 Alexander J. Davies
Alexander J. Davies Simon Rinaldi1
Simon Rinaldi1 Michael Costigan
Michael Costigan Seog Bae Oh
Seog Bae Oh
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 21 February 2020
Sec. Neuropharmacology
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.00142
This article is part of the Research Topic Advances in the Biology and Medicine of Pain View all 36 articles
Cytotoxicity and consequent cell death pathways are a critical component of the immune response to infection, disease or injury. While numerous examples of inflammation causing neuronal sensitization and pain have been described, there is a growing appreciation of the role of cytotoxic immunity in response to painful nerve injury. In this review we highlight the functions of cytotoxic immune effector cells, focusing in particular on natural killer (NK) cells, and describe the consequent action of these cells in the injured nerve as well as other chronic pain conditions and peripheral neuropathies. We describe how targeted delivery of cytotoxic factors via the immune synapse operates alongside Wallerian degeneration to allow local axon degeneration in the absence of cell death and is well-placed to support the restoration of homeostasis within the nerve. We also summarize the evidence for the expression of endogenous ligands and receptors on injured nerve targets and infiltrating immune cells that facilitate direct neuro-immune interactions, as well as modulation of the surrounding immune milieu. A number of chronic pain and peripheral neuropathies appear comorbid with a loss of function of cellular cytotoxicity suggesting such mechanisms may actually help to resolve neuropathic pain. Thus while the immune response to peripheral nerve injury is a major driver of maladaptive pain, it is simultaneously capable of directing resolution of injury in part through the pathways of cellular cytotoxicity. Our growing knowledge in tuning immune function away from inflammation toward recovery from nerve injury therefore holds promise for interventions aimed at preventing the transition from acute to chronic pain.
The role played by the immune system in the context of nerve injury and pain is extensive (Ren and Dubner, 2010; Totsch and Sorge, 2017; Raoof et al., 2018). The two key effector mechanisms of the cellular immune response are inflammation and cytotoxicity. The importance of inflammation has long been recognized. The cardinal signs of inflammation – calor (warmth), dolor (pain), tumor (swelling), and rubor (redness and hyperemia) – were first documented by the Roman physician Celsus in the 1st century AD (Scott et al., 2004). These symptoms are in large part the product of the actions of inflammatory mediators such as chemokines, cytokines and vasodilators released from immune cells in a process designed to protect and facilitate the repair of injured tissue. The role of cytotoxicity, on the other hand, is to kill living cells and/or degenerate tissues via apoptotic mechanisms and is a critical function of immunity against cancer – another term introduced to the latin lexicon by Celsus (Hajdu, 2016). Pathways leading from immune activation to inflammation and their contribution to the generation of neuropathic pain are relatively well documented (Calvo et al., 2012). However, recent evidence points to a role of cytotoxicity in the immune response to nerve injury, which can in turn affect pain outcomes (Davies et al., 2019).
Neuropathic pain is defined as that arising from a lesion or disease of the somatosensory nervous system (Treede et al., 2008; Costigan et al., 2009b). Peripheral neuropathies leading to pain almost invariably result from a partial loss of innervation. This may occur either from disconnection of nerve branches from their peripheral target by physical injury (Bennett and Xie, 1988; Seltzer et al., 1990; Kim and Chung, 1992; Decosterd and Woolf, 2000) or a dying-back of axons from their terminals due to metabolic (O’Brien et al., 2014) or chemical neurotoxicity (Hoke and Ray, 2014). Axonal loss after injury is characterized by the mechanisms of Wallerian degeneration: a neuron-autonomous (i.e., self-determined) process involving active metabolic signaling within the axon leading to cytoskeletal destabilization and fragmentation (Gerdts et al., 2016; Llobet Rosell and Neukomm, 2019). Additionally, dying back of axons utilizes apoptotic pathways, while protecting the cell body from death (Yaron and Schuldiner, 2016).
The cellular response to nerve injury involves dedifferentiation of resident Schwann cells, and dynamic infiltration of systemic immune cells (Rotshenker, 2011). Normally this process would clear the way for axonal regeneration and nerve repair, resulting in transient hypersensitivity followed by recovery. However, interruption of this complex cascade of degeneration and regeneration can lead to aberrant processing which in some cases can transition into chronic pain (Xie et al., 2017; Davies et al., 2019). Thus, peripheral neuropathies lie at the intersection of multiple degeneration, immune and cell death-related pathways, and how these interact influences pain outcome over time.
Natural killer cells are a key component of the innate immune response (Eberl et al., 2015), originally identified in the 1970’s as a population of large granular lymphocytes. NK cells were found to be spontaneously cytotoxic to freshly isolated cells and many tumor or immortalized cell lines (Timonen et al., 1981). Toxicity occurred despite the lymphocytes originating from healthy donors, and without the cells being deliberately sensitized. This non-adaptive, non-major histocompatibility complex (MHC)-restricted cytotoxicity was defined as ‘natural’ cytotoxicity (Trinchieri, 1989). Later, a system of classification of human cytotoxic lymphocytes was proposed by Lanier and Phillips based on CD3 expression, and whether or not cytotoxicity was MHC restricted (Lanier and Phillips, 1986), leading to CD8+ T cells (CTLs) and natural killer (NK) cells, with NK cells further divided into CD3 positive (NKT) and negative populations. A smaller population of ‘unconventional’ T cells defined by expression of the gamma and delta (γδ) T cell receptor chains are also capable of cytotoxic functions (Chien et al., 2014).
Natural killer cells develop from a lymphoid precursor common to B and T cells, but unlike their adaptive cousins, NK cells lack the machinery for somatic recombination and thus do not generate antigen-specific receptors. NK cells instead possess three main sets of receptors that control target cell recognition: natural killer group 2 (NKG2); killer-cell immunoglobulin-like receptors (KIR) and natural cytotoxicity receptors (NCR). These receptor families inhibit or activate NK cell function through a host of target cell surface ligands (Guia et al., 2018). Target cells can undergo loss of MHC-I ligands (known as ‘missing-self’), which normally are inhibitory to NK cells, or alternately NK cells can recognize stress-induced ligands expressed by the target cell (‘self-recognition’). The net result of these interactions is to allow NK cells to define and subsequently remove transformed or damaged cells (Chiossone et al., 2018).
Critical to NK cell cytotoxicity is the recognition and adhesion to the target cell, forming an ‘immune synapse’ (Davis et al., 1999). Target cell engagement is followed by exocytosis and fusion of lytic granules containing pore-forming perforin proteins and serine proteases such as the granzyme family (Henkart, 1985; Figure 1). NK cells can also induce target cell death signaling via activation of intracellular caspase cascades including tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and Fas (CD95/Apo 1)-mediated apoptosis (Zamai et al., 1998). In addition, NK cells can effect target cell removal through antibody-dependent cellular cytotoxicity (ADCC) (Wang et al., 2015; Figure 2).
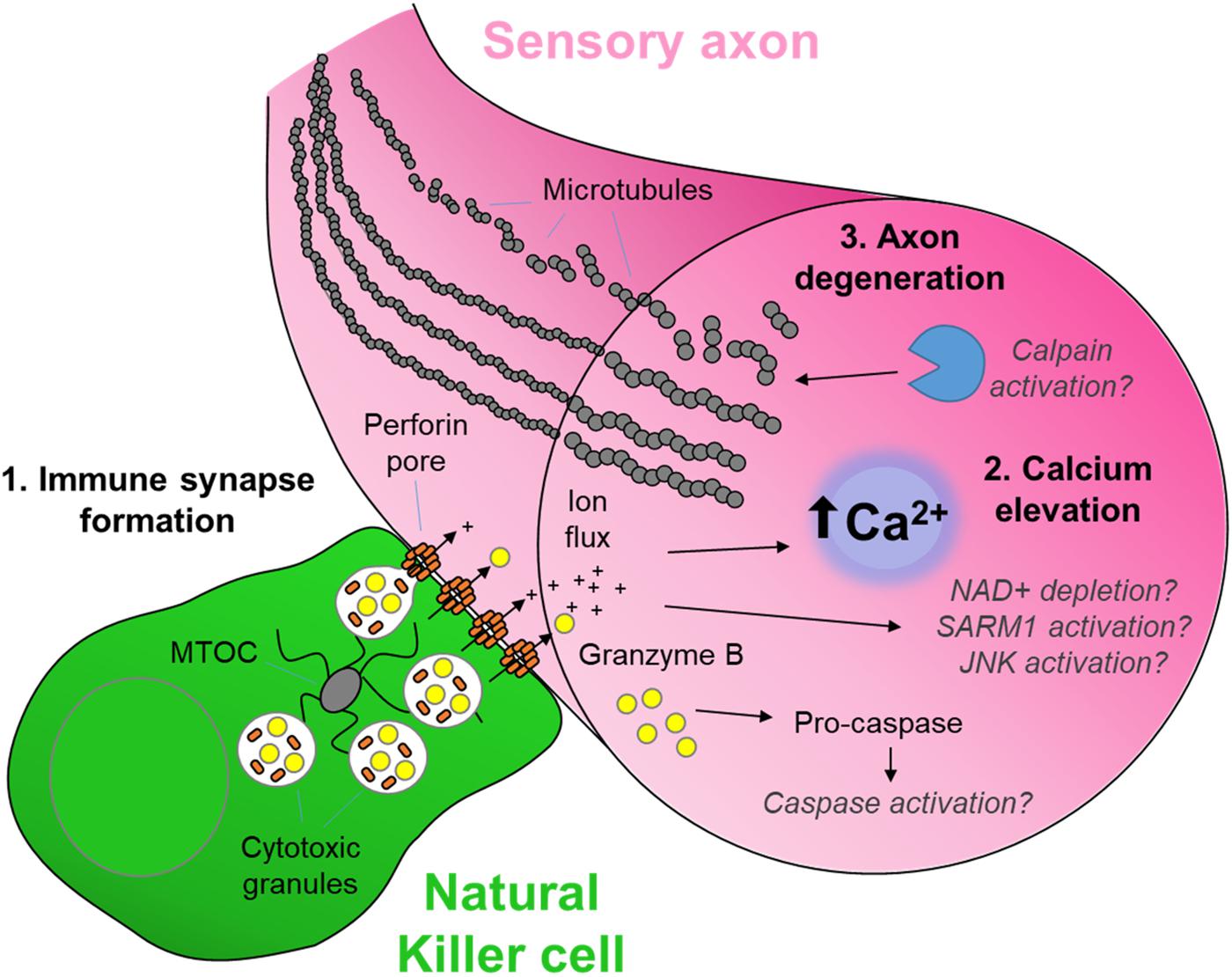
Figure 1. The cytotoxic neuro-immune synapse: Potential downstream intracellular pathways. Cytotoxic natural killer cells form an immunological synapse upon recognition and adhesion to a target, in this case the axon of a stressed sensory neuron. (1) Cytotoxic/lytic granules are held by the microtubule organizing center (MTOC) of the NK cell. Immune synapse formation triggers reorientation and polarization of the granules by the MTOC which directs the fusion of the granules to the synaptic membrane. This mechanism ensures the precision release of the cytotoxic contents directly through the targeted axonal membrane. (2) The assembly of perforin pore complexes in the axonal membrane from individual perforin subunits released to the extracellular space allows the flow of ions including Ca2+, and larger molecules such as the serine protease granzyme B, into the intracellular environment of the target neuron. (3) Ca2+ flux leads to rapid axon microtubule destabilization and axon degeneration. The full mechanism of cytotoxicity in the axon is unclear. However, in the case of neuron-autonomous Wallerian degeneration, the late-phase Ca2+ elevation and subsequent calpain activation required for microtubule destabilization is gated by a metabolic signaling cascade involving depletion of nicotinamide adenine dinucleotide (NAD+), activation of Sterile-alpha and armadillo motif containing protein (SARM) and C-Jun N-terminal kinase (JNK) (Llobet Rosell and Neukomm, 2019). Although caspase 3 is a major target of granzyme B protease activity (Adrain et al., 2005), its role in cytotoxic axon degeneration remains unclear. Pathways shown in gray italics are yet to be determined specifically in cytotoxicity-induced axonal degeneration.
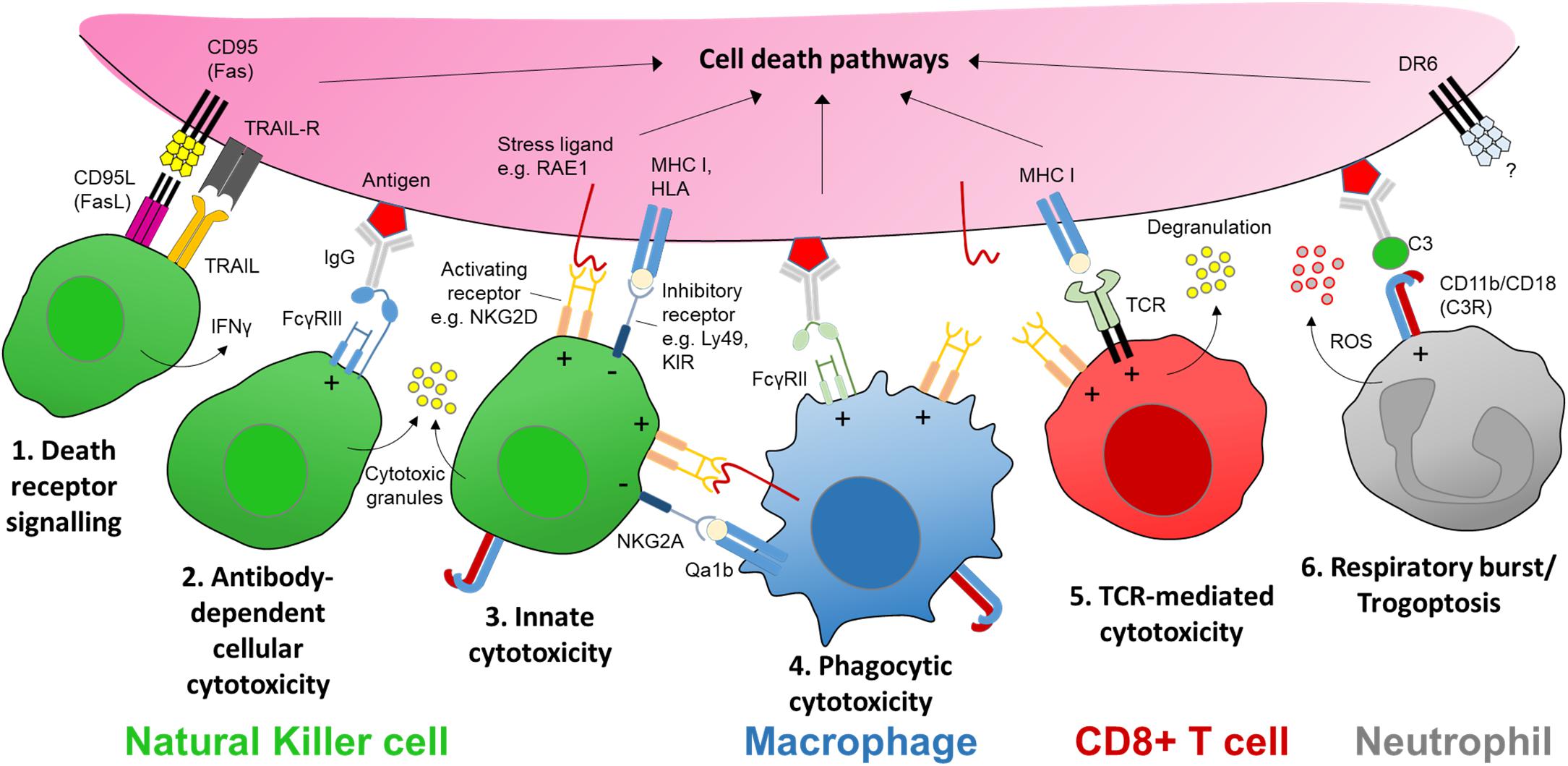
Figure 2. The receptors and ligands of cellular cytotoxicity. There are several key cellular effector mechanisms capable of evoking intracellular cell death pathways. In the case of nerve injury this could be a targeted sensory axon. Natural killer (NK) cells are specialist cytotoxic effector cells expressing multiple receptors and ligands for executing cellular cytotoxicity. (1) CD95 (Fas) is a transmembrane protein that belongs to the tumor necrosis factor (TNF) family. Engagement with its ligand CD95L (FasL) expressed on NK cells (as well as activated CD8+ T cells and plasma cells) triggers apoptotic signaling in the target cell. Apoptosis may also be induced by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) via its receptor on target axons. (2) Cross-linking of an antigen-antibody complex with the low affinity Fc receptor FcγRIIIa (CD16a) on human NK cells (FcγRIII/CD16 in mice) can trigger cytotoxic granule release (Rosales, 2017). (3) NK cells express a host of activating receptors such as NKG2D which recognize ligands expressed at times of neural stress, tumorigenesis or infection. Cytotoxic activity by either Fc or activator receptor function is kept in balance by inhibitory signaling from major histocompatibility complex (MHC) class I molecules or human leukocyte antigens (HLA), present on nearly all somatic cells; these molecules signal through inhibitory receptors expressed on NK cells, e.g., Ly49 family in mice, and killer immunoglobulin-like receptors (KIR) in humans. (4) Macrophages may elicit a form of antibody-dependent cellular phagocytosis (ADCP) via engagement with another low affinity Fc receptor FcγRIIa (CD32a) in humans. Macrophages/monocytes may also express natural killer receptors and ligands allowing them to recognize stressed target cells as well as interact with NK cells via natural killer receptor ligands (e.g., RAE1) while protecting themselves from killing by inhibitory MHC I molecules such as Qa1b (in mice) (Zhou et al., 2012). (5) Sensitized CD8+ T cells recognize MHC I-presented antigens at the target membrane surface via corresponding T cell receptors (TCR), which together with co-stimulation via receptors such as NKG2D, triggers cytotoxic granule release in a manner similar to NK cells. CD8+ T cells may also trigger apoptosis in target cells via FasL. (6) Neutrophils can recognize opsonized targets, such as antibody-antigen complexes, via complement receptors (e.g., C3R) or Fc receptors including FcγRIIa (Rosales, 2017). Complement or antibody Fc receptor signaling neutrophils can lead to respiratory burst and the release of reactive oxygen species (ROS), or an active mechanical disruption of the target cell membrane known as ‘trogoptosis’ (Matlung et al., 2018). Signaling via death receptor 6 (DR6) is thought to be involved in axon degeneration in the peripheral nervous system as well as neurodegeneration in the brain but the extrinsic signal for the receptor remains unknown (Gamage et al., 2017). ‘+’ and ‘−’ within the immune cell denotes stimulatory and inhibitory signaling, respectively.
Human NK cells can be broadly separated into cytotoxic CD56dimCD16+ cells and cytokine producing CD56brightCD16– (Poli et al., 2009); the equivalent populations in mice being CD27–CD11b+ and CD27+CD11b–, respectively (Crinier et al., 2018). In addition to cytotoxicity, CD56bright NK cells can shape the immune response in their vicinity by secreting a host of signaling molecules including interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNFα) and colony-stimulating factor 2 (CSF2) (Vivier et al., 2008; Poli et al., 2009) allowing functional interactions with T cells, dendritic cells and macrophages (Michel et al., 2012; Ferlazzo and Moretta, 2014; Pallmer and Oxenius, 2016). Unlike other innate immune cells, NK cells are not generally considered phagocytic, although phagocytosis of fungal pathogens by NK cells has been reported (Voigt et al., 2014).
Two major families of stress-induced ligands for the activatory receptor NKG2 member D (NKG2D) receptor have been identified in humans: MHC class I chain-related protein A (MICA) and B (MICB), and UL16-binding proteins (ULBP1-6), encoded by the family of retinoic acid early transcript 1 (RAET1) genes (Spear et al., 2013). The homologous ligands in the mouse are separated into three families: Histocompatibility 60 (H-60) (Diefenbach et al., 2001), murine UL16-binding protein-like transcript 1 (MULT1) (Carayannopoulos et al., 2002) and the RAE1 proteins family encoded by five Raet1 genes (α, β, γ, δ and ε) (Cerwenka et al., 2000). NKG2D ligands are often expressed by tumors or virally infected cells (Guia et al., 2018); for example, influenza infection has been shown to upregulate Raet1 gene expression in mouse sensory neurons in vitro (Backstrom et al., 2007). NKG2D ligands may also be expressed by other cell stressors such as during DNA damage or tissue injury (Raulet et al., 2013). The Raet1 gene family (not to be confused with ribonucleic acid export 1, Rae1) was originally identified by its expression in a mouse carcinoma cell line in response to retinoic acid treatment (Nomura et al., 1994).
One of the first descriptions of NK cell-mediated neurotoxicity was in sympathetic ganglion neurons following systemic treatment with guanethidine (Hickey et al., 1992). NK cells harvested from adult mice and stimulated ex vivo with the cytokine interleukin-2 (IL-2) were also cytotoxic to dissociated embryonic dorsal root ganglion (DRG) neurons (Backstrom et al., 2000). A clue to the molecular interactions involved was a reduction in DRG cell cytotoxicity by blockade of the NKG2D receptor on NK cells (Backstrom et al., 2003), as well as the high basal expression of Raet1 in the embryonic sensory neurons (Nomura et al., 1996), which is likely the result of downstream signaling from retinoic acid. Retinoic acid signaling is critical in neurodevelopment (Maden, 2007), providing neurotrophic effects on axonal outgrowth (Corcoran et al., 2000) and acting as a regeneration mediator after nerve injury in adult neurons (Puttagunta and Di Giovanni, 2011).
In contrast to embryonic neurons, Raet1 expression is minimal in uninjured adult sensory neurons (Backstrom et al., 2000; Davies et al., 2019). Transcripts for Raet1a, Raet1b and Raet1e, as well as Ulbp1 (encoding MULT1) and H60b, were identified in single DRG neurons isolated from 6 to 8 week old mice, although abundance was low (Usoskin et al., 2015). Raet1 transcripts are however significantly upregulated in DRG neurons after peripheral nerve injury as detected by whole tissue quantitative-PCR and in situ hybridization (Davies et al., 2019). The Raet1e transcript specifically was also identified by RNA sequencing of mouse DRG, though it did not reach significance as a differentially expressed gene, likely due to the low abundance at the early time points assessed after injury (<24 h) (Rozenbaum et al., 2018). Additionally, deep sequencing of the rat sciatic nerve showed significant upregulation of Raet1 4 days after crush injury (Yi et al., 2015), suggesting either local expression within the injured axon, or additional expression by resident cells within the nerve. Recruitment of NK cells into the injured peripheral nerve (Cui et al., 2000; Hu et al., 2007; Davies et al., 2019) allows for the targeting of RAE1–expressing injured axons for degeneration (Davies et al., 2019) as well as possibly targeting other cell types within the nerve (Yi et al., 2015).
The signaling process driving Raet1 expression in injured sensory neurons is currently unclear. RAE1 expression during herpes virus infection occurs via the inhibition of histone deacetylase 3 (HDAC3), which normally acts as constitutive repressor of NKG2D-ligand gene expression (Greene et al., 2016). HDAC3 is also exported from the nucleus of injured DRG neurons (Cho et al., 2013) contributing to the histone acetylation which is thought to be necessary for ‘regeneration associated gene’ expression (Cho and Cavalli, 2014). The potential for autoimmune neurodegeneration by NK cells raises the interesting question of epigenetic influences on NKG2D ligand expression as a possible cause of sensory autoimmune neuropathies (Schleinitz et al., 2010). This has been demonstrated in principle by conditional overexpression of Raet1e within a population of TRPV1 receptor-positive sensory neurons, which resulted in a loss of heat sensitivity compared to littermate controls, consistent with the absence of peripheral signaling from this important subset of heat-sensitive nociceptive fibers (Davies et al., 2019). Trpv1 expression in the cell bodies of these sensory nerves was preserved, however, suggesting that the effect of Raet1e overexpression occurred in the peripheral axons, much like after injury (Davies et al., 2019). Further work is required to examine the dynamics of the expression of immune ligands within sensory neurons in health and disease.
Peripheral neuropathy is a common side-effect of many chemotherapeutic agents. Axon degeneration occurs after treatment with oxaliplatin or vincristine, despite differences in the mode of action of these drugs (Starobova and Vetter, 2017). Oxaliplatin is a platinum-based chemotherapeutic, which blocks tumor cell replication through DNA binding thus leading to apoptosis, but is also associated with significant toxic side-effects including neurotoxicity (Oun et al., 2018). Accumulation of oxaliplatin in the DRG specifically is thought to contribute to the symptoms of peripheral sensory neuropathy (Sprowl et al., 2013) with functional changes to nerve excitability early in treatment correlating with the severity of neuropathy over time (Park et al., 2009). Vincristine is a vinca alkaloid drug which binds tubulin, resulting in microtubule destabilization and interruption of mitosis in dividing cells (Bates and Eastman, 2017). In sensory neurons, microtubule arrangement alterations are observed particularly in the large myelinated axons of rat sciatic nerves following vincristine treatment but prior to significant axon degeneration (Topp et al., 2000). The mechanisms of peripheral neuropathy by vincristine treatment in vivo are likely to involve mitochondrial toxicity and disruption of axonal transport (Fukuda et al., 2017). Neuronal microtubules are highly dynamic and participate in the transport of newly synthesized RNA, proteins and organelles required for nerve homeostasis (Prior et al., 2017). Disruption of axonal transport will therefore likely affect the local expression of RNA and protein that is critical for the efficient wiring and function of developing and regenerating axons (Poulopoulos et al., 2019) and likely explains why the longest axons tend to be affected, resulting in a ‘stocking and glove’ distribution of symptoms at the extremities (Starobova and Vetter, 2017). Non-neuron-autonomous effects from other cell types including immune cells may also contribute to the neuropathic effect of vincristine (Lees et al., 2017).
Chemotherapeutic agents upregulate stress-related proteins in tumor (as well as other) cells, including ligands for NK cells (Zingoni et al., 2017). For example, low-dose chemotherapeutic agents, including oxaliplatin, are capable of upregulating ligands for the activating receptors NKG2D and DNAX accessory molecule-1 (DNAM-1; CD226) and TRAIL in multiple tumor lines leading to enhanced susceptibility to NK cell cytotoxicity (Soriani et al., 2009; Siew et al., 2015). Vincristine and oxaliplatin themselves only mildly impair NK cell cytotoxicity (Markasz et al., 2007). Chemotherapeutic agents can also enhance NK cell function by down-regulation of inhibitory ‘self’ ligands on the target cell surface (Fine et al., 2010); therefore the sensitivity of tumor or other target cells to NK cell cytotoxicity can be initiated by modulation of activating and inhibitory ligands (Quirk and Ganapathy-Kanniappan, 2017). Pre-treatment with the cytokine interleukin 2 (IL-2) increases the anti-tumor response rate to oxaliplatin particularly in patients with low blood lymphocyte counts (Lissoni et al., 2005) but there are currently no data available on how this immune modulation may impact on the neuropathic side effects of chemotherapy.
Immunotherapies that recruit cytotoxic effectors cells (Cifaldi et al., 2017), such as tumor-targeted monoclonal antibodies (mAbs) and checkpoint inhibitors, are also prone to inducing peripheral neuropathic side-effects (Federico et al., 2017; Spain et al., 2017) and in rare cases the inflammatory polyneuropathy Guillain-Barré syndrome (GBS) (McNeill et al., 2019). Tumor targeting by mAbs is fast becoming a mainstay in immunotherapy. Both NK cells and macrophages contribute to ADCC. Binding of the Fc portion of immunoglobulins (IgG) to Fc receptors FcγRIIIa (CD16) or FcγRIIa (CD32) transmits a stimulatory signal to NK cells triggering cytotoxic attack, as well as the release of cytokines such as IFNγ (Cheung and Dyer, 2013; Figure 2). However, the increased use of mAbs has brought an appreciation of significant side effects, in particular autoimmune disease (Hansel et al., 2010) and neurological complications (Bosch et al., 2011). For example, dinutuximab (ch14.18) is a mAb licensed for the treatment for high-risk childhood neuroblastoma and targets the disialosyl glycolipid GD2. NK cells are thought to be the main driver of the anti-tumor effect of neuroblastoma immunotherapy (Main et al., 1985; Lode et al., 1998; Wang et al., 2015) such that gain of NK cell function is often sought in combination with antibody treatment (Koehn et al., 2012). The combination of anti-GD2 mAbs with IL-2 improves outcomes in high-risk neuroblastoma patients suggesting that cellular effectors, in particular NK cells, are key to this response (Yu et al., 2010). However, during anti-GD2 mAb immunotherapy patients experience significant neuropathic pain (Cheung et al., 1987) and in severe cases irreversible peripheral sensory neuropathy. Anti-GD2 mAbs recognize the GD2 antigen on axons and affect peripheral nerve function in vivo (Slart et al., 1997; Xiao et al., 1997) but how this occurs remains unclear. The anti-tumor effect of anti-GD2 antibodies involves activation of the complement system (complement-dependent cytotoxicity, CDC) in addition to ADCC (Imai et al., 2005). Research has until now focused on antibody modifications designed to reduce complement activation, which has had some success in reducing neuropathic side effects in rat models (Sorkin et al., 2010). However, trials with newer mAb isoforms with impaired complement activity still report significant peripheral nerve toxicities (Navid et al., 2014; Anghelescu et al., 2015; Federico et al., 2017), suggesting that other mechanisms of mAb-induced neuropathy are at play (Calvo et al., 2012). Although immune-mediated inflammation has been proposed as a possible cause of chemotherapy and immunotherapy-induced pain (Starobova and Vetter, 2017), the role of cellular cytotoxicity in these pathologies requires further investigation.
Wallerian degeneration, the process responsible for axonal degeneration following nerve axotomy in vivo and in the severed axons of cultured sensory neurons in vitro, is an active processes intrinsic to the neuron (Lunn et al., 1989; Perry et al., 1990). The discovery of a spontaneous mouse mutation with delayed or ‘slow’ Wallerian degeneration (WldS) demonstrated that over-activity of the nicotinamide adenine dinucleotide (NAD+) biosynthetic enzyme NMNAT1, is sufficient to protect the severed axon from fragmentation (Araki et al., 2004; Sasaki et al., 2009). A Drosophila screen of axon degeneration after olfactory neuron axotomy led to the identification of dSarm and its mammalian ortholog: Sterile alpha and TIR motif-containing 1 (SARM1) (Osterloh et al., 2012). SARM1 rapidly breaks down NAD+ and therefore acts as the key regulator of axonal degeneration after injury (Gerdts et al., 2015; Figure 1). SARM1 has been proposed to be a point of convergence for axonal degeneration associated with chemotherapy (Geisler et al., 2016) and metabolic neuropathies (Turkiew et al., 2017) but interestingly, not neuronal death (Geisler et al., 2019a).
Axonal loss may also occur in intact neurons by pruning or dying back. These processes share many features with Wallerian degeneration such as disruption of microtubules and cytoskeleton rearrangement (Luo and O’Leary, 2005). However, in contrast to the Wallerian degeneration of severed axons, axonal pruning during development uses the machinery of apoptosis while avoiding neuronal death via the endogenous caspase inhibitor, X-linked inhibitor of apoptosis protein (XIAP) (Unsain et al., 2013; Yaron and Schuldiner, 2016; Figure 3). The degree of peripheral axon pruning after injury may also be driven by a combination of trophic and axon guidance factors, which also co-opt apoptotic pathways (Vanderhaeghen and Cheng, 2010).
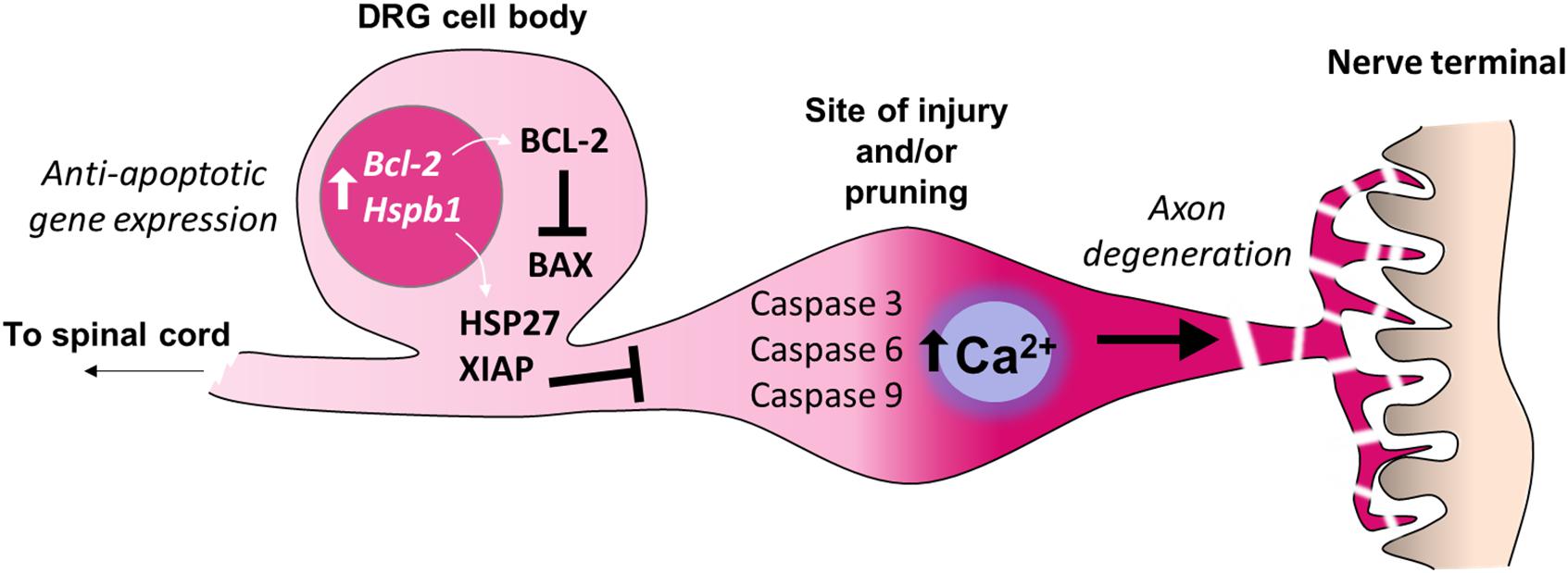
Figure 3. Inhibition of apoptosis in the cell bodies of sensory neurons. Adult sensory neurons are protected from cell death by expression of Bcl2 (B-cell lymphoma 2). The gene product BCL-2 suppresses the function of the pro-apoptotic protein BAX (BCL2 Associated X) which can be induced by cell death signaling. The expression of further anti-apoptotic genes in the cell bodies of sensory neurons prevent axon degeneration intracellular signaling cascades from causing neuronal death. For example, expression of Hspb1 (Heat shock protein family B1), also known as HSP27, prevents sensory neuron death after nerve injury. X-linked inhibitor of apoptosis protein (XIAP) prevents cell death during developmental axon pruning by inhibiting caspases in the cell body. Thus axonal activation of caspases 3, 6 and 9, which is sufficient for certain forms of axon-restricted degeneration, occurs in the absence of cell apoptosis (Simon et al., 2012; Cusack et al., 2013).
Genes related to programed cell death have long been known to be regulated within the dorsal root ganglia (DRG) of injured sensory neurons (Costigan et al., 2002). For example, the elevation of pro-apoptotic caspases occurs in injured DRG neurons as well as surrounding non-neuronal glial cells (Vigneswara et al., 2013; Wiberg et al., 2018), and single cell analysis of sensory ganglia shows that caspase 3 is upregulated in all sensory neuron subtypes after injury (Hu et al., 2016). In fact more than 10 cell death-related genes are among the top 438 genes commonly induced by nerve injury in more than 5 sensory neuron subtypes revealed by single cell gene ontology analysis (Renthal et al., 2019). Other cell death-related genes, such as programed cell death 2 (Pdcd2) are upregulated only in the non-peptidergic (NP) population of nociceptive neurons; conversely, NP neurons showed a down-regulation of the neuronal survival related genes ISL LIM homeobox 1 (Isl1) and oxidation resistance 1 (Oxr1) (Hu et al., 2016), suggesting a selective sensitivity of this nociceptive neuron subpopulation to cell-death mechanisms.
While substantial neuronal death can occur in the DRG in neonatal animals after nerve injury (Himes and Tessler, 1989), such sensory loss is limited in adults (Whiteside et al., 1998; Tandrup et al., 2000; Shi et al., 2001). Nerve injury in young animals (up to 3 weeks of age) results in a reduced expression of the neuroprotective anti-apoptotic gene Bcl-2 (Gillardon et al., 1994; Farlie et al., 1995); this developmental period correlates with the lack of neuropathic pain development following peripheral nerve injury (Howard et al., 2005; Costigan et al., 2009a; Fitzgerald and McKelvey, 2016). In adults, DRG expression of Bcl-2 remains high (Merry et al., 1994) regardless of nerve injury type (Gillardon et al., 1994). The upregulation of heat shock proteins such as HSP27 (encoded by Hspb1) also support neuronal survival after axotomy (Costigan et al., 1998; Lewis et al., 1999; Figure 3). Conversely, after peripheral nerve transection, adult mice deficient in the pro-apoptotic gene Bax display greater autotomy (self-mutilation) (Lyu et al., 2017), a sign of ongoing neuropathic pain (Bennett and Xie, 1988), suggesting that certain apoptotic or ‘cell death’-related pathways may in fact be pain-protective in the face of nerve injury. Similarly, cross-linking of the death receptor Fas (also known as CD95) on sensory neurons enhances neurite outgrowth and nerve regeneration after sciatic nerve crush (Desbarats et al., 2003). Upregulation of Fas ligand within the injured nerve (Li et al., 2018) is therefore likely to have an important bearing on the resolution of nerve injury via non-apoptotic signaling to neurons (Desbarats et al., 2003) and immune cells (Williams et al., 2018) in addition to apoptotic mechanisms (Li et al., 2018; Figure 2).
Despite the role of SARM1 as the gate-keeper of intrinsic axon degeneration after injury (DiAntonio, 2019), extrinsic factors also influence axon degeneration (Conforti et al., 2014). For example, recent evidence points to the potential of extracellular signaling driving Wallerian degeneration through the orphan death receptor 6 (DR6) (Gamage et al., 2017). The cytotoxic machinery of NK cells is also well-placed to initiate axon degeneration. Immune-synapse formation with a target cell, followed by perforin pore formation and granzyme entry to the cytoplasm, typically triggers a cascade of calcium-dependent signaling events which in many cell types then leads to apoptosis (Trapani and Smyth, 2002). In sensory neurons, increases in axonal calcium either through the perforin pore or released from intracellular stores, appears critical to the subsequent localized axon degeneration by NK cells (Davies et al., 2019; Figure 1). The ability of calpain inhibitors to prevent axonal degeneration after nerve injury (McGonigal et al., 2010; Ma et al., 2013), as well as injury by complement-mediated membrane attack complex (MAC) pore formation (O’Hanlon et al., 2003), suggests that calcium-dependent processes are sufficient for degeneration. Thus the formation of a ‘neuro-immune synapse’ and its consequent actions within the axon represents an important extrinsic mechanism of axon degeneration in the periphery (Figure 1). The exact mechanisms downstream from granzyme entry into the axoplasm and how these interact with the known intrinsic pathways of axon degeneration, including SARM1, are currently unknown.
The relationship between axon degeneration and neuropathic pain is complex. Experiments in Wallerian degeneration null mice show that while chronic constriction injury and chemotherapy-induced ‘pain’ does appear to require axon degeneration (Ramer et al., 1997; Geisler et al., 2016), other nerve injury models, such as spinal nerve transection, do not (Ramer and Bisby, 1998). On the other hand, complete nerve crush injury inducing Wallerian degeneration of the injured axons results in less long-term mechanical hypersensitivity than a partial or ‘moderate’ crush injury (Huang et al., 2012; Davies et al., 2019). Thus it appears that axon degeneration is neither necessary nor sufficient for neuropathic pain. Instead, the relative proportion of the injured and uninjured axon fibers and their functional subtypes or modalities is a more likely determining factor, reflective of the fact that most pre-clinical neuropathic pain models are based on a partial injury of the nerve (Seltzer et al., 1990; Kim and Chung, 1992; Decosterd and Woolf, 2000). In addition, neuropathies as a result of physical trauma, metabolic or chemical injury result in an intermediate state of injury between normal and axotomized, as evidenced by the pathological changes observed in axons the absence of complete Wallerian degeneration (Topp et al., 2000; Park et al., 2009; Huang et al., 2012; Davies et al., 2019). Axons in such a ‘metastable’ state of injury (Williams et al., 2014), may also be defined by the expression of cell-stress markers, including immune ligands, on their membranes (Davies et al., 2019). These findings suggest that peripheral nerve recovery may be achieved by either axonal stabilization (Geisler et al., 2019b) if intervention is performed early, or by accelerated degeneration if significant injury has already occurred (Davies et al., 2019).
Granzymes are a family of serine proteases produced by cytotoxic NK and T cells that have recently been linked to nerve injury and pain. Granzyme B is detected within the mouse peripheral nerve after transection and crush injuries, of which NK cells are a significant source (Davies et al., 2019). In vitro, the presence of granzyme B in the culture media is not sufficient to induce DRG axon toxicity, indicating that granzymes are delivered to the axon cytosol by direct contact between cytotoxic cells and sensory nerves (Davies et al., 2019). This suggests the formation of a neuro-immune synapse is critical in the neurodegenerative effect of cellular cytotoxicity by NK cells (Figure 1). In clinical neuropathies, granzymes have been labeled in the peripheral nerves (Noguchi et al., 2005; Ni Mhaolcatha et al., 2019), and granzyme B positive lymphocytes are seen in close apposition to neurons in the DRG in the late-stages of Guillain-Barré syndrome (GBS) in which axon degeneration is observed (Wanschitz et al., 2003). In the central nervous system (CNS) granzyme B-positive cells can be found next to apoptotic neurons following stroke (Chaitanya et al., 2011). Further studies are required to determine the exact role of these cytotoxic effector proteins following nerve injury.
Besides direct cytotoxicity, another potential target for granzymes are protease-activated receptors (PARs), a number of which have been implicated in pain pathogenesis (McDougall and Muley, 2015). For example, PAR-4 activation by the serine protease cathepsin G is capable of suppressing sensory neuron excitability (Sessenwein et al., 2017). Granzymes have been implicated in pain phenotypes more broadly, for example in a genome wide association study (GWAS) of healthy human twins the gene GZMM, encoding granzyme M, was identified as strongly associated to thermal nociception (Williams et al., 2012). Conversely, an inhibitor of granzyme B, serpinA3N, is produced by DRG neurons after peripheral nerve injury (Vega-Avelaira et al., 2009) where it is thought to attenuate mechanical allodynia in mice by inhibition of another serine protease, leukocyte elastase (Vicuna et al., 2015). Treatment of mice with small molecule inhibitors of leukocyte elastase after nerve injury temporarily reduced established neuropathic mechanical allodynia (Bali and Kuner, 2017). Furthermore, serpinA3N is capable of rescuing human fetal neuronal cell death mediated by anti-CD3 and anti-CD28 activated T cells (Haile et al., 2015). What role serpinA3N or other serine protease inhibitors may have in mitigating cytotoxic effector functions in the peripheral nerve after injury remains to be explored.
Natural killer cells now join macrophages as immune cells participating in the degeneration process after peripheral nerve injury (Chen et al., 2015). Macrophages are phagocytic cells that may be tissue resident or originate from the monocyte population of the blood and are an important component of the innate immune response to nerve injury (Zigmond and Echevarria, 2019). Macrophages play a key role in the removal and degradation of myelin debris during Wallerian degeneration (Stoll et al., 1989), and appear capable of degenerating both myelinated and unmyelinated fibers, as liposome depletion of macrophages prior to partial nerve ligation led to a preservation of both fiber types (Liu et al., 2000). However, macrophages do not appear capable of overriding the intrinsic delay in Wallerian degeneration in the WldS mouse despite their recruitment to the injured nerve (Rosenberg et al., 2012). In the absence of frank nerve injury, however, macrophages could play an active role in the peripheral axon degeneration that occurs in aged mice (Yuan et al., 2018).
Monocytes and macrophages exert cytotoxicity via cell-contact dependent phagocytosis (Munn and Cheung, 1990; Gul et al., 2014; Figure 2). Neuron morphology may be affected by the phagocytosis of viable axons and dendrites, in a process termed ‘neurophagy’ (Vilalta and Brown, 2018). After peripheral nerve injury for example, both central and peripheral projections of primary afferent neurons display signals for phagocytosis by microglia (Maeda et al., 2010) and macrophages (Wakatsuki et al., 2017), respectively. Macrophages can recognize the loss of sialic acid residues on membrane-bound glycolipids (desialylation) leading to the removal of neurites by phagocytosis (Linnartz-Gerlach et al., 2016). Neurite phagocytosis by macrophages requires the cell adhesion molecule CD11b, part of the complement C3 receptor (CR3), which recognizes deposition of the complement component C1 on desialyated glycolipids (Linnartz-Gerlach et al., 2016). Thus the complement system works in synergy with innate immunity to mark neuronal membranes for removal. In a model of simian immunodeficiency virus (SIV) infection, monocyte chemotaxis within the DRG led to neuronal loss which correlated the loss of intraepidermal nerve fibers (IENF) (Lakritz et al., 2015). Conversely, sialylation of DRG neuronal cell bodies after injury may help to prevent cytotoxic attack (Peng et al., 2004) by engaging inhibitory signaling via SIGLECs (sialic acid-binding immunoglobulin-type lectins) on innate immune cells, including NK cells and monocytes (Nicoll et al., 1999; Crocker and Varki, 2001).
Neutrophils respond to tissue injury within minutes by sensing necrotic cell debris and damage-associated molecular patterns (DAMPs) (Wang, 2018). Initial contact by patrolling neutrophils results in their death by apoptosis and recruitment of a neutrophil swarm (Kienle and Lammermann, 2016). Neutrophil swarming often leads to collateral damage, although cloaking of the damage site by tissue resident macrophage may help to limit the inflammatory cascade (Kienle and Lammermann, 2016; Uderhardt et al., 2019). Neutrophil accumulation requires IL-1 and TNF receptor signaling, probably via IL-1β and TNFα released from resident cells early after injury (Nadeau et al., 2011). Neutrophil depletion experiments confirm their importance in the subsequent development of hyperalgesia after nerve injury (Perkins and Tracey, 2000; Nadeau et al., 2011). Blockade of CXCL1 cytokine signaling led to a reduction in neutrophil recruitment, and also reduced mechanical hyperalgesia in a model of post-surgical pain (Carreira et al., 2013) although the source of CXCL1 in the nerve and DRG after injury remains unclear (Silva et al., 2017).
Although traditionally known for amplifying the inflammatory response to tissue injury, neutrophils also contribute to tissue repair by the release of growth factors, and polarizing phagocytic macrophages to release anti-inflammatory cytokines such as TGFβ and IL-10 (Wang, 2018). Neutrophils appear to be critical for regeneration of CNS axons, such as after optic nerve crush (Kurimoto et al., 2013), although in the peripheral nervous system no significant deficits in recovery from sciatic nerve injury were observed after neutrophil depletion (Nadeau et al., 2011). More recent evidence suggests that in the absence of infiltrating monocyte-derived macrophages, neutrophils also perform a significant phagocytic role in the injured peripheral nerve (Lindborg et al., 2017).
Neutrophils can release reactive oxygen species (ROS) in a metabolic event known as a respiratory burst, or release of neutrophil extracellular traps (NETs) in a cell death processes known as NETosis. These cytotoxic mechanisms are typically targeted at invading pathogens but may be dysregulated in immune-related disease (Papayannopoulos, 2018). A novel form of cytotoxicity enacted by neutrophils has recently been described. Clearance of antibody-coated or ‘opsonized’ cancer cells, termed ‘trogoptosis,’ results from target cell membrane disruption leading to necrotic death (Matlung et al., 2018). Opsonization of peripheral nerve components such as myelin may occur after injury either by coating with IgG (Vargas et al., 2010) or complement C3 (Bruck and Friede, 1991). The C3 receptor (also known as CD11b/CD18) is expressed by NK cells, macrophages, monocytes and granulocytes (Arnaout, 1990) as well as neutrophils, where it has been shown to be critical for immune synapse formation and ADCC (van Spriel et al., 2001; Figure 2). Thus the same processes known to mediate phagocytosis of cellular debris in the injured nerve may serve the dual purpose of engaging cell death pathways by infiltrating immune cells.
Schwann cells, which form the myelinating units of the peripheral nerve, respond dynamically within minutes of injury (Wong et al., 2017). Schwann cells collaborate with macrophages to phagocytose degenerated myelin after nerve injury utilizing both Tyro3-Axl-Mer (TAM) receptor-mediated and autophagic mechanisms (Brosius Lutz et al., 2017). Recent evidence suggests that Schwann cells may themselves be early participants in peripheral axon degeneration (Catenaccio et al., 2017), and delaying dedifferentiation of Schwann cells prolongs the survival of axons after crush injury (Catenaccio et al., 2017). The presence of newly discovered Schwann cells adjacent to free nerve endings in the skin are ideally placed to mediate in the dying back of nerve terminals that occurs in multiple peripheral neuropathies (Abdo et al., 2019). However, the role of Schwann cells in triggering Wallerian degeneration remains unclear.
Peripheral nerve injury recruits CD8+ T cells to the injured sciatic nerve (Hu et al., 2007; Bali and Kuner, 2017) where expression of antigen-presenting MHC class I molecules are increased (Bombeiro et al., 2016). Although sensory neurons express IFN-γ receptors (typically required for MHC I induction) they fail to upregulate MHC class I in response to IFN-γ signaling (Turnley et al., 2002). Correspondingly, MHC class I expression in axons of the injured nerve is low (Bombeiro et al., 2016). Therefore, the mechanism of CD8+ T cell activation by MHC class I induction and antigen presentation after acute nerve injury is unclear. In vitro, antigen-specific CD8+ T cells are known to cause axonal injury (Meuth et al., 2009; Sauer et al., 2013). Lentivirus infection of sensory neurons leads to damage by CD8+ T cells via CD40-CD154 mediated signaling (Zhu et al., 2006). Conversely, trigeminal ganglion neurons infected with herpes simplex virus type I are resistant to apoptosis induced by CD8+ T cells (Knickelbein et al., 2008), suggesting that the mechanisms controlling T cell mediated neurodegeneration are context dependent.
CD8+ T cells (as well as γδ T cells) express the receptor NKG2D and engagement with ligands such as RAE1 augments CD8+ T cell function, but only after prior stimulation by antigen-specific T cell receptors (TCRs) (Lanier, 2015), suggesting that NK cells are an important responder to ‘stressed-self’ in the non-immunized state. NK and CD8+ T cells exhibit mutually antagonistic responses to IL-2 immune stimulation (Alvarez et al., 2014) meaning that despite their overlapping cytotoxic roles one or other population will tend to dominate in any given immune response. An example of this is the aberrant expression of RAE1 in pulmonary epithelial cells, which has previously been shown to contribute to chronic obstructive pulmonary disease (COPD) via cytotoxic lymphocytes in an NK but not T-cell dependent manner (Borchers et al., 2009). Similarly, in the case of peripheral nerve injury NK cells are sufficient for degeneration of axons after IL-2/anti-IL-2 antibody complex stimulation (Davies et al., 2019) despite the simultaneous expansion of CD8+ T cells (Boyman et al., 2006). However, additional roles for CD8+ T cells within the nerve post-injury cannot be ruled out. Interestingly, resolution of neuropathic pain in mouse models of chemotherapy-induced neuropathy requires CD8+ T cells (Krukowski et al., 2016; Laumet et al., 2019a); in these studies the authors propose an indirect involvement of CD8+ T cells in suppressing DRG neuron activity by upregulation of IL-10 signaling (Krukowski et al., 2016). T cells are frequently identified in cellular and genetic analysis of peripheral neuropathies (Moalem et al., 2004; Costigan et al., 2009a; Vicuna et al., 2015; Cobos et al., 2018) and there is mounting evidence of their involvement in numerous pain states (Laumet et al., 2019b). Whether such an association between pain and T-cells require cytotoxic effector functions remains to be determined.
Acute pain (self-administered painful electrical stimulation) transiently increases functional NK cell activity as well as the proportion of NK cells in peripheral blood (Greisen et al., 1999); this effect was abolished by application of a local anesthetic to the skin, suggesting a neurogenic activation of NK cells (Greisen et al., 1999). NK cell function can also be stimulated by electro-acupuncture in rats (Kim et al., 2005). Peripheral nerve injury increased blood NK cytotoxicity in the 1st week after injury in mice (Kang et al., 2007), whereas splenic NK function assessed 3 weeks after nerve ligation was reduced (Sunagawa et al., 2000) suggesting a window of time after injury in which NK function is active followed by either recovery or exhaustion.
Gene ontology analysis of differentially expressed genes (DEG) shows significant upregulation of NK cell cytotoxicity-related genes after peripheral nerve injury (Costigan et al., 2009a) and during myelination in vitro (Wu, 2018). Cell senescence in the aging vertebral disc, a risk factor for low back pain, has also recently been associated with a differential expression of NK cell mediated cytotoxicity-related genes (Liu et al., 2018). Few studies have directly analyzed NK cells in pain conditions in humans. Low back pain patients presented with a lower percentage of NK cells in peripheral blood than asymptomatic subjects (Brennan et al., 1994), while recent studies in a similar cohort of chronic lower back pain patients identified an increase in anti-inflammatory T regulatory (Treg) cells (Luchting et al., 2014, 2015). In the context of tumor immunotherapy, the efficiency of NK cell cytotoxicity benefits from a simultaneous suppression of Treg cell function (Ni et al., 2012; Littwitz-Salomon et al., 2015). Similarly an imbalance in NK and Treg cells leading to loss of NK cell function by the release of immuno-suppressive cytokine such as TGF-β may contribute to low back pain (Luchting et al., 2014, 2015). On the other hand, treatment of radicular pain patients with pulsed radiofrequency to the DRG, which led to improved pain outcomes, reduced NK cell frequency in cerebrospinal fluid (CSF) (Das et al., 2018), although peripheral blood counts were not reported.
A number of studies have used mass cytometry to assess immune cell subsets, including NK cells, in patients after surgery or trauma. In patients undergoing hip arthroplasty for non-traumatic osteoarthritis NK cells increased 1.6-fold in the blood within an hour of surgical trauma, followed by contraction at 24 and 72 h (Gaudilliere et al., 2014). In a study of severe trauma patients, NK cells expressing the transcription factor T-bet peaked in the blood 1 day after injury suggesting the recruitment of immature NK cells from the bone marrow (Seshadri et al., 2017). Traumatic brain injury resulted in a higher frequency of inhibitory receptor (KIR2D+NKG2A+) expressing NK cells in the circulation compared to healthy or major surgery controls, which interestingly correlated with a loss of HLA class I in monocytes and impaired cytotoxicity to missing-self (Roquilly et al., 2017). While these studies suggest a significant alterations of NK cell function in the aftermath of injury, including to the central nervous system, the NK cell phenotype has yet to be explored in patients with defined injury to a peripheral nerve.
Recent studies with more broadly defined chronic pain conditions failed to find an association between NK cell frequencies and pain groups. The percentage of CD56brightCD16+ cells were slightly increased in patients with broadly defined severe chronic pain, however, generalized NK cell cytotoxic activity and the frequency of the major NK cell subsets were not significantly different (Yoon et al., 2018). In a study of 14 patients with complex regional pain syndrome (CRPS) and 14 controls, Russo and colleagues used mass cytometry to identify an expansion of both CD4+ and CD8+ memory T lymphocytes in patients compared to controls (Russo et al., 2019). NK cells (defined as CD56+ CD19–CD3–) were more than 50% elevated in CRPS patient blood but this did not reach significance. Thus results from human studies appear to show regulation of NK cells in chronic pain conditions, though the correlation between NK cell function and pain outcomes remains unclear. The inability to assign function from cell numbers alone is likely due to positive and negative effector functions and diverse cell subsets (Eberl et al., 2015). Appreciation of the differential NK cell subsets that are present in the blood, splenic compartments and injured nerves of mice and humans (Crinier et al., 2018) will aid further deep phenotyping to build a clearer picture of NK cell function in chronic pain.
There is no difference observed in the numbers of nerve-infiltrating NK cells in rats with mechanical allodynia after injury compared to those without (Cui et al., 2000). Furthermore, mice lacking NK cells after chronic depletion maintain a robust development of thermal and mechanical allodynia (Davies et al., 2019) in a well characterized model of neuropathic nerve injury (Kim and Chung, 1992). Therefore, NK cells do not appear to be necessary for the development of neuropathic pain. Instead, various factors may affect the pain-resolving potential of NK cells in the period after nerve injury (Davies et al., 2019). For example, long-term over-expression of NKG2D ligands such as Raet1 is known to cause a down-regulation of NK cell receptor expression and loss of function (Morvan et al., 2017). Additionally, activating ligands may be shed from damaged nerves, preventing their removal by NK cells. For example, cleavage of NKG2D ligands on tumor cells by disintegrins and metalloproteinases (ADAMs) (Waldhauer et al., 2008) helps tumors escape immuno-surveillance by NK cells (Ferrari de Andrade et al., 2018). As well as functions related to extracellular matrix remodeling (Yong, 2005), matrix metalloproteinases (MMPs) may affect the neuro-immune balance through the regulation of immune ligands on neurons and other cells of the nervous system. Specifically, MMP-9 and MMP-14 are capable of the cleavage of NKG2D ligands (Liu et al., 2010; Sun et al., 2011) are upregulated after nerve injury and contribute to neuropathic pain in preclinical models (Kawasaki et al., 2008; Tonello et al., 2019). Certain MMP gene variants have also been associated with the risk of low back pain in patients (Tegeder and Lotsch, 2009; Bjorland et al., 2017).
Natural killer cell function after nerve injury may also be compromised by morphine, which suppresses both spontaneous and cytokine-stimulated NK cell functions (Yeager et al., 1995). Immune suppression in response to chronic pain and neurological stress has long been known to affect tumor immunity and mortality (Liebeskind, 1991). Chronic stress paradigms, including surgical stress, reduces NK cell cytotoxic function in rodents (Aarstad et al., 1983; Pollock et al., 1987; Ben-Eliyahu et al., 1999) in an opioid-dependent manner (Shavit et al., 1984). Immune suppression after traumatic injury in patients, shown to be related to surgical stress (Blazar et al., 1986; Pollock et al., 1991), could have an important bearing on pain outcomes by reducing the anti-neuropathic function of cytotoxic effectors.
Macrophages have a recognized involvement in pain hypersensitivity with multiple subtypes persisting within the nerve many months after injury (Liang et al., 2019). Depletion of peripheral monocytes/macrophages in mice by systemic treatment with clodronate liposomes reduced degeneration of both myelinated and unmyelinated fibers following nerve injury (Liu et al., 2000) and these mice also showed a reduced incidence of pain (Liu et al., 2000; Cobos et al., 2018). Incidence of neuropathic mechanical allodynia was also delayed by macrophage depletion in a rat model of diabetes (Mert et al., 2009). Activation of the angiotensin type 2 receptor (AT2R) in macrophages can drive neuropathic pain (Shepherd et al., 2018b). The cellular functions of macrophages within the injured nerve may include the production of reactive oxygen species (ROS) leading to nociceptor sensation (Shepherd et al., 2018a) as well as inflammatory cytokine production (Arango Duque and Descoteaux, 2014). Inflammatory macrophages (M1) within the nerve are also implicated in pain associated with diabetic neuropathy (Saika et al., 2019). M1 skewed macrophages within and around the damaged nerve likely precipitate neuropathic symptoms (Kiguchi et al., 2017; Saika et al., 2019) and conversely, macrophage polarization to an anti-inflammatory ‘M2’ phenotype can attenuate neuropathy-induced mechanical hypersensitivity via the production of opioids (Kiguchi et al., 2015; Pannell et al., 2016). Patients with complex regional pain syndrome show an elevation of CD14+ CD16+ monocytes in their blood (Ritz et al., 2011) suggesting that altered macrophage function at the systemic level is functionally related to nociplastic pain, i.e., pain that arises from altered nociceptive function in the absence of obvious precipitating cause (Aydede and Shriver, 2018). Transcriptomic analysis of human DRG from neuropathic pain sufferers suggests that sex differences in pain susceptibility may be driven by differential gene expression in macrophages (North et al., 2019). Interestingly, the transcriptomic profile of macrophages in mice differs between nerve and DRG compartments (Liang et al., 2019), and recent experiments using tissue-targeted macrophage depletion suggest that macrophages in DRG but not the sciatic nerve are required for mechanical allodynia after nerve injury (Yu et al., 2020). The divergence of neuropathic pain development in young and adult animals may also be a function of differential functional interactions between macrophage and cells within the DRG (Vega-Avelaira et al., 2009) or microglia/T cell signaling in the CNS (Sorge et al., 2015).
In mice, activated macrophages express NKG2D (Zhou et al., 2012) suggesting that in addition to NK cells, macrophages may interact with damaged axons via the expression of the ligand RAE1 after traumatic injury (Davies et al., 2019; Figure 2). Macrophages and NK cells communicate through a wide variety of cell contact cues and soluble factors (Michel et al., 2012). Functional influence is two-way, with macrophages able to either activate or inhibit NK cells (Nedvetzki et al., 2007; Krneta et al., 2017), and NK cells capable of killing over-stimulated macrophages (Nedvetzki et al., 2007). Thus activated NK cells in the immune milieu of the injured nerve may additionally help resolve neuropathic pain by eliminating M1 macrophages (Nedvetzki et al., 2007) as well as damaged axons (Davies et al., 2019).
Endometriosis is another example of a disease that may be influenced by a functional interaction between NK cells and macrophages. Endometriosis is the ectopic growth of endometrial cells forming lesions within the peritoneal cavity surrounding the uterus (Symons et al., 2018) leading to chronic pelvic pain and infertility (Thiruchelvam et al., 2015). A higher density of nerve fibers surrounded by macrophages are observed in endometrial lesions relative to surrounding tissue (Tran et al., 2009) and growth factor production by these cells has recently been proposed as a key driver of neurogenesis and nerve fiber sensitization in these lesions (Forster et al., 2019). Although inflammatory cytokines such as TNFα are elevated in the peritoneal cavity in painful endometriosis (Scholl et al., 2009), TNFα antagonists are yet to show clinical efficacy (Lu et al., 2013). Endometriosis pathophysiology is thought to involve a failure of NK cells to eliminate endometrial lesions from the peritoneal cavity (Sikora et al., 2011). Peripheral blood and peritoneal NK cells show reduced cytotoxic function in women with endometriosis (Garzetti et al., 1993; Thiruchelvam et al., 2015), and downregulation of NK cell cytotoxic capability via inhibitory interactions with macrophages (Yang et al., 2017). Disease severity is correlated with a loss of NK cell cytotoxicity as well as a resistance of the endometrial lesions to NK mediated attack (Oosterlynck et al., 1991). A failure of NK cell cytotoxicity to target aberrantly sprouting peripheral nerves in the presence of activated macrophages could therefore contribute to the symptoms of this disease. Early clinical trials of recombinant IL-2 treatment in endometriosis showed lower recurrence of pain symptoms (Acien et al., 2003), suggesting the potential benefit of the anti-neuropathic function NK cells (Davies et al., 2019). A registered trial aimed at assessing the potential use of an inhibitor of programed cell death 1 (PD-1) could lead to an immunotherapeutic treatment for endometriosis1.
Interactions between NK cells and peripheral nerves precipitating neuropathy may also occur as a result of transformation of NK cells themselves. A case study in the 1990’s first reported an NK cell-specific leukemia with associated peripheral neuropathy presenting as tingling hands and back pain with progressive sensory loss in the extremities (Bobker and Deloughery, 1993); sural nerve biopsy revealed heavy lymphocytic infiltration although NK cell identity could not be confirmed (Bobker and Deloughery, 1993). Later, NK cell lymphoproliferative disease (NK-LPD) was reported as associated with peripheral neuropathy (Leitenberg et al., 1995), with steroid treatment improving neurological symptoms concurrent with a reduction in NK cell counts (Leitenberg et al., 1995). Prolonged F-wave latencies were consistent with a demyelinating sensorimotor polyneuropathy and sural nerve biopsies revealed an inflammatory neuropathy with myelin loss (Leitenberg et al., 1995). Subsequent cases of peripheral neuropathy coincidental to the NK lymphocytosis have been reported (Lackowski et al., 1998; de Boer et al., 1999; Noguchi et al., 2005). The effectiveness of steroids (Leitenberg et al., 1995; Sano et al., 2017) or an anti-CD52 antibody targeting a broad population of lymphocytes including NK cells (Chee et al., 2009) for the resolution of the peripheral lesions suggests the involvement of cellular immunity in the neurological aspect of the disease (Leitenberg et al., 1995). Nerve biopsies within these patients revealed lymphocytic infiltrates (Bobker and Deloughery, 1993; Rabbani et al., 1999) and labeling of granzyme and perforin suggest these cells possess cytotoxic activity (Noguchi et al., 2005). A recent observation of CD3– CD56+ cells in cutaneous nerve infiltration secondary to NK-LPD appears to confirm their NK cell identity (Ni Mhaolcatha et al., 2019). A longitudinal study of a severe polyneuropathy was additionally characterized by elevated serum levels of IL-2 and TNFα, as well as increased intrinsic NK cell cytotoxicity (Wex et al., 2005).
Overall, 3% of chronic NK-LPD cases appear to go on to develop peripheral neuropathy (Poullot et al., 2014). The reason for the development of neuropathy with NK cell proliferation in a minority of patients is currently unknown. NK cell leukemia is thought to be caused by a dysregulation of survival signals, including gain-of-function mutations of the transcription factor STAT3, leading to cell proliferation (Lamy et al., 2017). NK cells from lymphoproliferative disorders possess a skewed pattern of KIR gene expression (Hoffmann et al., 2000; Epling-Burnette et al., 2004) suggesting either a propensity for over-activation or loss of constitutive inhibition. Prior infection with Epstein-Barr virus (EBV) is also implicated in the pathogenesis of an aggressive form of NK-LPD (Hart et al., 1992; Hirose et al., 1997) and is itself a known risk-factor for peripheral neuropathy (Brizzi and Lyons, 2014). Prior transformation of the neurons or supporting cells of the nerve may therefore be a pre-requisite for target by NK cells.
Natural killer cells play an important role in the immune response to infections from EBV, cytomegalovirus (CMV), varicella zoster virus (VZV), and herpes simplex virus (HSV) (Orange, 2013), and each of these can precipitate peripheral neuropathy (Shin and Simpson, 2013; Brizzi and Lyons, 2014). GBS is a rapid-onset autoimmune peripheral neuropathy often preceded by bacterial or viral infection (Hartung, 1999). The most commonly identified prodromal infections are C. jejuni (32%), CMV (13%), EBV (10%) and M. pneumoniae (5%) (Jacobs et al., 1998). IL-2 is elevated in the serum of GBS patients, with levels returning to normal during the recovery phase (Hartung et al., 1991). However, NK cell activity is lower than baseline within a week of the onset of neuropathic symptoms in GBS (Yoshii and Shinohara, 1998), suggesting exhaustion or loss of NK cell function could exacerbate coincidental neuropathy. Analysis of KIR receptor and human leukocyte antigen (HLA) ligand gene combinations in GBS patients suggests a higher occurrence of inhibitory pairs that would in theory suppress NK cell function (Blum et al., 2014). Thus impairment of an otherwise beneficial innate immune response may contribute to the pathogenesis of GBS. This could explain why treatments designed to suppress the cellular immune response, such as corticosteroids, do not significantly reduce and may actually amplify disease severity (Hughes and van Doorn, 2012).
Chronic inflammatory demyelinating polyneuropathy (CIDP) is an inflammatory neuropathy typified by myelin loss and/or axonal damage of bilateral nerves of the extremities. The disease is characterized by a growing list of auto-antibodies found in patient sera that target peripheral nerve components (Fehmi et al., 2018). CIDP autoantibodies may exert a pathogenic effect by directly interrupting the axonal-myelination structure, or complement-dependent cytotoxicity (Rinaldi and Bennett, 2014). There is also an appreciation of cellular immune component to CIDP (Mathey et al., 2015). Both macrophage phagocytosis (Koike et al., 2018) and clonal expansion of auto-reactive T cells are thought to play a role in the disease, although direct evidence for a myelin antigen-specific auto-reactive T cell population is currently lacking (Schneider-Hohendorf et al., 2012). Reduced cellular cytotoxicity could also be a risk factor in CIDP. For example, missense loss-of-function mutations in perforin have been identified in some CIDP patients (Buttini et al., 2015), and a study of 22 CIDP patients and 22 healthy controls showed the proportion of CD3– CD56+ NK cells was lower in the blood of patients (Sanvito et al., 2009). CSF from inflammatory neuropathy patients contains elevated NK cells in association with GBS, whereas CD8+ T and CD3+ NKT cells levels were increased in CIDP patients (Heming et al., 2019). These findings suggest that heterogeneity in the cytotoxic cellular immune response could play role in differentiating the acute and chronic forms of inflammatory peripheral neuropathy.
Neurobiologists are increasingly aware of the critical role played by immune function and/or dysfunction in pain syndromes (Hore and Denk, 2019). Despite comprising a relatively minor proportion of peripheral blood cells, NK cells appear to play a unique role in the immune response to peripheral nerve activation and injury, enabled by the remarkable specificity of their innate receptor repertoire and the neuro-immune synapse. One beneficial function that these cells would be particularly suited to is the selective pruning of damaged or aberrantly sprouting peripheral axons following injury or disease. As the archetypal cytotoxic immune cell, NK cells have shone a light on the contribution of other immune cells, including macrophages, neutrophils and CD8+ T cells, whose cytotoxic potential in the response to nerve injury is yet to be fully elucidated. A number of chronic pain conditions, as well as pain risk-factors including chronic stress and opioid use, are co-morbid with loss of NK cell function, suggesting NK cell modulation as a potential therapy. Developments in the fields of cancer and immunotherapy are already providing tools to tune NK cells and therefore shift the immune response within the damaged nerve from maladaptive to adaptive. However, the potential role of cellular cytotoxicity in the resolution of neuropathic pain must be weighed against cytotoxic mechanisms themselves precipitating injury and therefore augmenting chronic pain. Thus, maintaining a homeostatic balance – a central tenant of immunology – remains the priority for immune function, including cellular cytotoxicity by NK and other cells, within the peripheral nerve.
AD wrote the draft and designed the figures. All authors contributed to writing, editing, and reviewing the manuscript.
This work was supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC) United Kingdom (AD), Medical Research Council United Kingdom (MR/P008399/1) (SR), and the National Research Foundation of Korea grant (NRF-2018R1A5A2024418) (SO).
AD, MC, and SO are named on a patent application for the use of immune therapy in the treatment of nerve injury (PCT/KR2019/00806).
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past co-authorship with one of the authors MC.
The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health, United Kingdom.
Aarstad, H. J., Gaudernack, G., and Seljelid, R. (1983). Stress causes reduced natural killer activity in mice. Scand. J. Immunol. 18, 461–464. doi: 10.1111/j.1365-3083.1983.tb00878.x
Abdo, H., Calvo-Enrique, L., Lopez, J. M., Song, J., Zhang, M.-D., Usoskin, D., et al. (2019). Specialized cutaneous schwann cells initiate pain sensation. Science 365:695. doi: 10.1126/science.aax6452
Acien, P., Quereda, F. J., Gomez-Torres, M. J., Bermejo, R., and Gutierrez, M. (2003). GnRH analogues, transvaginal ultrasound-guided drainage and intracystic injection of recombinant interleukin-2 in the treatment of endometriosis. Gynecol. Obstet. Invest. 55, 96–104. doi: 10.1159/000070181
Adrain, C., Murphy, B. M., and Martin, S. J. (2005). Molecular ordering of the caspase activation cascade initiated by the cytotoxic T lymphocyte/natural killer (CTL/NK) protease granzyme B. J. Biol. Chem. 280, 4663–4673. doi: 10.1074/jbc.m410915200
Alvarez, M., Bouchlaka, M. N., Sckisel, G. D., Sungur, C. M., Chen, M., and Murphy, W. J. (2014). Increased antitumor effects using IL-2 with anti-TGF-beta reveals competition between mouse NK and CD8 T cells. J. Immunol. 193, 1709–1716. doi: 10.4049/jimmunol.1400034
Anghelescu, D. L., Goldberg, J. L., Faughnan, L. G., Wu, J., Mao, S., Furman, W. L., et al. (2015). Comparison of pain outcomes between two anti-GD2 antibodies in patients with neuroblastoma. Pediatr. Blood Cancer 62, 224–228. doi: 10.1002/pbc.25280
Araki, T., Sasaki, Y., and Milbrandt, J. (2004). Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010–1013. doi: 10.1126/science.1098014
Arango Duque, G., and Descoteaux, A. (2014). Macrophage cytokines: involvement in immunity and infectious diseases. Front. Immunol. 5:491. doi: 10.3389/fimmu.2014.00491
Arnaout, M. A. (1990). Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood 75, 1037–1050. doi: 10.1182/blood.v75.5.1037.bloodjournal7551037
Aydede, M., and Shriver, A. (2018). Recently introduced definition of “nociplastic pain” by the international association for the study of pain needs better formulation. PAIN 159, 1176–1177. doi: 10.1097/j.pain.0000000000001184
Backstrom, E., Chambers, B. J., Ho, E. L., Naidenko, O. V., Mariotti, R., Fremont, D. H., et al. (2003). Natural killer cell-mediated lysis of dorsal root ganglia neurons via RAE1/NKG2D interactions. Eur. J. Immunol. 33, 92–100. doi: 10.1002/immu.200390012
Backstrom, E., Chambers, B. J., Kristensson, K., and Ljunggren, H. G. (2000). Direct NK cell-mediated lysis of syngenic dorsal root ganglia neurons in vitro. J. Immunol. 165, 4895–4900. doi: 10.4049/jimmunol.165.9.4895
Backstrom, E., Ljunggren, H. G., and Kristensson, K. (2007). NK cell-mediated destruction of influenza A virus-infected peripheral but not central neurones. Scand. J. Immunol. 65, 353–361. doi: 10.1111/j.1365-3083.2007.01912.x
Bali, K. K., and Kuner, R. (2017). Therapeutic potential for leukocyte elastase in chronic pain states harboring a neuropathic component. Pain 158, 2243–2258. doi: 10.1097/j.pain.0000000000001032
Bates, D., and Eastman, A. (2017). Microtubule destabilising agents: far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 83, 255–268. doi: 10.1111/bcp.13126
Ben-Eliyahu, S., Page, G. G., Yirmiya, R., and Shakhar, G. (1999). Evidence that stress and surgical interventions promote tumor development by suppressing natural killer cell activity. Int. J. Cancer 80, 880–888. doi: 10.1002/(sici)1097-0215(19990315)80:6<880::aid-ijc14>3.0.co;2-y
Bennett, G. J., and Xie, Y. K. (1988). A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33, 87–107. doi: 10.1016/0304-3959(88)90209-6
Bjorland, S., Roe, C., Moen, A., Schistad, E., Mahmood, A., and Gjerstad, J. (2017). Genetic predictors of recovery in low back and lumbar radicular pain. Pain 158, 1456–1460. doi: 10.1097/j.pain.0000000000000934
Blazar, B. A., Rodrick, M. L., O’Mahony, J. B., Wood, J. J., Bessey, P. Q., Wilmore, D. W., et al. (1986). Suppression of natural killer-cell function in humans following thermal and traumatic injury. J. Clin. Immunol. 6, 26–36. doi: 10.1007/bf00915361
Blum, S., Csurhes, P., Reddel, S., Spies, J., and McCombe, P. (2014). Killer immunoglobulin-like receptor and their HLA ligands in guillain-barre syndrome. J. Neuroimmunol. 267, 92–96. doi: 10.1016/j.jneuroim.2013.12.007
Bobker, D. H., and Deloughery, T. G. (1993). Natural killer cell leukemia presenting with a peripheral neuropathy. Neurology 43, 1853–1854.
Bombeiro, A. L., Thome, R., Oliveira Nunes, S. L., Monteiro Moreira, B., Verinaud, L., and Oliveira, A. L. (2016). MHC-I and PirB upregulation in the central and peripheral nervous system following sciatic nerve injury. PLoS One 11:e0161463. doi: 10.1371/journal.pone.0161463
Borchers, M. T., Wesselkamper, S. C., Curull, V., Ramirez-Sarmiento, A., Sanchez-Font, A., Garcia-Aymerich, J., et al. (2009). Sustained CTL activation by murine pulmonary epithelial cells promotes the development of COPD-like disease. J. Clin. Invest. 119, 636–649. doi: 10.1172/JCI34462
Bosch, X., Saiz, A., Ramos-Casals, M., and Group, B. S. (2011). Monoclonal antibody therapy-associated neurological disorders. Nat. Rev. Neurol. 7, 165–172. doi: 10.1038/nrneurol.2011.1
Boyman, O., Kovar, M., Rubinstein, M. P., Surh, C. D., and Sprent, J. (2006). Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 311, 1924–1927. doi: 10.1126/science.1122927
Brennan, P. C., Graham, M. A., Triano, J. J., Hondras, M. A., and Anderson, R. J. (1994). Lymphocyte profiles in patients with chronic low back pain enrolled in a clinical trial. J. Manip. Physiol. Ther. 17, 219–227.
Brizzi, K. T., and Lyons, J. L. (2014). Peripheral nervous system manifestations of infectious diseases. Neurohospitalist 4, 230–240. doi: 10.1177/1941874414535215
Brosius Lutz, A., Chung, W. S., Sloan, S. A., Carson, G. A., Zhou, L., Lovelett, E., et al. (2017). Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc. Natl. Acad. Sci. U.S.A. 114, E8072–E8080. doi: 10.1073/pnas.1710566114
Bruck, W., and Friede, R. L. (1991). The role of complement in myelin phagocytosis during PNS wallerian degeneration. J. Neurol. Sci. 103, 182–187. doi: 10.1016/0022-510x(91)90162-z
Buttini, S., Cappellano, G., Ripellino, P., Briani, C., Cocito, D., Osio, M., et al. (2015). Variations of the perforin gene in patients with chronic inflammatory demyelinating polyradiculoneuropathy. Genes Immun. 16, 99–102. doi: 10.1038/gene.2014.59
Calvo, M., Dawes, J. M., and Bennett, D. L. (2012). The role of the immune system in the generation of neuropathic pain. Lancet Neurol. 11, 629–642. doi: 10.1016/S1474-4422(12)70134-5
Carayannopoulos, L. N., Naidenko, O. V., Fremont, D. H., and Yokoyama, W. M. (2002). Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J. Immunol. 169, 4079–4083. doi: 10.4049/jimmunol.169.8.4079
Carreira, E. U., Carregaro, V., Teixeira, M. M., Moriconi, A., Aramini, A., and Verri, W. A. Jr., et al. (2013). Neutrophils recruited by CXCR1/2 signalling mediate post-incisional pain. Eur. J. Pain 17, 654–663. doi: 10.1002/j.1532-2149.2012.00240.x
Catenaccio, A., Llavero Hurtado, M., Diaz, P., Lamont, D. J., Wishart, T. M., and Court, F. A. (2017). Molecular analysis of axonal-intrinsic and glial-associated co-regulation of axon degeneration. Cell Death Dis. 8:e3166. doi: 10.1038/cddis.2017.489
Cerwenka, A., Bakker, A. B., McClanahan, T., Wagner, J., Wu, J., Phillips, J. H., et al. (2000). Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity 12, 721–727. doi: 10.1016/s1074-7613(00)80222-8
Chaitanya, G. V., Eeka, P., Munker, R., Alexander, J. S., and Babu, P. P. (2011). Role of cytotoxic protease granzyme-b in neuronal degeneration during human stroke. Brain Pathol. 21, 16–30. doi: 10.1111/j.1750-3639.2010.00426.x
Chee, C. E., Warrington, K. J., and Tefferi, A. (2009). Chronic natural killer-cell lymphocytosis successfully treated with alemtuzumab. Blood 114, 3500–3502. doi: 10.1182/blood-2009-07-233403
Chen, P., Piao, X., and Bonaldo, P. (2015). Role of macrophages in Wallerian degeneration and axonal regeneration after peripheral nerve injury. Acta Neuropathol. 130, 605–618. doi: 10.1007/s00401-015-1482-4
Cheung, N. K., and Dyer, M. A. (2013). Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 13, 397–411. doi: 10.1038/nrc3526
Cheung, N. K., Lazarus, H., Miraldi, F. D., Abramowsky, C. R., Kallick, S., Saarinen, U. M., et al. (1987). Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma. J. Clin. Oncol. 5, 1430–1440. doi: 10.1200/jco.1987.5.9.1430
Chien, Y. H., Meyer, C., and Bonneville, M. (2014). gammadelta T cells: first line of defense and beyond. Annu. Rev. Immunol. 32, 121–155. doi: 10.1146/annurev-immunol-032713-120216
Chiossone, L., Dumas, P. Y., Vienne, M., and Vivier, E. (2018). Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 18, 671–688. doi: 10.1038/s41577-018-0061-z
Cho, Y., and Cavalli, V. (2014). HDAC signaling in neuronal development and axon regeneration. Curr. Opin. Neurobiol 27, 118–126. doi: 10.1016/j.conb.2014.03.008
Cho, Y., Sloutsky, R., Naegle, K. M., and Cavalli, V. (2013). Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 155, 894–908. doi: 10.1016/j.cell.2013.10.004
Cifaldi, L., Locatelli, F., Marasco, E., Moretta, L., and Pistoia, V. (2017). Boosting natural killer cell-based immunotherapy with anticancer drugs: a perspective. Trends Mol. Med. 23, 1156–1175. doi: 10.1016/j.molmed.2017.10.002
Cobos, E. J., Nickerson, C. A., Gao, F., Chandran, V., Bravo-Caparros, I., Gonzalez-Cano, R., et al. (2018). Mechanistic differences in neuropathic pain modalities revealed by correlating behavior with global expression profiling. Cell Rep. 22, 1301–1312. doi: 10.1016/j.celrep.2018.01.006
Conforti, L., Gilley, J., and Coleman, M. P. (2014). Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 15, 394–409. doi: 10.1038/nrn3680
Corcoran, J., Shroot, B., Pizzey, J., and Maden, M. (2000). The role of retinoic acid receptors in neurite outgrowth from different populations of embryonic mouse dorsal root ganglia. J. Cell Sci. 113(Pt 14), 2567–2574.
Costigan, M., Befort, K., Karchewski, L., Griffin, R. S., D’Urso, D., Allchorne, A., et al. (2002). Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci. 3:16. doi: 10.1186/1471-2202-3-16
Costigan, M., Mannion, R. J., Kendall, G., Lewis, S. E., Campagna, J. A., Coggeshall, R. E., et al. (1998). Heat shock protein 27: developmental regulation and expression after peripheral nerve injury. J. Neurosci. 18, 5891–5900. doi: 10.1523/jneurosci.18-15-05891.1998
Costigan, M., Moss, A., Latremoliere, A., Johnston, C., Verma-Gandhu, M., Herbert, T. A., et al. (2009a). T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J. Neurosci. 29, 14415–14422. doi: 10.1523/JNEUROSCI.4569-09.2009
Costigan, M., Scholz, J., and Woolf, C. J. (2009b). Neuropathic pain: a maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 32, 1–32. doi: 10.1146/annurev.neuro.051508.135531
Crinier, A., Milpied, P., Escaliere, B., Piperoglou, C., Galluso, J., Balsamo, A., et al. (2018). High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity 49, 971–986.e5. doi: 10.1016/j.immuni.2018.09.009
Crocker, P. R., and Varki, A. (2001). Siglecs in the immune system. Immunology 103, 137–145. doi: 10.1046/j.0019-2805.2001.01241.x
Cui, J. G., Holmin, S., Mathiesen, T., Meyerson, B. A., and Linderoth, B. (2000). Possible role of inflammatory mediators in tactile hypersensitivity in rat models of mononeuropathy. Pain 88, 239–248. doi: 10.1016/s0304-3959(00)00331-6
Cusack, C. L., Swahari, V., Hampton Henley, W., Michael Ramsey, J., and Deshmukh, M. (2013). Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat. Commun. 4:1876. doi: 10.1038/ncomms2910
Das, B., Conroy, M., Moore, D., Lysaght, J., and McCrory, C. (2018). Human dorsal root ganglion pulsed radiofrequency treatment modulates cerebrospinal fluid lymphocytes and neuroinflammatory markers in chronic radicular pain. Brain Behav. Immun. 70, 157–165. doi: 10.1016/j.bbi.2018.02.010
Davies, A. J., Kim, H. W., Gonzalez-Cano, R., Choi, J., Back, S. K., Roh, S. E., et al. (2019). Natural killer cells degenerate intact sensory afferents following nerve injury. Cell 176, 716–728e.18. doi: 10.1016/j.cell.2018.12.022
Davis, D. M., Chiu, I., Fassett, M., Cohen, G. B., Mandelboim, O., and Strominger, J. L. (1999). The human natural killer cell immune synapse. Proc. Natl. Acad. Sci. U.S.A. 96, 15062–15067.
de Boer, R., Kiers, L., and Grigg, A. (1999). Natural killer cell lymphoproliferative disease associated with combined peripheral and autonomic neuropathy. J. Clin. Neurosci. 6, 61–63. doi: 10.1016/s0967-5868(99)90609-4
Decosterd, I., and Woolf, C. J. (2000). Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158. doi: 10.1016/s0304-3959(00)00276-1
Desbarats, J., Birge, R. B., Mimouni-Rongy, M., Weinstein, D. E., Palerme, J. S., and Newell, M. K. (2003). Fas engagement induces neurite growth through ERK activation and p35 upregulation. Nat. Cell Biol. 5, 118–125. doi: 10.1038/ncb916
DiAntonio, A. (2019). Axon degeneration: mechanistic insights lead to therapeutic opportunities for the prevention and treatment of peripheral neuropathy. Pain 160(Suppl. 1), S17–S22. doi: 10.1097/j.pain.0000000000001528
Diefenbach, A., Jensen, E. R., Jamieson, A. M., and Raulet, D. H. (2001). Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 413, 165–171. doi: 10.1038/35093109
Eberl, G., Colonna, M., Di Santo, J. P., and McKenzie, A. N. (2015). Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science 348:aaa6566. doi: 10.1126/science.aaa6566
Epling-Burnette, P. K., Painter, J. S., Chaurasia, P., Bai, F., Wei, S., Djeu, J. Y., et al. (2004). Dysregulated NK receptor expression in patients with lymphoproliferative disease of granular lymphocytes. Blood 103, 3431–3439. doi: 10.1182/blood-2003-02-0400
Farlie, P. G., Dringen, R., Rees, S. M., Kannourakis, G., and Bernard, O. (1995). bcl-2 transgene expression can protect neurons against developmental and induced cell death. Proc. Natl. Acad. Sci. U.S.A. 92, 4397–4401. doi: 10.1073/pnas.92.10.4397
Federico, S. M., McCarville, M. B., Shulkin, B. L., Sondel, P. M., Hank, J. A., Hutson, P., et al. (2017). A pilot trial of humanized anti-GD2 monoclonal antibody (hu14.18K322A) with chemotherapy and natural killer cells in children with recurrent/refractory neuroblastoma. Clin. Cancer Res. 23, 6441–6449. doi: 10.1158/1078-0432.CCR-17-0379
Fehmi, J., Scherer, S. S., Willison, H. J., and Rinaldi, S. (2018). Nodes, paranodes and neuropathies. J. Neurol. Neurosurg. Psychiatry 89, 61–71. doi: 10.1136/jnnp-2016-315480
Ferlazzo, G., and Moretta, L. (2014). Dendritic cell editing by natural killer cells. Crit. Rev. Oncogl. 19, 67–75. doi: 10.1615/critrevoncog.2014010827
Ferrari de Andrade, L., Tay, R. E., Pan, D., Luoma, A. M., Ito, Y., Badrinath, S., et al. (2018). Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 359, 1537–1542. doi: 10.1126/science.aao0505
Fine, J. H., Chen, P., Mesci, A., Allan, D. S., Gasser, S., Raulet, D. H., et al. (2010). Chemotherapy-induced genotoxic stress promotes sensitivity to natural killer cell cytotoxicity by enabling missing-self recognition. Cancer Res. 70, 7102–7113. doi: 10.1158/0008-5472.CAN-10-1316
Fitzgerald, M., and McKelvey, R. (2016). Nerve injury and neuropathic pain - A question of age. Exp. Neurol. 275(Pt 2), 296–302. doi: 10.1016/j.expneurol.2015.07.013
Forster, R., Sarginson, A., Velichkova, A., Hogg, C., Dorning, A., Horne, A. W., et al. (2019). Macrophage-derived insulin-like growth factor-1 is a key neurotrophic and nerve-sensitizing factor in pain associated with endometriosis. FASEB J. 33, 11210–11222. doi: 10.1096/fj.201900797R
Fukuda, Y., Li, Y., and Segal, R. A. (2017). A mechanistic understanding of axon degeneration in chemotherapy-induced peripheral neuropathy. Front. Neurosci. 11:481. doi: 10.3389/fnins.2017.00481
Gamage, K. K., Cheng, I., Park, R. E., Karim, M. S., Edamura, K., Hughes, C., et al. (2017). Death receptor 6 promotes wallerian degeneration in peripheral axons. Curr. Biol. 27, 890–896. doi: 10.1016/j.cub.2017.01.062
Garzetti, G. G., Ciavattini, A., Provinciali, M., Fabris, N., Cignitti, M., and Romanini, C. (1993). Natural killer cell activity in endometriosis: correlation between serum estradiol levels and cytotoxicity. Obstet. Gynecol. 81, 665–668.
Gaudilliere, B., Fragiadakis, G. K., Bruggner, R. V., Nicolau, M., Finck, R., Tingle, M., et al. (2014). Clinical recovery from surgery correlates with single-cell immune signatures. Sci. Transl. Med. 6:255ra131. doi: 10.1126/scitranslmed.3009701
Geisler, S., Doan, R. A., Cheng, G. C., Cetinkaya-Fisgin, A., Huang, S. X., Hoke, A., et al. (2019a). Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight 4:129920. doi: 10.1172/jci.insight.129920
Geisler, S., Doan, R. A., Strickland, A., Huang, X., Milbrandt, J., and DiAntonio, A. (2016). Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 139, 3092–3108. doi: 10.1093/brain/aww251
Geisler, S., Huang, S. X., Strickland, A., Doan, R. A., Summers, D. W., Mao, X., et al. (2019b). Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J. Exp. Med. 216, 294–303. doi: 10.1084/jem.20181040
Gerdts, J., Brace, E. J., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2015). SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 348, 453–457. doi: 10.1126/science.1258366
Gerdts, J., Summers, D. W., Milbrandt, J., and DiAntonio, A. (2016). Axon self-destruction: new links among SARM1, MAPKs, and NAD+ Metabolism. Neuron 89, 449–460. doi: 10.1016/j.neuron.2015.12.023
Gillardon, F., Wickert, H., and Zimmermann, M. (1994). Differential expression of bcl-2 and bax mRNA in axotomized dorsal root ganglia of young and adult rats. Eur. J. Neurosci. 6, 1641–1644. doi: 10.1111/j.1460-9568.1994.tb00555.x
Greene, T. T., Tokuyama, M., Knudsen, G. M., Kunz, M., Lin, J., Greninger, A. L., et al. (2016). A Herpesviral induction of RAE-1 NKG2D ligand expression occurs through release of HDAC mediated repression. eLife 5:e14749. doi: 10.7554/eLife.14749
Greisen, J., Hokland, M., Grofte, T., Hansen, P. O., Jensen, T. S., Vilstrup, H., et al. (1999). Acute pain induces an instant increase in natural killer cell cytotoxicity in humans and this response is abolished by local anaesthesia. Br. J. Anaesth. 83, 235–240. doi: 10.1093/bja/83.2.235
Guia, S., Fenis, A., Vivier, E., and Narni-Mancinelli, E. (2018). Activating and inhibitory receptors expressed on innate lymphoid cells. Semin. Immunopathol. 40, 331–341. doi: 10.1007/s00281-018-0685-x
Gul, N., Babes, L., Siegmund, K., Korthouwer, R., Bogels, M., Braster, R., et al. (2014). Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J. Clin. Invest. 124, 812–823. doi: 10.1172/JCI66776
Haile, Y., Carmine-Simmen, K., Olechowski, C., Kerr, B., Bleackley, R. C., and Giuliani, F. (2015). Granzyme B-inhibitor serpina3n induces neuroprotection in vitro and in vivo. J. Neuroinflamm. 12:157. doi: 10.1186/s12974-015-0376-7
Hajdu, S. I. (2016). Pathfinders in oncology from ancient times to the end of the middle ages. Cancer 122, 1638–1646. doi: 10.1002/cncr.29955
Hansel, T. T., Kropshofer, H., Singer, T., Mitchell, J. A., and George, A. J. T. (2010). The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 9, 325–338. doi: 10.1038/nrd3003
Hart, D. N., Baker, B. W., Inglis, M. J., Nimmo, J. C., Starling, G. C., Deacon, E., et al. (1992). Epstein-Barr viral DNA in acute large granular lymphocyte (natural killer) leukemic cells. Blood 79, 2116–2123. doi: 10.1182/blood.v79.8.2116.bloodjournal7982116
Hartung, H.-P. (1999). Infections and the guillain-barré syndrome. J. Neurol. Neurosurg. Amp. 66, 277–277.
Hartung, H. P., Reiners, K., Schmidt, B., Stoll, G., and Toyka, K. V. (1991). Serum interleukin-2 concentrations in guillain-barre syndrome and chronic idiopathic demyelinating polyradiculoneuropathy: comparison with other neurological diseases of presumed immunopathogenesis. Ann. Neurol. 30, 48–53. doi: 10.1002/ana.410300110
Heming, M., Schulte-Mecklenbeck, A., Brix, T., Wolbert, J., Ruland, T., Klotz, L., et al. (2019). Immune cell profiling of the cerebrospinal fluid provides pathogenetic insights into inflammatory neuropathies. Front. Immunol. 10:515. doi: 10.3389/fimmu.2019.00515
Henkart, P. A. (1985). Mechanism of lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 3, 31–58. doi: 10.1146/annurev.iy.03.040185.000335
Hickey, W. F., Ueno, K., Hiserodt, J. C., and Schmidt, R. E. (1992). Exogenously-induced, natural killer cell-mediated neuronal killing: a novel pathogenetic mechanism. J. Exp. Med. 176, 811–817. doi: 10.1084/jem.176.3.811
Himes, B. T., and Tessler, A. (1989). Death of some dorsal root ganglion neurons and plasticity of others following sciatic nerve section in adult and neonatal rats. J. Comp. Neurol. 284, 215–230. doi: 10.1002/cne.902840206
Hirose, Y., Masaki, Y., Yoshioka, R., and Takiguchi, T. (1997). Aggressive natural killer cell lymphoproliferative disorder associated with Epstein-Barr viral RNA. Am. J. Hematol. 54, 314–320. doi: 10.1002/(sici)1096-8652(199704)54:4<314::aid-ajh10>3.0.co;2-b
Hoffmann, T., De Libero, G., Colonna, M., Wodnar-Filipowicz, A., Passweg, J., Favre, G., et al. (2000). Natural killer-type receptors for HLA class I antigens are clonally expressed in lymphoproliferative disorders of natural killer and T-cell type. Br. J. Haematol. 110, 525–536.
Hoke, A., and Ray, M. (2014). Rodent models of chemotherapy-induced peripheral neuropathy. ILAR J. 54, 273–281. doi: 10.1093/ilar/ilt053
Hore, Z., and Denk, F. (2019). Neuroimmune interactions in chronic pain - An interdisciplinary perspective. Brain Behav. Immun. 79, 56–62. doi: 10.1016/j.bbi.2019.04.033
Howard, R. F., Walker, S. M., Mota, P. M., and Fitzgerald, M. (2005). The ontogeny of neuropathic pain: postnatal onset of mechanical allodynia in rat spared nerve injury (SNI) and chronic constriction injury (CCI) models. Pain 115, 382–389. doi: 10.1016/j.pain.2005.03.016
Hu, G., Huang, K., Hu, Y., Du, G., Xue, Z., Zhu, X., et al. (2016). Single-cell RNA-seq reveals distinct injury responses in different types of DRG sensory neurons. Sci. Rep. 6:31851. doi: 10.1038/srep31851
Hu, P., Bembrick, A. L., Keay, K. A., and McLachlan, E. M. (2007). Immune cell involvement in dorsal root ganglia and spinal cord after chronic constriction or transection of the rat sciatic nerve. Brain Behav. Immun. 21, 599–616. doi: 10.1016/j.bbi.2006.10.013
Huang, C., Zou, W., Lee, K., Wang, E., Zhu, X., and Guo, Q. (2012). Different symptoms of neuropathic pain can be induced by different degrees of compressive force on the C7 dorsal root of rats. Spine J. 12, 1154–1160. doi: 10.1016/j.spinee.2012.10.036
Hughes, R. A., and van Doorn, P. A. (2012). Corticosteroids for guillain-barre syndrome. Cochrane Database Syst. Rev. 10:CD001446.
Imai, M., Landen, C., Ohta, R., Cheung, N. K., and Tomlinson, S. (2005). Complement-mediated mechanisms in anti-GD2 monoclonal antibody therapy of murine metastatic cancer. Cancer Res. 65, 10562–10568. doi: 10.1158/0008-5472.can-05-1894
Jacobs, B. C., Rothbarth, P. H., van der Meche, F. G., Herbrink, P., Schmitz, P. I., de Klerk, M. A., et al. (1998). The spectrum of antecedent infections in Guillain-Barre syndrome: a case-control study. Neurology 51, 1110–1115. doi: 10.1212/wnl.51.4.1110
Kang, Y. J., Song, H. K., Chon, J. Y., and You, J. H. (2007). Alterations in NK Cell cytotoxicity induced by peripheral nerve injury in mice. Korean J. Anesthesiol. 52, 219–223.
Kawasaki, Y., Xu, Z. Z., Wang, X., Park, J. Y., Zhuang, Z. Y., Tan, P. H., et al. (2008). Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat. Med. 14, 331–336. doi: 10.1038/nm1723
Kienle, K., and Lammermann, T. (2016). Neutrophil swarming: an essential process of the neutrophil tissue response. Immunol. Rev. 273, 76–93. doi: 10.1111/imr.12458
Kiguchi, N., Kobayashi, D., Saika, F., Matsuzaki, S., and Kishioka, S. (2017). Pharmacological regulation of neuropathic pain driven by inflammatory macrophages. Int. J. Mol. Sci. 18:E2296. doi: 10.3390/ijms18112296
Kiguchi, N., Kobayashi, Y., Saika, F., Sakaguchi, H., Maeda, T., and Kishioka, S. (2015). Peripheral interleukin-4 ameliorates inflammatory macrophage-dependent neuropathic pain. Pain 156, 684–693. doi: 10.1097/j.pain.0000000000000097
Kim, C. K., Choi, G. S., Oh, S. D., Han, J. B., Kim, S. K., Ahn, H. J., et al. (2005). Electroacupuncture up-regulates natural killer cell activity Identification of genes altering their expressions in electroacupuncture induced up-regulation of natural killer cell activity. J. Neuroimmunol. 168, 144–153. doi: 10.1016/j.jneuroim.2005.07.005
Kim, S. H., and Chung, J. M. (1992). An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50, 355–363. doi: 10.1016/0304-3959(92)90041-9
Knickelbein, J. E., Khanna, K. M., Yee, M. B., Baty, C. J., Kinchington, P. R., and Hendricks, R. L. (2008). Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 322, 268–271. doi: 10.1126/science.1164164
Koehn, T. A., Trimble, L. L., Alderson, K. L., Erbe, A. K., McDowell, K. A., Grzywacz, B., et al. (2012). Increasing the clinical efficacy of NK and antibody-mediated cancer immunotherapy: potential predictors of successful clinical outcome based on observations in high-risk neuroblastoma. Front. Pharmacol. 3:91. doi: 10.3389/fphar.2012.00091
Koike, H., Nishi, R., Ikeda, S., Kawagashira, Y., Iijima, M., Katsuno, M., et al. (2018). Ultrastructural mechanisms of macrophage-induced demyelination in CIDP. Neurology 91, 1051–1060. doi: 10.1212/WNL.0000000000006625
Krneta, T., Gillgrass, A., Poznanski, S., Chew, M., Lee, A. J., Kolb, M., et al. (2017). M2-polarized and tumor-associated macrophages alter NK cell phenotype and function in a contact-dependent manner. J. Leukoc. Biol. 101, 285–295. doi: 10.1189/jlb.3A1215-552R
Krukowski, K., Eijkelkamp, N., Laumet, G., Hack, C. E., Li, Y., Dougherty, P. M., et al. (2016). CD8+ T cells and endogenous IL-10 are required for resolution of chemotherapy-induced neuropathic pain. J. Neurosci. 36, 11074–11083. doi: 10.1523/jneurosci.3708-15.2016
Kurimoto, T., Yin, Y., Habboub, G., Gilbert, H. Y., Li, Y., Nakao, S., et al. (2013). Neutrophils express oncomodulin and promote optic nerve regeneration. J. Neurosci. 33, 14816–14824. doi: 10.1523/JNEUROSCI.5511-12.2013
Lackowski, D., Koberda, J. L., DeLoughery, T. G., and So, Y. (1998). Natural killer cell leukemia as a cause of peripheral neuropathy and meningitis: case report. Neurology 51, 640–641. doi: 10.1212/wnl.51.2.640
Lakritz, J. R., Robinson, J. A., Polydefkis, M. J., Miller, A. D., and Burdo, T. H. (2015). Loss of intraepidermal nerve fiber density during SIV peripheral neuropathy is mediated by monocyte activation and elevated monocyte chemotactic proteins. J. Neuroinflamm. 12:237. doi: 10.1186/s12974-015-0456-8
Lamy, T., Moignet, A., and Loughran, T. P. Jr. (2017). LGL leukemia: from pathogenesis to treatment. Blood 129, 1082–1094. doi: 10.1182/blood-2016-08-692590
Lanier, L. L. (2015). NKG2D receptor and its ligands in host defense. Cancer Immunol. Res. 3, 575–582. doi: 10.1158/2326-6066.CIR-15-0098
Lanier, L. L., and Phillips, J. H. (1986). Evidence for three types of human cytotoxic lymphocyte. Immunol. Today 7, 132–134. doi: 10.1016/0167-5699(86)90076-9
Laumet, G., Edralin, J. D., Dantzer, R., Heijnen, C. J., and Kavelaars, A. (2019a). Cisplatin educates CD8+ T cells to prevent and resolve chemotherapy-induced peripheral neuropathy in mice. Pain 160, 1459–1468. doi: 10.1097/j.pain.0000000000001512
Laumet, G., Ma, J., Robison, A. J., Kumari, S., Heijnen, C. J., and Kavelaars, A. (2019b). T cells as an emerging target for chronic pain therapy. Front. Mol. Neurosci. 12:216. doi: 10.3389/fnmol.2019.00216
Lees, J. G., Makker, P. G., Tonkin, R. S., Abdulla, M., Park, S. B., Goldstein, D., et al. (2017). Immune-mediated processes implicated in chemotherapy-induced peripheral neuropathy. Eur. J. Cancer 73, 22–29. doi: 10.1016/j.ejca.2016.12.006
Leitenberg, D., Eisen, R. N., Goldstein, J. M., Busque, L., Rinder, H. M., Stys, P., et al. (1995). Natural killer cell lymphoproliferative disease associated with neuropathy. Am. J. Med. 99, 99–101. doi: 10.1016/s0002-9343(99)80112-0
Lewis, S. E., Mannion, R. J., White, F. A., Coggeshall, R. E., Beggs, S., Costigan, M., et al. (1999). A role for HSP27 in sensory neuron survival. J. Neurosci. 19, 8945–8953. doi: 10.1523/jneurosci.19-20-08945.1999
Li, Y., Sun, Y., Cai, M., Zhang, H., Gao, N., Huang, H., et al. (2018). Fas Ligand Gene (Faslg) plays an important role in nerve degeneration and regeneration after rat sciatic nerve injury. Front. Mol. Neurosci. 11:210. doi: 10.3389/fnmol.2018.00210
Liang, Z., Hore, Z., Harley, P., Stanley, F. U., Michrowska, A., Dahiya, M., et al. (2019). A transcriptional toolbox for exploring peripheral neuro-immune interactions. bioRxiv [Preprint]. Available at http://biorxiv.org/content/early/2019/10/22/813980.abstract (accessed December 26, 2019).
Lindborg, J. A., Mack, M., and Zigmond, R. E. (2017). Neutrophils are critical for myelin removal in a peripheral nerve injury model of wallerian degeneration. J. Neurosci. 37, 10258–10277. doi: 10.1523/JNEUROSCI.2085-17.2017
Linnartz-Gerlach, B., Schuy, C., Shahraz, A., Tenner, A. J., and Neumann, H. (2016). Sialylation of neurites inhibits complement-mediated macrophage removal in a human macrophage-neuron Co-culture system. Glia 64, 35–47. doi: 10.1002/glia.22901
Lissoni, P., Brivio, F., Fumagalli, L., Di Fede, G., and Brera, G. (2005). Enhancement of the efficacy of chemotherapy with oxaliplatin plus 5-fluorouracil by pretreatment with IL-2 subcutaneous immunotherapy in metastatic colorectal cancer patients with lymphocytopenia prior to therapy. Vivo 19, 1077–1080.
Littwitz-Salomon, E., Akhmetzyanova, I., Vallet, C., Francois, S., Dittmer, U., and Gibbert, K. (2015). Activated regulatory T cells suppress effector NK cell responses by an IL-2-mediated mechanism during an acute retroviral infection. Retrovirology 12:66. doi: 10.1186/s12977-015-0191-3
Liu, C., Chen, N., Huang, K., Jiang, M., Liang, H., Sun, Z., et al. (2018). Identifying hub genes and potential mechanisms associated with senescence in human annulus cells by gene expression profiling and bioinformatics analysis. Mol. Med. Rep. 17, 3465–3472. doi: 10.3892/mmr.2017.8322
Liu, G., Atteridge, C. L., Wang, X., Lundgren, A. D., and Wu, J. D. (2010). The membrane type matrix metalloproteinase MMP14 mediates constitutive shedding of MHC class I chain-related molecule A independent of A disintegrin and metalloproteinases. J. Immunol. 184, 3346–3350. doi: 10.4049/jimmunol.0903789
Liu, T., van Rooijen, N., and Tracey, D. J. (2000). Depletion of macrophages reduces axonal degeneration and hyperalgesia following nerve injury. Pain 86, 25–32. doi: 10.1016/s0304-3959(99)00306-1
Llobet Rosell, A., and Neukomm, L. J. (2019). Axon death signalling in Wallerian degeneration among species and in disease. Open Biol. 9, 190118. doi: 10.1098/rsob.190118
Lode, H. N., Xiang, R., Dreier, T., Varki, N. M., Gillies, S. D., and Reisfeld, R. A. (1998). Natural killer cell-mediated eradication of neuroblastoma metastases to bone marrow by targeted interleukin-2 therapy. Blood 91, 1706–1715. doi: 10.1182/blood.v91.5.1706.1706_1706_1715
Lu, D., Song, H., and Shi, G. (2013). Anti-TNF-alpha treatment for pelvic pain associated with endometriosis. Cochrane Database Syst. Rev. 28:CD008088.
Luchting, B., Rachinger-Adam, B., Heyn, J., Hinske, L. C., Kreth, S., and Azad, S. C. (2015). Anti-inflammatory T-cell shift in neuropathic pain. J. Neuroinflamm. 12:12. doi: 10.1186/s12974-014-0225-0
Luchting, B., Rachinger-Adam, B., Zeitler, J., Egenberger, L., Mohnle, P., Kreth, S., et al. (2014). Disrupted TH17/Treg balance in patients with chronic low back pain. PLoS One 9:e104883. doi: 10.1371/journal.pone.0104883
Lunn, E. R., Perry, V. H., Brown, M. C., Rosen, H., and Gordon, S. (1989). Absence of wallerian degeneration does not hinder regeneration in peripheral nerve. Eur. J. Neurosci. 1, 27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x
Luo, L., and O’Leary, D. D. (2005). Axon retraction and degeneration in development and disease. Annu. Rev. Neurosci. 28, 127–156. doi: 10.1146/annurev.neuro.28.061604.135632
Lyu, C., Lyu, G. W., Martinez, A., and Shi, T. S. (2017). Effect of nerve injury on the number of dorsal root ganglion neurons and autotomy behavior in adult Bax-deficient mice. J Pain Res. 10, 2079–2087. doi: 10.2147/JPR.S133087
Ma, M., Ferguson, T. A., Schoch, K. M., Li, J., Qian, Y., Shofer, F. S., et al. (2013). Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol. Dis. 56, 34–46. doi: 10.1016/j.nbd.2013.03.009
Maden, M. (2007). Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 8, 755–765. doi: 10.1038/nrn2212
Maeda, M., Tsuda, M., Tozaki-Saitoh, H., Inoue, K., and Kiyama, H. (2010). Nerve injury-activated microglia engulf myelinated axons in a P2Y12 signaling-dependent manner in the dorsal horn. Glia 58, 1838–1846. doi: 10.1002/glia.21053
Main, E. K., Lampson, L. A., Hart, M. K., Kornbluth, J., and Wilson, D. B. (1985). Human neuroblastoma cell lines are susceptible to lysis by natural killer cells but not by cytotoxic T lymphocytes. J. Immunol. 135, 242–246.
Markasz, L., Stuber, G., Vanherberghen, B., Flaberg, E., Olah, E., Carbone, E., et al. (2007). Effect of frequently used chemotherapeutic drugs on the cytotoxic activity of human natural killer cells. Mol. Cancer Ther. 6, 644–654. doi: 10.1158/1535-7163.mct-06-0358
Mathey, E. K., Park, S. B., Hughes, R. A., Pollard, J. D., Armati, P. J., Barnett, M. H., et al. (2015). Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J. Neurol. Neurosurg. Psychiatry 86, 973–985.
Matlung, H. L., Babes, L., Zhao, X. W., van Houdt, M., Treffers, L. W., van Rees, D. J., et al. (2018). Neutrophils kill antibody-opsonized cancer cells by trogoptosis. Cell Rep. 23, 3946–3959.e6. doi: 10.1016/j.celrep.2018.05.082
McDougall, J. J., and Muley, M. M. (2015). The role of proteases in pain. Handb. Exp. Pharmacol. 227, 239–260. doi: 10.1007/978-3-662-46450-2_12
McGonigal, R., Rowan, E. G., Greenshields, K. N., Halstead, S. K., Humphreys, P. D., Rother, R. P., et al. (2010). Anti-GD1a antibodies activate complement and calpain to injure distal motor nodes of Ranvier in mice. Brain 133, 1944–1960. doi: 10.1093/brain/awq119
McNeill, C. J., Fehmi, J., Gladwin, J., and Price, C. (2019). A rare case of Miller Fisher variant of Guillain-Barre syndrome (GBS) induced by a checkpoint inhibitor. BMJ Case Rep. 12:e229443. doi: 10.1136/bcr-2019-229443
Merry, D. E., Veis, D. J., Hickey, W. F., and Korsmeyer, S. J. (1994). bcl-2 protein expression is widespread in the developing nervous system and retained in the adult PNS. Development 120, 301–311.
Mert, T., Gunay, I., Ocal, I., Guzel, A. I., Inal, T. C., Sencar, L., et al. (2009). Macrophage depletion delays progression of neuropathic pain in diabetic animals. Naunyn Schmiedebergs Arch. Pharmacol. 379, 445–452. doi: 10.1007/s00210-008-0387-3
Meuth, S. G., Herrmann, A. M., Simon, O. J., Siffrin, V., Melzer, N., Bittner, S., et al. (2009). Cytotoxic CD8+ T cell-neuron interactions: perforin-dependent electrical silencing precedes but is not causally linked to neuronal cell death. J. Neurosci. 29, 15397–15409. doi: 10.1523/JNEUROSCI.4339-09.2009
Michel, T., Hentges, F., and Zimmer, J. (2012). Consequences of the crosstalk between monocytes/macrophages and natural killer cells. Front. Immunol. 3:403. doi: 10.3389/fimmu.2012.00403
Moalem, G., Xu, K., and Yu, L. (2004). T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience 129, 767–777. doi: 10.1016/j.neuroscience.2004.08.035
Morvan, M. G., Champsaur, M., Reizis, B., and Lanier, L. L. (2017). Chronic in vivo interaction of dendritic cells expressing the ligand Rae-1epsilon with NK cells impacts NKG2D expression and function. Immunohorizons 1, 10–19. doi: 10.4049/immunohorizons.1700004
Munn, D. H., and Cheung, N. K. (1990). Phagocytosis of tumor cells by human monocytes cultured in recombinant macrophage colony-stimulating factor. J. Exp. Med. 172, 231–237. doi: 10.1084/jem.172.1.231
Nadeau, S., Filali, M., Zhang, J., Kerr, B. J., Rivest, S., Soulet, D., et al. (2011). Functional recovery after peripheral nerve injury is dependent on the pro-inflammatory cytokines IL-1beta and TNF: implications for neuropathic pain. J. Neurosci. 31, 12533–12542. doi: 10.1523/JNEUROSCI.2840-11.2011
Navid, F., Sondel, P. M., Barfield, R., Shulkin, B. L., Kaufman, R. A., Allay, J. A., et al. (2014). Phase I trial of a novel anti-GD2 monoclonal antibody, Hu14.18K322A, designed to decrease toxicity in children with refractory or recurrent neuroblastoma. J. Clin. Oncol. 32, 1445–1452. doi: 10.1200/JCO.2013.50.4423
Nedvetzki, S., Sowinski, S., Eagle, R. A., Harris, J., Vely, F., Pende, D., et al. (2007). Reciprocal regulation of human natural killer cells and macrophages associated with distinct immune synapses. Blood 109, 3776–3785. doi: 10.1182/blood-2006-10-052977
Ni, J., Miller, M., Stojanovic, A., Garbi, N., and Cerwenka, A. (2012). Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J. Exp. Med. 209, 2351–2365. doi: 10.1084/jem.20120944
Ni Mhaolcatha, S., Flynn, A., Hayes, B., O’Shea, D., Bennett, M., and Fitzgibbon, J. (2019). A stab in the back: an unusual case of cutaneous neural infiltration as a manifestation of chronic lymphoproliferative disorder of natural killer cells. Am. J. Dermatopathol. 41, 378–381. doi: 10.1097/DAD.0000000000001280
Nicoll, G., Ni, J., Liu, D., Klenerman, P., Munday, J., Dubock, S., et al. (1999). Identification and characterization of a novel siglec, siglec-7, expressed by human natural killer cells and monocytes. J. Biol. Chem. 274, 34089–34095. doi: 10.1074/jbc.274.48.34089
Noguchi, M., Yoshita, M., Sakai, K., Matsumoto, Y., Arahata, M., Ontachi, Y., et al. (2005). Peripheral neuropathy associated with chronic natural killer cell lymphocytosis. J. Neurol. Sci. 232, 119–122. doi: 10.1016/j.jns.2005.01.013
Nomura, M., Takihara, Y., and Shimada, K. (1994). Isolation and characterization of retinoic acid-inducible cDNA clones in F9 cells: one of the early inducible clones encodes a novel protein sharing several highly homologous regions with a Drosophila polyhomeotic protein. Differentiation 57, 39–50. doi: 10.1046/j.1432-0436.1994.5710039.x
Nomura, M., Zou, Z., Joh, T., Takihara, Y., Matsuda, Y., and Shimada, K. (1996). Genomic structures and characterization of Rae1 family members encoding GPI-anchored cell surface proteins and expressed predominantly in embryonic mouse brain. J. Biochem. 120, 987–995. doi: 10.1093/oxfordjournals.jbchem.a021517
North, R. Y., Li, Y., Ray, P., Rhines, L. D., Tatsui, C. E., Rao, G., et al. (2019). Electrophysiological and transcriptomic correlates of neuropathic pain in human dorsal root ganglion neurons. Brain 142, 1215–1226. doi: 10.1093/brain/awz063
O’Brien, P. D., Sakowski, S. A., and Feldman, E. L. (2014). Mouse models of diabetic neuropathy. ILAR J. 54, 259–272. doi: 10.1093/ilar/ilt052
O’Hanlon, G. M., Humphreys, P. D., Goldman, R. S., Halstead, S. K., Bullens, R. W., Plomp, J. J., et al. (2003). Calpain inhibitors protect against axonal degeneration in a model of anti-ganglioside antibody-mediated motor nerve terminal injury. Brain 126, 2497–2509. doi: 10.1093/brain/awg254
Oosterlynck, D. J., Cornillie, F. J., Waer, M., Vandeputte, M., and Koninckx, P. R. (1991). Women with endometriosis show a defect in natural killer activity resulting in a decreased cytotoxicity to autologous endometrium. Fertil. Steril. 56, 45–51. doi: 10.1016/s0015-0282(16)54414-8
Orange, J. S. (2013). Natural killer cell deficiency. J. Allergy Clin. Immunol. 132, 515–525. doi: 10.1016/j.jaci.2013.07.020
Osterloh, J. M., Yang, J., Rooney, T. M., Fox, A. N., Adalbert, R., Powell, E. H., et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. doi: 10.1126/science.1223899
Oun, R., Moussa, Y. E., and Wheate, N. J. (2018). The side effects of platinum-based chemotherapy drugs: a review for chemists. Dalton Trans. 47, 6645–6653. doi: 10.1039/c8dt00838h
Pallmer, K., and Oxenius, A. (2016). Recognition and regulation of T cells by NK cells. Front. Immunol. 7:251. doi: 10.3389/fimmu.2016.00251
Pannell, M., Labuz, D., Celik, M. O., Keye, J., Batra, A., Siegmund, B., et al. (2016). Adoptive transfer of M2 macrophages reduces neuropathic pain via opioid peptides. J. Neuroinflamm. 13:262.
Papayannopoulos, V. (2018). Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 18, 134–147. doi: 10.1038/nri.2017.105
Park, S. B., Lin, C. S., Krishnan, A. V., Goldstein, D., Friedlander, M. L., and Kiernan, M. C. (2009). Oxaliplatin-induced neurotoxicity: changes in axonal excitability precede development of neuropathy. Brain 132, 2712–2723. doi: 10.1093/brain/awp219
Peng, X. Q., Zhang, X. L., Fang, Y., Xie, W. R., and Xie, Y. K. (2004). Sialic acid contributes to hyperexcitability of dorsal root ganglion neurons in rats with peripheral nerve injury. Brain Res. 1026, 185–193. doi: 10.1016/j.brainres.2004.07.075
Perkins, N. M., and Tracey, D. J. (2000). Hyperalgesia due to nerve injury: role of neutrophils. Neuroscience 101, 745–757. doi: 10.1016/s0306-4522(00)00396-1
Perry, V. H., Brown, M. C., Lunn, E. R., Tree, P., and Gordon, S. (1990). Evidence that very slow wallerian degeneration in C57BL/Ola mice is an intrinsic property of the peripheral nerve. Eur. J. Neurosci. 2, 802–808. doi: 10.1111/j.1460-9568.1990.tb00472.x
Poli, A., Michel, T., Theresine, M., Andres, E., Hentges, F., and Zimmer, J. (2009). CD56bright natural killer (NK) cells: an important NK cell subset. Immunology 126, 458–465. doi: 10.1111/j.1365-2567.2008.03027.x
Pollock, R. E., Lotzova, E., and Stanford, S. D. (1991). Mechanism of surgical stress impairment of human perioperative natural killer cell cytotoxicity. Arch. Surg. 126, 338–342.
Pollock, R. E., Lotzova, E., Stanford, S. D., and Romsdahl, M. M. (1987). Effect of surgical stress on murine natural killer cell cytotoxicity. J. Immunol. 138, 171–178.
Poullot, E., Zambello, R., Leblanc, F., Bareau, B., De March, E., Roussel, M., et al. (2014). Chronic natural killer lymphoproliferative disorders: characteristics of an international cohort of 70 patients. Ann. Oncol. 25, 2030–2035. doi: 10.1093/annonc/mdu369
Poulopoulos, A., Murphy, A. J., Ozkan, A., Davis, P., Hatch, J., Kirchner, R., et al. (2019). Subcellular transcriptomes and proteomes of developing axon projections in the cerebral cortex. Nature 565, 356–360. doi: 10.1038/s41586-018-0847-y
Prior, R., Van Helleputte, L., Benoy, V., and Van Den Bosch, L. (2017). Defective axonal transport: a common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiol. Dis. 105, 300–320. doi: 10.1016/j.nbd.2017.02.009
Puttagunta, R., and Di Giovanni, S. (2011). Retinoic acid signaling in axonal regeneration. Front. Mol. Neurosci. 4:59. doi: 10.3389/fnmol.2011.00059
Quirk, K., and Ganapathy-Kanniappan, S. (2017). Is there an opportunity for current chemotherapeutics to up-regulate MIC-A/B ligands? Front. Pharmacol. 8:732. doi: 10.3389/fphar.2017.00732
Rabbani, G. R., Phyliky, R. L., and Tefferi, A. (1999). A long-term study of patients with chronic natural killer cell lymphocytosis. Br. J. Haematol. 106, 960–966. doi: 10.1046/j.1365-2141.1999.01624.x
Ramer, M. S., and Bisby, M. A. (1998). Differences in sympathetic innervation of mouse DRG following proximal or distal nerve lesions. Exp. Neurol. 152, 197–207. doi: 10.1006/exnr.1998.6855
Ramer, M. S., French, G. D., and Bisby, M. A. (1997). Wallerian degeneration is required for both neuropathic pain and sympathetic sprouting into the DRG. Pain 72, 71–78. doi: 10.1016/s0304-3959(97)00019-5
Raoof, R., Willemen, H., and Eijkelkamp, N. (2018). Divergent roles of immune cells and their mediators in pain. Rheumatology 57, 429–440. doi: 10.1093/rheumatology/kex308
Raulet, D. H., Gasser, S., Gowen, B. G., Deng, W., and Jung, H. (2013). Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 31, 413–441. doi: 10.1146/annurev-immunol-032712-095951
Ren, K., and Dubner, R. (2010). Interactions between the immune and nervous systems in pain. Nat. Med. 16, 1267–1276. doi: 10.1038/nm.2234
Renthal, W., Tochitsky, I., Yang, L., Cheng, Y.-C., Li, E., Kawaguchi, R., et al. (2019). Transcriptional reprogramming of distinct peripheral sensory neuron subtypes after axonal injury. bioRxiv [Preprint], Available at: http://biorxiv.org/content/early/2019/11/16/838854.abstract (accessed November 30, 2019).
Rinaldi, S., and Bennett, D. L. (2014). Pathogenic mechanisms in inflammatory and paraproteinaemic peripheral neuropathies. Curr. Opin. Neurol. 27, 541–551. doi: 10.1097/WCO.0000000000000137
Ritz, B. W., Alexander, G. M., Nogusa, S., Perreault, M. J., Peterlin, B. L., Grothusen, J. R., et al. (2011). Elevated blood levels of inflammatory monocytes (CD14+ CD16+) in patients with complex regional pain syndrome. Clin. Exp. Immunol. 164, 108–117. doi: 10.1111/j.1365-2249.2010.04308.x
Roquilly, A., David, G., Cinotti, R., Vourc’h, M., Morin, H., Rozec, B., et al. (2017). Role of IL-12 in overcoming the low responsiveness of NK cells to missing self after traumatic brain injury. Clin. Immunol. 177, 87–94. doi: 10.1016/j.clim.2015.08.006
Rosales, C. (2017). Fcγ receptor heterogeneity in leukocyte functional responses. Front. Immunol. 8:280.
Rosenberg, A. F., Wolman, M. A., Franzini-Armstrong, C., and Granato, M. (2012). In vivo nerve-macrophage interactions following peripheral nerve injury. J. Neurosci. 32, 3898–3909. doi: 10.1523/JNEUROSCI.5225-11.2012
Rotshenker, S. (2011). Wallerian degeneration: the innate-immune response to traumatic nerve injury. J. Neuroinflamm. 8:109. doi: 10.1186/1742-2094-8-109
Rozenbaum, M., Rajman, M., Rishal, I., Koppel, I., Koley, S., Medzihradszky, K. F., et al. (2018). Translatome regulation in neuronal injury and axon regrowth. eNeuro 5:ENEURO.276-17.2018. doi: 10.1523/ENEURO.0276-17.2018
Russo, M. A., Fiore, N. T., van Vreden, C., Bailey, D., Santarelli, D. M., McGuire, H. M., et al. (2019). Expansion and activation of distinct central memory T lymphocyte subsets in complex regional pain syndrome. J. Neuroinflamm. 16:63. doi: 10.1186/s12974-019-1449-9
Saika, F., Kiguchi, N., Matsuzaki, S., Kobayashi, D., and Kishioka, S. (2019). Inflammatory macrophages in the sciatic nerves facilitate neuropathic pain associated with Type 2 Diabetes mellitus. J. Pharmacol. Exp. Ther. 368, 535–544. doi: 10.1124/jpet.118.252668
Sano, H., Maeda, T., Omoto, M., Ogasawara, J. I., Koga, M., Kawai, M., et al. (2017). Steroid-responsive demyelinating peripheral neuropathy associated with chronic lymphoproliferative disorders of natural killer cells. Rinsho Shinkeigaku 57, 573–578. doi: 10.5692/clinicalneurol.cn-001064
Sanvito, L., Makowska, A., Gregson, N., Nemni, R., and Hughes, R. A. (2009). Circulating subsets and CD4(+)CD25(+) regulatory T cell function in chronic inflammatory demyelinating polyradiculoneuropathy. Autoimmunity 42, 667–677. doi: 10.3109/08916930903140907
Sasaki, Y., Vohra, B. P., Lund, F. E., and Milbrandt, J. (2009). Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J. Neurosci. 29, 5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009
Sauer, B. M., Schmalstieg, W. F., and Howe, C. L. (2013). Axons are injured by antigen-specific CD8+ T cells through a MHC class I- and granzyme B-dependent mechanism. Neurobiol. Dis. 59, 194–205. doi: 10.1016/j.nbd.2013.07.010
Schleinitz, N., Vely, F., Harle, J. R., and Vivier, E. (2010). Natural killer cells in human autoimmune diseases. Immunology 131, 451–458. doi: 10.1111/j.1365-2567.2010.03360.x
Schneider-Hohendorf, T., Schwab, N., Uceyler, N., Gobel, K., Sommer, C., and Wiendl, H. (2012). CD8+ T-cell immunity in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology 78, 402–408. doi: 10.1212/WNL.0b013e318245d250
Scholl, B., Bersinger, N. A., Kuhn, A., and Mueller, M. D. (2009). Correlation between symptoms of pain and peritoneal fluid inflammatory cytokine concentrations in endometriosis. Gynecol. Endocrinol. 25, 701–706. doi: 10.3109/09513590903159680
Scott, A., Khan, K. M., Cook, J. L., and Duronio, V. (2004). What is “inflammation”? Are we ready to move beyond Celsus? Br. J. Sports Med. 38, 248–249. doi: 10.1136/bjsm.2003.011221
Seltzer, Z., Dubner, R., and Shir, Y. (1990). A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 43, 205–218. doi: 10.1016/0304-3959(90)91074-s
Seshadri, A., Brat, G. A., Yorkgitis, B. K., Keegan, J., Dolan, J., Salim, A., et al. (2017). Phenotyping the immune response to trauma: a multiparametric systems immunology approach. Crit. Care Med. 45, 1523–1530. doi: 10.1097/CCM.0000000000002577
Sessenwein, J. L., Baker, C. C., Pradhananga, S., Maitland, M. E., Petrof, E. O., Allen-Vercoe, E., et al. (2017). Protease-mediated suppression of DRG neuron excitability by commensal bacteria. J. Neurosci. 37, 11758–11768. doi: 10.1523/JNEUROSCI.1672-17.2017
Shavit, Y., Lewis, J. W., Terman, G. W., Gale, R. P., and Liebeskind, J. C. (1984). Opioid peptides mediate the suppressive effect of stress on natural killer cell cytotoxicity. Science 223, 188–190. doi: 10.1126/science.6691146
Shepherd, A. J., Copits, B. A., Mickle, A. D., Karlsson, P., Kadunganattil, S., Haroutounian, S., et al. (2018a). Angiotensin II triggers peripheral macrophage-to-sensory neuron redox crosstalk to elicit pain. J. Neurosci. 38, 7032–7057. doi: 10.1523/JNEUROSCI.3542-17.2018
Shepherd, A. J., Mickle, A. D., Golden, J. P., Mack, M. R., Halabi, C. M., de Kloet, A. D., et al. (2018b). Macrophage angiotensin II type 2 receptor triggers neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 115, E8057–E8066.
Shi, T. J., Tandrup, T., Bergman, E., Xu, Z. Q., Ulfhake, B., and Hokfelt, T. (2001). Effect of peripheral nerve injury on dorsal root ganglion neurons in the C57 BL/6J mouse: marked changes both in cell numbers and neuropeptide expression. Neuroscience 105, 249–263. doi: 10.1016/s0306-4522(01)00148-8
Shin, S., and Simpson, D. (2013). “Viral Neuropathies,” in Encyclopedia of Pain, eds G. F. Gebhart, and R. F. Schmidt (Heidelberg: Springer).
Siew, Y. Y., Neo, S. Y., Yew, H. C., Lim, S. W., Ng, Y. C., Lew, S. M., et al. (2015). Oxaliplatin regulates expression of stress ligands in ovarian cancer cells and modulates their susceptibility to natural killer cell-mediated cytotoxicity. Int. Immunol. 27, 621–632. doi: 10.1093/intimm/dxv041
Sikora, J., Mielczarek-Palacz, A., and Kondera-Anasz, Z. (2011). Role of natural killer cell activity in the pathogenesis of endometriosis. Curr. Med. Chem. 18, 200–208. doi: 10.2174/092986711794088416
Silva, R. L., Lopes, A. H., Guimaraes, R. M., and Cunha, T. M. (2017). CXCL1/CXCR2 signaling in pathological pain: role in peripheral and central sensitization. Neurobiol. Dis. 105, 109–116. doi: 10.1016/j.nbd.2017.06.001
Simon, D. J., Weimer, R. M., McLaughlin, T., Kallop, D., Stanger, K., Yang, J., et al. (2012). A caspase cascade regulating developmental axon degeneration. J. Neurosci. 32, 17540–17553. doi: 10.1523/JNEUROSCI.3012-12.2012
Slart, R., Yu, A. L., Yaksh, T. L., and Sorkin, L. S. (1997). An animal model of pain produced by systemic administration of an immunotherapeutic anti-ganglioside antibody. Pain 69, 119–125. doi: 10.1016/s0304-3959(96)03247-2
Sorge, R. E., Mapplebeck, J. C., Rosen, S., Beggs, S., Taves, S., Alexander, J. K., et al. (2015). Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 18, 1081–1083. doi: 10.1038/nn.4053
Soriani, A., Zingoni, A., Cerboni, C., Iannitto, M. L., Ricciardi, M. R., Di Gialleonardo, V., et al. (2009). ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 113, 3503–3511. doi: 10.1182/blood-2008-08-173914
Sorkin, L. S., Otto, M., Baldwin, W. M. III, Vail, E., Gillies, S. D., Handgretinger, R., et al. (2010). Anti-GD(2) with an FC point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain 149, 135–142. doi: 10.1016/j.pain.2010.01.024
Spain, L., Walls, G., Julve, M., O’Meara, K., Schmid, T., Kalaitzaki, E., et al. (2017). Neurotoxicity from immune-checkpoint inhibition in the treatment of melanoma: a single centre experience and review of the literature. Ann. Oncol. 28, 377–385. doi: 10.1093/annonc/mdw558
Spear, P., Wu, M. R., Sentman, M. L., and Sentman, C. L. (2013). NKG2D ligands as therapeutic targets. Cancer Immun. 13:8.
Sprowl, J. A., Ciarimboli, G., Lancaster, C. S., Giovinazzo, H., Gibson, A. A., Du, G., et al. (2013). Oxaliplatin-induced neurotoxicity is dependent on the organic cation transporter OCT2. Proc. Natl. Acad. Sci. U.S.A. 110, 11199–11204. doi: 10.1073/pnas.1305321110
Starobova, H., and Vetter, I. (2017). Pathophysiology of chemotherapy-induced peripheral neuropathy. Front. Mol. Neurosci. 10:174. doi: 10.3389/fnmol.2017.00174
Stoll, G., Griffin, J. W., Li, C. Y., and Trapp, B. D. (1989). Wallerian degeneration in the peripheral nervous system: participation of both Schwann cells and macrophages in myelin degradation. J. Neurocytol. 18, 671–683. doi: 10.1007/bf01187086
Sun, D., Wang, X., Zhang, H., Deng, L., and Zhang, Y. (2011). MMP9 mediates MICA shedding in human osteosarcomas. Cell Biol. Int. 35, 569–574. doi: 10.1042/CBI20100431
Sunagawa, M., Okada, M., Guo, S. Y., and Hisamitsu, T. (2000). Splenic natural killer cell activity is suppressed by ligation of unilateral mental nerve in rats. Masui 49, 250–254.
Symons, L. K., Miller, J. E., Kay, V. R., Marks, R. M., Liblik, K., Koti, M., et al. (2018). The immunopathophysiology of endometriosis. Trends Mol. Med. 24, 748–762. doi: 10.1016/j.molmed.2018.07.004
Tandrup, T., Woolf, C. J., and Coggeshall, R. E. (2000). Delayed loss of small dorsal root ganglion cells after transection of the rat sciatic nerve. J. Comp. Neurol. 422, 172–180. doi: 10.1002/(sici)1096-9861(20000626)422:2<172::aid-cne2>3.0.co;2-h
Tegeder, I., and Lotsch, J. (2009). Current evidence for a modulation of low back pain by human genetic variants. J. Cell Mol. Med. 13, 1605–1619. doi: 10.1111/j.1582-4934.2009.00703.x
Thiruchelvam, U., Wingfield, M., and O’Farrelly, C. (2015). Natural killer cells: key players in endometriosis. Am. J. Reprod. Immunol. 74, 291–301. doi: 10.1111/aji.12408
Timonen, T., Ortaldo, J. R., and Herberman, R. B. (1981). Characteristics of human large granular lymphocytes and relationship to natural killer and K cells. J. Exp. Med. 153, 569–582. doi: 10.1084/jem.153.3.569
Tonello, R., Lee, S. H., and Berta, T. (2019). Monoclonal antibody targeting the matrix metalloproteinase 9 prevents and reverses paclitaxel-induced peripheral neuropathy in mice. J. Pain 20, 515–527. doi: 10.1016/j.jpain.2018.11.003
Topp, K. S., Tanner, K. D., and Levine, J. D. (2000). Damage to the cytoskeleton of large diameter sensory neurons and myelinated axons in vincristine-induced painful peripheral neuropathy in the rat. J. Comp. Neurol. 424, 563–576. doi: 10.1002/1096-9861(20000904)424:4<563::aid-cne1>3.0.co;2-u
Totsch, S. K., and Sorge, R. E. (2017). Immune system involvement in specific pain conditions. Mol. Pain 13:1744806917724559. doi: 10.1177/1744806917724559
Tran, L. V., Tokushige, N., Berbic, M., Markham, R., and Fraser, I. S. (2009). Macrophages and nerve fibres in peritoneal endometriosis. Hum. Reprod. 24, 835–841. doi: 10.1093/humrep/den483
Trapani, J. A., and Smyth, M. J. (2002). Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2, 735–747. doi: 10.1038/nri911
Treede, R. D., Jensen, T. S., Campbell, J. N., Cruccu, G., Dostrovsky, J. O., Griffin, J. W., et al. (2008). Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 70, 1630–1635. doi: 10.1212/01.wnl.0000282763.29778.59
Turkiew, E., Falconer, D., Reed, N., and Hoke, A. (2017). Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J. Peripher. Nerv. Syst. 22, 162–171. doi: 10.1111/jns.12219
Turnley, A. M., Starr, R., and Bartlett, P. F. (2002). Failure of sensory neurons to express class I MHC is due to differential SOCS1 expression. J. Neuroimmunol. 123, 35–40. doi: 10.1016/s0165-5728(01)00480-5
Uderhardt, S., Martins, A. J., Tsang, J. S., Lammermann, T., and Germain, R. N. (2019). Resident macrophages cloak tissue microlesions to prevent neutrophil-driven inflammatory damage. Cell 177, 541–555e17. doi: 10.1016/j.cell.2019.02.028
Unsain, N., Higgins, J. M., Parker, K. N., Johnstone, A. D., and Barker, P. A. (2013). XIAP regulates caspase activity in degenerating axons. Cell Rep. 4, 751–763. doi: 10.1016/j.celrep.2013.07.015
Usoskin, D., Furlan, A., Islam, S., Abdo, H., Lonnerberg, P., Lou, D., et al. (2015). Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 18, 145–153. doi: 10.1038/nn.3881
van Spriel, A. B., Leusen, J. H., van Egmond, M., Dijkman, H. B., Assmann, K. J., Mayadas, T. N., et al. (2001). Mac-1 (CD11b/CD18) is essential for Fc receptor-mediated neutrophil cytotoxicity and immunologic synapse formation. Blood 97, 2478–2486. doi: 10.1182/blood.v97.8.2478
Vanderhaeghen, P., and Cheng, H. J. (2010). Guidance molecules in axon pruning and cell death. Cold Spring Harb. Perspect. Biol. 2:a001859. doi: 10.1101/cshperspect.a001859
Vargas, M. E., Watanabe, J., Singh, S. J., Robinson, W. H., and Barres, B. A. (2010). Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc. Natl. Acad. Sci. U.S.A. 107, 11993–11998. doi: 10.1073/pnas.1001948107
Vega-Avelaira, D., Geranton, S. M., and Fitzgerald, M. (2009). Differential regulation of immune responses and macrophage/neuron interactions in the dorsal root ganglion in young and adult rats following nerve injury. Mol. Pain 5:70. doi: 10.1186/1744-8069-5-70
Vicuna, L., Strochlic, D. E., Latremoliere, A., Bali, K. K., Simonetti, M., Husainie, D., et al. (2015). The serine protease inhibitor SerpinA3N attenuates neuropathic pain by inhibiting T cell-derived leukocyte elastase. Nat. Med. 21, 518–523. doi: 10.1038/nm.3852
Vigneswara, V., Berry, M., Logan, A., and Ahmed, Z. (2013). Caspase-2 is upregulated after sciatic nerve transection and its inhibition protects dorsal root ganglion neurons from apoptosis after serum withdrawal. PLoS One 8:e57861. doi: 10.1371/journal.pone.0057861
Vilalta, A., and Brown, G. C. (2018). Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 285, 3566–3575. doi: 10.1111/febs.14323
Vivier, E., Tomasello, E., Baratin, M., Walzer, T., and Ugolini, S. (2008). Functions of natural killer cells. Nat. Immunol. 9, 503–510. doi: 10.1038/ni1582
Voigt, J., Hunniger, K., Bouzani, M., Jacobsen, I. D., Barz, D., Hube, B., et al. (2014). Human natural killer cells acting as phagocytes against Candida albicans and mounting an inflammatory response that modulates neutrophil antifungal activity. J. Infect. Dis. 209, 616–626. doi: 10.1093/infdis/jit574
Wakatsuki, S., Tokunaga, S., Shibata, M., and Araki, T. (2017). GSK3B-mediated phosphorylation of MCL1 regulates axonal autophagy to promote Wallerian degeneration. J. Cell Biol. 216, 477–493. doi: 10.1083/jcb.201606020
Waldhauer, I., Goehlsdorf, D., Gieseke, F., Weinschenk, T., Wittenbrink, M., Ludwig, A., et al. (2008). Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 68, 6368–6376. doi: 10.1158/0008-5472.CAN-07-6768
Wang, J. (2018). Neutrophils in tissue injury and repair. Cell Tissue Res. 371, 531–539. doi: 10.1007/s00441-017-2785-7
Wang, W., Erbe, A. K., Hank, J. A., Morris, Z. S., and Sondel, P. M. (2015). NK Cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. immunol. 6:368. doi: 10.3389/fimmu.2015.00368
Wanschitz, J., Maier, H., Lassmann, H., Budka, H., and Berger, T. (2003). Distinct time pattern of complement activation and cytotoxic T cell response in guillain-barre syndrome. Brain 126, 2034–2042. doi: 10.1093/brain/awg207
Wex, H., Aumann, V., Hausler, M., Vorwerk, P., and Mittler, U. (2005). Chronic natural killer cell lymphocytosis is associated with elevated cytotoxic activity of natural killer cells. J. Pediatr. Hematol. Oncol. 27, 85–89. doi: 10.1097/01.mph.0000152571.06437.3f
Whiteside, G., Doyle, C. A., Hunt, S. P., and Munglani, R. (1998). Differential time course of neuronal and glial apoptosis in neonatal rat dorsal root ganglia after sciatic nerve axotomy. Eur. J. Neurosci. 10, 3400–3408. doi: 10.1046/j.1460-9568.1998.00346.x
Wiberg, R., Novikova, L. N., and Kingham, P. J. (2018). Evaluation of apoptotic pathways in dorsal root ganglion neurons following peripheral nerve injury. Neuroreport 29, 779–785. doi: 10.1097/WNR.0000000000001031
Williams, F. M., Scollen, S., Cao, D., Memari, Y., Hyde, C. L., Zhang, B., et al. (2012). Genes contributing to pain sensitivity in the normal population: an exome sequencing study. PLoS Genet 8:e1003095. doi: 10.1371/journal.pgen.1003095
Williams, J. W., Ferreira, C. M., Blaine, K. M., Rayon, C., Velazquez, F., Tong, J., et al. (2018). Non-apoptotic Fas (CD95) signaling on T cells regulates the resolution of Th2-mediated inflammation. Front, Immunol, 9:2521. doi: 10.3389/fimmu.2018.02521
Williams, P. R., Marincu, B. N., Sorbara, C. D., Mahler, C. F., Schumacher, A. M., Griesbeck, O., et al. (2014). A recoverable state of axon injury persists for hours after spinal cord contusion in vivo. Nat. Commun. 5:5683. doi: 10.1038/ncomms6683
Wong, K. M., Babetto, E., and Beirowski, B. (2017). Axon degeneration: make the Schwann cell great again. Neural Regen. Res. 12, 518–524. doi: 10.4103/1673-5374.205000
Wu, X. (2018). Genome expression profiling predicts the molecular mechanism of peripheral myelination. Int. J. Mol. Med. 41, 1500–1508. doi: 10.3892/ijmm.2017.3348
Xiao, W. H., Yu, A. L., and Sorkin, L. S. (1997). Electrophysiological characteristics of primary afferent fibers after systemic administration of anti-GD2 ganglioside antibody. Pain 69, 145–151. doi: 10.1016/s0304-3959(96)03280-0
Xie, W., Strong, J. A., and Zhang, J. M. (2017). Active nerve regeneration with failed target reinnervation drives persistent neuropathic pain. eNeuro 4:ENEURO.000817.2017. doi: 10.1523/ENEURO.0008-17.2017
Yang, H. L., Zhou, W. J., Chang, K. K., Mei, J., Huang, L. Q., Wang, M. Y., et al. (2017). The crosstalk between endometrial stromal cells and macrophages impairs cytotoxicity of NK cells in endometriosis by secreting IL-10 and TGF-beta. Reproduction 154, 815–825. doi: 10.1530/REP-17-0342
Yaron, A., and Schuldiner, O. (2016). Common and divergent mechanisms in developmental neuronal remodeling and dying back neurodegeneration. Curr. Biol. 26, R628–R639. doi: 10.1016/j.cub.2016.05.025
Yeager, M. P., Colacchio, T. A., Yu, C. T., Hildebrandt, L., Howell, A. L., Weiss, J., et al. (1995). Morphine inhibits spontaneous and cytokine-enhanced natural killer cell cytotoxicity in volunteers. Anesthesiology 83, 500–508. doi: 10.1097/00000542-199509000-00008
Yi, S., Zhang, H., Gong, L., Wu, J., Zha, G., Zhou, S., et al. (2015). Deep sequencing and bioinformatic analysis of lesioned sciatic nerves after crush injury. PLoS One 10:e0143491. doi: 10.1371/journal.pone.0143491
Yong, V. W. (2005). Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat. Rev. Neurosci. 6, 931–944. doi: 10.1038/nrn1807
Yoon, J. J., Song, J. A., Park, S. Y., and Choi, J. I. (2018). Cytotoxic activity and subset populations of peripheral blood natural killer cells in patients with chronic pain. Korean J. Pain 31, 43–49. doi: 10.3344/kjp.2018.31.1.43
Yoshii, F., and Shinohara, Y. (1998). Natural killer cells in patients with Guillain-Barre syndrome. J. Neurol. Sci. 157, 175–178. doi: 10.1016/s0022-510x(98)00065-3
Yu, A. L., Gilman, A. L., Ozkaynak, M. F., London, W. B., Kreissman, S. G., Chen, H. X., et al. (2010). Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 363, 1324–1334. doi: 10.1056/NEJMoa0911123
Yu, X., Liu, H., Hamel, K. A., Morvan, M. G., Yu, S., Leff, J., et al. (2020). Dorsal root ganglion macrophages contribute to both the initiation and persistence of neuropathic pain. Nat. Commun. 11:264. doi: 10.1038/s41467-019-13839-2
Yuan, X., Klein, D., Kerscher, S., West, B. L., Weis, J., Katona, I., et al. (2018). Macrophage depletion ameliorates peripheral neuropathy in aging mice. J. Neurosci. 38, 4610–4620. doi: 10.1523/JNEUROSCI.3030-17.2018
Zamai, L., Ahmad, M., Bennett, I. M., Azzoni, L., Alnemri, E. S., and Perussia, B. (1998). Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J. Exp. Med. 188, 2375–2380. doi: 10.1084/jem.188.12.2375
Zhou, Z., Zhang, C., Zhang, J., and Tian, Z. (2012). Macrophages help NK cells to attack tumor cells by stimulatory NKG2D ligand but protect themselves from NK killing by inhibitory ligand Qa-1. PLoS One 7:e36928. doi: 10.1371/journal.pone.0036928
Zhu, Y., Antony, J., Liu, S., Martinez, J. A., Giuliani, F., Zochodne, D., et al. (2006). CD8+ lymphocyte-mediated injury of dorsal root ganglion neurons during lentivirus infection: CD154-dependent cell contact neurotoxicity. J. Neurosci. 26, 3396–3403. doi: 10.1523/jneurosci.4767-05.2006
Zigmond, R. E., and Echevarria, F. D. (2019). Macrophage biology in the peripheral nervous system after injury. Prog. Neurobiol. 173, 102–121. doi: 10.1016/j.pneurobio.2018.12.001
Keywords: neuropathic pain, peripheral neuropathy, nerve injury, cellular cytotoxicity, innate immunity, natural killer cells, neuro-immunology, neuron-glia crosstalk
Citation: Davies AJ, Rinaldi S, Costigan M and Oh SB (2020) Cytotoxic Immunity in Peripheral Nerve Injury and Pain. Front. Neurosci. 14:142. doi: 10.3389/fnins.2020.00142
Received: 31 December 2019; Accepted: 04 February 2020;
Published: 21 February 2020.
Edited by:
Nick Andrews, Harvard Medical School, United StatesReviewed by:
Dara Bree, Cygnal Therapeutics, United StatesCopyright © 2020 Davies, Rinaldi, Costigan and Oh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander J. Davies, YWxleGFuZGVyLmRhdmllc0BuZGNuLm94LmFjLnVr; Michael Costigan, TWljaGFlbC5Db3N0aWdhbkBjaGlsZHJlbnMuaGFydmFyZC5lZHU=; Seog Bae Oh, b2RvbGJhZUBzbnUuYWMua3I=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.