 Francesca Binda
Francesca Binda Carla Pernaci
Carla Pernaci Smita Saxena
Smita Saxena
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurosci. , 02 April 2020
Sec. Neurodegeneration
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.00293
This article is part of the Research Topic Circuit Mechanisms of Neurodegenerative Diseases View all 10 articles
Spinocerebellar ataxias (SCAs) affect the cerebellum and its afferent and efferent systems that degenerate during disease progression. In the cerebellum, Purkinje cells (PCs) are the most vulnerable and their prominent loss in the late phase of the pathology is the main characteristic of these neurodegenerative diseases. Despite the constant advancement in the discovery of affected molecules and cellular pathways, a comprehensive description of the events leading to the development of motor impairment and degeneration is still lacking. However, in the last years the possible causal role for altered cerebellar development and neuronal circuit wiring in SCAs has been emerging. Not only wiring and synaptic transmission deficits are a common trait of SCAs, but also preventing the expression of the mutant protein during cerebellar development seems to exert a protective role. By discussing this tight relationship between cerebellar development and SCAs, in this review, we aim to highlight the importance of cerebellar circuitry for the investigation of SCAs.
Spinocerebellar ataxias (SCAs) are a large family of movement disorders characterized by the progressive loss of motor coordination, muscle tone and control, with a broad spectrum of severity ranging from mild gait and posture problems to death. Moreover, several comorbidities including pyramidal features, peripheral neuropathy, tremor, dystonia, Parkinsonian features, myoclonus, epilepsy, and dementia occur frequently in SCAs. The 47 as of now identified SCAs are dominantly inherited diseases, and a growing number of genes are implicated, revealing two major pathological alterations; (1) Microsatellite repeat expansions coding for CAG generating polyglutamine (PolyQ) repeats: SCA1, SCA2, SCA3, SCA6, SCA7, SCA17 and Dentatorubral-pallidoluysian atrophy DRPLA, and non-coding repeats: SCA8, SCA10, SCA12, SCA31, SCA36, SCA37). (2) Single-gene point mutations as observed in SCA5, SCA11, SCA13-16, SCA18, SCA19/22, SCA20-23, SCA26, SCA27-29, SCA34-35, SCA38, SCA40-46 (Ashizawa et al., 2018; Pilotto and Saxena, 2018).
Genes causing SCAs code for a large variety of proteins including ion channels (Riess et al., 1997; Zhuchenko et al., 1997; Waters et al., 2006; Lee et al., 2012; Coutelier et al., 2015; Fogel et al., 2015; Morino et al., 2015), transcription factors and repressors (Orr et al., 1993; Koide et al., 1999; Lin et al., 2018), scaffolding proteins (Ikeda et al., 2006; Tsoi et al., 2014), or signaling kinases (Chen et al., 2003; Houlden et al., 2007), phosphatases (Holmes et al., 1999), and receptors (Storey et al., 2001; Hara et al., 2004; Hara et al., 2008; Huang et al., 2012; Watson et al., 2017). Despite this heterogeneity, SCA patients share a slow dramatic degeneration of the cerebellum and its afferent and efferent systems, suggesting a common point of interception, wherein SCA causing mutations converge and promotes the onset of these neurodegenerative diseases. Interestingly, wiring defects of the cerebellar circuitry are a common finding in SCAs and they appear at the early asymptomatic phase (Ebner et al., 2013; Hansen et al., 2013; Dulneva et al., 2015; Jayabal et al., 2017). In preclinical rodent models of SCA1, inhibiting the expression of the mutant ATXN1 protein during cerebellar postnatal development exerted a protective effect on the ataxic phenotype (Serra et al., 2006) and ameliorated synaptic transmission in the cerebellar cortex (Zu et al., 2004; Barnes et al., 2011). Therefore, cerebellar development seems to be highly vulnerable to the expression of mutant SCA-causing molecules, and the resulting compromised cerebellar compartmentation, patterning and circuit wiring could have a direct impact on the severity of the disease. In this review, we focus on the possible role of SCA genes in cerebellar development in an attempt to define a new framework for SCAs.
The cerebellum is traditionally known for its role in motor control and more recently its involvement in higher cognitive functions has been recognized (Koziol et al., 2014; Adamaszek et al., 2017).
The cerebellum receives two major excitatory inputs: mossy fibers (MFs) and climbing fibers (CFs). MFs originate from several pre-cerebellar nuclei in the brainstem and spinal cord, and they carry vestibular, motor, sensory and proprioceptive information relayed by the granule cells (GCs). GC axons ascend into the molecular layer (ML), where they bifurcate into parallel fibers (PFs) that run longitudinally to the cerebellar lobule, contacting several Purkinje cells (PCs) on their way. Each PC receives up to 105–106 excitatory PF synaptic contacts, however, one PF makes only one to two synapses onto a single PC (Napper and Harvey, 1988). PF activity induces simple spike discharge in PCs; moreover, PCs spontaneously fire action potentials, and this pacemaker activity has been recorded in cerebellar slices during the pharmacological blockade of excitatory inputs (Hausser and Clark, 1997) and in cultured PCs (Nam and Hockberger, 1997). Notably, CFs only originate from the inferior olive nuclei (IO) in the brainstem and establish monoinnervation on mature PCs. CF activation signature in PCs is a high frequency burst of spikes known as complex spike (Eccles et al., 1966). CFs provide teaching signals to the cerebellum that drive cerebellar-mediated motor learning. The simultaneous activation of CF and PFs leads to long-term depression at the PF to PC synapses (Ito et al., 1982), while the sole activation of GCs promotes long-term potentiation at these synapses (Lev-Ram et al., 2002; Coesmans et al., 2004). Long-term plasticity at these excitatory synapses provides a major molecular platform for supporting cerebellar-mediated motor learning (De Zeeuw et al., 1998; Boyden et al., 2006; Hansel et al., 2006; Schonewille et al., 2010; Ly et al., 2013). Traditionally, CFs are known to carry sensory/motor errors but more recently, they have also been shown to mediate reward and predictive signals (Ohmae and Medina, 2015; Heffley et al., 2018; Kostadinov et al., 2019; Larry et al., 2019).
Anatomically, three parts are recognized in the cerebellum: the cerebellar cortex, the white matter and the deep cerebellar nuclei (DCN). The cerebellar cortex is the most superficial structure and organized into three layers (from the most superficial): the ML, the Purkinje cell layer (PCL), and the granule cell layer (GCL) (Figure 1).
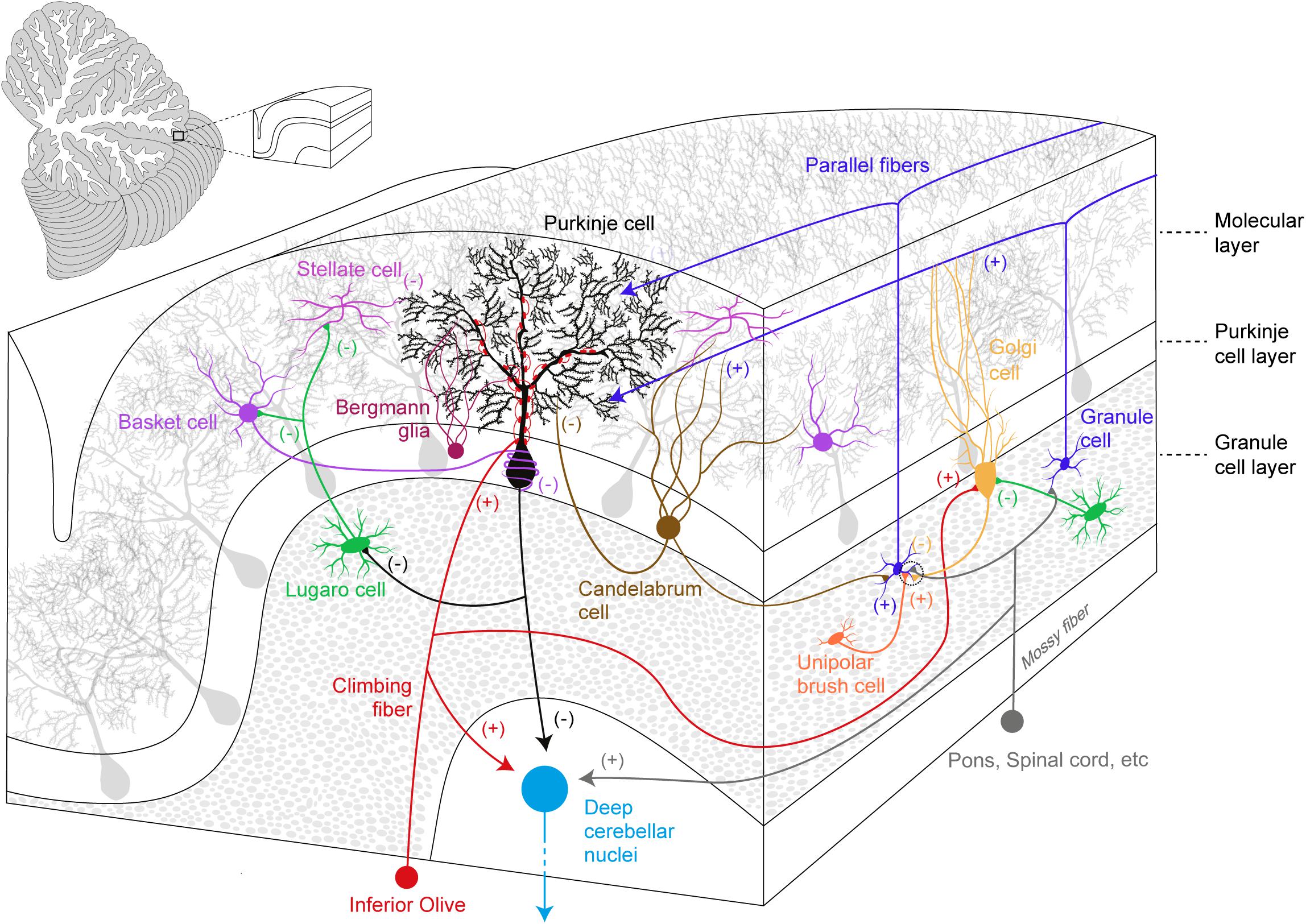
Figure 1. Simplified overview of the cerebellar cortical organization and its circuitry. Excitatory (+) and inhibitory (–) components are represented within the cerebellar cortex and deep cerebellar nuclei.
The ML accommodates the complex and ramified dendritic tree of PCs together with two types of GABAergic interneurons (molecular layer interneurons, MLIs): Basket cells (BCs) and Stellate cells (SCs). BCs synapse onto PC soma and its axon initial segment in a characteristic structure known as pinceau (Ango et al., 2004; Blot and Barbour, 2014), while SCs innervate PC dendrites. MLIs receive excitatory inputs from PFs and they provide feed-forward inhibition, which modulates PC spike output (Mittmann et al., 2005; Brown et al., 2019), calcium influx and long-term plasticity (Binda et al., 2016). Moreover, CF activation also recruits MLIs via a spillover mechanism (Szapiro and Barbour, 2007). PC somas define the PCL: PCs are the sole output of the cerebellar cortex and they send GABAergic projections to the DCN and vestibular nuclei. The PCL also hosts the small vertically oriented pear-shaped soma of Candelabrum cells (CaCs). CaCs have one or two long dendrites that enter the ML, and several short dendrites localized below the GCL. The axon of these neurons runs horizontally and is characterized by multiple vertical branches ascending to the ML (Laine and Axelrad, 1994). In the Macaca monkey, the immunohistochemical analysis of the cerebellum provided evidence for the release of glycine and GABA neurotransmitters by these neurons (Crook et al., 2006), however, CaCs targets and inputs still remain elusive.
Together with GCs, the GCL also contains glutamatergic interneurons, the Unipolar Brush cells (UBCs) that establish excitatory inputs onto GCs and on other UBCs (Nunzi et al., 2001). UBCs mainly receive excitatory inputs from a single MF carrying vestibular information and they are particularly enriched in the posterior cerebellum and flocculus (Rossi et al., 1995). UBCs also receive inhibitory synaptic inputs from Golgi Cells (GoCs) (Dugue et al., 2005). Glycinergic/GABAergic interneurons also populate the GCL and these include GoCs and Lugaro Cells (LCs). GoCs provide the sole inhibitory input to GCs, via feed-forward and a feedback inhibitory loops (Kanichay and Silver, 2008). LCs have spindle-shaped cell bodies (Laine and Axelrad, 1996) and they innervate the MLIs (Laine and Axelrad, 1998), GoCs (Dieudonne and Dumoulin, 2000; Dumoulin et al., 2001), and PCs (Dean et al., 2003). A prominent feature of these interneurons is their activation by serotonin, which induces firing in an otherwise silent cell (Dieudonne and Dumoulin, 2000; Dumoulin et al., 2001).
PCs originate in the ventricular zone around embryonic day E10.5-E13.5 in the mouse; by E14, they form a multicellular layer called the PC plate, a temporary structure which is replaced by a monolayer organization at early postnatal stage. PCs are characterized by an extensive dendritic tree, which develops during the first three postnatal weeks in the mouse (Sotelo and Dusart, 2009). The expression or the absence of specific molecular markers in PCs (Ex. ZebrinII (ZII)/Aldolase C) defines the compartmentation of the cerebellar cortex, organized in an alternate series of positive/negative parasagittal PC stripes (Brochu et al., 1990). The timing of PC birth determines their molecular fate, with early born (E10-E11.5) PCs becoming ZII+ cells and late-born (E11.5-E13.5) PCs destined to have a ZII− phenotype (Miale and Sidman, 1961). The spatial arrangement of CFs and MFs terminals follow PC patterning in the cerebellar cortex. Notably, immunolabelling of IO subnuclei in the rat revealed the preferential targeting of strongly ZII+ parasagittal bands by CFs originating from the subnucleus a of the caudal medial accessory olive (c-MAO), the rostral part of the MAO (r-MAO) and the dorsal and ventral lamellas of the principal olive (d-PO and v-PO, respectively). In contrast, ZII− bands of PCs are targeted by the subnucleus b of the c-MAO (Sugihara and Shinoda, 2004). Also, spinocerebellar and cuneocerebellar MFs terminate in the anterior cerebellum in complementary parasagittal bands with a defined distribution relative to the ZII± bands in this region (Ji and Hawkes, 1994; Valera et al., 2016). PC patterning plays a role in the definition of the final topographic map of cerebellar excitatory afferents and its modifications are mirrored by the altered spatial arrangement of CF and MF terminals (Blatt and Eisenman, 1988; Sillitoe et al., 2010; Reeber et al., 2013).
Activity-dependent mechanisms influence cerebellar compartmentation, the fine delineation of afferents topographic map and circuitry wiring. Of note, the targeted silencing of PC-specific neurotransmission affected the organization of ZII stripes, and altered spinocerebellar MF patterning (White et al., 2014). Furthermore, monoinnervation of PCs via CF relies on activity-dependent calcium influx as revealed by the impairment in the biased strengthening of a single CF in Cav2.1 (P/Q type) PC-targeted knockout (KO) mice (Hashimoto et al., 2011; Kawamura et al., 2013).
Purkinje cells are also involved in the final maturation of the cerebellum: during the early postnatal period, PC-mediated Sonic Hedgehog (SHH) signaling drives the proliferation of GC progenitors within the External Granule cell Layer (EGL) (Wechsler-Reya and Scott, 1999) and the level of SHH signaling is an important determinant for cerebellar foliation (Corrales et al., 2006).
Several SCA-linked mutations directly or indirectly affect PC activity, calcium dynamics and dendrite development. They are therefore potentially harmful to cerebellar compartmentation, wiring and afferent organization.
PC firing defects have been described in several SCA models. The expression of the mutant ATXN1 in SCA1 PCs or ATXN2 in SCA2 PCs reduced their firing frequency (Hansen et al., 2011; Dell’Orco et al., 2015) and this decrease was also associated with an increase in PC firing variability in the SCA2 mouse model (Kasumu et al., 2012). Furthermore, in vivo analysis of complex spike activity in SCA2 mice revealed a reduction in the complex spike frequency in response to pharmacological IO stimulation by systemic harmaline injection (Egorova et al., 2018).
Deletion or mutation in SPTBN2 gene (β-III spectrin) (Ikeda et al., 2006) causes SCA5, and β-III knockout in mice resulted in transient and resurgent sodium current reduction in PCs, leading to a decrease in PC spontaneous firing. Furthermore, PC single spike frequency was altered while no difference in complex spikes was detected (Perkins et al., 2010). Moreover, PCs presented reduced dendritic surface area, impaired mono-planar dendritic arborization and reduced spine density (Gao et al., 2011). An impaired dendritic development and spine density reduction is also a feature of SCA14 as demonstrated in cultured PCs expressing PKCγ harboring the SCA14-causing S119P mutation (Chen et al., 2003; Seki et al., 2009).
SCA6 is caused by a PolyQ tract expansion in the alternatively spliced exon 47 of the CACNA1A gene encoding for the α1A subunit of Cav2.1 voltage gated calcium channels (Zhuchenko et al., 1997). The PolyQ expansion alters Cav2.1 physiology via decreased channel expression in PCs, concomitantly reducing P/Q type calcium channel-mediated currents in SCA6 knock-in mice model. These modifications, however, were independent from the length of the PolyQ expansion (Watase et al., 2008). The presence of an internal ribosome entry site (IRES) within the Cav2.1 mRNA permits the expression of a C-terminus fragment (CT) containing the PolyQ tract. While the CT carrying the normal PolyQ tract serves as a transcription factor in PCs and promotes neurite outgrowth, the expression of the CT with the extended pathogenic PolyQ causes gait abnormalities and significant ML thinning (Du et al., 2013). Furthermore, in vivo recordings support a higher incidence of irregular PC firing in mice specifically expressing the pathogenic CT in PCs (Mark et al., 2015).
Three KCNC3 single mutations (R420H, R423H, and F448L) have been identified in families affected by SCA13 (Waters et al., 2006; Figueroa et al., 2011). KCNC3 encodes for Kv3.3 potassium channel, expressed in PCs where it participates in CF-mediated complex spike formation (Zagha et al., 2008). Additionally, based on computer modeling, Kv3.3-mediated conductance is proposed to interact with resurgent sodium currents in order to drive PC spontaneous firing (Akemann and Knopfel, 2006). R420H substitution in the S4 helix of Kv3.3 leads to the loss of the channel-mediated current via a dominant negative effect (Waters et al., 2006), while R423H and F448L mutations affect the gating properties of the channel. In the Kv3.3 KO SCA13 mouse model, PCs exhibited reduced spontaneous firing rate (Akemann and Knopfel, 2006) and lentiviral-mediated expression of Kv3.3 R424H (murine homolog of human Kv3.3 R423H) in cultured PCs led to impaired dendritic development (Irie et al., 2014). The missense mutation F145S in the Fibroblast Growth Factor 14 (FGF14) causes SCA27 in humans through a dominant negative effect (van Swieten et al., 2003; Brusse et al., 2006), and FGF14 ablation induces ataxia in mice (Wang et al., 2002). FGF14 modulates resurgent sodium currents in PCs (Yan et al., 2014) and FGF14 KO mice present a higher number of PCs lacking spontaneous firing when compared to WT (Shakkottai et al., 2009).
The cerebellum undergoes a profound change in terms of maturation and synaptic refinement during the first postnatal weeks, a process mediated by the activity of several molecules. During this period, CFs and PFs establish their final innervation territory onto PCs. Electrophysiological studies in mice revealed that at postnatal day 3 (P3), PC soma display multiple/polyinnervation by CFs. At this stage of development, these multiple CF synapses display similar synaptic strength, but thereafter they undergo functional differentiation. This diversification plateaus at P7 with PCs receiving both a strong CF input and several weaker ones. The weaker CFs are characterized by a lower multivesicular release probability. P/Q type voltage-gated calcium channel activity is required for CF diversification; Cav2.1 deletion in PCs prevents the selective biased strengthening of a single CF, and multiple CF innervation onto PCs persists throughout the cerebellar development (Hashimoto et al., 2011; Kawamura et al., 2013). All CFs at this stage synapse onto PC soma and only by P9-10, the strongest CF, starts to translocate from the soma to PC dendrites (Hashimoto et al., 2009). Following the functional differentiation and as cerebellar development proceeds, the strong/winner CF is further strengthened via an anterograde signaling involving the C1ql1–Bai3 (brain-specific angiogenesis Inhibitor 3) pathway (Kakegawa et al., 2015). Moreover, retrograde signaling mediated by the secreted semaphorin 3A (Sema3A)–Plexin A4 and Progranulin–Sortilin 1 pathways participate in the strengthening and/or the maintenance of CFs inputs (Uesaka et al., 2014; Uesaka et al., 2018). By the end of the second postnatal week, the majority of PCs (60%) show monoinnervation by a strong CF while the remaining PCs display polyinnervation by a strong CF and one or more weaker CFs (Hashimoto and Kano, 2003). At this time point, while the winner CF proceeds further onto the PC dendritic tree, somatic synapses of weak CFs are progressively eliminated (Hashimoto et al., 2009).
The establishment of CF monoinnervation on PCs requires extensive pruning of synapses. An early study in the x-irradiated agranular cerebellum in the rat supports two consecutive phases of elimination: an early GC independent phase (from P7 to around P11 in the mouse) and a late phase (from P12 to around P17 in the mouse) that relies on the correct establishment of PF synapses onto PCs (Crepel et al., 1981). Indeed, impairing PF to PC synapse formation by deleting the glutamate receptor δ2 (GluRδ2) caused a marked reduction in PF synaptic contacts on PC dendrites by the second postnatal week (Kurihara et al., 1997). Additionally, persistent CF-mediated PC polyinnervation (Hashimoto et al., 2001) and its invasion into PF territory i.e., the distal portion of PC dendritic tree was observed (Hashimoto et al., 2001; Ichikawa et al., 2002). Several molecules and signaling pathways participate in CF synapse elimination process. These involve Serotonin 3A receptors (5-HT3A) (Oostland et al., 2013), C1Ql1-Bai3 signaling (Kakegawa et al., 2015), IGF-I-mediated signaling (Kakizawa et al., 2003), mGluR1 (Kano et al., 1997; Levenes et al., 1997), PKCγ (Kano et al., 1995), phospholipase Cβ4 (Hashimoto et al., 2000), semaphorin 7A (Sema7A)-Plexin C1 signaling (Uesaka et al., 2014), and Sema7A-Integrin B1 (Uesaka et al., 2014). Ligand-receptor signaling such as BDNF-TrkB signaling (Bosman et al., 2006; Johnson et al., 2007; Sherrard et al., 2009; Choo et al., 2017), NMDA receptors (Rabacchi et al., 1992; Kakizawa et al., 2000), GluRδ2 receptors (Kurihara et al., 1997), and P/Q type voltage gated calcium channels (Miyazaki et al., 2004; Hashimoto et al., 2011) are also implicated. GABAergic transmission (Nakayama et al., 2012), the immediate early gene Arc (Mikuni et al., 2013), Ca/Calmodulin Kinase IV (Ribar et al., 2000), αCa/Calmodulin Kinase II (Hansel et al., 2006) plays an important role in this process. Lastly, glutamate transporter GLAST (Watase et al., 1998; Miyazaki et al., 2017), the brain-specific receptor-like proteins BSRPs (Miyazaki et al., 2006) and myosin Va (Takagishi et al., 2007) are also involved (see Figure 2 for complete list of molecules involved in this process). Molecular pathways and players determining CF strengthening and synaptic refinement are susceptible to SCA-causing molecules/mutations and several studies have identified problems in CF to PC synaptic transmission (Smeets and Verbeek, 2016). Physiological, morphological and developmental abnormalities of these synaptic inputs can precede PC degeneration (Duvick et al., 2010; Barnes et al., 2011; Ebner et al., 2013; Ruegsegger et al., 2016).
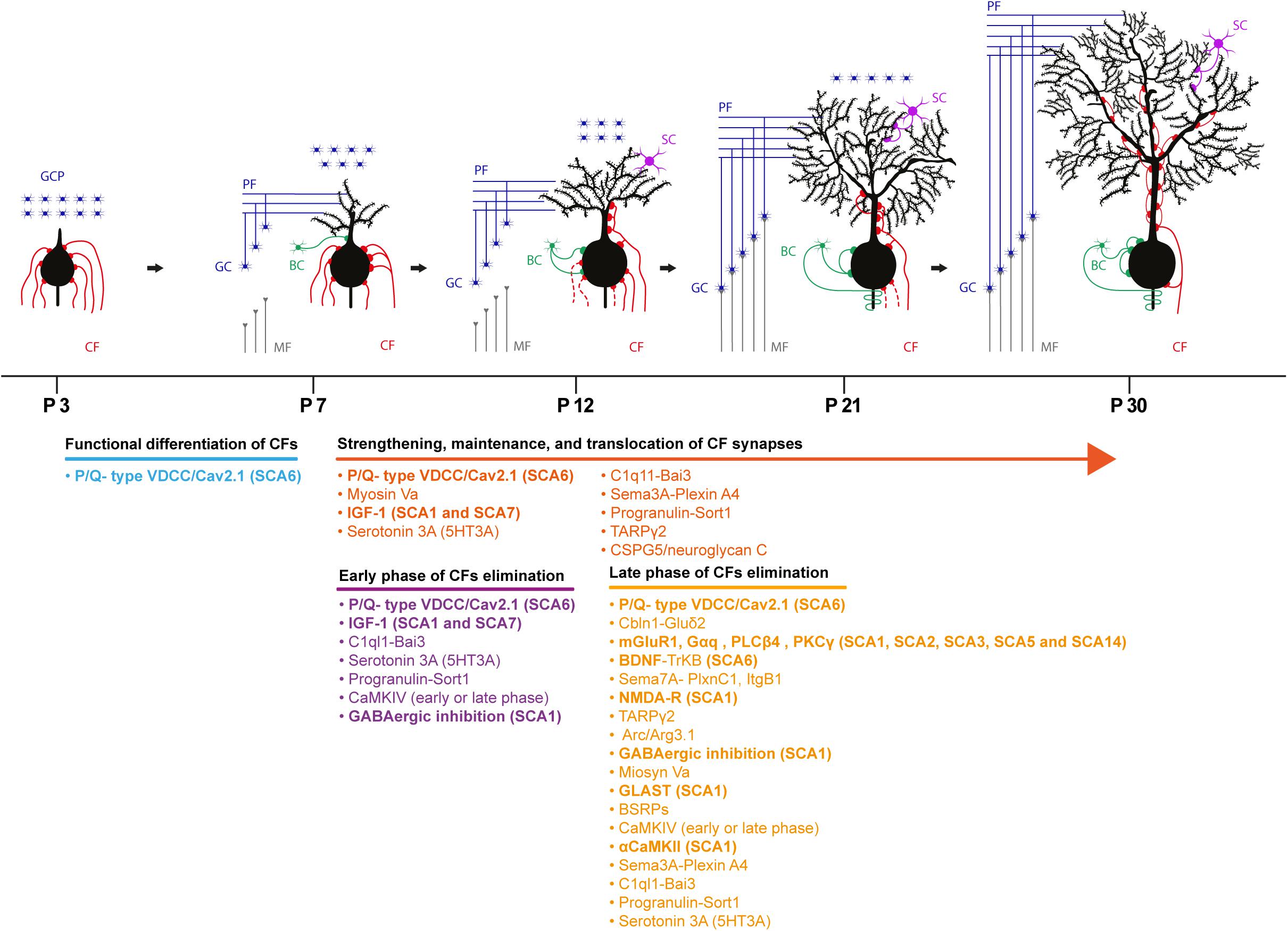
Figure 2. Schematic depicting the developmental synaptogenesis and refinement of CF to PC synapses. Four representative phases of the cerebellar circuit maturation are described with the molecular underpinnings involved in the process. Bold molecules indicate molecules implicated and identified in different SCAs in both human and rodent models. GCP, granule cell progenitor; CF, climbing fiber; MF, mossy fiber; GC, granule cell; PF, parallel fiber; BC, basket cell; SC, stellate cell.
The pharmacological manipulation of the Insulin-like growth factor I (IGF-I) pathway in vivo revealed the involvement of this hormone in the early phase of CF synapse elimination and strengthening. Application of exogenous IGF-I to the cerebellum at P8 caused an increase in the percentage of PCs displaying CF-mediated polyinnervation in young adult mice; and no significant effect when the IGF-I signaling was augmented during the late phase of synaptic pruning (P12). Moreover, together with the strengthening of the winner CF, the enhancement of weaker CF-mediated excitatory responses was also observed. In agreement with these results, inhibiting IGF-I signaling by local application of antisera against IGF-I and its receptor decreased EPSCs elicited by activation of the winner CF (Kakizawa et al., 2003). The analysis of transcriptional changes in murine models of SCA1 and SCA7 revealed the conserved downregulation of mRNA coding for the insulin-like growth factor binding protein 5 (IGFBP5) (Beattie et al., 2006), leading to enhanced IGF-I receptor activation probably via increased IGF-I availability (Gatchel et al., 2008). The modulation of the IGF-I signaling pathway by the mutant-ATXN1 therefore could interfere with CF strengthening and synapse elimination. Indeed, CF polyinnervation of young adult PCs and the impaired pruning of somatic CF synapse have been identified in SCA1 (Ebner et al., 2013). Nevertheless, a detailed analysis of the process of CF synapse elimination during cerebellar development is required to underpin SCA1- associated defects in CF wiring and function.
The retrograde BDNF to TrkB signaling in the developing cerebellum contributes to the late phase of CF synapse elimination. The PC-targeted knockout of BDNF resulted in a significant increase in the percentage of PCs showing polyinnervation by CFs at P16-P19, while no difference in innervation was detected prior to this time point. Similar results were obtained when the expression of TrkB was specifically downregulated in CFs (Choo et al., 2017). Accordingly, qRT-PCR analysis of post-mortem cerebellar samples from SCA6 patients revealed a significant decrease in the expression of BDNF transcripts (Takahashi et al., 2012), and an impairment in synaptic elimination of weaker CFs in the Cav2.1[84Q] SCA6 mouse model (Jayabal et al., 2017).
The mGluR1 cascade participates in postnatal maturation of CFs and mice lacking mGlur1 (Kano et al., 1997) or its downstream effectors PLCβ4 (Hashimoto et al., 2000) and PKCγ (Kano et al., 1995) displayed impairments in the late phase of synapse elimination. Importantly, the PC-specific expression of a splice variant of mGluR1 in mGluR1 knockout mice was effective in rescuing the late phase of CF synapse elimination (Ichise et al., 2000). Several lines of evidence indicate that the activation of mGluR1 receptors at PF to PC synapse is a major player at this stage of CF development (Hashimoto and Kano, 2013). mGluR1 signaling defects at PF to PC synapses have been identified in rodent models of SCA1 (Shuvaev et al., 2017), SCA3 (Konno et al., 2014), and SCA5 (Armbrust et al., 2014). The reduced amplitude of the mGluR1-dependent slow EPSCs component (Hartmann et al., 2008), the reduced dendritic calcium transients mediated by the mGluR1-PLCβ-IP3 receptor pathway activation and the impairment of short (Brown et al., 2004) and long-term plasticity (Ichise et al., 2000) have been demonstrated in these models. Furthermore, mGluR1 targeting to the PC dendritic spines was affected and a more diffuse expression of this metabotropic receptor was observed in SCA3 models (Konno et al., 2014) and SCA5 (Armbrust et al., 2014). On the contrary, the increased basal calcium concentration in PCs is likely to be responsible for the enhancement of mGluR1 signaling via a positive feedback loop in SCA2 (Meera et al., 2017).
mGluR1 signaling is a potential target for pharmacological interventions. Acute treatment of SCA1 mice via a single injection of the mGluR1 positive allosteric modulator Ro0711401 resulted in a significant long-lasting improvement of motor performance (Notartomaso et al., 2013). Exploiting mGluR1-GABAB functional coupling to enhance mGluR1 signaling (Tabata and Kano, 2006) was also proven effective in rescuing motor impairment in SCA1 mice (Shuvaev et al., 2017). Moreover, in the doxycycline-regulated conditional transgenic mouse model of SCA1, the improved motor performance of symptomatic mice due to the inhibition of mutant-ATXN1 expression was associated with restored mGluR1 expression levels in dendritic spines (Zu et al., 2004).
Missense mutations in PKCγ, the downstream mGluR1 effector, causes SCA14 (Chen et al., 2003). The expression of the SCA14-associated PKCγ-S119P mutant via in vivo lentiviral-mediated transduction of cerebellar neurons impaired CF synapse elimination, resulting in an increased number of PCs exhibiting polyinnervation. Importantly, this deficit in CF maturation appeared only by the expression of the PKCγ mutant during the time window of cerebellar development (Shuvaev et al., 2011). Among the proteins that contribute to the late phase of elimination, the GLAST glutamate transporter, NMDA receptors and αCamKII have been shown to be affected in SCA1. The decreased expression of the GLAST glutamate transporter in Bergmann glia (Cvetanovic, 2015), and the aberrant activation of extra synaptic NMDA receptors (Iizuka et al., 2015) in the cerebellum are involved in disease progression. Moreover, the decreased activation of αCamKII has been observed in SCA1 mouse model (Ruegsegger et al., 2016).
PF to PC synapse formation and transmission are also affected in SCAs. Abnormal PF invasion of CF innervation territory onto PCs is associated with the PC-specific expression of the pathogenic ATXN1-[30Q]-D766 mutant (Ebner et al., 2013); and the reduced strength of PF to PC synapse has also been reported in the SCA1[82Q] mouse model (Hourez et al., 2011). In mixed rat primary cerebellar cultures containing GCs and PCs, transfection of GCs with the SCA27 causing splice variant FGF14b-F150S mutant negatively modulated calcium currents in GCs and impaired PF to PC synaptic transmission, as demonstrated by the reduced EPSCs amplitude evoked in PCs by GCs (Yan et al., 2013). In agreement with these in vitro results, a reduced PC responsiveness to PF stimulation was also observed in acute cerebellar slices from the FGF14 knockout SCA27 mouse model, likely caused by a presynaptic deficit in glutamate release (Tempia et al., 2015).
The involvement of altered glutamatergic transmission in SCAs is well established, but the role of GABAergic transmission in these diseases remains poorly investigated. Interestingly, the GABA transporter 1 (GAT1) KO mouse (Chiu et al., 2005) and the vesicular GABA transporter (VGAT) KO mouse (Kayakabe et al., 2013) display poor motor coordination and an ataxic gait. Moreover, the AxJ cerebellar ataxia mouse model is characterized by PCs expressing high levels of the ionotropic GABAA receptor (GABAAR) and enlarged IPSCs (Lappe-Siefke et al., 2009). These findings therefore support a possible role of GABAergic transmission in ataxia onset and in disease progression. Indeed, postmortem analysis of brain tissue from SCA1 patients revealed a prominent increase in the BC to PC synapses (Edamakanti et al., 2018).
Key molecules and molecular pathways involved in PC development, activity and cerebellar afferent synaptic organization are affected in SCAs highlighting the possible role of cerebellar development and circuit wiring in the pathobiology of these neurodegenerative diseases. As the earliest manifestation of several SCAs, developmental defects characterize the conserved long silent phase of the pathology. However, their causative vs. protective role in the onset of ataxia and degeneration requires further investigation. Results from the conditional SCA1 mouse model supports their detrimental role, since motor impairment is partially rescued by delaying the expression of mutant ATXN1 until cerebellar development is complete (Serra et al., 2006). Nevertheless, if and how developmental deficits exacerbate the severity of the disease in other SCAs has still to be elucidated and further experiments are required.
A so far poorly explored area in the SCA research field is the investigation of the downstream effects of cortical wiring deficits on the final output stage of the cerebellum that generates the abnormal commands underlying motor impairments. The DCN is the main output of the cerebellum, and it receives GABAergic projections from PCs and excitatory inputs from CFs (Sugihara et al., 1999) and from MF collaterals (Shinoda et al., 1992; Matsushita and Gao, 1997; Matsushita and Xiong, 1997; Wu et al., 1999). Stimulation of the IO mediates the rebound increase in the firing of excitatory DCN neurons following an initial PC-mediated pause in their activity (Hoebeek et al., 2010). DCN activity is also effectively modulated by PC synchronicity that mediates time-locked spiking and rising of the firing frequency as well (Person and Raman, 2012a). Mechanisms of PC synchronizations are not fully elucidated, but experimental evidence supports the role of CF, GC to PC transmission and/or PC collaterals (Person and Raman, 2012b). By affecting the cortical synaptic organization of the cerebellum and PC activity, SCAs molecules could therefore strongly impact DCN activity and cerebellar neuronal coding; the so far identified defects in SCAs predict negative effects on the rebound firing properties of the DCN and PC synchronicity that require further and much needed investigation.
FB CP, and SS wrote the review. SS supervised the overall project. All authors read and commented on the final version of the review.
FB, CP, and SS were supported by the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program [Grant Number: 38-481 (ERC)].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Biagio Walter Farruggia for the realization of Figures 1, 2.
Adamaszek, M., D’agata, F., Ferrucci, R., Habas, C., Keulen, S., Kirkby, K. C., et al. (2017). Consensus paper: cerebellum and emotion. Cerebellum 16, 552–576. doi: 10.1007/s12311-016-0815-8
Akemann, W., and Knopfel, T. (2006). Interaction of Kv3 potassium channels and resurgent sodium current influences the rate of spontaneous firing of Purkinje neurons. J. Neurosci. 26, 4602–4612. doi: 10.1523/jneurosci.5204-05.2006
Ango, F., Di Cristo, G., Higashiyama, H., Bennett, V., Wu, P., and Huang, Z. J. (2004). Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at Purkinje axon initial segment. Cell 119, 257–272. doi: 10.1016/j.cell.2004.10.004
Armbrust, K. R., Wang, X., Hathorn, T. J., Cramer, S. W., Chen, G., Zu, T., et al. (2014). Mutant beta-III spectrin causes mGluR1alpha mislocalization and functional deficits in a mouse model of spinocerebellar ataxia type 5. J. Neurosci. 34, 9891–9904. doi: 10.1523/JNEUROSCI.0876-14.2014
Ashizawa, T., Oz, G., and Paulson, H. L. (2018). Spinocerebellar ataxias: prospects and challenges for therapy development. Nat. Rev. Neurol. 14, 590–605. doi: 10.1038/s41582-018-0051-6
Barnes, J. A., Ebner, B. A., Duvick, L. A., Gao, W., Chen, G., Orr, H. T., et al. (2011). Abnormalities in the climbing fiber-Purkinje cell circuitry contribute to neuronal dysfunction in ATXN1[82Q] mice. J. Neurosci. 31, 12778–12789. doi: 10.1523/JNEUROSCI.2579-11.2011
Beattie, J., Allan, G. J., Lochrie, J. D., and Flint, D. J. (2006). Insulin-like growth factor-binding protein-5 (IGFBP-5): a critical member of the IGF axis. Biochem. J. 395, 1–19. doi: 10.1042/bj20060086
Binda, F., Dorgans, K., Reibel, S., Sakimura, K., Kano, M., Poulain, B., et al. (2016). Inhibition promotes long-term potentiation at cerebellar excitatory synapses. Sci. Rep. 6:33561. doi: 10.1038/srep33561
Blatt, G. J., and Eisenman, L. M. (1988). Topographic and zonal organization of the olivocerebellar projection in the reeler mutant mouse. J. Comp. Neurol. 267, 603–615. doi: 10.1002/cne.902670412
Blot, A., and Barbour, B. (2014). Ultra-rapid axon-axon ephaptic inhibition of cerebellar Purkinje cells by the pinceau. Nat. Neurosci. 17, 289–295. doi: 10.1038/nn.3624
Bosman, L. W., Hartmann, J., Barski, J. J., Lepier, A., Noll-Hussong, M., Reichardt, L. F., et al. (2006). Requirement of TrkB for synapse elimination in developing cerebellar Purkinje cells. Brain Cell Biol. 35, 87–101. doi: 10.1007/s11068-006-9002-z
Boyden, E. S., Katoh, A., Pyle, J. L., Chatila, T. A., Tsien, R. W., and Raymond, J. L. (2006). Selective engagement of plasticity mechanisms for motor memory storage. Neuron 51, 823–834. doi: 10.1016/j.neuron.2006.08.026
Brochu, G., Maler, L., and Hawkes, R. (1990). Zebrin II: a polypeptide antigen expressed selectively by Purkinje cells reveals compartments in rat and fish cerebellum. J. Comp. Neurol. 291, 538–552. doi: 10.1002/cne.902910405
Brown, A. M., Arancillo, M., Lin, T., Catt, D. R., Zhou, J., Lackey, E. P., et al. (2019). Molecular layer interneurons shape the spike activity of cerebellar Purkinje cells. Sci. Rep. 9:1742. doi: 10.1038/s41598-018-38264-1
Brown, S. P., Safo, P. K., and Regehr, W. G. (2004). Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J. Neurosci. 24, 5623–5631. doi: 10.1523/jneurosci.0918-04.2004
Brusse, E., De Koning, I., Maat-Kievit, A., Oostra, B. A., Heutink, P., and van Swieten, J. C. (2006). Spinocerebellar ataxia associated with a mutation in the fibroblast growth factor 14 gene (SCA27): a new phenotype. Mov. Disord. 21, 396–401. doi: 10.1002/mds.20708
Chen, D. H., Brkanac, Z., Verlinde, C. L., Tan, X. J., Bylenok, L., Nochlin, D., et al. (2003). Missense mutations in the regulatory domain of PKC gamma: a new mechanism for dominant nonepisodic cerebellar ataxia. Am. J. Hum. Genet. 72, 839–849. doi: 10.1086/373883
Chiu, C. S., Brickley, S., Jensen, K., Southwell, A., Mckinney, S., Cull-Candy, S., et al. (2005). GABA transporter deficiency causes tremor, ataxia, nervousness, and increased GABA-induced tonic conductance in cerebellum. J. Neurosci. 25, 3234–3245. doi: 10.1523/jneurosci.3364-04.2005
Choo, M., Miyazaki, T., Yamazaki, M., Kawamura, M., Nakazawa, T., Zhang, J., et al. (2017). Retrograde BDNF to TrkB signaling promotes synapse elimination in the developing cerebellum. Nat. Commun. 8:195. doi: 10.1038/s41467-017-00260-w
Coesmans, M., Weber, J. T., De Zeeuw, C. I., and Hansel, C. (2004). Bidirectional parallel fiber plasticity in the cerebellum under climbing fiber control. Neuron 44, 691–700. doi: 10.1016/j.neuron.2004.10.031
Corrales, J. D., Blaess, S., Mahoney, E. M., and Joyner, A. L. (2006). The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development 133, 1811–1821. doi: 10.1242/dev.02351
Coutelier, M., Blesneac, J., Monteil, A., Monin, M. L., Ando, K., Mundwiller, E., et al. (2015). A recurrent mutation in CACNA1G alters cav3.1 T-type calcium-channel conduction and causes autosomal-dominant cerebellar ataxia. Am. J. Hum. Genet. 97, 726–737. doi: 10.1016/j.ajhg.2015.09.007
Crepel, F., Delhaye-Bouchaud, N., and Dupont, J. L. (1981). Fate of the multiple innervation of cerebellar Purkinje cells by climbing fibers in immature control, x-irradiated and hypothyroid rats. Brain Res. 227, 59–71. doi: 10.1016/0165-3806(81)90094-8
Crook, J., Hendrickson, A., and Robinson, F. R. (2006). Co-localization of glycine and gaba immunoreactivity in interneurons in Macaca monkey cerebellar cortex. Neuroscience 141, 1951–1959. doi: 10.1016/j.neuroscience.2006.05.012
Cvetanovic, M. (2015). Decreased expression of glutamate transporter GLAST in Bergmann glia is associated with the loss of Purkinje neurons in the spinocerebellar ataxia type 1. Cerebellum 14, 8–11. doi: 10.1007/s12311-014-0605-0
De Zeeuw, C. I., Hansel, C., Bian, F., Koekkoek, S. K., Van Alphen, A. M., Linden, D. J., et al. (1998). Expression of a protein kinase C inhibitor in Purkinje cells blocks cerebellar LTD and adaptation of the vestibulo-ocular reflex. Neuron 20, 495–508. doi: 10.1016/s0896-6273(00)80990-3
Dean, I., Robertson, S. J., and Edwards, F. A. (2003). Serotonin drives a novel GABAergic synaptic current recorded in rat cerebellar purkinje cells: a Lugaro cell to Purkinje cell synapse. J. Neurosci. 23, 4457–4469. doi: 10.1523/jneurosci.23-11-04457.2003
Dell’Orco, J. M., Wasserman, A. H., Chopra, R., Ingram, M. A., Hu, Y. S., Singh, V., et al. (2015). Neuronal atrophy early in degenerative ataxia is a compensatory mechanism to regulate membrane excitability. J. Neurosci. 35, 11292–11307. doi: 10.1523/JNEUROSCI.1357-15.2015
Dieudonne, S., and Dumoulin, A. (2000). Serotonin-driven long-range inhibitory connections in the cerebellar cortex. J. Neurosci. 20, 1837–1848. doi: 10.1523/jneurosci.20-05-01837.2000
Du, X., Wang, J., Zhu, H., Rinaldo, L., Lamar, K. M., Palmenberg, A. C., et al. (2013). Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell 154, 118–133. doi: 10.1016/j.cell.2013.05.059
Dugue, G. P., Dumoulin, A., Triller, A., and Dieudonne, S. (2005). Target-dependent use of co-released inhibitory transmitters at central synapses. J. Neurosci. 25, 6490–6498. doi: 10.1523/jneurosci.1500-05.2005
Dulneva, A., Lee, S., Oliver, P. L., Di Gleria, K., Kessler, B. M., Davies, K. E., et al. (2015). The mutant Moonwalker TRPC3 channel links calcium signaling to lipid metabolism in the developing cerebellum. Hum. Mol. Genet. 24, 4114–4125. doi: 10.1093/hmg/ddv150
Dumoulin, A., Triller, A., and Dieudonne, S. (2001). IPSC kinetics at identified GABAergic and mixed GABAergic and glycinergic synapses onto cerebellar Golgi cells. J. Neurosci. 21, 6045–6057. doi: 10.1523/jneurosci.21-16-06045.2001
Duvick, L., Barnes, J., Ebner, B., Agrawal, S., Andresen, M., Lim, J., et al. (2010). SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron 67, 929–935. doi: 10.1016/j.neuron.2010.08.022
Ebner, B. A., Ingram, M. A., Barnes, J. A., Duvick, L. A., Frisch, J. L., Clark, H. B., et al. (2013). Purkinje cell ataxin-1 modulates climbing fiber synaptic input in developing and adult mouse cerebellum. J. Neurosci. 33, 5806–5820. doi: 10.1523/JNEUROSCI.6311-11.2013
Eccles, J. C., Llinas, R., and Sasaki, K. (1966). The excitatory synaptic action of climbing fibres on the Purkinje cells of the cerebellum. J. Physiol. 182, 268–296. doi: 10.1113/jphysiol.1966.sp007824
Edamakanti, C. R., Do, J., Didonna, A., Martina, M., and Opal, P. (2018). Mutant ataxin1 disrupts cerebellar development in spinocerebellar ataxia type 1. J. Clin. Invest. 128, 2252–2265. doi: 10.1172/JCI96765
Egorova, P. A., Gavrilova, A. V., and Bezprozvanny, I. B. (2018). In vivo analysis of the climbing fiber-Purkinje cell circuit in SCA2-58Q transgenic mouse model. Cerebellum 17, 590–600. doi: 10.1007/s12311-018-0951-4
Figueroa, K. P., Waters, M. F., Garibyan, V., Bird, T. D., Gomez, C. M., Ranum, L. P. W., et al. (2011). Frequency of KCNC3 DNA variants as causes of spinocerebellar ataxia 13 (SCA13). PLoS One 6:e17811. doi: 10.1371/journal.pone.0017811
Fogel, B. L., Hanson, S. M., and Becker, E. B. E. (2015). Do mutations in the murine ataxia gene TRPC3 cause cerebellar ataxia in humans? Mov. Disord. 30, 284–286. doi: 10.1002/mds.26096
Gao, Y. Z., Perkins, E. M., Clarkson, Y. L., Tobia, S., Lyndon, A. R., Jackson, M., et al. (2011). beta-III spectrin is critical for development of Purkinje cell dendritic tree and spine morphogenesis. J. Neurosci. 31, 16581–16590. doi: 10.1523/JNEUROSCI.3332-11.2011
Gatchel, J. R., Watase, K., Thaller, C., Carson, J. P., Jafar-Nejad, P., Shaw, C., et al. (2008). The insulin-like growth factor pathway is altered in spinocerebellar ataxia type 1 and type 7. Proc. Natl. Acad. Sci. U.S.A. 105, 1291–1296. doi: 10.1073/pnas.0711257105
Hansel, C., De Jeu, M., Belmeguenai, A., Houtman, S. H., Buitendijk, G. H., Andreev, D., et al. (2006). alphaCaMKII Is essential for cerebellar LTD and motor learning. Neuron 51, 835–843. doi: 10.1016/j.neuron.2006.08.013
Hansen, S. T., Meera, P., Otis, T. S., and Pulst, S. M. (2011). Changes in Purkinje cell firing accompany biochemical and behavioral changes in a mouse model of SCA 2. Neurology 76, A346–A347.
Hansen, S. T., Meera, P., Otis, T. S., and Pulst, S. M. (2013). Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum. Mol. Genet. 22, 271–283. doi: 10.1093/hmg/dds427
Hara, K., Fukushima, T., Suzuki, T., Shimohata, T., Oyake, M., Ishiguro, H., et al. (2004). Japanese SCA families with an unusual phenotype linked to a locus overlapping with SCA15 locus. Neurology 62, 648–651. doi: 10.1212/01.wnl.0000110190.08412.25
Hara, K., Shiga, A., Nozaki, H., Mitsui, J., Takahashi, Y., Ishiguro, H., et al. (2008). Total deletion and a missense mutation of ITPR1 in Japanese SCA15 families. Neurology 71, 547–551. doi: 10.1212/01.wnl.0000311277.71046.a0
Hartmann, J., Dragicevic, E., Adelsberger, H., Henning, H. A., Sumser, M., Abramowitz, J., et al. (2008). TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 59, 392–398. doi: 10.1016/j.neuron.2008.06.009
Hashimoto, K., Ichikawa, R., Kitamura, K., Watanabe, M., and Kano, M. (2009). Translocation of a “winner” climbing fiber to the Purkinje cell dendrite and subsequent elimination of “losers” from the soma in developing cerebellum. Neuron 63, 106–118. doi: 10.1016/j.neuron.2009.06.008
Hashimoto, K., Ichikawa, R., Takechi, H., Inoue, Y., Aiba, A., Sakimura, K., et al. (2001). Roles of glutamate receptor delta 2 subunit (GluRdelta 2) and metabotropic glutamate receptor subtype 1 (mGluR1) in climbing fiber synapse elimination during postnatal cerebellar development. J. Neurosci. 21, 9701–9712. doi: 10.1523/jneurosci.21-24-09701.2001
Hashimoto, K., and Kano, M. (2003). Functional differentiation of multiple climbing fiber inputs during synapse elimination in the developing cerebellum. Neuron 38, 785–796. doi: 10.1016/s0896-6273(03)00298-8
Hashimoto, K., and Kano, M. (2013). Synapse elimination in the developing cerebellum. Cell Mol. Life. Sci. 70, 4667–4680. doi: 10.1007/s00018-013-1405-2
Hashimoto, K., Tsujita, M., Miyazaki, T., Kitamura, K., Yamazaki, M., Shin, H. S., et al. (2011). Postsynaptic P/Q-type Ca2+ channel in Purkinje cell mediates synaptic competition and elimination in developing cerebellum. Proc. Natl. Acad. Sci. U.S.A. 108, 9987–9992. doi: 10.1073/pnas.1101488108
Hashimoto, K., Watanabe, M., Kurihara, H., Offermanns, S., Jiang, H., Wu, Y., et al. (2000). Climbing fiber synapse elimination during postnatal cerebellar development requires signal transduction involving G alpha q and phospholipase C beta 4. Prog. Brain Res. 124, 31–48. doi: 10.1016/s0079-6123(00)24006-5
Hausser, M., and Clark, B. A. (1997). Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron 19, 665–678. doi: 10.1016/s0896-6273(00)80379-7
Heffley, W., Song, E. Y., Xu, Z., Taylor, B. N., Hughes, M. A., Mckinney, A., et al. (2018). Coordinated cerebellar climbing fiber activity signals learned sensorimotor predictions. Nat. Neurosci. 21, 1431–1441. doi: 10.1038/s41593-018-0228-8
Hoebeek, F. E., Witter, L., Ruigrok, T. J. H., and De Zeeuw, C. I. (2010). Differential olivo-cerebellar cortical control of rebound activity in the cerebellar nuclei. Proc. Natl. Acad. Sci. U.S.A. 107, 8410–8415. doi: 10.1073/pnas.0907118107
Holmes, S. E., O’hearn, E. E., Mcinnis, M. G., Gorelick-Feldman, D. A., Kleiderlein, J. J., Callahan, C., et al. (1999). Expansion of a novel CAG trinucleotide repeat in the 5’ region of PPP2R2B is associated with SCA12. Nat. Genet. 23, 391–392. doi: 10.1038/70493
Houlden, H., Johnson, J., Gardner-Thorpe, C., Lashley, T., Hernandez, D., Worth, P., et al. (2007). Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat. Genet. 39, 1434–1436. doi: 10.1038/ng.2007.43
Hourez, R., Servais, L., Orduz, D., Gall, D., Millard, I., De Kerchove D’exaerde, A., et al. (2011). Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J. Neurosci. 31, 11795–11807. doi: 10.1523/JNEUROSCI.0905-11.2011
Huang, L. J., Chardon, J. W., Carter, M. T., Friend, K. L., Dudding, T. E., Schwartzentruber, J., et al. (2012). Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J. Rare Dis. 7:67. doi: 10.1186/1750-1172-7-67
Ichikawa, R., Miyazaki, T., Kano, M., Hashikawa, T., Tatsumi, H., Sakimura, K., et al. (2002). Distal extension of climbing fiber territory and multiple innervation caused by aberrant wiring to adjacent spiny branchlets in cerebellar Purkinje cells lacking glutamate receptor delta 2. J. Neurosci. 22, 8487–8503. doi: 10.1523/jneurosci.22-19-08487.2002
Ichise, T., Kano, M., Hashimoto, K., Yanagihara, D., Nakao, K., Shigemoto, R., et al. (2000). mGluR1 in cerebellar Purkinje cells essential for long-term depression, synapse elimination, and motor coordination. Science 288, 1832–1835. doi: 10.1126/science.288.5472.1832
Iizuka, A., Nakamura, K., and Hirai, H. (2015). Long-term oral administration of the NMDA receptor antagonist memantine extends life span in spinocerebellar ataxia type 1 knock-in mice. Neurosci. Lett. 592, 37–41. doi: 10.1016/j.neulet.2015.02.055
Ikeda, Y., Dick, K. A., Weatherspoon, M. R., Gincel, D., Armbrust, K. R., Dalton, J. C., et al. (2006). Spectrin mutations cause spinocerebellar ataxia type 5. Nat. Genet. 38, 184–190.
Irie, T., Matsuzaki, Y., Sekino, Y., and Hirai, H. (2014). Kv3.3 channels harbouring a mutation of spinocerebellar ataxia type 13 alter excitability and induce cell death in cultured cerebellar Purkinje cells. J. Physiol. 592, 229–247. doi: 10.1113/jphysiol.2013.264309
Ito, M., Sakurai, M., and Tongroach, P. (1982). Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. J. Physiol. 324, 113–134. doi: 10.1113/jphysiol.1982.sp014103
Jayabal, S., Ljungberg, L., and Watt, A. J. (2017). Transient cerebellar alterations during development prior to obvious motor phenotype in a mouse model of spinocerebellar ataxia type 6. J. Physiol. (Lond). 595, 949–966. doi: 10.1113/JP273184
Ji, Z., and Hawkes, R. (1994). Topography of Purkinje cell compartments and mossy fiber terminal fields in lobules II and III of the rat cerebellar cortex: spinocerebellar and cuneocerebellar projections. Neuroscience 61, 935–954. doi: 10.1016/0306-4522(94)90414-6
Johnson, E. M., Craig, E. T., and Yeh, H. H. (2007). TrkB is necessary for pruning at the climbing fibre-Purkinje cell synapse in the developing murine cerebellum. J. Physiol. 582, 629–646. doi: 10.1113/jphysiol.2007.133561
Kakegawa, W., Mitakidis, N., Miura, E., Abe, M., Matsuda, K., Takeo, Y. H., et al. (2015). Anterograde C1ql1 signaling is required in order to determine and maintain a single-winner climbing fiber in the mouse cerebellum. Neuron 85, 316–329. doi: 10.1016/j.neuron.2014.12.020
Kakizawa, S., Yamada, K., Iino, M., Watanabe, M., and Kano, M. (2003). Effects of insulin-like growth factor I on climbing fibre synapse elimination during cerebellar development. Eur. J. Neurosci. 17, 545–554. doi: 10.1046/j.1460-9568.2003.02486.x
Kakizawa, S., Yamasaki, M., Watanabe, M., and Kano, M. (2000). Critical period for activity-dependent synapse elimination in developing cerebellum. J. Neurosci. 20, 4954–4961. doi: 10.1523/jneurosci.20-13-04954.2000
Kanichay, R. T., and Silver, R. A. (2008). Synaptic and cellular properties of the feedforward inhibitory circuit within the input layer of the cerebellar cortex. J. Neurosci. 28, 8955–8967. doi: 10.1523/JNEUROSCI.5469-07.2008
Kano, M., Hashimoto, K., Chen, C., Abeliovich, A., Aiba, A., Kurihara, H., et al. (1995). Impaired synapse elimination during cerebellar development in PKC gamma mutant mice. Cell 83, 1223–1231. doi: 10.1016/0092-8674(95)90147-7
Kano, M., Hashimoto, K., Kurihara, H., Watanabe, M., Inoue, Y., Aiba, A., et al. (1997). Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron 18, 71–79. doi: 10.1016/s0896-6273(01)80047-7
Kasumu, A. W., Liang, X., Egorova, P., Vorontsova, D., and Bezprozvanny, I. (2012). Chronic suppression of inositol 1,4,5-triphosphate receptor-mediated calcium signaling in cerebellar Purkinje cells alleviates pathological phenotype in spinocerebellar ataxia 2 mice. J. Neurosci. 32, 12786–12796. doi: 10.1523/JNEUROSCI.1643-12.2012
Kawamura, Y., Nakayama, H., Hashimoto, K., Sakimura, K., Kitamura, K., and Kano, M. (2013). Spike timing-dependent selective strengthening of single climbing fibre inputs to Purkinje cells during cerebellar development. Nat. Commun. 4:2732. doi: 10.1038/ncomms3732
Kayakabe, M., Kakizaki, T., Kaneko, R., Sasaki, A., Nakazato, Y., Shibasaki, K., et al. (2013). Motor dysfunction in cerebellar Purkinje cell-specific vesicular GABA transporter knockout mice. Front. Cell Neurosci. 7:286. doi: 10.3389/fncel.2013.00286
Koide, R., Kobayashi, S., Shimohata, T., Ikeuchi, T., Maruyama, M., Saito, M., et al. (1999). A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum. Mol. Genet. 8, 2047–2053. doi: 10.1093/hmg/8.11.2047
Konno, A., Shuvaev, A. N., Miyake, N., Miyake, K., Iizuka, A., Matsuura, S., et al. (2014). Mutant ataxin-3 with an abnormally expanded polyglutamine chain disrupts dendritic development and metabotropic glutamate receptor signaling in mouse cerebellar Purkinje cells. Cerebellum 13, 29–41. doi: 10.1007/s12311-013-0516-5
Kostadinov, D., Beau, M., Pozo, M. B., and Hausser, M. (2019). Predictive and reactive reward signals conveyed by climbing fiber inputs to cerebellar Purkinje cells. Nat. Neurosci. 22, 950–962. doi: 10.1038/s41593-019-0381-8
Koziol, L. F., Budding, D., Andreasen, N., D’arrigo, S., Bulgheroni, S., Imamizu, H., et al. (2014). Consensus paper: the cerebellum’s role in movement and cognition. Cerebellum 13, 151–177. doi: 10.1007/s12311-013-0511-x
Kurihara, H., Hashimoto, K., Kano, M., Takayama, C., Sakimura, K., Mishina, M., et al. (1997). Impaired parallel fiber– > Purkinje cell synapse stabilization during cerebellar development of mutant mice lacking the glutamate receptor delta2 subunit. J. Neurosci. 17, 9613–9623. doi: 10.1523/jneurosci.17-24-09613.1997
Laine, J., and Axelrad, H. (1994). The candelabrum cell: a new interneuron in the cerebellar cortex. J. Comp. Neurol. 339, 159–173. doi: 10.1002/cne.903390202
Laine, J., and Axelrad, H. (1996). Morphology of the Golgi-impregnated Lugaro cell in the rat cerebellar cortex: a reappraisal with a description of its axon. J. Comp. Neurol. 375, 618–640. doi: 10.1002/(sici)1096-9861(19961125)375:4<618::aid-cne5>3.0.co;2-4
Laine, J., and Axelrad, H. (1998). Lugaro cells target basket and stellate cells in the cerebellar cortex. Neuroreport 9, 2399–2403. doi: 10.1097/00001756-199807130-00045
Lappe-Siefke, C., Loebrich, S., Hevers, W., Waidmann, O. B., Schweizer, M., Fehr, S., et al. (2009). The ataxia (axJ) mutation causes abnormal GABAA receptor turnover in mice. PLoS Genet. 5:e1000631. doi: 10.1371/journal.pgen.1000631
Larry, N., Yarkoni, M., Lixenberg, A., and Joshua, M. (2019). Cerebellar climbing fibers encode expected reward size. eLife 8:e46870. doi: 10.7554/eLife.46870
Lee, Y. C., Durr, A., Majczenko, K., Huang, Y. H., Liu, Y. C., Lien, C. C., et al. (2012). Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann. Neurol. 72, 859–869. doi: 10.1002/ana.23701
Levenes, C., Daniel, H., Jaillard, D., Conquet, F., and Crepel, F. (1997). Incomplete regression of multiple climbing fibre innervation of cerebellar Purkinje cells in mGLuR1 mutant mice. Neuroreport 8, 571–574. doi: 10.1097/00001756-199701200-00038
Lev-Ram, V., Wong, S. T., Storm, D. R., and Tsien, R. Y. (2002). A new form of cerebellar long-term potentiation is postsynaptic and depends on nitric oxide but not cAMP. Proc. Natl. Acad. Sci. U.S.A. 99, 8389–8393. doi: 10.1073/pnas.122206399
Lin, P. F., Zhang, D., Xu, G. R., and Yan, C. Z. (2018). Identification of IFRD1 variant in a Han Chinese family with autosomal dominant hereditary spastic paraplegia associated with peripheral neuropathy and ataxia. J. Hum. Genet. 63, 521–524. doi: 10.1038/s10038-017-0394-7
Ly, R., Bouvier, G., Schonewille, M., Arabo, A., Rondi-Reig, L., Lena, C., et al. (2013). T-type channel blockade impairs long-term potentiation at the parallel fiber-Purkinje cell synapse and cerebellar learning. Proc. Natl. Acad. Sci. U.S.A. 110, 20302–20307. doi: 10.1073/pnas.1311686110
Mark, M. D., Krause, M., Boele, H. J., Kruse, W., Pollok, S., Kuner, T., et al. (2015). Spinocerebellar ataxia type 6 protein aggregates cause deficits in motor learning and cerebellar plasticity. J. Neurosci. 35, 8882–8895. doi: 10.1523/JNEUROSCI.0891-15.2015
Matsushita, M., and Gao, X. (1997). Projections from the thoracic cord to the cerebellar nuclei in the rat, studied by anterograde axonal tracing. J. Comp. Neurol. 386, 409–421. doi: 10.1002/(sici)1096-9861(19970929)386:3<409::aid-cne6>3.0.co;2-5
Matsushita, M., and Xiong, G. X. (1997). Projections from the cervical enlargement to the cerebellar nuclei in the rat, studied by anterograde axonal tracing. J. Comp. Neurol. 377, 251–261. doi: 10.1002/(sici)1096-9861(19970113)377:2<251::aid-cne7>3.0.co;2-2
Meera, P., Pulst, S., and Otis, T. (2017). A positive feedback loop linking enhanced mGluR function and basal calcium in spinocerebellar ataxia type 2. eLife 6:e26377. doi: 10.7554/eLife.26377
Miale, I. L., and Sidman, R. L. (1961). An autoradiographic analysis of histogenesis in the mouse cerebellum. Exp. Neurol. 4, 277–296. doi: 10.1016/0014-4886(61)90055-3
Mikuni, T., Uesaka, N., Okuno, H., Hirai, H., Deisseroth, K., Bito, H., et al. (2013). Arc/Arg3.1 is a postsynaptic mediator of activity-dependent synapse elimination in the developing cerebellum. Neuron 78, 1024–1035. doi: 10.1016/j.neuron.2013.04.036
Mittmann, W., Koch, U., and Hausser, M. (2005). Feed-forward inhibition shapes the spike output of cerebellar Purkinje cells. J. Physiol. 563, 369–378. doi: 10.1113/jphysiol.2004.075028
Miyazaki, T., Hashimoto, K., Shin, H. S., Kano, M., and Watanabe, M. (2004). P/Q-type Ca2+ channel alpha1A regulates synaptic competition on developing cerebellar Purkinje cells. J. Neurosci. 24, 1734–1743. doi: 10.1523/jneurosci.4208-03.2004
Miyazaki, T., Hashimoto, K., Uda, A., Sakagami, H., Nakamura, Y., Saito, S. Y., et al. (2006). Disturbance of cerebellar synaptic maturation in mutant mice lacking BSRPs, a novel brain-specific receptor-like protein family. FEBS Lett. 580, 4057–4064. doi: 10.1016/j.febslet.2006.06.043
Miyazaki, T., Yamasaki, M., Hashimoto, K., Kohda, K., Yuzaki, M., Shimamoto, K., et al. (2017). Glutamate transporter GLAST controls synaptic wrapping by Bergmann glia and ensures proper wiring of Purkinje cells. Proc. Natl. Acad. Sci. U.S.A. 114, 7438–7443. doi: 10.1073/pnas.1617330114
Morino, H., Matsuda, Y., Muguruma, K., Miyamoto, R., Ohsawa, R., Ohtake, T., et al. (2015). A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Mol. Brain 8:89. doi: 10.1186/s13041-015-0180-4
Nakayama, H., Miyazaki, T., Kitamura, K., Hashimoto, K., Yanagawa, Y., Obata, K., et al. (2012). GABAergic inhibition regulates developmental synapse elimination in the cerebellum. Neuron 74, 384–396. doi: 10.1016/j.neuron.2012.02.032
Nam, S. C., and Hockberger, P. E. (1997). Analysis of spontaneous electrical activity in cerebellar Purkinje cells acutely isolated from postnatal rats. J. Neurobiol. 33, 18–32. doi: 10.1002/(sici)1097-4695(199707)33:1<18::aid-neu3>3.0.co;2-g
Napper, R. M., and Harvey, R. J. (1988). Number of parallel fiber synapses on an individual Purkinje cell in the cerebellum of the rat. J. Comp. Neurol. 274, 168–177. doi: 10.1002/cne.902740204
Notartomaso, S., Zappulla, C., Biagioni, F., Cannella, M., Bucci, D., Mascio, G., et al. (2013). Pharmacological enhancement of mGlu1 metabotropic glutamate receptors causes a prolonged symptomatic benefit in a mouse model of spinocerebellar ataxia type 1. Mol. Brain 6:48. doi: 10.1186/1756-6606-6-48
Nunzi, M. G., Birnstiel, S., Bhattacharyya, B. J., Slater, N. T., and Mugnaini, E. (2001). Unipolar brush cells form a glutamatergic projection system within the mouse cerebellar cortex. J. Comp. Neurol. 434, 329–341. doi: 10.1002/cne.1180
Ohmae, S., and Medina, J. F. (2015). Climbing fibers encode a temporal-difference prediction error during cerebellar learning in mice. Nat. Neurosci. 18, 1798–1803. doi: 10.1038/nn.4167
Oostland, M., Buijink, M. R., and Van Hooft, J. A. (2013). Serotonergic control of Purkinje cell maturation and climbing fibre elimination by 5-HT3 receptors in the juvenile mouse cerebellum. J. Physiol. 591, 1793–1807. doi: 10.1113/jphysiol.2012.246413
Orr, H. T., Chung, M. Y., Banfi, S., Kwiatkowski, T. J. Jr., Servadio, A., Beaudet, A. L., et al. (1993). Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 4, 221–226. doi: 10.1038/ng0793-221
Perkins, E. M., Clarkson, Y. L., Sabatier, N., Longhurst, D. M., Millward, C. P., Jack, J., et al. (2010). Loss of beta-III spectrin leads to Purkinje cell dysfunction recapitulating the behavior and neuropathology of spinocerebellar ataxia type 5 in humans. J. Neurosci. 30, 4857–4867. doi: 10.1523/JNEUROSCI.6065-09.2010
Person, A. L., and Raman, I. M. (2012a). Purkinje neuron synchrony elicits time-locked spiking in the cerebellar nuclei. Nature 481, 502–505. doi: 10.1038/nature10732
Person, A. L., and Raman, I. M. (2012b). Synchrony and neural coding in cerebellar circuits. Front. Neural Circuits 6:97. doi: 10.3389/fncir.2012.00097
Pilotto, F., and Saxena, S. (2018). Epidemiology of inherited cerebellar ataxias and challenges in clinical research. Clin. Transl. Neurosci. 2:12.
Rabacchi, S., Bailly, Y., Delhaye-Bouchaud, N., and Mariani, J. (1992). Involvement of the N-methyl D-aspartate (n.d.) receptor in synapse elimination during cerebellar development. Science 256, 1823–1825. doi: 10.1126/science.1352066
Reeber, S. L., Loeschel, C. A., Franklin, A., and Sillitoe, R. V. (2013). Establishment of topographic circuit zones in the cerebellum of scrambler mutant mice. Front. Neural Circuits 7:122. doi: 10.3389/fncir.2013.00122
Ribar, T. J., Rodriguiz, R. M., Khiroug, L., Wetsel, W. C., Augustine, G. J., and Means, A. R. (2000). Cerebellar defects in Ca2+/calmodulin kinase IV-deficient mice. J. Neurosci. 20:RC107.
Riess, O., Schols, L., Bottger, H., Nolte, D., Vieira-Saecker, A. M., Schimming, C., et al. (1997). SCA6 is caused by moderate CAG expansion in the alpha1A-voltage-dependent calcium channel gene. Hum. Mol. Genet. 6, 1289–1293. doi: 10.1093/hmg/6.8.1289
Rossi, D. J., Alford, S., Mugnaini, E., and Slater, N. T. (1995). Properties of transmission at a giant glutamatergic synapse in cerebellum: the mossy fiber-unipolar brush cell synapse. J. Neurophysiol. 74, 24–42. doi: 10.1152/jn.1995.74.1.24
Ruegsegger, C., Stucki, D. M., Steiner, S., Angliker, N., Radecke, J., Keller, E., et al. (2016). Impaired mTORC1-Dependent Expression of Homer-3 Influences SCA1 Pathophysiology. Neuron 89, 129–146. doi: 10.1016/j.neuron.2015.11.033
Schonewille, M., Belmeguenai, A., Koekkoek, S. K., Houtman, S. H., Boele, H. J., Van Beugen, B. J., et al. (2010). Purkinje cell-specific knockout of the protein phosphatase PP2B impairs potentiation and cerebellar motor learning. Neuron 67, 618–628. doi: 10.1016/j.neuron.2010.07.009
Seki, T., Shimahara, T., Yamamoto, K., Abe, N., Amano, T., Adachi, N., et al. (2009). Mutant gamma PKC found in spinocerebellar ataxia type 14 induces aggregate-independent maldevelopment of dendrites in primary cultured Purkinje cells. Neurobiol. Dis. 33, 260–273. doi: 10.1016/j.nbd.2008.10.013
Serra, H. G., Duvick, L., Zu, T., Carlson, K., Stevens, S., Jorgensen, N., et al. (2006). RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 127, 697–708. doi: 10.1016/j.cell.2006.09.036
Shakkottai, V. G., Xiao, M., Xu, L., Wong, M., Nerbonne, J. M., Ornitz, D. M., et al. (2009). FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol. Dis. 33, 81–88. doi: 10.1016/j.nbd.2008.09.019
Sherrard, R. M., Dixon, K. J., Bakouche, J., Rodger, J., Lemaigre-Dubreuil, Y., and Mariani, J. (2009). Differential expression of TrkB isoforms switches climbing fiber-Purkinje cell synaptogenesis to selective synapse elimination. Dev. Neurobiol. 69, 647–662. doi: 10.1002/dneu.20730
Shinoda, Y., Sugiuchi, Y., Futami, T., and Izawa, R. (1992). Axon collaterals of mossy fibers from the pontine nucleus in the cerebellar dentate nucleus. J. Neurophysiol. 67, 547–560. doi: 10.1152/jn.1992.67.3.547
Shuvaev, A. N., Horiuchi, H., Seki, T., Goenawan, H., Irie, T., Iizuka, A., et al. (2011). Mutant PKCgamma in spinocerebellar ataxia type 14 disrupts synapse elimination and long-term depression in Purkinje cells in vivo. J. Neurosci. 31, 14324–14334. doi: 10.1523/JNEUROSCI.5530-10.2011
Shuvaev, A. N., Hosoi, N., Sato, Y., Yanagihara, D., and Hirai, H. (2017). Progressive impairment of cerebellar mGluR signalling and its therapeutic potential for cerebellar ataxia in spinocerebellar ataxia type 1 model mice. J. Physiol. 595, 141–164. doi: 10.1113/JP272950
Sillitoe, R. V., Vogel, M. W., and Joyner, A. L. (2010). Engrailed homeobox genes regulate establishment of the cerebellar afferent circuit map. J. Neurosci. 30, 10015–10024. doi: 10.1523/JNEUROSCI.0653-10.2010
Smeets, C. J., and Verbeek, D. S. (2016). Climbing fibers in spinocerebellar ataxia: a mechanism for the loss of motor control. Neurobiol. Dis. 88, 96–106. doi: 10.1016/j.nbd.2016.01.009
Sotelo, C., and Dusart, I. (2009). Intrinsic versus extrinsic determinants during the development of Purkinje cell dendrites. Neuroscience 162, 589–600. doi: 10.1016/j.neuroscience.2008.12.035
Storey, E., Gardner, R. J. M., Knight, M. A., Kennerson, M. L., Tuck, R. R., Forrest, S. M., et al. (2001). A new autosomal dominant pure cerebellar ataxia. Neurology 57, 1913–1915. doi: 10.1212/wnl.57.10.1913
Sugihara, I., and Shinoda, Y. (2004). Molecular, topographic, and functional organization of the cerebellar cortex: a study with combined aldolase C and olivocerebellar labeling. J. Neurosci. 24, 8771–8785. doi: 10.1523/jneurosci.1961-04.2004
Sugihara, I., Wu, H. S., and Shinoda, Y. (1999). Morphology of single olivocerebellar axons labeled with biotinylated dextran amine in the rat. J. Comp. Neurol. 414, 131–148. doi: 10.1002/(sici)1096-9861(19991115)414:2<131::aid-cne1>3.0.co;2-f
Szapiro, G., and Barbour, B. (2007). Multiple climbing fibers signal to molecular layer interneurons exclusively via glutamate spillover. Nat. Neurosci. 10, 735–742. doi: 10.1038/nn1907
Tabata, T., and Kano, M. (2006). GABA(B) receptor-mediated modulation of glutamate signaling in cerebellar Purkinje cells. Cerebellum 5, 127–133. doi: 10.1080/14734220600788911
Takagishi, Y., Hashimoto, K., Kayahara, T., Watanabe, M., Otsuka, H., Mizoguchi, A., et al. (2007). Diminished climbing fiber innervation of Purkinje cells in the cerebellum of myosin Va mutant mice and rats. Dev. Neurobiol. 67, 909–923. doi: 10.1002/dneu.20375
Takahashi, M., Ishikawa, K., Sato, N., Obayashi, M., Niimi, Y., Ishiguro, T., et al. (2012). Reduced brain-derived neurotrophic factor (BDNF) mRNA expression and presence of BDNF-immunoreactive granules in the spinocerebellar ataxia type 6 (SCA6) cerebellum. Neuropathology 32, 595–603. doi: 10.1111/j.1440-1789.2012.01302.x
Tempia, F., Hoxha, E., Negro, G., Alshammari, M. A., Alshammari, T. K., Panova-Elektronova, N., et al. (2015). Parallel fiber to Purkinje cell synaptic impairment in a mouse model of spinocerebellar ataxia type 27. Front. Cell Neurosci. 9:205. doi: 10.3389/fncel.2015.00205
Tsoi, H., Yu, A. C. S., Chen, Z. F. S., Ng, N. K. N., Chan, A. Y. Y., Yuen, L. Y. P., et al. (2014). A novel missense mutation in CCDC88C activates the JNK pathway and causes a dominant form of spinocerebellar ataxia. J. Med. Genet. 51, 590–595. doi: 10.1136/jmedgenet-2014-102333
Uesaka, N., Abe, M., Konno, K., Yamazaki, M., Sakoori, K., Watanabe, T., et al. (2018). Retrograde signaling from progranulin to Sort1 counteracts synapse elimination in the developing cerebellum. Neuron 97:e795. doi: 10.1016/j.neuron.2018.01.018
Uesaka, N., Uchigashima, M., Mikuni, T., Nakazawa, T., Nakao, H., Hirai, H., et al. (2014). Retrograde semaphorin signaling regulates synapse elimination in the developing mouse brain. Science 344, 1020–1023. doi: 10.1126/science.1252514
Valera, A. M., Binda, F., Pawlowski, S. A., Dupont, J. L., Casella, J. F., Rothstein, J. D., et al. (2016). Stereotyped spatial patterns of functional synaptic connectivity in the cerebellar cortex. eLife 5:e09862. doi: 10.7554/eLife.09862
van Swieten, J. C., Brusse, E., De Graaf, B. M., Krieger, E., Van De Graaf, R., De Koning, I., et al. (2003). A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebral ataxia. Am. J. Hum. Genet. 72, 191–199. doi: 10.1086/345488
Wang, Q., Bardgett, M. E., Wong, M., Wozniak, D. F., Lou, J. Y., Mcneil, B. D., et al. (2002). Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron 35, 25–38. doi: 10.1016/s0896-6273(02)00744-4
Watase, K., Barrett, C. F., Miyazaki, T., Ishiguro, T., Ishikawa, K., Hu, Y., et al. (2008). Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc. Natl. Acad. Sci. U.S.A. 105, 11987–11992. doi: 10.1073/pnas.0804350105
Watase, K., Hashimoto, K., Kano, M., Yamada, K., Watanabe, M., Inoue, Y., et al. (1998). Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur. J. Neurosci. 10, 976–988. doi: 10.1046/j.1460-9568.1998.00108.x
Waters, M. F., Minassian, N. A., Stevanin, G., Figueroa, K. P., Bannister, J. P., Nolte, D., et al. (2006). Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat. Genet. 38, 447–451. doi: 10.1038/ng1758
Watson, L. M., Bamber, E., Schnekenberg, R. P., Williams, J., Bettencourt, C., Lickiss, J., et al. (2017). Dominant mutations in GRM1 cause spinocerebellar ataxia type 44 (vol 101, pg 451, 2017). Am. J. Hum. Genet. 101, 866–866.
Wechsler-Reya, R. J., and Scott, M. P. (1999). Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog. Neuron 22, 103–114. doi: 10.1016/s0896-6273(00)80682-0
White, J. J., Arancillo, M., Stay, T. L., George-Jones, N. A., Levy, S. L., Heck, D. H., et al. (2014). Cerebellar zonal patterning relies on Purkinje cell neurotransmission. J. Neurosci. 34, 8231–8245. doi: 10.1523/JNEUROSCI.0122-14.2014
Wu, H. S., Sugihara, I., and Shinoda, Y. (1999). Projection patterns of single mossy fibers originating from the lateral reticular nucleus in the rat cerebellar cortex and nuclei. J. Comp. Neurol. 411, 97–118. doi: 10.1002/(sici)1096-9861(19990816)411:1<97::aid-cne8>3.0.co;2-o
Yan, H., Pablo, J. L., and Pitt, G. S. (2013). FGF14 regulates presynaptic Ca2+ channels and synaptic transmission. Cell Rep. 4, 66–75. doi: 10.1016/j.celrep.2013.06.012
Yan, H. D., Pablo, J. L., Wang, C. J., and Pitt, G. S. (2014). FGF14 modulates resurgent sodium current in mouse cerebellar Purkinje neurons. eLife 3:e04193. doi: 10.7554/eLife.04193
Zagha, E., Lang, E. J., and Rudy, B. (2008). Kv3.3 channels at the Purkinje cell soma are necessary for generation of the classical complex spike waveform. J. Neurosci. 28, 1291–1300. doi: 10.1523/JNEUROSCI.4358-07.2008
Zhuchenko, O., Bailey, J., Bonnen, P., Ashizawa, T., Stockton, D. W., Amos, C., et al. (1997). Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 15, 62–69. doi: 10.1038/ng0197-62
Keywords: cerebellum, spinocerebellar ataxia, Purkinje cell degeneration, cerebellar circuits, circuit maturation
Citation: Binda F, Pernaci C and Saxena S (2020) Cerebellar Development and Circuit Maturation: A Common Framework for Spinocerebellar Ataxias. Front. Neurosci. 14:293. doi: 10.3389/fnins.2020.00293
Received: 22 December 2019; Accepted: 13 March 2020;
Published: 02 April 2020.
Edited by:
Patrizia Longone, Santa Lucia Foundation (IRCCS), ItalyReviewed by:
Filippo Tempia, University of Turin, ItalyCopyright © 2020 Binda, Pernaci and Saxena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Smita Saxena, c21pdGEuc2F4ZW5hQGRibXIudW5pYmUuY2g=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.