
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Neurosci., 21 April 2020
Sec. Neurodegeneration
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.00256
 Maja Malmberg1,2
Maja Malmberg1,2 Tarja Malm3
Tarja Malm3 Oskar Gustafsson4
Oskar Gustafsson4 Andrea Sturchio5Caroline Graff6,7
Andrea Sturchio5Caroline Graff6,7 Alberto J. Espay5Anthony P. Wright4Samir El Andaloussi4,8
Alberto J. Espay5Anthony P. Wright4Samir El Andaloussi4,8 Anders Lindén9,10
Anders Lindén9,10 Kariem Ezzat4*
Kariem Ezzat4*Amyloids are fibrillar protein aggregates associated with diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), type II diabetes and Creutzfeldt–Jakob disease. The process of amyloid polymerization involves three pathological protein transformations; from natively folded conformation to the cross-β conformation, from biophysically soluble to insoluble, and from biologically functional to non-functional. While amyloids share a similar cross-β conformation, the biophysical transformation can either take place spontaneously via a homogeneous nucleation mechanism (HON) or catalytically on an exogenous surface via a heterogeneous nucleation mechanism (HEN). Here, we postulate that the different nucleation pathways can serve as a mechanistic basis for an etiological classification of amyloidopathies, where hereditary forms generally follow the HON pathway, while sporadic forms follow seed-induced (prions) or surface-induced (including microbially induced) HEN pathways. Critically, the conformational and biophysical amyloid transformation results in loss-of-function (LOF) of the original natively folded and soluble protein. This LOF can, at least initially, be the mechanism of amyloid toxicity even before amyloid accumulation reaches toxic levels. By highlighting the important role of non-protein species in amyloid formation and LOF mechanisms of toxicity, we propose a generalized mechanistic framework that could help better understand the diverse etiology of amyloid diseases and offer new opportunities for therapeutic interventions, including replacement therapies.
The term amyloid refers to a particular conformational state of proteins where they transform from being soluble and natively folded into insoluble aggregates of fibrillar nature. More than 35 peptides and proteins are known to form amyloids in different human diseases (Chiti and Dobson, 2017). Nearly all the proteins that form amyloids have biological functions in their normal, natively folded state. Some proteins such as antibodies, lipoproteins and serum amyloid (SAA) lead to systemic amyloidosis including light-chain amyloidosis, Apo-AI amyloidosis and AA amyloidosis, respectively (Chiti and Dobson, 2017). Other proteins accumulate in specific organs leading to localized amyloid pathology. These amyloidopathies include thyroid medullary carcinoma, pulmonary alveolar proteinosis and atrial amyloidosis resulting from the amyloid accumulation of calcitonin, surfactant protein C and atrial natriuretic factor, respectively (Chiti and Dobson, 2017). Localized amyloidopathies also include type II diabetes and neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). Type II diabetes is characterized by the amyloid accumulation of the peptide hormone islet amyloid polypeptide (IAPP), while AD and PD are characterized by the accumulation of the amyloid beta (Aβ) and alpha synuclein (α-syn) peptides, respectively (Eisenberg and Jucker, 2012). Moreover, AD and other neurodegenerative diseases such as frontal temporal dementia with Parkinsonism and Pick’s disease involve amyloid aggregates of the microtubule-associated protein tau (Gendron, 2009). In addition, the amyloid aggregation of the infamous tumor suppressor transcription factor p53 is involved in many cancers (Navalkar et al., 2019). While some amyloids were shown to have beneficial biological function, for example acting as storage for peptide hormones in secretory granules (Jackson and Hewitt, 2017), the vast majority of amyloids are pathological. This explains the existence of several biological protective mechanisms that ensure that proteins are correctly folded such as the presence of chaperones, or degraded when incorrectly folded via processes such as autophagy, ubiquitin–proteasome mediated degradation, and the unfolded protein response (Knowles et al., 2014). Moreover, specific sequence patterns that tend to easily form amyloids, such as alternating hydrophilic-hydrophobic stretches, appear to have been selected against during evolution (Broome and Hecht, 2000; Shoulders and Raines, 2009).
A small proportion of amyloidopathies is of genetic hereditary origin; however, the majority of amyloid diseases are sporadic (Chiti and Dobson, 2017). Hereditary forms of amyloidopathies are caused by mutations in the genes encoding the amyloidogenic proteins, either via gene duplication or mutations that facilitate protein aggregation leading to early onset of the disease (more details below). For sporadic forms, a small subset of amyloidopathies, termed transmissible spongiform encephalopathies (TSEs), are caused by infectious protein particles called prions (Cobb and Surewicz, 2009). Prions transfer from one organism to another inducing neurodegeneration in the recipient host in diseases such as Creutzfeldt–Jakob disease and Kuru. For the vast majority of other sporadic forms, the causes remain unclear. However, several environmental factors are known to increase disease risk, including infections (Itzhaki et al., 2016), lipid dysregulation (Mesa-Herrera et al., 2019), pollution (Kilian and Kitazawa, 2018), and traumatic brain injury (Johnson et al., 2010).
The term amyloid describes a unique class of protein conformation, where proteins adopt elongated fibrillar morphology. While this is a characteristic feature of pathological protein aggregates, it has been demonstrated that even normal non-pathogenic proteins can be forced to adopt the amyloid conformation under certain denaturing conditions (Fändrich et al., 2001). This led to the “generic hypothesis” suggesting that amyloid formation originates from the fundamental properties of proteins, based on the ability of backbone groups to form hydrogen bonds and the ability of side-chain groups to interact via hydrophobic and van der Waals interactions (Auer et al., 2007). To obtain their characteristic morphology, amyloids share a similar core cross-β conformation (Eisenberg and Sawaya, 2017). In this conformation, the protein molecules are arranged in the form of two oppositely stacked β-sheets, excluding the water molecules in-between and interdigitating their side chains forming a dry steric zipper (Eisenberg and Jucker, 2012). Such an elongated cross-β spine constitutes the basic amyloid fibrillar subunit, the protofilament (Figure 1). Apart from the fixed cross-β conformation, protein stacking in the spine can come in a variety of forms. For example, the cross-β architecture can consist of one folded molecule or two separate molecules and the β-sheets can stack in parallel, anti-parallel, face-to-face or face-to-back orientations (Eisenberg and Sawaya, 2017). Moreover, the protofilament length varies depending on the number strands forming each sheet. Once protofilaments are formed, they can associate in a variety of ways leading to different superstructural polymorphs. These polymorphs include flat fibrillar structures with varied number of horizontally stacked protofilaments, which can evolve to amyloid crystals, or different twisted ribbon structures (of single or multiple intertwined protofilaments), and these can further evolve into nanotubes (Mezzenga and Adamcik, 2018). Such superstructural polymorphism depends on many factors including the protein side chain arrangements, the nucleation mechanism (see below) and environmental conditions such as pH, temperature and ion concentration (Morel et al., 2010; Tycko and Wickner, 2013). In addition, this biochemical structural transformation is accompanied by a biophysical phase transformation that is dominated by nucleation-growth kinetics as described below.
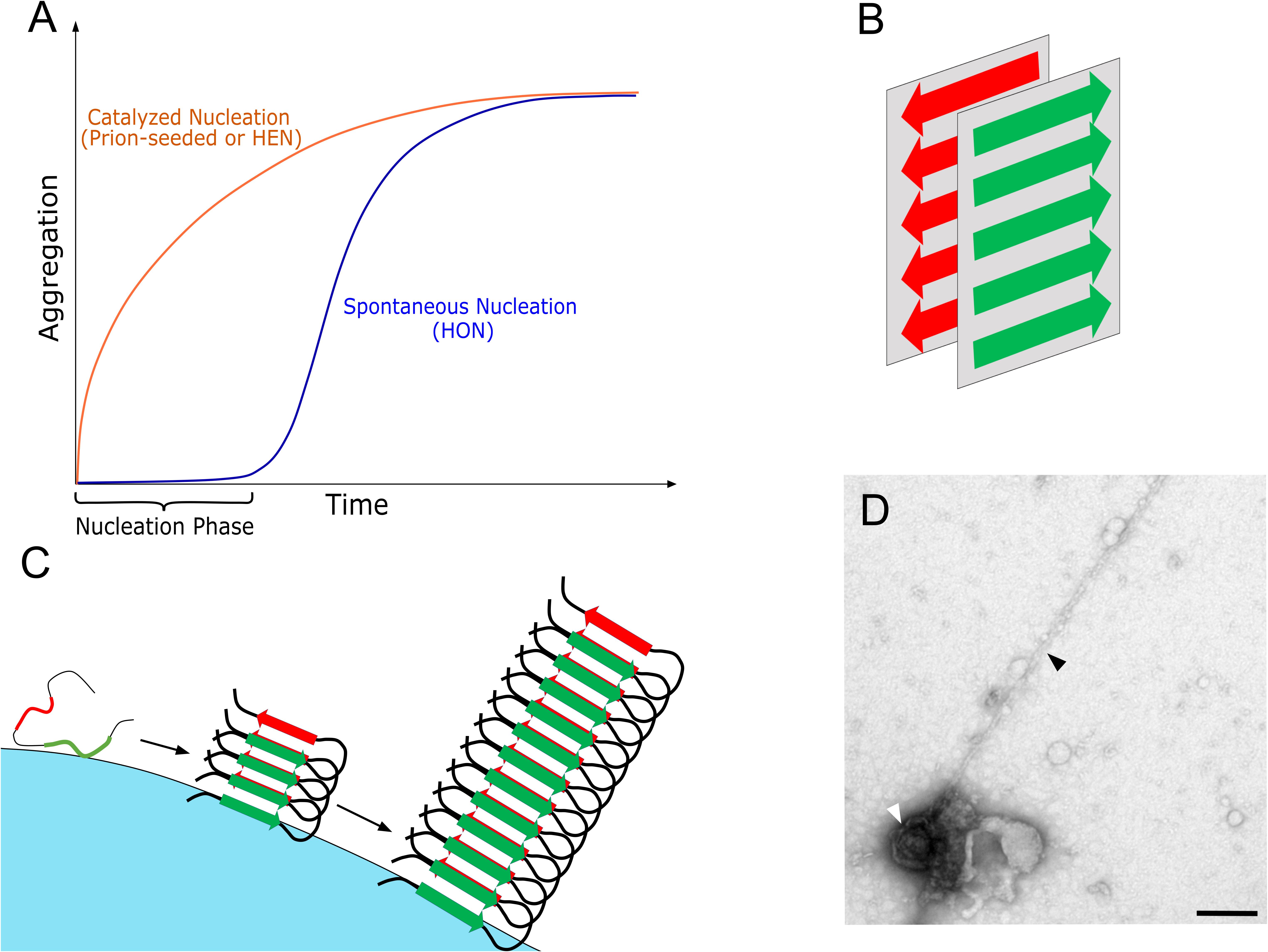
Figure 1. (A) A schematic representation of the kinetics of amyloid aggregation with the rate-limiting nucleation phase that can be bypassed either by adding a preformed seed (prion) or by surface catalysis via HEN. (B) A schematic representation of the cross-β conformation, which is the core conformation of amyloids where two β-sheets are stacked opposite to each other forming the protofilament with the characteristic elongated amyloid morphology. While the cross-β conformation remains constant, variable β-sheet orientations or protofilament associations lead to different amyloid polymorphs. (C) A schematic representation of HEN where an exogenous surface catalyze amyloid nucleation via binding, concentrating, and inducing conformational changes in the bound peptides/proteins, which facilitate amyloid transformation. (D) A negatively stained transmission electron microscopy picture demonstrating direct interaction between the surface of a virus (HSV-1) and a growing amyloid protofilament of Aβ 1–42 peptide (unpublished image from the viral catalyzed nucleation experiments performed in the study of Ezzat et al. (2019), where HSV-1 was incubated with 50 μM Aβ 1–42 for 100 min. at 37°C). In the same publication, we demonstrated that HSV-1 accelerated amyloid aggregation in vitro and in vivo. The viral particle is indicated by a white arrow and the protofilament with a black arrow, bar = 200 nm.
Phase transformation is the process involving transitioning from one state of matter to another, such as liquid to solid or gas to liquid or gas to solid transformations. These transformations are very common in nature, including phenomena such as crystallization and amyloid aggregation (liquid to solid transformation), rain precipitation (gas to liquid transformation) and planet formation (gas to solid transformation) (Karthika et al., 2016; Ros et al., 2019). That is why both the thermodynamics and kinetics of phase transformation have been widely studied. Thermodynamically, the process involves the transition from a less stable (higher free energy) to a more stable (lower free energy) phase under specified conditions (Mezzenga and Adamcik, 2018). Kinetically, the mechanism of phase transformation involves two steps that are well described by the classical nucleation theory (Karthika et al., 2016). Initially, a rate-limiting nucleation step takes place, where an energy barrier needs to be overcome to create the initial molecular assembly (nucleus) of the new phase. Once the nucleus is formed, this is followed by a growth step where the system rapidly transforms into the new phase. The nucleation-growth mechanism accurately describes the kinetics of amyloid formation as studied experimentally, with the distinctive sigmoidal kinetics curve involving a nucleation lag phase followed by a rapid growth or elongation phase (Knowles et al., 2014; Figure 1).
While nucleus formation is necessary, it is both thermodynamically unfavorable and rare, as it depends on the unlikely event of the spontaneous formation of a stable nucleus of the new phase within the bulk of the transforming phase (Vekilov, 2010; Eisenberg and Jucker, 2012). In the case of amyloid aggregation, this involves the spontaneous conformational change of the protein followed by spontaneous association of protein molecules into cross-β sheet rich nucleus. This pathway to nucleation is called homogenous nucleation (HON) (Buell, 2017), and with all the protective mechanisms that are in place to maintain proper protein folding (see above), it is not surprising that proteins do not normally reach the amyloid state. In contrast to the slow and rare HON pathway, catalyzed nucleation pathways exist that are faster and more common. One catalytic pathway is called seeding, where adding a preformed nucleus (seed/prion) enables the system to completely bypass the nucleation step and move directly to the growth or elongation step (Eisenberg and Jucker, 2012). The other important catalytic pathway is the heterogeneous nucleation mechanism (HEN), where an exogenous surface catalyzes the nucleation process (Buell, 2017; Srivastava et al., 2019). In HEN, the surface lowers the energy barrier to nucleation and acts as a scaffold that facilitates nucleus formation via binding, concentrating and enabling conformational changes in the bound proteins (Auer et al., 2009; John et al., 2018; Figure 1). Like seeding, HEN usually eradicates the lag phase completely from the kinetics. As an efficient nucleation mechanism, the polymerization of functional protein filaments such as actin or tubulin is dependent on HEN mechanisms, that are tightly controlled via sophisticated nucleator protein complexes such as γ-tubulin ring complex (Kollman et al., 2011) and the actin-related protein 2/3 complex (Campellone and Welch, 2010).
In the case of amyloids, many biological and non-biological surfaces have been shown to be capable of inducing amyloid aggregation via HEN. This includes microbial surfaces (Bantle et al., 2019; Ezzat et al., 2019), lipid vesicles (Habchi et al., 2018), and nanoparticles (Linse et al., 2007). Additionally, polymer surfaces such as glycosaminoglycans (GAGs) (Iannuzzi et al., 2015) and nucleic acids (Liu and Zhang, 2011) have been shown to accelerate amyloid aggregation. Moreover, the growing fibril surface itself can serve as a site for HEN, in a phenomenon termed secondary nucleation (Tö et al., 2018). Furthermore, the concentration of proteins in intracellular droplets that form via a liquid-liquid phase separation (LLPS) process can sometimes lead to amyloid formation (Brangwynne, 2017). Very recently, Yuan et al. (2019) have demonstrated that the interfaces created by LLPS can act as sites of HEN for amyloids (Yuan et al., 2019). While a variety of surfaces were shown to induce amyloid aggregation via HEN, the exact properties of a particular surface that mediate HEN of amyloids remain poorly understood. However, the main differences between HEN in normal protein polymerization and amyloid aggregation are the lack of controlled nucleation via nucleator complexes. Additionally, in normal processes, protein subunits assemble in their native rather than unfolded cross-β conformation.
Amyloid aggregation is a process of pathological protein transformation at three levels, a biochemical conformational transformation, a biophysical phase transformation and a biological functional transformation. At the biochemical structural level, amyloids share a similar cross-β conformation across different pathologies and different polymorphs. At the biophysical level however, there are distinct nucleation-dependent pathways to amyloid formation that are well-defined in thermodynamic and kinetic terms. The nucleation barrier is what separates the soluble and insoluble states of a protein; and thus, the pathways to nucleation are the decisive mechanisms in the biophysical transformation process. The nucleation barrier dictates whether a protein would spontaneously form an amyloid via HON or whether it requires a catalytic event, which can be a preformed seed/prion or an exogenous surface (HEN). In addition, together with other environmental factors such as pH and ion concentration, it affects the final polymorphic superstructure (Srivastava et al., 2019).
Here, we propose that different nucleation pathways could also serve as the mechanistic basis for an etiological classification of amyloid pathologies (Figure 2). In this framework, HON is facilitated by mutations that render the protein more prone for spontaneous self-assembly; and hence, is expected to be the dominant mechanism in hereditary amyloidopathies. This includes structural mutations that facilitate spontaneous nucleus formation (Chiti et al., 2002; Kim and Hecht, 2008) or gene duplication and/or triplication, where the increased concentration of the protein would lead to lowering of the nucleation barrier increasing the probability of spontaneous nucleus formation (Auer et al., 2007). Such mutations include amyloid precursor protein (APP) gene duplication in Down syndrome and synuclein gene (SNCA) duplication in familial PD, both leading to early AD and PD pathologies, respectively (Konno et al., 2016; Lott and Head, 2019).
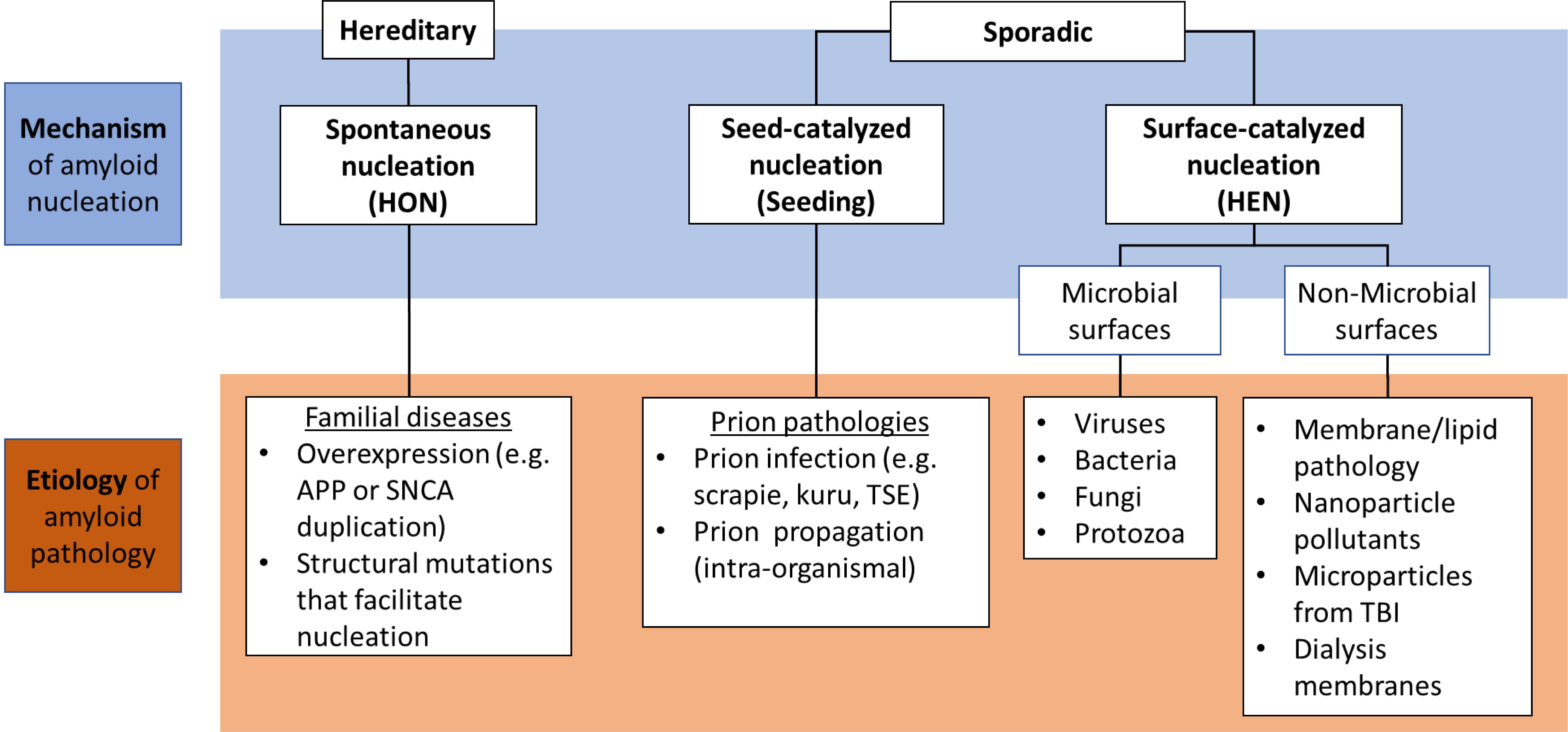
Figure 2. Classification of different amyloid etiologies in relation to the nucleation mechanisms where hereditary forms are usually caused by genetic mutations that facilitate HON, while sporadic forms are mainly catalyzed via prions or pathological nucleating surfaces (PNSs) which can be of microbial or non-microbial origin. APP, amyloid precursor protein; SNCA, α-synuclein gene; TSE, transmissible spongiform encephalopathy; TBI, traumatic brain injury.
However, in the normal state in absence of mutations, the protein retains its native structure protected from aggregation by the nucleation barrier together with the other biological mechanisms that prevent proteins from going down the amyloid pathway (such as the chaperone and proteasome machinery). In this case, pathogenic catalytic mechanisms are required for the pathological transformation. This can take place via seeding by a preformed amyloid fragment (seed/prion) whose source can be the same organism due to amyloid fragmentation leading to prion propagation (intra-organismal), or a different organism resulting from a prion infection (Jucker and Walker, 2013). Alternatively, nucleation can be induced by aberrant surfaces that catalyze HEN. Such pathological nucleating surfaces (PNSs) can either be of endogenous origin due to a membrane or lipid pathology, or from an exogenous source such as microbes or nanoparticulate pollutants. These HEN catalysts can be responsible for the non-hereditary sporadic disease forms.
BOX 1. In further support for the importance of LOF diseases mechanisms, in many disease models of amyloid diseases, knocking out/down the pathogenic protein leads to disease phenotypes in absence of plaques or aggregates.
• α-synuclein (Abeliovich et al., 2000; Chandra et al., 2005; Gorbatyuk et al., 2010; Greten-Harrison et al., 2010; Collier et al., 2016).
• Amyloid Precursor Protein (APP) (Senechal et al., 2007; Tyan et al., 2012; Wang et al., 2012; Liu et al., 2019; Martinsson et al., 2019; Southam et al., 2019; Truong et al., 2019).
• TDP-43 (Kabashi et al., 2009; Yang et al., 2014; Prpar Mihevc et al., 2016).
• Tau (Kimura et al., 2014; Lei et al., 2014; Biundo et al., 2018; Velazquez et al., 2018).
• Superoxide dismutase 1 (SOD-1) (Saccon et al., 2013)∗.
• PrP (Sakaguchi et al., 1996; Criado et al., 2005; Bremer et al., 2010; Küffer et al., 2016).
• IAPP (Gebre-Medhin et al., 1998; Mulder et al., 2000).
• P53 (Donehower et al., 1992; Ano Bom et al., 2012; Lasagna-Reeves et al., 2013; Ghosh et al., 2017).
∗A comprehensive review of the evidence of LOF toxicity of SOD-1 from animal models and clinical data.
While non-protein factors have usually been considered “co-factors” to a “protein-only” driven process based on prion seeding (Soto, 2011), we emphasize that surfaces are independent causal factors as they are able to induce amyloid aggregation in the absence protein seeds/prions via the distinct and separate pathway of HEN. Lipid vesicles (Habchi et al., 2018), nanoparticles (Linse et al., 2007) and viruses (Ezzat et al., 2019) have been shown to induce amyloid aggregation via HEN in absence of preformed seeds. Additionally, being thermodynamically more favorable, HEN is more likely to be the prevalent pathway of amyloid aggregation in-vivo in the absence of genetic mutations that can facilitate HON. In this regard, microbes such as viruses and bacteria, which are capable of invading and reproducing in tissues, can be potent mediators of HEN in sporadic amyloidopathies. We have recently shown that viruses such as respiratory syncytial virus (RSV) and herpes simplex virus type 1 (HSV-1) are able to induce amyloid formation by catalyzing HEN of IAPP and Aβ, respectively (Ezzat et al., 2019). In vivo, HSV-1 intracranial infection in an AD animal model resulted in amyloid accumulation within 48 h post-infection (Ezzat et al., 2019). Similar observations were demonstrated for other pathogens such as bacteria and fungi (Kumar et al., 2016; Dominy et al., 2019). This shows that microbes are potent HEN inducers of amyloid aggregation. On the other hand, PNSs may arise from endogenous sources. These can be the result of lipid dysregulation involving lipoproteins such as ApoE ε4, which is a known genetic risk factor for AD (Potter and Wisniewski, 2012), or membrane components such as cholesterol, gangliosides and GAGs (Iannuzzi et al., 2015; Penke et al., 2018). Furthermore, membrane fragment microparticles from brain injury (Zhao et al., 2017) can potentially act as catalytic surfaces for HEN mediated amyloid aggregation in traumatic brain injury. Moreover, as has been reported for the amyloid aggregation of insulin (Nayak et al., 2008), synthetic membranes can act as sites for HEN mediated aggregation of some plasma proteins such as β2 microglobulin in dialysis-related amyloidosis (Scarpioni et al., 2016).
It can also be postulated that in some cases HON and HEN mechanisms can overlap, where mutations that would facilitate spontaneous amyloid aggregation via HON can also render the protein more vulnerable for surface-catalyzed amyloid transformation via HEN. Furthermore, HEN mechanisms could lead to distinctive amyloid superstructural polymorphs based on the properties of the catalyzing surface. Virus-induced amyloid aggregation, for example, can be expected to result in particularly deformed polymorphs due to HEN occurring on an acutely curved nanosurface. Crystalline deformation has been demonstrated before when crystallization takes place on a curved surface (Meng et al., 2014; Gómez et al., 2015). In the case of amyloids, horizontal stacking of protofilaments will be limited by the surface curvature. This, together with the possible existence of multiple nucleation sites on the same viral particle would lead to distinct polymorphic features that can act as histopathological hallmarks for viral-induced amyloidopathies, and can help trace back the etiology. Moreover, the conformational and phase transformations would result in pathogenic functional transformations that are described in the section “Gain or Loss of Function?”
From a functional point-of-view, it has been difficult to correlate the pathogenicity of amyloids with particular structural features (Eisenberg and Jucker, 2012; Collinge, 2016). Here we postulate that while the gain-of-function (GOF) toxicity becomes more likely with increased amyloid accumulation in a tissue (especially in systemic forms of amyloidosis), a loss-of-function (LOF) toxicity likely constitutes the initial cytotoxic mechanism. Nearly all amyloid-forming proteins have known functions in their native folded state. Since any protein needs to adopt an appropriate conformation in order to perform its function, protein unfolding into the cross-β conformation accompanied by phase transformation into solid fibrils generally abolishes the native function of the protein. Proteins such as lipoproteins, antibodies and IAPP (also called amylin) are not able to perform their homeostatic, immunological, or hormonal functions in their pathological amyloid forms in Apo-AI amyloidosis, light-chain amyloidosis and diabetes, respectively (Hieronymus and Griffin, 2015; Muchtar et al., 2016; Lu et al., 2017). The same applies to Aβ, prion protein (PrP) and α-syn, which are the most studied in the context of neurodegenerative disorders. Soluble Aβ was shown to be important for synaptic plasticity and memory (Parihar and Brewer, 2010), soluble PrP on the other hand is involved in myelin maintenance and cellular proliferation processes (Castle and Gill, 2017), while α-syn is important for the regulation of neurotransmission and response to cellular stress (Benskey et al., 2016). Thus, it is expected that these original functions will be lost even before amyloid accumulation reaches substantial toxic GOF levels, and that LOF can at least initially be the neurodegenerative mechanism, as has been suggested previously (Saccon et al., 2013; Vanden Broeck et al., 2014; Benskey et al., 2016; Kepp, 2016).
The importance of LOF is further supported by the fact that in amyloid disease models, knocking out/down the protein results in disease phenotypes in the absence of the amyloidogenic protein and its aggregated forms (Box 1). Moreover, in AD for example, it has been repeatedly demonstrated that there is not always a correlation between the plaque load and disease severity. This has been shown in animal models (Hsia et al., 1999) and in healthy subjects with significant plaque load but without significant cognitive impairment (Aizenstein et al., 2008). One way to explain this paradox within a GOF framework has been to postulate that toxicity comes from a not-very-well-defined species called the amyloid oligomers, and not from the plaques (Selkoe, 2002). However, an additional explanation can be the fact that proteins that lose their native conformation will instantly lose their function even if they do not become particularly more toxic, and that such LOF contributes to neuronal degeneration. This is further illustrated by neuronal phenotypes in knockout animal models of several different amyloid pathologies (Box 1). Moreover, in many amyloidopathies, the CSF levels of the soluble peptides/proteins that eventually form amyloid aggregates decrease rather than increase (Kang, 2016; Hu et al., 2017). In AD and PD, the decrease in CSF levels of soluble Aβ (Fagan et al., 2009; Bateman et al., 2012; Buchhave et al., 2012) and α-syn (Tokuda et al., 2006; Wang et al., 2015; Mollenhauer et al., 2019; Parnetti et al., 2019) is among the early biochemical markers of the disease. A similar downward trend is observed for the CSF levels of soluble PrP in CJD (Meyne et al., 2009; Dorey et al., 2015). This indicates that the soluble to insoluble transformation, which would be accompanied by decreased levels and consequential LOF of the soluble fraction, might be among the earliest pathological changes. Notably, lower levels of soluble Aβ and α-syn are present even in patients with APP gene duplication in Down syndrome (Tapiola et al., 2001; Portelius et al., 2014) and SNCA gene duplication in familial PD (Kasuga et al., 2010). This suggests that pathological overexpression, which would facilitate HON by lowering the nucleation barrier, can also lead to decreased levels and LOF of the soluble proteins, which will be sequestered in the amyloid form. Taken together with data from knockout and knock down studies and the poor correlation between the amyloid plaque load and disease severity, these different lines of evidence strongly suggest an critical role of LOF mechanisms in the pathophysiology of amyloid diseases.
Furthermore, it has been shown that amyloid fragments (seeds/prions) can propagate pathology from one region to another within the same organism in diseases such as AD and PD (Brundin et al., 2010; Steiner et al., 2011). In this case, seeds/prions will induce LOF phase transformation when they encounter a new protein pool. The same can be true for HEN induced by microbes, where the ability of a pathogen to infect a particular area would lead to LOF amyloid transformation in that area. This may explain why in some neurodegenerative diseases the spread of the pathology follows the anatomical connections, which are the same routes for both prion and microbial propagation (Hawkes et al., 2007; Brettschneider et al., 2015). The LOF framework might also explain the failure of therapeutic approaches aiming only to reduce the amyloid forming proteins and open new directions in treatment that include restoring protein homeostasis via replacement therapy with functional, non-aggregating forms of the protein (Mockett et al., 2017). Indeed, synthetic IAPP (amylin) analogs such as pramlintide are clinically used as replacement therapy in diabetes (Hieronymus and Griffin, 2015). Furthermore, overexpression of soluble amyloid precursor protein alpha (APPsα) has been shown to restore synaptic plasticity, and rescue spatial memory in an AD mouse model with preexisting pathology and amyloidosis (Fol et al., 2016). This demonstrates that replacement therapy within a LOF framework is a promising approach; one that can be extended to other amyloidopathies.
The amyloid aggregation phenomenon, especially in the context of prions, is sometimes referred to as a process of protein “self-replication” that is dominated by a “protein-only” species leading to different prion “strains” that possess different pathogenic potentials (Collinge, 2016). Here, we argue that the phenomenon of amyloid aggregation is better described in physical terms rather than biological terms that imply preservation and transfer of biological information via replication and strain diversity. Amyloid aggregation is a process of nucleation-dependent phase transformation that is very common in nature similar to crystal growth or snow formation in non-biological systems. Moreover, other normal biological processes such as biomineralization of hard tissues (Veis and Dorvee, 2013) and the assembly of actin or tubulin (Job et al., 2003; Firat-Karalar and Welch, 2011) are also dominated by nucleation-dependent mechanisms. While many of these non-organic and organic phenomena share similar features with amyloids such as self-assembly, repeated patterns and superstructural polymorphism, in none of these cases is the process referred to as “self-replication” in the biological sense of the word. Moreover, polymorphic heterogeneity is dependent on the nucleation mechanism and environmental factors (such as pH, concentration, and temperature); factors that are not encoded in the core molecular conformation; and hence, cannot be faithfully replicated. Importantly, nucleation reactions take place via HEN, where no information is transferred from the catalyzing surface to the growing fiber, while still affecting superstructural polymorphism. This lack of information preservation or transfer indicates that the amyloid/prion phenomenon cannot be compared to the nucleic acids in terms of biological replication; which in the case of nucleic acids, is dominated by well-controlled mechanisms and machinery that ensure preservation and faithful replication of the genetic information (Table 1). In that sense, amyloid/prion heterogeneity cannot also be compared to biological strains in terms of the fidelity of storage and transfer of biological information, and thus, we opt for the term polymorphs instead, which is accordance with nomenclature for similar phenomena, such as crystalline polymorphs for example.
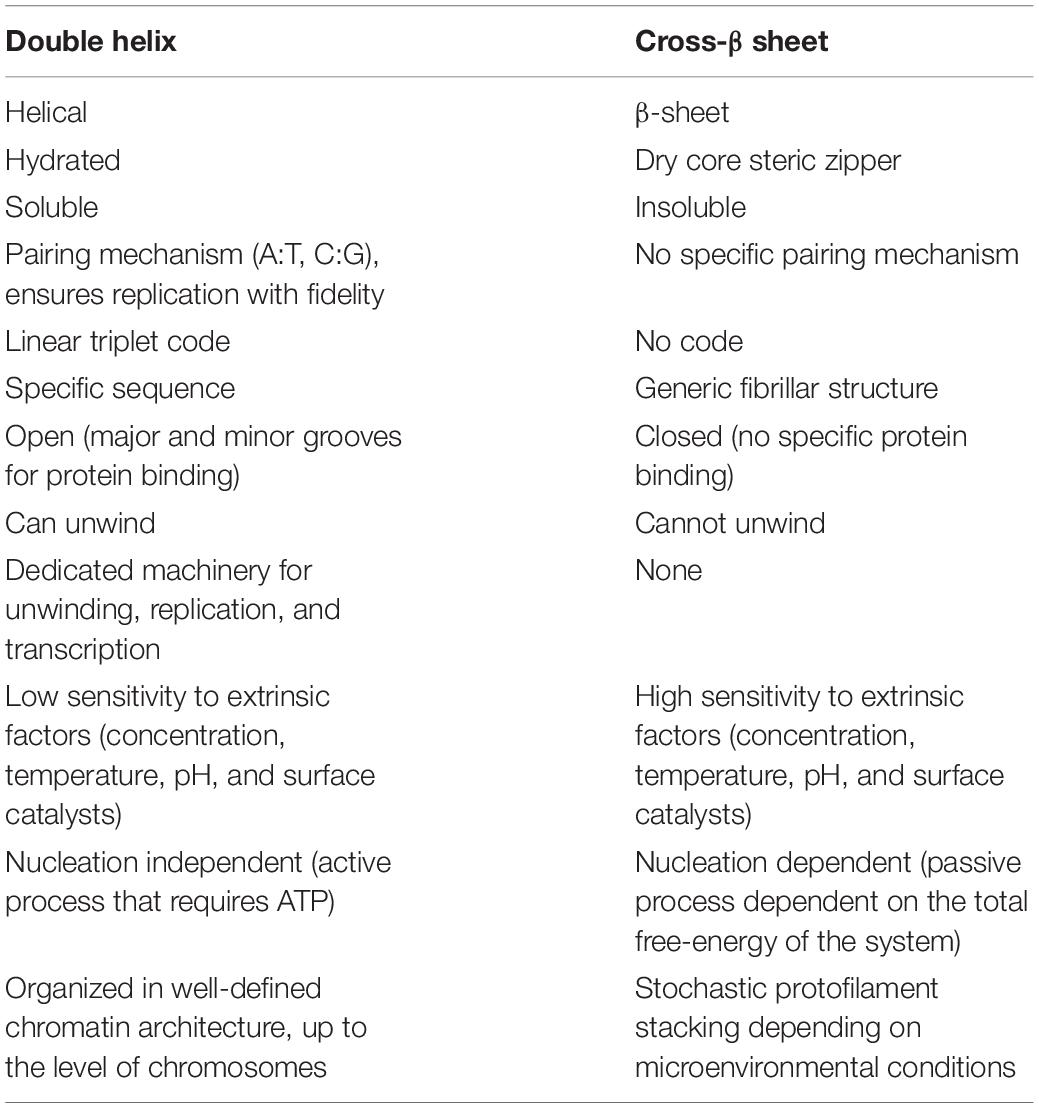
Table 1. The differences between the double helical structural architecture of DNA and the cross-β sheet architecture of amyloids in terms of the capability of both architectures to hold and transmit biological information.
We are aware that the “protein-only” hypothesis of protein “self-replication” was initially introduced to distinguish amyloids from viral infections based on the absence of nucleic acids within amyloids (Zabel and Reid, 2015). Despite the historical importance of such distinction, it does not imply that the amyloid phenomenon should always be understood within the bounds of this historical dichotomy. Structural and biophysical studies of amyloids in recent years have uncovered important details about the common structural features of amyloids and the different physical pathways of their aggregation, as discussed above. Many of these new advances do not fit easily within the “protein-only” paradigm. This is particularly apparent in relation to HEN phenomena, which can be mediated by viral and other microbial surfaces; the very species the “protein-only” hypothesis was supposed to exclude from the pathology. HEN also clearly demonstrates the lack of information transfer during the amyloid aggregation process. That is why a new synthesis of the available data was necessary to accommodate for these findings. In that sense, we think that the nucleation-based classification of amyloid pathologies that we describe here does offer a more accurate and inclusive way to describe the multifactorial nature of amyloid aggregation using a well-defined physical framework.
One advantage of this physical classification is that it provides a mechanistic explanation of phenomena that are currently unaccounted for within the “protein-only” paradigm, including the sporadic amyloidopathies. It allows the integration of risk factors (such as lipid pathology, infections, and pollution) into the core of the pathogenesis via a well-defined mechanism; HEN. Furthermore, by highlighting the common physical foundations of the amyloid aggregation process, it becomes much easier to find correlations and common mechanisms between different amyloid pathologies that have been studied separately in isolated disease contexts. This creates a logical framework where data from different diseases can be integrated into a more general understanding. One outcome of such general understanding is that HEN and LOF mechanisms assume a more clear and prominent role in disease etiology and pathophysiology, opening new opportunities for novel diagnostic and therapeutic modalities. This is particularly important at a time where the failure of previous therapeutic interventions calls for new ways to understand amyloid pathologies.
In relation to novel diagnostics, HEN pathways are expected to contribute to amyloid polymorphism (see above), which can help in the differential histopathological diagnosis by relating particular amyloid polymorphs to certain HEN interactions. This may enable the development of new therapeutic interventions to specifically target these interactions, or preventive measures such as vaccines targeting specific microbes involved in HEN-mediated amyloid induction. In addition, highlighting the LOF angle of the pathology can lead to new treatments that aim to restore the original protein functions via different replacement therapy approaches.
While a number of proteins polymerize into filaments for functional purposes (actin and collagen for example), most proteins perform their functions in a soluble state. However, under certain circumstances, soluble proteins are pathologically nucleated to form fibrillar amyloid polymers. This uncontrolled nucleation leads to a conformational transformation from native conformation into the cross-β conformation and a phase transformation into solid fibrils. Such biochemical and biophysical transformations would lead to loss of biological function even if the resulting aggregates are not particularly more toxic. Within this framework, different etiologies of amyloid diseases can be linked to different mechanisms of nucleation. We point out that familial mutations facilitate spontaneous nucleation, leading to HON dominated mechanisms. The sporadic forms on the other hand may rely more on catalytic nucleation mechanisms, either via prion seeding or HEN. HEN mechanisms, in turn, can be mediated by a plethora of PNSs, among which microbial membranes such as viruses and bacteria may be of critical importance due to their ability to invade and replicate in various tissues. Furthermore, we propose that amyloids are more precisely described in physical terms similar to other organic and non-organic phase transformations, rather than in biological terms that invoke self-replication and biological strains. Such a generalized framework for a mechanistic-based understanding can open new avenues for the exploration of new measures to diagnose, prevent, and treat amyloidopathies.
KE conceived the concept and wrote the manuscript. MM, TM, OG, AS, CG, AE, AW, SE, and AL read and critically contributed to the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abeliovich, A., Schmitz, Y., Fariñ, I., Choi-Lundberg, D., Ho, W.-H., Castillo, P. E., et al. (2000). Mice Lacking -Synuclein Display Functional Deficits in the Nigrostriatal Dopamine System. Neuron 25, 239–252.
Aizenstein, H. J., Nebes, R. D., Saxton, J. A., Price, J. C., Mathis, C. A., Tsopelas, N. D., et al. (2008). Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol. 65, 1509–1517. doi: 10.1001/archneur.65.11.1509
Ano Bom, A. P. D., Rangel, L. P., Costa, D. C. F., De Oliveira, G. A. P., Sanches, D., Braga, C. A., et al. (2012). Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. J. Biol. Chem. 287, 28152–28162. doi: 10.1074/jbc.M112.340638
Auer, S., Dobson, C. M., and Vendruscolo, M. (2007). Characterization of the nucleation barriers for protein aggregation and amyloid formation. HFSP J. 1, 137–146. doi: 10.2976/1.2760023
Auer, S., Trovato, A., and Vendruscolo, M. (2009). A Condensation-Ordering Mechanism in Nanoparticle- Catalyzed Peptide Aggregation. PLoS Comput. Biol 5:e1000458. doi: 10.1371/journal.pcbi.1000458
Bantle, C. M., Phillips, A. T., Smeyne, R. J., Rocha, S. M., Olson, K. E., and Tjalkens, R. B. (2019). Infection with mosquito-borne alphavirus induces selective loss of dopaminergic neurons, neuroin fl ammation and widespread protein aggregation. NPJ Parkinsons Dis 5, 20. doi: 10.1038/s41531-019-0090-8
Bateman, R. J., Xiong, C., Benzinger, T. L. S., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Benskey, M. J., Perez, R. G., and Manfredsson, F. P. (2016). The contribution of alpha synuclein to neuronal survival and function - Implications for Parkinson’s disease. J. Neurochem. 137, 331–359. doi: 10.1111/jnc.13570
Biundo, F., Del Prete, D., Zhang, H., Arancio, O., and D’Adamio, L. (2018). A role for tau in learning, memory and synaptic plasticity. Sci. Rep. 8, 1–13. doi: 10.1038/s41598-018-21596-3
Brangwynne, C. P. (2017). Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382. doi: 10.1126/science.aaf4382
Bremer, J., Baumann, F., Tiberi, C., Wessig, C., Fischer, H., Schwarz, P., et al. (2010). Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 13, 310–318. doi: 10.1038/nn.2483
Brettschneider, J., Del Tredici, K., Lee, V. M.-Y., and Trojanowski, J. Q. (2015). Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 16, 109–120. doi: 10.1038/nrn3887
Broome, B. M., and Hecht, M. H. (2000). Nature disfavors sequences of alternating polar and non-polar amino acids: Implications for amyloidogenesis. J. Mol. Biol. 296, 961–968. doi: 10.1006/jmbi.2000.3514
Brundin, P., Melki, R., and Kopito, R. (2010). Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11, 301–307. doi: 10.1038/nrm2873
Buchhave, P., Minthon, L., Zetterberg, H., Wallin, ÅK., Blennow, K., and Hansson, O. (2012). Cerebrospinal fluid levels of β-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 69, 98–106. doi: 10.1001/archgenpsychiatry.2011.155
Buell, A. K. (2017). The Nucleation of Protein Aggregates - From Crystals to Amyloid Fibrils, 1st Edn. Amsterdam: Elsevier Inc., doi: 10.1016/bs.ircmb.2016.08.014
Campellone, K. G., and Welch, M. D. (2010). A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 11, 237–251. doi: 10.1038/nrm2867
Castle, A. R., and Gill, A. C. (2017). Physiological functions of the cellular prion protein. Front. Mol. Biosci. 4:19. doi: 10.3389/fmolb.2017.00019
Chandra, S., Gallardo, G., Fernández-Chacón, R., Schlüter, O. M., and Südhof, T. C. (2005). α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell 123, 383–396. doi: 10.1016/j.cell.2005.09.028
Chiti, F., Calamai, M., Taddei, N., Stefani, M., Ramponi, G., and Dobson, C. M. (2002). Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc. Natl. Acad. Sci. U. S. A. 99, 16419–16426. doi: 10.1073/pnas.212527999
Chiti, F., and Dobson, C. M. (2017). Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 86, 27–68. doi: 10.1146/annurev-biochem-061516-045115
Cobb, N. J., and Surewicz, W. K. (2009). Prion diseases and their biochemical mechanisms. Biochemistry 48, 2574–2585. doi: 10.1021/bi900108v
Collier, T. J., Eugene Redmond, D., Steece-Collier, K., Lipton, J. W., and Manfredsson, F. P. (2016). Is alpha-synuclein loss-of-function a contributor to parkinsonian pathology? Evidence from non-human primates. Front. Neurosci. 10:12. doi: 10.3389/fnins.2016.00012
Collinge, J. (2016). Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 539, 217–226. doi: 10.1038/nature20415
Criado, J. R., Sánchez-Alavez, M., Conti, B., Giacchino, J. L., Wills, D. N., Henriksen, S. J., et al. (2005). Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiol. Dis. 19, 255–265. doi: 10.1016/j.nbd.2005.01.001
Dominy, S. S., Lynch, C., Ermini, F., Benedyk, M., Marczyk, A., Konradi, A., et al. (2019). Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 5, eaau3333. doi: 10.1126/sciadv.aau3333
Donehower, L. A., Harvey, M., Slagle, B. L., McArthur, M. J., Montgomery, C. A., Butel, J. S., et al. (1992). Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221. doi: 10.1038/356215a0
Dorey, A., Tholance, Y., Vighetto, A., Perret-Liaudet, A., Lachman, I., Krolak-Salmon, P., et al. (2015). Association of cerebrospinal fluid prion protein levels and the distinction between Alzheimer disease and Creutzfeldt-Jakob disease. JAMA Neurol. 72, 267–275. doi: 10.1001/jamaneurol.2014.4068
Eisenberg, D., and Jucker, M. (2012). The amyloid state of proteins in human diseases. Cell 148, 1188–1203. doi: 10.1016/j.cell.2012.02.022
Eisenberg, D. S., and Sawaya, M. R. (2017). Structural Studies of Amyloid Proteins at the Molecular Level. Annu. Rev. Biochem 86, 69–95. doi: 10.1146/annurev-biochem-061516-045104
Ezzat, K., Pernemalm, M., Pålsson, S., Roberts, T. C., Järver, P., Dondalska, A., et al. (2019). The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat. Commun. 10, 2331. doi: 10.1038/s41467-019-10192-2
Fagan, A. M., Head, D., Shah, A. R., Marcus, D., Mintun, M., Morris, J. C., et al. (2009). Decreased cerebrospinal fluid Aβ 42 correlates with brain atrophy in cognitively normal elderly. Ann. Neurol. 65, 176–183. doi: 10.1002/ana.21559
Fändrich, M., Fletcher, M. A., and Dobson, C. M. (2001). Amyloid fibrils from muscle myoglobin. Nature 410, 165–166. doi: 10.1038/35065514
Firat-Karalar, E. N., and Welch, M. D. (2011). New mechanisms and functions of actin nucleation. Curr. Opin. Cell Biol. 23, 4–13. doi: 10.1016/j.ceb.2010.10.007
Fol, R., Braudeau, J., Ludewig, S., Abel, T., Weyer, S. W., Roederer, J. P., et al. (2016). Viral gene transfer of APPsα rescues synaptic failure in an Alzheimer’s disease mouse model. Acta Neuropathol. 131, 247–266. doi: 10.1007/s00401-015-1498-9
Gebre-Medhin, S., Mulder, H., Pekny, M., Westermark, G., Törnell, J., Westermark, P., et al. (1998). Increased insulin secretion and glucose tolerance in mice lacking islet amyloid polypeptide (amylin). Biochem. Biophys. Res. Commun 250, 271–277. doi: 10.1006/bbrc.1998.9308
Gendron, T. F. (2009). The role of tau in neurodegeneration. Mol. Neurodegener. 4, 1–19. doi: 10.1186/1750-1326-4-13
Ghosh, S., Salot, S., Sengupta, S., Navalkar, A., Ghosh, D., Jacob, R., et al. (2017). P53 amyloid formation leading to its loss of function: Implications in cancer pathogenesis. Cell Death Differ. 24, 1784–1798. doi: 10.1038/cdd.2017.105
Gómez, L. R., García, N. A., Vitelli, V., Lorenzana, J., and Vega, D. A. (2015). Phase nucleation in curved space. Nat. Commun. 6, 6856. doi: 10.1038/ncomms7856
Gorbatyuk, O. S., Li, S., Nash, K., Gorbatyuk, M., Lewin, A. S., Sullivan, L. F., et al. (2010). In vivo RNAi-mediated α-synuclein silencing induces nigrostriatal degeneration. Mol. Ther. 18, 1450–1457. doi: 10.1038/mt.2010.115
Greten-Harrison, B., Polydoro, M., Morimoto-Tomita, M., Diao, L., Williams, A. M., Nie, E. H., et al. (2010). αβγ-Synuclein triple knockout mice revealage-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. U. S. A. 107, 19573–19578. doi: 10.1073/pnas.1005005107
Habchi, J., Chia, S., Galvagnion, C., Michaels, T. C. T., Bellaiche, M. M. J., Ruggeri, F. S., et al. (2018). Cholesterol catalyses Aβ42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes. Nat. Chem 10, 673–683. doi: 10.1038/s41557-018-0031-x
Hawkes, C. H., Del Tredici, K., and Braak, H. (2007). Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 33, 599–614. doi: 10.1111/j.1365-2990.2007.00874.x
Hieronymus, L., and Griffin, S. (2015). Role of Amylin in Type 1 and Type 2 Diabetes. Diabetes Educ. 41, 47S–56S. doi: 10.1177/0145721715607642
Hsia, A. Y., Masliah, E., Mcconlogue, L., Yu, G. Q., Tatsuno, G., Hu, K., et al. (1999). Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. U. S. A. 96, 3228–3233. doi: 10.1073/pnas.96.6.3228
Hu, X., Yang, Y., and Gong, D. (2017). Changes of cerebrospinal fluid Aβ42, t-tau, and p-tau in Parkinson’s disease patients with cognitive impairment relative to those with normal cognition: a meta-analysis. Neurol. Sci. 38, 1953–1961. doi: 10.1007/s10072-017-3088-1
Iannuzzi, C., Irace, G., and Sirangelo, I. (2015). The effect of glycosaminoglycans (GAGs) on amyloid aggregation and toxicity. Molecules 20, 2510–2528. doi: 10.3390/molecules20022510
Itzhaki, R. F., Lathe, R., Balin, B. J., Ball, M. J., Bearer, E. L., Braak, H., et al. (2016). Microbes and Alzheimer’s disease. J. Alzheimer’s Dis. 51, 979–984. doi: 10.3233/JAD-160152
Jackson, M. P., and Hewitt, E. W. (2017). Why are functional amyloids non−toxic in humans? Biomolecules 7, 1–13. doi: 10.3390/biom7040071
Job, D., Valiron, O., and Oakley, B. (2003). Microtubule nucleation. Curr. Opin. Cell Biol. 15, 111–117. doi: 10.1016/S0955-0674(02)00003-0
John, T., Gladytz, A., Kubeil, C., Martin, L. L., Risselada, H. J., and Abel, B. (2018). Impact of nanoparticles on amyloid peptide and protein aggregation: A review with a focus on gold nanoparticles. Nanoscale 10, 20894–20913. doi: 10.1039/c8nr04506b
Johnson, V. E., Stewart, W., and Smith, D. H. (2010). Traumatic brain injury and amyloid-β pathology: A link to alzheimer’s disease? Nat. Rev. Neurosci 11, 361–370.
Jucker, M., and Walker, L. C. (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. doi: 10.1038/nature12481
Kabashi, E., Lin, L., Tradewell, M. L., Dion, P. A., Bercier, V., Bourgouin, P., et al. (2009). Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 19, 671–683. doi: 10.1093/hmg/ddp534
Kang, J.-H. (2016). Cerebrospinal Fluid Amyloid β1-42, Tau, and Alpha-Synuclein Predict the Heterogeneous Progression of Cognitive Dysfunction in Parkinson’s Disease. J. Mov. Disord. 9, 89–96. doi: 10.14802/jmd.16017
Karthika, S., Radhakrishnan, T. K., and Kalaichelvi, P. (2016). A Review of Classical and Nonclassical Nucleation Theories. Cryst. Growth Des. 16, 6663–6681. doi: 10.1021/acs.cgd.6b00794
Kasuga, K., Tokutake, T., Ishikawa, A., Uchiyama, T., Tokuda, T., Onodera, O., et al. (2010). Differential levels of α-synuclein, β-amyloid42 and tau in CSF between patients with dementia with Lewy bodies and Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 81, 608–610. doi: 10.1136/jnnp.2009.197483
Kepp, K. P. (2016). Alzheimer’s disease due to loss of function: A new synthesis of the available data. Prog. Neurobiol. 143, 36–60. doi: 10.1016/j.pneurobio.2016.06.004
Kilian, J., and Kitazawa, M. (2018). The emerging risk of exposure to air pollution on cognitive decline and Alzheimer’s disease – Evidence from epidemiological and animal studies. Biomed. J. 41, 141–162. doi: 10.1016/j.bj.2018.06.001
Kim, W., and Hecht, M. H. (2008). Mutations Enhance the Aggregation Propensity of the Alzheimer’s Aβ Peptide. J. Mol. Biol. 377, 565–574. doi: 10.1016/j.jmb.2007.12.079
Kimura, T., Whitcomb, D. J., Jo, J., Regan, P., Piers, T., Heo, S., et al. (2014). Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. B Biol. Sci 369, 20130144. doi: 10.1098/rstb.2013.0144
Knowles, T. P. J., Vendruscolo, M., and Dobson, C. M. (2014). The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396. doi: 10.1038/nrm3810
Kollman, J. M., Merdes, A., Mourey, L., and Agard, D. A. (2011). Microtubule nucleation by γ-tubulin complexes. Nat. Rev. Mol. Cell Biol. 12, 709–721. doi: 10.1038/nrm3209
Konno, T., Ross, O. A., Puschmann, A., Dickson, D. W., and Wszolek, Z. K. (2016). Autosomal dominant Parkinson’s disease caused by SNCA duplications. Park. Relat. Disord. 22, S1–S6. doi: 10.1016/j.parkreldis.2015.09.007
Küffer, A., Lakkaraju, A. K. K., Mogha, A., Petersen, S. C., Airich, K., Doucerain, C., et al. (2016). The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 536, 464–468. doi: 10.1038/nature19312
Kumar, D. K. V., Choi, S. H., Washicosky, K. J., Eimer, W. A., Tucker, S., Ghofrani, J., et al. (2016). Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimers disease. Sci. Transl. Med. 8, 34072–34072. doi: 10.1126/scitranslmed.aaf1059
Lasagna-Reeves, C. A., Clos, A. L., Castillo-Carranza, D., Sengupta, U., Guerrero-Muñoz, M., Kelly, B., et al. (2013). Dual role of p53 amyloid formation in cancer; loss of function and gain of toxicity. Biochem. Biophys. Res. Commun. 430, 963–968. doi: 10.1016/j.bbrc.2012.11.130
Lei, P., Ayton, S., Moon, S., Zhang, Q., Volitakis, I., Finkelstein, D. I., et al. (2014). Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol. Neurodegener. 9, 1–12. doi: 10.1186/1750-1326-9-29
Linse, S., Cabaleiro-Lago, C., Xue, W.-F., Lynch, I., Lindman, S., Thulin, E., et al. (2007). Nucleation of protein fibrillation by nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 104, 8691–8696. doi: 10.1073/pnas.0701250104
Liu, A., Zhang, Y., Han, L., He, G., Xie, W., Zhou, Z., et al. (2019). Regulation of Neurotransmitter Release by Amyloid Precursor Protein Through Synapsin Phosphorylation. Neurochem. Res. 44, 683–691. doi: 10.1007/s11064-017-2418-2
Liu, C., and Zhang, Y. (2011). Nucleic acid-mediated protein aggregation and assembly, 1st Edn. Amsterdam: Elsevier Inc., doi: 10.1016/B978-0-12-386483-3.00005-7
Lott, I. T., and Head, E. (2019). Dementia in Down syndrome: unique insights for Alzheimer disease research. Nat. Rev. Neurol. 15, 135–147. doi: 10.1038/s41582-018-0132-6
Lu, C., Zuo, K., Lu, Y., Liang, S., Huang, X., Zeng, C., et al. (2017). Apolipoprotein A-1-related amyloidosis 2 case reports and review of the literature. Medicine (Baltimore) 96, e8148. doi: 10.1097/MD.0000000000008148
Martinsson, I., Capetillo-Zarate, E., Faideau, M., Willén, K., Esteras, N., Frykman, S., et al. (2019). APP depletion alters selective pre- and post-synaptic proteins. Mol. Cell. Neurosci. 95, 86–95. doi: 10.1016/j.mcn.2019.02.003
Meng, G., Paulose, J., Nelson, D. R., and Manoharan, V. N. (2014). Elastic instability of a crystal growing on a curved surface. Science 343, 634–637. doi: 10.1126/science.1244827
Mesa-Herrera, F., Taoro-González, L., Valdés-Baizabal, C., Diaz, M., and Marín, R. (2019). Lipid and Lipid Raft Alteration in Aging and Neurodegenerative Diseases: A Window for the Development of New Biomarkers. Int. J. Mol. Sci. 20, 3810. doi: 10.3390/ijms20153810
Meyne, F., Gloeckner, S. F., Ciesielczyk, B., Heinemann, U., Krasnianski, A., Meissner, B., et al. (2009). Total prion protein levels in the cerebrospinal fluid are reduced in patients with various neurological disorders. J. Alzheimer’s Dis. 17, 863–873. doi: 10.3233/JAD-2009-1110
Mezzenga, R., and Adamcik, J. (2018). The Amyloid Polymorphism in the Protein Folding and Aggregation Energy Landscape. Angew Chem Int Ed Engl 57, 8370–8382. doi: 10.1002/ange.201713416
Mockett, B. G., Richter, M., Abraham, W. C., and Mülle, U. C. (2017). Therapeutic potential of secreted amyloid precursor protein APPsα. Front. Mol. Neurosci. 10:30. doi: 10.3389/fnmol.2017.00030
Mollenhauer, B., Caspell-Garcia, C. J., Coffey, C. S., Taylor, P., Singleton, A., Shaw, L. M., et al. (2019). Longitudinal analyses of cerebrospinal fluid α-Synuclein in prodromal and early Parkinson’s disease. Mov. Disord. 34, 1354–1364. doi: 10.1002/mds.27806
Morel, B., Varela, L., Azuaga, A. I., and Conejero-Lara, F. (2010). Environmental conditions affect the kinetics of nucleation of amyloid fibrils and determine their morphology. Biophys. J. 99, 3801–3810. doi: 10.1016/j.bpj.2010.10.039
Muchtar, E., Buadi, F. K., Dispenzieri, A., and Gertz, M. A. (2016). Immunoglobulin light-chain amyloidosis: From basics to new developments in diagnosis, prognosis and therapy. Acta Haematol. 135, 172–190. doi: 10.1159/000443200
Mulder, H., Gebre-Medhin, S., Betsholtz, C., Sundler, F., and Ahrén, B. (2000). Islet amyloid polypeptide (amylin)-deficient mice develop a more severe form of alloxan-induced diabetes. Am. J. Physiol. - Endocrinol. Metab 278, E684–E691. doi: 10.1152/ajpendo.2000.278.4.e684
Navalkar, A., Ghosh, S., Pandey, S., Paul, A., Datta, D., and Maji, S. K. (2019). Prion-like p53 Amyloids in Cancer. Biochemistry 59, 146–155. doi: 10.1021/acs.biochem.9b00796
Nayak, A., Dutta, A. K., and Belfort, G. (2008). Surface-enhanced nucleation of insulin amyloid fibrillation. Biochem. Biophys. Res. Commun. 369, 303–307. doi: 10.1016/j.bbrc.2008.01.159
Parihar, M. S., and Brewer, G. J. (2010). Amyloid-β as a modulator of synaptic plasticity. J. Alzheimer’s Dis. 22, 741–763. doi: 10.3233/JAD-2010-101020
Parnetti, L., Paciotti, S., Farotti, L., Bellomo, G., Sepe, F. N., and Eusebi, P. (2019). Parkinson’s and Lewy body dementia CSF biomarkers. Clin. Chim. Acta 495, 318–325. doi: 10.1016/j.cca.2019.04.078
Penke, B., Paragi, G., Gera, J., Berkecz, R., Kovács, Z., Crul, T., et al. (2018). The Role of Lipids and Membranes in the Pathogenesis of Alzheimer’s Disease: A Comprehensive View. Curr. Alzheimer Res. 15, 1191–1212. doi: 10.2174/1567205015666180911151716
Portelius, E., Hölttä, M., Soininen, H., Bjerke, M., Zetterberg, H., Westerlund, A., et al. (2014). Altered cerebrospinal fluid levels of amyloid β and amyloid precursor-like protein 1 peptides in Down’s syndrome. NeuroMolecular Med. 16, 510–516. doi: 10.1007/s12017-014-8302-1
Potter, H., and Wisniewski, T. (2012). Apolipoprotein E: essential catalyst of the Alzheimer amyloid cascade. Int. J. Alzheimers. Dis. 2012, 489428. doi: 10.1155/2012/489428
Prpar Mihevc, S., Baralle, M., Buratti, E., and Rogelj, B. (2016). TDP-43 aggregation mirrors TDP-43 knockdown, affecting the expression levels of a common set of proteins. Sci. Rep. 6, 1–9. doi: 10.1038/srep33996
Ros, K., Johansen, A., Riipinen, I., and Schlesinger, D. (2019). The effect of nucleation on icy pebble growth in protoplanetary discs. arXiv[Preprint] Available online at: http://arxiv.org/abs/1907.08471
Saccon, R. A., Bunton-Stasyshyn, R. K. A., Fisher, E. M. C., and Fratta, P. (2013). Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136, 2342–2358. doi: 10.1093/brain/awt097
Sakaguchi, S., Katamine, S., Nishida, N., Moriuchi, R., Shigematsu, K., Sugimoto, T., et al. (1996). Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature 380, 528–531. doi: 10.1038/380528a0
Scarpioni, R., Ricardi, M., Albertazzi, V., De Amicis, S., Rastelli, F., and Zerbini, L. (2016). Dialysis-related amyloidosis: Challenges and solutions. Int. J. Nephrol. Renovasc. Dis 9, 319–328. doi: 10.2147/IJNRD.S84784
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science (80-.). 298, 789–791. doi: 10.1126/science.1074069
Senechal, Y., Kelly, P. H., Cryan, J. F., Natt, F., and Dev, K. K. (2007). Amyloid precursor protein knockdown by siRNA impairs spontaneous alternation in adult mice. J. Neurochem. 102, 1928–1940. doi: 10.1111/j.1471-4159.2007.04672.x
Shoulders, M. D., and Raines, R. T. (2009). Collagen Structure and Stability. Annu. Rev. Biochem. 78, 929–958. doi: 10.1146/annurev.biochem.77.032207.120833
Soto, C. (2011). Prion hypothesis: the end of the controversy? Trends Biochem. Sci. 36, 151–158. doi: 10.1016/j.tibs.2010.11.001
Southam, K. A., Stennard, F., Pavez, C., and Small, D. H. (2019). Knockout of Amyloid β Protein Precursor (APP) Expression Alters Synaptogenesis, Neurite Branching and Axonal Morphology of Hippocampal Neurons. Neurochem. Res. 44, 1346–1355. doi: 10.1007/s11064-018-2512-0
Srivastava, A. K., Pittman, J. M., Zerweck, J., Venkata, B. S., Moore, P. C., Sachleben, J. R., et al. (2019). β−Amyloid Aggregation and Heterogeneous Nucleation. Protein Sci 28, 1567–1581. doi: 10.1002/pro.3674
Steiner, J. A., Angot, E., and Brundin, P. (2011). A deadly spread: cellular mechanisms of α-synuclein transfer. Cell Death Differ. 18, 1425–1433. doi: 10.1038/cdd.2011.53
Tapiola, T., Soininen, H., and Pirttilä, T. (2001). CSF tau and Aβ42 levels in patients with Down’s syndrome. Neurology 56, 979. doi: 10.1212/WNL.56.7.979
Tö, M., Michaels, T. C. T., Sanagavarapu, K., Yang, X., Meisl, G., Cohen, S. I. A., et al. (2018). Secondary nucleation in amyloid formation. Biophys Rev. 9, 329–338. doi: 10.1039/c8cc02204f
Tokuda, T., Salem, S. A., Allsop, D., Mizuno, T., Nakagawa, M., Qureshi, M. M., et al. (2006). Decreased α-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem. Biophys. Res. Commun. 349, 162–166. doi: 10.1016/j.bbrc.2006.08.024
Truong, P. H., Ciccotosto, G. D., and Cappai, R. (2019). Analysis of Motor Function in Amyloid Precursor-Like Protein 2 Knockout Mice: The Effects of Ageing and Sex. Neurochem. Res. 44, 1356–1366. doi: 10.1007/s11064-018-2669-6
Tyan, S. H., Shih, A. Y. J., Walsh, J. J., Maruyama, H., Sarsoza, F., Ku, L., et al. (2012). Amyloid precursor protein (APP) regulates synaptic structure and function. Mol. Cell. Neurosci. 51, 43–52. doi: 10.1016/j.mcn.2012.07.009
Tycko, R., and Wickner, R. B. (2013). Molecular structures of amyloid and prion fibrils: Consensus versus controversy. Acc. Chem. Res. 46, 1487–1496. doi: 10.1021/ar300282r
Vanden Broeck, L., Callaerts, P., and Dermaut, B. (2014). TDP-43-mediated neurodegeneration: Towards a loss-of-function hypothesis? Trends Mol. Med. 20, 66–71. doi: 10.1016/j.molmed.2013.11.003
Veis, A., and Dorvee, J. R. (2013). Biomineralization mechanisms: A new paradigm for crystal nucleation in organic matrices. Calcif. Tissue Int. 93, 307–315. doi: 10.1007/s00223-012-9678-2
Velazquez, R., Ferreira, E., Tran, A., Turner, E. C., Belfiore, R., Branca, C., et al. (2018). Acute tau knockdown in the hippocampus of adult mice causes learning and memory deficits. Aging Cell 17, 1–12. doi: 10.1111/acel.12775
Wang, H., Megill, A., He, K., Kirkwood, A., and Lee, H. K. (2012). Consequences of inhibiting amyloid precursor protein processing enzymes on synaptic function and plasticity. Neural Plast 2012, 272374. doi: 10.1155/2012/272374
Wang, L., Gao, L., Tang, H., Nie, K., Wang, L., Zhao, J., et al. (2015). Cerebrospinal fluid alpha-synuclein as a biomarker for Parkinson’s disease diagnosis: A systematic review and meta-analysis. Int. J. Neurosci. 125, 645–654. doi: 10.3109/00207454.2014.961454
Yang, C., Wang, H., Qiao, T., Yang, B., Aliaga, L., Qiu, L., et al. (2014). Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A. 111, 1121–1129. doi: 10.1073/pnas.1322641111
Yuan, C., Levin, A., Chen, W., Xing, R., Zou, Q., Herling, T. W., et al. (2019). Nucleation and Growth of Amino Acid and Peptide Supramolecular Polymers through Liquid – Liquid Phase Separation. Angew. Chem. Int. Ed. Engl. 100190, 2–10. doi: 10.1002/anie.201911782
Zabel, M. D., and Reid, C. (2015). A brief history of prions. Pathog. Dis. 73, 1–8. doi: 10.1093/femspd/ftv087
Keywords: amyloid, nucleation, Alzheiemr’s, Parkinson’s, virus, prion, protein-only hypothesis
Citation: Malmberg M, Malm T, Gustafsson O, Sturchio A, Graff C, Espay AJ, Wright AP, El Andaloussi S, Lindén A and Ezzat K (2020) Disentangling the Amyloid Pathways: A Mechanistic Approach to Etiology. Front. Neurosci. 14:256. doi: 10.3389/fnins.2020.00256
Received: 18 September 2019; Accepted: 06 March 2020;
Published: 21 April 2020.
Edited by:
Vincenzo La Bella, University of Palermo, ItalyReviewed by:
Herbert Budka, Medical University of Vienna, AustriaCopyright © 2020 Malmberg, Malm, Gustafsson, Sturchio, Graff, Espay, Wright, El Andaloussi, Lindén and Ezzat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kariem Ezzat, a2FyaWVtLmV6emF0LmFobWVkQGtpLnNl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.