 Mingzhu Huang1,2†
Mingzhu Huang1,2† Changhe Wang
Changhe Wang Xinjiang Kang
Xinjiang Kang- 1School of Life Sciences, Liaocheng University, Liaocheng, China
- 2Center for Mitochondrial Biology and Medicine, The Key Laboratory of Biomedical Information Engineering of Ministry of Education, School of Life Science and Technology and Frontier Institute of Science and Technology, Xi’an Jiaotong University, Xi’an, China
- 3Key Laboratory of Medical Electrophysiology of Ministry of Education and Medical Electrophysiological Key Laboratory of Sichuan Province, Institute of Cardiovascular Research, Southwest Medical University, Luzhou, China
α-synuclein (α-Syn) is a presynaptic enriched protein involved in the pathogenesis of Parkinson’s disease. However, the physiological roles of α-Syn remain poorly understood. Recent studies have indicated a critical role of α-Syn in the sensing and generation of membrane curvature during vesicular exocytosis and endocytosis. It has been known to modulate the assembly of SNARE complex during exocytosis including vesicle docking, priming and fusion steps. Growing evidence suggests that α-Syn also plays critical roles in the endocytosis of synaptic vesicles. It also modulates the availability of releasable vesicles by promoting synaptic vesicles clustering. Here, we provide an overview of recent progresses in understanding the function of α-Syn in the regulation of exocytosis, endocytosis, and vesicle recycling under physiological and pathological conditions.
Introduction
α-synuclein (α-Syn), encoded by SNCA1/PARK1 gene, consists of 140 amino acids that structurally contains three major domains: an amphipathic helix-containing N-terminal membrane binding domain (residues 1–60), a hydrophobic non-amyloid-beta component (NAC) domain (residues 61–95) relevant to α-Syn aggregation, and a hydrophilic C-terminal domain (residues 96–140) with chaperone-like activity (Recchia et al., 2004). α-Syn is an intrinsically disordered monomeric protein in aqueous solution (Fauvet et al., 2012; Theillet et al., 2016). However, the N-terminal domain of α-Syn forms an amphipathic α-helix following the interaction with negatively charged membrane lipids (Davidson et al., 1998; Jao et al., 2008; Maltsev et al., 2013), while the C-terminal domain is still unstructured (Bodner et al., 2009). In cytosol, α-Syn monomers stepwise form into amyloid fibrils (Conway et al., 2000; Nonaka et al., 2010; Giehm et al., 2011; Horvath et al., 2012; Lashuel et al., 2013). However, recent studies reveal that N-terminally acetylated α-Syn is preserved in disordered monomer conditions against oligomerization in non-neuronal and neuronal cells under physiological conditions (Theillet et al., 2016; Medeiros et al., 2017). Whereas, the membrane-bound monomers undergo conformational change to form β-sheet-rich intermediates (protofibrils) upon accumulation, and then self-associate to ring-like oligomers or amyloid fibrils on the membrane (Wang et al., 2016). Both of cytosol and membrane-bound ring-like oligomers can form transmembrane pores which disrupt the integrity of the membrane, intracellular calcium homeostasis and signaling (Lashuel et al., 2002; Lashuel et al., 2013). Lewy bodies are intracellular inclusions mainly composed by α-Syn fibrils.
α-synuclein is abundant in presynapse and interacts with synaptic vesicles (SVs) to modulate vesicle recycling physiologically (Maroteaux et al., 1988; Jensen et al., 1998). α-Syn also plays critical roles in PD pathogenesis. Both duplication/triplication (Singleton et al., 2003; Ibanez et al., 2004) and point mutations (e.g., A30P, E46K, and A53T) in SNCA1 (Polymeropoulos et al., 1997; Kruger et al., 1998; Zarranz et al., 2004) are associated with autosomal dominate familial PD. Among the PD-linked mutations, A53T is located at the dimer interface of the fibril and E46K is related to the stabilization of the protofilament. The structure of fibril carrying both mutations is distinct from that of wild-type, such as reduced helical periodicity. In addition, the E46K fibril shows a right-handed chirality that is distinct from the left-handed chirality of the wild-type and A53T fibrils (Li et al., 2018). Recent study also indicates that peptides derived from α-Syn act as antigenic epitopes presented by major histocompatibility complex protein to be recognized by T cells in PD patients and hints that PD may be a kind of autoimmune disease (Sulzer et al., 2017). Some studies show that oligomers (Fusco et al., 2017; Mor et al., 2017), protofibrils (Stefanis, 2012), intermediates and fibrils (Peelaerts et al., 2015; Wong and Krainc, 2017; Li et al., 2018) of α-Syn are toxic agents, of note, α-Syn fibrils are transmissible to induce the aggregation of endogenous α-Syn in primary neurons (Li et al., 2018). Thus, the toxic gain-of-function effect of α-Syn mutants may mediate PD pathogenesis (Tsika et al., 2010; Colla et al., 2012). Whereas, recent evidence shows that mature fibrils are non-toxic, and even the toxicity of the oligomers depends on the amount of β-sheet content in the rigid regions of them (Bartels et al., 2011; Cremades et al., 2012; Fusco et al., 2017). Here we mainly focus on the physiological and pathological roles of α-Syn in membrane remodeling and vesicle recycling at nerve terminals.
α-Syn Is a Curvature Sensing and Deforming Protein
α-synuclein, a curvature sensing and generating protein (Varkey et al., 2010; Braun et al., 2012; Shen et al., 2012; Westphal and Chandra, 2013), functions as an amphipathic lipid packing sensor (ALPS). It is reported that the binding affinity of α-Syn with small unilamellar vesicles is 15-fold higher than that with large unilamellar vesicles, suggesting that α-Syn may be a curvature-sensing protein (Middleton and Rhoades, 2010). Alternatively, Hatzakis and coworkers propose that the intrinsic curvature selective binding is mediated by higher density of binding sites on highly curved membranes (Hatzakis et al., 2009; Bhatia et al., 2010). The amphipathic N-terminus of α-Syn is composed of hydrophobic and polar amino acids, which are segregated at opposite sides of the α-helix. These helices are arranged parallel to the membrane bilayer, with their hydrophobic faces inserting into the outer leaflet of the bilayer, creating the asymmetry curvature of the two leaflets of the bilayer and thus causing the membrane curvature (Shen et al., 2012; Westphal and Chandra, 2013). Braun et al. (2012) also report that α-Syn induces both positive mean curvature and negative gaussian curvature in Membranes. Compared to α-Syn, most of the other ALPS proteins, e.g., endophilin and amphiphysin, are structurally different, containing both highly curved scaffolding BAR (Bin-amphiphysin/rvs) domain (Peter et al., 2004; McMahon and Gallop, 2005; Frost et al., 2009; Mim et al., 2012) and membrane-inserting N-terminal amphipathic helix (McMahon and Gallop, 2005; Campelo et al., 2008) for membrane curvature. It is reported that α-Syn tubulation activity (a form of membrane curvature) is about 2.5-fold less than that of endophilin A1, probably attributed to the fact that α-Syn only wedges the amphipathic helix into the membrane to induce membrane curvature without the assistance of BAR domain (Westphal and Chandra, 2013). Moreover, not all of the α-Syn species are involved in the generation of membrane curvature, and only monomeric α-Syn can take the activities. Pathologically, regarding the PD-related α-Syn mutations, A30P mutant that shows decreased phospholipid binding activity is deficient in membrane bending (Westphal and Chandra, 2013). Whereas E46K and A53T mutations, which have increased or comparable phospholipid binding activity, show similar membrane tubulation activities to that of wild type α-Syn (Westphal and Chandra, 2013). α-Syn overexpression results in impaired membrane curvature and tubulation, which might be one of the mechanisms underlying the membrane disruption in PD (Varkey et al., 2010). Golgi fragmentation (Gosavi et al., 2002; Fujita et al., 2006), lysosome dysfunction (Meredith et al., 2002; Xilouri et al., 2016), mitochondrial degeneration (Martin et al., 2006; Wong and Krainc, 2017), and endoplasmic reticulum functional defects (Wong and Krainc, 2017) have been reported in cell lines and PD animal models with α-Syn overexpression. Both exocytosis and endocytosis are related to the membrane deforming. Due to the curvature-sensing and -forming property of α-Syn, it may participate in exocytosis, endocytosis and vesicle recycling physiologically and pathologically (Lautenschlager et al., 2017).
α-Syn and Exocytosis
Neurotransmitter release from pre-synapse through exocytosis is crucial for efficient neuronal communication (Chapman, 2002; Sudhof, 2004; Wu et al., 2014). With the arrival of action potentials, extracellular calcium flows into the nerve terminals through voltage-gated calcium channels and triggers the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-dependent vesicular exocytosis (Sudhof and Rothman, 2009; Rizo and Xu, 2015). SNARE complex, a key component for membrane fusion between vesicles and presynaptic terminals during neurotransmission (Weber et al., 1998), is composed of synaptobrevin (or vesicle associated membrane protein-2, VAMP2), syntaxin and synaptosome-associated protein 25 kD (SNAP25). VAMP2 is a synaptic vesicle protein (v-SNARE), whereas Syntaxin and SNAP25 (t-SNAREs) are localized in target membrane (Sollner et al., 1993). Each SNARE protein contains one (VAMP2 and syntaxin) or two (SNAP25) coiled-coil SNARE motifs, thus the assembly of t-SNARE and v-SNARE proteins forms a trans-SNARE complex containing a twisted parallel four-helix bundle that pulls the opposing membranes together and eventually initiates membrane fusion. Following exocytosis, the strained trans-SNARE complex relaxes into the fully zippered cis-SNARE, then is disassembled and recycled for another round of exocytosis (Sutton et al., 1998; Jahn and Scheller, 2006; Sudhof and Rothman, 2009). The molecular mechanism of SNARE-mediated membrane fusion has been summarized in recent reviews (Rizo and Xu, 2015; Baker and Hughson, 2016; Brunger et al., 2018; Tian et al., 2019). Here we mainly focus on the critical role of α-Syn in SNARE assembly and vesicular exocytosis.
Exocytosis is divided into three sequential steps, docking, priming and vesicle fusion. Synaptic vesicles are tethered to specialized regions on the plasma membrane whereby they become stably bound. After docking, the vesicles are primed or matured through a series of ATP-dependent reactions. When an action potential invades, the voltage-gated Ca2+ channel-mediated calcium influx triggers the fusion pore opening of the primed vesicles (Sudhof, 1995). α-Syn participates all the three stages (Wang et al., 2016). (1) It is well-established that α-Syn facilitates the vesicles docking to the plasma membrane probably by increasing the SNARE complex assembly (Figure 1; Spillantini and Goedert, 2000; Burre et al., 2010, 2014; Georgieva et al., 2010; Lou et al., 2017). Interestingly, some studies show that α-Syn may function to bridge secretory vesicles and plasma membrane. One working model is that the N terminus of α-Syn binds to the plasma membrane and concurrently the C terminus interacts with VAMP2, thereby cross-bridging the plasma membrane and vesicle to facilitate the vesicle docking (Lou et al., 2017). The other model is that the broken alpha-helix conformation of α-Syn spanning between synaptic vesicles and the plasma membrane can also help to dock the vesicles to the plasma membrane (Georgieva et al., 2010). On the contrary, some studies show that α-Syn inhibits vesicle docking to the plasma membrane in a VAMP2-dependent or -independent manner. Specifically, larger α-Syn oligomers preferentially bind to the N terminus of VAMP2, inhibiting SNARE complex formation and thereby blocking the vesicle docking (Choi et al., 2013), which seems to contradict the fact that higher-order α-Syn multimers binds to membrane to promote SNARE complex formation (Burre et al., 2014). The divergence here needs to be clarified in the future study. Moreover, in vitro studies indicate that the inhibition is independent of VAMP2 (DeWitt and Rhoades, 2013) at the docking stage (Lai et al., 2014). (2) α-Syn is also involved in vesicle priming stage. The overexpression of WT or A30P α-Syn in PC-12 and chromaffine cells impairs catecholamine release probably by inhibiting vesicle priming. α-Syn overexpression increases the number of docked dense core vesicles, whereas the fusion machinery and the dynamics of amperometric quantal events remain unchanged (Larsen et al., 2006). (3) α-Syn is related to the dilation of fusion pore during vesicle fusion. α-Syn and other isoforms (β-Syn and γ-Syn) share the conserved N terminus, which is responsible for the pore dilation. Endogenous and overexpressed synuclein promote fusion pore dilation and cargo discharge, and slow down fusion pore re-closure in neurons and adrenal chromaffin cells (Logan et al., 2017). Moreover, PD-linked mutants, including A30P and A53T, which impair exocytosis, abrogate the property of α-Syn on pore dilation, suggesting that the mechanisms of α-Syn on release frequency and fusion pore dilation are different (Logan et al., 2017).

Figure 1. α-Syn promotes the assembly of SNARE complex. The disordered monomeric α-Syn in cytosol forms an amphipathic α-helix following the interaction with the membrane of vesicles. α-Syn binds to the N terminus of VAMP2 via its C terminus and to phospholipids via its N terminus to promotes the assembly of SNARE complex.
Collectively, α-Syn plays a critical role in the docking, priming, and fusion steps of exocytosis, probably by serving as a non-classical chaperone that facilitate SNARE assembly (Chandra et al., 2005; Burre et al., 2010, 2014). The mechanism by which α-Syn maintains the presynaptic SNARE complex assembly is the simultaneous binding of α-Syn to the N terminus of VAMP2 via its C terminus and to phospholipids via its N terminus (Burre et al., 2010). Whereas, only the membrane-bound α-Syn, which forms high-order multimers larger than octamers in a defined orientation, can function to chaperon SNARE complex assembly (Burre et al., 2014). It is proposed that α-Syn may also cooperate with cysteine string protein-α (CSPα) to maintain SNARE complex assembly. CSPα can form a chaperone complex with HSP70 (Heat-shock protein 70) and SGT (small glutamine-rich tetratricopeptide repeat-containing protein) to stabilize monomeric SNAP-25 and thus facilitate SNARE complex formation (Chamberlain and Burgoyne, 1997; Nemani et al., 2010). SNAP-25 undergoes deformation without the chaperone of CSPα, and subjects to ubiquitination and proteasome-dependent degradation, leading to the reduction of SNAP-25 and SNARE complex. CSPα knockout in mice leads to the reduction of SNAP-25 and SNARE complex, and thus the impaired exocytosis in pre-synapse. Overexpression of wild-type α-Syn mostly rescues CSPα deficiency-mediated neurodegeneration and SNARE complex assembly (Sharma et al., 2011), indicating a compensatory effect of α-Syn with CSPα in mediating SNARE protein assembly. Moreover, overexpression of α-Syn mutant A30P, but not A53T, fails to rescue the CSPα deficiency, indicating that the efficient binding to phospholipid is necessary for this compensating function of α-Syn (Chandra et al., 2005). However, not all of studies support the opinion that α-Syn facilitates SNARE complex assembly. Darios et al. report that there is no direct interaction between endogenous α-Syn and SNARE proteins (Darios et al., 2010). α-Syn may inhibit SNARE complex assembly and exocytosis by decreasing the levels of arachidonic acid (Darios et al., 2010), which is proposed to regulate SNARE complex formation and exocytosis in neurons and endocrine cells (Connell et al., 2007; Latham et al., 2007). Future research is needed to unveil the mechanisms underlying the divergence in the role of α-Syn in SNARE complex assembly and exocytosis.
α-Syn and Endocytosis
To sustain the high rate of neurotransmission without depleting the recycling vesicle pools and to prevent the unlimited expansion of pre-synapse plasma membrane, endocytosis is tightly coupled to exocytosis to retrieve the fused synaptic vesicle components from the neuronal surface (Xie et al., 2017; Song et al., 2018). The tight coupling of endocytosis to exocytosis is critical for the maintenance of presynaptic structural integrity, the replenishing of the releasable vesicle pools and the sustained neurotransmission (Saheki and De Camilli, 2012; Wu et al., 2014; Leitz and Kavalali, 2016). α-Syn is reported to mediate membrane curvature and tabulation, a critical step in endocytosis. Clathrin-mediated endocytosis (CME) is the best characterized endocytic pathway, and is the predominant route for vesicle retrieval (McMahon and Boucrot, 2011). α-Syn mutants or overexpression impair endocytosis. In the calyx of held of A53T transgenic mice, both slow and fast vesicle endocytosis are inhibited (Xu et al., 2016). Furthermore, overexpression of human α-Syn induces a loss of synaptic vesicles and an expansion of the plasma membrane by inhibiting CME and activity-dependent bulk endocytosis. The phenomenon only occurs at 20 Hz, but not 5 Hz, stimulation, indicating that the defect is activity-dependent. Moreover, the N-terminal α-helix is also necessary for the regulatory role of α-Syn in endocytosis, as proved by PD-related mutant A30P with disrupted N-terminus (Busch et al., 2014). It is generally accepted that α-Syn oligomers co-exist with monomers under physiological conditions (Wang et al., 2014). Both monomers and oligomers impair CME, but with distinct mechanisms. Acute injection of α-Syn monomers into the lamprey reticulospinal synapse increases clathrin intermediates and clathrin-coated vesicles, indicating that the clathrin uncoating mechanism is impaired by α-Syn monomers. Whereas introduction of dimeric α-Syn increases clathrin-coated pits along the plasma membrane, suggesting a defect in vesicle fission (Medeiros et al., 2017). Furthermore, the inhibitory role of synuclein functions temporally before dynamin, indicating that synucleins act at early steps of vesicular endocytosis (Vargas et al., 2014). Along with endocytosis, receptor internalization is also mediated by α-Syn, i.e., α-Syn plays critical roles in regulating the homeostasis of dopamine transporter and NMDA receptor through CME (Cheng et al., 2011; Kisos et al., 2014).
α-Syn and Vesicle Clustering
Endogenous α-Syn also promotes vesicle clustering (Diao et al., 2013; Fusco et al., 2016). By using a single-vesicle optical microscopy, Diao et al. (2013) show that α-Syn induces vesicle clustering, which is critically dependent on the interactions of α-Syn with VAMP2 and negatively charged lipids. Whereas only A30P mutant disrupts vesicles clustering due to the defects in lipid binding among three familial PD-related point mutants (A30P, E46K, A53T) (Diao et al., 2013). Another independent study shows that residues 1–25 of N terminus and residues 65–97 of central segment function as dual membrane anchors, by which α-Syn can simultaneously bind two different vesicles, thereby prompting vesicle clustering (Fusco et al., 2016). Physiologically, α-Syn regulates the trafficking of synaptic vesicles in distal reserve pool to control the amount of vesicles docked at the synapses for neurotransmitter release (Wislet-Gendebien et al., 2006; Auluck et al., 2010). The physiological role of α-Syn is to bind to and induce the clustering of synaptic vesicles in vitro and in vivo (Kwasny et al., 1991; Gitler et al., 2008; Soper et al., 2008; Diao et al., 2013). In cultured hippocampal slices, α-Syn knockdown decreases the distal reserve synaptic vesicle pool by 50% with the intact docked vesicle pool (Murphy et al., 2000). Modest overexpression of α-Syn in hippocampal neurons displays a reduction in the number of docked vesicles and increase in the number of vesicles in the distal reserve pool, indicating a defect in the re-clustering of synaptic vesicles after endocytosis (Nemani et al., 2010).
Conclusion
α-synuclein is a membrane curvature sensing and deforming protein, functioning in exocytosis and endocytosis (Figure 2). The exact roles of α-Syn are still ambiguous, far from understood and still need to be clarified for a better understanding of the physiological and pathological roles of α-Syn. Moreover, the deficiency in exocytosis, endocytosis, and vesicles recycling may underlie a common pathogenic pathway shared by other PD risky genes (i.e., synaptotagmin-11, parkin, LRRK2, DJ-1, synaptojanin1, and TMEM230) (Abeliovich and Gitler, 2016; Pan et al., 2018; Wang et al., 2018). Advances in super resolution microscopy, single-molecule FRET imaging, cryo electron microscopy offer new opportunities for further studies about the physiological role of α-Syn in exocytosis, endocytosis, and vesicle recycling, while in vivo genetic modulation, optogenetic/chemical genetic stimulation, in vivo optometric recording or two photon imaging will benefit the pathogenic connections between α-Syn, vesicle recycling, and neurodegeneration. It will reveal novel therapeutic targets for PD in gaining insight into the mechanisms of these PD risk genes in exocytosis, endocytosis, and vesicles recycling.
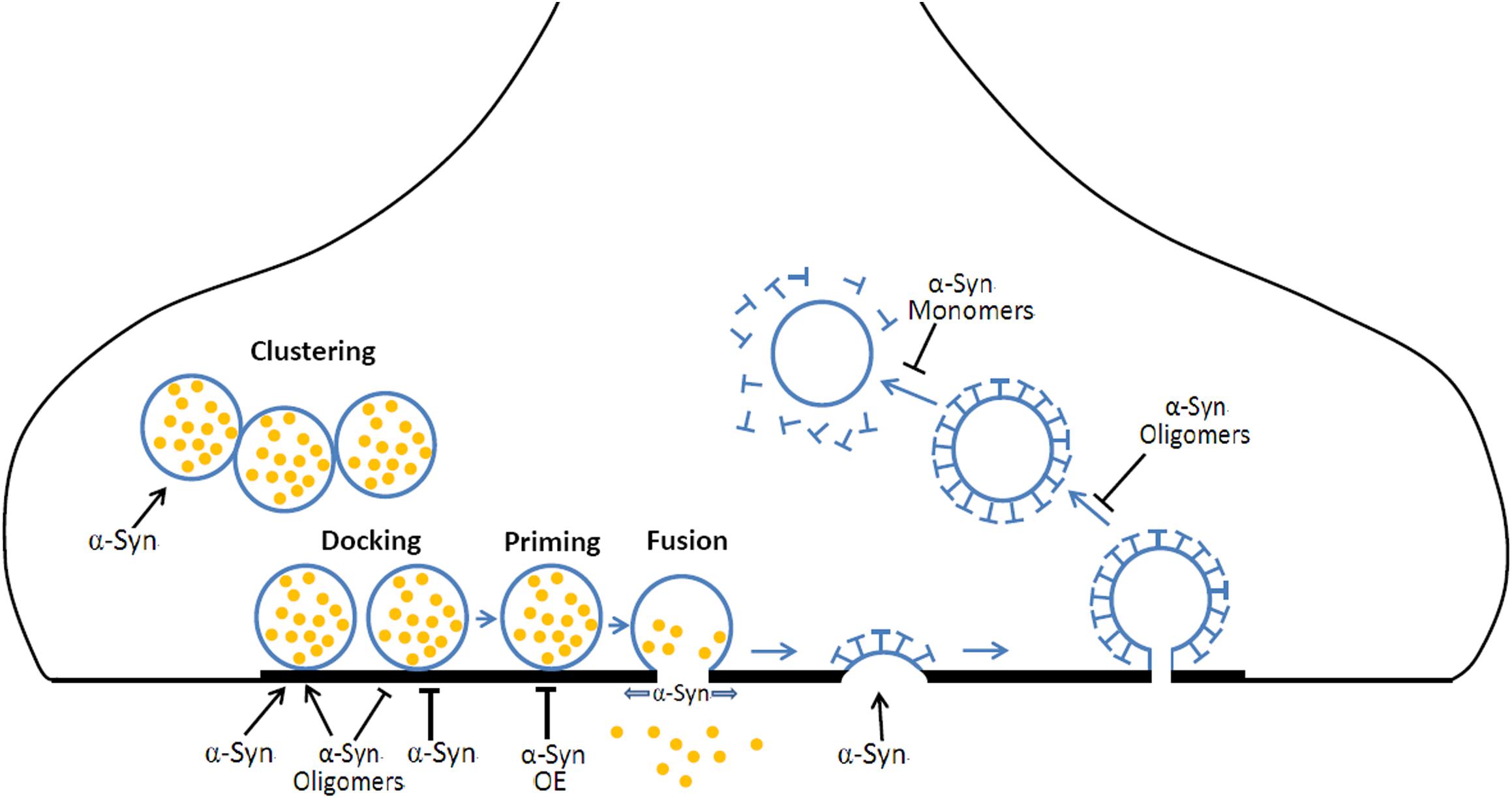
Figure 2. The roles of α-Syn in vesicular exocytosis and endocytosis. During neurotransmission, (1) α-Syn mediates the clustering of synaptic vesicles, resulting in a local increase of releasable vesicles. (2) α-Syn and α-Syn oligomers prompt or inhibit vesicle docking at the active zone. (3) α-Syn overexpression (OE) inhibits the priming of synaptic vesicles. (4) α-Syn promotes the fusion pore opening and slows fusion pore reclosing. (5) α-Syn is required for CME at early steps. (6) α-Syn oligomers OE results in a defect in vesicle fission while α-Syn monomers OE induces a defect in uncoating mechanism during CME.
Author Contributions
MH, BW, and XL drafted the manuscript with help from CF, CW, and XK. All authors coordinated, revised, and approved the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81571235, 31670843, 31400708, 21790390, and 21790394), the Key Research and Development Program of Shaanxi Province, China (2017SF-113). Fundamental Research Funds for the Central Universities (2017qngz10), and the National Natural Science Foundation of Shandong Province of China (ZR2016CM16).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abeliovich, A., and Gitler, A. D. (2016). Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539, 207–216. doi: 10.1038/nature20414
Auluck, P. K., Caraveo, G., and Lindquist, S. (2010). Alpha-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 26, 211–233. doi: 10.1146/annurev.cellbio.042308.113313
Baker, R. W., and Hughson, F. M. (2016). Chaperoning SNARE assembly and disassembly. Nat. Rev. Mol. Cell Biol. 17, 465–479. doi: 10.1038/nrm.2016.65
Bartels, T., Choi, J. G., and Selkoe, D. J. (2011). Alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. doi: 10.1038/nature10324
Bhatia, V. K., Hatzakis, N. S., and Stamou, D. (2010). A unifying mechanism accounts for sensing of membrane curvature by BAR domains, amphipathic helices and membrane-anchored proteins. Semin. Cell Dev. Biol. 21, 381–390. doi: 10.1016/j.semcdb.2009.12.004
Bodner, C. R., Dobson, C. M., and Bax, A. (2009). Multiple tight phospholipid-binding modes of alpha-synuclein revealed by solution NMR spectroscopy. J. Mol. Biol. 390, 775–790. doi: 10.1016/j.jmb.2009.05.066
Braun, A. R., Sevcsik, E., Chin, P., Rhoades, E., Tristram-Nagle, S., and Sachs, J. N. (2012). Alpha-Synuclein induces both positive mean curvature and negative Gaussian curvature in membranes. J. Am. Chem. Soc. 134, 2613–2620. doi: 10.1021/ja208316h
Brunger, A. T., Leitz, J., Zhou, Q., Choi, U. B., and Lai, Y. (2018). Ca(2+)-triggered synaptic vesicle fusion initiated by release of inhibition. Trends Cell Biol. 28, 631–645. doi: 10.1016/j.tcb.2018.03.004
Burre, J., Sharma, M., and Sudhof, T. C. (2014). Alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. U.S.A. 111, E4274–E4283. doi: 10.1073/pnas.1416598111
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Busch, D. J., Oliphint, P. A., Walsh, R. B., Banks, S. M., Woods, W. S., George, J. M., et al. (2014). Acute increase of alpha-synuclein inhibits synaptic vesicle recycling evoked during intense stimulation. Mol. Biol. Cell 25, 3926–3941. doi: 10.1091/mbc.E14-02-0708
Campelo, F., McMahon, H. T., and Kozlov, M. M. (2008). The hydrophobic insertion mechanism of membrane curvature generation by proteins. Biophys. J. 95, 2325–2339. doi: 10.1529/biophysj.108.133173
Chamberlain, L. H., and Burgoyne, R. D. (1997). The molecular chaperone function of the secretory vesicle cysteine string proteins. J. Biol. Chem. 272, 31420–31426. doi: 10.1074/jbc.272.50.31420
Chandra, S., Gallardo, G., Fernandez-Chacon, R., Schluter, O. M., and Sudhof, T. C. (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. doi: 10.1016/j.cell.2005.09.028
Chapman, E. R. (2002). Synaptotagmin: a Ca(2+) sensor that triggers exocytosis? Nat. Rev. Mol. Cell Biol. 3, 498–508. doi: 10.1038/nrm855
Cheng, F., Li, X., Li, Y., Wang, C., Wang, T., Liu, G., et al. (2011). Alpha-Synuclein promotes clathrin-mediated NMDA receptor endocytosis and attenuates NMDA-induced dopaminergic cell death. J. Neurochem. 119, 815–825. doi: 10.1111/j.1471-4159.2011.07460.x
Choi, B. K., Choi, M. G., Kim, J. Y., Yang, Y., Lai, Y., Kweon, D. H., et al. (2013). Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. U.S.A. 110, 4087–4092. doi: 10.1073/pnas.1218424110
Colla, E., Jensen, P. H., Pletnikova, O., Troncoso, J. C., Glabe, C., and Lee, M. K. (2012). Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J. Neurosci. 32, 3301–3305. doi: 10.1523/JNEUROSCI.5368-11.2012
Connell, E., Darios, F., Broersen, K., Gatsby, N., Peak-Chew, S. Y., Rickman, C., et al. (2007). Mechanism of arachidonic acid action on syntaxin-Munc18. EMBO Rep. 8, 414–419. doi: 10.1038/sj.embor.7400935
Conway, K. A., Lee, S. J., Rochet, J. C., Ding, T. T., Williamson, R. E., and Lansbury, P. T. Jr. (2000). Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U.S.A. 97, 571–576. doi: 10.1073/pnas.97.2.571
Cremades, N., Cohen, S. I., Deas, E., Abramov, A. Y., Chen, A. Y., Orte, A., et al. (2012). Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell 149, 1048–1059. doi: 10.1016/j.cell.2012.03.037
Darios, F., Ruiperez, V., Lopez, I., Villanueva, J., Gutierrez, L. M., and Davletov, B. (2010). Alpha-synuclein sequesters arachidonic acid to modulate SNARE-mediated exocytosis. EMBO Rep. 11, 528–533. doi: 10.1038/embor.2010.66
Davidson, W. S., Jonas, A., Clayton, D. F., and George, J. M. (1998). Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. doi: 10.1074/jbc.273.16.9443
DeWitt, D. C., and Rhoades, E. (2013). alpha-Synuclein can inhibit SNARE-mediated vesicle fusion through direct interactions with lipid bilayers. Biochemistry 52, 2385–2387. doi: 10.1021/bi4002369
Diao, J., Burre, J., Vivona, S., Cipriano, D. J., Sharma, M., Kyoung, M., et al. (2013). Native alpha-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. eLife 2:e00592. doi: 10.7554/eLife.00592
Fauvet, B., Mbefo, M. K., Fares, M. B., Desobry, C., Michael, S., Ardah, M. T., et al. (2012). alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 287, 15345–15364. doi: 10.1074/jbc.M111.318949
Frost, A., Unger, V. M., and De Camilli, P. (2009). The BAR domain superfamily: membrane-molding macromolecules. Cell 137, 191–196. doi: 10.1016/j.cell.2009.04.010
Fujita, Y., Ohama, E., Takatama, M., Al-Sarraj, S., and Okamoto, K. (2006). Fragmentation of Golgi apparatus of nigral neurons with alpha-synuclein-positive inclusions in patients with Parkinson’s disease. Acta Neuropathol. 112, 261–265. doi: 10.1007/s00401-006-0114-4
Fusco, G., Chen, S. W., Williamson, P. T. F., Cascella, R., Perni, M., Jarvis, J. A., et al. (2017). Structural basis of membrane disruption and cellular toxicity by alpha-synuclein oligomers. Science 358, 1440–1443. doi: 10.1126/science.aan6160
Fusco, G., Pape, T., Stephens, A. D., Mahou, P., Costa, A. R., Kaminski, C. F., et al. (2016). Structural basis of synaptic vesicle assembly promoted by alpha-synuclein. Nat. Commun. 7:12563. doi: 10.1038/ncomms12563
Georgieva, E. R., Ramlall, T. F., Borbat, P. P., Freed, J. H., and Eliezer, D. (2010). The lipid-binding domain of wild type and mutant alpha-synuclein: compactness and interconversion between the broken and extended helix forms. J. Biol. Chem. 285, 28261–28274. doi: 10.1074/jbc.M110.157214
Giehm, L., Svergun, D. I., Otzen, D. E., and Vestergaard, B. (2011). Low-resolution structure of a vesicle disrupting & α-synuclein oligomer that accumulates during fibrillation. Proc. Natl. Acad. Sci. U.S.A. 108, 3246–3251. doi: 10.1073/pnas.1013225108
Gitler, A. D., Bevis, B. J., Shorter, J., Strathearn, K. E., Hamamichi, S., Su, L. J., et al. (2008). The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 145–150. doi: 10.1073/pnas.0710685105
Gosavi, N., Lee, H. J., Lee, J. S., Patel, S., and Lee, S. J. (2002). Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 277, 48984–48992. doi: 10.1074/jbc.M208194200
Hatzakis, N. S., Bhatia, V. K., Larsen, J., Madsen, K. L., Bolinger, P. Y., Kunding, A. H., et al. (2009). How curved membranes recruit amphipathic helices and protein anchoring motifs. Nat. Chem. Biol. 5, 835–841. doi: 10.1038/nchembio.213
Horvath, I., Weise, C. F., Andersson, E. K., Chorell, E., Sellstedt, M., Bengtsson, C., et al. (2012). Mechanisms of protein oligomerization: inhibitor of functional amyloids templates alpha-synuclein fibrillation. J. Am. Chem. Soc. 134, 3439–3444. doi: 10.1021/ja209829m
Ibanez, P., Bonnet, A. M., Debarges, B., Lohmann, E., Tison, F., Pollak, P., et al. (2004). Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364, 1169–1171. doi: 10.1016/S0140-6736(04)17104-3
Jahn, R., and Scheller, R. H. (2006). SNAREs–engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643. doi: 10.1038/nrm2002
Jao, C. C., Hegde, B. G., Chen, J., Haworth, I. S., and Langen, R. (2008). Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U.S.A. 105, 19666–19671. doi: 10.1073/pnas.0807826105
Jensen, P. H., Nielsen, M. S., Jakes, R., Dotti, C. G., and Goedert, M. (1998). Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 273, 26292–26294. doi: 10.1074/jbc.273.41.26292
Kisos, H., Ben-Gedalya, T., and Sharon, R. (2014). The clathrin-dependent localization of dopamine transporter to surface membranes is affected by alpha-synuclein. J. Mol. Neurosci. 52, 167–176. doi: 10.1007/s12031-013-0118-1
Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Kwasny, O., Schabus, R., and Fuchs, M. (1991). Corrective osteotomy for the treatment of carpal tunnel syndrome in malaligned healed fractures of the distal radius. Unfallchirurg 94, 478–481.
Lai, Y., Kim, S., Varkey, J., Lou, X., Song, J. K., Diao, J., et al. (2014). Nonaggregated alpha-synuclein influences SNARE-dependent vesicle docking via membrane binding. Biochemistry 53, 3889–3896. doi: 10.1021/bi5002536
Larsen, K. E., Schmitz, Y., Troyer, M. D., Mosharov, E., Dietrich, P., Quazi, A. Z., et al. (2006). Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 26, 11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006
Lashuel, H. A., Hartley, D., Petre, B. M., Walz, T., and Lansbury, P. T. Jr. (2002). Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature 418:291. doi: 10.1038/418291a
Lashuel, H. A., Overk, C. R., Oueslati, A., and Masliah, E. (2013). The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48. doi: 10.1038/nrn3406
Latham, C. F., Osborne, S. L., Cryle, M. J., and Meunier, F. A. (2007). Arachidonic acid potentiates exocytosis and allows neuronal SNARE complex to interact with Munc18a. J. Neurochem. 100, 1543–1554.
Lautenschlager, J., Kaminski, C. F., and Kaminski Schierle, G. S. (2017). Alpha-Synuclein - regulator of exocytosis, endocytosis, or both? Trends Cell Biol. 27, 468–479. doi: 10.1016/j.tcb.2017.02.002
Leitz, J., and Kavalali, E. T. (2016). Ca2+ dependence of synaptic vesicle endocytosis. Neuroscientist 22, 464–476. doi: 10.1177/1073858415588265
Li, Y., Zhao, C., Luo, F., Liu, Z., Gui, X., Luo, Z., et al. (2018). Amyloid fibril structure of alpha-synuclein determined by cryo-electron microscopy. Cell Res. 28, 897–903. doi: 10.1038/s41422-018-0075-x
Logan, T., Bendor, J., Toupin, C., Thorn, K., and Edwards, R. H. (2017). Alpha-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci. 20, 681–689. doi: 10.1038/nn.4529
Lou, X., Kim, J., Hawk, B. J., and Shin, Y. K. (2017). Alpha-Synuclein may cross-bridge v-SNARE and acidic phospholipids to facilitate SNARE-dependent vesicle docking. Biochem. J. 474, 2039–2049. doi: 10.1042/BCJ20170200
Maltsev, A. S., Chen, J., Levine, R. L., and Bax, A. (2013). Site-specific interaction between alpha-synuclein and membranes probed by NMR-observed methionine oxidation rates. J. Am. Chem. Soc. 135, 2943–2946. doi: 10.1021/ja312415q
Maroteaux, L., Campanelli, J. T., and Scheller, R. H. (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988
Martin, L. J., Pan, Y., Price, A. C., Sterling, W., Copeland, N. G., Jenkins, N. A., et al. (2006). Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 26, 41–50. doi: 10.1523/JNEUROSCI.4308-05.2006
McMahon, H. T., and Boucrot, E. (2011). Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 12, 517–533. doi: 10.1038/nrm3151
McMahon, H. T., and Gallop, J. L. (2005). Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 438, 590–596. doi: 10.1038/nature04396
Medeiros, A. T., Soll, L. G., Tessari, I., Bubacco, L., and Morgan, J. R. (2017). Alpha-Synuclein dimers impair vesicle fission during clathrin-mediated synaptic vesicle recycling. Front. Cell. Neurosci. 11:388. doi: 10.3389/fncel.2017.00388
Meredith, G. E., Totterdell, S., Petroske, E., Santa, Cruz K, Callison, R. C. Jr., and Lau, Y. S. (2002). Lysosomal malfunction accompanies alpha-synuclein aggregation in a progressive mouse model of Parkinson’s disease. Brain Res. 956, 156–165. doi: 10.1016/S0006-8993(02)03514-X
Middleton, E. R., and Rhoades, E. (2010). Effects of curvature and composition on alpha-synuclein binding to lipid vesicles. Biophys. J. 99, 2279–2288. doi: 10.1016/j.bpj.2010.07.056
Mim, C., Cui, H., Gawronski-Salerno, J. A., Frost, A., Lyman, E., Voth, G. A., et al. (2012). Structural basis of membrane bending by the N-BAR protein endophilin. Cell 149, 137–145. doi: 10.1016/j.cell.2012.01.048
Mor, D. E., Tsika, E., Mazzulli, J. R., Gould, N. S., Kim, H., Daniels, M. J., et al. (2017). Dopamine induces soluble alpha-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 20, 1560–1568. doi: 10.1038/nn.4641
Murphy, D. D., Rueter, S. M., Trojanowski, J. Q., and Lee, V. M. (2000). Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 20, 3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000
Nemani, V. M., Lu, W., Berge, V., Nakamura, K., Onoa, B., Lee, M. K., et al. (2010). Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79. doi: 10.1016/j.neuron.2009.12.023
Nonaka, T., Watanabe, S. T., Iwatsubo, T., and Hasegawa, M. (2010). Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases. J. Biol. Chem. 285, 34885–34898. doi: 10.1074/jbc.M110.148460
Pan, P. Y., Zhu, Y., Shen, Y., and Yue, Z. (2018). Crosstalk between presynaptic trafficking and autophagy in Parkinson’s disease. Neurobiol. Dis. doi: 10.1016/j.nbd.2018.04.020 [Epub ahead of print].
Peelaerts, W., Bousset, L., Van der Perren, A., Moskalyuk, A., Pulizzi, R., Giugliano, M., et al. (2015). alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344. doi: 10.1038/nature14547
Peter, B. J., Kent, H. M., Mills, I. G., Vallis, Y., Butler, P. J., Evans, P. R., et al. (2004). BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science 303, 495–499. doi: 10.1126/science.1092586
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Recchia, A., Debetto, P., Negro, A., Guidolin, D., Skaper, S. D., and Giusti, P. (2004). Alpha-synuclein and Parkinson’s disease. FASEB J. 18, 617–626. doi: 10.1096/fj.03-0338rev
Rizo, J., and Xu, J. (2015). The synaptic vesicle release machinery. Annu. Rev. Biophys. 44, 339–367. doi: 10.1146/annurev-biophys-060414-034057
Saheki, Y., and De Camilli, P. (2012). Synaptic vesicle endocytosis. Cold Spring Harb. Perspect. Biol. 4:a005645. doi: 10.1101/cshperspect.a005645
Sharma, M., Burre, J., and Sudhof, T. C. (2011). CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 13, 30–39. doi: 10.1038/ncb2131
Shen, H., Pirruccello, M., and De Camilli, P. (2012). SnapShot: membrane curvature sensors and generators. Cell 150, 1300, 1300.e1–1300.e2. doi: 10.1016/j.cell.2012.08.017
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi: 10.1126/science.1090278
Sollner, T., Whiteheart, S. W., Brunner, M., Erdjument-Bromage, H., Geromanos, S., Tempst, P., et al. (1993). SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318–324. doi: 10.1038/362318a0
Song, Q., Huang, M., Wang, B., Kang, X., and Wang, C. (2018). Bidirectional regulation of Ca(2+) in exo-endocytosis coupling. Sci. China Life Sci. 61, 1583–1585. doi: 10.1007/s11427-018-9429-6
Soper, J. H., Roy, S., Stieber, A., Lee, E., Wilson, R. B., Trojanowski, J. Q., et al. (2008). Alpha-synuclein-induced aggregation of cytoplasmic vesicles in Saccharomyces cerevisiae. Mol. Biol. Cell 19, 1093–1103. doi: 10.1091/mbc.E07-08-0827
Spillantini, M. G., and Goedert, M. (2000). The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann. N. Y. Acad. Sci. 920, 16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x
Stefanis, L. (2012). Alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a009399. doi: 10.1101/cshperspect.a009399
Sudhof, T. C. (1995). The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature 375, 645–653. doi: 10.1038/375645a0
Sudhof, T. C. (2004). The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509–547. doi: 10.1146/annurev.neuro.26.041002.131412
Sudhof, T. C., and Rothman, J. E. (2009). Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477. doi: 10.1126/science.1161748
Sulzer, D., Alcalay, R. N., Garretti, F., Cote, L., Kanter, E., Agin-Liebes, J., et al. (2017). T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 546, 656–661. doi: 10.1038/nature22815
Sutton, R. B., Fasshauer, D., Jahn, R., and Brunger, A. T. (1998). Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395, 347–353. doi: 10.1038/26412
Theillet, F. X., Binolfi, A., Bekei, B., Martorana, A., Rose, H. M., Stuiver, M., et al. (2016). Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 530, 45–50. doi: 10.1038/nature16531
Tian, Z., Gong, J., Crowe, M., Lei, M., Li, D., Ji, B., et al. (2019). Biochemical studies of membrane fusion at the single-particle level. Prog. Lipid Res. 73, 92–100. doi: 10.1016/j.plipres.2019.01.001
Tsika, E., Moysidou, M., Guo, J., Cushman, M., Gannon, P., Sandaltzopoulos, R., et al. (2010). Distinct region-specific alpha-synuclein oligomers in A53T transgenic mice: implications for neurodegeneration. J. Neurosci. 30, 3409–3418. doi: 10.1523/JNEUROSCI.4977-09.2010
Vargas, K. J., Makani, S., Davis, T., Westphal, C. H., Castillo, P. E., and Chandra, S. S. (2014). Synucleins regulate the kinetics of synaptic vesicle endocytosis. J. Neurosci. 34, 9364–9376. doi: 10.1523/JNEUROSCI.4787-13.2014
Varkey, J., Isas, J. M., Mizuno, N., Jensen, M. B., Bhatia, V. K., Jao, C. C., et al. (2010). Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J. Biol. Chem. 285, 32486–32493. doi: 10.1074/jbc.M110.139576
Wang, C., Kang, X., Zhou, L., Chai, Z., Wu, Q., Huang, R., et al. (2018). Synaptotagmin-11 is a critical mediator of parkin-linked neurotoxicity and Parkinson’s disease-like pathology. Nat. Commun. 9:81. doi: 10.1038/s41467-017-02593-y
Wang, C., Zhao, C., Li, D., Tian, Z., Lai, Y., Diao, J., et al. (2016). Versatile structures of alpha-synuclein. Front. Mol. Neurosci. 9:48. doi: 10.3389/fnmol.2016.00048
Wang, L., Das, U., Scott, D. A., Tang, Y., McLean, P. J., and Roy, S. (2014). alpha-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr. Biol. 24, 2319–2326. doi: 10.1016/j.cub.2014.08.027
Weber, T., Zemelman, B. V., McNew, J. A., Westermann, B., Gmachl, M., Parlati, F., et al. (1998). SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772. doi: 10.1016/S0092-8674(00)81404-X
Westphal, C. H., and Chandra, S. S. (2013). Monomeric synucleins generate membrane curvature. J. Biol. Chem. 288, 1829–1840. doi: 10.1074/jbc.M112.418871
Wislet-Gendebien, S., D’Souza, C., Kawarai, T., St George-Hyslop, P., Westaway, D., Fraser, P., et al. (2006). Cytosolic proteins regulate alpha-synuclein dissociation from presynaptic membranes. J. Biol. Chem. 281, 32148–32155. doi: 10.1074/jbc.M605965200
Wong, Y. C., and Krainc, D. (2017). alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat. Med. 23, 1–13. doi: 10.1038/nm.4269
Wu, L. G., Hamid, E., Shin, W., and Chiang, H. C. (2014). Exocytosis and endocytosis: modes, functions, and coupling mechanisms. Annu. Rev. Physiol. 76, 301–331. doi: 10.1146/annurev-physiol-021113-170305
Xie, Z., Long, J., Liu, J., Chai, Z., Kang, X., and Wang, C. (2017). Molecular mechanisms for the coupling of endocytosis to exocytosis in neurons. Front. Mol. Neurosci. 10:47. doi: 10.3389/fnmol.2017.00047
Xilouri, M., Brekk, O. R., and Stefanis, L. (2016). Autophagy and alpha-synuclein: relevance to Parkinson’s disease and related synucleopathies. Mov. Disord. 31, 178–192. doi: 10.1002/mds.26477
Xu, J., Wu, X. S., Sheng, J., Zhang, Z., Yue, H. Y., Sun, L., et al. (2016). Alpha-Synuclein mutation inhibits endocytosis at mammalian central nerve terminals. J. Neurosci. 36, 4408–4414. doi: 10.1523/JNEUROSCI.3627-15.2016
Keywords: α-synuclein, Parkinson’s disease, exocytosis, endocytosis, vesicle recycling
Citation: Huang M, Wang B, Li X, Fu C, Wang C and Kang X (2019) α-Synuclein: A Multifunctional Player in Exocytosis, Endocytosis, and Vesicle Recycling. Front. Neurosci. 13:28. doi: 10.3389/fnins.2019.00028
Received: 29 November 2018; Accepted: 14 January 2019;
Published: 28 January 2019.
Edited by:
Jiajie Diao, University of Cincinnati, United StatesReviewed by:
Dechang Li, Zhejiang University, ChinaChuchu Wang, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, China
Copyright © 2019 Huang, Wang, Li, Fu, Wang and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chongluo Fu, ZnVjaG9uZ2x1b0BsY3UuZWR1LmNu Changhe Wang, Y2hhbmdoZXdhbmdAeGp0dS5lZHUuY24= Xinjiang Kang, a3hqMzM1QDE2My5jb20=
†These authors have contributed equally to this work