and Prinz, A. A. (2003). Current compensation in neuronal homeostasis. Neuron 37, 2–4. doi: 10.1016/s0896-6273(02)01173-x
Mee, C. J., Pym, E. C., Moffat, K. G., and Baines, R. A. (2004). Regulation of neuronal excitability through pumilio-dependent control of a sodium channel gene. J. Neurosci. 24, 8695–8703. doi: 10.1523/jneurosci.2282-04.2004
Menon, K. P., Sanyal, S., Habara, Y., Sanchez, R., Wharton, R. P., Ramaswami, M., et al. (2004). The translational repressor Pumilio regulates presynaptic morphology and controls postsynaptic accumulation of translation factor eIF-4E. Neuron 44, 663–676. doi: 10.1016/j.neuron.2004.10.028
Muraro, N. I., Weston, A. J., Gerber, A. P., Luschnig, S., Moffat, K. G., and Baines, R. A. (2008). Pumilio binds para mRNA and requires Nanos and Brat to regulate sodium current in Drosophila motorneurons. J. Neurosci. 28, 2099–2109. doi: 10.1523/jneurosci.5092-07.2008
Ng, F. S., Sengupta, S., Huang, Y., Yu, A. M., You, S., Roberts, M. A., et al. (2016). TRAP-seq profiling and RNAi-based genetic screens identify conserved glial genes required for adult Drosophila behavior. Front. Mol. Neurosci. 9:146. doi: 10.3389/fnmol.2016.00146
Nguyen, J., Gibbons, C., Dykstra-Aiello, C., Ellingsen, R., Koh, K., Taishi, P., et al. (2019). Interleukin-1 receptor accessory proteins are required for normal homeostatic responses to sleep deprivation. J. Appl. Physiol. 127, 770–780. doi: 10.1152/japplphysiol.00366.2019
Ni, J. D., Gurav, A. S., Liu, W., Ogunmowo, T. H., Hackbart, H., Elsheikh, A., et al. (2019). Differential regulation of the Drosophila sleep homeostat by circadian and arousal inputs. eLife 8:e40487.
Nitz, D. A., van Swinderen, B., Tononi, G., and Greenspan, R. J. (2002). Electrophysiological correlates of rest and activity in Drosophila melanogaster. Curr. Biol. 12, 1934–1940. doi: 10.1016/s0960-9822(02)01300-3
Parisky, K. M., Agosto, J., Pulver, S. R., Shang, Y., Kuklin, E., Hodge, J., et al. (2008). PDF cells are a GABA-responsive wake-promoting component of the Drosophila sleep circuit. Neuron 61, 672–682. doi: 10.1016/j.neuron.2008.10.042
Pfeiffenberger, C., and Allada, R. (2012). Cul3 and the BTB adaptor insomniac are key regulators of sleep homeostasis and a dopamine arousal pathway in Drosophila. PLoS Genet. 8:e1003003. doi: 10.1371/journal.pgen.1003003
Pimentel, D., Donlea, J. M., Talbot, C. B., Song, S. M., Thurston, A. J. F., and Miesenböck, G. (2016). Operation of a homeostatic sleep switch. Nature 536, 333–337. doi: 10.1038/nature19055
Pozo, K., and Goda, Y. (2010). Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 66, 337–351. doi: 10.1016/j.neuron.2010.04.028
Rogulja, D., and Young, M. W. (2012). Control of sleep by cyclin A and its regulator. Science 335, 1617–1621. doi: 10.1126/science.1212476
Schweers, B. A., Walters, K. J., and Stern, M. (2002). The Drosophila melanogaster translational repressor pumilio regulates neuronal excitability. Genetics 161, 1177–1185.
Sehgal, A., Joiner, W., and Crocker, A. (2007). Molecular analysis of sleep: wake cycles in Drosophila. Cold. Spring Harb. Symp. Quant. Biol. 72, 557–564.
Seidner, G., Robinson, J., Wu, M., Roberts, S. W., Keene, A. C., and Joiner, W. J. (2015). Identification of neurons with a privileged role in sleep homeostasis in Drosophila Melanogaster. Curr. Biol. 25, 2928–2938. doi: 10.1016/j.cub.2015.10.006
Seluzicki, A., Flourakis, M., Kula-Eversole, E., Zhang, L., Kilman, V., and Allada, R. (2014). Dual PDF signaling pathways reset clocks via TIMELESS and acutely excite target neurons to control circadian behavior. PLoS Biol. 12:e1001810. doi: 10.1371/journal.pbio.1001810
Seugnet, L., Suzuki, Y., Merlin, G., Gottschalk, L., Duntley, S. P., and Shaw, P. J. (2011). Notch signaling modulates sleep homeostasis and learning after sleep deprivation in Drosophila. Curr. Biol. 21, 835–840. doi: 10.1016/j.cub.2011.04.001
Shaw, P. J., Cirelli, C., Greenspan, R. J., and Tononi, G. (2000). Correlates of sleep and waking in Drosophila melanogaster. Science 287, 1834–1837. doi: 10.1126/science.287.5459.1834
Shaw, P. J., Tononi, G., Greenspan, R. J., and Robinson, D. F. (2002). Stress response genes protect against lethal effects of sleep deprivation in Drosophila. Nature 417, 287–291. doi: 10.1038/417287a
Siegel, J. M. (2008). Do all animals sleep? Trends Neurosci. 31, 208–213. doi: 10.1016/j.tins.2008.02.001
Sitaraman, D., Aso, Y., Jin, X., Chen, N., Felix, M., Rubin, G. M., et al. (2015). Propagation of homeostatic sleep signals by segregated synaptic microcircuits of the Drosophila mushroom body. Curr. Biol. 25, 2915–2927. doi: 10.1016/j.cub.2015.09.017
Tabuchi, M., Lone, S. R., Liu, S., Liu, Q., Zhang, J., Spira, A. P., et al. (2015). Sleep interacts with aβ to modulate intrinsic neuronal excitability. Curr. Biol. 25, 702–712. doi: 10.1016/j.cub.2015.01.016
Tan, H. L., Kheirandish-Gozal, L., and Gozal, D. (2019). Sleep, Sleep Disorders, and Immune Function. Allergy and Sleep, 2019. Cham: Springer.
Thimgan, M. S., Gottschalk, L., Toedebusch, C., McLeland, J., Rechtschaffen, A., Gilliland-Roberts, M., et al. (2013). Cross-translational studies in human and Drosophila identify markers of sleep loss. PLoS One 8:e61016. doi: 10.1371/journal.pone.0061016
Turrigiano, G. G. (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435. doi: 10.1016/j.cell.2008.10.008
Turrigiano, G. G. (2012). Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 4:a005736. doi: 10.1101/cshperspect.a005736
Turrigiano, G. G., Leslie, K. R., Desai, N. S., Rutherford, L. C., and Nelson, S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896. doi: 10.1038/36103
Turrigiano, G. G., and Nelson, S. B. (2004). Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 5, 97–107. doi: 10.1038/nrn1327
Van Swinderen, B., Nitz, D. A., and Greenspan, R. J. (2004). Uncoupling of brain activity from movement defines arousal States in Drosophila. Curr. Biol. 14, 81–87. doi: 10.1016/j.cub.2003.12.057
Vanderheyden, W. M., Goodman, A. G., Taylor, R. H., Frank, M. G., Van Dongen, H. P. A., and Gerstner, J. R. (2018). Astrocyte expression of the Drosophila TNF-alpha homologue, Eiger, regulates sleep in flies. PLoS Genet. 14:e1007724. doi: 10.1371/journal.pgen.1007724
Vanderheyden, W. M., Van Dongen, H. P. A., Frank, M. G., and Gerstner, J. R. (2019). Sleep pressure regulates mushroom body neural-glial interactions in Drosophila. Matters Sel. 209:8.
Vessey, J. P., Vaccani, A., Xie, Y., Dahm, R., Karra, D., Kiebler, M. A., et al. (2006). Dendritic localization of the translational repressor Pumilio 2 and its contribution to dendritic stress granules. J. Neurosci. 26, 6496–6508. doi: 10.1523/jneurosci.0649-06.2006
Vyazovskiy, V. V., Olcese, U., Lazimy, Y. M., Faraguna, U., Esser, S., Cirelli, C., et al. (2009). Cortical firing and sleep homeostasis. Neuron 63, 865–878. doi: 10.1016/j.neuron.2009.08.024
Wang, M., Ogé, L., Perez-Garcia, M. D., Hamama, L., and Sakr, S. (2018). The PUF protein family: overview on PUF RNA targets, biological functions, and post transcriptional regulation. Int. J. Mol. Sci. 19:410. doi: 10.3390/ijms19020410
Weston, A. J., and Baines, R. A. (2007). Translational regulation of neuronal electrical properties. Invert. Neurosci. 7, 75–86. doi: 10.1007/s10158-006-0037-8
Wharton, R. P., Sonoda, J., Lee, T., Patterson, M., and Yoshihiko, M. (1998). The pumilio RNA-binding domain is also a translational regulator. Mol. Cell. 1, 863–872. doi: 10.1016/s1097-2765(00)80085-4
Keywords: sleep homeostasis, neuronal homeostasis, Pumilio, pum, Drosophila, chronic sleep deprivation, synaptic proteins, neuronal excitability
Citation: De Jesús-Olmo LA, Rodríguez N, Francia M, Alemán-Rios J, Pacheco-Agosto CJ, Ortega-Torres J, Nieves R, Fuenzalida-Uribe N, Ghezzi A and Agosto JL (2020) Pumilio Regulates Sleep Homeostasis in Response to Chronic Sleep Deprivation in Drosophila melanogaster. Front. Neurosci. 14:319. doi: 10.3389/fnins.2020.00319
Received: 05 November 2019; Accepted: 18 March 2020;
Published: 17 April 2020.
Edited by:
Steven Brown, University of Zurich, SwitzerlandReviewed by:
Elzbieta M. Pyza, Jagiellonian University, PolandRichard A. Baines, University of Manchester, United Kingdom
Copyright © 2020 De Jesús-Olmo, Rodríguez, Francia, Alemán-Rios, Pacheco-Agosto, Ortega-Torres, Nieves, Fuenzalida-Uribe, Ghezzi and Agosto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis A. De Jesús-Olmo, bHVpcy5kZWplc3VzM0B1cHIuZWR1; José L. Agosto, am9zZS5hZ29zdG8xQHVwci5lZHU=