
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 05 February 2020
Sec. Neurogenesis
Volume 14 - 2020 | https://doi.org/10.3389/fnins.2020.00023
This article is part of the Research TopicNeurogenesis Editor's Pick 2021View all 10 articles
 Juliana Magdalon1,2
Juliana Magdalon1,2 Fernanda Mansur1
Fernanda Mansur1 André Luiz Teles e Silva1,3
André Luiz Teles e Silva1,3 Vitor Abreu de Goes1,2
Vitor Abreu de Goes1,2 Orly Reiner4
Orly Reiner4 Andréa Laurato Sertié1*
Andréa Laurato Sertié1*Current evidence indicates that certain immune molecules such as components of the complement system are directly involved in neurobiological processes related to brain development, including neurogenesis, neuronal migration, synaptic remodeling, and response to prenatal or early postnatal brain insults. Consequently, complement system dysfunction has been increasingly implicated in disorders of neurodevelopmental origin, such as schizophrenia, autism spectrum disorder (ASD) and Rett syndrome. However, the mechanistic evidence for a causal relationship between impaired complement regulation and these disorders varies depending on the disease involved. Also, it is still unclear to what extent altered complement expression plays a role in these disorders through inflammation-independent or -dependent mechanisms. Furthermore, pathogenic mutations in specific complement components have been implicated in the etiology of 3MC syndrome, a rare autosomal recessive developmental disorder. The aims of this review are to discuss the current knowledge on the roles of the complement system in sculpting brain architecture and function during normal development as well as after specific inflammatory insults, such as maternal immune activation (MIA) during pregnancy, and to evaluate the existing evidence associating aberrant complement with developmental brain disorders.
The complement system has been increasingly implicated in multiple physiological and homeostatic functions, including development and maintenance of the central nervous system (CNS) (Orsini et al., 2014; Coulthard et al., 2018b), in addition to its well documented roles in immune surveillance and host defense against pathogens and injured cells (Kolev et al., 2014). Virtually all complement components can be locally produced in the brain, where they play critical roles in almost every aspect of normal brain development, including neurogenesis (Rahpeymai et al., 2006; Coulthard et al., 2017; Gorelik et al., 2017a, b), neuronal migration (Gorelik et al., 2017a, b), and synaptic refinement (Schafer et al., 2012; Stephan et al., 2012). Moreover, the complement system plays an important role in the maintenance of uninjured brain homeostasis, protecting from infection and inflammation, eliminating damaged cells and supporting regeneration (Alawieh et al., 2015; Hammad et al., 2018). However, in the injured, aged or diseased CNS, the synthesis of components of the complement pathway markedly increases and contributes to local inflammation and tissue damage, which may lead to blood brain barrier injury (Orsini et al., 2014; Hammad et al., 2018). Consequently, the brain parenchyma can be invaded by a number of peripheral blood-derived inflammatory cells and molecules, including complement proteins, which amplify local damage and brain malfunction (Brennan et al., 2012).
Given the multi-faceted functions of complement in CNS development, dysfunction of specific components of the complement system has been increasingly linked to developmental brain disorders, including 3MC syndrome, a rare autosomal recessive disorder with facial dysmorphism, growth deficiency and cognitive deficit (Sirmaci et al., 2010; Rooryck et al., 2011; Munye et al., 2017), as well as to more prevalent and genetically complex disorders, such as schizophrenia (Sekar et al., 2016) and autism spectrum disorder (ASD) (Warren et al., 1991; Fagan et al., 2017). However, despite recent advances, several aspects of the involvement of the complement system in the pathogenesis of these complex neurodevelopmental disorders are still unclear. It is not yet fully established, for example, whether aberrant complement expression produced locally during brain development plays an etiological role in these disorders independently of any role in inflammation, or whether aberrant complement activation both systemically and in the CNS, as a result of inflammatory insults during prenatal or early postnatal neurodevelopment, also plays a part in the pathophysiology of these disorders as a secondary event.
In support of the first possibility are, as abovementioned, recent findings reporting multiple emerging novel non-inflammatory roles of complement in every stage of brain development (Coulthard et al., 2018b), as well as data from genetic studies showing an association between risk variants in complement genes and neurodevelopmental disorders (Warren et al., 1991; Odell et al., 2005; Sekar et al., 2016). In addition, there are studies suggesting that the neuropathological and behavioral phenotypes in genetically modified mouse models with aberrant complement expression in the brain parallel known features of these human developmental brain disorders (Chu et al., 2010; Perez-Alcazar et al., 2014; Sekar et al., 2016; Comer et al., 2019).
In support of the latter possibility are data from different studies describing the presence of activated astrocytes and microglia in brains as well as the presence of altered expression of immune molecules, including complement components, in peripheral blood and/or cerebrospinal fluid obtained from individuals with schizophrenia and ASD (Mayilyan et al., 2006; Corbett et al., 2007; Morgan et al., 2010; Ashwood et al., 2011; Ishii et al., 2018). In addition, recent studies using animal models have suggested a role for the complement system in brain and behavioral abnormalities in offspring associated with prenatal maternal immune activation (MIA) (Pedroni et al., 2014; McDonald et al., 2015). In humans, severe maternal infections during pregnancy have been highlighted as a potential risk factor for these neurodevelopmental disorders (Patterson, 2009, 2011; Malkova et al., 2012; Knuesel et al., 2014; Coiro et al., 2015). One possibility is that immune molecules released by the maternal immune response can cross the placenta and enter the fetal brain, where they contribute to the pathological and behavioral changes. The ongoing immune dysregulation in the brain and the peripheral immune system of individuals with these neurodevelopmental diseases suggest that MIA or other inflammatory insults during prenatal or early postnatal development may induce an irregular immune phenotype that persists into adulthood.
This review aims to highlight the importance of the complement system in regulating the development of the healthy and diseased brain. First, we provide an overview of the well-known concepts of complement system activation in the immune system context. Then, we discuss recent progress in understanding the roles of the complement system in important physiological processes of normal brain development, as well as initial findings suggesting a potential role for complement in neuropathological and behavioral abnormalities in MIA offspring. Finally, we evaluate the current evidence for the involvement of the complement system dysfunction in disorders that trace their origin to abnormal brain development, including schizophrenia, ASD, Rett syndrome, and 3MC syndrome.
The complement cascade is composed of several soluble and membrane-bound proteins that are mainly secreted by the liver, but also by leukocytes, adipocytes, cells in the CNS (such as neurons, astrocytes, and microglia), among others (Veerhuis et al., 2011; Bajic et al., 2015). Complement is activated through the classical, lectin and alternative pathways, which are initiated by different stimuli and result in the generation of: (1) opsonins (such as C3b and C4b), which recognize and bind to target cells to facilitate their removal by phagocytic cells that express complement receptors (such as complement receptor type 1, CR1); (2) anaphylatoxin proteins (such as C3a and C5a), which are proinflammatory peptides that interact with and activate immune cells through interaction with their receptors (C3a receptor, C3aR, and C5a receptor, C5aR); (3) terminal membrane attack complex (MAC), which are pores that disrupt lipid bilayers and lyses targeted (opsonized) pathogens or self-damaged cells (Ricklin et al., 2010; Bajic et al., 2015; Figure 1).
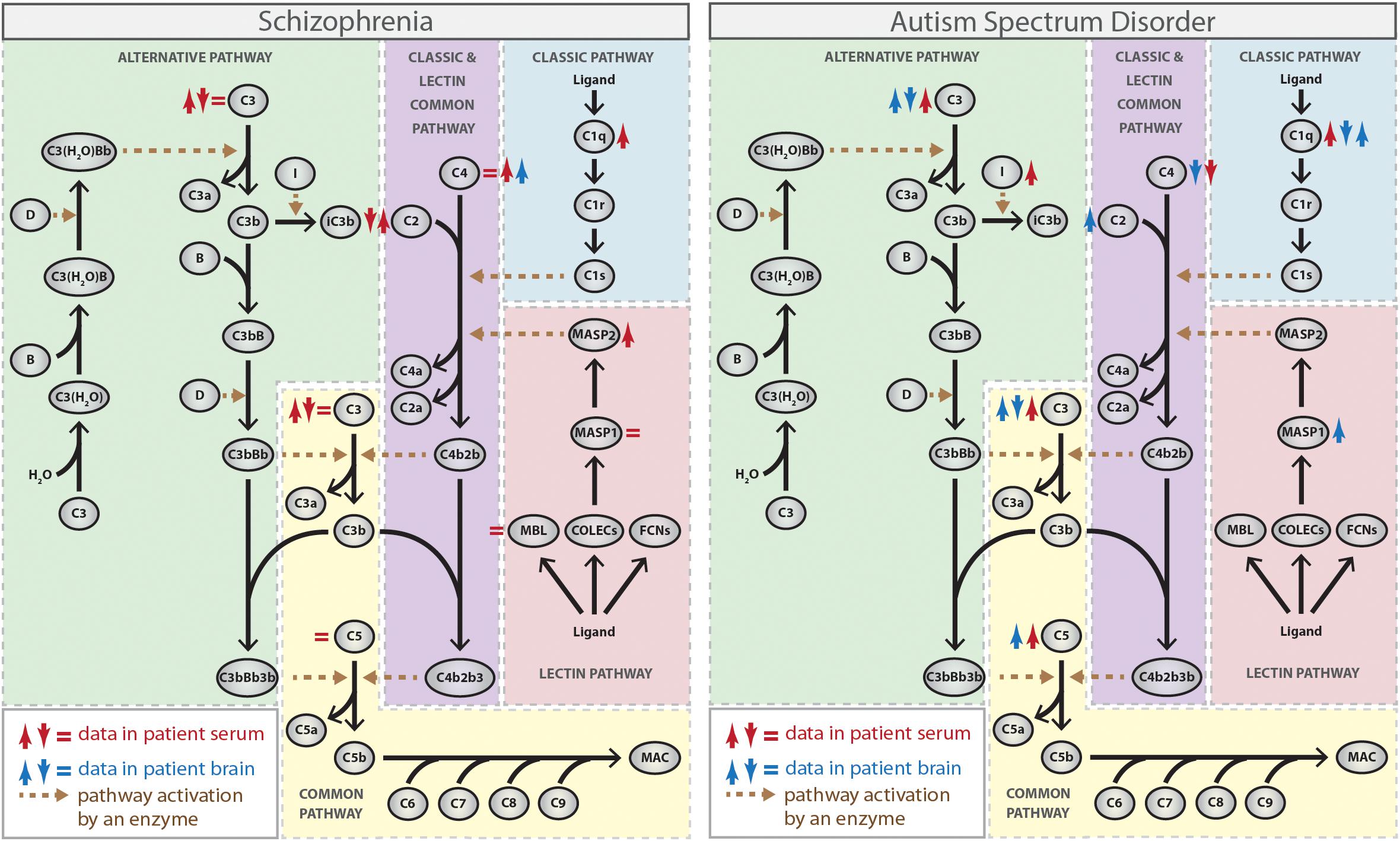
Figure 1. Complement activation pathways and the activity or expression of their individual components (either protein or mRNA) in schizophrenia and autism spectrum disorder (ASD). ↑ red, increased protein activity or expression in blood from patients; ↓ red, decreased protein activity or expression in blood from patients; ↑ blue, increased RNA expression in brain tissues from patients; ↓ blue, decreased RNA expression in brain tissues from patients (Spivak et al., 1989, 1993; Warren et al., 1994; Wong et al., 1996; Maes et al., 1997; Shcherbakova et al., 1999; Hakobyan et al., 2005; Mayilyan et al., 2006; Corbett et al., 2007; Boyajyan et al., 2010; Momeni et al., 2012; Nardone et al., 2014; Li et al., 2016; Sekar et al., 2016; Fagan et al., 2017; Shen et al., 2018). The Figure does not distinguish between strong and weak evidence.
The classical pathway is initiated by C1 complex activation, which consists of a recognition molecule C1q and two copies of each of the homologous C1r and C1s serine proteases. Initially, C1q binds to one of its ligands, such as antibodies, C-reactive protein and some structures on invading microorganisms or apoptotic cells, which triggers a conformational change within the C1 complex resulting in the activation of C1r, which subsequently cleaves and activates C1s (Gaboriaud et al., 2004). The activated C1s then cleaves the complement protein C4 into the fragments C4a and C4b. C4b may then opsonize the activator and facilitates phagocytosis. Additionally, C4b binds to the complement protein C2, which is subsequently cleaved by C1s into the fragments C2a and C2b. C2a remains attached to C4b and forms the C3 convertase C4b2b (previously termed C4b2a) of the classical pathway, which cleaves the complement protein C3 into the fragments C3a and C3b, triggering the final common part of the complement cascade (Muller-Eberhard et al., 1967; Dunkelberger and Song, 2010; Figure 1).
The lectin pathway is functionally similar to the classical pathway and also leads to formation of the C3 convertase C4b2b. However, the lectin pathway is initiated by binding of collectins [mannose-binding lectin (MBL), collectin-10 (COLEC10), and collectin-11 (COLEC11)] or ficolins (ficolin-1, ficolin-2, and ficolin-3) to sugar moieties or certain acetyl groups present on the surface of a large variety of pathogens, which leads to activation of the MBL-associated serine proteases (MASP) 1 and 2, structurally and functionally similar to C1r and C1s (Garred et al., 2016). While both enzymes cleave C2, only MASP2 cleaves C4, generating the C3 convertase C4b2b in a reaction analogous to the classical pathway (Chen and Wallis, 2004; Heja et al., 2012; Figure 1).
In contrast to the other two pathways, the alternative pathway does not depend on the recognition of exogenous materials and is constitutively active at low levels through a process called “tickover.” This pathway is activated by spontaneous hydrolysis of plasma C3 to form C3(H2O), which binds to factor B. Factor D cleaves factor B to form Ba and Bb, which then generates C3(H2O)Bb, the initial alternative pathway C3 convertase that can cleave C3 into C3a and C3b (Bexborn et al., 2008; Lachmann, 2018). Subsequently, C3b generated from any of the pathways can bind to factor B, which is cleaved by factor D, generating C3bBb, the main C3 convertase of the alternative pathway. C3bBb produces more C3b molecules, promoting an amplification loop for the entire system. Also, factor I cleaves C3b to iC3b, allowing iC3b to interact with leukocyte complement receptors (CR3 and CR4) and trigger the inflammatory response (Lachmann, 2009; Figure 1).
Therefore, the terminal phase of the complement cascade is similar for the classical, lectin, and alternative pathways and leads to the generation of C3 convertases. The incorporation of C3b into the C3 convertases generates C5 convertases (C4b2b3b for the classical and lectin pathways, and C3bBbC3b for the alternative pathway) that cleave C5 into C5a, which binds to its receptors on phagocytic cells, and C5b, which associates sequentially to other complement proteins (C6, C7, C8, and C9) forming the MAC and leading to cell lysis (Muller-Eberhard, 1985; Bajic et al., 2015; Figure 1).
It should be noted that the activity of the complement system is tightly regulated to protect host cells from indiscriminate attack and limit the deposition of complement molecules on pathogen and host surfaces. Among the complement regulators is SERPING1 (or C1-inhibitor), which binds to and inactivates C1r, C1s, and MASP-1/2 proteases, thus leading to inhibition of the classical and lectin pathways (Ricklin et al., 2010). Other regulators include: factor H, which acts as a cofactor for complement factor I (CFI) in the C3b cleavage and also favors the dissociation of factor Bb from C3b (Lachmann, 2009); CSMD1, which acts as a cofactor to CFI-mediated degradation of C4b and C3b and also inhibits MAC assembly by preventing binding of C7 to the C5b-6 complex (Escudero-Esparza et al., 2013); and CD59, which inhibits MAC formation by preventing binding of C9 to the C5b-8 complex (Farkas et al., 2002).
Besides playing a critical role during embryogenesis, the generation of new neurons is sustained throughout adulthood in the mammalian brain through the proliferation and differentiation of neural progenitor cells (NPC) present in neurogenic niches, mainly the subventricular zone of lateral ventricles and the subgranular zone of the hippocampal dentate gyrus (Fuentealba et al., 2012). Studies using animal and in vitro cell models have shown important roles for specific complement components in the regulation of neurogenesis both in the embryonic and adult brain under normal physiological conditions.
It has recently been shown that mouse embryos deficient for C3, Masp2 or treated with C3aR antagonist exhibit increased proliferation of NPC in the brain ventricular or subventricular zones, suggesting that these complement components inhibit NPC proliferation at early stages of cortical development (Gorelik et al., 2017a; Coulthard et al., 2018a). It is noteworthy, however, that an opposite trend was observed for C3aR knockout mouse embryos, that display decreased proliferation of NPC within the ventricular zone (Coulthard et al., 2018a; Table 1). This discrepancy between the use of C3aR pharmacological blocker and C3aR knockout may be attributed in part to combinatorial modulation of other signaling pathways in the absence of C3aR during the entire developmental period (Coulthard et al., 2018a). In the context of adult mouse brain, previous studies have shown that young adult mice lacking C3, C3aR or treated with C3aR antagonist exhibit reduced neurogenesis from NPC in the neurogenic niches, possibly due to impaired NPC differentiation rather than decreased proliferation of these cells (Rahpeymai et al., 2006). These findings were further corroborated by an in vitro study using NPC isolated from adult mouse brain showing that C3a stimulates their neuronal differentiation without altering their survival and proliferation (Shinjyo et al., 2009; Table 1). Consistent with the findings that C3a/C3aR signaling regulates neurogenesis, adult C3aR knockout mice show deficits in memory (Coulthard et al., 2018a).
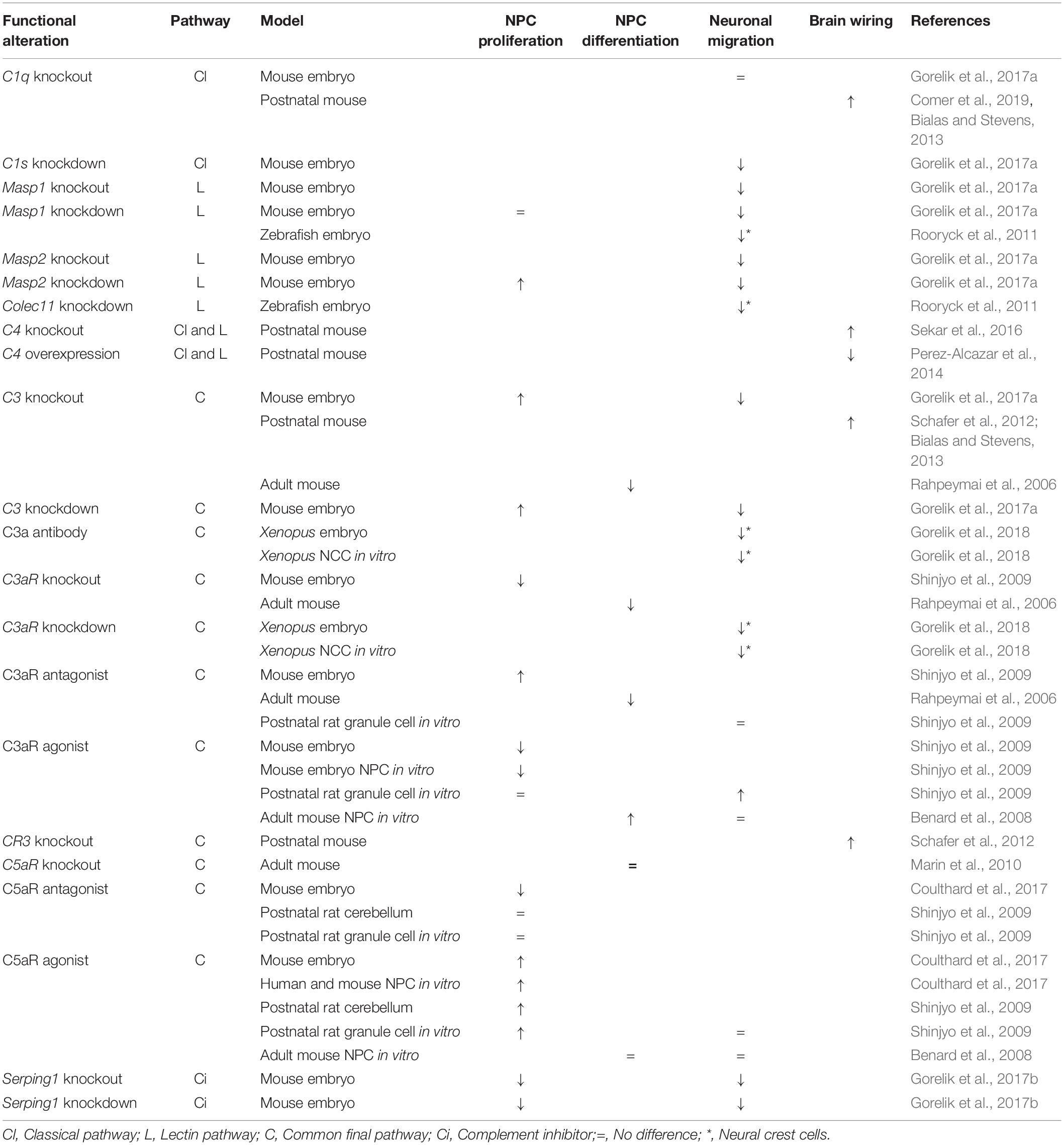
Table 1. Summary of the phenotypes observed after disturbances in the expression of individual components of the complement pathway.
Constitutive deficiency of C5aR and acute pharmacological blockade of C5aR during neurogenesis also caused opposing phenotypes of NPC proliferation. While the use of C5aR antagonist inhibits NPC proliferation in the ventricular zone of mouse embryos and lead to brain microstructural alterations and behavioral deficits (such as heightened anxiety, impaired coordination, and short-term memory) later in life (Coulthard et al., 2017), C5aR knockout mice exhibit increased proliferation of NPC within the ventricular zone (Coulthard et al., 2018a; Table 1). In addition, while in the postnatal rat cerebellar cortex a C5aR agonist was shown to stimulate proliferation of immature granule neurons, which suggests a role for the C5a-C5aR axis in the cerebellar histogenesis (Benard et al., 2008), C5a-C5aR1 signaling seems not to be involved in NPC proliferation and differentiation in the neurogenic niches of the adult brain (Bogestal et al., 2007; Shinjyo et al., 2009; Table 1).
Interestingly, it has recently been shown that mouse embryos deficient in the Serping1 gene, a known inhibitor of the classical and lectin pathways of the complement system, display decreased proliferation of both ventricular zone (radial) and intermediate (basal) progenitors during development of the cortex, suggesting that SERPING1 stimulates proliferation of NPC at early stages of cortical development (Gorelik et al., 2017b; Table 1). However, it is still unknown whether this function of SERPING1 is either dependent or independent on downstream activation of the complement system.
Together, the abovementioned studies suggest a role mostly for the anaphylatoxins in NPC proliferation and differentiation in the absence of other factors of the canonical pathogen-initiated complement activation routes. Also, these studies suggest that the spatiotemporal expression pattern of these complement components in different subsets of NPC seems to determine their role in progenitor neurogenesis.
Neuronal migration is an essential phenomenon for proper brain formation and establishment of neural circuit since most neurons must move from their birth position to their final location in the brain. During development, excitatory neurons arising from the proliferative neuroepithelium surface (the ventricular zone) exhibit mainly radial migration, in which early postmitotic neurons migrate along the processes of radial glial progenitors to their correct laminar position within the cortical plate. Inhibitory neurons are born in the ganglionic eminences and exhibit, initially, tangential migration, in which nascent neurons move in trajectories that are parallel to the ventricular surface (Marin et al., 2010). There is mounting evidence showing that the complement system plays an important role in the radial migration of pyramidal neurons during normal brain development.
A recent study has uncovered a direct role for the lectin arm of the complement system in radial neuronal migration in the developing cerebral cortex. C3-, Masp1- or Masp2-deficient mice exhibit impairments in radial migration resulting in improper positioning of neurons and disorganized cortical layers (Gorelik et al., 2017a). Importantly, the migration deficits observed in C3- or Masp2-deficient mice were partially rescued by addition of polypeptides that mimic C3 cleavage products, C3aR agonist or a dual C3aR/C5aR agonist, suggesting that activation of the lectin cascade leading to C3 cleavage, C3a and C5a generation and activation of both C3aR and C5aR are necessary for proper radial neuronal migration and cortical development (Gorelik et al., 2017a; Table 1).
It is also noteworthy that components of the lectin complement pathway, including MASP1 and CL-K1 (encoded by the Colec11 gene) proteins, as well as the C3a-C3aR axis, were shown to behave as early guidance cues to direct the migration of neural crest cells during embryonic vertebrate development, since deficiency of these components in zebrafish or Xenopus causes craniofacial abnormalities (Rooryck et al., 2011) or disorganized collective cell migration (Carmona-Fontaine et al., 2011; Table 1).
Abnormal radial neuronal migration was also observed in Serping1 knockout and knockdown mice but, unexpectedly, deficiency of SERPING1 resulted in a small but significant decrease in C3b levels, suggesting that the complement pathways are not being activated (Gorelik et al., 2017b). Moreover, addition of C3 mimicry cleavage products, or addition of a dual C3aR/C5aR agonist, but not a C3a peptide or a specific C3aR agonist, significantly improved impaired neuronal migration (Table 1). These findings corroborate a role for C3 cleavage and downstream complement activation at the level of C5a in controlling normal migration of newly born neurons to the cortical plate (Gorelik et al., 2017b). Interestingly, it has been recently shown that C3 knockout mouse embryos display reduced activity of the small GTPase Rac1 and reduced phosphorylation of Cofilin, a cytoskeleton protein, in migrating neurons entering the cortical plate. This suggests that Rac1 may be one of the key downstream mediators of the complement activity to control cytoskeletal remodeling required for proper neuronal migration in the developing brain (Gorelik et al., 2018).
In agreement with the results obtained with mouse embryos, previous studies have shown that adult mice deficient for C3a/C3aR signaling show impaired migration of both neuroblasts and newly formed neurons from the brain neurogenic niches (Rahpeymai et al., 2006), and that C3a stimulates migration of in vitro mouse adult brain NPC in response to low concentrations of the chemokine stromal cell-derived factor 1 alpha (Shinjyo et al., 2009). Also, the C3a-C3aR signaling was shown to regulate migration of immature granule neurons in the postnatal rat cerebellar cortex, suggesting that this signaling also plays a role in the cerebellum ontogenesis (Benard et al., 2008; Table 1).
Taken together, the existing findings indicate that functional activation of the lectin complement pathway and consequent production of anaphylatoxins are important for proper neuronal migration and correct positioning of neurons during brain development.
Formation of precise neural circuitry during development is essential for proper functions of the CNS. Synaptic contacts are generated in excess during early phases of development, and postnatally unnecessary synapses are eliminated while functionally important synapses are strengthened to construct appropriate neural circuits (Rakic et al., 1986; Riccomagno and Kolodkin, 2015). Much of the current understanding about the mechanisms underlying synapse refinement during CNS development has been studied using the mouse retinogeniculate pathway. Early in development, axons from retinal ganglion cells form transient connections with neurons of dorsal lateral geniculate nucleolus (dLGN) of the thalamus and, during early post-natal development, these connections are sculpted through the pruning of redundant synapses (Hooks and Chen, 2006). Important studies have revealed that mice deficient for complement C1q, C3, C4 or microglia-specific CR3 exhibit impaired elimination of retinogeniculate synapses and defects in eye-specific segregation (Stevens et al., 2007; Schafer et al., 2012; Sekar et al., 2016), suggesting that the classical complement system plays a crucial role in synapse pruning during development (Table 1). Mechanistically, it has been proposed that transforming growth factor (TGF)-β released by retinal astrocytes induces the expression of C1q in retinal ganglion cells, which is transported from cell bodies along axons to the dLGN, where it is released to bind weak synapses (Bialas and Stevens, 2013). Similar to the immune system, the binding of C1q and formation of C1 complex results in activation of the classical complement pathway, cleavage of C4, formation of C3 convertase and then production of C3a and C3b/iC3b fragments. Activated C3 (iC3b) binds to CR3 in microglia ultimately promoting the engulfment of overlapping and weaker synapses (Schafer et al., 2012).
It is noteworthy that, in accordance with the abovementioned data using the mouse retinogeniculate pathway, C1q- or C3-deficient mice exhibit increased number of excitatory synapses in the cortex and hippocampus, respectively (Chu et al., 2010; Perez-Alcazar et al., 2014), epilepsy (Chu et al., 2010) or abnormal hippocampus-dependent learning (Perez-Alcazar et al., 2014; Table 1). Also, C3 knockdown specifically in the prefrontal cortex lead to repetitive behavior and impaired social interaction in mice, possibly due to reduced synaptic pruning (Fagan et al., 2017). In addition, a study using cortical wild type neurons co-cultured with astrocytes derived from IkBα knockout mice, which overexpress the transcription factor NFkB and consequently complement C3, has shown that C3 released by astrocytes acts through neuronal C3aR reducing excitatory synaptic density and dendritic length and complexity (Lian et al., 2015). Moreover, it was shown that overexpression of C4 in mouse prefrontal cortex leads to dendritic spine dysgenesis, reduced connectivity in cortical neurons, enhanced microglia-mediated engulfment of synapses and deficits in social interactions (Comer et al., 2019; Table 1).
Although the mechanisms that drives the opsonization of weaker synapses are not yet fully understood, a recent study using whole cortical tissue from newborn and adult mice has shown that C1q is predominantly localized to the presynaptic region of the labeled synapses, and that C1q-tagged synapses display downregulation of proteins associated with synaptic transmission and increased expression of apoptotic markers, suggesting that weaker synapses induce apoptotic-like mechanisms that attracts C1q and then triggers synaptic elimination by microglia (Gyorffy et al., 2018).
Altogether, the findings abovementioned suggest that microglia- and astroglia-mediated complement-dependent synaptic refinement occurs in different regions of the developing CNS, and that alterations in this process affect behavior in animal models.
The developing brain is particularly vulnerable to environmental insults, such as ischemic and inflammatory insults, that can cause injury and long-term neurodevelopmental abnormalities manifesting as cognitive difficulties or behavioral problems (Meyer et al., 2006; Bilbo and Schwarz, 2009). Although the link between environmental risk factors, the immune response, and neurological dysfunction is not completely clear at present, accumulating evidence suggests that MIA via infection during pregnancy alters brain development and increases the risk for neurodevelopmental disorders in the offspring (Patterson, 2009, 2011; Malkova et al., 2012; Coiro et al., 2015). In this regard, some studies using animal models have reported a role for the complement system in brain abnormalities in the offspring following MIA.
Using a mouse model of inflammation-induced preterm birth and brain injury, a study has shown that treatment of pregnant mice with lipopolysaccharide (LPS) induced an increase in the levels of C5a in both the amniotic fluid and the fetal brain, as well as cortical abnormalities in the preterm fetuses, characterized by decreased expression of neuronal markers and increased cell death (Pedroni et al., 2014). Interestingly, these fetal cortical brain alterations associated with LPS-induced preterm birth were not observed in fetuses deficient for C5aR or born from mice treated with anti-C5 antibody (Pedroni et al., 2014). Furthermore, the neurotoxic effect of C5a was confirmed in vitro by treating isolated fetal cortical neurons with this anaphylatoxin, which inhibited the growth of neurites and increased cell death, phenotypes that were blocked by C5aR antagonist (Pedroni et al., 2014). Similar results were observed in another study using a mouse model of malaria in pregnancy, in which the offspring from infected mice has shown impaired learning and memory and depressive-like behavior compared to non-infected controls. The neurocognitive impairments observed in the malaria-exposed offspring were rescued by deletion of C5aR in the fetuses or by treating infected pregnant mice with anti-C5 antibody (McDonald et al., 2015). Together, these findings suggest a new role for the anaphylatoxin C5a in the cortical brain damages and behavioral disturbances observed in fetuses exposed to prenatal inflammation.
More recent studies have shown that MIA induced by synthetic dsRNA (polyI:C) in pregnant rodents, which acts to initiate an inflammatory response similar to that caused by viral infection, caused long-term increase in C1q (Han et al., 2017) and C4 (Duchatel et al., 2018) expression in the cortex of their offspring. The involvement of C1q and C4 in synaptic pruning (Stevens et al., 2007; Schafer et al., 2012; Sekar et al., 2016) suggests that these complement molecules may be involved in MIA-associated brain abnormalities in the offspring, but additional studies are necessary to confirm this possibility.
Schizophrenia (MIM 181500) is a chronic and disabling mental disorder that affects about 1% of the world population (Hafner and der Heiden, 1997; McGrath et al., 2008). The symptoms of schizophrenia include psychosis and deficits in cognition and social interaction, which most commonly emerge in late adolescence or early adulthood (Howes and Murray, 2014). Neuropathological findings in individuals with schizophrenia include abnormal cortical organization possibly due to altered neuronal migration (Akbarian et al., 1996; Arnold et al., 1997; Shenton et al., 2001) and reduced cortical gray matter thickness (Selemon and Goldman-Rakic, 1999; Cannon et al., 2002, 2015) and diminished synaptic density possibly due to excessive pruning of cortical synapses, thus producing hypoconnectivity of prefrontal cortex (Glantz and Lewis, 2000; Glausier and Lewis, 2013). Although the exact mechanisms underlying schizophrenia are not yet fully understood, both complex genetic and environmental risk factors have been implicated in its pathogenesis. Importantly, accumulating evidence suggests that complement dysregulation is among the risk factors for the disorder.
The earliest studies of the complement system in schizophrenia have focused on the complement hemolytic activity to measure both the overall function of the classical pathway and the function of its specific components in blood. Although the results from the different studies were diverse with some studies reporting either a decrease (Spivak et al., 1989, 1993) or no difference (Sasaki et al., 1994) in complement total hemolytic activity in patients compared to control individuals, the majority of data, mainly concerning individual complement components (such as C1, C2, C3, and C4), points toward higher activity of the classical pathway in patients (Maes et al., 1997; Shcherbakova et al., 1999; Hakobyan et al., 2005; Mayilyan et al., 2006; Figure 1). Accordingly, increased levels of C1q attached to circulating immune complexes (C1q-CIC) and increased expression of CR1 on blood cells, which can bind to C1q-CIC and mediate clearance of immune-complexes, were found in patients with schizophrenia (Arakelyan et al., 2011; Figure 1). Also, increased circulating C1q levels were observed in mothers of infants who later developed schizophrenia, which suggest that exposure of the fetal brain to maternally derived C1q might be a contributory factor for the disease (Severance et al., 2014), as suggested by rodent models of MIA. In addition, it is noteworthy that decreased expression of CSMD1, which codes for an inhibitor of the classical complement pathway, was observed in serum from patients with schizophrenia (Liu et al., 2019).
Curiously, a negative correlation was recently found between superior frontal cortical thickness and the expression levels of C5 and SERPING1 genes in peripheral blood mononuclear cells from a sample of adult Swedish twins enriched for schizophrenia patients, who are known to show reduced cortical gray matter thickness (Allswede et al., 2018). Whereas C5 is an activator of the complement system, SERPING1 codes for a protease involved in the inhibition of the classical and lectin complement cascades, and its increased expression could reflect a compensatory mechanism for higher complement activity. Although this finding cannot establish causality and further studies are clearly needed, it is tempting to speculate that the relationship between enhanced expression of these specific complement components in the blood and cortical thickness may represent a trace of earlier immune dysregulation during development.
While several lines of evidence support the involvement of the classical complement pathway in schizophrenia susceptibility, only a few studies have addressed specifically the role of the lectin and alternative pathways in the disease. Data on the involvement of the lectin cascade show higher MBL-bound MASP2 activity in serum from individuals with schizophrenia, suggesting increased activity of this pathway in the disorder (Mayilyan et al., 2006; Figure 1). On the other hand, conflicting results exist regarding the involvement of the alternative cascade, because a hemolysis-based assay indicated an upregulation of the alternative pathway (Boyajyan et al., 2010), while an ELISA-based assay identified suppression of the same cascade in the peripheral blood from patients with schizophrenia (Li et al., 2012). Therefore, further studies are required to confirm whether dysfunction of lectin and alternative complement cascades contribute somehow to schizophrenia.
As mentioned above, all of the complement pathways converge at the point of C3 and data related to C3 levels in blood from schizophrenia patients are also controversial. Some studies found no alteration (Spivak et al., 1993) or increased levels of C3 in patients compared to controls (Maes et al., 1997; Shcherbakova et al., 1999; Hakobyan et al., 2005; Boyajyan et al., 2010), while others found a decrease and suggest a negative correlation between the serum levels of C3 and the severity of the symptoms (Wong et al., 1996; Li et al., 2016; Figure 1). The reasons for these differences are unknown, but may include either population-specific genetics and environmental risk factors or the stage of illness (acute vs. chronic). Thus, additional studies using larger groups are needed to elucidate the precise nature of the relationship between C3 levels and schizophrenia susceptibility.
Genetic studies evaluating the association between complement gene polymorphisms and schizophrenia have also been conducted. Initial studies have yielded unreliable or conflicting results due to lack of replication or small sample sizes (Rudduck et al., 1985; Fananas et al., 1992; Wang et al., 1992; Schroers et al., 1997; Mayilyan et al., 2008; Zakharyan et al., 2011). However, more recent large-scale genome-wide association studies evaluating very large sample collections for hundreds of thousands of single-nucleotide polymorphisms (SNPs) have shown variants significantly associated with higher risk of schizophrenia at CSMD1 (Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium, 2011) and C4 genes (Sekar et al., 2016). CSMD1 is highly expressed in the CNS (Kraus et al., 2006) and its schizophrenia risk allele was associated with poor performance on neuropsychological measures of general cognitive ability and memory function (Donohoe et al., 2013; Koiliari et al., 2014). C4 is encoded by two different genes, C4A and C4B, which vary in structure and copy number leading to a wide range of expression levels of each isotype. Interestingly, the strongest association with schizophrenia was found with alleles that increase expression of C4A (Sekar et al., 2016). Accordingly, higher C4A expression was observed in brain samples from patients with schizophrenia compared to controls (Sekar et al., 2016) and a positive correlation was found between the copy numbers of C4 and neuropil contraction in different brain regions in patients (Prasad et al., 2018; Figure 1), which strongly suggest that increased C4 levels constitute a risk factor for schizophrenia.
Whereas the abnormal complement expression in the peripheral blood of both patients with schizophrenia and their mothers suggests that complement dysfunction as part of standard immune pathways may contribute to schizophrenia in a subset of patients, findings from several of the abovementioned studies also suggest that the clinical and neuropathological phenotypes of schizophrenia may also be, at least in part, due to dysfunction of locally synthesized complement in the brain during specific periods of neural development. The strong association between increased C4A expression with schizophrenia (Sekar et al., 2016), the involvement of classical complement cascade in synapse elimination (Stevens et al., 2007; Schafer et al., 2012; Bialas and Stevens, 2013; Sekar et al., 2016; Comer et al., 2019), and the decreased brain connectivity and sociability in mice overexpressing C4 in the prefrontal cortex (Comer et al., 2019) strongly suggest that enhanced complement-mediated synaptic pruning contributes directly to reduction in cortical gray matter thickness and to schizophrenia pathogenesis. On the other hand, while it is highly attractive to speculate that complement-mediated dysregulation in neuronal migration may contribute to the pathogenesis of schizophrenia, further detailed studies are still required to directly establish a causal link.
Autism spectrum disorder (MIM 209850) comprises a heterogeneous group of early onset neurodevelopmental diseases characterized by impairments in social-communicative skills and repetitive behaviors (APA, 2013) that affects at least 1.5% of the population worldwide (Christensen et al., 2016). The most consistent neuropathological findings in patients with ASD include increased cortical surface area during early childhood (Miles et al., 2000; Hazlett et al., 2011) and reduced number of Purkinje cells in the cerebellum (Allen, 2005) possibly due to abnormal progenitor cell neurogenesis, altered cortical organization (presence of heterotopias and more frequent and narrower minicolumns) suggestive of abnormal neuronal migration (Casanova et al., 2002; Whitney et al., 2008; Stoner et al., 2014), and increased cortical dendritic spine densities possibly due to defective synapse elimination during brain development (Hutsler and Zhang, 2010; Tang et al., 2014). Recent molecular genetic studies have identified a specific cause for ASD in almost 30% of the cases, while in the remaining cases the underlying pathogenic mechanisms may involve complex genetic and environmental risk factors (Bourgeron, 2015). Preliminary evidence suggests a possible role for the complement system in the pathogenesis of ASD.
Genetic association studies have reported that the complement C4B gene null allele has increased frequency in patients with ASD compared to control individuals (Warren et al., 1991; Odell et al., 2005; Mostafa and Shehab, 2010) and, accordingly, a significant decrease in the plasma levels of C4B protein was observed in ASD patients (Warren et al., 1994; Figure 1). On the other hand, proteomic analyses have suggested that the levels of other complement system proteins, such as C1q, C3, and C5, are elevated in plasma from patients with ASD (Corbett et al., 2007; Shen et al., 2018; Figure 1). In addition, a significantly increased activity of CFI, a negative regulatory component of the alternative pathway responsible for cleavage and inactivation of C3b and C4b, has been observed in plasma from ASD patients (Momeni et al., 2012).
The expression of complement system components in brain tissues from patients with ASD was also investigated and some conflicting results were obtained. While a genome-wide DNA methylation profiling of prefrontal cortex (Brodmann areas BA10 and BA24) has shown hypomethylation and, consequently, overexpression of C1q, C3, and CR3 genes in the ASD brains (Nardone et al., 2014), a more recent study found decreased mRNA levels of C1q, C3, and C4, and increased mRNA levels of C2, C5, and MASP1 in the middle frontal gyrus from patients compared to controls (Fagan et al., 2017; Figure 1). The discrepancies obtained in the expression of C1q and C3 genes in brain tissues from patients with ASD could be due to the analysis of different brain regions and/or analysis of different subgroups of patients combined with small sample sizes and need to be clarified in further studies.
Collectively, the evidence regarding association between complement dysfunction and ASD is far weaker than the evidence of complement dysregulation in schizophrenia susceptibility, and a recent large-scale genome-wide association study did not report common variants in complement genes significantly associated with ASD (Grove et al., 2019). However, the altered complement system expression in peripheral blood and in brain from patients with ASD might suggest that an aberrant activity of this system may somehow contribute to ASD. Based on decreased expression of some complement components in post-mortem brain of ASD patients (Fagan et al., 2017), the increased number of dendritic spines and glutamate synapses in ASD brains (Hutsler and Zhang, 2010; Tang et al., 2014) as well as in C1q- and C3-deficient mouse brains (Chu et al., 2010; Perez-Alcazar et al., 2014), the reduced elimination of retinogeniculate synapses in mice lacking C4 (Sekar et al., 2016) and the ASD-like phenotypes observed in C3-deficient mice (Perez-Alcazar et al., 2014; Fagan et al., 2017), it is tempting to speculate that diminished complement-mediated synaptic pruning, among other known mechanisms (Tang et al., 2014), may contribute to the cortical hyperconnectivity and behavioral phenotypes in ASD. Nevertheless, additional studies are clearly necessary to further explore the possible role of complement dysregulation in key aspects of ASD neuropathology.
Finally, it is noteworthy that, as occurs with schizophrenia and ASD, immune dysregulation may also contribute to some of the neuropathological features of Rett syndrome (RTT; MIM 312750) (Cortelazzo et al., 2014; De Felice et al., 2014; O’Driscoll et al., 2015), an X-linked progressive neurodevelopmental disorder primarily affecting girls at a frequency of 1:10,000 live female births. Although RTT is not classified as an ASD and is recognized as a distinct pathological entity (APA, 2013) (DSM-5), RTT may show overlapping symptoms with ASD, and is characterized by typical early development until age 6–18 months followed by a rapid deceleration in growth associated with progressive loss of acquired motor and language skills, severe cognitive impairment, intractable seizures, spasticity and stereotypic hand movements (Chahrour and Zoghbi, 2007). More than 95% of classic RTT cases are caused by sporadic loss-of-function mutations in the gene encoding MECP2 (Amir et al., 1999), a transcriptional regulator of gene expression that acts through epigenetic mechanisms on chromatin structure (Nan et al., 1998; Bienvenu and Chelly, 2006). Interestingly, a proteomic study has shown that the levels of several proteins involved in the immune system are altered in plasma from patients with RTT, including overexpression of complement factor B of the alternative complement pathway (Cortelazzo et al., 2014). Also, a recent genome-wide transcriptome (RNA-seq) analysis of post-mortem brain samples (from both frontal and temporal cortex) from young RTT patients showed that all three genes encoding complement C1q complex (C1QA, C1QB, C1QC) were downregulated in RTT human brains as they are in Mecp2 knockout mice (Lin et al., 2016), which suggest that the expression of these genes is regulated by MECP2. Although the involvement of C1q in RTT pathogenesis has not yet been clarified and additional studies are required, the role of C1q in regulating dendritic spine density (Chu et al., 2010; Perez-Alcazar et al., 2014) may contribute somehow to RTT.
3MC syndrome (MIM 257920; 265050; 248340) comprises a rare developmental disorder that unifies four autosomal recessive diseases with overlapping features previously known as Mingarelli, Malpeuch, Michels, and Carnevale syndromes (Titomanlio et al., 2005). This syndrome is characterized by a spectrum of developmental anomalies that include facial dysmorphism, cleft lip and/or palate, postnatal growth deficiency, learning disability and hearing impairment. Less often, individuals with 3MC syndrome may also show craniosynostosis, genital, limb, and vesicorenal anomalies (Titomanlio et al., 2005; Leal et al., 2008).
Several different potentially deleterious mutations in genes of the lectin arm of the complement system - such as MASP1, COLEC10, and COLEC11 genes – have been described in patients with 3MC syndrome (Sirmaci et al., 2010; Rooryck et al., 2011; Atik et al., 2015; Urquhart et al., 2016; Gardner et al., 2017; Munye et al., 2017; Graul-Neumann et al., 2018; Basdemirci et al., 2019). It has recently been shown that disease-associated mutations in COLEC10 and COLEC11 inhibit either production or secretion of encoded proteins in mammalian cells (Rooryck et al., 2011; Venkatraman Girija et al., 2015), which may explain the undetectable levels of CL-K1 in the serum of affected individuals (Rooryck et al., 2011). However, it is noteworthy that although at least some of these mutations impair the normal function of the encoded proteins, normal levels of downstream reaction cascade components (C2, C3, and C4) were found in serum from patients, suggesting a possible direct role of these lectin cascade proteins in 3MC syndrome pathogenesis independently of standard lectin pathway activation (Rooryck et al., 2011).
Studies using zebrafish (Rooryck et al., 2011) and an in vitro cell model (Munye et al., 2017) have shown that these components of the lectin pathway act as chemoattractants to guide cell migration and suggest that the craniofacial abnormalities in 3MC syndrome are the result of deficient migration of neural crest cells during development (Rooryck et al., 2011). Also, the involvement of components of the lectin pathway in regulating neuronal migration in the developing cerebral cortex (Gorelik et al., 2017a) also suggests that the cognitive impairment observed in 3MC patients may be explained in part by deficits in neuronal migration. Further studies, however, are necessary to further understand the mechanisms by which dysfunctional MASP1, COLEC10, and COLEC11 genes may lead to 3MC syndrome.
Here, we have highlighted the emerging functions of the complement system as a key regulator of normal CNS development. Components of lectin and terminal complement pathways, mainly the C3a and C5a anaphylatoxins, have been shown to regulate neural progenitor cell proliferation, neurogenesis and neuronal migration. Components of classical and terminal complement pathways, such as C1q, C3, and C4, have been shown to tag weaker synapses for removal by microglia, which sculpts brain connectivity. Consequently, alterations in the expression of complement components in the brain may lead to long-lasting changes in brain development and function. The severe autosomal recessive 3MC syndrome originates from rare pathogenic mutations in genes of the lectin pathway that regulate cell migration during embryonic development. In both schizophrenia and ASD, evidence is growing that abnormal complement signaling owing to genetic mutations or as a result of inflammatory insults during prenatal or early postnatal development may lead to changes in brain connectivity and may contribute to disease pathophysiology. Although research into the molecular mechanisms downstream from complement components has just begun to be explored, the progress in this field holds tremendous promise not only for increasing our understanding of neurodevelopment, but also for elucidating and potentially even treating neurodevelopmental disorders.
All authors have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by São Paulo Research Foundation (FAPESP) (Grant No. 2015/50138-4), Helen and Martin Kimmel Institute for Stem Cell Research, Nella and Leon Benoziyo Center for Neurological Diseases, David and Fela Shapell Family Center for Genetic Disorders Research, Brenden-Mann Women’s Innovation Impact Fund, Richard F. Goodman Yale/Weizmann Exchange Program, The Irving B. Harris Fund for New Directions in Brain Research, Irving Bieber, M.D. and Toby Bieber, M.D. Memorial Research Fund, Leff Family Barbara & Roberto Kaminitz, Sergio & Sônia Lozinsky Debbie Koren Jack and Lenore Lowenthal, and Dears Foundation. OR was supported by the Israel Science Foundation (Grant No. 347/15) and the ISF-NSFC Joint Research Program (Grant No. 2449/16). OR is an Incumbent of the Bernstein-Mason Professorial Chair of Neurochemistry and Head of the M. Judith Ruth Institute for Preclinical Brain Research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Akbarian, S., Kim, J. J., Potkin, S. G., Hetrick, W. P., Bunney, W. E. Jr., and Jones, E. G. (1996). Maldistribution of interstitial neurons in prefrontal white matter of the brains of schizophrenic patients. Arch. Gen. Psychiatry 53, 425–436. doi: 10.1001/archpsyc.1996.01830050061010
Alawieh, A., Elvington, A., and Tomlinson, S. (2015). Complement in the homeostatic and ischemic brain. Front. Immunol. 6:417. doi: 10.3389/fimmu.2015.00417
Allswede, D. M., Zheutlin, A. B., Chung, Y., Anderson, K., Hultman, C. M., Ingvar, M., et al. (2018). Complement gene expression correlates with superior frontal cortical thickness in humans. Neuropsychopharmacology 43, 525–533. doi: 10.1038/npp.2017.164
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
APA, (2013). Diagnostic and Statistical Manual of Mental Disorders. 5th Edn. Washington, DC: American Psychiatric Association.
Arakelyan, A., Zakharyan, R., Khoyetsyan, A., Poghosyan, D., Aroutiounian, R., Mrazek, F., et al. (2011). Functional characterization of the complement receptor type 1 and its circulating ligands in patients with schizophrenia. BMC Clin. Pathol. 11:10. doi: 10.1186/1472-6890-11-10
Arnold, S. E., Ruscheinsky, D. D., and Han, L. Y. (1997). Further evidence of abnormal cytoarchitecture of the entorhinal cortex in schizophrenia using spatial point pattern analyses. Biol. Psychiatry 42, 639–647. doi: 10.1016/s0006-3223(97)00142-x
Ashwood, P., Krakowiak, P., Hertz-Picciotto, I., Hansen, R., Pessah, I., and Van de Water, J. (2011). Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 25, 40–45. doi: 10.1016/j.bbi.2010.08.003
Atik, T., Koparir, A., Bademci, G., Foster, J. III, Altunoglu, U., Mutlu, G. Y., et al. (2015). Novel MASP1 mutations are associated with an expanded phenotype in 3MC1 syndrome. Orphanet J. Rare Dis. 10:128. doi: 10.1186/s13023-015-0345-343
Bajic, G., Degn, S. E., Thiel, S., and Andersen, G. R. (2015). Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J. 34, 2735–2757. doi: 10.15252/embj.201591881
Basdemirci, M., Sen, A., and Ceylaner, S. (2019). Novel mutation in MASP1 gene in a new family with 3MC syndrome. Clin. Dysmorphol. 28, 91–93. doi: 10.1097/MCD.0000000000000256
Benard, M., Raoult, E., Vaudry, D., Leprince, J., Falluel-Morel, A., Gonzalez, B. J., et al. (2008). Role of complement anaphylatoxin receptors (C3aR, C5aR) in the development of the rat cerebellum. Mol. Immunol. 45, 3767–3774. doi: 10.1016/j.molimm.2008.05.027
Bexborn, F., Andersson, P. O., Chen, H., Nilsson, B., and Ekdahl, K. N. (2008). The tick-over theory revisited: formation and regulation of the soluble alternative complement C3 convertase (C3(H2O)Bb). Mol. Immunol. 45, 2370–2379. doi: 10.1016/j.molimm.2007.11.003
Bialas, A. R., and Stevens, B. (2013). TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat. Neurosci. 16, 1773–1782. doi: 10.1038/nn.3560
Bienvenu, T., and Chelly, J. (2006). Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet. 7, 415–426. doi: 10.1038/nrg1878
Bilbo, S. D., and Schwarz, J. M. (2009). Early-life programming of later-life brain and behavior: a critical role for the immune system. Front. Behav. Neurosci. 3:14. doi: 10.3389/neuro.08.014.2009
Bogestal, Y. R., Barnum, S. R., Smith, P. L., Mattisson, V., Pekny, M., and Pekna, M. (2007). Signaling through C5aR is not involved in basal neurogenesis. J. Neurosci. Res. 85, 2892–2897. doi: 10.1002/jnr.21401
Bourgeron, T. (2015). From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 16, 551–563. doi: 10.1038/nrn3992
Boyajyan, A., Khoyetsyan, A., and Chavushyan, A. (2010). Alternative complement pathway in schizophrenia. Neurochem. Res. 35, 894–898. doi: 10.1007/s11064-010-0126-122
Brennan, F. H., Anderson, A. J., Taylor, S. M., Woodruff, T. M., and Ruitenberg, M. J. (2012). Complement activation in the injured central nervous system: another dual-edged sword? J. Neuroinflam. 9:137. doi: 10.1186/1742-2094-9-137
Cannon, T. D., Chung, Y., He, G., Sun, D., Jacobson, A., van Erp, T. G., et al. (2015). Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol. Psychiatry 77, 147–157. doi: 10.1016/j.biopsych.2014.05.023
Cannon, T. D., Thompson, P. M., van Erp, T. G., Toga, A. W., Poutanen, V. P., Huttunen, M., et al. (2002). Cortex mapping reveals regionally specific patterns of genetic and disease-specific gray-matter deficits in twins discordant for schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 99, 3228–3233. doi: 10.1073/pnas.052023499
Carmona-Fontaine, C., Theveneau, E., Tzekou, A., Tada, M., Woods, M., Page, K. M., et al. (2011). Complement fragment C3a controls mutual cell attraction during collective cell migration. Dev. Cell 21, 1026–1037. doi: 10.1016/j.devcel.2011.10.012
Casanova, M. F., Buxhoeveden, D. P., Switala, A. E., and Roy, E. (2002). Minicolumnar pathology in autism. Neurology 58, 428–432. doi: 10.1212/wnl.58.3.428
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. doi: 10.1016/j.neuron.2007.10.001
Chen, C. B., and Wallis, R. (2004). Two mechanisms for mannose-binding protein modulation of the activity of its associated serine proteases. J. Biol. Chem. 279, 26058–26065. doi: 10.1074/jbc.M401318200
Christensen, D. L., Bilder, D. A., Zahorodny, W., Pettygrove, S., Durkin, M. S., Fitzgerald, R. T., et al. (2016). Prevalence and characteristics of autism spectrum disorder among 4-year-old children in the autism and developmental disabilities monitoring network. J. Dev. Behav. Pediatr. 37, 1–8. doi: 10.1097/DBP.0000000000000235
Chu, Y., Jin, X., Parada, I., Pesic, A., Stevens, B., Barres, B., et al. (2010). Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc. Natl. Acad. Sci. U.S.A. 107, 7975–7980. doi: 10.1073/pnas.0913449107
Coiro, P., Padmashri, R., Suresh, A., Spartz, E., Pendyala, G., Chou, S., et al. (2015). Impaired synaptic development in a maternal immune activation mouse model of neurodevelopmental disorders. Brain Behav. Immun. 50, 249–258. doi: 10.1016/j.bbi.2015.07.022
Comer, A. L. J., Kretsge, L. N., Nguyen, T. P. H., Lee, J., Newmark, E. R., Hausmann, F. S., and Rosenthal, S. (2019). Increased expression of schizophrenia-associated gene C4 leads to hypoconnectivity of prefrontal cortex and reduced social interaction. bioRxiv[Preprint] doi: 10.1101/598342
Corbett, B. A., Kantor, A. B., Schulman, H., Walker, W. L., Lit, L., Ashwood, P., et al. (2007). A proteomic study of serum from children with autism showing differential expression of apolipoproteins and complement proteins. Mol. Psychiatry 12, 292–306. doi: 10.1038/sj.mp.4001943
Cortelazzo, A., De Felice, C., Guerranti, R., Signorini, C., Leoncini, S., Pecorelli, A., et al. (2014). Subclinical inflammatory status in Rett syndrome. Mediators Inflamm. 2014:480980. doi: 10.1155/2014/480980
Coulthard, L. G., Hawksworth, O. A., Conroy, J., Lee, J. D., and Woodruff, T. M. (2018a). Complement C3a receptor modulates embryonic neural progenitor cell proliferation and cognitive performance. Mol. Immunol. 101, 176–181. doi: 10.1016/j.molimm.2018.06.271
Coulthard, L. G., Hawksworth, O. A., Li, R., Balachandran, A., Lee, J. D., Sepehrband, F., et al. (2017). Complement C5aR1 signaling promotes polarization and proliferation of embryonic neural progenitor cells through PKCzeta. J. Neurosci. 37, 5395–5407. doi: 10.1523/JNEUROSCI.0525-17.2017
Coulthard, L. G., Hawksworth, O. A., and Woodruff, T. M. (2018b). Complement: the emerging architect of the developing brain. Trends Neurosci. 41, 373–384. doi: 10.1016/j.tins.2018.03.009
De Felice, C., Della Ragione, F., Signorini, C., Leoncini, S., Pecorelli, A., Ciccoli, L., et al. (2014). Oxidative brain damage in Mecp2-mutant murine models of Rett syndrome. Neurobiol. Dis. 68, 66–77. doi: 10.1016/j.nbd.2014.04.006
Donohoe, G., Walters, J., Hargreaves, A., Rose, E. J., Morris, D. W., Fahey, C., et al. (2013). Neuropsychological effects of the CSMD1 genome-wide associated schizophrenia risk variant rs10503253. Genes Brain Behav. 12, 203–209. doi: 10.1111/gbb.12016
Duchatel, R. J., Meehan, C. L., Harms, L. R., Michie, P. T., Bigland, M. J., Smith, D. W., et al. (2018). Increased complement component 4 (C4) gene expression in the cingulate cortex of rats exposed to late gestation immune activation. Schizophr. Res. 199, 442–444. doi: 10.1016/j.schres.2018.03.035
Dunkelberger, J. R., and Song, W. C. (2010). Complement and its role in innate and adaptive immune responses. Cell Res. 20, 34–50. doi: 10.1038/cr.2009.139
Escudero-Esparza, A., Kalchishkova, N., Kurbasic, E., Jiang, W. G., and Blom, A. M. (2013). The novel complement inhibitor human CUB and Sushi multiple domains 1 (CSMD1) protein promotes factor I-mediated degradation of C4b and C3b and inhibits the membrane attack complex assembly. FASEB J. 27, 5083–5093. doi: 10.1096/fj.13-230706
Fagan, K., Crider, A., Ahmed, A. O., and Pillai, A. (2017). Complement C3 expression is decreased in autism spectrum disorder subjects and contributes to behavioral deficits in rodents. Mol. Neuropsychiatry 3, 19–27. doi: 10.1159/000465523
Fananas, L., Moral, P., Panadero, M. A., and Bertranpetit, J. (1992). Complement genetic markers in schizophrenia: C3, BF and C6 polymorphisms. Hum. Hered. 42, 162–167. doi: 10.1159/000154060
Farkas, I., Baranyi, L., Ishikawa, Y., Okada, N., Bohata, C., Budai, D., et al. (2002). CD59 blocks not only the insertion of C9 into MAC but inhibits ion channel formation by homologous C5b-8 as well as C5b-9. J. Physiol. 539(Pt 2), 537–545. doi: 10.1113/jphysiol.2001.013381
Fuentealba, L. C., Obernier, K., and Alvarez-Buylla, A. (2012). Adult neural stem cells bridge their niche. Cell Stem Cell 10, 698–708. doi: 10.1016/j.stem.2012.05.012
Gaboriaud, C., Thielens, N. M., Gregory, L. A., Rossi, V., Fontecilla-Camps, J. C., and Arlaud, G. J. (2004). Structure and activation of the C1 complex of complement: unraveling the puzzle. Trends Immunol. 25, 368–373. doi: 10.1016/j.it.2004.04.008
Gardner, O. K., Haynes, K., Schweitzer, D., Johns, A., Magee, W. P., Urata, M. M., et al. (2017). Familial recurrence of 3MC syndrome in consanguineous families: a clinical and molecular diagnostic approach with review of the literature. Cleft Palate Craniofac. J. 54, 739–748. doi: 10.1597/15-151
Garred, P., Genster, N., Pilely, K., Bayarri-Olmos, R., Rosbjerg, A., Ma, Y. J., et al. (2016). A journey through the lectin pathway of complement-MBL and beyond. Immunol. Rev. 274, 74–97. doi: 10.1111/imr.12468
Glantz, L. A., and Lewis, D. A. (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57, 65–73. doi: 10.1001/archpsyc.57.1.65
Glausier, J. R., and Lewis, D. A. (2013). Dendritic spine pathology in schizophrenia. Neuroscience 251, 90–107. doi: 10.1016/j.neuroscience.2012.04.044
Gorelik, A., Sapir, T., Ben-Reuven, L., and Reiner, O. (2018). Complement C3 affects rac1 activity in the developing brain. Front. Mol. Neurosci. 11:150. doi: 10.3389/fnmol.2018.00150
Gorelik, A., Sapir, T., Haffner-Krausz, R., Olender, T., Woodruff, T. M., and Reiner, O. (2017a). Developmental activities of the complement pathway in migrating neurons. Nat. Commun. 8:15096. doi: 10.1038/ncomms15096
Gorelik, A., Sapir, T., Woodruff, T. M., and Reiner, O. (2017b). Serping1/C1 Inhibitor affects cortical development in a cell autonomous and non-cell autonomous manner. Front. Cell. Neurosci. 11:169. doi: 10.3389/fncel.2017.00169
Graul-Neumann, L.M., Mensah, M.A., Klopocki, E., Uebe, S., Ekici, A. B., Thiel, C. T., et al. (2018). Biallelic intragenic deletion in MASP1 in an adult female with 3MC syndrome. Eur. J. Med. Genet. 61, 363–368. doi: 10.1016/j.ejmg.2018.01.016
Grove, J., Ripke, S., Als, T. D., Mattheisen, M., Walters, R. K., Won, H., et al. (2019). Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444. doi: 10.1038/s41588-019-0344-348
Gyorffy, B. A., Kun, J., Torok, G., Bulyaki, E., Borhegyi, Z., Gulyassy, P., et al. (2018). Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proc. Natl. Acad. Sci. U.S.A. 115, 6303–6308. doi: 10.1073/pnas.1722613115
Hafner, H., and der Heiden, W. (1997). Epidemiology of schizophrenia. Can. J. Psychiatry 42, 139–151. doi: 10.1177/070674379704200204
Hakobyan, S., Boyajyan, A., and Sim, R. B. (2005). Classical pathway complement activity in schizophrenia. Neurosci. Lett. 374, 35–37. doi: 10.1016/j.neulet.2004.10.024
Hammad, A., Westacott, L., and Zaben, M. (2018). The role of the complement system in traumatic brain injury: a review. J. Neuroinflam. 15:24. doi: 10.1186/s12974-018-1066-z
Han, M., Zhang, J. C., and Hashimoto, K. (2017). Increased levels of C1q in the prefrontal cortex of adult offspring after maternal immune activation: prevention by 7,8-Dihydroxyflavone. Clin. Psychopharmacol. Neurosci. 15, 64–67. doi: 10.9758/cpn.2017.15.1.64
Hazlett, H. C., Poe, M. D., Gerig, G., Styner, M., Chappell, C., Smith, R. G., et al. (2011). Early brain overgrowth in autism associated with an increase in cortical surface area before age 2 years. Arch. Gen. Psychiatry 68, 467–476. doi: 10.1001/archgenpsychiatry.2011.39
Heja, D., Kocsis, A., Dobo, J., Szilagyi, K., Szasz, R., Zavodszky, P., et al. (2012). Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proc. Natl Acad. Sci. U.S.A. 109, 10498–10503. doi: 10.1073/pnas.1202588109
Hooks, B. M., and Chen, C. (2006). Distinct roles for spontaneous and visual activity in remodeling of the retinogeniculate synapse. Neuron 52, 281–291. doi: 10.1016/j.neuron.2006.07.007
Howes, O. D., and Murray, R. M. (2014). Schizophrenia: an integrated sociodevelopmental-cognitive model. Lancet 383, 1677–1687. doi: 10.1016/S0140-6736(13)62036-X
Hutsler, J. J., and Zhang, H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. doi: 10.1016/j.brainres.2009.09.120
Ishii, T., Hattori, K., Miyakawa, T., Watanabe, K., Hidese, S., Sasayama, D., et al. (2018). Increased cerebrospinal fluid complement C5 levels in major depressive disorder and schizophrenia. Biochem. Biophys. Res. Commun. 497, 683–688. doi: 10.1016/j.bbrc.2018.02.131
Knuesel, I., Chicha, L., Britschgi, M., Schobel, S. A., Bodmer, M., Hellings, J. A., et al. (2014). Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 10, 643–660. doi: 10.1038/nrneurol.2014.187
Koiliari, E., Roussos, P., Pasparakis, E., Lencz, T., Malhotra, A., Siever, L. J., et al. (2014). The CSMD1 genome-wide associated schizophrenia risk variant rs10503253 affects general cognitive ability and executive function in healthy males. Schizophr. Res. 154, 42–47. doi: 10.1016/j.schres.2014.02.017
Kolev, M., Le Friec, G., and Kemper, C. (2014). Complement–tapping into new sites and effector systems. Nat. Rev. Immunol. 14, 811–820. doi: 10.1038/nri3761
Kraus, D. M., Elliott, G. S., Chute, H., Horan, T., Pfenninger, K. H., Sanford, S. D., et al. (2006). CSMD1 is a novel multiple domain complement-regulatory protein highly expressed in the central nervous system and epithelial tissues. J. Immunol. 176, 4419–4430. doi: 10.4049/jimmunol.176.7.4419
Lachmann, P. J. (2009). The amplification loop of the complement pathways. Adv. Immunol. 104, 115–149. doi: 10.1016/S0065-2776(08)04004-4002
Lachmann, P. J. (2018). Looking back on the alternative complement pathway. Immunobiology 223, 519–523. doi: 10.1016/j.imbio.2018.02.001
Leal, G. F., Silva, E. O., Duarte, A. R., and Campos, J. F. (2008). Blepharophimosis, blepharoptosis, defects of the anterior chamber of the eye, caudal appendage, radioulnar synostosis, hearing loss and umbilical anomalies in sibs: 3MC syndrome? Am. J. Med. Genet. A 146A, 1059–1062. doi: 10.1002/ajmg.a.32252
Li, H., Zhang, Q., Li, N., Wang, F., Xiang, H., Zhang, Z., et al. (2016). Plasma levels of Th17-related cytokines and complement C3 correlated with aggressive behavior in patients with schizophrenia. Psychiatry Res. 246, 700–706. doi: 10.1016/j.psychres.2016.10.061
Li, Y., Zhou, K., Zhang, Z., Sun, L., Yang, J., Zhang, M., et al. (2012). Label-free quantitative proteomic analysis reveals dysfunction of complement pathway in peripheral blood of schizophrenia patients: evidence for the immune hypothesis of schizophrenia. Mol. Biosyst. 8, 2664–2671. doi: 10.1039/c2mb25158b
Lian, H., Yang, L., Cole, A., Sun, L., Chiang, A. C., Fowler, S. W., et al. (2015). NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85, 101–115. doi: 10.1016/j.neuron.2014.11.018
Lin, P., Nicholls, L., Assareh, H., Fang, Z., Amos, T. G., Edwards, R. J., et al. (2016). Transcriptome analysis of human brain tissue identifies reduced expression of complement complex C1Q Genes in Rett syndrome. BMC Genomics 17:427. doi: 10.1186/s12864-016-2746-2747
Liu, Y., Fu, X., Tang, Z., Li, C., Xu, Y., Zhang, F., et al. (2019). Altered expression of the CSMD1 gene in the peripheral blood of schizophrenia patients. BMC Psychiatry 19:113. doi: 10.1186/s12888-019-2089-2084
Maes, M., Delange, J., Ranjan, R., Meltzer, H. Y., Desnyder, R., Cooremans, W., et al. (1997). Acute phase proteins in schizophrenia, mania and major depression: modulation by psychotropic drugs. Psychiatry Res. 66, 1–11. doi: 10.1016/s0165-1781(96)02915-2910
Malkova, N. V., Yu, C. Z., Hsiao, E. Y., Moore, M. J., and Patterson, P. H. (2012). Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav. Immun. 26, 607–616. doi: 10.1016/j.bbi.2012.01.011
Marin, O., Valiente, M., Ge, X., and Tsai, L. H. (2010). Guiding neuronal cell migrations. Cold Spring Harb. Perspect. Biol. 2:a001834. doi: 10.1101/cshperspect.a001834
Mayilyan, K. R., Arnold, J. N., Presanis, J. S., Soghoyan, A. F., and Sim, R. B. (2006). Increased complement classical and mannan-binding lectin pathway activities in schizophrenia. Neurosci. Lett. 404, 336–341. doi: 10.1016/j.neulet.2006.06.051
Mayilyan, K. R., Dodds, A. W., Boyajyan, A. S., Soghoyan, A. F., and Sim, R. B. (2008). Complement C4B protein in schizophrenia. World J. Biol. Psychiatry 9, 225–230. doi: 10.1080/15622970701227803
McDonald, C. R., Cahill, L. S., Ho, K. T., Yang, J., Kim, H., Silver, K. L., et al. (2015). Experimental Malaria in pregnancy induces neurocognitive injury in uninfected offspring via a C5a-C5a receptor dependent pathway. PLoS Pathog. 11:e1005140. doi: 10.1371/journal.ppat.1005140
McGrath, J., Saha, S., Chant, D., and Welham, J. (2008). Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol. Rev. 30, 67–76. doi: 10.1093/epirev/mxn001
Meyer, U., Nyffeler, M., Engler, A., Urwyler, A., Schedlowski, M., Knuesel, I., et al. (2006). The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J. Neurosci. 26, 4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006
Miles, J. H., Hadden, L. L., Takahashi, T. N., and Hillman, R. E. (2000). Head circumference is an independent clinical finding associated with autism. Am. J. Med. Genet. 95, 339–350.
Momeni, N., Brudin, L., Behnia, F., Nordstrom, B., Yosefi-Oudarji, A., Sivberg, B., et al. (2012). High complement factor I activity in the plasma of children with autism spectrum disorders. Autism Res. Treat. 2012:868576. doi: 10.1155/2012/868576
Morgan, J. T., Chana, G., Pardo, C. A., Achim, C., Semendeferi, K., Buckwalter, J., et al. (2010). Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 68, 368–376. doi: 10.1016/j.biopsych.2010.05.024
Mostafa, G. A., and Shehab, A. A. (2010). The link of C4B null allele to autism and to a family history of autoimmunity in Egyptian autistic children. J. Neuroimmunol. 223, 115–119. doi: 10.1016/j.jneuroim.2010.03.025
Muller-Eberhard, H. J. (1985). Transmembrane channel-formation by five complement proteins. Biochem. Soc Symp. 50, 235–246.
Muller-Eberhard, H. J., Polley, M. J., and Calcott, M. A. (1967). Formation and functional significance of a molecular complex derived from the second and the fourth component of human complement. J. Exp. Med. 125, 359–380. doi: 10.1084/jem.125.2.359
Munye, M. M., Diaz-Font, A., Ocaka, L., Henriksen, M. L., Lees, M., Brady, A., et al. (2017). COLEC10 is mutated in 3MC patients and regulates early craniofacial development. PLoS Genet. 13:e1006679. doi: 10.1371/journal.pgen.1006679
Nan, X., Ng, H. H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N., et al. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389. doi: 10.1038/30764
Nardone, S., Sams, D. S., Reuveni, E., Getselter, D., Oron, O., Karpuj, M., et al. (2014). DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 4:e433. doi: 10.1038/tp.2014.70
Odell, D., Maciulis, A., Cutler, A., Warren, L., McMahon, W. M., Coon, H., et al. (2005). Confirmation of the association of the C4B null allelle in autism. Hum. Immunol. 66, 140–145. doi: 10.1016/j.humimm.2004.11.002
O’Driscoll, C. M., Lima, M. P., Kaufmann, W. E., and Bressler, J. P. (2015). Methyl CpG binding protein 2 deficiency enhances expression of inflammatory cytokines by sustaining NF-kappaB signaling in myeloid derived cells. J. Neuroimmunol. 283, 23–29. doi: 10.1016/j.jneuroim.2015.04.005
Orsini, F., De Blasio, D., Zangari, R., Zanier, E. R., and De Simoni, M. G. (2014). Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front. Cell. Neurosci. 8:380. doi: 10.3389/fncel.2014.00380
Patterson, P. H. (2009). Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav. Brain Res. 204, 313–321. doi: 10.1016/j.bbr.2008.12.016
Patterson, P. H. (2011). Maternal infection and immune involvement in autism. Trends Mol. Med. 17, 389–394. doi: 10.1016/j.molmed.2011.03.001
Pedroni, S. M., Gonzalez, J. M., Wade, J., Jansen, M. A., Serio, A., Marshall, I., et al. (2014). Complement inhibition and statins prevent fetal brain cortical abnormalities in a mouse model of preterm birth. Biochim. Biophys. Acta 1842, 107–115. doi: 10.1016/j.bbadis.2013.10.011
Perez-Alcazar, M., Daborg, J., Stokowska, A., Wasling, P., Bjorefeldt, A., Kalm, M., et al. (2014). Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Exp. Neurol. 253, 154–164. doi: 10.1016/j.expneurol.2013.12.013
Prasad, K. M., Chowdari, K. V., D’Aiuto, L. A., Iyengar, S., Stanley, J. A., and Nimgaonkar, V. L. (2018). Neuropil contraction in relation to Complement C4 gene copy numbers in independent cohorts of adolescent-onset and young adult-onset schizophrenia patients-a pilot study. Transl. Psychiatry 8:134. doi: 10.1038/s41398-018-0181-z
Rahpeymai, Y., Hietala, M. A., Wilhelmsson, U., Fotheringham, A., Davies, I., Nilsson, A. K., et al. (2006). Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J. 25, 1364–1374. doi: 10.1038/sj.emboj.7601004
Rakic, P., Bourgeois, J. P., Eckenhoff, M. F., Zecevic, N., and Goldman-Rakic, P. S. (1986). Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science 232, 232–235. doi: 10.1126/science.3952506
Riccomagno, M. M., and Kolodkin, A. L. (2015). Sculpting neural circuits by axon and dendrite pruning. Annu. Rev. Cell Dev. Biol. 31, 779–805. doi: 10.1146/annurev-cellbio-100913-113038
Ricklin, D., Hajishengallis, G., Yang, K., and Lambris, J. D. (2010). Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797. doi: 10.1038/ni.1923
Rooryck, C., Diaz-Font, A., Osborn, D. P., Chabchoub, E., Hernandez-Hernandez, V., Shamseldin, H., et al. (2011). Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat. Genet. 43, 197–203. doi: 10.1038/ng.757
Rudduck, C., Beckman, L., Franzen, G., and Lindstrom, L. (1985). C3 and C6 complement types in schizophrenia. Hum. Hered. 35, 255–258. doi: 10.1159/000153555
Sasaki, T., Nanko, S., Fukuda, R., Kawate, T., Kunugi, H., and Kazamatsuri, H. (1994). Changes of immunological functions after acute exacerbation in schizophrenia. Biol. Psychiatry 35, 173–178. doi: 10.1016/0006-3223(94)91149-91145
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium, (2011). Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 43, 969–976. doi: 10.1038/ng.940
Schroers, R., Nothen, M. M., Rietschel, M., Albus, M., Maier, W., Schwab, S., et al. (1997). Investigation of complement C4B deficiency in schizophrenia. Hum. Hered. 47, 279–282. doi: 10.1159/000154424
Sekar, A., Bialas, A. R., de Rivera, H., Davis, A., Hammond, T. R., Kamitaki, N., et al. (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. doi: 10.1038/nature16549
Selemon, L. D., and Goldman-Rakic, P. S. (1999). The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol. Psychiatry 45, 17–25. doi: 10.1016/s0006-3223(98)00281-289
Severance, E. G., Gressitt, K. L., Buka, S. L., Cannon, T. D., and Yolken, R. H. (2014). Maternal complement C1q and increased odds for psychosis in adult offspring. Schizophr. Res. 159, 14–19. doi: 10.1016/j.schres.2014.07.053
Shcherbakova, I., Neshkova, E., Dotsenko, V., Platonova, T., Shcherbakova, E., and Yarovaya, G. (1999). The possible role of plasma kallikrein-kinin system and leukocyte elastase in pathogenesis of schizophrenia. Immunopharmacology 43, 273–279.
Shen, L., Zhang, K., Feng, C., Chen, Y., Li, S., Iqbal, J., et al. (2018). iTRAQ-based proteomic analysis reveals protein profile in plasma from children with autism. Proteomics Clin. Appl. 12:e1700085. doi: 10.1002/prca.201700085
Shenton, M. E., Dickey, C. C., Frumin, M., and McCarley, R. W. (2001). A review of MRI findings in schizophrenia. Schizophr. Res. 49, 1–52. doi: 10.1016/s0920-9964(01)00163-163
Shinjyo, N., Stahlberg, A., Dragunow, M., Pekny, M., and Pekna, M. (2009). Complement-derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells. Stem Cells 27, 2824–2832. doi: 10.1002/stem.225
Sirmaci, A., Walsh, T., Akay, H., Spiliopoulos, M., Sakalar, Y. B., Hasanefendioglu-Bayrak, A., et al. (2010). MASP1 mutations in patients with facial, umbilical, coccygeal, and auditory findings of Carnevale, Malpuech, OSA, and Michels syndromes. Am. J. Hum. Genet. 87, 679–686. doi: 10.1016/j.ajhg.2010.09.018
Spivak, B., Radwan, M., Brandon, J., Baruch, Y., Stawski, M., Tyano, S., et al. (1993). Reduced total complement haemolytic activity in schizophrenic patients. Psychol. Med. 23, 315–318. doi: 10.1017/s0033291700028397
Spivak, B., Radwan, M., Elimelech, D., Baruch, Y., Avidan, G., and Tyano, S. (1989). A study of the complement system in psychiatric patients. Biol. Psychiatry 26, 640–642. doi: 10.1016/0006-3223(89)90091-90097
Stephan, A. H., Barres, B. A., and Stevens, B. (2012). The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 35, 369–389. doi: 10.1146/annurev-neuro-061010-113810
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Stoner, R., Chow, M. L., Boyle, M. P., Sunkin, S. M., Mouton, P. R., Roy, S., et al. (2014). Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med. 370, 1209–1219. doi: 10.1056/NEJMoa1307491
Tang, G., Gudsnuk, K., Kuo, S. H., Cotrina, M. L., Rosoklija, G., Sosunov, A., et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. doi: 10.1016/j.neuron.2014.07.040
Titomanlio, L., Bennaceur, S., Bremond-Gignac, D., Baumann, C., Dupuy, O., and Verloes, A. (2005). Michels syndrome, Carnevale syndrome, OSA syndrome, and Malpuech syndrome: variable expression of a single disorder (3MC syndrome)? Am. J. Med. Genet. A 137A, 332–335. doi: 10.1002/ajmg.a.30878
Urquhart, J., Roberts, R., de Silva, D., Shalev, S., Chervinsky, E., Nampoothiri, S., et al. (2016). Exploring the genetic basis of 3MC syndrome: findings in 12 further families. Am. J. Med. Genet. A 170A, 1216–1224. doi: 10.1002/ajmg.a.37564
Veerhuis, R., Nielsen, H. M., and Tenner, A. J. (2011). Complement in the brain. Mol. Immunol. 48, 1592–1603. doi: 10.1016/j.molimm.2011.04.003
Venkatraman Girija, U., Furze, C. M., Gingras, A. R., Yoshizaki, T., Ohtani, K., Marshall, J. E., et al. (2015). Molecular basis of sugar recognition by collectin-K1 and the effects of mutations associated with 3MC syndrome. BMC Biol. 13:27. doi: 10.1186/s12915-015-0136-132
Wang, C., Jian, B. X., Jiang, X. D., and Zhao, X. Z. (1992). Investigation of allotypes of HLA class III (C2, BF and C4) in patients with schizophrenia. Chin. Med. J. (Engl) 105, 316–318.
Warren, R. P., Singh, V. K., Cole, P., Odell, J. D., Pingree, C. B., Warren, W. L., et al. (1991). Increased frequency of the null allele at the complement C4b locus in autism. Clin. Exp. Immunol. 83, 438–440. doi: 10.1111/j.1365-2249.1991.tb05657.x
Warren, R. P., Burger, R. A., Odell, D., Torres, A. R., and Warren, W. L. (1994). Decreased plasma concentrations of the C4B complement protein in autism. Arch. Pediatr. Adolesc. Med. 148, 180–183. doi: 10.1001/archpedi.1994.02170020066011
Whitney, E. R., Kemper, T. L., Bauman, M. L., Rosene, D. L., and Blatt, G. J. (2008). Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum 7, 406–416. doi: 10.1007/s12311-008-0043-y
Wong, C. T., Tsoi, W. F., and Saha, N. (1996). Acute phase proteins in male Chinese schizophrenic patients in Singapore. Schizophr. Res. 22, 165–171. doi: 10.1016/s0920-9964(96)00037-30
Keywords: complement system, neural progenitor proliferation, neurogenesis, neuronal migration, synapse refinement, neurodevelopmental disorders
Citation: Magdalon J, Mansur F, Teles e Silva AL, de Goes VA, Reiner O and Sertié AL (2020) Complement System in Brain Architecture and Neurodevelopmental Disorders. Front. Neurosci. 14:23. doi: 10.3389/fnins.2020.00023
Received: 23 October 2019; Accepted: 10 January 2020;
Published: 05 February 2020.
Edited by:
Alfonso Represa, INSERM U901 Institut de Neurobiologie de la Méditerranée, FranceReviewed by:
Corentin Le Magueresse, INSERM U839 Institut du Fer à Moulin, FranceCopyright © 2020 Magdalon, Mansur, Teles e Silva, de Goes, Reiner and Sertié. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andréa Laurato Sertié, YW5kcmVhLnNlcnRpZUBlaW5zdGVpbi5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.