 Francisco Melo Mouro
Francisco Melo Mouro Catarina Miranda-Lourenço
Catarina Miranda-Lourenço Ana Maria Sebastião
Ana Maria Sebastião Maria José Diógenes
Maria José Diógenes
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 02 July 2019
Sec. Neuropharmacology
Volume 13 - 2019 | https://doi.org/10.3389/fnins.2019.00680
This article is part of the Research Topic Meeting of the Portuguese Society for Neurosciences SPN2019 View all 10 articles
Rett syndrome (RTT) is an X-linked neurodevelopmental disorder caused mainly by mutations in the MECP2 gene, being one of the leading causes of mental disability in females. Mutations in the MECP2 gene are responsible for 95% of the diagnosed RTT cases and the mechanisms through which these mutations relate with symptomatology are still elusive. Children with RTT present a period of apparent normal development followed by a rapid regression in speech and behavior and a progressive deterioration of motor abilities. Epilepsy is one of the most common symptoms in RTT, occurring in 60 to 80% of RTT cases, being associated with worsening of other symptoms. At this point, no cure for RTT is available and there is a pressing need for the discovery of new drug candidates to treat its severe symptoms. However, despite being a rare disease, in the last decade research in RTT has grown exponentially. New and exciting evidence has been gathered and the etiopathogenesis of this complex, severe and untreatable disease is slowly being unfolded. Advances in gene editing techniques have prompted cure-oriented research in RTT. Nonetheless, at this point, finding a cure is a distant reality, highlighting the importance of further investigating the basic pathological mechanisms of this disease. In this review, we focus our attention in some of the newest evidence on RTT clinical and preclinical research, evaluating their impact in RTT symptomatology control, and pinpointing possible directions for future research.
In 1966, the neuro pediatrician Andreas Rett described and published a report concerning a neurodevelopmental disorder affecting females (Rett, 1966; Percy, 2016). In this report, Dr. Rett described a disease characterized as having an early onset of developmental delay followed by a rapid regression, loss of communication ability and fine motor capabilities, as well as, the occurrence of stereotypic hand movements and periodic breathing during wakefulness (Percy, 2016). Today, this disease is known as Rett syndrome (RTT). RTT is a severe neurodevelopmental X-linked disorder affecting almost exclusively female patients, with prevalence of approximately 1:10000 live births (Segatto et al., 2014) and with a high rate of sporadic mutations (Patankar, 2014). Despite its rareness, RTT is still the second most common cause of severe mental retardation in females (De Felice et al., 2012). The majority of RTT cases are characterized by an archetypal clinical scenario, comprised by loss of acquired cognitive, social, and motor skills, in an usual four-stage neurologic regression, occurring simultaneously with the development of autistic behavior (Hagberg, 2002; Neul et al., 2010; De Felice et al., 2012; Braat and Kooy, 2015). According to Hagberg, stage I is named “early onset stagnation,” occurs 6 months to 1.5 years after birth and is characterized by delays on the developmental progress. Subsequently, stage II, or “developmental regression,” occurs from year 1 to 4, being characterized by rapid loss of acquired skills and communication and manifestation of mental deficiency. Stage III is termed “pseudostationary period,” being characterized by apparent preserved ambulatory ability and restitution of some communicative abilities, while a slow neuromotor regression occurs. Lastly, stage IV or “later motor deterioration,” onsets when the ambulation of stage III ceases and its characterized by complete wheelchair dependence, severe disability, wasting and distal distortion. RTT patients can also experience gastrointestinal problems, hypoplasia, early-onset osteoporosis, bruxism and screaming spells (Hagberg, 2002). Reductions in brain weight and cortical atrophy/microencephaly, resulting in a smaller head circumference, were early reported as a common feature occurring in children diagnosed with RTT (Jellinger et al., 1988). As thoroughly reviewed in Kyle et al. (2018), metabolic complications are also known as a common feature in RTT (Justice et al., 2013; Segatto et al., 2014). Also, as revised in Shulyakova et al. (2017), important anomalies in mitochondrial structure and function (altered electron transport chain complex function, increased oxidative stress and elevated levels of lactate and pyruvate in blood and cerebrospinal fluid) have been demonstrated in RTT. The clinical diagnosis of RTT is performed following a battery of co-existing and well-defined set of inclusion and exclusion criteria, which were recently revised (Neul et al., 2010; Kyle et al., 2018).
In Amir et al. (1999), using a systematic gene screening approach, identified mutations in the gene MECP2 as cause of some cases of RTT. Today, several mutations in the X-linked MECP2 gene are identified, being acknowledged as the cause of 95% of the classical RTT cases and of 40–50% of the atypical RTT cases (Neul et al., 2010; Zhang et al., 2017), resulting in a wide genetic and phenotypic heterogeneity of this disease (De Felice et al., 2012). MECP2 is a globally expressed pleiotropic factor, assuming a key role in maintaining homeostasis in different cells and systems (Shulyakova et al., 2017). As expected for a neurological condition (Neul and Zoghbi, 2004), in RTT the higher levels of MECP2 in the body are expressed in the brain (Shahbazian, 2002). Confirming the importance of MECP2 presence in the brain, the selective elimination of this gene from neurons (Guy et al., 2007) and oligodendrocytes (Nguyen et al., 2012) triggers RTT-like phenotype in mice (Shulyakova et al., 2017). In atypical RTT variants, mutations in other loci rather than the MECP2 have been identified (Neul et al., 2010).
A diversity of distinctive variant forms of RTT has been proposed, each of which presents different clinical features. Some of these variants have been identified and described in relatively small populations, which leads to difficulties in the definition of clear clinical characteristics (Neul et al., 2010). Nevertheless, according to Neul et al. (2010), there are three distinctive variants of RTT that have been amply identified and that are well characterized: (1) the preserved speech variant (Zappella, 1992), (2) the congenital variant (Rolando, 1985), and (3) the early seizure variant (Hanefeld, 1985). From these three atypical forms of RTT, the preserved speech variant, or Zappella variant, is the most common one. This variant has clearly defined clinical features and mutations on the MECP2 gene are identified in most of the diagnosed cases (Zappella, 1992; Renieri et al., 2009; Neul et al., 2010). On the contrary, in diagnosed cases belonging to the congenital and early seizure variants, mutations in the MECP2 gene are rarely found, while mutations in different genes are reported (Huppke et al., 2000; Archer et al., 2006; Neul et al., 2010; De Felice et al., 2012). Namely, mutations in the CDKL5 gene have been described in both males and females diagnosed with early-seizure-onset RTT variant (Mari et al., 2005; Scala et al., 2005; Zhao et al., 2014; Zhang et al., 2017). On the other hand, mutations in the FOXG1 gene have been identified in the congenital RTT variant (Zhang et al., 2017), first described by Rolando (1985). The congenital variant of RTT is clinically characterized by hypotonia and developmental delay occurring earlier than in classical RTT variant (Rolando, 1985; Jacob et al., 2009). Importantly, the majority of children diagnosed with the congenital variant of RTT do not present mutations in the MECP2 or CDKL5 mutations (Jacob et al., 2009).
The available treatment for RTT is mainly symptomatic. With appropriate social and familiar care, attention to orthopedic complications, physiotherapy to treat and ease muscle rigidity, control of epileptic episodes, and a balanced and healthy nutrition, women with RTT can survive until middle age and older age (Kyle et al., 2018). There is a sudden death rate of 26% in RTT and patients mainly perish due to cardiac complications, respiratory infection and respiratory failure (Kyle et al., 2018). Most of the preclinical and clinical studies in RTT aims at finding ways to prevent or control epileptic episodes, due to the importance that it has for the outcome of RTT prognosis (Krajnc, 2015; Clarke and Abdala Sheikh, 2018). Therefore, preclinical research in RTT is majorly focused on finding ways of correcting detrimental modifications in neurotransmission occurring in this disease, using Mecp2-null mouse models (Clarke and Abdala Sheikh, 2018). Recently, however, a paradigm shift has been proposed. Through gene therapy it was proven that it is possible to reverse RTT symptoms in diseased adult mice by re-activating Mecp2 expression (Guy et al., 2007). Also, a few years later, it was shown that switching-off the production of Mecp2 protein in adult mice leads to the development of symptoms equivalent to those of mice born with the mutation (McGraw et al., 2011; Clarke and Abdala Sheikh, 2018). Considering that, by rule, neurodevelopmental disorders tend to be non-reversible, such results were received with considerable enthusiasm (Clarke and Abdala Sheikh, 2018). Thus, these studies showed that gene therapy would have to necessarily deliver a working MECP2 gene throughout the patient life (it would not be sufficient to do it just on child development) and that treatment could be administered regardless of disease stage and/or age (Clarke and Abdala Sheikh, 2018). Although exciting, the above-mentioned discoveries have to be taken carefully. Indeed, in order to work, MECP2 gene therapy would have to deliver the precise amount of MECP2 protein in each cell of the body, as too much or too few MECP2 protein can trigger RTT-like symptoms (Clarke and Abdala Sheikh, 2018). As a particularly harsh example, in MECP2 duplication syndrome, an overproduction of MECP2 protein leads to mental disability and autistic-like behavior (Moretti and Zoghbi, 2006; Ramocki et al., 2009). Moreover, even small deviations from the necessary MECP2 protein levels are related with modifications in brain function, which can lead to cognitive and mental disability (Chao and Zoghbi, 2012). Therefore, as mentioned in Clarke and Abdala Sheikh (2018), gene therapy in RTT delivers two major challenges: (1) it potentially means that it would be necessary to deliver the exact right amount of MECP2 to every cell and (2) in females, it would be necessary to avoid delivering additional copies of the gene to the cells that already express a healthy copy; two pitfalls that are extremely difficult to overcome with the available technology.
At this point, although possible, finding a cure for RTT is a distant reality. In the meanwhile, it is more important than ever to find new treatments to alleviate symptoms, reduce pain and discomfort and increase the quality of life of both the patient and the caregiver. Therefore, in this review we will turn our attention to new discoveries which show potential in RTT preclinical investigation.
Rett syndrome is characterized by structural and molecular deficiencies in synaptic transmission. Evidence displays important modifications in basal transmission, in short and long-term plasticity processes and in neurotransmitter release (Boggio et al., 2010). Studies using different Mecp2 genetic mutated mice have made it possible to understand that individual phenotypic characteristics of RTT can be associated with dysfunctions in very specific neuronal populations. Indeed, the selective deletion of the Mecp2 gene from different neuronal populations, in different ages and in distinctive brain regions accounts for specific phenotypical traits of the disease. Thus, such data has led authors to claim that the phenotypical consequences of Mecp2 deletion are time and region-dependent (Chao et al., 2010). In this regard, dysfunctional GABAergic signaling constitutes a vital element in RTT symptomatology.
The GABAergic signaling system comprises a key pathway commonly disturbed in neurodevelopmental diseases, such as RTT, fragile X syndrome, Dravet Syndrome, neurofibromatosis type I, Tourette syndrome, Down Syndrome and in Autism Spectrum Disorders (Lee and Tierney, 2011; El-Ansary and Al-Ayadhi, 2014; Braat and Kooy, 2015; Kim and Yoon, 2017). GABA is the main inhibitory neurotransmitter in the brain, exerting its actions via the activation of two subtypes of GABA receptors, the ionotropic GABAAR and the metabotropic GABABR (Chebib and Johnston, 1999; Rombo et al., 2016). GABAAR are ligand-gated receptors responsible for mediating the majority of inhibitory synaptic transmission in the CNS (Reddy, 2010). Structurally, GABAAR are heteropentamers with five protein subunits that form the chloride ion channel (Reddy, 2010). GABAAR can be assembled by seven different classes of subunits, some of which comprised by different homologous variants (α1–6, β1–3, γ1–3, σ1–3, δ, θ, ε). Usually, most GABAAR are composed by α, β, and γ or δ-subunits (Sieghart, 2006). The binding site for the endogenous modulator GABA is located in the cleft between the α and β subunits. Apart from the GABA binding site, there are several other binding sites in the GABAAR which constitute targets for benzodiazepines and barbiturates (Smith, 2002). Furthermore, each pharmacological effect appears to be directly related with a specific binding site on the receptor surface, which depends on the subunit composition of the receptor (Cai et al., 2018). Post-synaptic GABAAR are ubiquitously distributed, being responsible for generating the phasic currents in response to presynaptic GABA release. On the other hand, extrasynaptic GABAAR are preferentially activated when GABA levels are low, being highly sensitive to extracellular GABA concentrations and responsible for generating non-desensitizing tonic inhibition (Wang, 2011; Cai et al., 2018). Despite being exclusively present in a minority of neurons, several clusters of GABAergic interneurons are responsible for regulating and controlling the majority of excitatory neurons. Thus, if an imbalance in the inhibitory system occurs, the following excessive excitatory output leads to disturbances on the excitation/inhibition balance, ultimately resulting in dysfunction of cognitive processes (Braat and Kooy, 2015). Importantly, genetic alterations in GABAergic signaling system has been shown to be involved in several neurodevelopmental diseases (for a detailed review see Braat and Kooy, 2015).
Evidence regarding dysfunctional GABAergic signaling in RTT has been mainly obtained in studies using RTT-mouse models. Such studies have been gathering substantial evidence confirming the critical involvement of GABAergic transmission in RTT symptomatology, highlighting the clinical relevance of this signaling system. Indeed, an incorrect balance between excitation and inhibition reflecting a dysfunction in GABAergic and glutamatergic signaling systems have been described in Mecp2-knockout mice (Dani et al., 2005; Chao et al., 2007; Dani and Nelson, 2009; Calfa et al., 2011). Also, as mentioned before regarding other neuromodulatory systems, the relationship between RTT phenotypical expression and GABAergic signaling is region, time and neuronal population dependent (El-Khoury et al., 2014). Confirming the critical involvement of the GABAergic system in RTT symptomatology, it has been shown that mice with Mecp2-deficiency exclusively in GABAergic neurons rapidly develop forepaw stereotyped movements, compulsive grooming, learning and memory deficits, abnormal social behavior, electroencephalography hyperexcitability, lack of motor coordination, severe respiratory dysrhythmias and premature lethality. Additionally, the results of this work also showed that the selective deletion of the Mecp2 gene in a specific subset of forebrain GABAergic neurons was sufficient to trigger many of the aforementioned symptoms. Thus, these data strongly suggests that the loss of Mecp2 in GABAergic neurons acts as a pivotal mediator for some characteristic RTT phenotypes (Chao et al., 2010).
The critical relevance of MECP2 in GABAergic signaling was also shown in other brain regions, such as the hippocampus. Indeed, patch-clamp recordings performed in hippocampal slices obtained from Mecp2 mutant mice, revealed that hippocampal circuits in CA3 neurons display diminished basal inhibitory rhythmic activity, which, consequently, leaves the circuitry susceptible to hyperexcitability (Zhang et al., 2008). Likewise, in the brain stem, an imbalance between inhibitory and excitatory transmission was reported to occur early at postnatal day 7 in Mecp2 mutant mice. Additionally, GABAergic transmission was found to be significantly depressed in the same animals, which may be related with deficient presynaptic GABA release and diminished expression levels of subunits α2 and α4 on the post-synaptic GABAAR (Medrihan et al., 2008). In the thalamus, Mecp2 differentially regulates the development of GABAergic synapses in excitatory or inhibitory neurons (Zhang Z.W. et al., 2010). Also, in Mecp2 mutant mice, electrophysiological deficiencies have been reported in norepinephrinergic neurons of the locus coeruleus which receive GABAAergic inhibitory inputs (Jin et al., 2013a; Zhang X. et al., 2010). In the locus coeruleus, Mecp2 deficiency leads to simultaneous abnormalities in the pre- and post-synaptic GABAergic component, impairing GABAAergic and GABABergic post-synaptic inhibitory currents and reducing the presynaptic release of GABA (Jin et al., 2013a). Indeed, it has been described that Mecp2 deficiency abnormally increases extrasynaptic GABAAR activity, an effect suggested to occur as a compensatory response to the deficient GABA-mediate synaptic inhibition (Zhong et al., 2015).
The above-mentioned evidence highlights the critical role of MECP2 gene for correct GABAergic signaling in different brain regions. Besides brain region and synaptic location of the receptor, in a recent study it was also shown that selective deletion of Mecp2 from either parvalbumin-positive neurons or somatostatin-positive neurons leads to different phenotypical outcomes, that together comprise almost the full range of RTT-like phenotypes. Specifically, Mecp2 deletion from parvalbumin-positive neurons leads to motor, sensory, cognitive and social deficits, while ablation of Mecp2 from somatostatin-positive neurons originated stereotyped behavior and seizures (Ito-Ishida et al., 2015). Recently, reductions in GABAergic transmission in the nucleus tractus solitarius, a key brain region involved in the integration of respiratory sensory information, were reported (Chen et al., 2018).
Although scarce, there is available data collected from human samples supporting findings in rodent models of RTT (Braat and Kooy, 2015). GABAAR binding was found to be significantly altered in the basal ganglia (Blue et al., 1999) and in the frontotemporal cortex (Yamashita et al., 1998) in RTT patients. Additionally, a disturbed process of GABAergic neuronal maturation was described in the cerebrospinal fluid of RTT patients (Duarte et al., 2013) (for a more details see Braat and Kooy, 2015). Also, an analysis performed in post-mortem human brains, has shown defects in the expression of the GABR gene GABRB3, contained in the human chromosome 15q11-13, in RTT patients (Samaco et al., 2005). Congruently, subsequent evidence obtained with mice models, suggested that Mecp2 gene is vital for the correct expression of both alleles of GABRB3 in neurons (Hogart et al., 2007).
Considering the anomalies observed in GABAergic signaling in RTT and their crucial role in symptomatology and disease progression, correction of these modifications has been proposed as a therapeutic strategy. Indeed, adjustment of the GABAergic system activity in Mecp2 mutant mice leads to improvements in several phenotypical features (Braat and Kooy, 2015). In accordance, in a recent study, it was found that an in vivo treatment with the inhibitor of GABA reuptake, Tiagabine, significantly increased the life-spam of Mecp2 knockout mice, although it did not ameliorate motor deficits (El-Khoury et al., 2014). In another study, the benzodiazepine Midazolam transiently abolished breathing abnormalities in Mecp2 mutant mice (Voituron and Hilaire, 2011). Also, the enhancement of GABAAR-mediated signaling, either through blockade of GABA reuptake or through positive allosteric modulation of the GABAAR, resulted in a significant reduction on respiratory problems (Abdala et al., 2010). Recently, respiratory dysrhythmia has been related with deficient GABAergic signaling in the Kölliker-Fuse area, or subparabrachial nucleus, a brain area responsible for the regulation of breathing rate. Congruently, boosting GABA transmission reduced respiratory arrhythmia in a RTT-mouse model (Abdala et al., 2016). Also, early exposure to the extrasynaptic GABAAR agonist THIP was shown to regulate neuron hyperexcitability on the locus coeruleus, known to be involved in the regulation of breathing (Zhong et al., 2017). Finally, genetic re-expression of Mecp2 only in GABAergic neurons of male and female Mecp2 null mice enhanced inhibitory signaling. Also, it extended animal lifespan, mitigated ataxia, apraxia, and social withdrawal, displaying that the restoration of Mecp2 expression in GABAergic neurons significantly improves the symptomatology of RTT (Ure et al., 2016; Figure 1).
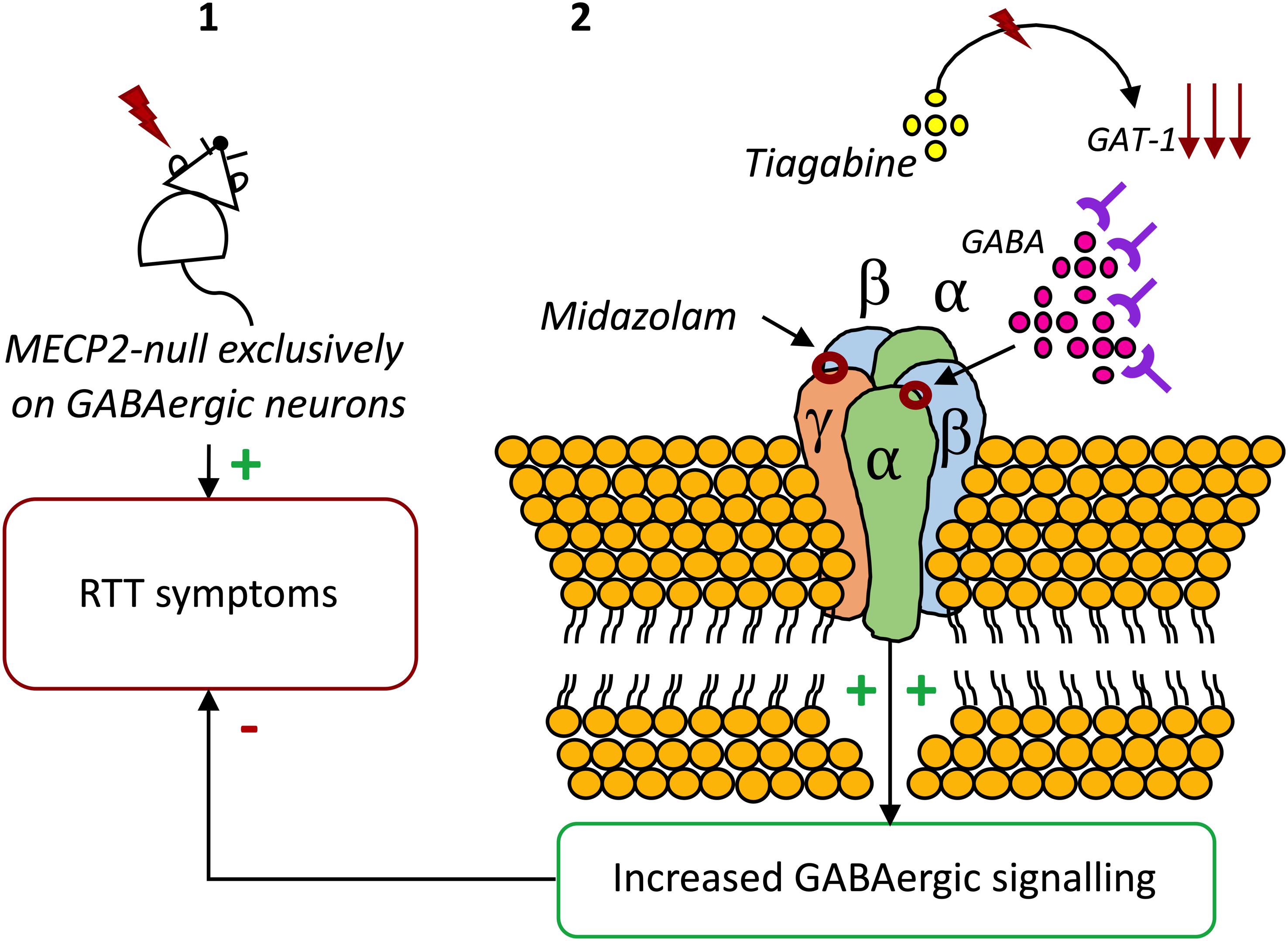
Figure 1. The potential of GABAAR modulators on RTT. (1) Exclusive deletion of Mecp2 from GABAergic neurons results in almost the full range of neuropsychiatric symptoms of RTT (Chao et al., 2010). (2) Blocking GABA reuptake using tiagabine (El-Khoury et al., 2014) or enhancing GABAAR activity with the benzodiazepine Midazolam increases the life-spam and reduces symptoms in Mecp2 mutant mice (Voituron and Hilaire, 2011).
Taken together, these results emphasize the crucial role of MECP2 in GABAergic neurons. Also, they demonstrate a clear relationship between modifications in GABAergic signaling and symptomatology in RTT, highlining the potential of drug agents which correct these modifications to improve RTT symptoms. Therefore, on the following sections we will focus our attention on new findings that, directly or indirectly, involve the modulation of GABAergic signaling in RTT (Figure 2).
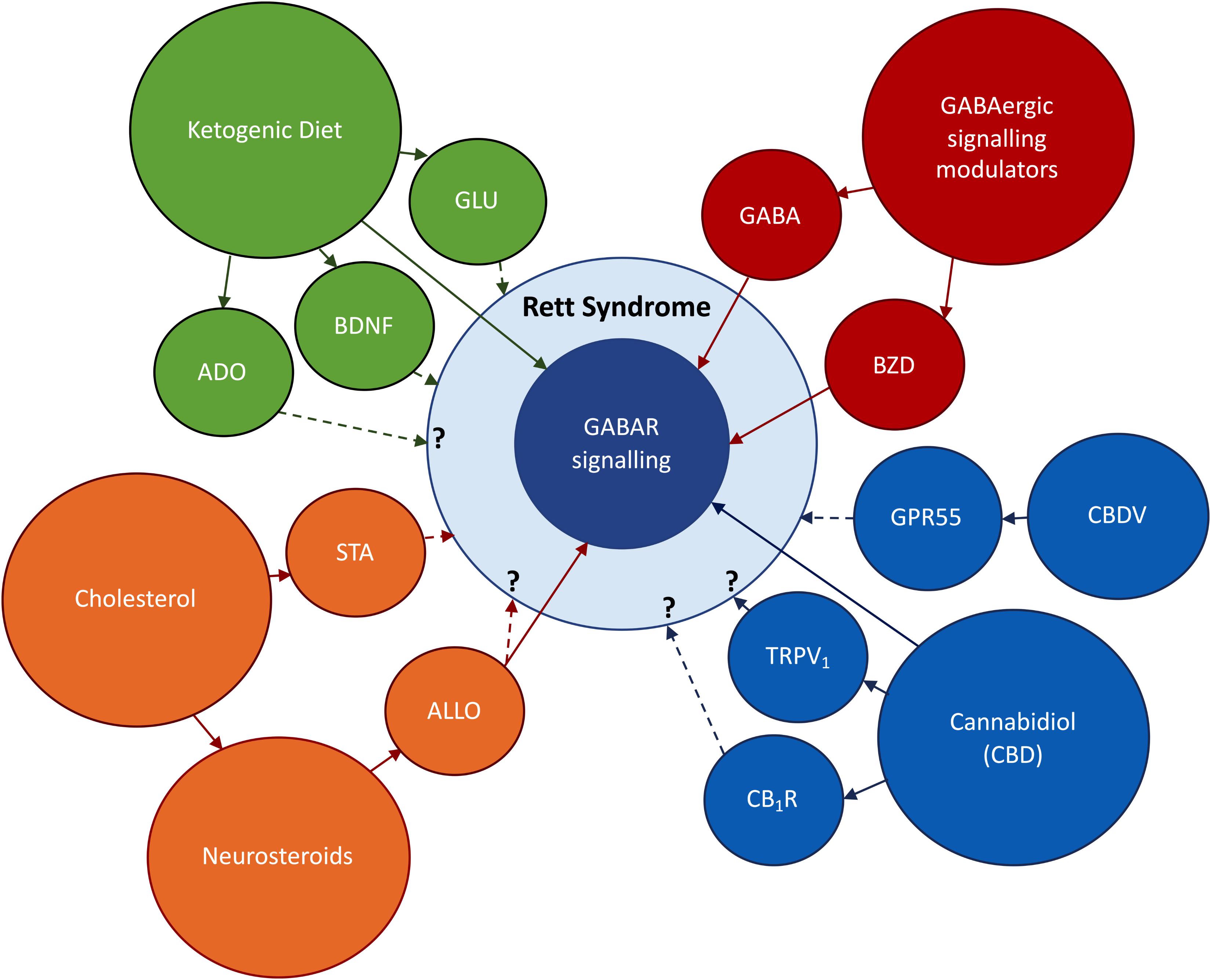
Figure 2. Research topics to be addressed in this review. In red: the use of direct and indirect modulators of GABAergic signaling, respectively, benzodiazepines (BZD) and GABA enhancers. In orange: Abnormal cholesterol metabolism occurring in RTT, the use of statins (STA) to improve RTT symptoms; the impact of deficits on cholesterol uptake on neurosteroidogenesis; the possible therapeutic actions of the neurosteroid allopregnanolone (ALLO). In blue: the use of derivatives of the cannabis plant to ameliorate RTT symptoms; cannabidiol (CBD) has shown promising anti-epileptic effects via GABA-dependent mechanisms; the effects of CBD on CB1R and TRPV1 and its impact on RTT symptoms are still undisclosed; Cannabidivarin (CBDV) has been identified as a promising therapeutic drug in an RTT-mice model. In green: Ketogenic Diet (KD) has potent anticonvulsive actions via GABAergic and glutamatergic (GLU) signaling systems; the KD can also improve RTT symptoms via BDNF-mediated effects or through increases in adenosine production. BNDF signaling is known to be impaired in RTT (Li and Pozzo-Miller, 2014), while unpublished data obtained in our lab points to deregulations in adenosinergic signaling. Direct arrows denote direct interaction with GABAergic signaling system, while doted arrows denote known or undisclosed involvement on RTT pathophysiology.
Rett syndrome was initially considered to be a metabolic disease (Justice et al., 2013). Indeed, originally, Dr. Rett, convinced of being in the presence of a disease with a clear metabolic component, named it “cerebroatrophic hyperammonaemia” (Kyle et al., 2018). Hyperammonemia is a clinical condition characterized by increased ammonia levels, which manifests itself in a variety of symptoms and signs, including significant CNS abnormalities (Auron and Brophy, 2012). The fact that hyperammonaemia has been detected only in a minority of RTT patients led to the abandonment of the hypothesis of cerebroatrophic hyperammonaemia as the central cause for RTT (Campos-Castelló et al., 1988; Justice et al., 2013). Recently, however, the metabolic components of RTT and its importance for disease progression have been brought again into light.
Cholesterol is a vital component of the cellular membrane structure and a precursor for numerous signaling molecules (Nagy and Ackerman, 2013). Cholesterol does not cross the BBB and therefore brain cholesterol has to be generated locally, which directly contrasts with other organs that are able of acquiring it from circulating lipoproteins (Pfrieger and Ungerer, 2011). In the brain, the production of cholesterol is a highly compartmentalized process presenting a very delicate balance. Its production takes place in the endoplasmic reticulum and requires, as an energy source, the correct functioning of the mitochondria (Justice et al., 2013). Importantly, mitochondrial function and structure is compromised in both RTT patients and Mecp2 mutant mouse models (Shulyakova et al., 2017). After synthesis, brain cholesterol must be quickly renewed and turned-over, as it can be rapidly oxidized by reactive oxygen species (ROS), known to accumulate whenever mitochondria dysfunction occurs (Justice et al., 2013). To maintain homeostasis, cholesterol must be converted into the 24S-OHC by the neuron-specific enzyme Cyp46a1 (Lund et al., 2003; Kyle et al., 2018). Thus, a tight regulation of cholesterol homeostasis in the brain assumes particular importance: too much or too little cholesterol is detrimental, negatively impacting on cognitive processes, memory and motor skills (Kyle et al., 2018). Concordantly, a vast number of neurological diseases present abnormal lipid synthesis, storage and recycling (Waterham, 2006), namely, SLOS and Niemann–Pick type C disease, Alzheimer’s, Parkinson’s, and Huntington’s diseases, Amyotrophic lateral sclerosis, Fragile X syndrome, and, as recently discovered, RTT (Justice et al., 2013).
Using Mecp2 mutant mice, a mutagenesis screening to identify genes that influence RTT-linked phenotypes was performed (Buchovecky et al., 2013; Nagy and Ackerman, 2013). To do so, descendent of MECP2 mice were screened for limp gasping, tremors and activity, and mice that displayed reduced phenotypic features were bred to establish the hereditability of putative suppressor genes (Buchovecky et al., 2013). This screening highlighted that a mutation in the Sqle gene, encoding squalene epoxidase, which is a rate-limiting enzyme in cholesterol biosynthesis, was sufficient to restore function and longevity in Mecp2 mutant mice. The authors also showed that cholesterol metabolism is perturbed in brains and livers of Mecp2 mutant male mice, revealing profound and complex dysregulations in cholesterol metabolism. Furthermore, HMG-CoA reductase inhibitors, or statins, were able to ameliorate the systemic imbalance of lipid profile, alleviate motor symptoms and increase longevity in Mecp2 mutant mice (Buchovecky et al., 2013). Studies with samples collected from human RTT patients have also shown modifications in lipid profile. Directly comparing with age-matched healthy donors, imbalances in both high (HDL) and low (LDL) density lipoprotein levels were found in RTT patients. Accompanying these abnormalities in plasma lipid profile, a marked reduction in SRB1 was detected. SRB1 is ubiquitously expressed, playing vital roles in cellular lipid uptake, mediating, for instance, the uptake of HDL-derived cholesterol in the liver (Sticozzi et al., 2013). Congruently, using freshly isolated human fibroblasts, it has been shown that total cholesterol and LDL levels are significantly increased in RTT patients, while SRB1 expression was quantified has being 70% lower in RTT patients compared with healthy controls (Segatto et al., 2014). On the other hand, and in accordance with data from Buchovecky et al. (2013), cholesterol synthesis was found to be reduced in RTT fibroblasts (Segatto et al., 2014). Therefore, these data provided strong evidence toward the importance of cholesterol homeostasis in RTT. Also, imbalances in cholesterol metabolism can affect the synthesis of steroid hormones, among which neurosteroids can have an important role in several brain diseases, as it will be approached in the following section.
Neurosteroids were described, for the first time, has having anesthetic and anticonvulsive actions in the late 1940s (Selye, 1941; Selye and Masson, 1942; Clarke, 1973; Reddy and Estes, 2016). Almost half a century later, alphaxolone, a synthetic neurosteroid, was found to increase synaptic inhibition through GABAAR activation (Harrison and Simmonds, 1984), which constituted a major advanced in neurosteroid research (Majewska et al., 1986).
Neuroactive steroids are steroid molecules synthesized in the brain that modulate neuronal excitability by rapid non-genomic actions (Reddy, 2010). Cholesterol constitutes the raw material for the biosynthesis of all steroid hormones (Hu et al., 2010). De novo synthesis of neurosteroids is a sequential and highly compartmentalized process involving, as first step, the translocation of cholesterol from the cytoplasm to the inner mitochondrial membrane. Once inside the mitochondria, cholesterol is converted into pregnenolone, the precursor of all steroid hormones (Reddy, 2010; Melcangi et al., 2014). Pregnenolone is then transformed into progesterone by the action of the enzyme 3β-HSD (Melcangi et al., 2008). The conversion of cholesterol to pregnenolone in the mitochondria is, consequently, the first rate-limiting step in the biosynthesis of all steroid hormones (Stoffel-Wagner, 2003). ALLO is derived from progesterone by 5α-reductase and 3α-HSD (Liu and Wong-Riley, 2010), and is viewed as the most potent endogenous modulator of the GABAergic system via interaction with the GABAAR (Hosie et al., 2006; Reddy, 2010; Figure 3).
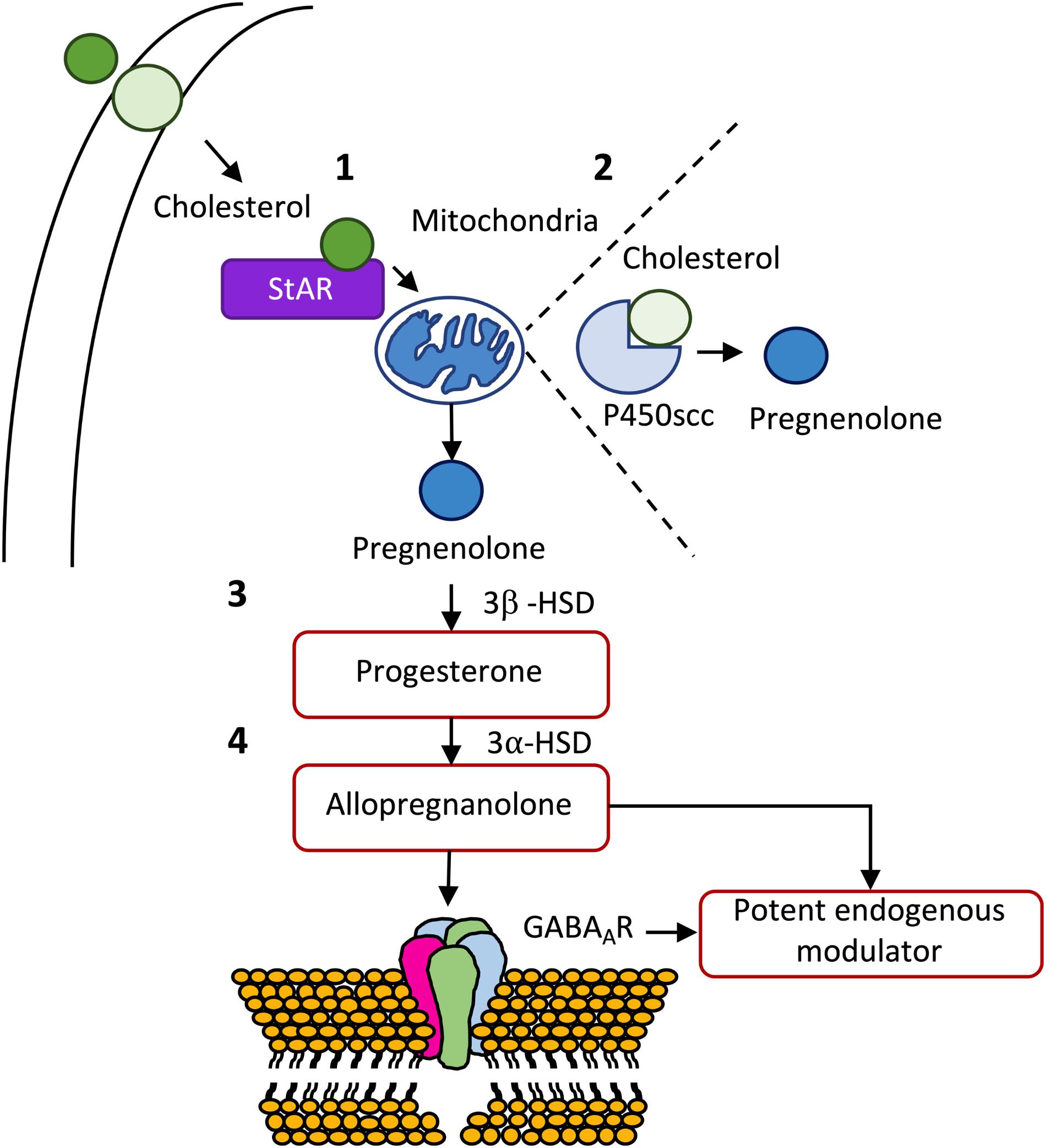
Figure 3. De novo synthesis of some important neurosteroids. (1) After being transported from the outer to the inner membrane, cholesterol is transported from the outer to the inner membrane of the mitochondria by steroidogenic acute regulatory protein (StAR). (2) Once inside the mitochondria, cholesterol is transformed in pregnenolone, the mother of all steroid hormones, by cytochrome P450 cholesterol side-chain cleavage (P450scc) enzyme (CYP11A1). (3) Pregnenolone can be, subsequently, transformed into progesterone by 3β-hydroxysteroid dehydrogenase (3β-HSD), which in turn, can originate allopregnanolone through the action of 3α-hydroxysteroid dehydrogenase (3α-HSD). (4) Allopregnanolone is viewed as the most potent endogenous modulator of the GABAergic system via interaction with the GABAA receptor (for a detailed review see Melcangi et al., 2014).
Recent studies indicate that neurosteroids have anxiolytic, antidepressant and antipsychotic properties. Furthermore, neurosteroids stimulate neurogenesis, facilitate regeneration of neurons after injury, promote myelinization, and improve cognition (Reddy and Estes, 2016; Cai et al., 2018). The development of new analogous neurosteroid agents with promising therapeutic properties has resulted in four compounds tested in clinical trials for epilepsy, traumatic brain injury, status epilepticus and Fragile X syndrome. Also, several synthetic neurosteroids have been prepared for therapeutic use in the past decades, among which, the best-known are, the aforementioned alphaxolone, and also alphadolone, minaxolone, and ganaxolone (Reddy and Estes, 2016).
Neurosteroids have been implicated in the behavioral profile of some neurologic diseases. In SLOS, an autosomal recessive disorder which develops due to an inborn error in cholesterol metabolism (Tint et al., 1994; Aneja and Tierney, 2008; Lee and Tierney, 2011), impaired neurosteroid synthesis or the synthesis of an inhibitory analog form of neurosteroids on the brain have been proposed to occur (Porter, 2002). Congruently, urinary analysis has allowed the identification of neurosteroid-like compounds in SLOS patients, being foreseeable that this abnormal synthesis also occurs in the brain (Marcos et al., 2004; Lee and Tierney, 2011). In Fragile X syndrome, patients show autism-like phenotypes characterized by cognitive impairment, anxiety, mood swings and behavioral and learning difficulties (Reddy and Estes, 2016). A clinical trial using ganaxolone was found to be safe but produced no significant effects on the outcome measures in the overall population of the study. However, specific subsets of the children with Fragile X syndrome enrolled on the clinical trial, particularly the ones with higher anxiety, lower cognitive abilities and who have frequent seizure episodes, might benefit from treatment with this neurosteroid (Ligsay et al., 2017).
Recently, it has been described that ALLO (Meletti et al., 2017) and progesterone (Meletti et al., 2018) are decreased in cerebrospinal fluid (CSF) of patients affected by status epilepticus. Progesterone levels were found to be 64% lower than in healthy controls (Meletti et al., 2018). Interestingly, neurosteroids are also being proposed for the treatment of schizophrenia, as neurosteroids show promising anti-psychotic potential (Cai et al., 2018).
The therapeutic potential of neurosteroids is mainly related with their ability to rapidly modulate the activity of the GABAergic neurons, known to be involved in the pathophysiology of several psychiatric disorders (Mellédo and Baker, 2002; Dubrovsky, 2005; Marx et al., 2006) and neurodegenerative diseases (Carunchio et al., 2008; Li et al., 2016; Kim and Yoon, 2017; Lozovaya et al., 2018), and commonly disturbed in neurodevelopmental diseases (see section “Dysfunctional GABAR Signalling in RTT: Implications for Symptomatology”). The modulatory effects of neurosteroids on GABAAR are unique and extremely complex, depending on several factors, such as, the subunit composition of the receptor, the receptor localization, concentration and the structure of the neurosteroid (Sieghart, 2006; Reddy, 2010; Reddy and Jian, 2010; Wang, 2011; Reddy and Estes, 2016; Cai et al., 2018). Previous studies have shown that specific combinations of subunits in the GABAAR confer more or less affinity for neurosteroids, but that ALLO enhances GABAAergic transmission whenever the GABAAR are composed by any α subunit (Puia et al., 1993, 2003; Maitra and Reynolds, 1999; Belelli et al., 2002). Importantly, both post-synaptic GABAAR and extrasynaptic GABAAR are highly sensible to neurosteroid modulation. In particular, extrasynaptic GABAAR containing the δ subunit located in specific brain regions as the hypothalamus, hippocampal dentate gyrus and cerebellum, are highly sensitive to neurosteroids, which may open a therapeutic window for the use of neurosteroids in several brain diseases, including RTT (Smith, 2002; Reddy and Estes, 2016). Indeed, neurosteroids have been proposed as a viable alternative to overcome benzodiazepine tolerance, as they can bind and enhance the activity of all GABAAR isoforms, including extrasynaptic GABAAR containing the highly neurosteroid sensitive δ subunit (Brown et al., 2002; Uusi-Oukari and Korpi, 2010; Meera et al., 2011; Rogawski et al., 2013; Carver and Reddy, 2016).
Also, unlike benzodiazepines, neurosteroids are able of modulating the activity of GABAAR that lack the mandatory γ subunit, which confers benzodiazepine sensitivity to the GABAAR (Bianchi and Macdonald, 2003). In this regard, neurosteroids have been tested in super refractory status epilepticus with mixed-results (Broomall et al., 2014; Rosenthal et al., 2017; Vaitkevicius et al., 2017). Analogs of ALLO were found to be protective against partial seizures induced by electrical stimulation in animals (Kaminski et al., 2004; Figure 4).
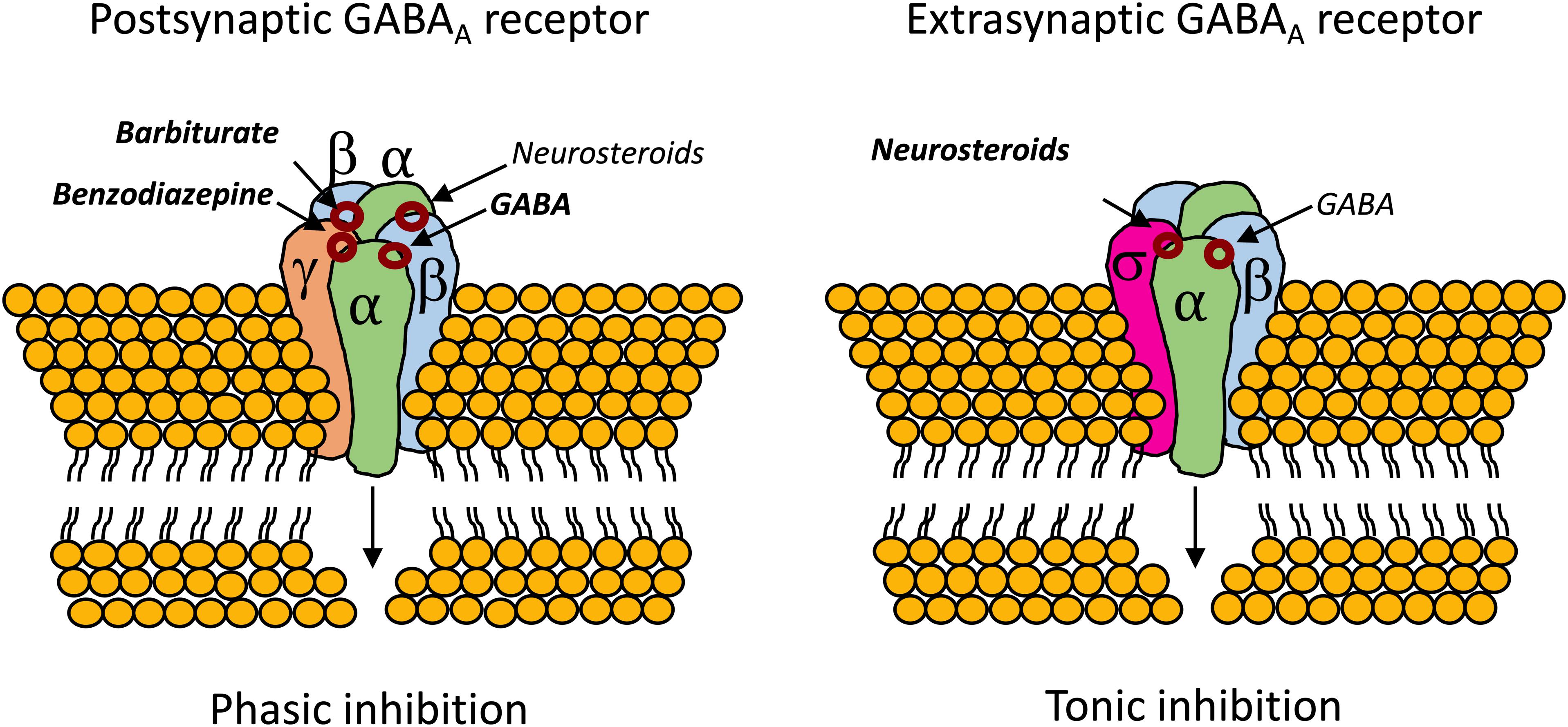
Figure 4. Binding sites for neurosteroids on the postsynaptic and extrasynaptic GABAAR (Reddy, 2011; Cai et al., 2018). Contrary to benzodiazepines, which require the mandatory γ subunit, neurosteroids can act on GABAAR whenever they are composed by α subunits (Bianchi and Macdonald, 2003). The extrasynaptic GABAAR usually contains the highly neurosteroid sensitive δ subunit, which opens an exploitable therapeutic window, especially in cases of benzodiazepine tolerance (Brown et al., 2002; Uusi-Oukari and Korpi, 2010; Meera et al., 2011; Rogawski et al., 2013; Carver and Reddy, 2016).
The role of ALLO as a neuroprotective agent starts before birth. Indeed, ALLO levels fluctuate during perinatal periods (Bernardi et al., 1998; Concas et al., 1998; Genazzani et al., 1998; Grobin and Morrow, 2001; Jin et al., 2013b) and its actions are known to contribute to neuroprotection of the fetal brain (Liu and Wong-Riley, 2010). In rats, ALLO levels drop immediately after birth, keeping a steady decline until a temporary elevation at 10 to 14 days postnatal, then decrease to a steady low level 3 weeks after birth (Grobin and Morrow, 2001). The drop in ALLO levels seems to coincide with drastic reductions in progesterone levels immediately after birth (Paoletti et al., 2006). Relevantly, in MECP2 mutant mice, the reductions in ALLO levels coincide with the onset of RTT symptoms in such animals (Jin et al., 2013b).
There are very few examples of the use of neurosteroids in RTT available on literature. Jin et al. (2013b), using an optogenetic approach to directly stimulate GABAAR in the locus coeruleus, found that ALLO increases the amplitude, frequency and decay time of GABAAergic inhibitory post-synaptic currents (IPSCs). In this study, the authors described a time-dependent modulation of GABAAergic transmission by ALLO in Mecp2 mutant mice. Like in wild-type mice, during the first 2 weeks of the postnatal period, ALLO increases GABAAergic inhibitory post-synaptic potentials and suppresses neuronal excitability in locus coeruleus neurons of Mecp2 mutant mice. However, such effect abruptly deteriorates at 3 weeks of age in Mecp2 deficient mice, which coincides with the onset of RTT symptoms. Therefore, the authors hypothesize that, in MECP2 mutant mice, declines in progesterone levels after birth may lead to decreases in ALLO biosynthesis which in turn, becomes insufficient to potentiate GABAAergic transmission through the GABAAR (Jin et al., 2013b). There is, also, the possibility that the subunit composition of the GABAAR shifts during development (Rissman and Mobley, 2011; Datta et al., 2015) or in pathophysiological conditions (Drexel et al., 2013; Govindpani et al., 2017; Kwakowsky et al., 2018) and also that it varies accordingly to temporal and local constrains (Walton et al., 2017). Thus, these modifications in GABAAR composition may change the receptor sensibility to neurosteroids in Mecp2 mutant mice (Jin et al., 2013b) (see Figure 5). Also, GABAAR sensitivity toward ALLO can shift due to neurosteroid withdrawal (Gulinello et al., 2001; Mascia et al., 2002; Boggio et al., 2010), long-term steroid administration (Follesa et al., 2002) and age (Shen et al., 2007), amongst other factors [reviewed in Mellon (2007)]. Other studies have shown that ALLO stimulates GABAAergic currents in the hippocampus (Park et al., 2011), cerebellum (Cooper et al., 1999), and neocortex (Puia et al., 2003).
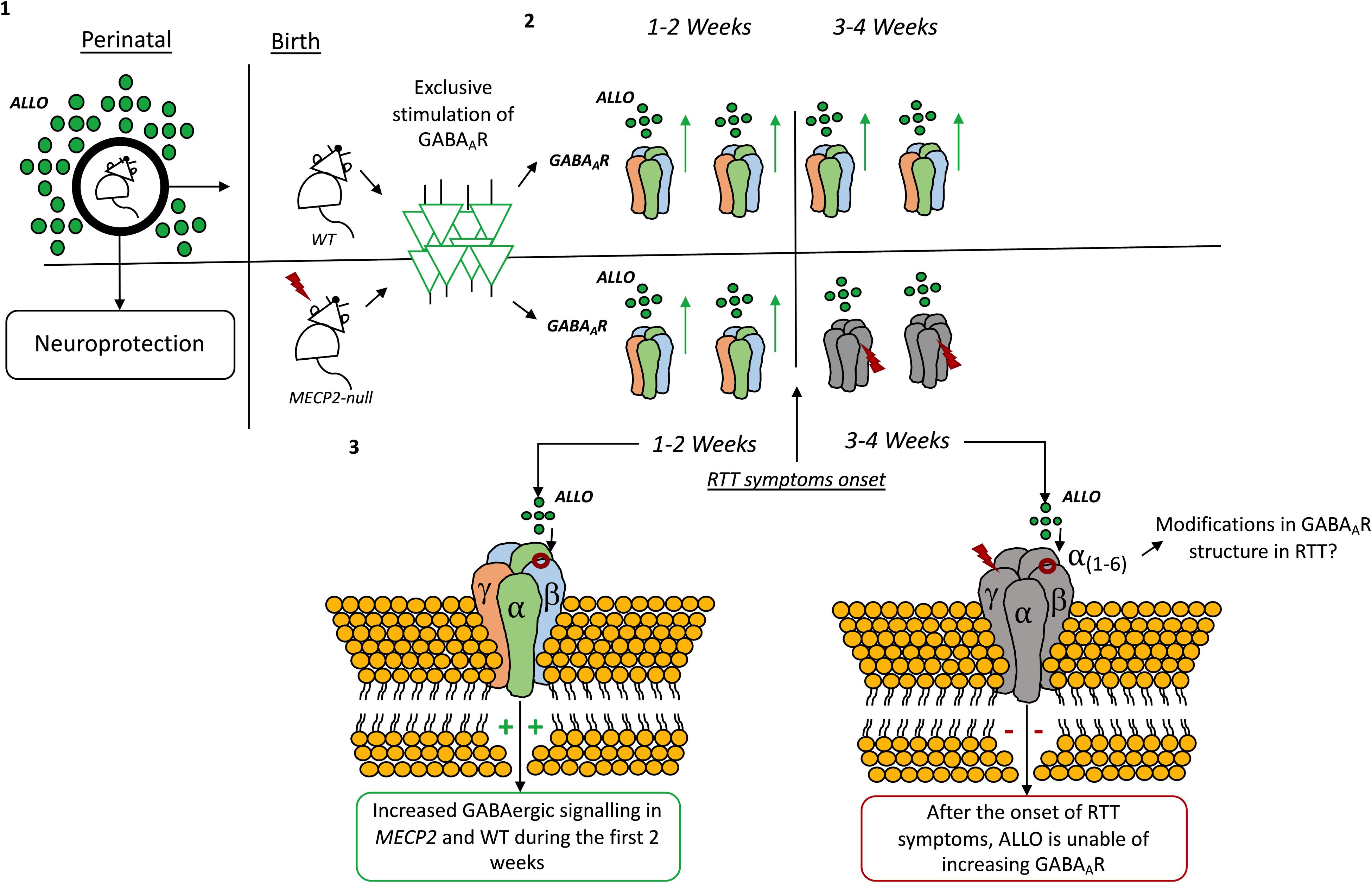
Figure 5. Time-dependent modulation by allopregnanolone on GABAAR signaling. (1) Allopregnanolone actions are known to contribute to neuroprotection of the fetal brain (Liu and Wong-Riley, 2010). In rats, allopregnanolone levels change immediately after birth and during the first weeks of life (Grobin and Morrow, 2001). (2) By exclusively stimulating the GABAA-ergic neurons in the locus coeruleus it was found that allopregnanolone increases the amplitude, frequency and decay time of GABAAR inhibitory post-synaptic currents (IPSCs) both in wild-type and Mecp2 mutant animals. (3) However, after the first 2 weeks, allopregnanolone effects are lost in Mecp2-null animals, coinciding with the onset of RTT symptoms in such animals (Jin et al., 2013b). The drop in allopregnanolone levels seems to coincide with drastic reductions in progesterone levels immediately after birth (Paoletti et al., 2006). Modifications in the composition of the GABAAR can also be responsible for the effect (Jin et al., 2013b).
Evidence displaying the existing of a time-dependent modulation of GABAAergic transmission by ALLO in Mecp2 mutant mice is certainly important, as it shows how the GABAergic system is vulnerable and mutable in RTT. However, there is an ample bulk of evidence displaying that, even in wild-type animals, changes in GABAAR sensitivity toward neurosteroids occur during embryonic development and even later in puberty. These changes can potentially lead to different effects of ALLO and other neurosteroids during development (Follesa et al., 2002). Thus, the GABAAR subunit composition may shift accordingly to the developmental stage. Therefore, the existing time-dependent GABAAR sensitivity to neurosteroids can reflect a changeable requirement of neurosteroid actions by the GABAergic system, which, in turn, may be related with the hormonal milieu of the specific period of life (Colciago and Magnaghi, 2016). Also, either physiological or pathophysiological conditions can change the way neurosteroids act on the GABAergic system. Understanding how the subunit composition of the GABAAR shifts in wild-type animals comparing to RTT mouse models, may offer important insight to elucidate the role and therapeutic potential of neurosteroids in RTT. Also, it would be relevant to study how the existent imbalances in cholesterol metabolism can impact the biosynthesis of neurosteroids in RTT.
The effects of ALLO on hippocampal-dependent memory are a great example of how the actions of neurosteroids can shift according to physiological/pathological conditions. GABAAR in the dentate gyrus, where ALLO is actively metabolized, display low sensitivity to this neurosteroid. In turn, lower ALLO metabolism in the CA1 region of the hippocampus suggests that local steroid metabolism plays a crucial role in the regulation of GABAAR-mediated inhibition in a regionally dependent manner (Belelli and Herd, 2003), specifically in brain regions devoted to memory formation and consolidation (Colciago and Magnaghi, 2016). This is strengthened by observations that ALLO increases neurogenesis, neuronal survival and reduces apoptosis in the hippocampus (Charalampopoulos et al., 2008; Rossetti et al., 2015). ALLO also increases the density of dendritic spines, the number of mature excitatory synapses and induces the formation of clusters of the actin binding protein debrin (Shimizu et al., 2015), which is known to be decreased in the brain of Alzheimer’s disease patients and individuals with mild cognitive impairment (Harigaya et al., 1996; Counts et al., 2012). Congruently, using an Alzheimer’s disease mice model, ALLO restored hippocampal-dependent learning and memory deficits and induced survival of newly generated cells in 3xTgAD mice and non-Tg aged mice (Singh et al., 2012). Thus, in 3xTgAD mice it has been proposed that an early deficit in neurosteroid synthesis may contribute to the cognitive phenotype of AD and that ALLO shows the potential to function as a regenerative therapeutic tool to delay or even prevent characteristic neurogenic and cognitive deficits of Alzheimer’s disease (Wang et al., 2010; Singh et al., 2012). In addition, ALLO was found to significantly reduce amyloid-β accumulation in the hippocampus, cortex and amygdala, to reduce microglia activation and increase expression of liver-X-receptor, pregnane-X-receptor and HMG-CoA-R, three vital proteins involved in cholesterol homeostasis and clearance from the brain (Chen et al., 2011). Despite the clear effects of ALLO on the molecular mechanisms of hippocampal memory, on the other hand, it has been described that ALLO impairs recognition memory consolidation and contextual fear memory in wild-type mice (Misane et al., 2013; Rabinowitz et al., 2014), and spatial learning in rats (Johansson et al., 2002; Matthews et al., 2002; Turkmen et al., 2004). Importantly, Mecp2 mutant mice display learning and memory impairments, besides the classic breathing abnormalities, hypoactivity, motor deficits, anxiety alterations and stereotypies. Likewise, Mecp2 mutant mice have reduced hippocampus, amygdala and striatum volumes and an overall reduction in brain volume (Stearns et al., 2007). Therefore, direct research on the possible restorative effects of neurosteroids in RTT could constitute an interesting and potentially successful approach.
Epilepsy is fairly a common comorbidity in RTT, with reports showing its presence in 60–80% of patients. In RTT, there is no unambiguous pattern of seizure episodes, and almost all seizure types are reported (Krajnc, 2015). The manifestations of seizures seems to constitute an age-related event (Chapleau et al., 2013), with onset usually in stage II or III (see section “Introduction”), around 4 years of age, but with possibility of maximal onset age between 7 and 12 years (Krajnc, 2015). In RTT, due to limited experience of epilepsy treatment, there are no definitive recommendations regarding treatment approach with the anticonvulsants currently available. Therefore, the choice of treatment for epilepsy in RTT can be challenging (Krajnc, 2015), being considered a major problem in RTT treatment (Chapleau et al., 2013). Additionally, evidence suggests that the prevalence of drug resistance epilepsy in patients with RTT is about the same as in other newly diagnosed epilepsies, in which 20 to 40% of the patients become refractory to treatment (French, 2007). However, prevention and efficient management of epileptic episodes is of major importance in RTT, as the severity and frequency of epileptic episodes are strong determinants to the clinical severity of RTT phenotype (Krajnc, 2015; Clarke and Abdala Sheikh, 2018).
A number of different AED are used in treatment of epilepsy in RTT, either in monotherapy or combined therapy, and its selection varies from case to case (Krajnc, 2015). Regardless of their many mechanisms of action, one of the main targets of several AEDs is the GABAergic signaling system (Greenfield, 2013). For instance, the effects of valproate acid, one of the most commonly prescribed AEDs, on the GABA signaling system (such as, increased GABA turnover and consequent enhancement of synaptic and extrasynaptic inhibition) are important for its antiseizure actions (Löscher, 1989; Greenfield, 2013; Rogawski et al., 2016). On the other hand, the effects of AEDs on GABAergic signaling can be more direct, like the ones of benzodiazepines, which act as direct agonists of GABAAR, known to have a specific binding site for this type of drugs. Benzodiazepines are known for augmenting the amplitude (Macdonald and Barker, 1978) or decay time (Tietz et al., 1999) of GABAAR-evoked IPSCs (Greenfield, 2013). However, in order to mediate increases in GABAAR currents, the receptor must have in its composition a γ subunit and the selectivity of each benzodiazepine depends on the specific α subunit (for detailed review see Riss et al., 2008). The clinical advantages of benzodiazepines relate with its efficacy against many seizure types (Riss et al., 2008), rapidity of onset, minimal toxicity, possibility of administration via several routes and high efficacy track-record (Macdonald and Barker, 1978). However, its use is limited by adverse side effects, such as cognitive impairment and sedation, tolerance, withdrawal and drug interactions (Riss et al., 2008). Tolerance to benzodiazepine treatment has been associated with time-dependent modifications in GABAAR subunit composition (Betjemann and Lowenstein, 2015). Namely, the internalization of subunits β1–3 and γ2 on the GABAAR, may lead do decreased inhibition of synaptic transmission (Goodkin et al., 2008). Although benzodiazepines are not commonly prescribed as AED in RTT cases, different studies using RTT-mice models have shown the promising effects of benzodiazepines midazolam (Voituron and Hilaire, 2011) and diazepam (Abdala et al., 2010), particularly attenuating respiratory defects.
Recently, natural constituents of the cannabis plant have been emerging as promising AEDs. Importantly, the ECBS stands as a promising pharmacological target in RTT (Vigli et al., 2018; Zamberletti et al., 2019). The ECBS is a complex neuromodulatory system that regulates important physiological processes (Kano et al., 2009), such as, anxiety and stress (Jenniches et al., 2016), social behavior (Wei et al., 2017), motor coordination (El Manira and Kyriakatos, 2010) and memory/learning (Marsicano and Lafenêtre, 2009). Importantly, these are behaviors usually compromised in RTT (Vigli et al., 2018). Cannabinoids modulate neuronal activity mainly via the CB1R and CB2R. CB1R activation is linked with neuroprotection by controlling excitotoxicity via inhibition of excessive excitatory transmission and consequent calcium release (Vendel and de Lange, 2014). Indeed, the control of excitotoxicity constitutes a major physiological role of the ECBS (Kano et al., 2009). Although the CB1R is classically denoted as a Gi/o protein coupled receptor, mainly linked with inhibitory effects, CB1R coupling to G proteins is mutable. CB1R is now believed to have very few “intrinsic” properties, being the cellular consequences of its activation likely a reflex of cell type and/or location in which the receptor is situated, their activity in different brain regions and due to temporal constrains (Busquets-Garcia et al., 2018). Contrary to initial belief, which placed CB2R exclusively in the immune system (Kano et al., 2009), CB2R are now known to be present in neurons, microglia and astrocytes (for a review see Solymosi and Kofalvi, 2017). CB2R are involved in the control of glial activation and inflammation and on the regulation of neuronal excitability (Solymosi and Kofalvi, 2017). Moreover, CB2R levels can increase in neurons and astrocytes under very specific circumstances, such as, in situations of neuroinflammation and in certain diseases (Onaivi et al., 2012; Di Marzo et al., 2015; Fernández-Ruiz et al., 2015). Related with its physiological roles, the lack of psychoactivity associated with CB2R activation, has made this receptor a particular promising therapeutic target for cannabis-based therapies (Cassano et al., 2017). For thorough reviews on the ECBS and its actions, natural cannabinoids and their molecular targets, and the clinical applications of cannabinoids (see Kano et al., 2009; Morales et al., 2017; Solymosi and Kofalvi, 2017).
Today, more than 120 phytocannabinoids (natural occurring cannabinoids) have been identified as constituents of the cannabis plant. The most abundant cannabinoids in the cannabis plant are delta-9-tetrahydrocannabinoid, or Δ9-THC, followed by Δ8-THC, CBN, CBD, CBG, CBC, THCV, CBV, and CBDV (Morales et al., 2017). From all the natural constituents of the cannabis plant, CBD, a non-psychoactive cannabinoid (Loewe, 1946; Pertwee, 2009), is being widely studied given its high therapeutic value. CBD has been shown, both in humans and rodents, to have antiseizure (Jones et al., 2010; Pamplona and Coan, 2017; Perucca, 2017; Zaheer et al., 2018), anti-inflammatory (Malfait et al., 2000; Nagarkatti et al., 2009; Petrosino et al., 2018), antioxidant (Hampson et al., 2000), and anti-psychotic properties (Zuardi et al., 1991, 2012; Iseger and Bossong, 2015), to have neuroprotective effects (Iuvone et al., 2009; Fernández-Ruiz et al., 2013), to reduce nausea (Parker et al., 2011; Mersiades et al., 2018) and to work as an anxiolytic and anti-depressive drug (de Mello Schier et al., 2014; Blessing et al., 2015; Zuardi et al., 2017). Furthermore, CBD is known to potentiate the clinical efficacy of Δ9-THC, increasing the durability of its beneficial effects, while preventing its psychoactive effects (Russo and Guy, 2006; Solymosi and Kofalvi, 2017). CBD acts as a surprisingly high potent antagonist at both the CB1R and the CB2R (Thomas et al., 2009). Recently, it has also been proposed that CBD acts as a negative allosteric modulator (NAM) at the CB1R (Laprairie et al., 2015). Many of CBD therapeutic properties have been suggested to occur due to its effect at the TRPV1 (Morales et al., 2017).
The medical use of cannabis has been increasing and the full potential of the cannabis plant is yet to be unfold. Cannabis-based drugs are been used, or proposed for use, in neuropathic pain and muscle spasticity associated with Multiple Sclerosis (Fitzpatrick and Downer, 2017; Solymosi and Kofalvi, 2017; Rice and Cameron, 2018), neurodegenerative diseases (Fagan and Campbell, 2014; Basavarajappa et al., 2017; Navarro et al., 2018), chronic pain (Carter et al., 2015; Pascual et al., 2018), to alleviate AIDS-related weight loss (Badowski and Perez, 2016; Rock and Parker, 2016) and to reduce vomiting and nausea associated with chemotherapy (Badowski and Yanful, 2018). Most relevantly for this work, cannabinoid use is being increasingly proposed as a very promising anti-epileptic drug, even in cases of treatment-resistant epilepsy (Maa and Figi, 2014; O’Connell et al., 2017; Pamplona and Coan, 2017). In a recent clinical trial, CBD significantly reduced convulsive-seizure frequency in Dravet syndrome patients (Devinsky et al., 2017). In a double-blind placebo-controlled trial, 120 children suffering with Dravet syndrome and drug-resistance epilepsy received either CBD (oral solution 20 mg/kg per day) or placebo over 14 weeks. In the CBD group, 43% of the patients had a reduction of 50% in seizure frequency, as opposed to a 27% decrease in the placebo group. Furthermore, 5% of the patients treated with CBD became seizure-free compared with a 0% incident in the placebo group. On the CGIC, a Likert-like scale questionnaire which allows assessing improvement after a treatment comparing with a baseline period (ranging from 1, very much improved to 7 very much worse), in the CBD group, 62% of the patients increased at least one category on the CGIC compared with 27% on the placebo group. Treatment with CBD provoked more adverse side effects reflected on diarrhea, vomiting, fatigue, pyrexia, somnolence, and some abnormalities on liver-function tests (Devinsky et al., 2017). About 1 year after this clinical trial, the FDA approved Epidiolex® (CBD) oral solution for treatment in Dravet and Lennox–Gastaut syndromes, two diseases that, in resemblance with RTT, present refractory epilepsy and high mortality rates. Also, Nabiximols (Sativex®), a combination of CBD and Δ9-THC, is approved to treat muscle spasticity in Multiple Sclerosis (Novotna et al., 2011), an abnormality in muscle tone present in RTT (Kyle et al., 2018).
The mechanism by which CBD exerts anticonvulsive effects is still not completely clarified. Recently, however, using human recombinant GABAAR, CBD was shown to act as a positive allosteric modulator of the GABAAR, with the effects being selective for the β-subunit (higher in receptors with β2/β3 over β1 subunits) and the maximum effect being detected in receptors which included the α2-subunit (Bakas et al., 2017). The authors hypothesized that these results may explain the anticonvulsant and anxiolytic properties of this compound (Bakas et al., 2017). Indeed, previously, it was reported that CBD attenuated seizures in two well characterized seizure models, the acute pilocarpine TLE and the penicillin model of partial seizure (Jones et al., 2012). Curiously, in the acute epilepsy model using pilocarpine, a muscarinic acetylcholine receptor agonist, the antiseizure effects of CBD were modest and the drug was not able of reducing mortality and severity of episodes. On the other hand, using the penicillin model, a selective antagonist of the GABAAR, CBD had a powerful antiseizure effect, significantly reducing the number of animals experiencing tonic-clonic episodes and their mortality. Importantly, CBD exerted only minor negligible motor effects (Jones et al., 2012). At the time, the authors speculated that the antiseizure effect of CBD could be occurring via a GABAergic-mediated mechanism (Jones et al., 2012). In support of this hypothesis, a previous study had shown an anticonvulsive action of CBD on several seizure models mediated via disinhibition of GABA release (Consroe et al., 1982; Jones et al., 2012). In a very recent study, CBD was shown to have antiseizure effects in several models of acute epilepsy (Patra et al., 2019). Additionally, this study showed, for the first time, that chronic CBD administration improves seizure burden ratio, attenuates cognitive impairment and reduces motor comorbidities, well after the onset of symptoms in the RISE-SRS TLE (Patra et al., 2019). Thus, the results of this study are particularly relevant for research in RTT, considering the positive effects of a chronic administration of CBD on both seizures and motor impairment, which constitute two common comorbidities in RTT. Additionally, cognitive impairment is also characteristic of RTT and, therefore, finding that CBD improves cognition in models of acute and chronic epilepsy in also relevant for cannabinoid-related research in RTT.
Direct evidence of cannabinoid use in RTT mice models is objectively scarce in literature. To the best of our knowledge, there are only two reports evaluating the impact of a chronic treatment with a phytocannabinoid in an RTT-mice model. In both these reports (Vigli et al., 2018; Zamberletti et al., 2019), the authors used CBDV, a propyl analog of CBD devoid of psychoactive actions (Morales et al., 2017). CBDV has very weak activity at the CB1R and CB2R (Hill et al., 2013; Rosenthaler et al., 2014; Morales et al., 2017) and it has been proposed to activate the TRPV1 (Iannotti et al., 2014). Additionally, an effect of CBDV on the deorphanized cannabinoid receptor GPR55 has been described. Besides some cannabinoids, GPR55 is also activated by LPI. Notably, it has been found that CBDV is a potent inhibitor of LPI-induced GPR55 signaling (Lauckner et al., 2008; Anavi-Goffer et al., 2012). As stated in Vigli et al. (2018), GPR55 has been implied in important cognitive processes, such as, spatial memory regulation (Marichal-Cancino et al., 2018), social behavior (Kramar et al., 2017) and motor function (Bjursell et al., 2016). Moreover, in a recent work, CBDV was shown to attenuate seizures and social withdrawal in a mice model of Dravet syndrome (Kaplan et al., 2017). Concerning cannabinoids and RTT, the administration of CBDV by intraperitoneal injection (escalating dose ranging from 2 to 100 mg/kg of body weight) to Mecp2-308 male mice during 14 days, rescued sociability impairments, improved the general health status and increased the brain weight of these animals. Furthermore, a molecular analysis of GPR55 levels, revealed an up-regulation of this receptor in the hippocampus (Vigli et al., 2018). These results showed, for the first time, the clinical potential of non-psychoactive constituents of the cannabis plant and also the important role of the GPR55 as a promising drug-target on RTT. Very recently, a new study using CBDV has confirmed the potential of this particular phytocannabinoid in RTT. In this study, chronic administration of CBDV (0.2 to 2 mg/kg) was shown to completely rescue cognitive deficits, delaying neurological (although transiently) and motor impairments in male MECP2 mutant mice (Zamberletti et al., 2019). Importantly, CBDV administration normalized BDNF and insulin growth factor 1 (IGF1) levels and restored normal signaling in the PI3K/AKT/mTOR pathway at an advanced stage of the disease. Interestingly, Mecp2 deletion in this RTT mouse model provoked upregulation of CB1R and CB2R and CBDV treatment led to a normalization of these changes (Zamberletti et al., 2019). Although these two independent studies suggest a promising role of CBDV as a therapeutic drug in RTT, the fact that different molecular modifications in the ECBS were detected in each model, highlight the necessity of further studies.
The promising antiseizure effects of CBD, even in cases of refractory-epilepsy, observed in both clinical trials with humans and in laboratory animals, the effects of combinations of CBD and Δ9-THC in controlling muscle spasticity and motor symptoms, and the positive results of CBDV administration in two different mouse models of RTT, place cannabinoids as a viable therapeutic strategy in RTT. Moreover, CBD positively modifies impairments in motor, cognitive and social processes in animal models, further highlighting the potential of cannabinoid molecules to tackle RTT-symptomology.
Around 100 years ago, it was noted that a high fat diet could have long-lasting effects in preventing seizures without inducing significant caloric deprivation (Rogawski et al., 2016). These findings resulted on the creation of the KD, trademarked by an increase in the production in the liver of ketones bodies, such as BHB, acetoacetate and acetone (Peterman, 1924; Freeman and Kossoff, 2010; Rogawski et al., 2016). The fact that the KD was created in a time where the available AEDs were very limited, contributed for a rapid gain in popularity of this diet (Rogawski et al., 2016). However, the development of the antiepileptic drug phenytoin substantially decreased the use of the KD. In the last 30 years, interest in using the KD resurged, especially in children presenting refractory epilepsy (Rogawski et al., 2016).
An abnormal increase of ketone bodies in the blood was early suggested as the underlaying mechanism of the anticonvulsive properties of low carbohydrate KD (Talbot et al., 1927; Huttenlocher, 1976). Subsequently, many other possible mechanisms were enounced, such as, pH changes, anticonvulsive effects of hyperlipidemia, effects of the KD directly on sodium and potassium balance, and also an effect on cerebral metabolism, reflected on the change of glucose, as the main source of energy, to BHB, as a consequence of carbohydrate restriction (for details see Huttenlocher, 1976). To clarify these questions, Peter Huttenlocher designed a new KD in which ketonemia was induced by feeding of MCT. In his report of 1976, the MCT diet was administered to 18 children and he observed no differences in hyperlipidemia, no significant changes in pH, considerable reductions in blood glucose levels in one-third of the children and a gradual increase in plasma BHB and acetoacetate levels during a diet that was kept from 3 months to 4 years. Furthermore, plasma levels of BHB showed a significant correlation with the anticonvulsive effects of the MCT diet and, along with ketonemia, were rapidly abolished by intravenous infusion of glucose (Huttenlocher, 1976). Today, the mechanisms behind the anticonvulsive actions of the KD are fairly well characterized (for a detailed review see Rogawski et al., 2016).
One of the proposed mechanisms of action of the KD are increases in the synthesis of GABA and decreases in the synthesis of excitatory neurotransmitters, such as glutamate (Rogawski et al., 2016). Indeed, it has been suggested that one of the anticonvulsive mechanisms of the KD relates with modifications in the way glutamate is processed in the brain. Particularly, due to a more active astrocytic metabolism, the conversion of glutamate to glutamine is increased, resulting in a more efficient removal of glutamate and increased GABA synthesis (Yudkoff et al., 2008). A study with 26 children suffering from refractory epilepsy, revealed that the KD increases levels of GABA in the cerebrospinal fluid, without affecting glutamate concentration. In this study, the results showed higher GABA levels in the responders to the diet compared with non-responders. Furthermore, the anticonvulsive action of the diet was more evident (90% reduction in seizures) in children who had higher levels of GABA at the beginning of the treatment and in whom a gradual increase in GABA levels during treatment was registered. Also, the anticonvulsive effects of the diet seem to be age-dependent, with younger children being the best responders (Dahlin et al., 2005). However, studies with animal models performed to confirm a GABAergic-mediated mechanism of anticonvulsive actions of the KD have produced mixed-results (Hartman et al., 2007). If, on one hand, the KD administered to mice fails to confer protection against the clonic seizures induced by GABAAR antagonist pentylenetetrazol, on the other hand, it is much more effective protecting against seizures induced by the maximal electroshock test, a test in which the classical anticonvulsive drugs targeting the GABAergic system only confer weak protection (Uhlemann and Neims, 1972; Hartman et al., 2007). On the contrary, the KD was found to protect against seizures induced by GABAAR antagonists pentylenetetrazol, bicuculline, and picrotoxin in rats (for a detailed review see Hartman et al., 2007). It has also been suggested that ketone bodies, such as acetoacetate, compete with chloride for the allosteric binding site at the vesicular glutamate transporter, thus inhibiting exocytotic glutamate release (Juge et al., 2010). Additionally, the authors of this work showed that acetoacetate protected against seizure-like activity induced by 4-aminopyridine (Juge et al., 2010). Recently, using the spontaneously epileptic Kcna1-null mice, the ketone body BHB was shown to exert anti-seizure effects, restore impairments in synaptic plasticity processes and raise the threshold of mitochondrial permeability transition (Kim et al., 2015; Rogawski et al., 2016).
Concerning the clinical use of the KD in RTT, Haas et al. (1986), reported a clinical study were the MCT KD was used in 7 girls diagnosed with RTT who presented anticonvulsive resistant seizures. Among the 5 girls capable of tolerating the diet, the KD improved seizure control, slightly ameliorated behavior and motor skills and increased bodyweight. The results from this work led the authors to suggest that, considering the defects in the carbohydrate metabolism occurring in RTT, the KD stands as a logical choice in this disease (Haas et al., 1986). Surprisingly, until very recently, this was the only clinical report of the use of KD in RTT. Indeed, evidence of the application of the KD in RTT is extremely hard to find in the literature. In Liebhaber et al. (2003) reported a case of an RTT patient who was treated with the KD for 4 years. The treatment started when the patient was 8 years old and presented a refractory epilepsy. The diet led to a 70% reduction in seizure frequency and improved contact and behavior. In Giampietro et al. (2006) reported a case with a female patient suffering from an atypical RTT variant in which the KD significantly reduced seizure occurrence. In 2011, a long-term follow-up of the use of the KD in 226 patients with refractory epilepsy was reported. This report included one patient with RTT, which presented a 75–99% seizure reduction after KD (Caraballo et al., 2011). Evidence on the use of the KD in RTT-mouse models is also scarce. In the only direct approach available in the literature, it has been described that a restricted KD positively impacts anxiety and motor measures in a male mutant Mecp2 mice. However, the authors also showed that the results of the KD were very similar to the ones from a calorie restriction diet, concluding that, most likely, it was the caloric restriction that produced the positive outcomes, rather than the composition of the KD by itself (Mantis et al., 2009).
Reports on the use of the KD in autism spectrum disorders and other brain diseases are also available in literature. With relevance for the phenotypic expression of RTT, it has been described that 4 weeks of a KD regime increases the social behavior of wild-type rats, registered in three different social interaction protocols. On the other hand, the exogenous administration of ketone bodies had no effects on social interaction in the same paradigms (Kasprowska-Liśkiewicz et al., 2017). Also, in an EL mice model (Meidenbauer et al., 2011), which presents autism spectrum disorders phenotype and comorbid epilepsy, the KD was shown to improve sociability and reduce repetitive behavior (Ruskin et al., 2017). Using the same animal model, the KD was shown to induce a 1 month delay in epileptogenesis (Todorova et al., 2000) and to have anticonvulsive actions (Mantis et al., 2004). In an Huntington’s disease mice model, the KD was shown to produce positive outcomes on weight lose without affecting cognitive processes (Ruskin et al., 2011). More recently, in an animal model of autism induced by prenatal exposure to valproic acid, the KD was, once again, associated with improvement in social behavior (Castro et al., 2017).
Another potential mechanism of action underlying the anticonvulsive effects of the KD involves the modulation of adenosine receptor activity (Masino et al., 2013; Rogawski et al., 2016). Adenosine is an ubiquitous neuromodulator affecting the action and activity of several neurotransmitter receptors (Sebastião and Ribeiro, 2015). A1R activity mediates the inhibitory effects of adenosine, including inhibition of neurotransmitter release and changes in post-synaptic membrane conductance (Dunwiddie and Masino, 2001). On the other hand, the A2AR mediates the excitatory effects of adenosine by coupling to Gs proteins, consequently leading to stimulation of adenylate cyclase, increases in intracellular cAMP levels and PKA activation (Sebastião and Ribeiro, 2015). Relevantly, caffeine, the most widely consumed legal drug in the world, is a non-selective antagonist at the A1R and A2AR (Fredholm et al., 1999). Recently, the KD was demonstrated to suppress seizures in a transgenic mice model with spontaneous seizures by activation of the A1R (Masino et al., 2011) and similar results were shown in an in vitro mimic of the KD (Kawamura et al., 2010). Adenosine is directly synthetized from ATP and given that ATP levels are increased in the KD it is feasible that it results in an increase in adenosine synthesis (Rogawski et al., 2016). Adenosine receptor activity, particularly A2AR activity, is also known to directly facilitate the actions of the important BDNF (Diógenes et al., 2004, 2007; Fernandes et al., 2008; Fontinha et al., 2008; Vaz et al., 2015). Importantly, BDNF expression is impaired in RTT and it has been suggested that it can have important therapeutic actions in RTT (reviewed in Katz, 2014; Li and Pozzo-Miller, 2014).
In conclusion, if, on one hand, it is particularly well accepted and proven that the KD has powerful antiseizure actions (although the mechanisms of action are still not completely described) (Vining et al., 1998; Hassan et al., 1999; Neal et al., 2008), on the other hand, reports on the application of this diet to RTT cases and in RTT-mice models are scarce. The lack of research is even more surprising considering the positive results shown in both clinical studies with humans and in RTT mouse-models. Relevantly, a prospective study with 145 children aged between 2 and 16 years, suffering from intractable epilepsy and who had never experienced the KD, showed that 3 months of diet resulted in a 75% seizure reduction rate, with 38% of the children in the experimental group experiencing a reduction of 50% in seizures. Also, five children experienced reductions of 90% in seizures and one child became seizure free (Neal et al., 2008). Importantly, in children allocated to the control group who continued their usual treatment with AED, some of them experienced worsening of symptoms and none became seizure-free (Neal et al., 2008; Masino et al., 2013).
The present review summarizes important recent evidence concerning metabolic, synaptic, functional and molecular dysfunctions occurring in RTT (see Table 1). Due to the ubiquitous role of MECP2 gene, it has been a tremendous challenge to comprehend and characterize the mechanisms through which MECP2 mutations lead to symptomatology in RTT. With the development and improvement of gene-editing technologies, research in RTT has drifted from “therapy-directed” to “cure-oriented” investigation. However, the path for a definitive cure is paved with many challenges and, most likely, this achievement in not around the corner. Meanwhile, it is paramount to better understand this disease and to improve the available treatment by exploring new therapeutic avenues. In this work, we reviewed promising evidence on different research lines that may have an important impact in the field. From modulators of GABAergic signaling, to cannabinoids and the KD, and cleverly exploiting the metabolic features of this disease, an ample bulk of evidence has been gathered, creating a plethora of research lines to be followed in the future. RTT is a devastating disease, both for the patient and for the caregivers, and clinical and preclinical research directed at finding new therapeutic approaches are more important than ever, now that we are beginning to understand some of the mechanisms of this disease.
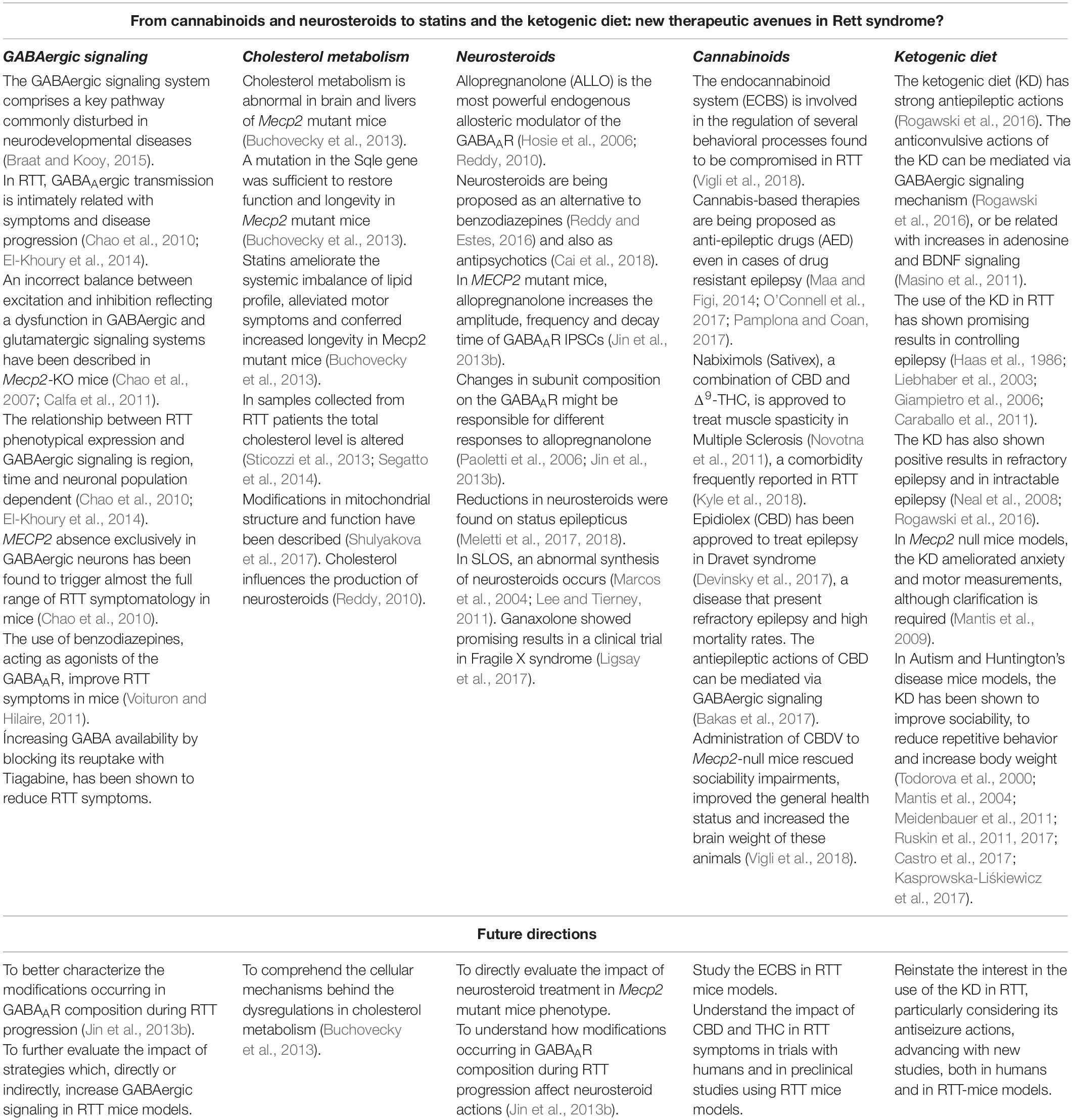
Table 1. Summary of the main topics of this review and future directions.
Summarizing, in the future it would be interesting to: (1) better characterize the modifications occurring in GABAAR composition during RTT disease progression; (2) study how cholesterol imbalances impact on neurosteroid production in RTT and their potential therapeutic use in this disease; (3) disclose the cellular mechanisms behind the dysregulations in cholesterol metabolism; (4) directly study the immense potentiality of CBD and other cannabinoids on RTT mouse-models and in controlled clinical trials with humans; (5) reinstate the interest in the use of the KD in RTT, particularly considering its antiseizure actions, advancing with new studies, both in humans and in RTT-mouse models; (6) to design further studies on the therapeutic properties of statins in RTT, and (7) continue to research mitochondria dynamics in RTT and its role on the disease progression and symptomatology.
FM and MD wrote the manuscript. CM-L revised and assisted on manuscript writing. AS revised the manuscript. All authors contributed to the final version of the manuscript.
FM was in receipt of a fellowship (IMM/CT/8-2018). The authors would like to thank the following organizations for their funding: AdoRett – LISBOA-01-0145-FEDER-031929; the Association Française du Syndrome de Rett Program “Educação pela Ciência” | Bolsas CHLN/FMUL; GAPIC. Project No. 20190017; Twinning action (SynaNet) from the EU H2020 Programme; and the UID/BIM/50005/2019, project financed by the FCT/Ministério da Ciência, Tecnologia e Ensino Superior (MCTES), through the Fundos do Orçamento de Estado.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
24S-OHC, oxysterol 24(S)-hydroxycholesterol; 3xTg-AD, triple-transgenic mouse model of AD; 3 α-HSD, 3 α-hydroxysteroid dehydrogenase; 3 β-HSD, 3 β-hydroxysteroid dehydrogenase; A1R, adenosine A1 receptor; A2AR, adenosine A2A receptor; AED, anti-epileptic drugs; ALLO, allopregnanolone; ATP, adenosine triphosphate; BBB, blood brain barrier; BDNF, brain-derived neurotrophic factor; BHB, β-hydroxybutyrate; BZD, benzodiazepines; CA1, cornu ammonis 1; cAMP, cyclic adenosine monophosphate; CB1R, cannabinoid receptor 1; CB2R, cannabinoid receptor 2; CBC, cannabichromene; CBD, cannabidiol; CBDV, cannabidivarin; CBG, cannabigerol; CBN, cannabinol; CBV, cannabivarin; CDKL5, cyclin-dependent kinase-like 5; CGIC, seven-category caregiver global impression of change questionnaire; CNS, central nervous system; CYP11A1, cytochrome P450 cholesterol side-chain cleavage (P450scc) enzyme; Cyp46a1, cholesterol 24-hydroxylase; ECBS, endocannabinoid system; FDA, United States Food and Drug Administration; FOXG1, forkhead box protein G1; GABA, gamma-aminobutyric acid; GABAAR, GABAA receptor; GABABR, GABAB receptor; GABR, GABAA receptor subunit; GLU, glutamate; GPR55, G-protein coupled receptor 55; HDL, high density lipoprotein; HMG-CoA, 3-hydroxy-3-methylglutaryl-coA; HMG-CoA-R, 3-hydroxy-3-methylglutaryl-coA reductase; IPSCs, post-synaptic inhibitory potentials; KD, ketogenic diet; LDL, low density lipoprotein; LPI, l- α-lysophosphatidylinositol; MCT, medium chain triglycerides; MECP2, methyl-CpG-binding protein 2; PKA, protein kinase A; RISE-SRS, reduced intensity status epilepticus–spontaneous recurrent seizures; RTT, Rett syndrome; SLOS, Smith–Lemli–Opitz syndrome; Sqle, squalene monooxygenase; SRB1, scavenger protein B1; STA, statins; StAR, steroidogenic acute regulatory protein; Tg, transgenic; THCV, Δ 9-tetrahydrocannabivarin; THIP, GABAAR agonist gaboxadol; TLE, model of temporal lobe epilepsy; TRPV1, transient receptor potential cation channel subfamily V member 1; Δ 8-THC, Δ 8-tetrahydrocannabinol; Δ 9-THC, Δ 9-tetrahydrocannabino.
Abdala, A. P., Toward, M. A., Dutschmann, M., Bissonnette, J. M., and Paton, J. F. R. (2016). Deficiency of GABAergic synaptic inhibition in the kölliker-fuse area underlies respiratory dysrhythmia in a mouse model of Rett syndrome. J. Physiol. 594, 223–237. doi: 10.1113/JP270966
Abdala, A. P. L., Dutschmann, M., Bissonnette, J. M., and Paton, J. F. R. (2010). Correction of respiratory disorders in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 18208–18213. doi: 10.1073/pnas.1012104107
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Anavi-Goffer, S., Baillie, G., Irving, A. J., Gertsch, J., Greig, I. R., Pertwee, R. G., et al. (2012). Modulation of l-α-lysophosphatidylinositol/GPR55 mitogen-activated protein kinase (MAPK) signaling by Cannabinoids. J. Biol. Chem. 287, 91–104. doi: 10.1074/jbc.M111.296020
Aneja, A., and Tierney, E. (2008). Autism: the role of cholesterol in treatment. Int. Rev. Psychiatry 20, 165–170. doi: 10.1080/09540260801889062
Archer, H. L., Evans, J., Edwards, S., Colley, J., Newbury-Ecob, R., O’Callaghan, F., et al. (2006). CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J. Med. Genet. 43, 729–734. doi: 10.1136/jmg.2006.041467
Auron, A., and Brophy, P. D. (2012). Hyperammonemia in review: pathophysiology, diagnosis, and treatment. Pediatr. Nephrol. 27, 207–222. doi: 10.1007/s00467-011-1838-5
Badowski, M. E., and Perez, S. E. (2016). Clinical utility of dronabinol in the treatment of weight loss associated with HIV and AIDS. HIV AIDS 8, 37–45. doi: 10.2147/HIV.S81420
Badowski, M. E., and Yanful, P. K. (2018). Dronabinol oral solution in the management of anorexia and weight loss in AIDS and cancer. Ther. Clin. Risk Manag. 14, 643–651. doi: 10.2147/TCRM.S126849
Bakas, T., van Nieuwenhuijzen, P. S., Devenish, S. O., McGregor, I. S., Arnold, J. C., and Chebib, M. (2017). The direct actions of cannabidiol and 2-arachidonoyl glycerol at GABA A receptors. Pharmacol. Res. 119, 358–370. doi: 10.1016/j.phrs.2017.02.022
Basavarajappa, B. S., Shivakumar, M., Joshi, V., and Subbanna, S. (2017). Endocannabinoid system in neurodegenerative disorders. J. Neurochem. 142, 624–648. doi: 10.1111/jnc.14098
Belelli, D., Casula, A., Ling, A., and Lambert, J. J. (2002). The influence of subunit composition on the interaction of neurosteroids with GABA(A) receptors. Neuropharmacology 43, 651–661.
Belelli, D., and Herd, M. B. (2003). The contraceptive agent Provera enhances GABA(A) receptor-mediated inhibitory neurotransmission in the rat hippocampus: evidence for endogenous neurosteroids? J. Neurosci. 23, 10013–10020.
Bernardi, F., Salvestroni, C., Casarosa, E., Nappi, R. E., Lanzone, A., Luisi, S., et al. (1998). Aging is associated with changes in allopregnanolone concentrations in brain, endocrine glands and serum in male rats. Eur. J. Endocrinol. 138, 316–321.
Betjemann, J. P., and Lowenstein, D. H. (2015). Status epilepticus in adults. Lancet Neurol. 14, 615–624. doi: 10.1016/S1474-4422(15)00042-3
Bianchi, M. T., and Macdonald, R. L. (2003). Neurosteroids shift partial agonist activation of GABA(A) receptor channels from low- to high-efficacy gating patterns. J. Neurosci. 23, 10934–10943.
Bjursell, M., Ryberg, E., Wu, T., Greasley, P. J., Bohlooly-Y, M., and Hjorth, S. (2016). Deletion of Gpr55 results in subtle effects on energy metabolism, motor activity and thermal pain sensation. PLoS One 11:e0167965. doi: 10.1371/journal.pone.0167965
Blessing, E. M., Steenkamp, M. M., Manzanares, J., and Marmar, C. R. (2015). Cannabidiol as a potential treatment for anxiety disorders. Neurotherapeutics 12, 825–836. doi: 10.1007/s13311-015-0387-1
Blue, M. E., Naidu, S., and Johnston, M. V. (1999). Altered development of glutamate and GABA receptors in the basal ganglia of girls with Rett syndrome. Exp. Neurol. 156, 345–352. doi: 10.1006/exnr.1999.7030
Boggio, E. M., Lonetti, G., Pizzorusso, T., and Giustetto, M. (2010). Synaptic determinants of Rett syndrome. Front. Synapt. Neurosci. 2:28. doi: 10.3389/fnsyn.2010.00028
Braat, S., and Kooy, R. F. (2015). The GABAA receptor as a therapeutic target for neurodevelopmental disorders. Neuron 86, 1119–1130. doi: 10.1016/j.neuron.2015.03.042
Broomall, E., Natale, J. E., Grimason, M., Goldstein, J., Smith, C. M., Chang, C., et al. (2014). Pediatric super-refractory status epilepticus treated with allopregnanolone. Ann. Neurol. 76, 911–915. doi: 10.1002/ana.24295
Brown, N., Kerby, J., Bonnert, T. P., Whiting, P. J., and Wafford, K. A. (2002). Pharmacological characterization of a novel cell line expressing human alpha(4)beta(3)delta GABA(A) receptors. Br. J. Pharmacol. 136, 965–974. doi: 10.1038/sj.bjp.0704795
Buchovecky, C. M., Turley, S. D., Brown, H. M., Kyle, S. M., McDonald, J. G., Liu, B., et al. (2013). A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat. Genet. 45, 1013–1020. doi: 10.1038/ng.2714
Busquets-Garcia, A., Bains, J., and Marsicano, G. (2018). CB1 receptor signaling in the brain: extracting specificity from ubiquity. Neuropsychopharmacology 43, 4–20. doi: 10.1038/npp.2017.206
Cai, H., Cao, T., Zhou, X., and Yao, J. K. (2018). Neurosteroids in schizophrenia: pathogenic and therapeutic implications. Front. Psychiatry 9:73. doi: 10.3389/fpsyt.2018.00073
Calfa, G., Hablitz, J. J., and Pozzo-Miller, L. (2011). Network hyperexcitability in hippocampal slices from Mecp2 mutant mice revealed by voltage-sensitive dye imaging. J. Neurophysiol. 105, 1768–1784. doi: 10.1152/jn.00800.2010
Campos-Castelló, J., Peral Guerra, M., Riviere Gómez, A., Oliete García, F., Herranz Tanarro, J., Toledano Barrero, M., et al. (1988). [Rett’s syndrome: study of 15 cases]. An. Esp. Pediatr. 28, 286–292.
Caraballo, R., Vaccarezza, M., Cersósimo, R., Rios, V., Soraru, A., Arroyo, H., et al. (2011). Long-term follow-up of the ketogenic diet for refractory epilepsy: multicenter Argentinean experience in 216 pediatric patients. Seizure 20, 640–645. doi: 10.1016/j.seizure.2011.06.009
Carter, G. T., Javaher, S. P., Nguyen, M. H., Garret, S., and Carlini, B. H. (2015). Re-branding cannabis: the next generation of chronic pain medicine? Pain Manag. 5, 13–21. doi: 10.2217/pmt.14.49
Carunchio, I., Mollinari, C., Pieri, M., Merlo, D., and Zona, C. (2008). GABA A receptors present higher affinity and modified subunit composition in spinal motor neurons from a genetic model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 28, 1275–1285. doi: 10.1111/j.1460-9568.2008.06436.x
Carver, C. M., and Reddy, D. S. (2016). Neurosteroid structure-activity relationships for functional activation of extrasynaptic δGABA(A) receptors. J. Pharmacol. Exp. Ther. 357, 188–204. doi: 10.1124/jpet.115.229302
Cassano, T., Calcagnini, S., Pace, L., De Marco, F., Romano, A., and Gaetani, S. (2017). Cannabinoid receptor 2 signaling in neurodegenerative disorders: from pathogenesis to a promising therapeutic target. Front. Neurosci. 11:30. doi: 10.3389/fnins.2017.00030
Castro, K., Baronio, D., Perry, I. S., Riesgo, R. D. S., and Gottfried, C. (2017). The effect of ketogenic diet in an animal model of autism induced by prenatal exposure to valproic acid. Nutr. Neurosci. 20, 343–350. doi: 10.1080/1028415X.2015.1133029
Chao, H.-T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Chao, H.-T., and Zoghbi, H. Y. (2012). MeCP2: only 100% will do. Nat. Neurosci. 15, 176–177. doi: 10.1038/nn.3027
Chao, H.-T., Zoghbi, H. Y., and Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56, 58–65. doi: 10.1016/j.neuron.2007.08.018
Chapleau, C. A., Lane, J., Pozzo-Miller, L., and Percy, A. K. (2013). Evaluation of current pharmacological treatment options in the management of Rett syndrome: from the present to future therapeutic alternatives. Curr. Clin. Pharmacol. 8, 358–369.
Charalampopoulos, I., Remboutsika, E., Margioris, A. N., and Gravanis, A. (2008). Neurosteroids as modulators of neurogenesis and neuronal survival. Trends Endocrinol. Metab. 19, 300–307. doi: 10.1016/j.tem.2008.07.004
Chebib, M., and Johnston, G. A. R. (1999). The “ABC” of gaba receptors: a brief review. Clin. Exp. Pharmacol. Physiol. 26, 937–940. doi: 10.1046/j.1440-1681.1999.03151.x
Chen, C.-Y., Di Lucente, J., Lin, Y.-C., Lien, C.-C., Rogawski, M. A., Maezawa, I., et al. (2018). Defective GABAergic neurotransmission in the nucleus tractus solitarius in Mecp2-null mice, a model of Rett syndrome. Neurobiol. Dis. 109, 25–32. doi: 10.1016/j.nbd.2017.09.006
Chen, S., Wang, J. M., Irwin, R. W., Yao, J., Liu, L., and Brinton, R. D. (2011). Allopregnanolone promotes regeneration and reduces β-amyloid burden in a preclinical model of Alzheimer’s disease. PLoS One 6:e24293. doi: 10.1371/journal.pone.0024293
Clarke, A. J., and Abdala Sheikh, A. P. (2018). A perspective on “cure” for Rett syndrome. Orphanet J. Rare Dis. 13:44. doi: 10.1186/s13023-018-0786-6
Colciago, A., and Magnaghi, V. (2016). Neurosteroids involvement in the epigenetic control of memory formation and storage. Neural Plast. 2016:5985021. doi: 10.1155/2016/5985021
Concas, A., Mostallino, M. C., Porcu, P., Follesa, P., Barbaccia, M. L., Trabucchi, M., et al. (1998). Role of brain allopregnanolone in the plasticity of gamma-aminobutyric acid type A receptor in rat brain during pregnancy and after delivery. Proc. Natl. Acad. Sci. U.S.A. 95, 13284–13289.
Consroe, P., Benedito, M. A., Leite, J. R., Carlini, E. A., and Mechoulam, R. (1982). Effects of cannabidiol on behavioral seizures caused by convulsant drugs or current in mice. Eur. J. Pharmacol. 83, 293–298.
Cooper, E. J., Johnston, G. A., and Edwards, F. A. (1999). Effects of a naturally occurring neurosteroid on GABAA IPSCs during development in rat hippocampal or cerebellar slices. J. Physiol. 521(Pt 2), 437–449.
Counts, S. E., He, B., Nadeem, M., Wuu, J., Scheff, S. W., and Mufson, E. J. (2012). Hippocampal drebrin loss in mild cognitive impairment. Neurodegener. Dis. 10, 216–219. doi: 10.1159/000333122
Dahlin, M., Elfving, A., Ungerstedt, U., and Amark, P. (2005). The ketogenic diet influences the levels of excitatory and inhibitory amino acids in the CSF in children with refractory epilepsy. Epilepsy Res. 64, 115–125. doi: 10.1016/j.eplepsyres.2005.03.008
Dani, V. S., Chang, Q., Maffei, A., Turrigiano, G. G., Jaenisch, R., and Nelson, S. B. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 12560–12565. doi: 10.1073/pnas.0506071102
Dani, V. S., and Nelson, S. B. (2009). Intact long-term potentiation but reduced connectivity between neocortical layer 5 pyramidal neurons in a mouse model of Rett syndrome. J. Neurosci. 29, 11263–11270. doi: 10.1523/JNEUROSCI.1019-09.2009
Datta, D., Arion, D., and Lewis, D. A. (2015). Developmental expression patterns of GABAA receptor subunits in layer 3 and 5 pyramidal cells of monkey prefrontal cortex. Cereb. Cortex 25, 2295–2305. doi: 10.1093/cercor/bhu040
De Felice, C., Signorini, C., Leoncini, S., Pecorelli, A., Durand, T., Valacchi, G., et al. (2012). The role of oxidative stress in Rett syndrome: an overview: oxidative stress and Rett syndrome. Ann. N.Y. Acad. Sci. 1259, 121–135. doi: 10.1111/j.1749-6632.2012.06611.x
de Mello Schier, A. R., de Oliveira Ribeiro, N. P., Coutinho, D. S., Machado, S., Arias-Carrión, O., Crippa, J. A., et al. (2014). Antidepressant-like and anxiolytic-like effects of cannabidiol: a chemical compound of Cannabis sativa. CNS Neurol Disord. Drug Targets 13, 953–960.
Devinsky, O., Cross, J. H., Laux, L., Marsh, E., Miller, I., Nabbout, R., et al. (2017). Trial of cannabidiol for drug-resistant seizures in the dravet syndrome. N. Engl. J. Med. 376, 2011–2020. doi: 10.1056/NEJMoa1611618
Di Marzo, V., Stella, N., and Zimmer, A. (2015). Endocannabinoid signalling and the deteriorating brain. Nat. Rev. Neurosci. 16, 30–42. doi: 10.1038/nrn3876
Diógenes, M. J., Assaife-Lopes, N., Pinto-Duarte, A., Ribeiro, J. A., and Sebastião, A. M. (2007). Influence of age on BDNF modulation of hippocampal synaptic transmission: interplay with adenosine A2A receptors. Hippocampus 17, 577–585. doi: 10.1002/hipo.20294
Diógenes, M. J., Fernandes, C. C., Sebastião, A. M., and Ribeiro, J. A. (2004). Activation of adenosine A2A receptor facilitates brain-derived neurotrophic factor modulation of synaptic transmission in hippocampal slices. J. Neurosci. 24, 2905–2913. doi: 10.1523/JNEUROSCI.4454-03.2004
Drexel, M., Kirchmair, E., and Sperk, G. (2013). Changes in the expression of GABAA receptor subunit mRNAs in parahippocampal areas after kainic acid induced seizures. Front. Neural Circ. 7:142. doi: 10.3389/fncir.2013.00142
Duarte, S. T., Armstrong, J., Roche, A., Ortez, C., Pérez, A., O’Callaghan, M., et al. (2013). Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PLoS One 8:e068851. doi: 10.1371/journal.pone.0068851
Dubrovsky, B. O. (2005). Steroids, neuroactive steroids and neurosteroids in psychopathology. Progr. Neuro Psychopharmacol. Biol. Psychiatry 29, 169–192. doi: 10.1016/j.pnpbp.2004.11.001
Dunwiddie, T. V., and Masino, S. A. (2001). The role and regulation of adenosine in the central nervous system. Annu. Rev. Neurosci. 24, 31–55. doi: 10.1146/annurev.neuro.24.1.31
El Manira, A., and Kyriakatos, A. (2010). The role of endocannabinoid signaling in motor control. Physiology 25, 230–238. doi: 10.1152/physiol.00007.2010
El-Ansary, A., and Al-Ayadhi, L. (2014). GABAergic/glutamatergic imbalance relative to excessive neuroinflammation in autism spectrum disorders. J. Neuroinflam. 11:189. doi: 10.1186/s12974-014-0189-0
El-Khoury, R., Panayotis, N., Matagne, V., Ghata, A., Villard, L., and Roux, J.-C. (2014). GABA and glutamate pathways are spatially and developmentally affected in the brain of Mecp2-deficient mice. PLoS One 9:e92169. doi: 10.1371/journal.pone.0092169
Fagan, S. G., and Campbell, V. A. (2014). The influence of cannabinoids on generic traits of neurodegeneration: cannabinoids and neurodegeneration. Br. J. Pharmacol. 171, 1347–1360. doi: 10.1111/bph.12492
Fernandes, C. C., Pinto-Duarte, A., Ribeiro, J. A., and Sebastião, A. M. (2008). Postsynaptic action of brain-derived neurotrophic factor attenuates alpha7 nicotinic acetylcholine receptor-mediated responses in hippocampal interneurons. J. Neurosci. 28, 5611–5618. doi: 10.1523/JNEUROSCI.5378-07.2008
Fernández-Ruiz, J., Moro, M. A., and Martínez-Orgado, J. (2015). Cannabinoids in neurodegenerative disorders and stroke/brain trauma: from preclinical models to clinical applications. Neurotherapeutics 12, 793–806. doi: 10.1007/s13311-015-0381-7
Fernández-Ruiz, J., Sagredo, O., Pazos, M. R., García, C., Pertwee, R., Mechoulam, R., et al. (2013). Cannabidiol for neurodegenerative disorders: important new clinical applications for this phytocannabinoid? Br. J. Clin. Pharmacol. 75, 323–333. doi: 10.1111/j.1365-2125.2012.04341.x
Fitzpatrick, J.-M. K., and Downer, E. J. (2017). Toll-like receptor signalling as a cannabinoid target in multiple sclerosis. Neuropharmacology 113, 618–626. doi: 10.1016/j.neuropharm.2016.04.009
Follesa, P., Porcu, P., Sogliano, C., Cinus, M., Biggio, F., Mancuso, L., et al. (2002). Changes in GABAA receptor gamma 2 subunit gene expression induced by long-term administration of oral contraceptives in rats. Neuropharmacology 42, 325–336.
Fontinha, B. M., Diógenes, M. J., Ribeiro, J. A., and Sebastião, A. M. (2008). Enhancement of long-term potentiation by brain-derived neurotrophic factor requires adenosine A2A receptor activation by endogenous adenosine. Neuropharmacology 54, 924–933. doi: 10.1016/j.neuropharm.2008.01.011
Fredholm, B. B., Bättig, K., Holmén, J., Nehlig, A., and Zvartau, E. E. (1999). Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol. Rev. 51, 83–133.
Freeman, J. M., and Kossoff, E. H. (2010). Ketosis and the ketogenic diet, 2010: advances in treating epilepsy and other disorders. Adv. Pediatr. 57, 315–329. doi: 10.1016/j.yapd.2010.08.003
French, J. A. (2007). Refractory epilepsy: clinical overview. Epilepsia 48, 3–7. doi: 10.1111/j.1528-1167.2007.00992.x
Genazzani, A. R., Petraglia, F., Bernardi, F., Casarosa, E., Salvestroni, C., Tonetti, A., et al. (1998). Circulating levels of allopregnanolone in humans: gender, age, and endocrine influences. J. Clin. Endocrinol. Metab. 83, 2099–2103. doi: 10.1210/jcem.83.6.4905
Giampietro, P. F., Schowalter, D. B., Merchant, S., Campbell, L. R., Swink, T., and Roa, B. B. (2006). Widened clinical spectrum of the Q128P MECP2 mutation in Rett syndrome. Childs Nerv. Syst. 22, 320–324. doi: 10.1007/s00381-005-1155-z
Goodkin, H. P., Joshi, S., Mtchedlishvili, Z., Brar, J., and Kapur, J. (2008). Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J. Neurosci. 28, 2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008
Govindpani, K., Calvo-Flores Guzmán, B., Vinnakota, C., Waldvogel, H. J., Faull, R. L., and Kwakowsky, A. (2017). Towards a better understanding of GABAergic remodeling in alzheimer’s disease. Int. J. Mol. Sci. 18:1813. doi: 10.3390/ijms18081813
Greenfield, L. J. (2013). Molecular mechanisms of antiseizure drug activity at GABAA receptors. Seizure 22, 589–600. doi: 10.1016/j.seizure.2013.04.015
Grobin, A. C., and Morrow, A. L. (2001). 3Alpha-hydroxy-5alpha-pregnan-20-one levels and GABA(A) receptor-mediated 36Cl(-) flux across development in rat cerebral cortex. Brain Res. Dev. Brain Res. 131, 31–39.
Gulinello, M., Gong, Q. H., Li, X., and Smith, S. S. (2001). Short-term exposure to a neuroactive steroid increases alpha4 GABA(A) receptor subunit levels in association with increased anxiety in the female rat. Brain Res. 910, 55–66.
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147. doi: 10.1126/science.1138389
Haas, R. H., Rice, M. A., Trauner, D. A., Merritt, T. A., Opitz, J. M., and Reynolds, J. F. (1986). Therapeutic effects of a ketogenic diet in Rett syndrome. Am. J. Med. Genet. 25, 225–246. doi: 10.1002/ajmg.1320250525
Hagberg, B. (2002). Clinical manifestations and stages of Rett syndrome. Ment. Retardat. Dev. Disabil. Res. Rev. 8, 61–65. doi: 10.1002/mrdd.10020
Hampson, A. J., Grimaldi, M., Lolic, M., Wink, D., Rosenthal, R., and Axelrod, J. (2000). Neuroprotective antioxidants from marijuana. Ann. N.Y. Acad. Sci. 899, 274–282.
Harigaya, Y., Shoji, M., Shirao, T., and Hirai, S. (1996). Disappearance of actin-binding protein, drebrin, from hippocampal synapses in Alzheimer’s disease. J. Neurosci. Res. 43, 87–92. doi: 10.1002/jnr.490430111
Harrison, N. L., and Simmonds, M. A. (1984). Modulation of the GABA receptor complex by a steroid anaesthetic. Brain Res. 323, 287–292.
Hartman, A. L., Gasior, M., Vining, E. P. G., and Rogawski, M. A. (2007). The neuropharmacology of the ketogenic diet. Pediatr. Neurol. 36, 281–292. doi: 10.1016/j.pediatrneurol.2007.02.008
Hassan, A. M., Keene, D. L., Whiting, S. E., Jacob, P. J., Champagne, J. R., and Humphreys, P. (1999). Ketogenic diet in the treatment of refractory epilepsy in childhood. Pediatr. Neurol. 21, 548–552.
Hill, T. D. M., Cascio, M.-G., Romano, B., Duncan, M., Pertwee, R. G., Williams, C. M., et al. (2013). Cannabidivarin-rich cannabis extracts are anticonvulsant in mouse and rat via a CB1 receptor-independent mechanism. Br. J. Pharmacol. 170, 679–692. doi: 10.1111/bph.12321
Hogart, A., Nagarajan, R. P., Patzel, K. A., Yasui, D. H., and LaSalle, J. M. (2007). 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum. Mol. Genet. 16, 691–703. doi: 10.1093/hmg/ddm014
Hosie, A. M., Wilkins, M. E., da Silva, H. M. A., and Smart, T. G. (2006). Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444, 486–489. doi: 10.1038/nature05324
Hu, J., Zhang, Z., Shen, W.-J., and Azhar, S. (2010). Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr. Metab. 7:47. doi: 10.1186/1743-7075-7-47
Huppke, P., Laccone, F., Krämer, N., Engel, W., and Hanefeld, F. (2000). Rett syndrome: analysis of MECP2 and clinical characterization of 31 patients. Hum. Mol. Genet. 9, 1369–1375.
Huttenlocher, P. R. (1976). Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatr. Res. 10, 536–540. doi: 10.1203/00006450-197605000-00006
Iannotti, F. A., Hill, C. L., Leo, A., Alhusaini, A., Soubrane, C., Mazzarella, E., et al. (2014). Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: potential for the treatment of neuronal hyperexcitability. ACS Chem. Neurosci. 5, 1131–1141. doi: 10.1021/cn5000524
Iseger, T. A., and Bossong, M. G. (2015). A systematic review of the antipsychotic properties of cannabidiol in humans. Schizophr Res. 162, 153–161. doi: 10.1016/j.schres.2015.01.033
Ito-Ishida, A., Ure, K., Chen, H., Swann, J. W., and Zoghbi, H. Y. (2015). Loss of MeCP2 in parvalbumin-and somatostatin-expressing neurons in mice leads to distinct Rett syndrome-like phenotypes. Neuron 88, 651–658. doi: 10.1016/j.neuron.2015.10.029
Iuvone, T., Esposito, G., De Filippis, D., Scuderi, C., and Steardo, L. (2009). Cannabidiol: a promising drug for neurodegenerative disorders? CNS Neurosci. Ther. 15, 65–75. doi: 10.1111/j.1755-5949.2008.00065.x
Jacob, F. D., Ramaswamy, V., Andersen, J., and Bolduc, F. V. (2009). Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature. Eur. J. Hum. Genet. 17, 1577–1581. doi: 10.1038/ejhg.2009.95
Jellinger, K., Armstrong, D., Zoghbi, H. Y., and Percy, A. K. (1988). Neuropathology of Rett syndrome. Acta Neuropathol. 76, 142–158. doi: 10.1007/BF00688098
Jenniches, I., Ternes, S., Albayram, O., Otte, D. M., Bach, K., Bindila, L., et al. (2016). Anxiety, stress, and fear response in mice with reduced endocannabinoid levels. Biol. Psychiatry 79, 858–868. doi: 10.1016/j.biopsych.2015.03.033
Jin, X., Cui, N., Zhong, W., Jin, X.-T., and Jiang, C. (2013a). GABAergic synaptic inputs of locus coeruleus neurons in wild-type and Mecp2-null mice. Am. J. Physiol. Cell Physiol. 304, C844–C857. doi: 10.1152/ajpcell.00399.2012
Jin, X., Zhong, W., and Jiang, C. (2013b). Time-dependent modulation of GABAA-ergic synaptic transmission by allopregnanolone in locus coeruleus neurons of Mecp2-null mice. Am. J. Physiol. Cell Physiol. 305, C1151–C1160. doi: 10.1152/ajpcell.00195.2013
Johansson, I.-M., Birzniece, V., Lindblad, C., Olsson, T., and Bäckström, T. (2002). Allopregnanolone inhibits learning in the morris water maze. Brain Res. 934, 125–131. doi: 10.1016/S0006-8993(02)02414-9
Jones, N. A., Glyn, S. E., Akiyama, S., Hill, T. D. M., Hill, A. J., Weston, S. E., et al. (2012). Cannabidiol exerts anti-convulsant effects in animal models of temporal lobe and partial seizures. Seizure 21, 344–352. doi: 10.1016/j.seizure.2012.03.001
Jones, N. A., Hill, A. J., Smith, I., Bevan, S. A., Williams, C. M., Whalley, B. J., et al. (2010). Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo. J. Pharmacol. Exp. Ther. 332, 569–577. doi: 10.1124/jpet.109.159145
Juge, N., Gray, J. A., Omote, H., Miyaji, T., Inoue, T., Hara, C., et al. (2010). Metabolic control of vesicular glutamate transport and release. Neuron 68, 99–112. doi: 10.1016/j.neuron.2010.09.002
Justice, M. J., Buchovecky, C. M., Kyle, S. M., and Djukic, A. (2013). A role for metabolism in Rett syndrome pathogenesis: new clinical findings and potential treatment targets. Rare Dis. 1:e27265. doi: 10.4161/rdis.27265
Kaminski, R. M., Livingood, M. R., and Rogawski, M. A. (2004). Allopregnanolone analogs that positively modulate GABA receptors protect against partial seizures induced by 6-Hz electrical stimulation in mice. Epilepsia 45, 864–867. doi: 10.1111/j.0013-9580.2004.04504.x
Kano, M., Ohno-Shosaku, T., Hashimotodani, Y., Uchigashima, M., and Watanabe, M. (2009). Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 89, 309–380. doi: 10.1152/physrev.00019.2008
Kaplan, J. S., Stella, N., Catterall, W. A., and Westenbroek, R. E. (2017). Cannabidiol attenuates seizures and social deficits in a mouse model of dravet syndrome. PNAS 114, 11229–11234. doi: 10.1073/pnas.1711351114
Kasprowska-Liśkiewicz, D., Liśkiewicz, A. D., Nowacka-Chmielewska, M. M., Nowicka, J., Małecki, A., and Barski, J. J. (2017). The ketogenic diet affects the social behavior of young male rats. Physiol. Behav. 179, 168–177. doi: 10.1016/j.physbeh.2017.06.007
Katz, D. M. (2014). “Brain-Derived Neurotrophic Factor and Rett Syndrome,” in Neurotrophic Factors Handbook of Experimental Pharmacology, eds G. R. Lewin and B. D. Carter (Heidelberg: Springer), 481–495.
Kawamura, M., Ruskin, D. N., and Masino, S. A. (2010). Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J. Neurosci. 30, 3886–3895. doi: 10.1523/JNEUROSCI.0055-10.2010
Kim, D. Y., Simeone, K. A., Simeone, T. A., Pandya, J. D., Wilke, J. C., Ahn, Y., et al. (2015). Ketone bodies mediate anti-seizure effects through mitochondrial permeability transition. Ann. Neurol. 78, 77–87. doi: 10.1002/ana.24424
Kim, Y. S., and Yoon, B.-E. (2017). Altered GABAergic signaling in brain disease at various stages of life. Exp. Neurobiol. 26:122. doi: 10.5607/en.2017.26.3.122
Krajnc, N. (2015). Management of epilepsy in patients with Rett syndrome: perspectives and considerations. Therapeu. Clin. Risk Manag. 11, 925–932. doi: 10.2147/TCRM.S55896
Kramar, C., Loureiro, M., Renard, J., and Laviolette, S. R. (2017). Palmitoylethanolamide modulates GPR55 receptor signaling in the ventral hippocampus to regulate mesolimbic dopamine activity, social interaction, and memory processing. Cannabis Cannabinoid Res. 2, 8–20. doi: 10.1089/can.2016.0030
Kwakowsky, A., Calvo-Flores Guzmán, B., Pandya, M., Turner, C., Waldvogel, H. J., and Faull, R. L. (2018). GABAA receptor subunit expression changes in the human Alzheimer’s disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. J. Neurochem. 145, 374–392. doi: 10.1111/jnc.14325
Kyle, S. M., Vashi, N., and Justice, M. J. (2018). Rett syndrome: a neurological disorder with metabolic components. Open Biol. 8:170216. doi: 10.1098/rsob.170216
Laprairie, R. B., Bagher, A. M., Kelly, M. E. M., and Denovan-Wright, E. M. (2015). Cannabidiol is a negative allosteric modulator of the cannabinoid CB 1 receptor: negative allosteric modulation of CB 1 by cannabidiol. Br. J. Pharmacol. 172, 4790–4805. doi: 10.1111/bph.13250
Lauckner, J. E., Jensen, J. B., Chen, H.-Y., Lu, H.-C., Hille, B., and Mackie, K. (2008). GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. U.S.A. 105, 2699–2704. doi: 10.1073/pnas.0711278105
Lee, R. W. Y., and Tierney, E. (2011). Hypothesis: the role of sterols in autism spectrum disorder. Autism Res. Treat. 2011, 1–7. doi: 10.1155/2011/653570
Li, W., and Pozzo-Miller, L. (2014). BDNF deregulation in Rett syndrome. Neuropharmacology 76(Pt C), 737–746. doi: 10.1016/j.neuropharm.2013.03.024
Li, Y., Sun, H., Chen, Z., Xu, H., Bu, G., and Zheng, H. (2016). Implications of GABAergic Neurotransmission in Alzheimer’s Disease. Front Aging Neurosci. 8:31. doi: 10.3389/fnagi.2016.00031
Liebhaber, G. M., Riemann, E., and Matthias Baumeister, F. A. (2003). Ketogenic diet in Rett syndrome. J. Child Neurol. 18, 74–75. doi: 10.1177/08830738030180011801
Ligsay, A., Van Dijck, A., Nguyen, D. V., Lozano, R., Chen, Y., Bickel, E. S., et al. (2017). A randomized double-blind, placebo-controlled trial of ganaxolone in children and adolescents with fragile X syndrome. J. Neurodev. Disord. 9:26. doi: 10.1186/s11689-017-9207-8
Liu, Q., and Wong-Riley, M. T. T. (2010). Postnatal development of N-methyl-D-aspartate receptor subunits 2A, 2B, 2C, 2D, and 3B immunoreactivity in brain stem respiratory nuclei of the rat. Neuroscience 171, 637–654. doi: 10.1016/j.neuroscience.2010.09.055
Loewe, S. (1946). Studies on the pharmacology and acute toxicity of compounds with marihuana activity. J. Pharmacol. Exp. Ther. 88, 154–161.
Löscher, W. (1989). Valproate enhances GABA turnover in the substantia nigra. Brain Res. 501, 198–203.
Lozovaya, N., Eftekhari, S., Cloarec, R., Gouty-Colomer, L. A., Dufour, A., Riffault, B., et al. (2018). GABAergic inhibition in dual-transmission cholinergic and GABAergic striatal interneurons is abolished in Parkinson disease. Nat. Commun. 9:1422. doi: 10.1038/s41467-018-03802-y
Lund, E. G., Xie, C., Kotti, T., Turley, S. D., Dietschy, J. M., and Russell, D. W. (2003). Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 278, 22980–22988. doi: 10.1074/jbc.M303415200
Maa, E., and Figi, P. (2014). The case for medical marijuana in epilepsy. Epilepsia 55, 783–786. doi: 10.1111/epi.12610
Macdonald, R., and Barker, J. L. (1978). Benzodiazepines specifically modulate GABA-mediated postsynaptic inhibition in cultured mammalian neurones. Nature 271, 563–564.
Maitra, R., and Reynolds, J. N. (1999). Subunit dependent modulation of GABAA receptor function by neuroactive steroids. Brain Res. 819, 75–82.
Majewska, M. D., Harrison, N. L., Schwartz, R. D., Barker, J. L., and Paul, S. M. (1986). Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science 232, 1004–1007.
Malfait, A. M., Gallily, R., Sumariwalla, P. F., Malik, A. S., Andreakos, E., Mechoulam, R., et al. (2000). The nonpsychoactive cannabis constituent cannabidiol is an oral anti-arthritic therapeutic in murine collagen-induced arthritis. PNAS 97, 9561–9566. doi: 10.1073/pnas.160105897
Mantis, J. G., Centeno, N. A., Todorova, M. T., McGowan, R., and Seyfried, T. N. (2004). Management of multifactorial idiopathic epilepsy in EL mice with caloric restriction and the ketogenic diet: role of glucose and ketone bodies. Nutr. Metab. 1:11. doi: 10.1186/1743-7075-1-11
Mantis, J. G., Fritz, C. L., Marsh, J., Heinrichs, S. C., and Seyfried, T. N. (2009). Improvement in motor and exploratory behavior in Rett syndrome mice with restricted ketogenic and standard diets. Epilepsy Behav. 15, 133–141. doi: 10.1016/j.yebeh.2009.02.038
Marcos, J., Guo, L.-W., Wilson, W. K., Porter, F. D., and Shackleton, C. (2004). The implications of 7-dehydrosterol-7-reductase deficiency (smith-lemli-opitz syndrome) to neurosteroid production. Steroids 69, 51–60.
Mari, F., Azimonti, S., Bertani, I., Bolognese, F., Colombo, E., Caselli, R., et al. (2005). CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 14, 1935–1946. doi: 10.1093/hmg/ddi198
Marichal-Cancino, B. A., Fajardo-Valdez, A., Ruiz-Contreras, A. E., Méndez-Díaz, M., and Prospéro-García, O. (2018). Possible role of hippocampal GPR55 in spatial learning and memory in rats. Acta Neurobiol. Exp. 78, 41–50. doi: 10.21307/ane-2018-001
Marsicano, G., and Lafenêtre, P. (2009). “Roles of the Endocannabinoid System in Learning and Memory,” in Behavioral Neurobiology of the Endocannabinoid System, eds D. Kendall and S. Alexander (Berlin: Springer), 201–230.
Marx, C. E., Stevens, R. D., Shampine, L. J., Uzunova, V., Trost, W. T., Butterfield, M. I., et al. (2006). Neuroactive steroids are altered in schizophrenia and bipolar disorder: relevance to pathophysiology and therapeutics. Neuropsychopharmacology 31, 1249–1263. doi: 10.1038/sj.npp.1300952
Mascia, M. P., Biggio, F., Mancuso, L., Cabras, S., Cocco, P. L., Gorini, G., et al. (2002). Changes in GABA(A) receptor gene expression induced by withdrawal of, but not by long-term exposure to, ganaxolone in cultured rat cerebellar granule cells. J. Pharmacol. Exp. Ther. 303, 1014–1020. doi: 10.1124/jpet.102.040063
Masino, S. A., Kawamura, M., Cote, J. L., Williams, R. B., and Ruskin, D. N. (2013). Adenosine and autism: a spectrum of opportunities. Neuropharmacology 68, 116–121. doi: 10.1016/j.neuropharm.2012.08.013
Masino, S. A., Li, T., Theofilas, P., Sandau, U. S., Ruskin, D. N., Fredholm, B. B., et al. (2011). A ketogenic diet suppresses seizures in mice through adenosine A1 receptors. J. Clin. Invest. 121, 2679–2683. doi: 10.1172/JCI57813
Matthews, D. B., Morrow, A. L., Tokunaga, S., and McDaniel, J. R. (2002). Acute ethanol administration and acute allopregnanolone administration impair spatial memory in the Morris water task. Alcohol. Clin. Exp. Res. 26, 1747–1751. doi: 10.1097/01.ALC.0000037219.79257.17
McGraw, C. M., Samaco, R. C., and Zoghbi, H. Y. (2011). Adult neural function requires MeCP2. Science 333, 186–186. doi: 10.1126/science.1206593
Medrihan, L., Tantalaki, E., Aramuni, G., Sargsyan, V., Dudanova, I., Missler, M., et al. (2008). Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J. Neurophysiol. 99, 112–121. doi: 10.1152/jn.00826.2007
Meera, P., Wallner, M., and Otis, T. S. (2011). Molecular basis for the high THIP/gaboxadol sensitivity of extrasynaptic GABA(A) receptors. J. Neurophysiol. 106, 2057–2064. doi: 10.1152/jn.00450.2011
Meidenbauer, J. J., Mantis, J. G., and Seyfried, T. N. (2011). The EL mouse: a natural model of autism and epilepsy. Epilepsia 52, 347–357. doi: 10.1111/j.1528-1167.2010.02898.x
Melcangi, R. C., Garcia-Segura, L. M., and Mensah-Nyagan, A. G. (2008). Neuroactive steroids: state of the art and new perspectives. Cell. Mol. Life Sci. 65, 777–797. doi: 10.1007/s00018-007-7403-5
Melcangi, R. C., Giatti, S., Calabrese, D., Pesaresi, M., Cermenati, G., Mitro, N., et al. (2014). Levels and actions of progesterone and its metabolites in the nervous system during physiological and pathological conditions. Prog. Neurobiol. 113, 56–69. doi: 10.1016/j.pneurobio.2013.07.006
Meletti, S., Lucchi, C., Monti, G., Giovannini, G., Bedin, R., Trenti, T., et al. (2017). Decreased allopregnanolone levels in cerebrospinal fluid obtained during status epilepticus. Epilepsia 58, e16–e20. doi: 10.1111/epi.13625
Meletti, S., Lucchi, C., Monti, G., Giovannini, G., Bedin, R., Trenti, T., et al. (2018). Low levels of progesterone and derivatives in cerebrospinal fluid of patients affected by status epilepticus. J. Neurochem. 147, 275–284. doi: 10.1111/jnc.14550
Mellédo, J.-M. L., and Baker, G. B. (2002). Neuroactive steroids and anxiety disorders. J. Psychiatry Neurosci. 27, 161–165.
Mellon, S. H. (2007). Neurosteroid regulation of central nervous system development. Pharmacol. Ther. 116, 107–124. doi: 10.1016/j.pharmthera.2007.04.011
Mersiades, A. J., Tognela, A., Haber, P. S., Stockler, M., Lintzeris, N., Simes, J., et al. (2018). Oral cannabinoid-rich THC/CBD cannabis extract for secondary prevention of chemotherapy-induced nausea and vomiting: a study protocol for a pilot and definitive randomised double-blind placebo-controlled trial (CannabisCINV). BMJ Open 8:e020745. doi: 10.1136/bmjopen-2017-020745
Misane, I., Kruis, A., Pieneman, A. W., Ögren, S. O., and Stiedl, O. (2013). GABA(A) receptor activation in the CA1 area of the dorsal hippocampus impairs consolidation of conditioned contextual fear in C57BL/6J mice. Behav. Brain Res. 238, 160–169. doi: 10.1016/j.bbr.2012.10.027
Morales, P., Hurst, D. P., and Reggio, P. H. (2017). “Molecular Targets of the Phytocannabinoids: A Complex Picture,” in Phytocannabinoids, eds A. D. Kinghorn, H. Falk, S. Gibbons, and J. Kobayashi (Cham: Springer International Publishing), 103–131.
Moretti, P., and Zoghbi, H. Y. (2006). MeCP2 dysfunction in Rett syndrome and related disorders. Curr. Opin. Genet. Dev. 16, 276–281. doi: 10.1016/j.gde.2006.04.009
Nagarkatti, P., Pandey, R., Rieder, S. A., Hegde, V. L., and Nagarkatti, M. (2009). Cannabinoids as novel anti-inflammatory drugs. Future Med. Chem. 1, 1333–1349. doi: 10.4155/fmc.09.93
Nagy, G., and Ackerman, S. L. (2013). Cholesterol metabolism and Rett syndrome pathogenesis. Nat. Genet. 45, 965–967. doi: 10.1038/ng.2738
Navarro, G., Borroto-Escuela, D., Angelats, E., Etayo, Í., Reyes-Resina, I., Pulido-Salgado, M., et al. (2018). Receptor-heteromer mediated regulation of endocannabinoid signaling in activated microglia. role of CB1 and CB2 receptors and relevance for Alzheimer’s disease and levodopa-induced dyskinesia. Brain Behav. Immun. 67, 139–151. doi: 10.1016/j.bbi.2017.08.015
Neal, E. G., Chaffe, H., Schwartz, R. H., Lawson, M. S., Edwards, N., Fitzsimmons, G., et al. (2008). The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 7, 500–506. doi: 10.1016/S1474-4422(08)70092-9
Neul, J. L., Kaufmann, W. E., Glaze, D. G., Christodoulou, J., Clarke, A. J., Bahi-Buisson, N., et al. (2010). Rett syndrome: revised diagnostic criteria and nomenclature. Ann. Neurol. 68, 944–950. doi: 10.1002/ana.22124
Neul, J. L., and Zoghbi, H. Y. (2004). Rett syndrome: a prototypical neurodevelopmental disorder. Neuroscientist 10, 118–128. doi: 10.1177/1073858403260995
Nguyen, M. V. C., Du, F., Felice, C. A., Shan, X., Nigam, A., Mandel, G., et al. (2012). MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J. Neurosci. 32, 10021–10034. doi: 10.1523/JNEUROSCI.1316-12.2012
Novotna, A., Mares, J., Ratcliffe, S., Novakova, I., Vachova, M., Zapletalova, O., et al. (2011). A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (sativex ), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis: Sativex for refractory spasticity in MS. Eur. J. Neurol. 18, 1122–1131. doi: 10.1111/j.1468-1331.2010.03328.x
O’Connell, B. K., Gloss, D., and Devinsky, O. (2017). Cannabinoids in treatment-resistant epilepsy: a review. Epilepsy Behav. 70, 341–348. doi: 10.1016/j.yebeh.2016.11.012
Onaivi, E. S., Ishiguro, H., Gu, S., and Liu, Q.-R. (2012). CNS effects of CB2 cannabinoid receptors: beyond neuro-immuno-cannabinoid activity. J. Psychopharmacol. 26, 92–103. doi: 10.1177/0269881111400652
Pamplona, F. A., and Coan, A. C. (2017). Potential clinical benefits of CBD-rich cannabis extracts over purified CBD in treatment-resistant epilepsy: observational data meta-analysis. bioRxiv doi: 10.1101/212662
Paoletti, A. M., Romagnino, S., Contu, R., Orrù, M. M., Marotto, M. F., Zedda, P., et al. (2006). Observational study on the stability of the psychological status during normal pregnancy and increased blood levels of neuroactive steroids with GABA-A receptor agonist activity. Psychoneuroendocrinology 31, 485–492. doi: 10.1016/j.psyneuen.2005.11.006
Park, H.-M., Choi, I.-S., Nakamura, M., Cho, J.-H., Lee, M.-G., and Jang, I.-S. (2011). Multiple effects of allopregnanolone on GABAergic responses in single hippocampal CA3 pyramidal neurons. Eur. J. Pharmacol. 652, 46–54. doi: 10.1016/j.ejphar.2010.10.097
Parker, L. A., Rock, E. M., and Limebeer, C. L. (2011). Regulation of nausea and vomiting by cannabinoids. Br. J. Pharmacol. 163, 1411–1422. doi: 10.1111/j.1476-5381.2010.01176.x
Pascual, D., Sánchez-Robles, E. M., García, M. M., and Goicoechea, C. (2018). Chronic pain and cannabinoids. great expectations or a christmas carol. Biochem. Pharmacol. 157, 33–42. doi: 10.1016/j.bcp.2018.07.033
Patankar, J. V. (2014). Cholesterol metabolism is a potential therapeutic target for Rett syndrome. Clin. Genet. 85, 229–230. doi: 10.1111/cge.12284
Patra, P. H., Barker-Haliski, M., White, H. S., Whalley, B. J., Glyn, S., Sandhu, H., et al. (2019). Cannabidiol reduces seizures and associated behavioral comorbidities in a range of animal seizure and epilepsy models. Epilepsia 60, 303–314. doi: 10.1111/epi.14629
Percy, A. K. (2016). Progress in Rett Syndrome: from discovery to clinical trials. Wien. Med. Wochensch. 166, 325–332. doi: 10.1007/s10354-016-0491-9
Pertwee, R. G. (2009). Cannabinoid pharmacology: the first 66 years: cannabinoid pharmacology. Br. J. Pharmacol. 147, S163–S171. doi: 10.1038/sj.bjp.0706406
Perucca, E. (2017). Cannabinoids in the treatment of epilepsy: hard evidence at last? J Epilepsy Res. 7, 61–76. doi: 10.14581/jer.17012
Peterman, M. G. (1924). The ketogenic diet in the treatment of epilepsy: a preliminary report. Am. J. Dis. Child 28, 28–33. doi: 10.1001/archpedi.1924.04120190031004
Petrosino, S., Verde, R., Vaia, M., Allarà, M., Iuvone, T., and Di Marzo, V. (2018). Anti-inflammatory properties of cannabidiol, a nonpsychotropic cannabinoid, in experimental allergic contact dermatitis. J. Pharmacol. Exp. Ther. 365, 652–663. doi: 10.1124/jpet.117.244368
Pfrieger, F. W., and Ungerer, N. (2011). Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 50, 357–371. doi: 10.1016/j.plipres.2011.06.002
Porter, F. D. (2002). Malformation syndromes due to inborn errors of cholesterol synthesis. J. Clin. Invest. 110, 715–724. doi: 10.1172/JCI16386
Puia, G., Duciæ, I., Vicini, S., and Costa, E. (1993). Does neurosteroid modulatory efficacy depend on GABAA receptor subunit composition? Recept. Channels 1, 135–142.
Puia, G., Mienville, J.-M., Matsumoto, K., Takahata, H., Watanabe, H., Costa, E., et al. (2003). On the putative physiological role of allopregnanolone on GABA(A) receptor function. Neuropharmacology 44, 49–55.
Rabinowitz, A., Cohen, S. J., Finn, D. A., and Stackman, R. W. (2014). The neurosteroid allopregnanolone impairs object memory and contextual fear memory in male C57BL/6J mice. Horm Behav. 66, 238–246. doi: 10.1016/j.yhbeh.2014.05.005
Ramocki, M. B., Peters, S. U., Tavyev, Y. J., Zhang, F., Carvalho, C. M. B., Schaaf, C. P., et al. (2009). Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann. Neurol. 66, 771–782. doi: 10.1002/ana.21715
Reddy, D. S. (2011). Role of anticonvulsant and antiepileptogenic neurosteroids in the pathophysiology and treatment of epilepsy. Front. Endocrinol. 2:38. doi: 10.3389/fendo.2011.00038
Reddy, D. S., and Estes, W. A. (2016). Clinical potential of neurosteroids for CNS disorders. Trends Pharmacol. Sci. 37, 543–561. doi: 10.1016/j.tips.2016.04.003
Reddy, D. S., and Jian, K. (2010). The testosterone-derived neurosteroid androstanediol is a positive allosteric modulator of GABAA receptors. J. Pharmacol. Exp. Ther. 334, 1031–1041. doi: 10.1124/jpet.110.169854
Renieri, A., Mari, F., Mencarelli, M. A., Scala, E., Ariani, F., Longo, I., et al. (2009). Diagnostic criteria for the Zappella variant of Rett syndrome (the preserved speech variant). Brain Dev. 31, 208–216. doi: 10.1016/j.braindev.2008.04.007
Rett, A. (1966). [On a unusual brain atrophy syndrome in hyperammonemia in childhood]. Wien. Med. Wochenschr. 116, 723–726.
Rice, J., and Cameron, M. (2018). Cannabinoids for treatment of MS symptoms: state of the evidence. Curr. Neurol. Neurosci. Rep. 18:50. doi: 10.1007/s11910-018-0859-x
Riss, J., Cloyd, J., Gates, J., and Collins, S. (2008). Benzodiazepines in epilepsy: pharmacology and pharmacokinetics. Acta Neurol. Scand. 118, 69–86. doi: 10.1111/j.1600-0404.2008.01004.x
Rissman, R. A., and Mobley, W. C. (2011). Implications for treatment: GABAA receptors in aging, down syndrome and Alzheimer’s disease. J. Neurochem. 117, 613–622. doi: 10.1111/j.1471-4159.2011.07237.x
Rock, E. M., and Parker, L. A. (2016). Cannabinoids as potential treatment for chemotherapy-induced nausea and vomiting. Front. Pharmacol. 7:221. doi: 10.3389/fphar.2016.00221
Rogawski, M. A., Löscher, W., and Rho, J. M. (2016). Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb. Perspect. Med. 6:a022780. doi: 10.1101/cshperspect.a022780
Rogawski, M. A., Loya, C. M., Reddy, K., Zolkowska, D., and Lossin, C. (2013). Neuroactive steroids for the treatment of status epilepticus. Epilepsia 54(Suppl. 6), 93–98. doi: 10.1111/epi.12289
Rombo, D. M., Ribeiro, J. A., and Sebastião, A. M. (2016). Hippocampal GABAergic transmission: a new target for adenosine control of excitability. J. Neurochem. 139, 1056–1070. doi: 10.1111/jnc.13872
Rosenthal, E. S., Claassen, J., Wainwright, M. S., Husain, A. M., Vaitkevicius, H., Raines, S., et al. (2017). Brexanolone as adjunctive therapy in super-refractory status epilepticus. Ann. Neurol. 82, 342–352. doi: 10.1002/ana.25008
Rosenthaler, S., Pöhn, B., Kolmanz, C., Huu, C. N., Krewenka, C., Huber, A., et al. (2014). Differences in receptor binding affinity of several phytocannabinoids do not explain their effects on neural cell cultures. Neurotoxicol. Teratol. 46, 49–56. doi: 10.1016/j.ntt.2014.09.003
Rossetti, M. F., Varayoud, J., Moreno-Piovano, G. S., Luque, E. H., and Ramos, J. G. (2015). Environmental enrichment attenuates the age-related decline in the mRNA expression of steroidogenic enzymes and reduces the methylation state of the steroid 5α-reductase type 1 gene in the rat hippocampus. Mol. Cell. Endocrinol. 412, 330–338. doi: 10.1016/j.mce.2015.05.024
Ruskin, D. N., Fortin, J. A., Bisnauth, S. N., and Masino, S. A. (2017). Ketogenic diets improve behaviors associated with autism spectrum disorder in a sex-specific manner in the EL mouse. Physiol. Behav. 168, 138–145. doi: 10.1016/j.physbeh.2016.10.023
Ruskin, D. N., Ross, J. L., Kawamura, M., Ruiz, T. L., Geiger, J. D., and Masino, S. A. (2011). A ketogenic diet delays weight loss and does not impair working memory or motor function in the R6/2 1J mouse model of huntington’s disease. Physiol. Behav. 103, 501–507. doi: 10.1016/j.physbeh.2011.04.001
Russo, E., and Guy, G. W. (2006). A tale of two cannabinoids: The therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med. Hypoth. 66, 234–246. doi: 10.1016/j.mehy.2005.08.026
Samaco, R. C., Hogart, A., and LaSalle, J. M. (2005). Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 14, 483–492. doi: 10.1093/hmg/ddi045
Scala, E., Ariani, F., Mari, F., Caselli, R., Pescucci, C., Longo, I., et al. (2005). CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J. Med. Genet. 42, 103–107. doi: 10.1136/jmg.2004.026237
Sebastião, A. M., and Ribeiro, J. A. (2015). Neuromodulation and metamodulation by adenosine: impact and subtleties upon synaptic plasticity regulation. Brain Res. 1621, 102–113. doi: 10.1016/j.brainres.2014.11.008
Segatto, M., Trapani, L., Di Tunno, I., Sticozzi, C., Valacchi, G., Hayek, J., et al. (2014). Cholesterol metabolism is altered in Rett syndrome: a study on plasma and primary cultured fibroblasts derived from patients. PLoS One 9:e104834. doi: 10.1371/journal.pone.0104834
Selye, H. (1941). On the hormonal activity of a steroid compound. Science 94:94. doi: 10.1126/science.94.2430.94
Selye, H., and Masson, G. (1942). Additional steroids with luteoid activity. Science 96:358. doi: 10.1126/science.96.2494.358
Shahbazian, M. D. (2002). Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 11, 115–124. doi: 10.1093/hmg/11.2.115
Shen, H., Gong, Q. H., Aoki, C., Yuan, M., Ruderman, Y., Dattilo, M., et al. (2007). Reversal of neurosteroid effects at alpha4beta2delta GABAA receptors triggers anxiety at puberty. Nat. Neurosci. 10, 469–477. doi: 10.1038/nn1868
Shimizu, H., Ishizuka, Y., Yamazaki, H., and Shirao, T. (2015). Allopregnanolone increases mature excitatory synapses along dendrites via protein kinase A signaling. Neuroscience 305, 139–145. doi: 10.1016/j.neuroscience.2015.07.079
Shulyakova, N., Andreazza, A. C., Mills, L. R., and Eubanks, J. H. (2017). Mitochondrial dysfunction in the pathogenesis of Rett syndrome: implications for mitochondria-targeted therapies. Front. Cell. Neurosci. 11:58. doi: 10.3389/fncel.2017.00058
Sieghart, W. (2006). Structure, pharmacology, and function of GABAA receptor subtypes. Adv. Pharmacol. 54, 231–263.
Singh, C., Liu, L., Wang, J. M., Irwin, R. W., Yao, J., Chen, S., et al. (2012). Allopregnanolone restores hippocampal-dependent learning and memory and neural progenitor survival in aging 3xTgAD and nonTg mice. Neurobiol. Aging 33, 1493–1506. doi: 10.1016/j.neurobiolaging.2011.06.008
Smith, S. S. (2002). Withdrawal properties of a neuroactive steroid: implications for GABAA receptor gene regulation in the brain and anxiety behavior. Steroids 67, 519–528. doi: 10.1016/S0039-128X(01)00170-2
Solymosi, K., and Kofalvi, A. (2017). Cannabis: a treasure trove or pandora’s box? Mini Rev. Med. Chem. 17, 1223–1291. doi: 10.2174/1389557516666161004162133
Stearns, N. A., Schaevitz, L. R., Bowling, H., Nag, N., Berger, U. V., and Berger-Sweeney, J. (2007). Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neuroscience 146, 907–921. doi: 10.1016/j.neuroscience.2007.02.009
Sticozzi, C., Belmonte, G., Pecorelli, A., Cervellati, F., Leoncini, S., Signorini, C., et al. (2013). Scavenger receptor B1 post-translational modifications in Rett syndrome. FEBS Lett. 587, 2199–2204. doi: 10.1016/j.febslet.2013.05.042
Stoffel-Wagner, B. (2003). Neurosteroid biosynthesis in the human brain and its clinical implications. Ann. N. Y. Acad. Sci. 1007, 64–78.
Talbot, F. B., Metcalf, K. M., and Moriarty, M. E. (1927). Epilepsy: chemical investigations of rational treatment by production of ketosis. Am. J. Dis. Child. 33, 218–225. doi: 10.1001/archpedi.1927.04130140038005
Thomas, A., Baillie, G. L., Phillips, A. M., Razdan, R. K., Ross, R. A., and Pertwee, R. G. (2009). Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro: Cannabinoid antagonism by cannabidiol. Br. J. Pharmacol. 150, 613–623. doi: 10.1038/sj.bjp.0707133
Tietz, E. I., Zeng, X. J., Chen, S., Lilly, S. M., Rosenberg, H. C., and Kometiani, P. (1999). Antagonist-induced reversal of functional and structural measures of hippocampal benzodiazepine tolerance. J. Pharmacol. Exp. Ther. 291, 932–942.
Tint, G. S., Irons, M., Elias, E. R., Batta, A. K., Frieden, R., Chen, T. S., et al. (1994). Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med. 330, 107–113. doi: 10.1056/NEJM199401133300205
Todorova, M. T., Tandon, P., Madore, R. A., Stafstrom, C. E., and Seyfried, T. N. (2000). The ketogenic diet inhibits epileptogenesis in EL mice: a genetic model for idiopathic epilepsy. Epilepsia 41, 933–940.
Turkmen, S., Lundgren, P., Birzniece, V., Zingmark, E., Backstrom, T., and Johansson, I.-M. (2004). 3β-20β-dihydroxy-5α-pregnane (UC1011) antagonism of the GABA potentiation and the learning impairment induced in rats by allopregnanolone. Eur. J. Neurosci. 20, 1604–1612. doi: 10.1111/j.1460-9568.2004.03610.x
Uhlemann, E. R., and Neims, A. H. (1972). Anticonvulsant properties of the ketogenic diet in mice. J. Pharmacol. Exp. Ther. 180, 231–238.
Ure, K., Lu, H., Wang, W., Ito-Ishida, A., Wu, Z., He, L., et al. (2016). Restoration of Mecp2 expression in GABAergic neurons is sufficient to rescue multiple disease features in a mouse model of Rett syndrome. eLife 5:e14198. doi: 10.7554/eLife.14198
Uusi-Oukari, M., and Korpi, E. R. (2010). Regulation of GABAA receptor subunit expression by pharmacological agents. Pharmacol. Rev. 62, 97–135. doi: 10.1124/pr.109.002063
Vaitkevicius, H., Husain, A. M., Rosenthal, E. S., Rosand, J., Bobb, W., Reddy, K., et al. (2017). First-in-man allopregnanolone use in super-refractory status epilepticus. Ann. Clin. Transl. Neurol. 4, 411–414. doi: 10.1002/acn3.408
Vaz, S. H., Lérias, S. R., Parreira, S., Diógenes, M. J., and Sebastião, A. M. (2015). Adenosine A2A receptor activation is determinant for BDNF actions upon GABA and glutamate release from rat hippocampal synaptosomes. Purinerg. Signal 11, 607–612. doi: 10.1007/s11302-015-9476-1
Vendel, E., and de Lange, E. C. M. (2014). Functions of the CB1 and CB2 receptors in neuroprotection at the level of the blood–brain barrier. NeuroMol. Med. 16, 620–642. doi: 10.1007/s12017-014-8314-x
Vigli, D., Cosentino, L., Raggi, C., Laviola, G., Woolley-Roberts, M., and De Filippis, B. (2018). Chronic treatment with the phytocannabinoid cannabidivarin (CBDV) rescues behavioural alterations and brain atrophy in a mouse model of Rett syndrome. Neuropharmacology 140, 121–129. doi: 10.1016/j.neuropharm.2018.07.029
Vining, E. P., Freeman, J. M., Ballaban-Gil, K., Camfield, C. S., Camfield, P. R., Holmes, G. L., et al. (1998). A multicenter study of the efficacy of the ketogenic diet. Arch. Neurol. 55, 1433–1437.
Voituron, N., and Hilaire, G. (2011). The benzodiazepine Midazolam mitigates the breathing defects of Mecp2-deficient mice. Respir. Physiol. Neurobiol. 177, 56–60. doi: 10.1016/j.resp.2011.02.002
Walton, J. C., McNeill, J. K., Oliver, K. A., and Albers, H. E. (2017). Temporal regulation of GABAA receptor subunit expression: role in synaptic and extrasynaptic communication in the suprachiasmatic nucleus. eNeuro 4:ENEURO.0352-16.2017. doi: 10.1523/ENEURO.0352-16.2017
Wang, J. M., Singh, C., Liu, L., Irwin, R. W., Chen, S., Chung, E. J., et al. (2010). Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 107, 6498–6503. doi: 10.1073/pnas.1001422107
Wang, M. (2011). Neurosteroids and GABA-A receptor function. Front. Endocrinol. 2:44. doi: 10.3389/fendo.2011.00044
Waterham, H. R. (2006). Defects of cholesterol biosynthesis. FEBS Lett. 580, 5442–5449. doi: 10.1016/j.febslet.2006.07.027
Wei, D., Allsop, S., Tye, K., and Piomelli, D. (2017). Endocannabinoid signaling in the control of social behavior. Trends Neurosci. 40, 385–396. doi: 10.1016/j.tins.2017.04.005
Yamashita, Y., Matsuishi, T., Ishibashi, M., Kimura, A., Onishi, Y., Yonekura, Y., et al. (1998). Decrease in benzodiazepine receptor binding in the brains of adult patients with Rett syndrome. J. Neurol. Sci. 154, 146–150.
Yudkoff, M., Daikhin, Y., Horyn, O., Nissim, I., and Nissim, I. (2008). Ketosis and brain handling of glutamate, glutamine, and GABA. Epilepsia 49(Suppl. 8), 73–75. doi: 10.1111/j.1528-1167.2008.01841.x
Zaheer, S., Kumar, D., Khan, M. T., Giyanwani, P. R., and Kiran, F. N. U. (2018). Epilepsy and cannabis: a literature review. Cureus 10:e3278. doi: 10.7759/cureus.3278
Zamberletti, E., Gabaglio, M., Piscitelli, F., Brodie, J. S., Woolley-Roberts, M., Barbiero, I., et al. (2019). Cannabidivarin completely rescues cognitive deficits and delays neurological and motor defects in male Mecp2 mutant mice. J. Psychopharmacol. doi: 10.1177/0269881119844184 [Epub ahead of print].
Zhang, L., He, J., Jugloff, D. G. M., and Eubanks, J. H. (2008). The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus 18, 294–309. doi: 10.1002/hipo.20389
Zhang, Q., Wang, J., Li, J., Bao, X., Zhao, Y., Zhang, X., et al. (2017). Novel FOXG1 mutations in Chinese patients with Rett syndrome or Rett-like mental retardation. BMC Med. Genet. 18:96. doi: 10.1186/s12881-017-0455-y
Zhang, X., Cui, N., Wu, Z., Su, J., Tadepalli, J. S., Sekizar, S., et al. (2010). Intrinsic membrane properties of locus coeruleus neurons in Mecp2-null mice. Am. J. Physiol. Cell Physiol. 298, C635–C646. doi: 10.1152/ajpcell.00442.2009
Zhang, Z.-W., Zak, J. D., and Liu, H. (2010). MeCP2 is required for normal development of GABAergic circuits in the thalamus. J. Neurophysiol. 103, 2470–2481. doi: 10.1152/jn.00601.2009
Zhao, Y., Zhang, X., Bao, X., Zhang, Q., Zhang, J., Cao, G., et al. (2014). Clinical features and gene mutational spectrum of CDKL5-related diseases in a cohort of Chinese patients. BMC Med. Genet. 15:24. doi: 10.1186/1471-2350-15-24
Zhong, W., Cui, N., Jin, X., Oginsky, M. F., Wu, Y., Zhang, S., et al. (2015). Methyl CpG binding protein 2 gene disruption augments tonic currents of γ-aminobutyric acid receptors in locus coeruleus neurons: impact on neuronal excitability and breathing. J. Biol. Chem. 290, 18400–18411. doi: 10.1074/jbc.M115.650465
Zhong, W., Johnson, C. M., Cui, N., Oginsky, M. F., Wu, Y., and Jiang, C. (2017). Effects of early-life exposure to THIP on brainstem neuronal excitability in the Mecp2-null mouse model of Rett syndrome before and after drug withdrawal. Physiol. Rep. 5:e13110. doi: 10.14814/phy2.13110
Zuardi, A. W., Antunes Rodrigues, J., and Cunha, J. M. (1991). Effects of cannabidiol in animal models predictive of antipsychotic activity. Psychopharmacology 104, 260–264. doi: 10.1007/BF02244189
Zuardi, A. W., Crippa, J. A. S., Hallak, J. E. C., Bhattacharyya, S., Atakan, Z., Martin-Santos, R., et al. (2012). A critical review of the antipsychotic effects of cannabidiol: 30 years of a translational investigation. Curr. Pharm. Des. 18, 5131–5140.
Keywords: Rett syndrome, GABAAR, epilepsy, cholesterol, ketogenic diet, cannabinoids, neurosteroids
Citation: Mouro FM, Miranda-Lourenço C, Sebastião AM and Diógenes MJ (2019) From Cannabinoids and Neurosteroids to Statins and the Ketogenic Diet: New Therapeutic Avenues in Rett Syndrome? Front. Neurosci. 13:680. doi: 10.3389/fnins.2019.00680
Received: 12 April 2019; Accepted: 13 June 2019;
Published: 02 July 2019.
Edited by:
Francisco Ciruela, University of Barcelona, SpainReviewed by:
Maurizio Giustetto, University of Turin, ItalyCopyright © 2019 Mouro, Miranda-Lourenço, Sebastião and Diógenes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francisco Melo Mouro, Zm1vdXJvQG1lZGljaW5hLnVsaXNib2EucHQ=; Maria José Diógenes, ZGlvZ2VuZXNAbWVkaWNpbmEudWxpc2JvYS5wdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.