
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 18 February 2019
Sec. Neurodegeneration
Volume 13 - 2019 | https://doi.org/10.3389/fnins.2019.00075
This article is part of the Research Topic Iron and Neurodegeneration View all 30 articles
 José Vicente Llorens1,2*Sirena Soriano3,4Pablo Calap-Quintana1,2Pilar Gonzalez-Cabo5,6,7
José Vicente Llorens1,2*Sirena Soriano3,4Pablo Calap-Quintana1,2Pilar Gonzalez-Cabo5,6,7 María Dolores Moltó1,2,8*
María Dolores Moltó1,2,8*Friedreich’s ataxia (FRDA) is a rare early-onset degenerative disease that affects both the central and peripheral nervous systems, and other extraneural tissues, mainly the heart and endocrine pancreas. This disorder progresses as a mixed sensory and cerebellar ataxia, primarily disturbing the proprioceptive pathways in the spinal cord, peripheral nerves and nuclei of the cerebellum. FRDA is an inherited disease with an autosomal recessive pattern caused by an insufficient amount of the nuclear-encoded mitochondrial protein frataxin, which is an essential and highly evolutionary conserved protein whose deficit results in iron metabolism dysregulation and mitochondrial dysfunction. The first experimental evidence connecting frataxin with iron homeostasis came from Saccharomyces cerevisiae; iron accumulates in the mitochondria of yeast with deletion of the frataxin ortholog gene. This finding was soon linked to previous observations of iron deposits in the hearts of FRDA patients and was later reported in animal models of the disease. Despite advances made in the understanding of FRDA pathophysiology, the role of iron in this disease has not yet been completely clarified. Some of the questions still unresolved include the molecular mechanisms responsible for the iron accumulation and iron-mediated toxicity. Here, we review the contribution of the cellular and animal models of FRDA and relevance of the studies using FRDA patient samples to gain knowledge about these issues. Mechanisms of mitochondrial iron overload are discussed considering the potential roles of frataxin in the major mitochondrial metabolic pathways that use iron. We also analyzed the effect of iron toxicity on neuronal degeneration in FRDA by reactive oxygen species (ROS)-dependent and ROS-independent mechanisms. Finally, therapeutic strategies based on the control of iron toxicity are considered.
Nicholaus Friedreich, a German neurologist among a family of professors of medicine, had described a new disease entity in successive papers published between 1863 and 1877 (Koeppen, 2013). The new disease, named later Friedreich’s ataxia (FRDA), was characterized by degenerative atrophy of the posterior columns of the spinal cord, leading to the development of progressive ataxia, sensory loss and muscle weakness in patients and was associated with cardiomyopathy in many cases. Clear diagnostic criteria for FRDA were established by Anita Harding in the 1980s (Harding, 1981), who specified the hereditary nature of the disease with an autosomal recessive pattern, an onset during puberty, progressive ataxia of the limbs and gait with the absence of tendon reflexes in the legs, extensor plantar responses, and dysarthria. In 1996, the identification of the causative gene of FRDA and expansion of a trinucleotide repeat as the most frequent mutation (Campuzano et al., 1996) led to the development of a molecular tool for a definitive diagnosis of the disease. It was crucial to address the differential diagnosis with respect to other peripheral neuropathies and ataxias with similar clinical features, as well as the clinical heterogeneity in FRDA, including atypical phenotypes (Bidichandani and Delatycki, 2017). Moreover, the availability of this molecular approach allowed for appropriate genetic counseling.
Friedreich’s ataxia (OMIM #229300; ORPHA:95) is a rare neurodegenerative disease affecting 1:20,000–1:50,000 people of Indo-European and North African descendent (Vankan, 2013), and with a carrier frequency of 1:60–1:100, FRDA is the most frequently inherited ataxia in these populations. The neurological signs and symptoms in FRDA are the results of pathological changes that disturb both the central and peripheral nervous systems (NSs) (reviewed in Marmolino, 2011; Koeppen and Mazurkiewicz, 2013). The dorsal root ganglia (DRG) are particularly vulnerable and have been identified as the first site of neuropathology with the degeneration of the large sensory neurons and posterior columns, leading to the loss of position and vibration senses. Next, affectation of the corticospinal and spinocerebellar tracts of the spinal cord occurs, with loss of myelinated fibers. In the cerebellum, dentate nuclei (DN) also show progressive atrophy. Cerebral abnormalities are being progressively reported with the application of neuroimaging techniques (Selvadurai et al., 2018). Non-neurological phenotypes of FRDA mainly include hypertrophic cardiomyopathy, which is the leading cause of mortality (Raman R. et al., 2011; Payne and Wagner, 2012; Weidemann et al., 2013; Koeppen et al., 2015). The heart typically maintains adequate systolic function (Kipps et al., 2009) in FRDA patients who develop a severe hypertrophic cardiomyopathy until shortly before death (Rajagopalan et al., 2010). Additionally, diabetes mellitus and skeletal abnormalities are also present in this disease (Parkinson et al., 2013; Bidichandani and Delatycki, 2017).
Friedreich’s ataxia is caused by loss-of-function mutations in the frataxin gene (FXN) (Campuzano et al., 1996). The most common mutation appears in homozygosis in most patients and consists of an abnormally expanded GAA trinucleotide repeat (usually between 600 and 900 repeats) in the first intron of the gene FXN (Campuzano et al., 1996; Monrós et al., 1997). The remaining patients (approximately 4%) are compound heterozygous with the expanded GAA repeat on one allele and another pathogenic variant (point mutation, deletion or insertion) on the other allele (Galea et al., 2016). The pathological GAA expansion affects the expression of FXN by blocking the transcription of mRNA through the formation of sticky DNA triplexes and R-loop structures (reviewed in Kumari and Usdin, 2012) and/or inducing abnormal heterochromatinization (Saveliev et al., 2003). Thus, the level of frataxin is strongly reduced from 5 to 30% of the physiological level (Campuzano et al., 1997). This reduction is related to the disease severity and length of the GAA repeat. In general, longer expansions are present in patients with a more severe phenotype such as earlier onset, faster progression and/or presence of non-neurological features (Parkinson et al., 2013). Although most of the pathogenic point mutations also decrease the level of functional protein, some lead to the production of a less active protein (De Castro et al., 2000; Bridwell-Rabb et al., 2011).
Frataxin is a small acidic protein, synthetized in the cytoplasm as a 210-residue polypeptide that is then imported into the mitochondria, where it is proteolytically processed to the mature and most abundant form of 130 residues [frataxin (81–210)] (Condò et al., 2007). Although frataxin has historically been considered a protein exclusively confined to the mitochondrial matrix, several studies have reported the existence of an extramitochondrial pool of frataxin in different human cell types (Acquaviva et al., 2005; Condò et al., 2006; Lu and Cortopassi, 2007). Extramitochondrial frataxin corresponds to the (81–210) mature form of the protein (Condò et al., 2010) and earlier studies suggested that after the initial transport and processing in the mitochondria, mature frataxin was transported back to the cytosol (Acquaviva et al., 2005; Condò et al., 2006). However, a recent study describes an N-terminal acetylated extramitochondrial form of frataxin in erythrocytes. This (76–210) isoform does not contain the mitochondrial targeting sequence, and remains in the cytosol where it is cleaved to produce an (81–210) protein identical to the mitochondrial mature form (Guo et al., 2018).
The three-dimensional structure of frataxin shows seven antiparallel β-sheets flanked by two α-helices, producing a characteristic globular αβ fold (Dhe-Paganon et al., 2000). The protein is ubiquitously expressed (Campuzano et al., 1996, 1997), but different cells types show distinct susceptibility to its deficiency. This could be explained by frataxin reduction in different tissues being associated with distinct transcriptomic profiles, as described recently (Chandran et al., 2017). Frataxin is a highly conserved protein through evolution, and its cellular function is critical for life in multicellular organisms. A strong reduction of frataxin in Drosophila melanogaster seriously affects viability (Anderson et al., 2005; Llorens et al., 2007). Knockout of the frataxin gene causes embryonic lethality in mice (Cossée et al., 2000) and in the plant Arabidopsis thaliana (Vazzola et al., 2007). In line with this, FRDA patients with non-GAA-repeat mutations in both frataxin alleles, resulting in totally deficient frataxin function, have not been reported.
At the time of FXN identification, there was no evidence about the function of frataxin. Experiments in yeast Saccharomyces cerevisiae promptly suggested a potential role for frataxin in iron homeostasis regulation. Deletion of the ortholog of frataxin in yeast (Yfh1) causes iron accumulation in the mitochondria (Babcock et al., 1997; Foury and Cazzalini, 1997) that was initially suggested at the expense of the cytosolic iron pool (Knight et al., 1998). However, measurements of cytosolic iron in frataxin-deficient yeast cells show that the cytosolic iron levels are not affected by the reduction of the frataxin synthesis (Li et al., 2012). Also associated with frataxin deficiency are reduced activities of mitochondrial iron-sulfur cluster (ISC) enzymes and aconitase, mitochondrial dysfunction and oxidative stress (reviewed in Calabrese et al., 2005; Lupoli et al., 2018). Since then, efforts have been made to answer several questions about iron in FRDA, including whether its accumulation is a primary or secondary event in the disease, what is the mechanism causing the deregulation and buildup of iron, and the role of this metal in the neuropathology of FRDA. Here, we review the contribution of the cellular and animal models of this disease and relevance of the studies using patient samples to gain knowledge about these issues.
Iron accumulation is a hallmark in FRDA and initially was suggested to be a primary pathogenic event triggered by frataxin deficiency. Tissues in which iron overload has been reported in FRDA patients are showed in Supplementary Table S1. The first observations linking iron and FRDA were made in postmortem heart samples from a few patients, where dense intracytoplasmic deposits of iron particles were detected in the cardiac fibers (Sanchez-Casis et al., 1976; Lamarche et al., 1980). These findings were later corroborated in subsequent studies that use different techniques for iron quantitation, such as Perls Prussian blue staining for histological detection or atomic absorption spectroscopy to measure total iron levels in tissue. The excess iron in the heart of FRDA patients appears as multifocal aggregates highly localized rather than as a diffuse pattern (Ramirez et al., 2012; Koeppen et al., 2015). This regional distribution of the excess of iron could be responsible for the findings of iron levels equivalent to healthy controls in bulk extracts or from random biopsied cardiac samples (Michael et al., 2006; Koeppen et al., 2015; Kruger et al., 2016). In addition to the heart, positive iron staining was observed in the liver and spleen of patients, showing a distribution consistent with a mitochondrial location (Bradley et al., 2000) as has been described in the frataxin-deficient yeast model (Babcock et al., 1997; Foury and Cazzalini, 1997).
In regions of the NS, such as DN and DRG, several scenarios regarding the iron content in FRDA have been reported: normal levels (Bradley et al., 2000; Koeppen et al., 2007, 2013; Solbach et al., 2014), accumulation (Bonilha da Silva et al., 2014; Harding et al., 2016), and limited and regional increases of this metal (Koeppen et al., 2009). These diverse results could be partly due to the technique used for iron measurements and their capability to discriminate between the accumulation and redistribution of iron. In most of these studies, the iron content has been quantified from a small number of patient samples, a situation that could also contribute to the different results reported. A more recent study with a higher number of FRDA subjects analyzed used magnetic resonance imaging, which allows in vivo iron quantitation (Harding et al., 2016). The authors found a significant increase in the iron concentration in the FRDA cohort compared with that in controls, in the DN and Red nucleus in the midbrain. However, the technical approach used in this study did not allow the authors to discriminate between deposition and redistribution or whether the location of the iron was extracellular or intracellular. Changes in the distribution of iron and other metals, such as copper and zinc, in the DN of FRDA patients have also been reported (Koeppen et al., 2012), although the total amount of these metals was similar to that of controls. It is worth highlighting that copper can also generate oxygen radicals and, in the context of FRDA, may exacerbate the oxidative injury caused by the increased amount of iron. Mitochondrial iron accumulation has also been documented in fibroblasts and lymphoblasts of FRDA patients. Although some authors have found an increase in the total iron content (Delatycki et al., 1999) or labile iron fraction (Tan et al., 2001), normal levels of iron have been reported in other studies (Wong et al., 1999; Sturm et al., 2005).
Several mouse models of FRDA have been obtained using different experimental strategies, and most of them show iron overload because of frataxin reduction (Supplementary Table S2). However, no iron accumulation was detected in embryonic samples of the frataxin knockout (KO) mice (Cossée et al., 2000), and the conditional model of frataxin deficiency in striated muscle (MCK-KO mice) showed heart intramitochondrial iron buildup only in late stages of the disease (Puccio et al., 2001). This led to the hypothesis that the increase in iron is not the causative pathological mechanism in FRDA and is a secondary effect.
Iron accumulation has been studied, particularly in muscle creatine kinase (MCK) conditional Fxn knockout mice (MCK-KO). This mutant develops a severe cardiac phenotype associated with almost ablation of frataxin in heart and skeletal muscle (Puccio et al., 2001). Different studies on this mouse line have reported distinct ages of onset for the iron overload despite using the same iron detection methodology (Supplementary Table S2). Some authors (Puccio et al., 2001) found iron deposits in 10-week-old mice, whereas animals 7 weeks of age did not present iron accumulation but showed deficits in ISC enzyme activities. By contrast, other authors (Huang et al., 2013) observed progressive iron accumulation from 5 to 10 weeks of age, with the presence of the iron deposits being the first observable histological change in the MCK-KO hearts. Therefore, there is no consensus about when iron starts to accumulate and whether the presence of iron deposits is relevant for the development of heart pathology in FRDA.
An interesting result found in the MCK-KO model is that iron levels increase in other tissues besides the heart (Supplementary Table S2), such as the liver, kidney and spleen, as well as serum (Huang et al., 2009; Whitnall et al., 2012). Because the amount of frataxin in these non-muscular tissues is within the normal range in the MCK-KO model (Huang et al., 2009; Whitnall et al., 2012), the iron increase was explained as an alteration of systemic iron metabolism orchestrated by the frataxin reduction in heart and skeletal muscle. It could be operated through a signaling mechanism involving hepcidin (Huang et al., 2009; Whitnall et al., 2012), a key regulator of systemic iron metabolism in mammals. These explanations indicate that excess iron plays a significant role in the disease. The mitochondrial iron in the heart of MCK-KO mutants is in the form of biomineral iron, which aggregates with phosphorus and sulfur (Whitnall et al., 2012). This is important because excess of iron that is not bound to ferritin, the main cellular protein that stores iron, can be very toxic by generating reactive oxygen species (ROS).
In the liver of mice with hepatic Fxn deficiency (FxnAlb mice), a fivefold increase in the mitochondrial iron level has been observed at 28 days of age compared with that in control mice. Meanwhile, 14-day-old mutant showed deficits in ISC enzymes in the liver but normal levels of iron (Martelli et al., 2015), suggesting that iron accumulation is a secondary event also in the liver. As in FRDA patients, iron deposition in the NS of FRDA mouse models has not been clarified yet. In conditional knockout models with frataxin reduction in neurons and other tissues (Puccio et al., 2001) or in parvalbumin-positive cells in the DRG, cerebellum and brain (Piguet et al., 2018), no iron accumulation has been observed. Other studies have reported few iron deposits in NS late in disease progression (Simon et al., 2004) or that iron accumulates both intracellularly and extracellularly in the brain following the loss of Fxn (Chen et al., 2016a). To understand these diverse findings, the authors have indicated that the different sensitivities of the techniques used to measure iron could be an explanation, or that, in some mouse models, cells loss may occur before sufficient iron is accumulated to be detected.
Other animal models of FRDA have also shown an increase in iron levels in the mitochondria (Supplementary Table S2), as is the case of D. melanogaster (Soriano et al., 2013). In this species, severe loss of frataxin function results in iron accumulation in multiple tissues, including the brain (Chen et al., 2016b). This was the first FRDA model clearly demonstrating that iron can accumulate in the NS. Additionally, frataxin-deficient flies also showed increased levels of other transition metals, such as zinc, copper and manganese, suggesting that frataxin reduction affects the homeostasis of other metals besides iron (Soriano et al., 2016). In the plant A. thaliana, frataxin is localized in both the mitochondria and chloroplasts, where it is suggested to have similar functions; its deficiency alters the iron content, and this metal accumulates in the two organelles (Martin et al., 2009; Turowski et al., 2015). These results indicate that frataxin-defective organisms show a common propensity to accumulate Fe into organelles where this protein is located. Although many studies have suggested that iron deposits are not instrumental and represent a late event in the pathogenesis of FRDA, the role of Fe in the disease pathophysiology is not fully understood. Recently, frataxin function has been nicely discussed (Alsina et al., 2018), and it can offer some insight into the mechanisms underlying iron accumulation in FRDA tissues and in model organisms. We review this subject in the following section.
Iron is vital for the cell because it participates in essential biological processes such as DNA synthesis, gene regulation, cellular respiration, and energy production. The mitochondria play a major role in the cellular metabolism of this element and synthesis of heme and ISCs, which are needed for the function of many proteins involved in such cellular processes (Napier et al., 2005; Richardson et al., 2010). Iron homeostasis must be highly controlled because the depletion of its level can produce cellular death, and an increased level of iron promotes the production of ROS. In its Fe+2 state, iron can interact with oxygen via the Fenton and Haber-Weiss reactions to catalyze the production of ROS, which oxidize and damage proteins, lipids and nucleic acids (Eaton and Qian, 2002). Therefore, precise regulatory systems are needed to ensure the proper amount and oxidative state of iron in the cell and its different compartments.
Although frataxin has been proven to be a mitochondrial iron-binding protein (Foury et al., 2007) involved in the homeostasis of iron, its role remains unclear. Many studies have indicated that this protein could play roles related to iron storage (Park et al., 2003; Huang et al., 2011), heme (Yoon and Cowan, 2004; Napier et al., 2005) and ISC synthesis (Foury et al., 2007; Richardson et al., 2010; Lane et al., 2015) and could function as an iron sensor (Adinolfi et al., 2009) and a metabolic switch (Becker et al., 2002). Some of these possible roles have been more thoroughly tested and proven that others, with the involvement in ISC synthesis being the most accepted one thus far. We discuss these functions next and its possible relationship with mitochondrial iron accumulation in FRDA.
The observation that yeast frataxin (Yfh1), under aerobic conditions and an excess of iron, can form spheroidal oligomers that have ferroxidase activity (Adamec et al., 2000; Gakh et al., 2002; Park et al., 2002) promptly suggested that frataxin could function as an iron storage protein, similar to ferritin (Pandolfo and Pastore, 2009). These Yfh1 multimers have the capacity to bind up to 50 atoms of iron per monomer and maintain it in a redox-inactive state through the ferroxidase activity, which converts redox-active Fe+2 to Fe+3 (Gakh et al., 2006). On this line, frataxin could store iron in a bioavailable form in the mitochondria, thus its deficit would lead to iron loading in this organelle.
Frataxins are proteins capable of binding iron. Nuclear magnetic resonance (NMR) studies predominantly mapped the iron-binding sites in the conserved acidic α1 and β1 regions, which contain a clustering of negatively charged aspartate and glutamate residues (reviewed in Bencze et al., 2006; Pandolfo and Pastore, 2009). However, the hypothesis of frataxin as an iron storage protein presents several problems. Perhaps, one of most serious questions is that, in the case of human frataxin (FXN), the mature form of the protein is not prone to oligomerization. Only the precursor and intermediate forms of FXN can form oligomers and in an iron-independent manner (Adinolfi et al., 2002; O’Neill et al., 2005). Nevertheless, it has lately reported that the mature form of FXN, under specific experimental conditions can form oligomers, but they are unstable (Ahlgren et al., 2017). Other important caveat to this hypothesis is the redundant role that FXN would play with mitochondrial ferritin. In addition, it has been reported that oligomerization-deficient mutant Yfh1 is able to substitute functionally wild-type Yfh1 (Aloria et al., 2004), indicating that oligomerization is not required for frataxin function.
Frataxin can bind iron directly without forming oligomers (Bencze et al., 2006; Cook et al., 2006). Depending on the organism (bacteria, yeast or human) and state of iron (Fe+2 or Fe+3), different possible iron-binding sites in the protein have been described (Yoon and Cowan, 2003; Bou-Abdallah et al., 2004; Cook et al., 2006; Yoon et al., 2007; Huang et al., 2008). The primary site that binds Fe+2 involves the residues 18, 19 and 22 of CyaY (and corresponding ones in Yfh1 and FXN) at the acidic ridge in the α1 helix of frataxin. This raised the possibility of new functions for frataxin in iron-dependent processes, in which some of the involved machinery components seem to interact with FXN. Frataxin interactions have been reported with mitochondrial aconitase, ferrochelatase (Fech), succinate dehydrogenase and the components of the ISC synthesis machinery (Gerber et al., 2003; Yoon and Cowan, 2003; Bulteau et al., 2004; Bencze et al., 2006). Among these interactions, the binding of frataxin with components of the ISC complex has been reproduced and characterized in independent studies (reviewed in Martelli and Puccio, 2014).
The heme biosynthetic pathway is carried out in mitochondria. This pathway includes several enzymes (Sassa, 2006; Furuyama et al., 2007; Chung et al., 2012; Dailey and Meissner, 2013; Chiabrando et al., 2014), with the 5-aminolevulinic acid synthase (ALAS) being the first one in that pathway and the rate-limiting enzyme. ALAS has two isoenzymes, ALAS1 and ALAS2. Whereas ALAS1 expression is negatively regulated by heme directly bound to a cysteine-proline dipeptide motif (Furuyama et al., 2007), the ALAS2 gene contains an iron-responsive element (IRE) that interacts with iron regulatory proteins (IRPs) regulating its expression at the posttranscriptional level. Under iron deficiency conditions, the binding of IRP to ALAS2 mRNA inhibits its translation; by contrast, under iron sufficiency conditions, IRPs detach from the IRE, resulting in increased ALAS2 translation. In this manner, the cytosolic iron availability regulates the rate of heme synthesis (Ponka, 1997).
The final enzyme in the heme biosynthetic pathway is Fech, which inserts ferrous iron into protoporphyrin IX. This enzyme contains a [2Fe-2S] cluster that is necessary for enzymatic activity (Wu et al., 2001). The disruption in ISC synthesis could adversely affect the rate of heme synthesis (Richardson et al., 2010; Huang et al., 2011). It has been shown that mitoferrin (Mfrn1) is responsible for iron transport into mitochondria, allowing the direct transfer of ferrous iron for heme and/or ISC synthesis (Fleming, 2011). The role of frataxin in heme synthesis is not clear, although a reduction of heme synthesis was observed in yeast and mouse frataxin mutants (Lesuisse et al., 2003; Huang et al., 2009). Different studies have suggested a direct role of frataxin in the heme synthesis pathway by binding directly to Fech and acting as an iron donor (Yoon and Cowan, 2004; Napier et al., 2005). Thus, this metal would accumulate because of frataxin deficiency.
Iron-sulfur clusters are protein cofactors present in almost all living organisms. They play a critical role in several cellular functions, from electron transport in mitochondrial respiratory complexes to DNA repair or metabolism. The de novo synthesis of ISC occurs in the mitochondria (Lill, 2009; Beilschmidt and Puccio, 2014). The components of the ISC synthesis machinery are the cysteine desulfurase (NFS1), which converts L-cysteine to L-alanine with the generation of sulfur, an accessory protein (ISD11), a reductant (ferredoxin), a scaffold protein (ISCU) and an iron delivery protein that is at the core of the debate (Lill and Mühlenhoff, 2006; Py and Barras, 2010). Besides ISD11, recent findings showed that human NFS1 needs another accessory protein, the mitochondrial acyl carrier protein (ACP), making the cysteine desulfurase complex (NFS1-ISD11-ACP) (Van Vranken et al., 2016). In addition, NFS1 requires FXN for its full function (Tsai and Barondeau, 2010; Fox et al., 2015). Regarding the role of FXN in the ISC synthesis, there are two main hypothesis. It was first suggested that frataxin could be the iron donor (Gerber et al., 2003; Yoon and Cowan, 2003; Layer et al., 2006). Using a combination of different techniques based on isothermal titration calorimetry and multinuclear NMR spectroscopy, Cai et al. (2018) have recently showed that FXN can function as an iron donor in vitro. They found that FXN-Fe2+ interacts weakly to the NFS1-ISD11-ACP complex, but the presence of ISCU largely improves this interaction and that dissociation of Fe2+ from FXN occurs when the L-cysteine and ferredoxin enters into the equation. These results suggest that FXN is the proximal source of iron for ISC assembly, but in vivo characterization is required to confirm this role for frataxin.
Other studies have proposed that frataxin could actually act as a regulator in the synthesis of ISC (Adinolfi et al., 2009; Tsai and Barondeau, 2010; Bridwell-Rabb et al., 2012). The binding of frataxin to the ISCU/NFS1/ISD11 complex can stabilize it, activates the cysteine desulfurase activity and control the entry of iron to the complex (Tsai and Barondeau, 2010; Colin et al., 2013; Pandey et al., 2013).
Mutation of specific aspartate and glutamate residues in the acidic ridge of frataxin, where iron-binding sites were previously mapped, alters the interaction of frataxin with ISCU. Unexpectedly, these mutations does not reduce the total number of iron-binding sites (Schmucker et al., 2011). These results suggested that some of these residues might be critical for frataxin interaction with the core components of the ISC complex, but not for binding iron. Consequently, some authors (Gentry et al., 2013) proposed that frataxin might have unknown binding sites for this metal. Using different spectroscopic and structural methodologies, a new iron-binding site was identified at His86 in the N-terminus of the mature FXN form, therefore not positioned in the acidic ridge. Interestingly, this residue was essential for iron binding and for in vitro productive synthesis of ISC by ISCU, without showing a direct interaction with the scaffold protein (Gentry et al., 2013). These results support the proposal that frataxin is the iron donor for ISC assembly.
Extramitochondrial frataxin has been shown to physically interact with IscU1, the cytosolic isoform of the human ISC, indicating a role for frataxin in the Fe/S cluster assembly in the cytosol (Acquaviva et al., 2005). The cytosolic form of frataxin has also been reported to interact and possibly regulate the function of the cytosolic aconitase/iron regulatory protein-1 (IRP1) (Condò et al., 2010). Other functions suggested for frataxin in the cytosol include preventing oxidative stress and apoptosis (Condò et al., 2006); and in the nucleus, frataxin could promote DNA repair and telomere stability (Lill et al., 2015).
As indicate above, frataxin could act as an iron sensor to tightly regulate ISC synthesis (Adinolfi et al., 2009). In E. coli, CyaY, far from facilitating iron delivery, can inhibit the ISC synthesis process. With normal iron levels, CyaY has a low affinity for the IscS-IscU system (the bacterial ISC synthesis machinery), and ISC is loaded to the final acceptor. However, under conditions of iron excess, the affinity of CyaY for IscS would increase, slowing the rate of ISC synthesis and avoiding an overproduction of ISC. This would avoid the degradation of ISC not inserted into an acceptor protein and the release of free iron that could catalyze the production of ROS. However, the consequence of frataxin deficiency in FRDA patients is a decrease in ISC protein function, suggesting that FXN might operate as an activator of the ISC synthesis. In vitro studies also support the role of FXN as an allosteric activator of the ISC assembly (Tsai and Barondeau, 2010). In contrast, both FXN and CyaY can complement a yeast strain with deletion of Yfh1, indicating that eukaryotic and bacterial frataxins can at least partially conserve their functions (Cavadini et al., 2000; Bedekovics et al., 2007). To clarify this controversial results, Bridwell-Rabb et al. (2012) conducted enzyme kinetic assays in which analogous components of the human and E. coli ISC complex were interchanged. Interestingly, they found that CyaY could substitute FXN as an activator of the cysteine desulfurase in the human ISC complex. Conversely, FXN could also replace CyaY as an inhibitor in the assembly of the bacterial ISC. Therefore, both frataxin proteins have the property to modulate the ISC formation, but the mechanism of action of any of them, as an activator or inhibitor of ISC biogenesis, depends on another component of the ISC assembly machinery. This component was found to be the cysteine desulfurase, which controls how the frataxin protein operates in the ISC complex (Bridwell-Rabb et al., 2012). Differences between the human and bacteria cysteine desulfurase might explain the different role of frataxin in humans with respect to E. coli and by extension in eukaryotes vs prokaryotes. Further studies are needed to define the nature of these differences that dictate how frataxin acts in the process of the ISC synthesis.
Examination of the role of frataxin in ISC and heme metabolism in differentiating erythroid cells has suggested that frataxin acts as a metabolic switch in the diversion of iron from one metabolic pathway to another. This was supported by the observation that an increased level of protoporphyrin IX (PPIX) leads to the downregulation of frataxin expression and potential diversion of iron from ISC synthesis toward heme synthesis (Becker et al., 2002). The molar ratio between frataxin and Fech affects the rate of heme synthesis (Yoon and Cowan, 2004), and these findings have suggested that frataxin could function as a metabolic switch in regulating mitochondrial iron metabolism, independently of the expression levels of PPIX or Fech. This metabolic switch allows the mitochondria to favor heme or ISC syntheses, depending on the frataxin levels. The possible mechanism behind this switching may be the relative affinities of frataxin for Fech vs ISCU. This may result in the preferential binding of frataxin to Fech over ISCU at relatively lower frataxin levels (Richardson et al., 2010).
When the frataxin level is reduced, there would be no control in the rate of ISC formation, causing iron accumulation (Adinolfi et al., 2009). Regarding the metabolic switch role of frataxin, its depletion would provoke the enhancement in the reduction of ISC formation at the ISCU machinery level and at the end, subjecting the cell to increased iron accumulation and ROS production (Yoon and Cowan, 2004).
Although the mechanisms responsible for mitochondrial iron loading in FRDA are not fully understood, there are several reasonable hypotheses that could explain this alteration. In the MCK-KO mouse, Huang et al. reported altered heme and ISC synthesis and mitochondrial iron storage. Significantly diminished activity and expression of ISC enzymes, such as NFS1 and ISCU1/2, were observed that agreed with the reduction in the expression of succinate dehydrogenase complex, subunit A (Sdha) (Huang et al., 2009). At the same time, other studies have shown a downregulation in the number of key heme synthesis enzymes and decreased heme levels in the heart of the MCK KO model (Schoenfeld et al., 2005; Huang et al., 2009). A decrease in the synthesis of different products that require iron in the mitochondria could explain the accumulation of this metal because it cannot be exported from the organelle being part of those products. However, frataxin deficiency can also affect the expression of several other proteins, some of them critical for the proper control of iron metabolism (Figure 1). In DN, Koeppen et al. (2007) observed modification of the expression of proteins such as transferrin receptor 1 (TFR1), ferritins (FRTs) and ferroportin (FPN), alterations shared among animal models of FRDA. In the MCK-KO mouse (Martelli and Puccio, 2014), has been described marked TFR1 upregulation, leading to increased iron uptake, and the downregulation of FPN, decreasing cellular iron release from the cytosol. There are also decreased levels of ferritin light chain 1 (FRTL1) and heavy chain 1 (FRTH), implying reduced cytosolic iron incorporation and storage. Finally, an increased expression of Sec15l1 (an exocyst complex protein) and the mitochondrial iron importer mitoferrin-2 (MFRN2) in the mutant could facilitate mitochondrial iron influx, which was seen to be enhanced in this model (Whitnall et al., 2008).
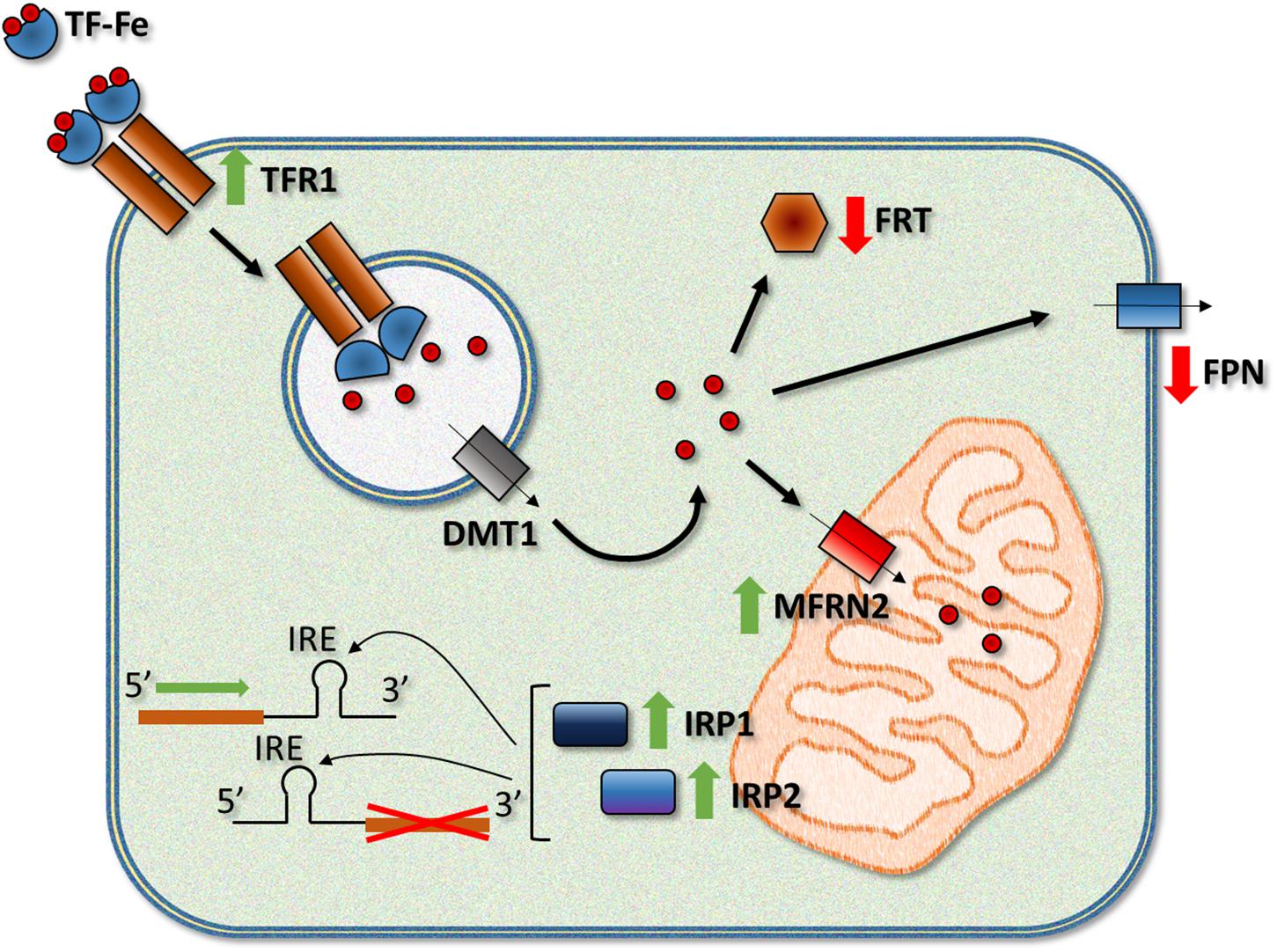
Figure 1. Frataxin deficiency leads to a series of alterations in the activity and/or expression of different iron-related proteins. Among the most relevant changes observed in humans and FRDA models, there is an increase in the activity of the iron regulatory proteins IRP1 and IRP2, upregulation of TFR1 (transferrin receptor 1) and the mitochondrial iron importer MFRN2 (mitoferrin-2), and downregulation of the iron exporter FPN (ferroportin) and iron storage proteins FRTs (ferritins, light and heavy chains). TF, transferrin; DMT1, divalent metal transporter 1.
The link between frataxin deficiency and alteration in the expression of some of these proteins could be IRP1 and IRP2. Both proteins bind to IRE elements in the 5′ and 3′ UTR of the mRNAs of iron metabolism-related proteins (Chiang et al., 2016). When bound to a 5′ IRE, IRP1 and IRP2 repress the translation of the mRNAs while binding of the IRPs to a 3′ IRE increases the stability and translation of the mRNAs. The activity of both IRPs is increased by the low levels of cellular iron by different mechanisms. Interestingly, IRP1 activity is controlled by the insertion of an ISC, which converts the protein into a cytosolic aconitase and makes it unable to bind to IRE elements. Since frataxin depletion reduces ISC synthesis, it is reasonable to suggest a role for IRP1 in the deregulation of iron metabolism. Nevertheless, the activity of IRP2, which is controlled by iron-dependent controlled ubiquitination and degradation, is also increased in MCK-KO mouse, possibly due to cytosolic iron depletion (Whitnall et al., 2008). More recently, Martelli et al. (2015) studied a double KO mouse with complete ablation of Irp1 and liver-specific deletion of Fxn. In these mice, the authors observed that alterations in the expression of proteins such as TFR1 and divalent metal transporter 1 (DMT1) were indeed due, a least partially, to IRP1, while the regulatory protein seemed to exert little effect on the expression of other proteins such as FRTL and FPN. Other models of the disease also manifest similar alterations in the expression of proteins involved in iron metabolism. Frataxin depletion in D. melanogaster produces alterations in the expression of mitoferrin, IRP-1A and ferritin, while genetic modifiers for Malvolio, Tsf1 and Tsf3 (Drosophila homologs of DMT1 and transferrins) and IRP-1A and IRP-1B could correct the motor deficiency phenotype of frataxin-deficient flies (Soriano et al., 2016; Calap-Quintana et al., 2018; Monnier et al., 2018). Although some hints point to the possible causes of iron dysregulation in FRDA, more research is needed to propose a more precise and final hypothesis.
Initially, it was postulated that iron overload in FRDA activates the formation of ROS via the Fenton reaction, which would arbitrate the pathogenesis of the disease. The highly redox active environment in the mitochondria could promote ROS generation when the free iron content increases. Nevertheless, an ROS independent mechanism mediated by iron has recently been proposed for FRDA neurodegeneration. These two mechanisms are described in the following paragraphs (Figure 2).
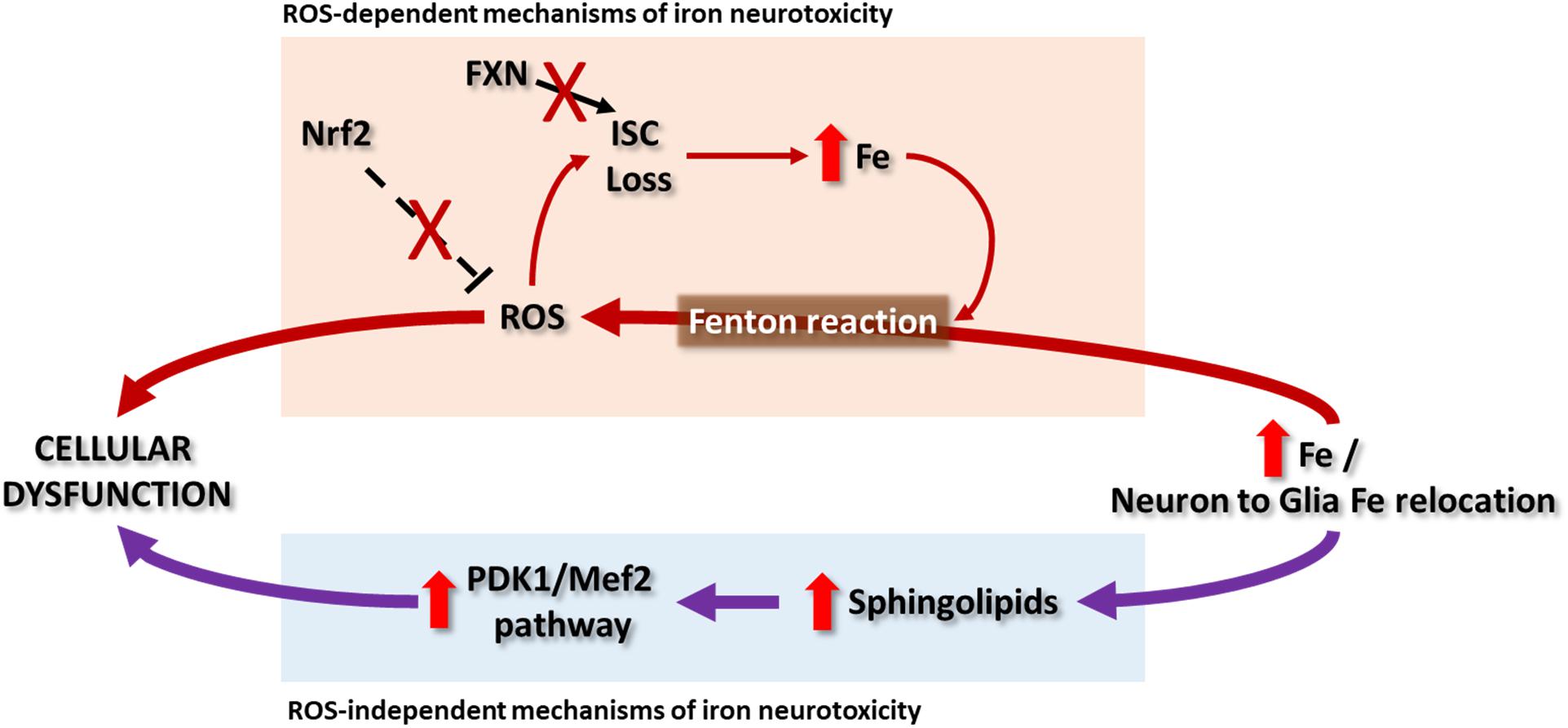
Figure 2. In Friedreich ataxia patients, deposits of iron in NS have not been clearly reported and it remains a controversial topic, while some evidences point to a relocation of iron from neurons to glial cells. So far there are some hypotheses that explain the possible mechanisms of iron toxicity that lead to cellular dysfunction. The ROS-dependent hypothesis proposes the iron-mediated catalysis of ROS by Fenton reaction. Besides the impairment in ISC synthesis due to frataxin deficiency, the radical species can in turn damage the ISC of proteins generating new free iron that will participate in the Fenton reaction. Contributing to this cycle is the observation that the Nrf2 signaling is defective in some cells and models of the disease. Another recent hypothesis, independent of ROS production, was explored in Drosophila and mouse models. It suggests that the cellular dysfunction can be due to the activation of the PDK1/Mef2 (3-phosphoinositide dependent protein kinase-1/myocyte enhancer factor-2) pathway by means of an increase in sphingolipids synthesis. Both hypothesis as well as the relevance of ROS in the pathology of FRDA have partial support, since the evidence pointing to them vary depending on the study.
Iron accumulation in the NS is a common feature in neurodegenerative diseases, including Alzheimer’s disease (Connor et al., 1992), Parkinson’s disease (Gorell et al., 1995), Huntington’s disease (Dumas et al., 2012) or amyotrophic lateral sclerosis (Kwan et al., 2012). In these disorders, iron deposits are observed in specific regions of the NS. As indicated previously, in Friedreich ataxia patients, deposits of iron have not been clearly reported in NS, and the increment of iron levels in the DN remains controversial (Waldvogel et al., 1999; Koeppen et al., 2007; Harding et al., 2016). This region is interesting because the patients develop neuronal atrophy and abnormal proliferation of the synaptic terminal, called grumose degeneration. The Koeppen’s group confirmed the presence of iron metabolism alteration in the NS of FRDA patients by changes in the expression of proteins related to iron, such as FPN, FRT and TFR1, in the DN (Koeppen et al., 2012) and DRG (Koeppen et al., 2009). They observed a relocation of iron from the affected neurons to the microglia, in the case of DN, or from neurons to the satellite cells in the DRGs. The increment of iron levels correlates with the production of ROS in these areas, suggesting that alterations of iron homeostasis could contribute to the pathogenesis of the disease through ROS production. Additionally, frataxin deficiency is related to low antioxidant defenses (Chantrel-Groussard et al., 2001; Paupe et al., 2009), making the nervous cells have an oxidative stress-prone phenotype.
The toxicity of iron may be due to its localization, biochemical form or cell ability to prevent the generation and propagation of iron. In living organisms, iron can be found in its reduced form, ferrous iron (Fe2+), and its oxidized form, ferric iron (Fe3+). Most iron binds, as a prosthetic group, to proteins such as hemoglobin, myoglobin, cytochromes or storage proteins; only less than 5% of iron in the cell is in the labile iron pool (LIP) (Li and Reichmann, 2016). The cellular LIP is defined as a pool of chelatable and redox-active iron complexes (Kakhlon and Cabantchik, 2002). LIP has the capacity to participate in the formation of harmful free radicals including the hydroxyl radical. Fe2+ is catalyzed to Fe3+ in a chemical reaction mediated by hydrogen peroxide (H2O2), which leads to the formation of hydroxide radical (OH∙). Cytotoxic events are started in the cell via the Fenton reaction. To avoid the negative effects of excess free iron, it is coupled to proteins such as FRT, which is responsible for iron storage inside the cell. In the satellite cells of DRG from FRDA patients, an increment of cytosolic FRT (Koeppen et al., 2009) was observed; in DN, ferritin is expressed mostly in oligodendrocytes, satellite cells around the neurons. As the disease progresses, the neurons begin to undergo atrophy, and these cells disappear, producing microglia FRT-positive cells (Koeppen et al., 2007). Noteworthy, mitochondrial FRT has not been detected in DRG neurons, a protein responsible to store free mitochondrial iron and with a very important role in antioxidant activity (Koeppen et al., 2007; Campanella et al., 2009). FPN is a transmembrane protein that exports iron and is expressed in the central NS and DRG. Interestingly, in the DRG from FRDA patients, the expression of FPN is exclusive to satellite cells (Koeppen et al., 2009), probably as a response to the iron release from DRG neurons to these cells (Koeppen et al., 2016). In the synaptic terminals of neurons from the DN of FRDA patients, is observed an increment of the ferroportin levels (Koeppen et al., 2007). The nerve endings present an elevated number of mitochondria, suggesting a response in the biosynthesis of this protein due to the dyshomeostasis of iron. Thus, the toxicity of this metal could be an important factor in grumose degeneration.
Because mitochondria are an important generator of ROS, the continuous presence of iron in the mitochondrial matrix makes it susceptible to new production of ROS. Proteins with ISC clusters, such as complex I, II and III of the electron transport chain, can be damaged by ROS. These complexes show reduced activity in FRDA patients (Rötig et al., 1997), probably because of iron toxicity. Moreover, these proteins will generate new free iron that will participate in the Fenton reaction and carry out new oxidative processes. In plasma and urine from FRDA patients (Emond et al., 2000; Schulz et al., 2000), similar to in all the disease models generated (González-Cabo and Palau, 2013), oxidative stress markers have been detected.
In the last years, neuronal models of the disease have been developed to better understand the neurodegenerative process. Oxidative stress, mitochondrial dysfunction, and iron dyshomeostasis are features shared among the neuronal models. For example, the granulate cells of the cerebellum from YG8R mice show an increment of ROS, lipid peroxidation and reduced glutathione levels (Bradshaw et al., 2016); the neurons of DRG from YG8R mice present an increment of ROS, dysfunctional mitochondria, and calcium dyshomeostasis (Mollá et al., 2017); the motor neuron cell line (NSC34) with reduced expression of the frataxin gene shows an increment in the GSSG/GSH ratio and more production of ROS (Piermarini et al., 2016) and alteration in complex I activity of the electron transport chain (Carletti et al., 2014); reduction of Drosophila frataxin (fh) expression in glial cells produces massive accumulation of fatty acids and increased sensitivity to oxidative stress (Navarro et al., 2010).
Under oxidative stress, the transcription factor Nrf2 binds to antioxidant response element (ARE) and modulates the expression of many genes involved in the antioxidant defense. It has been reported that Nrf2 signaling is defective in fibroblasts from FRDA patients (Paupe et al., 2009), the DRG and cerebellum of YG8R mice (Shan et al., 2013) and motor neuron and Schwann cell models (D’Oria et al., 2013; Shan et al., 2013), making these cells and tissues more vulnerable to oxidative stress damage and the neurodegeneration process. Concretely, frataxin deficiency in neurons promotes axonal dystrophy with multiple axonal spheroid formation mainly due to Ca2+ imbalance and oxidative stress (Mollá et al., 2017). The axonal swellings are associated with specific cytoskeletal changes (Mincheva-Tasheva et al., 2014; Piermarini et al., 2016; Mollá et al., 2017), improper axonal transport and autophagic flux (Shidara and Hollenbeck, 2010; Mollá et al., 2017). Moreover, the oxidative stress damage effects to the neurites growth leads to shorter dendrites due to alterations in the dynamics of the microtubules (Shan et al., 2013; Carletti et al., 2014; Piermarini et al., 2016). When oxidative stress is reduced in these neurons, either by direct addition of antioxidants (Piermarini et al., 2016) or through drugs that increase the expression of Nrf2 (Petrillo et al., 2017), axonal growth is improved. In any case, these alterations affect the cellular viability of both neurons and glia, triggering cell death processes either by apoptosis (Mincheva-Tasheva et al., 2014; Igoillo-Esteve et al., 2015; Katsu-Jiménez et al., 2016) or autophagy (Bolinches-Amorós et al., 2014; Edenharter et al., 2018). It has been shown that iron is a key element in the induction of cell death (Dixon et al., 2012). This new process, called ferroptosis, is dependent on iron and is accompanied by an increase in ROS and lipid peroxidation and a decrease in GSH. As we have reviewed previously, these characteristics are observed in frataxin-deficient cells; therefore, new studies are needed to determine the possible cell death due to a ferroptosis process.
The extent to which oxidative stress is necessary for the pathogenesis of FRDA is, however, controversial. The deficit in ISC enzymes and increase in iron levels were not paired with an increase in ROS in the MCK-KO mouse (Seznec et al., 2005), in FxnAlb mice with specific depletion of Fxn in liver (Martelli et al., 2015), and in the inducible and reversible FRDAkd mouse model (Chandran et al., 2017). Similarly, increased oxidative stress was not detected in several Drosophila knockdown models, and overexpression of ROS scavengers did not rescue the viability, heart dysfunction or neurodegeneration phenotypes (Anderson et al., 2005; Shidara and Hollenbeck, 2010; Tricoire et al., 2014; Chen et al., 2016b). Moreover, FRDA neural tissue did not show differences in the susceptibility to oxidative stress in a study using patient samples (Macevilly and Muller, 1997). The absence of ROS markers in these studies might be attributed to differences in methodology or the distinctive features of each of the animal models or tissues employed. Alternatively, it was suggested that the discrepancies in the presence of ROS would be explained by differences in the timepoints studied, in a scenario where oxidative stress would be a secondary effect rather than a primary cause, occurring later in the progression of FRDA (discussed in Lupoli et al., 2018). Recent findings in the fly and mouse models of FRDA actually support a mechanism that is independent of ROS that links neurodegeneration and iron accumulation via the sphingolipid pathway (Chen et al., 2016a,b).
Alterations in the homeostasis of lipids are not exclusive of FRDA (Huang et al., 1978; Filla et al., 1980; Thurman and Draper, 1989; Raman S.V. et al., 2011; Worth et al., 2014) and have been reported in neurodegenerative disorders, including Parkinson’s, Alzheimer’s, Huntington’s, multiple sclerosis and Niemann–Pick diseases (reviewed in Shamim et al., 2018). In the case of FRDA, lipid droplet accumulation was first described in the cardiac muscle of the NSE-KO mouse model (Puccio et al., 2001) and was further characterized as pathological in a glial frataxin knockdown model in Drosophila (Navarro et al., 2010). The accumulation and dyshomeostasis of lipids have been reported as well in several other cellular and animal models of the disease (Martelli et al., 2012; Schiavi et al., 2013; Obis et al., 2014). More recently, the expression of a mutant allele of the Drosophila fh in mosaic clones produced an age-dependent degeneration of photoreceptor neurons with impaired mitochondrial function, increased levels of Fe2+ and Fe3+ in multiple tissues and dramatic lipid droplet accumulation in glial cells (Chen et al., 2016b), consistent with the work from Navarro et al. (2010). Because the Fenton reaction can induce lipid peroxidation, oxidative stress has been classically proposed to be the link between abnormal lipid homeostasis and neurodegeneration in FRDA. However, Chen et al. did not observe an increase in free radical production and, neither providing the antioxidant AD4 nor overexpressing the ROS scavengers catalase, SOD1 and SOD2, suppressed neurodegeneration (Chen et al., 2016b). Moreover, reducing the number of lipid droplets by overexpressing brummer lipase or lipase 4 was insufficient to improve the neurodegeneration defect in the knockout photoreceptors. Because high iron conditions increase sphingolipid synthesis in wild-type yeast (Lee et al., 2012), Chen et al. (2016b) pursued the characterization of this pathway in their fly model. The synthesis of sphingolipids, including dihydrosphingosine, dihydroceramide and sphingosine, was actually increased in the fh mutants. Both the genetic knockdown and chemical inhibition of the fly serine palmitoyltransferase, an enzyme essential for the de novo synthesis of sphingolipids, rescued neurodegeneration. Additionally, they found that downstream targets of the transcription factor Mef2 were upregulated in response to the increase in sphingolipids in fh mutants and knocking down the Pdk1/Mef2 pathway led to the suppression of photoreceptor degeneration. To ask whether this novel pathway described in Drosophila was conserved in vertebrates, Chen et al. (2016a) developed a mouse model with reduced Fxn levels in the NS. Both ferrous and ferric iron were found to be increased in the cerebral cortex and, consistent with their observation in the fly model, they could not find increased ROS production measured as levels of lipid peroxidation. Loss of Fxn in the NS led to the activation of the PDK1/Mef2 pathway in the mouse before the appearance of the neurological manifestations. Sphingolipids and PDK1 activation were also found to be increased in cardiac tissue from FRDA patients (Chen et al., 2016a). Nevertheless, alterations in the PDK1/Mef2 pathway were not replicated in the FRDAkd-inducible mouse model of FRDA (Chandran et al., 2017). In this study, the authors did not find changes in PDK phosphorylation in the brain, muscle, heart or liver in the chronic adult knockdown mice. The levels in the cerebellum and heart of five target genes of Mef2 were analyzed as well and displayed changes that were inconsistent across tissues. Alternatively, other biochemical pathways could be altered in relation to iron under frataxin-deficient conditions. In line, several genes involved in hemochromatosis, an iron-overload disorder, are upregulated in the FRDAkd-inducible mouse model, in which no increase in ROS and no strong evidence of Pdk1/Mef2 activation were observed (Chandran et al., 2017).
An optimal therapy in FRDA could consist of providing patients with sufficient frataxin amount to restore the functions of this protein. Several strategies are being developed to obtain this objective, such as gene therapy (Perdomini et al., 2014; Piguet et al., 2018), transplanting hematopoietic stem and progenitor cells (Rocca et al., 2017) or improving FXN transcription (Soragni and Gottesfeld, 2016). While advancing the development of such strategies for its application to patients, rational treatment could be considered to remove excess iron.
The utilization of iron chelators is one of the first strategies proposed to treat FRDA patients to eliminate excess iron accumulation in the mitochondria. To that end, an iron chelator should be able to cross the cell membranes, access mitochondria and redistribute the excess labile iron. Deferiprone (DFP, 1, 2 dimethyl-3-hydroxy-pyrid-4-one), an orally administered lipid soluble iron chelator can easily cross the blood–brain barrier and cellular membranes. DFP shows relatively low affinity for iron and can transfer this metal to circulating transferrin, as well as to cellular acceptors—e.g., for heme synthesis (Sohn et al., 2008). Therefore, its use is less probable to produce iron depletion than other known chelators. For all these reasons, DFP has been considered as the prototypic compound for iron redeployment (Cabantchik et al., 2013). It is also used to treat iron-overloaded patients with β-thalassemia who need continuous blood transfusions (Totadri et al., 2015), and their properties make it an interesting therapeutic compound for FRDA.
The use of DFP in cellular and animal models of FRDA has beneficial results, e.g., frataxin-deficient HEK-293 cells could restore mitochondrial dysfunction after DFP treatment. It was attributed to the ability of this compound to make available the iron accumulated into mitochondria (Kakhlon et al., 2008). These conclusions were also supported by our group using a Drosophila FRDA model, in which DFP improved the survival and climbing ability phenotypes because of the removal of excess free mitochondrial iron, thereby preventing its toxic effects (Soriano et al., 2013). Recently, it has been reported that DFP is effective to reduce ROS production and improve calcium handling kinetics in an FRDA human-induced pluripotent stem cell-derived cardiomyocyte model (Lee et al., 2016).
The efficacy of DFP has already been assessed in clinical trials in FRDA patients, but the results are inconclusive. However, considering these studies, DFP has shown beneficial effects on cardiac parameters achieved at low doses of the compound, while treatment with higher doses of DFP could aggravate the disorder by affecting the ataxia phenotype (Pandolfo et al., 2014). A high dose of DFP could remove too much iron, provoking a deficiency of this metal, which could affect the cellular functions that require iron. On the other hand, the positive action of this drug on the heart compared with NS has also been related to the initiation of treatment before cardiac dysfunction is present (Elincx-Benizri et al., 2016). Overall, DFP seems to have therapeutic value in FRDA and is quite safe at lower doses, but systematic, large scale, placebo controlled longer studies are needed to evaluate its usefulness (Strawser et al., 2017). In line with these issues, our results using a Drosophila model of the disease indicate that DFP treatments at the larval stage recover FRDA-like phenotypes in flies, but no improvement was observed when the drug is administered to adults (Soriano et al., 2013). Additionally, we found that reducing the excess of iron accumulating into mitochondria through a genetic strategy has benefit in this FRDA model (Navarro et al., 2015; Soriano et al., 2016). These results show a causal relationship between iron and frataxin deficiency. Thus, iron is a critical factor in the pathophysiology of FRDA and appears not be a simple epiphenomenal event in the disease. Consequently, research on how to deal with the iron excess and eradicate the iron toxicity is still an area of great interest.
In the last decades, some small molecules with pharmacophore characteristics for iron chelation are being explored for treatment in neurodegenerative diseases with regional increase of iron levels (reviewed in Singh et al., 2018). These compounds are derivatives of the 8-hydroxyquinoline, hydroxypyridone, and arylhydrazones, and some of them are able to reduce selectively the iron excess in the mitochondria (Mena et al., 2015). The use of pro-chelating compounds, which are activated in the target region, have the benefit to remove the excess of iron in the affected regions. Thus, this strategy would protect cells in non-affected areas from the risk of iron subtraction. Novel pro-chelators triggered by H2O2 were obtained as promising candidates for management of Parkinson’s disease (Perez et al., 2008). Similarly, this approach might be useful in FRDA since H2O2 is a critical pathogenic agent associated to frataxin deficiency (Anderson et al., 2008). Finally, new compounds with multifunctional actions, as previously indicated for DFP, are also being examined for effective treatment of neurodegenerative diseases (Singh et al., 2018). Several of these diseases, including FRDA, share some pathological features such as regional iron increase, oxidative stress and dysregulation of calcium signaling. Recently, a novel compound, named CT51 (N-(1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl)-2-(7-hydroxy-2-oxo-2H-chromen-4 yl) acetamide), has been synthesized that seems to suppress these cellular impairments (García-Beltrán et al., 2017). CT51 can easily permeate the cell membrane and distribute to both mitochondria and the cytoplasm. It works as a highly selective iron chelator and possesses significant capability for free radical quenching, protecting cells against oxidative damage. The use of CT51 in primary hippocampal neurons decreases the continuous release of calcium provoked by an agonist of ryanodine receptor-calcium channels, supporting a protective role of this compound in neuronal function. Although developments of this kind of drugs are not a curative strategy, because the frataxin deficiency will persist, they might improve the current pharmacological treatments and might produce increases in quality of life and slowing of disease progression.
The relationship between neurodegeneration and iron accumulation is well established. However, in Friedreich’s ataxia, it remains unclear whether iron loading is the cause or consequence of the pathophysiology. The studies in model organisms suggested both hypotheses in relation to the role of iron in this disease: a key role in the pathophysiologic mechanisms or a secondary event that appeared late in the disease progression. It was also debated the extent to which oxidative stress is crucial to explain the pathogenesis of FRDA. Indeed, additional pathways might be involved in the toxicity of iron independent of ROS production. Differences in the distinctive features of each of the FRDA models studied, vulnerability of the different tissues and cells to frataxin deficit and timepoints analyzed could partially explain the contradictory results. Nevertheless, from the therapeutic point of view, it is interesting to explore whether new iron chelators showing protective properties on cellular function could improve the mitochondrial dysfunction in FRDA.
JL and MM planned the manuscript. JL, SS, PC-Q, PG-C, and MM wrote the manuscript and agreed on the finally submitted version of the manuscript.
Financial support of this work came from Generalitat Valenciana, Spain (PROMETEOII/2014/067) to MM, the Spanish Ministry of Economy and Competitiveness [SAF2015-66625-R] within the framework of the National R+D+I Plan to PG-C, Fundación Ramón Areces, Spain (CIVP18A3899) to PG-C and MM. PC-Q was a recipient of a fellowship from Generalitat Valenciana, Spain, and JL is supported by a research contract from FARA and FARA Ireland.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2019.00075/full#supplementary-material
Acquaviva, F., De Biase, I., Nezi, L., Ruggiero, G., Tatangelo, F., Pisano, C., et al. (2005). Extra-mitochondrial localisation of frataxin and its association with IscU1 during enterocyte-like differentiation of the human colon adenocarcinoma cell line Caco-2. J. Cell Sci. 118, 3917–3924. doi: 10.1242/jcs.02516
Adamec, J., Rusnak, F., Owen, W. G., Naylor, S., Benson, L. M., Gacy, A. M., et al. (2000). Iron-dependent self-assembly of recombinant yeast frataxin: implications for Friedreich ataxia. Am. J. Hum. Genet. 67, 549–562. doi: 10.1086/303056
Adinolfi, S., Iannuzzi, C., Prischi, F., Pastore, C., Iametti, S., Martin, S. R., et al. (2009). Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 16, 390–396. doi: 10.1038/nsmb.1579
Adinolfi, S., Trifuoggi, M., Politou, A. S., Martin, S., and Pastore, A. (2002). A structural approach to understanding the iron-binding properties of phylogenetically different frataxins. Hum. Mol. Genet. 11, 1865–1877. doi: 10.1093/hmg/11.16.1865
Ahlgren, E.-C., Fekry, M., Wiemann, M., Söderberg, C. A., Bernfur, K., Gakh, O., et al. (2017). Iron-induced oligomerization of human FXN81-210 and bacterial CyaY frataxin and the effect of iron chelators. PLoS One 12:e0188937. doi: 10.1371/journal.pone.0188937
Aloria, K., Schilke, B., Andrew, A., and Craig, E. A. (2004). Iron-induced oligomerization of yeast frataxin homologue Yfh1 is dispensable in vivo. EMBO Rep. 5, 1096–1101. doi: 10.1038/sj.embor.7400272
Alsina, D., Purroy, R., Ros, J., and Tamarit, J. (2018). Iron in Friedreich ataxia: a central role in the pathophysiology or an epiphenomenon? Pharmaceuticals 11:E89. doi: 10.3390/ph11030089
Anderson, P. R., Kirby, K., Hilliker, A. J., and Phillips, J. P. (2005). RNAi-mediated suppression of the mitochondrial iron chaperone, frataxin, in Drosophila. Hum. Mol. Genet. 14, 3397–3405. doi: 10.1093/hmg/ddi367
Anderson, P. R., Kirby, K., Orr, W. C., Hilliker, A. J., and Phillips, J. P. (2008). Hydrogen peroxide scavenging rescues frataxin deficiency in a Drosophila model of Friedreich’s ataxia. Proc. Natl. Acad. Sci. U.S.A. 105, 611–616. doi: 10.1073/pnas.0709691105
Babcock, M., de Silva, D., Oaks, R., Davis-Kaplan, S., Jiralerspong, S., Montermini, L., et al. (1997). Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 276, 1709–1712. doi: 10.1126/science.276.5319.1709
Becker, E. M., Greer, J. M., Ponka, P., and Richardson, D. R. (2002). Erythroid differentiation and protoporphyrin IX down-regulate frataxin expression in Friend cells: characterization of frataxin expression compared to molecules involved in iron metabolism and hemoglobinization. Blood 99, 3813–3822. doi: 10.1182/blood.V99.10.3813
Bedekovics, T., Gajdos, G. B., Kispal, G., and Isaya, G. (2007). Partial conservation of functions between eukaryotic frataxin and the Escherichia coli frataxin homolog CyaY. FEMS Yeast Res. 7, 1276–1284. doi: 10.1111/j.1567-1364.2007.00296.x
Beilschmidt, L. K., and Puccio, H. M. (2014). Mammalian Fe-S cluster biogenesis and its implication in disease. Biochimie 100, 48–60. doi: 10.1016/j.biochi.2014.01.009
Bencze, K. Z., Kondapalli, K. C., Cook, J. D., McMahon, S., Millán-Pacheco, C., Pastor, N., et al. (2006). The structure and function of frataxin. Crit. Rev. Biochem. Mol. Biol. 41, 269–291. doi: 10.1080/10409230600846058
Bidichandani, S. I., and Delatycki, M. B. (2017). Friedreich Ataxia. Available at: http://www.ncbi.nlm.nih.gov/pubmed/20301458
Bolinches-Amorós, A., Mollá, B., Pla-Martín, D., Palau, F., and González-Cabo, P. (2014). Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front. Cell. Neurosci. 8:124. doi: 10.3389/fncel.2014.00124
Bonilha da Silva, C., Bergo, F. P. G., D’Abreu, A., Cendes, F., Lopes-Cendes, I., and França, M. C. (2014). Dentate nuclei T2 relaxometry is a reliable neuroimaging marker in Friedreich’s ataxia. Eur. J. Neurol. 21, 1131–1136. doi: 10.1111/ene.12448
Bou-Abdallah, F., Adinolfi, S., Pastore, A., Laue, T. M., and Dennis Chasteen, N. (2004). Iron binding and oxidation kinetics in frataxin CyaY of Escherichia coli. J. Mol. Biol. 341, 605–615. doi: 10.1016/j.jmb.2004.05.072
Bradley, J. L., Blake, J. C., Chamberlain, S., Thomas, P. K., Cooper, J. M., and Schapira, A. H. (2000). Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet. 9, 275–282. doi: 10.1093/hmg/9.2.275
Bradshaw, T. Y., Romano, L. E. L., Duncan, E. J., Nethisinghe, S., Abeti, R., Michael, G. J., et al. (2016). A reduction in Drp1-mediated fission compromises mitochondrial health in autosomal recessive spastic ataxia of Charlevoix Saguenay. Hum. Mol. Genet. 25, 3232–3244. doi: 10.1093/hmg/ddw173
Bridwell-Rabb, J., Iannuzzi, C., Pastore, A., and Barondeau, D. P. (2012). Effector role reversal during evolution: the case of frataxin in Fe-S cluster biosynthesis. Biochemistry 51, 2506–2514. doi: 10.1021/bi201628j
Bridwell-Rabb, J., Winn, A. M., and Barondeau, D. P. (2011). Structure-function analysis of Friedreich’s ataxia mutants reveals determinants of frataxin binding and activation of the Fe-S assembly complex. Biochemistry 50, 7265–7274. doi: 10.1021/bi200895k
Bulteau, A.-L., O’Neill, H. A., Kennedy, M. C., Ikeda-Saito, M., Isaya, G., and Szweda, L. I. (2004). Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 305, 242–245. doi: 10.1126/science.1098991
Cabantchik, Z. I., Munnich, A., Youdim, M. B., and Devos, D. (2013). Regional siderosis: a new challenge for iron chelation therapy. Front. Pharmacol. 4:167. doi: 10.3389/fphar.2013.00167
Cai, K., Frederick, R. O., Tonelli, M., and Markley, J. L. (2018). Interactions of iron-bound frataxin with ISCU and ferredoxin on the cysteine desulfurase complex leading to Fe-S cluster assembly. J. Inorg. Biochem. 183, 107–116. doi: 10.1016/j.jinorgbio.2018.03.007
Calabrese, V., Lodi, R., Tonon, C., D’Agata, V., Sapienza, M., Scapagnini, G., et al. (2005). Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J. Neurol. Sci. 233, 145–162. doi: 10.1016/j.jns.2005.03.012
Calap-Quintana, P., Navarro, J. A., González-Fernández, J., Martínez-Sebastián, M. J., Moltó, M. D., and Llorens, J. V. (2018). Drosophila melanogaster models of Friedreich’s ataxia. Biomed. Res. Int. 2018:5065190. doi: 10.1155/2018/5065190
Campanella, A., Rovelli, E., Santambrogio, P., Cozzi, A., Taroni, F., and Levi, S. (2009). Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: hypothesis for a protective role in Friedreich ataxia. Hum. Mol. Genet. 18, 1–11. doi: 10.1093/hmg/ddn308
Campuzano, V., Montermini, L., Lutz, Y., Cova, L., Hindelang, C., Jiralerspong, S., et al. (1997). Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 6, 1771–1780. doi: 10.1093/hmg/6.11.1771
Campuzano, V., Montermini, L., Moltò, M. D., Pianese, L., Cossée, M., Cavalcanti, F., et al. (1996). Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427. doi: 10.1126/science.271.5254.1423
Carletti, B., Piermarini, E., Tozzi, G., Travaglini, L., Torraco, A., Pastore, A., et al. (2014). Frataxin silencing inactivates mitochondrial Complex I in NSC34 motoneuronal cells and alters glutathione homeostasis. Int. J. Mol. Sci. 15, 5789–5806. doi: 10.3390/ijms15045789
Cavadini, P., Gellera, C., Patel, P. I., and Isaya, G. (2000). Human frataxin maintains mitochondrial iron homeostasis in Saccharomyces cerevisiae. Hum. Mol. Genet. 9, 2523–2530. doi: 10.1093/hmg/9.17.2523
Chandran, V., Gao, K., Swarup, V., Versano, R., Dong, H., Jordan, M. C., et al. (2017). Inducible and reversible phenotypes in a novel mouse model of Friedreich’s Ataxia. eLife 6:e30054. doi: 10.7554/eLife.30054
Chantrel-Groussard, K., Geromel, V., Puccio, H., Koenig, M., Munnich, A., Rötig, A., et al. (2001). Disabled early recruitment of antioxidant defenses in Friedreich’s ataxia. Hum. Mol. Genet. 10, 2061–2067. doi: 10.1093/hmg/10.19.2061
Chen, K., Ho, T. S.-Y., Lin, G., Tan, K. L., Rasband, M. N., and Bellen, H. J. (2016a). Loss of Frataxin activates the iron/sphingolipid/PDK1/Mef2 pathway in mammals. eLife 5:e20732. doi: 10.7554/eLife.20732
Chen, K., Lin, G., Haelterman, N. A., Ho, T. S.-Y., Li, T., Li, Z., et al. (2016b). Loss of Frataxin induces iron toxicity, sphingolipid synthesis, and Pdk1/Mef2 activation, leading to neurodegeneration. eLife 5:e16043. doi: 10.7554/eLife.16043
Chiabrando, D., Mercurio, S., and Tolosano, E. (2014). Heme and erythropoieis: more than a structural role. Haematologica 99, 973–983. doi: 10.3324/haematol.2013.091991
Chiang, S., Kovacevic, Z., Sahni, S., Lane, D. J. R., Merlot, A. M., Kalinowski, D. S., et al. (2016). Frataxin and the molecular mechanism of mitochondrial iron-loading in Friedreich’s ataxia. Clin. Sci. 130, 853–870. doi: 10.1042/CS20160072
Chung, J., Chen, C., and Paw, B. H. (2012). Heme metabolism and erythropoiesis. Curr. Opin. Hematol. 19, 156–162. doi: 10.1097/MOH.0b013e328351c48b
Colin, F., Martelli, A., Clémancey, M., Latour, J.-M., Gambarelli, S., Zeppieri, L., et al. (2013). Mammalian frataxin controls sulfur production and iron entry during de novo Fe4S4 cluster assembly. J. Am. Chem. Soc. 135, 733–740. doi: 10.1021/ja308736e
Condò, I., Malisan, F., Guccini, I., Serio, D., Rufini, A., and Testi, R. (2010). Molecular control of the cytosolic aconitase/IRP1 switch by extramitochondrial frataxin. Hum. Mol. Genet. 19, 1221–1229. doi: 10.1093/hmg/ddp592
Condò, I., Ventura, N., Malisan, F., Rufini, A., Tomassini, B., and Testi, R. (2007). In vivo maturation of human frataxin. Hum. Mol. Genet. 16, 1534–1540. doi: 10.1093/hmg/ddm102
Condò, I., Ventura, N., Malisan, F., Tomassini, B., and Testi, R. (2006). A pool of extramitochondrial frataxin that promotes cell survival. J. Biol. Chem. 281, 16750–16756. doi: 10.1074/jbc.M511960200
Connor, J. R., Snyder, B. S., Beard, J. L., Fine, R. E., and Mufson, E. J. (1992). Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. J. Neurosci. Res. 31, 327–335. doi: 10.1002/jnr.490310214
Cook, J. D., Bencze, K. Z., Jankovic, A. D., Crater, A. K., Busch, C. N., Bradley, P. B., et al. (2006). Monomeric yeast frataxin is an iron-binding protein. Biochemistry 45, 7767–7777. doi: 10.1021/bi060424r
Cossée, M., Puccio, H., Gansmuller, A., Koutnikova, H., Dierich, A., LeMeur, M., et al. (2000). Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum. Mol. Genet. 9, 1219–1226. doi: 10.1093/hmg/9.8.1219
Dailey, H. A., and Meissner, P. N. (2013). Erythroid heme biosynthesis and its disorders. Cold Spring Harb. Perspect. Med. 3:a011676. doi: 10.1101/cshperspect.a011676
De Castro, M., García-Planells, J., Monrós, E., Cañizares, J., Vázquez-Manrique, R., Vílchez, J. J., et al. (2000). Genotype and phenotype analysis of Friedreich’s ataxia compound heterozygous patients. Hum. Genet. 106, 86–92. doi: 10.1007/s004399900201
Delatycki, M. B., Camakaris, J., Brooks, H., Evans-Whipp, T., Thorburn, D. R., Williamson, R., et al. (1999). Direct evidence that mitochondrial iron accumulation occurs in Friedreich ataxia. Ann. Neurol. 45, 673–675. doi: 10.1002/1531-8249(199905)45:5<673::AID-ANA20>3.0.CO;2-Q
Dhe-Paganon, S., Shigeta, R., Chi, Y. I., Ristow, M., and Shoelson, S. E. (2000). Crystal structure of human frataxin. J. Biol. Chem. 275, 30753–30756. doi: 10.1074/jbc.C000407200
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
D’Oria, V., Petrini, S., Travaglini, L., Priori, C., Piermarini, E., Petrillo, S., et al. (2013). Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci. 14, 7853–7865. doi: 10.3390/ijms14047853
Dumas, E. M., Versluis, M. J., van den Bogaard, S. J. A., van Osch, M. J. P., Hart, E. P., van Roon-Mom, W. M. C., et al. (2012). Elevated brain iron is independent from atrophy in Huntington’s Disease. Neuroimage 61, 558–564. doi: 10.1016/j.neuroimage.2012.03.056
Eaton, J. W., and Qian, M. (2002). Molecular bases of cellular iron toxicity. Free Radic. Biol. Med. 32, 833–840. doi: 10.1016/S0891-5849(02)00772-4
Edenharter, O., Schneuwly, S., and Navarro, J. A. (2018). Mitofusin-dependent ER stress triggers glial dysfunction and nervous system degeneration in a drosophila model of Friedreich’s ataxia. Front. Mol. Neurosci. 11:38. doi: 10.3389/fnmol.2018.00038
Elincx-Benizri, S., Glik, A., Merkel, D., Arad, M., Freimark, D., Kozlova, E., et al. (2016). Clinical experience with deferiprone treatment for friedreich ataxia. J. Child Neurol. 31, 1036–1040. doi: 10.1177/0883073816636087
Emond, M., Lepage, G., Vanasse, M., and Pandolfo, M. (2000). Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 55, 1752–1753. doi: 10.1212/WNL.55.11.1752
Filla, A., Postiglione, A., Rubba, P., Patti, L., De Michele, G., Palma, V., et al. (1980). Plasma lipoprotein concentration and erythrocyte membrane lipids in patients with Friedreich’s ataxia. Acta Neurol. 2, 382–389.
Fleming, M. D. (2011). Congenital sideroblastic anemias: iron and heme lost in mitochondrial translation. Hematol. Am. Soc. Hematol. Educ. Progr. 2011, 525–531. doi: 10.1182/asheducation-2011.1.525
Foury, F., and Cazzalini, O. (1997). Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett. 411, 373–377. doi: 10.1016/S0014-5793(97)00734-5
Foury, F., Pastore, A., and Trincal, M. (2007). Acidic residues of yeast frataxin have an essential role in Fe-S cluster assembly. EMBO Rep. 8, 194–199. doi: 10.1038/sj.embor.7400881
Fox, N. G., Das, D., Chakrabarti, M., Lindahl, P. A., and Barondeau, D. P. (2015). Frataxin accelerates [2Fe-2S] cluster formation on the human Fe-S assembly complex. Biochemistry 54, 3880–3889. doi: 10.1021/bi5014497
Furuyama, K., Kaneko, K., and Vargas, P. D. (2007). Heme as a magnificent molecule with multiple missions: heme determines its own fate and governs cellular homeostasis. Tohoku J. Exp. Med. 213, 1–16. doi: 10.1620/tjem.213.1
Gakh, O., Adamec, J., Gacy, A. M., Twesten, R. D., Owen, W. G., and Isaya, G. (2002). Physical evidence that yeast frataxin is an iron storage protein. Biochemistry 41, 6798–6804. doi: 10.1021/bi025566+
Gakh, O., Park, S., Liu, G., Macomber, L., Imlay, J. A., Ferreira, G. C., et al. (2006). Mitochondrial iron detoxification is a primary function of frataxin that limits oxidative damage and preserves cell longevity. Hum. Mol. Genet. 15, 467–479. doi: 10.1093/hmg/ddi461
Galea, C. A., Huq, A., Lockhart, P. J., Tai, G., Corben, L. A., Yiu, E. M., et al. (2016). Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann. Neurol. 79, 485–495. doi: 10.1002/ana.24595
García-Beltrán, O., Mena, N. P., Aguirre, P., Barriga-González, G., Galdámez, A., Nagles, E., et al. (2017). Development of an iron-selective antioxidant probe with protective effects on neuronal function. PLoS One 12:e0189043. doi: 10.1371/journal.pone.0189043
Gentry, L. E., Thacker, M. A., Doughty, R., Timkovich, R., and Busenlehner, L. S. (2013). His86 from the N-terminus of frataxin coordinates iron and is required for Fe-S cluster synthesis. Biochemistry 52, 6085–6096. doi: 10.1021/bi400443n
Gerber, J., Mühlenhoff, U., and Lill, R. (2003). An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 4, 906–911. doi: 10.1038/sj.embor.embor918
González-Cabo, P., and Palau, F. (2013). Mitochondrial pathophysiology in Friedreich’s ataxia. J. Neurochem. 126(Suppl.), 53–64. doi: 10.1111/jnc.12303
Gorell, J. M., Ordidge, R. J., Brown, G. G., Deniau, J. C., Buderer, N. M., and Helpern, J. A. (1995). Increased iron-related MRI contrast in the Substantia nigra in Parkinson’s disease. Neurology 45, 1138–1143. doi: 10.1212/WNL.45.6.1138
Guo, L., Wang, Q., Weng, L., Hauser, L. A., Strawser, C. J., Mesaros, C., et al. (2018). Characterization of a new N-terminally acetylated extra-mitochondrial isoform of frataxin in human erythrocytes. Sci. Rep. 8:17043. doi: 10.1038/s41598-018-35346-y
Harding, A. E. (1981). Early onset cerebellar ataxia with retained tendon reflexes: a clinical and genetic study of a disorder distinct from Friedreich’s ataxia. J. Neurol. Neurosurg. Psychiatry 44, 503–508. doi: 10.1136/jnnp.44.6.503
Harding, I. H., Raniga, P., Delatycki, M. B., Stagnitti, M. R., Corben, L. A., Storey, E., et al. (2016). Tissue atrophy and elevated iron concentration in the extrapyramidal motor system in Friedreich ataxia: the IMAGE-FRDA study. J. Neurol. Neurosurg. Psychiatry 87, 1261–1263. doi: 10.1136/jnnp-2015-312665
Huang, J., Dizin, E., and Cowan, J. A. (2008). Mapping iron binding sites on human frataxin: implications for cluster assembly on the ISU Fe-S cluster scaffold protein. J. Biol. Inorg. Chem. 13, 825–836. doi: 10.1007/s00775-008-0369-4
Huang, M. L.-H., Becker, E. M., Whitnall, M., Suryo Rahmanto, Y., Ponka, P., and Richardson, D. R. (2009). Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc. Natl. Acad. Sci. U.S.A. 106, 16381–16386. doi: 10.1073/pnas.0906784106
Huang, M. L.-H., Lane, D. J. R., and Richardson, D. R. (2011). Mitochondrial mayhem: the mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid. Redox Signal. 15, 3003–3019. doi: 10.1089/ars.2011.3921
Huang, M. L.-H., Sivagurunathan, S., Ting, S., Jansson, P. J., Austin, C. J. D., Kelly, M., et al. (2013). Molecular and functional alterations in a mouse cardiac model of Friedreich ataxia: activation of the integrated stress response, eIF2α phosphorylation, and the induction of downstream targets. Am. J. Pathol. 183, 745–757. doi: 10.1016/j.ajpath.2013.05.032
Huang, Y. S., Nestruck, A. C., Barbeau, A., Bouchard, J. P., and Davignon, J. (1978). Plasma lipids and lipoproteins in Friedreich’s ataxia and familial spastic ataxia–evidence for an abnormal composition of high density lipoproteins. Can. J. Neurol. Sci. 5, 149–156.
Igoillo-Esteve, M., Gurgul-Convey, E., Hu, A., Romagueira Bichara Dos Santos, L., Abdulkarim, B., Chintawar, S., et al. (2015). Unveiling a common mechanism of apoptosis in β-cells and neurons in Friedreich’s ataxia. Hum. Mol. Genet. 24, 2274–2286. doi: 10.1093/hmg/ddu745
Kakhlon, O., and Cabantchik, Z. I. (2002). The labile iron pool: characterization, measurement, and participation in cellular processes(1). Free Radic. Biol. Med. 33, 1037–1046. doi: 10.1016/S0891-5849(02)01006-7
Kakhlon, O., Manning, H., Breuer, W., Melamed-Book, N., Lu, C., Cortopassi, G., et al. (2008). Cell functions impaired by frataxin deficiency are restored by drug-mediated iron relocation. Blood 112, 5219–5227. doi: 10.1182/blood-2008-06-161919
Katsu-Jiménez, Y., Loría, F., Corona, J. C., and Díaz-Nido, J. (2016). Gene transfer of brain-derived neurotrophic factor (BDNF) prevents neurodegeneration triggered by FXN deficiency. Mol. Ther. 24, 877–889. doi: 10.1038/mt.2016.32
Kipps, A., Alexander, M., Colan, S. D., Gauvreau, K., Smoot, L., Crawford, L., et al. (2009). The longitudinal course of cardiomyopathy in Friedreich’s ataxia during childhood. Pediatr. Cardiol. 30, 306–310. doi: 10.1007/s00246-008-9305-1
Knight, S. A., Sepuri, N. B., Pain, D., and Dancis, A. (1998). Mt-Hsp70 homolog, Ssc2p, required for maturation of yeast frataxin and mitochondrial iron homeostasis. J. Biol. Chem. 273, 18389–18393. doi: 10.1074/jbc.273.29.18389
Koeppen, A. H. (2013). Nikolaus Friedreich and degenerative atrophy of the dorsal columns of the spinal cord. J. Neurochem. 126, 4–10. doi: 10.1111/jnc.12218
Koeppen, A. H., Kuntzsch, E. C., Bjork, S. T., Ramirez, R. L., Mazurkiewicz, J. E., and Feustel, P. J. (2013). Friedreich ataxia: metal dysmetabolism in dorsal root ganglia. Acta Neuropathol. Commun. 1:26. doi: 10.1186/2051-5960-1-26
Koeppen, A. H., and Mazurkiewicz, J. E. (2013). Friedreich ataxia: neuropathology revised. J. Neuropathol. Exp. Neurol. 72, 78–90. doi: 10.1097/NEN.0b013e31827e5762
Koeppen, A. H., Michael, S. C., Knutson, M. D., Haile, D. J., Qian, J., Levi, S., et al. (2007). The dentate nucleus in Friedreich’s ataxia: the role of iron-responsive proteins. Acta Neuropathol. 114, 163–173. doi: 10.1007/s00401-007-0220-y
Koeppen, A. H., Morral, J. A., Davis, A. N., Qian, J., Petrocine, S. V., Knutson, M. D., et al. (2009). The dorsal root ganglion in Friedreich’s ataxia. Acta Neuropathol. 118, 763–776. doi: 10.1007/s00401-009-0589-x
Koeppen, A. H., Ramirez, R. L., Becker, A. B., Bjork, S. T., Levi, S., Santambrogio, P., et al. (2015). The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS One 10:e0116396. doi: 10.1371/journal.pone.0116396
Koeppen, A. H., Ramirez, R. L., Becker, A. B., and Mazurkiewicz, J. E. (2016). Dorsal root ganglia in Friedreich ataxia: satellite cell proliferation and inflammation. Acta Neuropathol. Commun. 4:46. doi: 10.1186/s40478-016-0288-5
Koeppen, A. H., Ramirez, R. L., Yu, D., Collins, S. E., Qian, J., Parsons, P. J., et al. (2012). Friedreich’s ataxia causes redistribution of iron, copper, and zinc in the dentate nucleus. Cerebellum 11, 845–860. doi: 10.1007/s12311-012-0383-5
Kruger, P. C., Yang, K. X., Parsons, P. J., Becker, A. B., Feustel, P. J., and Koeppen, A. H. (2016). Abundance and significance of iron, zinc, copper, and calcium in the hearts of patients with friedreich ataxia. Am. J. Cardiol. 118, 127–131. doi: 10.1016/j.amjcard.2016.04.024
Kumari, D., and Usdin, K. (2012). Is Friedreich ataxia an epigenetic disorder? Clin. Epigenetics 4:2. doi: 10.1186/1868-7083-4-2
Kwan, J. Y., Meoded, A., Danielian, L. E., Wu, T., and Floeter, M. K. (2012). Structural imaging differences and longitudinal changes in primary lateral sclerosis and amyotrophic lateral sclerosis. Neuroimage Clin. 2, 151–160. doi: 10.1016/j.nicl.2012.12.003
Lamarche, J. B., Côté, M., and Lemieux, B. (1980). The cardiomyopathy of Friedreich’s ataxia morphological observations in 3 cases. Can. J. Neurol. Sci. 7, 389–396. doi: 10.1017/S0317167100022927
Lane, D. J. R., Merlot, A. M., Huang, M. L.-H., Bae, D.-H., Jansson, P. J., Sahni, S., et al. (2015). Cellular iron uptake, trafficking and metabolism: key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta 1853, 1130–1144. doi: 10.1016/j.bbamcr.2015.01.021
Layer, G., Ollagnier-de Choudens, S., Sanakis, Y., and Fontecave, M. (2006). Iron-sulfur cluster biosynthesis: characterization of Escherichia coli CYaY as an iron donor for the assembly of [2Fe-2S] clusters in the scaffold IscU. J. Biol. Chem. 281, 16256–16263. doi: 10.1074/jbc.M513569200
Lee, Y.-J., Huang, X., Kropat, J., Henras, A., Merchant, S. S., Dickson, R. C., et al. (2012). Sphingolipid signaling mediates iron toxicity. Cell Metab. 16, 90–96. doi: 10.1016/j.cmet.2012.06.004
Lee, Y.-K., Lau, Y.-M., Ng, K.-M., Lai, W.-H., Ho, S.-L., Tse, H.-F., et al. (2016). Efficient attenuation of Friedreich’s ataxia (FRDA) cardiomyopathy by modulation of iron homeostasis-human induced pluripotent stem cell (hiPSC) as a drug screening platform for FRDA. Int. J. Cardiol. 203, 964–971. doi: 10.1016/j.ijcard.2015.11.101
Lesuisse, E., Santos, R., Matzanke, B. F., Knight, S. A. B., Camadro, J.-M., and Dancis, A. (2003). Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1). Hum. Mol. Genet. 12, 879–889. doi: 10.1093/hmg/ddg096
Li, K., and Reichmann, H. (2016). Role of iron in neurodegenerative diseases. J. Neural Transm. 123, 389–399. doi: 10.1007/s00702-016-1508-7
Li, L., Miao, R., Bertram, S., Jia, X., Ward, D. M., and Kaplan, J. (2012). A role for iron-sulfur clusters in the regulation of transcription factor Yap5-dependent high iron transcriptional responses in yeast. J. Biol. Chem. 287, 35709–35721. doi: 10.1074/jbc.M112.395533
Lill, R. (2009). Function and biogenesis of iron-sulphur proteins. Nature 460, 831–838. doi: 10.1038/nature08301
Lill, R., Dutkiewicz, R., Freibert, S. A., Heidenreich, T., Mascarenhas, J., Netz, D. J., et al. (2015). The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron–sulfur proteins. Eur. J. Cell Biol. 94, 280–291. doi: 10.1016/j.ejcb.2015.05.002
Lill, R., and Mühlenhoff, U. (2006). Iron-sulfur protein biogenesis in eukaryotes: components and mechanisms. Annu. Rev. Cell Dev. Biol. 22, 457–486. doi: 10.1146/annurev.cellbio.22.010305.104538
Llorens, J. V., Navarro, J. A., Martínez-Sebastián, M. J., Baylies, M. K., Schneuwly, S., Botella, J. A., et al. (2007). Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. FASEB J. 21, 333–344. doi: 10.1096/fj.05-5709com
Lu, C., and Cortopassi, G. (2007). Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts. Arch. Biochem. Biophys. 457, 111–122. doi: 10.1016/j.abb.2006.09.010
Lupoli, F., Vannocci, T., Longo, G., Niccolai, N., and Pastore, A. (2018). The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 592, 718–727. doi: 10.1002/1873-3468.12928
Macevilly, C. J., and Muller, D. P. (1997). Oxidative stress does not appear to be involved in the aetiology of Friedreich’s ataxia. Restor. Neurol. Neurosci. 11, 131–137. doi: 10.3233/RNN-1997-11303
Marmolino, D. (2011). Friedreich’s ataxia: past, present and future. Brain Res. Rev. 67, 311–330. doi: 10.1016/j.brainresrev.2011.04.001
Martelli, A., Friedman, L. S., Reutenauer, L., Messaddeq, N., Perlman, S. L., Lynch, D. R., et al. (2012). Clinical data and characterization of the liver conditional mouse model exclude neoplasia as a non-neurological manifestation associated with Friedreich’s ataxia. Dis. Model. Mech. 5, 860–869. doi: 10.1242/dmm.009829
Martelli, A., and Puccio, H. (2014). Dysregulation of cellular iron metabolism in Friedreich ataxia: from primary iron-sulfur cluster deficit to mitochondrial iron accumulation. Front. Pharmacol. 5:130. doi: 10.3389/fphar.2014.00130
Martelli, A., Schmucker, S., Reutenauer, L., Mathieu, J. R. R., Peyssonnaux, C., Karim, Z., et al. (2015). Iron regulatory protein 1 sustains mitochondrial iron loading and function in frataxin deficiency. Cell Metab. 21, 311–323. doi: 10.1016/j.cmet.2015.01.010
Martin, M., Colman, M. J. R., Gómez-Casati, D. F., Lamattina, L., and Zabaleta, E. J. (2009). Nitric oxide accumulation is required to protect against iron-mediated oxidative stress in frataxin-deficient Arabidopsis plants. FEBS Lett. 583, 542–548. doi: 10.1016/j.febslet.2008.12.039
Mena, N. P., García-Beltrán, O., Lourido, F., Urrutia, P. J., Mena, R., Castro-Castillo, V., et al. (2015). The novel mitochondrial iron chelator 5-((methylamino)methyl)-8-hydroxyquinoline protects against mitochondrial-induced oxidative damage and neuronal death. Biochem. Biophys. Res. Commun. 463, 787–792. doi: 10.1016/j.bbrc.2015.06.014
Michael, S., Petrocine, S. V., Qian, J., Lamarche, J. B., Knutson, M. D., Garrick, M. D., et al. (2006). Iron and iron-responsive proteins in the cardiomyopathy of Friedreich’s ataxia. Cerebellum 5, 257–267. doi: 10.1080/14734220600913246
Mincheva-Tasheva, S., Obis, E., Tamarit, J., and Ros, J. (2014). Apoptotic cell death and altered calcium homeostasis caused by frataxin depletion in dorsal root ganglia neurons can be prevented by BH4 domain of Bcl-xL protein. Hum. Mol. Genet. 23, 1829–1841. doi: 10.1093/hmg/ddt576
Mollá, B., Muñoz-Lasso, D. C., Riveiro, F., Bolinches-Amorós, A., Pallardó, F. V., Fernandez-Vilata, A., et al. (2017). Reversible axonal dystrophy by calcium modulation in frataxin-deficient sensory neurons of YG8R mice. Front. Mol. Neurosci. 10:264. doi: 10.3389/fnmol.2017.00264
Monnier, V., Llorens, J. V., and Navarro, J. A. (2018). Impact of Drosophila models in the study and treatment of Friedreich’s Ataxia. Int. J. Mol. Sci. 19:E1989. doi: 10.3390/ijms19071989
Monrós, E., Moltó, M. D., Martínez, F., Cañizares, J., Blanca, J., Vílchez, J. J., et al. (1997). Phenotype correlation and intergenerational dynamics of the Friedreich ataxia GAA trinucleotide repeat. Am. J. Hum. Genet. 61, 101–110. doi: 10.1086/513887
Napier, I., Ponka, P., and Richardson, D. R. (2005). Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood 105, 1867–1874. doi: 10.1182/blood-2004-10-3856
Navarro, J. A., Botella, J. A., Metzendorf, C., Lind, M. I., and Schneuwly, S. (2015). Mitoferrin modulates iron toxicity in a Drosophila model of Friedreich’s ataxia. Free Radic. Biol. Med. 85, 71–82. doi: 10.1016/j.freeradbiomed.2015.03.014
Navarro, J. A., Ohmann, E., Sanchez, D., Botella, J. A., Liebisch, G., Moltó, M. D., et al. (2010). Altered lipid metabolism in a Drosophila model of Friedreich’s ataxia. Hum. Mol. Genet. 19, 2828–2840. doi: 10.1093/hmg/ddq183
Obis,È., Irazusta, V., Sanchís, D., Ros, J., and Tamarit, J. (2014). Frataxin deficiency in neonatal rat ventricular myocytes targets mitochondria and lipid metabolism. Free Radic. Biol. Med. 73, 21–33. doi: 10.1016/j.freeradbiomed.2014.04.016
O’Neill, H. A., Gakh, O., Park, S., Cui, J., Mooney, S. M., Sampson, M., et al. (2005). Assembly of human frataxin is a mechanism for detoxifying redox-active iron. Biochemistry 44, 537–545. doi: 10.1021/bi048459j
Pandey, A., Gordon, D. M., Pain, J., Stemmler, T. L., Dancis, A., and Pain, D. (2013). Frataxin directly stimulates mitochondrial cysteine desulfurase by exposing substrate-binding sites, and a mutant Fe-S cluster scaffold protein with frataxin-bypassing ability acts similarly. J. Biol. Chem. 288, 36773–36786. doi: 10.1074/jbc.M113.525857
Pandolfo, M., Arpa, J., Delatycki, M. B., Le Quan Sang, K. H., Mariotti, C., Munnich, A., et al. (2014). Deferiprone in Friedreich ataxia: a 6-month randomized controlled trial. Ann. Neurol. 76, 509–521. doi: 10.1002/ana.24248
Pandolfo, M., and Pastore, A. (2009). The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J. Neurol. 256(Suppl.), 9–17. doi: 10.1007/s00415-009-1003-2
Park, S., Gakh, O., Mooney, S. M., and Isaya, G. (2002). The ferroxidase activity of yeast frataxin. J. Biol. Chem. 277, 38589–38595. doi: 10.1074/jbc.M206711200
Park, S., Gakh, O., O’Neill, H. A., Mangravita, A., Nichol, H., Ferreira, G. C., et al. (2003). Yeast frataxin sequentially chaperones and stores iron by coupling protein assembly with iron oxidation. J. Biol. Chem. 278, 31340–31351. doi: 10.1074/jbc.M303158200
Parkinson, M. H., Boesch, S., Nachbauer, W., Mariotti, C., and Giunti, P. (2013). Clinical features of Friedreich’s ataxia: classical and atypical phenotypes. J. Neurochem. 126(Suppl.), 103–117. doi: 10.1111/jnc.12317
Paupe, V., Dassa, E. P., Goncalves, S., Auchère, F., Lönn, M., Holmgren, A., et al. (2009). Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS One 4:e4253. doi: 10.1371/journal.pone.0004253
Payne, R. M., and Wagner, G. R. (2012). Cardiomyopathy in Friedreich ataxia: clinical findings and research. J. Child Neurol. 27, 1179–1186. doi: 10.1177/0883073812448535
Perdomini, M., Belbellaa, B., Monassier, L., Reutenauer, L., Messaddeq, N., Cartier, N., et al. (2014). Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat. Med. 20, 542–547. doi: 10.1038/nm.3510
Perez, C. A., Tong, Y., and Guo, M. (2008). Iron chelators as potential therapeutic agents for Parkinson’s disease. Curr. Bioact. Compd. 4, 150–158. doi: 10.2174/157340708786305952
Petrillo, S., Piermarini, E., Pastore, A., Vasco, G., Schirinzi, T., Carrozzo, R., et al. (2017). Nrf2-inducers counteract neurodegeneration in frataxin-silenced motor neurons: disclosing new therapeutic targets for Friedreich’s ataxia. Int. J. Mol. Sci. 18:E2173. doi: 10.3390/ijms18102173
Piermarini, E., Cartelli, D., Pastore, A., Tozzi, G., Compagnucci, C., Giorda, E., et al. (2016). Frataxin silencing alters microtubule stability in motor neurons: implications for Friedreich’s ataxia. Hum. Mol. Genet. 25, 4288–4301. doi: 10.1093/hmg/ddw260
Piguet, F., de Montigny, C., Vaucamps, N., Reutenauer, L., Eisenmann, A., and Puccio, H. (2018). Rapid and complete reversal of sensory ataxia by gene therapy in a novel model of friedreich ataxia. Mol. Ther. 26, 1940–1952. doi: 10.1016/j.ymthe.2018.05.006
Ponka, P. (1997). Tissue-specific regulation of iron metabolism and heme synthesis: distinct control mechanisms in erythroid cells. Blood 89, 1–25.
Puccio, H., Simon, D., Cossée, M., Criqui-Filipe, P., Tiziano, F., Melki, J., et al. (2001). Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 27, 181–186. doi: 10.1038/84818
Py, B., and Barras, F. (2010). Building Fe-S proteins: bacterial strategies. Nat. Rev. Microbiol. 8, 436–446. doi: 10.1038/nrmicro2356
Rajagopalan, B., Francis, J. M., Cooke, F., Korlipara, L. V. P., Blamire, A. M., Schapira, A. H. V., et al. (2010). Analysis of the factors influencing the cardiac phenotype in Friedreich’s ataxia. Mov. Disord. 25, 846–852. doi: 10.1002/mds.22864
Raman, R., Vaitheeswaran, K., Vinita, K., and Sharma, T. (2011). Is prevalence of retinopathy related to the age of onset of diabetes? Sankara nethralaya diabetic retinopathy epidemiology and molecular genetic report No. 5. Ophthalmic Res. 45, 36–41. doi: 10.1159/000314720
Raman, S. V., Phatak, K., Hoyle, J. C., Pennell, M. L., McCarthy, B., Tran, T., et al. (2011). Impaired myocardial perfusion reserve and fibrosis in Friedreich ataxia: a mitochondrial cardiomyopathy with metabolic syndrome. Eur. Heart J. 32, 561–567. doi: 10.1093/eurheartj/ehq443
Ramirez, R. L., Qian, J., Santambrogio, P., Levi, S., and Koeppen, A. H. (2012). Relation of cytosolic iron excess to cardiomyopathy of Friedreich’s ataxia. Am. J. Cardiol. 110, 1820–1827. doi: 10.1016/j.amjcard.2012.08.018
Richardson, D. R., Lane, D. J. R., Becker, E. M., Huang, M. L.-H., Whitnall, M., Suryo Rahmanto, Y., et al. (2010). Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. U.S.A. 107, 10775–10782. doi: 10.1073/pnas.0912925107
Rocca, C. J., Goodman, S. M., Dulin, J. N., Haquang, J. H., Gertsman, I., Blondelle, J., et al. (2017). Transplantation of wild-type mouse hematopoietic stem and progenitor cells ameliorates deficits in a mouse model of Friedreich’s ataxia. Sci. Transl. Med. 9:eaaj2347. doi: 10.1126/scitranslmed.aaj2347
Rötig, A., de Lonlay, P., Chretien, D., Foury, F., Koenig, M., Sidi, D., et al. (1997). Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 17, 215–217. doi: 10.1038/ng1097-215
Sanchez-Casis, G., Cote, M., and Barbeau, A. (1976). Pathology of the heart in Friedreich’s ataxia: review of the literature and report of one case. Can. J. Neurol. Sci. 3, 349–354. doi: 10.1017/S0317167100025580
Sassa, S. (2006). Modern diagnosis and management of the porphyrias. Br. J. Haematol. 135, 281–292. doi: 10.1111/j.1365-2141.2006.06289.x
Saveliev, A., Everett, C., Sharpe, T., Webster, Z., and Festenstein, R. (2003). DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature 422, 909–913. doi: 10.1038/nature01596
Schiavi, A., Torgovnick, A., Kell, A., Megalou, E., Castelein, N., Guccini, I., et al. (2013). Autophagy induction extends lifespan and reduces lipid content in response to frataxin silencing in C. elegans. Exp. Gerontol. 48, 191–201. doi: 10.1016/j.exger.2012.12.002
Schmucker, S., Martelli, A., Colin, F., Page, A., Wattenhofer-Donzé, M., Reutenauer, L., et al. (2011). Mammalian frataxin: an essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS One 6:e16199. doi: 10.1371/journal.pone.0016199
Schoenfeld, R. A., Napoli, E., Wong, A., Zhan, S., Reutenauer, L., Morin, D., et al. (2005). Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum. Mol. Genet. 14, 3787–3799. doi: 10.1093/hmg/ddi393
Schulz, J. B., Dehmer, T., Schöls, L., Mende, H., Hardt, C., Vorgerd, M., et al. (2000). Oxidative stress in patients with Friedreich ataxia. Neurology 55, 1719–1721. doi: 10.1212/WNL.55.11.1719
Selvadurai, L. P., Harding, I. H., Corben, L. A., and Georgiou-Karistianis, N. (2018). Cerebral abnormalities in Friedreich ataxia: a review. Neurosci. Biobehav. Rev. 84, 394–406. doi: 10.1016/j.neubiorev.2017.08.006
Seznec, H., Simon, D., Bouton, C., Reutenauer, L., Hertzog, A., Golik, P., et al. (2005). Friedreich ataxia: the oxidative stress paradox. Hum. Mol. Genet. 14, 463–474. doi: 10.1093/hmg/ddi042
Shamim, A., Mahmood, T., Ahsan, F., Kumar, A., and Bagga, P. (2018). Lipids: an insight into the neurodegenerative disorders. Clin. Nutr. Exp. 20, 1–19. doi: 10.1016/j.yclnex.2018.05.001
Shan, Y., Schoenfeld, R. A., Hayashi, G., Napoli, E., Akiyama, T., Iodi Carstens, M., et al. (2013). Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal. 19, 1481–1493. doi: 10.1089/ars.2012.4537
Shidara, Y., and Hollenbeck, P. J. (2010). Defects in mitochondrial axonal transport and membrane potential without increased reactive oxygen species production in a Drosophila model of Friedreich ataxia. J. Neurosci. 30, 11369–11378. doi: 10.1523/JNEUROSCI.0529-10.2010
Simon, D., Seznec, H., Gansmuller, A., Carelle, N., Weber, P., Metzger, D., et al. (2004). Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J. Neurosci. 24, 1987–1995. doi: 10.1523/JNEUROSCI.4549-03.2004
Singh, Y. P., Pandey, A., Vishwakarma, S., and Modi, G. (2018). A review on iron chelators as potential therapeutic agents for the treatment of Alzheimer’s and Parkinson’s diseases. Mol. Divers. doi: 10.1007/s11030-018-9878-4 [Epub ahead of print].
Sohn, Y.-S., Breuer, W., Munnich, A., and Cabantchik, Z. I. (2008). Redistribution of accumulated cell iron: a modality of chelation with therapeutic implications. Blood 111, 1690–1699. doi: 10.1182/blood-2007-07-102335
Solbach, K., Kraff, O., Minnerop, M., Beck, A., Schöls, L., Gizewski, E. R., et al. (2014). Cerebellar pathology in Friedreich’s ataxia: atrophied dentate nuclei with normal iron content. Neuroimage Clin. 6, 93–99. doi: 10.1016/j.nicl.2014.08.018
Soragni, E., and Gottesfeld, J. M. (2016). Translating HDAC inhibitors in Friedreich’s ataxia. Expert Opin. Orphan Drugs 4, 961–970. doi: 10.1080/21678707.2016.1215910
Soriano, S., Calap-Quintana, P., Llorens, J. V., Al-Ramahi, I., Gutiérrez, L., Martínez-Sebastián, M. J., et al. (2016). Metal homeostasis regulators suppress FRDA phenotypes in a Drosophila model of the disease. PLoS One 11:e0159209. doi: 10.1371/journal.pone.0159209
Soriano, S., Llorens, J. V., Blanco-Sobero, L., Gutiérrez, L., Calap-Quintana, P., Morales, M. P., et al. (2013). Deferiprone and idebenone rescue frataxin depletion phenotypes in a Drosophila model of Friedreich’s ataxia. Gene 521, 274–281. doi: 10.1016/j.gene.2013.02.049
Strawser, C., Schadt, K., Hauser, L., McCormick, A., Wells, M., Larkindale, J., et al. (2017). Pharmacological therapeutics in Friedreich ataxia: the present state. Expert Rev. Neurother. 17, 895–907. doi: 10.1080/14737175.2017.1356721
Sturm, B., Bistrich, U., Schranzhofer, M., Sarsero, J. P., Rauen, U., Scheiber-Mojdehkar, B., et al. (2005). Friedreich’s ataxia, no changes in mitochondrial labile iron in human lymphoblasts and fibroblasts: a decrease in antioxidative capacity? J. Biol. Chem. 280, 6701–6708. doi: 10.1074/jbc.M408717200
Tan, G., Chen, L. S., Lonnerdal, B., Gellera, C., Taroni, F. A., and Cortopassi, G. A. (2001). Frataxin expression rescues mitochondrial dysfunctions in FRDA cells. Hum. Mol. Genet. 10, 2099–2107. doi: 10.1093/hmg/10.19.2099
Thurman, P., and Draper, P. (1989). Biosynthesis of phenolic glycolipids in M. microti. Acta Leprol. 7(Suppl. 1):74.
Totadri, S., Bansal, D., Bhatia, P., Attri, S. V., Trehan, A., and Marwaha, R. K. (2015). The deferiprone and deferasirox combination is efficacious in iron overloaded patients with β-thalassemia major: a prospective, single center, open-label study. Pediatr. Blood Cancer 62, 1592–1596. doi: 10.1002/pbc.25533
Tricoire, H., Palandri, A., Bourdais, A., Camadro, J.-M., and Monnier, V. (2014). Methylene blue rescues heart defects in a Drosophila model of Friedreich’s ataxia. Hum. Mol. Genet. 23, 968–979. doi: 10.1093/hmg/ddt493
Tsai, C.-L., and Barondeau, D. P. (2010). Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry 49, 9132–9139. doi: 10.1021/bi1013062
Turowski, V. R., Aknin, C., Maliandi, M. V., Buchensky, C., Leaden, L., Peralta, D. A., et al. (2015). Frataxin is localized to both the chloroplast and mitochondrion and is involved in chloroplast Fe-S protein function in Arabidopsis. PLoS One 10:e0141443. doi: 10.1371/journal.pone.0141443
Van Vranken, J. G., Jeong, M.-Y., Wei, P., Chen, Y.-C., Gygi, S. P., Winge, D. R., et al. (2016). The mitochondrial acyl carrier protein (ACP) coordinates mitochondrial fatty acid synthesis with iron sulfur cluster biogenesis. eLife 5:e17828. doi: 10.7554/eLife.17828
Vankan, P. (2013). Prevalence gradients of Friedreich’s ataxia and R1b haplotype in Europe co-localize, suggesting a common Palaeolithic origin in the Franco-Cantabrian ice age refuge. J. Neurochem. 126(Suppl.), 11–20. doi: 10.1111/jnc.12215
Vazzola, V., Losa, A., Soave, C., and Murgia, I. (2007). Knockout of frataxin gene causes embryo lethality in Arabidopsis. FEBS Lett. 581, 667–672. doi: 10.1016/j.febslet.2007.01.030
Waldvogel, D., van Gelderen, P., and Hallett, M. (1999). Increased iron in the dentate nucleus of patients with Friedrich’s ataxia. Ann. Neurol. 46, 123–125. doi: 10.1002/1531-8249(199907)46:1<123::AID-ANA19>3.0.CO;2-H
Weidemann, F., Störk, S., Liu, D., Hu, K., Herrmann, S., Ertl, G., et al. (2013). Cardiomyopathy of Friedreich ataxia. J. Neurochem. 126(Suppl.), 88–93. doi: 10.1111/jnc.12217
Whitnall, M., Suryo Rahmanto, Y., Huang, M. L.-H., Saletta, F., Lok, H. C., Gutiérrez, L., et al. (2012). Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia. Proc. Natl. Acad. Sci. U.S.A. 109, 20590–20595. doi: 10.1073/pnas.1215349109
Whitnall, M., Suryo Rahmanto, Y., Sutak, R., Xu, X., Becker, E. M., Mikhael, M. R., et al. (2008). The MCK mouse heart model of Friedreich’s ataxia: alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation. Proc. Natl. Acad. Sci. U.S.A. 105, 9757–9762. doi: 10.1073/pnas.0804261105
Wong, A., Yang, J., Cavadini, P., Gellera, C., Lonnerdal, B., Taroni, F., et al. (1999). The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 8, 425–430. doi: 10.1093/hmg/8.3.425
Worth, A. J., Basu, S. S., Snyder, N. W., Mesaros, C., and Blair, I. A. (2014). Inhibition of neuronal cell mitochondrial complex I with rotenone increases lipid β-oxidation, supporting acetyl-coenzyme A levels. J. Biol. Chem. 289, 26895–26903. doi: 10.1074/jbc.M114.591354
Wu, C. K., Dailey, H. A., Rose, J. P., Burden, A., Sellers, V. M., and Wang, B. C. (2001). The 2.0 A structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat. Struct. Biol. 8, 156–160. doi: 10.1038/84152
Yoon, T., and Cowan, J. A. (2003). Iron-sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe-2S] clusters in ISU-type proteins. J. Am. Chem. Soc. 125, 6078–6084. doi: 10.1021/ja027967i
Yoon, T., and Cowan, J. A. (2004). Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 279, 25943–25946. doi: 10.1074/jbc.C400107200
Keywords: Friedreich’s ataxia, frataxin, iron, animal models, oxidative stress, lipid deregulation, iron chelators
Citation: Llorens JV, Soriano S, Calap-Quintana P, Gonzalez-Cabo P and Moltó MD (2019) The Role of Iron in Friedreich’s Ataxia: Insights From Studies in Human Tissues and Cellular and Animal Models. Front. Neurosci. 13:75. doi: 10.3389/fnins.2019.00075
Received: 15 November 2018; Accepted: 23 January 2019;
Published: 18 February 2019.
Edited by:
Giorgio Biasiotto, Università degli Studi di Brescia, ItalyReviewed by:
Jordi Tamarit, Universitat de Lleida, SpainCopyright © 2019 Llorens, Soriano, Calap-Quintana, Gonzalez-Cabo and Moltó. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José Vicente Llorens, ai52aWNlbnRlLmxsb3JlbnNAdXYuZXM= María Dolores Moltó, ZG1vbHRvQHV2LmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.