 Trevor Rajchgot1
Trevor Rajchgot1 Sini Christine Thomas1
Sini Christine Thomas1 Jo-Chiao Wang2Maryam Ahmadi1Mohammad Balood1Théo Crosson1Jenny Pena Dias3
Jo-Chiao Wang2Maryam Ahmadi1Mohammad Balood1Théo Crosson1Jenny Pena Dias3 Réjean Couture1Audrey Claing1
Réjean Couture1Audrey Claing1 Sébastien Talbot1*
Sébastien Talbot1*- 1Département de Pharmacologie et Physiologie, Faculté de Médecine, Université de Montréal, Montréal, QC, Canada
- 2Graduate Institute of Microbiology, College of Medicine, National Taiwan University, Taipei, Taiwan
- 3Johns Hopkins University School of Medicine, Division of Endocrinology, Diabetes and Metabolism, Baltimore, MD, United States
Diabetes is a common condition characterized by persistent hyperglycemia. High blood sugar primarily affects cells that have a limited capacity to regulate their glucose intake. These cells include capillary endothelial cells in the retina, mesangial cells in the renal glomerulus, Schwann cells, and neurons of the peripheral and central nervous systems. As a result, hyperglycemia leads to largely intractable complications such as retinopathy, nephropathy, hypertension, and neuropathy. Diabetic pain neuropathy is a complex and multifactorial disease that has been associated with poor glycemic control, longer diabetes duration, hypertension, advanced age, smoking status, hypoinsulinemia, and dyslipidemia. While many of the driving factors involved in diabetic pain are still being investigated, they can be broadly classified as either neuron -intrinsic or -extrinsic. In neurons, hyperglycemia impairs the polyol pathway, leading to an overproduction of reactive oxygen species and reactive nitrogen species, an enhanced formation of advanced glycation end products, and a disruption in Na+/K+ ATPase pump function. In terms of the extrinsic pathway, hyperglycemia leads to the generation of both overactive microglia and microangiopathy. The former incites a feed-forward inflammatory loop that hypersensitizes nociceptor neurons, as observed at the onset of diabetic pain neuropathy. The latter reduces neurons' access to oxygen, glucose and nutrients, prompting reductions in nociceptor terminal expression and losses in sensation, as observed in the later stages of diabetic pain neuropathy. Overall, microglia can be seen as potent and long-lasting amplifiers of nociceptor neuron activity, and may therefore constitute a potential therapeutic target in the treatment of diabetic pain neuropathy.
Introduction
Pain is defined as an unpleasant sensation triggered by noxious stimuli, inflammation, or damage to the nervous system. It is an evolutionarily-conserved defensive mechanism that prevents excessive tissue damage and preserves homeostasis by generating defensive withdrawal reflexes (Scholz and Woolf, 2002). Nociception is initiated by the detection of mechanical, chemical, or thermal noxious stimuli by specialized ion channel receptors present on sensory neurons (Scholz and Woolf, 2002). The activation of these ion channels triggers an influx of various cations, depolarizing the neurons' membrane potentials, which, in turn, activate voltage-gated sodium channels (NaVs). This leads to an influx of sodium, and the subsequent firing of action potentials. In the peripheral nervous system (first order fiber), painful sensations are relayed by small, myelinated Aδ-fibers (fast pain transmission) and unmyelinated C-fibers (slow pain transmission) to the spinal cord (second order fiber) (Tesfaye and Kempler, 2005). These action potentials then trigger defensive reflexes, and travel up to the brain (third order fiber) where pain information is integrated and its emotional perception occurs.
Chronic Pain
Chronic pain is a highly debilitating condition and it is the most common reasons for visits to health care providers (Scholz and Woolf, 2002). The most incapacitating type of chronic pain is peripheral neuropathic pain. This pain is unique in regard to its constancy, the severity of its symptoms, and its resistance to current pharmacological treatment (Zimmermann, 2001; Woolf, 2004). Neuropathic pain is usually generated by peripheral nerve damage resulting from neuronal or spinal cord injuries, surgery, cancer, infection, or diabetes (Scholz and Woolf, 2002; Woolf, 2004; Tsuda et al., 2005). In pathological states, this pain often persists after the disappearance of its causal stimulus. In some cases, pain can be perceived more severely, a phenomenon known as hyperalgesia, or can be generated by normally innocuous stimuli, a condition known as allodynia. Tactile allodynia originates from afferent Aβ fibers (light touch/pressure transmission) that gain the ability to release pro-inflammatory neuropeptides (SP and CGRP) in the synaptic cleft, and/or the sprouting of these fibers to the dorsal superficial laminae IIb, a zone normally restricted to the projection of C fibers (Woolf et al., 1992; Miki et al., 1998). In the CNS, thalamic higher-order neurons often become hyperexcitable and act as pain generators or amplifiers (Fischer and Waxman, 2010). For example, increasing N-Methyl-D-Aspartate receptor (NMDAR) phosphorylation reduces its endogenous blockade by magnesium, thereby enhancing calcium (Ca+2) and sodium (Na+) influx. This ultimately promotes the establishment of spinal windup, which is an increase in the excitability of spinal neurons (Haigh and Blake, 2001).
Sensitization of Sensory Neurons
Most drugs targeted to alleviate neuropathic pain are designed to block neurotransmission, and as such, only bring temporary relief (Ji and Suter, 2007). Neuropathic pain is often accompanied by persistent inflammation, as evidenced by the high levels of oxidative substances (Pabreja et al., 2011), inflammatory cytokines (Pabreja et al., 2011), and mediators (Tsuda et al., 2005) present in the neuronal micro-environment. Unlike classical neurotransmitters, these inflammatory molecules are mainly produced by peripheral immunocytes and central glial cells (Marchand et al., 2005; Tsuda et al., 2005). The transcriptomic data of nociceptor neurons notably revealed the expression of specific receptors for immunoglobulins, cytokines and chemokines (Chiu et al., 2014). This evidences the role of nociceptors in directly detecting and responding to interleukins (IL)-1β (Samad et al., 2001; Binshtok et al., 2008) and IL-6 (Opree and Kress, 2000), activin (TGFβ member) (Zhu et al., 2007), TNF-α (Wagner and Myers, 1996), CCL3 (Zhang et al., 2005), GDNF (Malin et al., 2006), histamine (Shim et al., 2007), kinin (Talbot et al., 2012; De Brito Gariepy et al., 2013), and PGE2 (Samad et al., 2001, 2002) released in the context of pain. Nociceptors can also sense IL-5 produced during allergic airway inflammation (Talbot et al., 2015), IL-31 produced during lymphoma-associated itch (Cevikbas et al., 2014), thymic stromal lymphopoietin (TSLP) and IL-4 produced during atopic dermatitis (Wilson et al., 2013; Oetjen et al., 2017), and IL-33 derived from contact with poison ivy (Liu et al., 2016). Additionally, nociceptors can drive IL-23 production during psoriasis (Riol-Blanco et al., 2014). Intracellular kinases and transcription factors downstream of these tyrosine kinase receptors, include PI3K (Pereira et al., 2015), MAP kinases (Ji et al., 2002a), p38 (Ji et al., 2002b), JAK1 (Ludbrook et al., 2016; Oetjen et al., 2017), and STAT3 (Mori et al., 2011) and their activation can lead to pain. Thus, these kinases lead to the post-translational modifications of ion channel transducers or voltage gated sodium channels (Julius, 2013). Nociceptor sensitization is thereby largely due to a decrease in the activation threshold of transient receptor potential vannilloid-1 (TRPV1) or transient receptor potential ankyrin-1 (TRPA1) (Davis et al., 2000; Bautista et al., 2006), and NaV1.7, NaV1.8, and NaV1.9 (Kerr et al., 2001; Nassar et al., 2004). In short, lowering the activation threshold of nociceptors results in pain hypersensitivity. For example, prostaglandin PGE2 is a well-known neuron sensitizer (Samad et al., 2001), which partly explains why non-steroidal anti-inflammatory drugs exhibit analgesic effects in inflammatory conditions (Vardeh et al., 2009). Nerve growth factor (NGF) has also been recognized as a major neuronal sensitizer (Ji et al., 2002b), which has led to the development of neutralizing monoclonal anti-NGF antibodies as a treatment for chronic inflammatory pain conditions (Hefti et al., 2006). These changes in hypersensitivity are limited to sites where sensitizing mediators are produced, which are known as zones of primary hyperalgesia. Outside of these zones, pain hypersensitivity usually results from central sensitization, which involves changes in the CNS (Woolf et al., 1992; Woolf, 2007; von Hehn et al., 2012). During inflammation, many sensitizing mediators are likely to be released simultaneously; therefore, the targeted pharmacological blockade of only one of these agents will have a limited effect. Conversely, targeting the sensitized nerve or convergent signaling mediators or enzymes may have broader and more durable effects as to treating inflammatory pain by stopping it at its source (Khoutorsky and Price, 2017).
Diabetes
Diabetes, derived from the Greek word diabanein, means “to pass through,” in reference to the symptomatic excessive urine production observed in patients (Kumar et al., 2005). The term diabetes, without qualification, usually refers to diabetes mellitus, which roughly translates to “excessive production of sweet urine,” known clinically as glycosuria (Kumar et al., 2005). According to the World Health Organization, at least 422 million people worldwide suffered from diabetes in 2014, representing 8.5% of the world's adult population (WHO, 2016). In 2012, diabetes was responsible for 1.5 million deaths, and its incidence is increasing by more than 8% per year (WHO, 2016). In some regions, such as in Eastern Mediterranean countries, prevalence is increasing by nearly 14%. The National Diabetes Information Clearinghouse estimates the yearly costs of diabetes to more than $132 billion in the United States. In terms of pathology, diabetes is the result of chronic high blood sugar stemming from either low insulin production, as observed in type 1 diabetes; or to a severe reduction in the response of insulin receptors (IR) to insulin, as observed in type 2 diabetes (Kumar et al., 2005). Chronic hyperglycemia causes the classical symptoms of diabetes, including polyuria (frequent urination), polydipsia and polyphagia (Kumar et al., 2005). While both types of diabetes share similar symptoms, they can be distinguished by measuring endogenous insulin production (Kumar et al., 2005).
Type 1 Diabetes
Formerly known as juvenile diabetes, type 1 diabetes (T1D) represents approximately 10% of diabetes cases in North America and Europe. There is currently no known preventive measure against type 1 diabetes, which is considered immune-mediated or idiopathic (Kumar et al., 2005). Insulin-dependent diabetes mellitus is characterized by the auto-immune, T-cell mediated (Rother, 2007) destruction of insulin-producing beta cells of the pancreatic islets of Langerhans. The destruction of β cells triggers insulin deficiency, which leads to increases of glucose in the patient's blood and urine. Evidence indicates that type I diabetes is induced by a combination of genetic susceptibility [mutation(s) to iddm1, drb1, dqa, and dqb1 gene locus], environmental factors [diet, vitamin D deficiency (Mathieu et al., 2005)], or exposure to a driving antigen (exposure to wheat protein (Knip and Siljander, 2008), antibody from cow's milk protein (Virtanen et al., 1994). There is no current preventive measure against T1D, which can be highly pathogenic, or even fatal, if left untreated. Emerging treatments such as pancreas (Noguchi, 2010) and islet transplants (Noguchi, 2009) have shown relatively positive outcomes in pre-clinical models, and are currently being studied in clinical trials. However, drawbacks to implantation include the necessity for immunosuppressant administration, which increased susceptibility to infection and cancer, graft rejection of the implanted pancreas/islets, hypoglycemia, and a current lack of suitable donors (Balamurugan et al., 2014).
Type 2 Diabetes
Type 2 diabetes (T2D), also known as non-insulin dependent diabetes mellitus, is a metabolic disorder characterized by chronic high blood glycemia and insulin receptor resistance, sometimes in combination with relative insulin deficiency (Kumar et al., 2005). This type of diabetes can be initially managed by increasing exercise and dietary modification. It represents almost 90% of Western countries' diabetic populations (Kumar et al., 2005). The onset of T2D is related to genetic and environmental factors. The environmental detrimental factors can include smoking, obesity, diet, alcoholism, low physical activity, high cholesterol, hypertension, metabolic syndrome, and Cushing syndrome (Kumar et al., 2005). In recent years, compelling research and efforts have been made to genetically identify mutant or polymorphic genes that predispose individuals to develop type 2 diabetes. These have been found to include tcf7l2, pparγ, fto, kcnj11, notch2, wfs1, cdkal1, igf2bp2, slc30a8, jazf1, hhex (Groop and Lyssenko, 2008; Lyssenko, 2008) and mody genes, which themselves can account for up to 5% of T2D cases (Billings and Florez, 2010). Mutations in both human leptin production and the human leptin receptor gene can cause severe obesity and pituitary dysfunction, which can in turn engender T2D (Clement et al., 1998; Wabitsch et al., 2015).
Complications of Diabetes
The chronic impairment of glucose metabolism associated with both types of diabetes has been associated with severe macrovascular (cardiovascular) disease and microvascular complications including retinopathy, nephropathy and sensory poly-neuropathy (Schemmel et al., 2009). Neuropathy is the most common complication seen in ambulatory care of type 2 diabetes patients (Schemmel et al., 2009). Overall, the aforementioned complications can result in debilitating and/or life-threatening conditions such as renal failure, erectile dysfunction, blindness, macular edema, impaired wound healing, hypertension, obesity, coronary artery disease, cerebrovascular accidents, heart failure, allodynia, hyperalgesia, nerve degeneration, insensitivity, and limb amputation.
Diabetic Pain Neuropathy
Diabetic pain neuropathy (DPN) is defined as the presence of signs and symptoms of peripheral nerve dysfunction in people with diabetes after having excluded other potential causes (Crofford, 1995). DPN is considered the principal cause of mortality, morbidity (Ziegler, 2008), and amputation (Molines et al., 2010) in diabetic patients, as well as the most common cause of neuropathy (Obrosova, 2009). The prevalence of DPN is thought to be proportional to disease duration and seems to be potentiated by an improper control of blood glycemia (Kumar et al., 2005). Ten percentage of 1-year diabetes patients suffer from neuropathy; this number increases to 50% amongst 25-year diabetes patients. Overall, 30% of diabetic patients suffer from DPN (Guastella and Mick, 2009). Interestingly, 39% of diabetic patients either receive no treatment for their symptoms or remain unmanaged (Daousi et al., 2004). While the prevalence of poorly-managed blood glycemia makes a significant proportion of diabetic patients highly susceptible to developing DPN, glycemic management in clinical care is slowly improving (Aschner et al., 2018). There is emerging evidence that genetic factors may play an important role in DPN pathogenesis (Prabodha et al., 2018).
DPN symptoms include paresthesia, numbness, and burning (Schemmel et al., 2009), which vary in nature and severity depending on the particular subpopulation of neurons being affected (Kumar et al., 2005). Certain patients with DPN do not present any symptoms; however, most report pain and/or loss of function in distal regions such as in their toes, feet, fingers, hands, or arms (Ziegler, 2008). Thus, at the onset of DPN, peripheral nerves often act as pulse generators, maintaining distal terminals of sensory nerve fibers in a state of hyperexcitability (Obrosova, 2009). When these fibers undergo active degeneration or impaired regeneration, they can begin to generate ectopic discharges, which induce positive pain symptoms. Later stages of DPN are characterized by a progressive loss of neuronal fibers, which is associated with a loss of sensation, and can ultimately cause diabetic foot syndrome (Yagihashi et al., 2007). The specific clinical diagnosis of DPN involves both electrophysiological and electromyography testing, respectively, assessing nerve conduction and muscular responses to electric stimulation (Kumar et al., 2005; Guastella and Mick, 2009). The metrics of blood glycemia, arterial pressure, heart rate, muscle force, reflex quality, and sensitivity to spatiotemporal changes can be used to indirectly help diagnose diabetic neuropathy in a more general sense (Guastella and Mick, 2009).
A Focus on the Molecular Drivers of Diabetic Pain Neuropathy
The origins of DPN are multifactorial (Figure 1), and result from neuron intrinsic (Figure 2) and extrinsic factors (Figure 3). This review will examine pre-clinical evidence supporting how chronic hyperglycemia dysregulate neurons' biochemical pathways, activates glia and how such impairments trigger DPN. Current theories (Brownlee, 2001, 2005) regarding neurons intrinsic factor driving the development of DPN include: uncontrolled oxidative stress (section Reactive Oxygen Species) (Nishikawa et al., 2000; Pop-Busui et al., 2006), the formation of reactive nitrogen species (section Reactive Nitrogen Species) (Zochodne and Levy, 2005), the formation of advanced glycation end products (section Advanced Glycation End Products) (Brownlee, 2005; Sugimoto et al., 2008), impaired Na+/K+ ATPase activity (section Ion Imbalance) (Vague et al., 1997; Gerbi et al., 1998; Raccah, 1998), an imbalance in the polyol pathway (section Polyol Pathway) and/or to the activity of the aldol reductase (section Aldose Reductase) (Oates, 2002). Extrinsically, it is believed that, at the spinal synapse, the neuro-immune interplay occurring between activated microglia and pain-sensing neurons maintains DPN (section Painful Glia to Microglia, an Emerging Target in DPN). The neuronal loss of energy supply occurring through microangiopathy (see Microangiopathy section) appears to be responsible for the loss of sensation observed in later stages of DPN. Finally, future therapeutic avenues will be discussed in the Conclusion and Future Therapeutic Directions section.
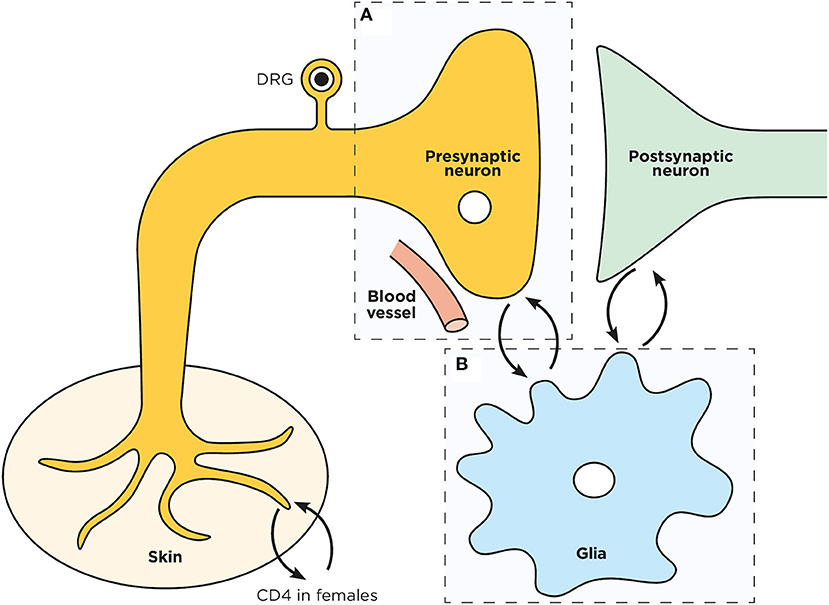
Figure 1. Schematic representation of a spinal dorsal horn tripartite synapse. Overview of the pre (A) and post- synaptic neurons interplay with microglia (B) in the spinal cord dorsal horn.
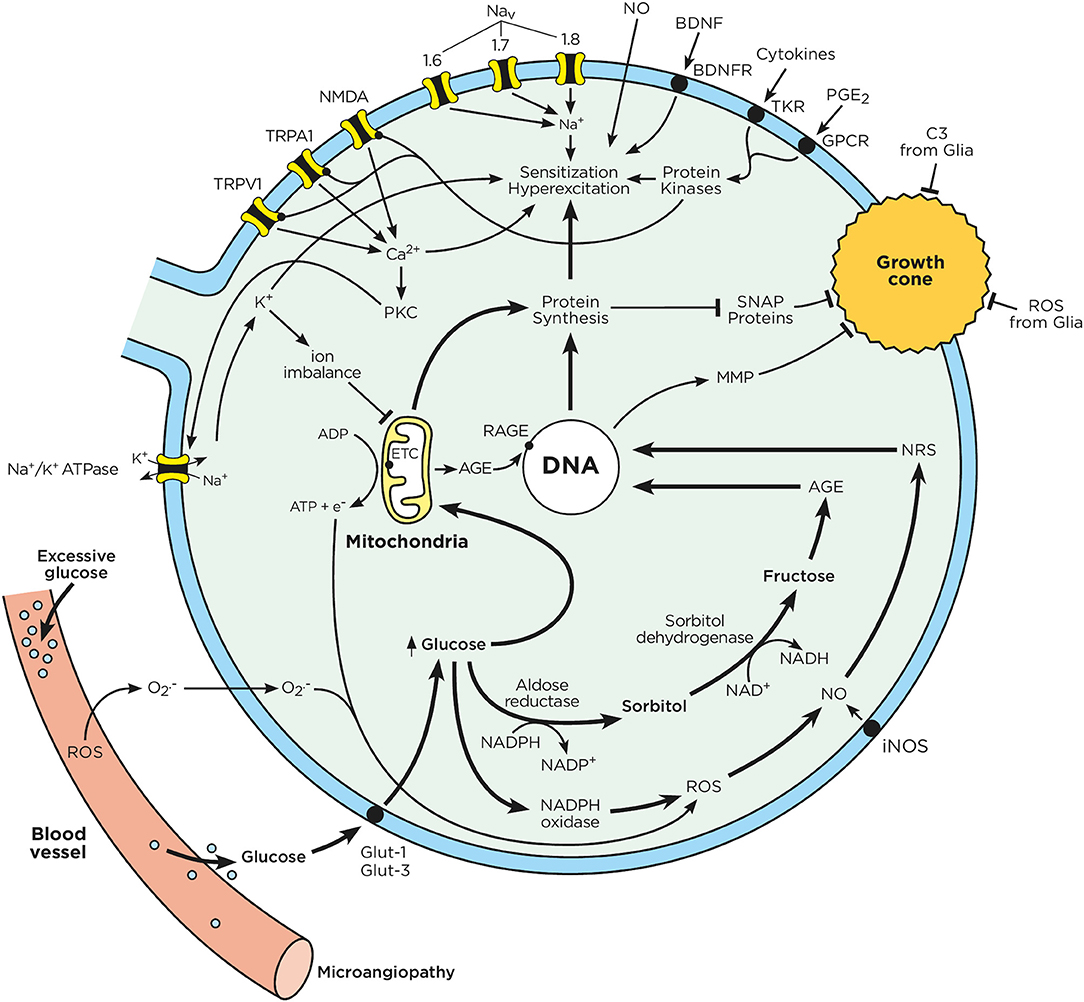
Figure 2. Chronic hyperglycemia impairs neuron function. Sensory neurons have a limited capacity to regulate their uptake of glucose. In the context of chronic hyperglycemia, such as in diabetes, high glucose concentrations drive mitochondria to produce ATP and transfer electrons. Excess glucose is also metabolized through the polyol pathway, leading to the production of advanced glycation end products. The electrons from the mitochondrial respiratory chain combine with intracellular oxygen and nitric oxide to produce ROS and RNS. Consequently, RNS, ROS, and AGE activate nuclear transcription factors, which enhance the expression of ion channel transducers (TRP and NaV channels) in addition to impairing neurons' capacity to self-repair. At the same time, microglia-released mediators (cytokines, ATP, BDNF, NO) stimulate GPCR and tyrosine kinase receptors, triggering downstream signaling cascades, which lead to the phosphorylation of TRP and NaV channels. A decrease in the activation threshold of these ion channel transducers can augment the influx of cations, which ultimately results in action potential firing and ectopic discharges. These effects enhance pain perception and signaling to the CNS. Chronic hyperglycemia also increases oxidative stress in the blood vessels that supply oxygen and nutrients to neuron terminals. This oxidative stress can cause microangiopathy, a phenomenon characterized by the loss of capillaries, which starves neuronal energy supplies. These phenomena are responsible for the loss of neuron terminals and pain insensitivity, as typically observed in later stage of DPN.
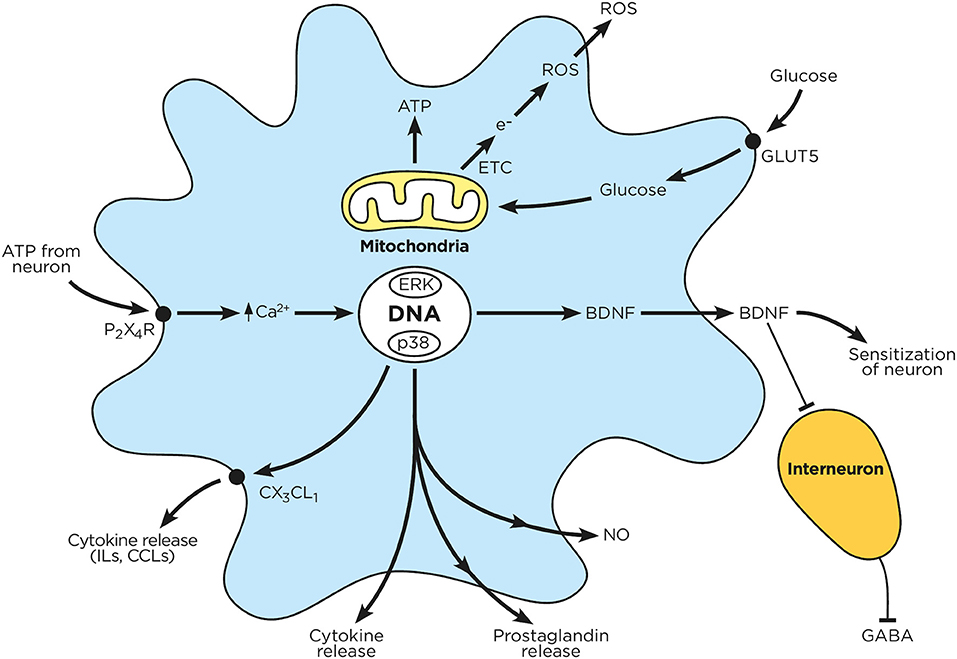
Figure 3. Chronic hyperglycemia impairs microglial function. Circulating glucose is taken up by microglia, which enhances mitochondrial ATP production and electron transfer. The released electrons combine with intracellular oxygen to produce reactive oxygen species. Sensory neurons release ATP, which in turn activates microglial P2X4R; this drives microglial calcium influx, MAPK activation, receptor phosphorylation and protein transduction (cytokines, prostaglandins, BDNF) as well as NO production. These mediators are subsequently released by microglia, and either block inhibitory interneurons or enhance neuronal activation.
Neuron Intrinsic Factors Driving Diabetic Pain Neuropathy
Reactive Oxygen Species
Cellular aerobic respiration generates the majority of intracellular free radicals (Kumar et al., 2005; Marieb et al., 2009), which are implicated in normal aging processes (Kumar et al., 2005). Cells are said to be in an oxidative stress state when their levels of reactive oxygen species (ROS) exceed their antioxidant capacity (Kumar et al., 2005). ROS are characterized by their high reactivity stemming from their unpaired valance electrons. These electrons can damage or modify the function of RNA, DNA and proteins. Given that neurons are unable to limit their glucose uptake (Brownlee, 2001), possess numerous mitochondria and long axons make them highly sensitive to oxidative damage. Excessive glucose metabolism by the mitochondrial respiratory chains increases the generation of superoxide anions (Nishikawa et al., 2000).
As seen in Figure 2, mitochondrial superoxide anions reduce glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity, which in turn reduces cells' anti-oxidative capacity (Du et al., 2000). The exposure of mitochondria to ROS progressively induces mitochondrial dysfunction, which in turn promotes energy deficiency, axonal degeneration and DPN. Mitochondria are key regulators of cell survival and apoptosis. Damaged mitochondria trigger axon degeneration through caspase activation and cycles of fusion and fission (Green and Reed, 1998). The fission of mitochondria is partly regulated by dynamin related protein 1 (DRP1) (Twig et al., 2008). Increased DRP1 levels have been associated with mitochondria dysfunction, reduced ATP production, and axonal degeneration (Leinninger et al., 2006). DRP1 is notably upregulated in the axons of diabetic patients (Leinninger et al., 2006). Overall, an excess of ROS generation along with the inability of neurons to metabolize free radicals can promote the progressive loss of organelles and dysfunction in nuclear cell membranes (Figueroa-Romero et al., 2008).
ROS also reduce axon neurotrophic factor (IGF-1, IGF-II, NGF, and NT-3) production levels, thereby impairing neurons' ability to regenerate (Ishii, 1995; Tomlinson et al., 1997). High glucose levels stimulate the generation of pro-oxidant and highly reactive advanced glycation end products (AGE; section Advanced Glycation End Products) (Baynes and Thorpe, 1999). AGEs and ROS appear to be interdependent (Metz et al., 2003; Monnier, 2003), and central to the etiology of neurovascular dysfunction (Cameron et al., 2001). AGE generation is enhanced by oxygen and ROS; AGE formation can trigger ROS generation and oxidative damage (Monnier, 2003). Finally, hyperglycemia promotes the over-activation of polyol pathways (section Polyol Pathway), reducing cells' NADPH/NADP+ ratios and neurons' antioxidant capacities (Figueroa-Romero et al., 2008). Chronic hyperglycemia also enhances PKC activity, either through PLC-DAG pathways, or by reducing DAG-kinase activity (Xia et al., 1994) (King and Loeken, 2004). Enhanced PKC activity also increases mitochondrial NADPH oxidase activity (Inoguchi et al., 2000), further enhancing ROS levels (Balbi et al., 2018). A visual summary of ROS effects on neurons and glia can be found in Figures 2, 3.
ROS are normally metabolized by endogenous antioxidant enzymes such as superoxide dismutase, catalase and glutathione peroxidase, and by certain vitamins such as A, C (ascorbic acid) and E (tocopherol) (Figueroa-Romero et al., 2008; Midaoui et al., 2015; Talbot et al., 2016a). Nutritional supplementation with antioxidants was shown to reduce DPN in rodents (Pop-Busui et al., 2006). While alpha-lipoic acid and superoxide dismutase improved symptoms and electroneurographic parameters among subjects with diabetic neuropathy (Bertolotto and Massone, 2012), clinical studies generally have shown mixed results in terms of antioxidant efficacy (Oyenihi et al., 2015). Overall, it is believed that increasing the bioavailability of antioxidant, as well as associated reductive stress, have limited impact on the patients' health outcome.
Reactive Nitrogen Species
Nitric oxide (NO) is a potent vasoactive gas formed by three nitric oxide synthase (NOS) isoenzymes: neuronal (nNOS), endothelial (eNOS), and inducible (iNOS) (Kumar et al., 2005). In physiology, eNOS releases NO, which dilates vascular endothelial cells (Kumar et al., 2005) and reduces platelet aggregation (Riddell and Owen, 1999). In the context of inflammation, as is seen in diabetes, iNOS is overexpressed/activated, producing large amounts of NO (~100-fold than other NOS) (Vareniuk et al., 2008; Haddad and Couture, 2016). In DPN, iNOS hyperactivation is found in keratinocytes, macrophages, leukocytes, sensory neurons, and microglia (Zochodne et al., 2000). Excess NO from nNOS acts as a pro-nociceptive mediator in sensory C fibers (Matsui et al., 2010). It can also overactivate neuronal NADPH oxidase and mitochondrial xanthine oxidase. These effects reduce the antioxidant capacity of cells and contribute to ROS overproduction. Hyperglycemia-induced ROS react with cellular and/or circulating NO to form reactive nitrogen species (RNS) and peroxynitrite (Zochodne et al., 2000). RNS react with the thiol groups of SNAP proteins (Di Stasi et al., 2002), impairing the formation of neuronal regeneration cones. Consequently, RNS severely impact the capacity of neurons to repair themselves after oxidative damage (Kennedy and Zochodne, 2005). Currently, no therapy aims to reduce RNS generation, as it is still considered to be a contributing factor to DPN rather than an inducer. Please refer to Figures 2, 3 for visual summaries of NO interactions with neurons and glia.
Advanced Glycation end Products
Advanced glycation end products (AGE) comprise a heterogeneous group of molecules formed by the non-enzymatic reaction of a sugar with an amino acid, a protein, a lipid, or a nucleic acid (Marieb et al., 2009). AGE precursors pass through several dehydration and redox reactions and molecular rearrangements to form AGEs (Sugimoto et al., 2008). The initial reaction leading to AGE formation is reversible, and depends on the available quantity of substrate (glucose) (Brownlee, 2005). However, in cases of chronic hyperglycemia (diabetes), AGE precursors are not degraded, but rather build in numbers, thereby generating AGEs (Sugimoto et al., 2008). As seen in Figure 2, AGEs are highly reactive, and can affect any type of protein, including matrix, basal membrane and structural proteins (Sugimoto et al., 2008). For example, AGEs bind to and modify the myelin of nervous fibers, prompting their phagocytosis by circulating macrophages or microglia (Bruck and Friede, 1990). This process contributes to classical DPN segmental demyelination (Said, 2007). AGEs can also directly interact with tubulin and actin neurofilaments found in neurons' axonal cytoskeletons (Sugimoto et al., 2008). AGE-directed modification of these proteins impairs axonal transport, and promotes axonal atrophy and/or degeneration (Sugimoto et al., 2008). The glycation of extracellular matrix (laminin) membranes is AGE-mediated and can counteract the innate ability of neurons to self-repair (Duran-Jimenez et al., 2009; Singh et al., 2014). AGE additionally binds to specific membrane receptors known as RAGE (Haslbeck et al., 2005), driving the transcription of pro-inflammatory mediators (Brownlee, 2005). RAGE stimulation increases matrix metalloproteinase production, which can further exacerbate nerve fiber damage (King, 2001). AGEs demonstrably accumulate in hyperglycemic patients experiencing retinopathy, nephropathy, hypertension and neuropathy (Brownlee, 2005). Environmental pollutants, smoking, and poor nutrition also enhance AGE formation (Sugimoto et al., 2008). Avoiding these factors can help control AGE formation and its associated damage (Sugimoto et al., 2008; Singh et al., 2014). Specific inhibitors such as aminoguanidine improve patients' nerve conduction velocity and neuronal blood flow, in addition to mitigating apoptosis and oxidative stress (Sugimoto et al., 2008; Orman et al., 2015).
Ion Imbalance
Na+/K+ ATPase Pump
The Na+/K+ ATPase pump is a ubiquitous, energy-dependent enzyme implicated in the cellular membrane transport of ions. Using ATP, it transfers three sodium ions outside the cell in exchange for two potassium ions transported into the cell (Kumar et al., 2005). In doing so, it maintains the membrane's electric potential and nerve conductance (Creange et al., 2006). During hyperglycemia, impairments to polyol pathways (section Polyol Pathway) and PKC activity alter the function of the Na+/K+ ATPase pump leading to faulty nerve conduction (Creange et al., 2006). Reduced intracellular potassium levels curbed nodal potassium conductance, thereby affecting axonal excitability (Misawa et al., 2005). Hypokalemia can also alter Ca2+/K+ pump function, leading to neuronal hypocalcaemia.
Calcium
In early stages of DPN, the elevated levels of intracellular calcium disrupt nerve conductance (Kostyuk et al., 2001) and can, through calcium cytotoxicity, cause irreversible damage to the nerve fibers (Creange et al., 2006). Conversely, the hypocalcaemia observed in later stages of DPN mediates axonal degeneration, as seen in Figure 2 (Gispen and Hamers, 1994). Overall, impaired neurotrophic factor levels in DPN-afflicted neurons drive mitochondrial depolarization and Ca2+ concentration impairment, which in turn negatively impacts the TCA cycle and ATP production (Fernyhough and Calcutt, 2010). ATP dysregulation impairs cellular calcium homeostasis, reducing levels of endoplasmic reticulum (ER) and plasma membrane Ca2+ pumps (PMCA), as evidenced in streptozotocin (STZ)-treated rats. The impairment of ER calcium homeostasis disrupts protein synthesis, post-translational modification, and trafficking, all of which may contribute to distal axonal degeneration (Fernyhough and Calcutt, 2010). T-type calcium channel blockers notably improve thermal and mechanical hypersensitivity in T2D mice (Misawa et al., 2009).
Sodium
The DRG neurons of STZ-treated rats show an increased expression of sodium channels (NaV1.3, NaV1.6, and NaV1.9) (Craner et al., 2002; Hong et al., 2004), which contributes to ectopic impulse generation and neuronal hypersensitivity (Fischer and Waxman, 2010). The groups of Howe (Howe et al., 1977) and Wall (Wall and Gutnick, 1974) demonstrated that thermal, mechanical, and chemical stimuli thresholds are reduced following spontaneous electrical activity. Uninjured axons proximal to the affected neurons also exhibit ectopic discharge. Both of these phenomena result in increased electrical impulses in the spinal dorsal horn. The association between ectopic discharges and increased sodium channel expression can partly account for the therapeutic efficacy of anticonvulsant and tricyclic antidepressants in the treatment of DPN (Spruce et al., 2003).
Polyol Pathway
Cellular glucose is converted into pyruvate by the actions of diverse enzymes implicated in glycolysis. In hyperglycemic conditions, excess glucose is not oxidized, but rather directed to the polyol pathway (Oates, 2002). Firstly, aldose reductase (AR) metabolizes glucose into sorbitol, which is later transformed into fructose by sorbitol dehydrogenase. Fructose is, notably, ten times more potent than glucose in generating AGE (Oka and Kato, 2001) (section Advanced Glycation End Products). Aldose reductase and sorbitol dehydrogenase are characterized by their lowered substrate affinity (elevated Km); the concentration of available substrate is therefore the limiting factor of this reaction (Oates, 2002).
Elevated sorbitol levels have been associated with cellular and organ damage (Oyama et al., 2006). It is believed that sorbitol directly depletes bioavailable myoinositol (MI) and increases its expulsion from the cell (Oka and Kato, 2001; Oates, 2002). Elevated blood sugar also prevents sorbitol's cellular reuptake by saturating its membrane transporter. A deficit in MI alters the metabolism of phospho-inositides, reducing diacylglycerol (DAG) and inositol triphosphate (IP3) production. This results in a lesser activation of PKC, which is itself a key activator of the Na+/K+ ATPase pump (Oka and Kato, 2001). A reduction in Na+/K+ ATPase pump activity triggers an intracellular reduction in K+, combined with increases in the concentration of Na+. In neurons, imbalances in ionic charges directly contribute to DPN by generating conductance anomalies (Oka and Kato, 2001). In this context, decreases in intracellular sodium concentrations affect sodium-dependent membrane transport. This transport is implicated in the reuptake of several amino acids and MI, contributing to a retro-positive feedback loop (Das Evcimen and King, 2007; Oates, 2008). Disruptions in the polyol pathway also increase ROS levels (section Reactive Oxygen Species) by reducing the production of glutathione, in addition to intracellular levels of antioxidants (NADPH). Polyol pathway pathologies also reduce the production of NO, which can in turn enhance vessel constriction. This impairs endothelial cell function can lead to the onset of microangiopathy (section Microangiopathy) (Oka and Kato, 2001); the effects of polyol pathways in DPN can be seen in Figure 1.
Aldose Reductase
In patients with T1D, polymorphisms in the genes coding for aldose reductase (AR) can impair thermal nociceptive thresholds (Thamotharampillai et al., 2006). Higher AR levels also correlate with a higher severity of intra-epidermal nerve fiber loss (Hirai et al., 2000). AR specific inhibitors (ARi) have been shown to reverse or delay the onset of DPN in diabetic animals (Schemmel et al., 2009). While ARis are unavailable on the US market, they are currently being used clinically in Japan (Hotta et al., 1996). Epalrestat is currently the only commercially available inhibitor (Singh Grewal et al., 2016); and prevented the progression of diabetic neuropathy and retinopathy/nephropathy in neuropathic patients as compared to a control group (Hotta et al., 2012).
Neuron Extrinsic Factors Promoting Diabetic Pain Neuropathy
Several neuron-extrinsic factors contribute to the onset and maintenance of neuropathic pain. Recent data has highlighted the key contribution of immune cells, acting as extrinsic factors, in driving DPN. Normally, the immune and sensory nervous systems work in concert to preserve homeostasis. They do so via interactions and exchanges between receptors, cytokines and neuropeptides (Talbot et al., 2016b; Veiga-Fernandes and Mucida, 2016). While this bidirectional communication helps to protect humans from danger, it can also contribute to disease pathophysiology (Chiu et al., 2013; Wilson et al., 2013; Talbot et al., 2015, 2016b; Foster et al., 2017). In fact, the somatosensory nervous system is anatomically positioned within primary and secondary lymphoid tissues and mucosa so as to interact with the cells of the immune system (Downing and Miyan, 2000; Rosas-Ballina et al., 2011; McMahon et al., 2015; Talbot et al., 2015; Veiga-Fernandes and Mucida, 2016; HD iPSC Consortium., 2017). Nociceptors, when sensing immunocyte-released cytokines, lower their firing thresholds; in doing so, they incite pain hypersensitivity (Wilson et al., 2013). While various immunocytes contribute to this phenomenon, this review will focus on the crucial role of microglia. Finally, we will review the role of blood vessels, which supply oxygen and glucose to the nerve, in generating DPN.
Microglia
The central nervous system (CNS) is primarily composed of afferent and efferent neuron fibers that transport electrical signals to and from the periphery, oligodendrocytes that form and repair myelin, and astrocytes and microglia that support and protect neurons (Marieb et al., 2009). Microglial cells represent 10–15% of all cells found within the human brain (Foster et al., 2015). Glia act as resident macrophage-related cells of the CNS, comprising the first line of defense against pathogen invasion, generating innate immune responses by recognizing, sequestering and processing antigens (Lawson et al., 1992). While there are two major types of microglia: resident, and perivascular (Gosselin et al., 2010), which express receptors for most inflammatory neurotransmitters (Hickey and Kimura, 1988), it seems that glia exists in nine distinct subtypes with different functions, appearance, and presence. Resident microglial cells are bone marrow-derived hematopoietic cells that invade the CNS during embryonic development (Pocock and Kettenmann, 2007). These are very rarely replaced, and rapidly proliferate while activated (Milligan and Watkins, 2009). Conversely, perivascular microglial cells are continuously replenished by bone marrow-derived hematopoietic precursors (Gosselin et al., 2010), particularly during CNS inflammation (Romero-Sandoval et al., 2008). Perivascular microglia can alter the blood-brain barrier's permeability, and exert anti-inflammatory effects, while resident microglial cells exert both pro- and anti-inflammatory effects (Milligan and Watkins, 2009).
While on a polarization continuum, microglia activation can be classified as either resting or activated. A resting microglial cell possesses a small soma with thin and ramified processes (Tsuda et al., 2005), express immunoreceptors (Lawson et al., 1992), and perform immune roles to maintain CNS homeostasis (Tsuda et al., 2005). Upon activation due to trauma, inflammation, or infection, microglia undergo several stereotypic changes in morphology, gene expression, function, and number (Tsuda et al., 2005). They upregulate various transmitters and receptors, including the complement receptor 3 (CR3) (Eriksson et al., 1993; Lassmann et al., 1993), major histocompatibility complex 2 (MHC2) (Shi et al., 2017), TLR4 (Sweitzer et al., 2002), and CD14 (Sweitzer et al., 2002). The intracellular events promoting glial cell activation remain unclear, but are known to involve the activation of cannabinoid CB2 receptors (Tanga et al., 2004), kinin B1 receptors (Li and Kim, 2017), P2X4R (Noda et al., 2007), NK-1R (Inoue, 2008), CX3CR-1 (Abbadie et al., 2009; Gao and Ji, 2010; Zhou et al., 2010), CCR-2 (Zhang and De Koninck, 2006; Milligan et al., 2008), MMP9 (Thacker et al., 2009), BDNF-R (Kawasaki et al., 2008), TLR3 (Tender et al., 2010) and TLR4 (Kim et al., 2007); leading to the phosphorylation of p38 MAPK (Jin et al., 2003; Tsuda et al., 2004; Tanga et al., 2005; Daulhac et al., 2006; Ji et al., 2009). For more information on microglia intracellular signaling refer to Popiolek-Barczyk and Mika (Chang et al., 2010). Furthermore, microglial activation can occur via endogenous pro-inflammatory signals (IL-1β, TNFα, IL-6, and NO), opioids (Popiolek-Barczyk and Mika, 2016) or heat shock protein (Hutchinson et al., 2008). Activated microglial soma increase in size and their long and thin ramifications withdraw, ultimately resulting in an amoeboid shape with few ramifications (Tsuda et al., 2005). They have implicated microglia in reward behavior (Costigan et al., 1998) as well as in the onset of chronic neurodegenerative diseases (Taylor et al., 2015; Hammond et al., 2018), such as lupus erythematosus (Salter and Stevens, 2017), Huntington's chorea (Nestor et al., 2018), and Alzheimer's disease (Eriksson et al., 1993).
Painful Glia
Microglial cells produce and release various excitatory peptides, including PG, SP, EAA, NO, and ATP, and express selective receptors for immunomodulatory neurotransmitters. Both peptides and receptors allow microglia to detect, and respond to, neuronal signals, thereby generating autocrine or paracrine feed forward inflammatory loops (McMahon et al., 2005). For example, TNFα and MMPs activate microglial p38 MAPK in the spinal cord dorsal horn during peripheral neuropathic pain (Svensson et al., 2005). MMP9-induced pro-IL-1β cleavage leads to p38 MAPK phosphorylation in microglia during the onset and early stages of neuropathic pain. MMP2-induced pro-IL-1β cleavage leads to astrocyte activation in later disease stages (Kawasaki et al., 2008). ATP-stimulated microglial P2X4R enhances the levels of intracellular Ca2+. Such influx activates various transcription factors, including NF-κB, p38, and ERK-MAPK (Tsuda et al., 2005), leading to the synthesis of pro-inflammatory cytokines (IL-1β, TNFα, and IL-6) (Watkins et al., 2001a; Marchand et al., 2005) or neuroexcitatory substances such as D-serine (Petrenko et al., 2003). This transcriptomic profile can initiate and maintain neuropathic pain by facilitating neuron-glial interactions (Hickey and Kimura, 1988; Abbadie et al., 2009). The interplay between neurons and glia can therefore sustain neuronal stimulation and sensitization by increasing glutamatergic stimulation and by reducing GABAergic inhibitory signals (Tsuda et al., 2005; Inoue, 2006; Scholz and Woolf, 2007; Biggs et al., 2010); the process can be seen in Figure 3.
In current pre-clinical literature, activated microglia have emerged as key drivers of pathological pain in chemotherapy-induced neuropathy and peripheral nerve and spinal cord injuries (Watkins et al., 2001b; Marchand et al., 2005; Tsuda et al., 2005; Daulhac et al., 2006; Pocock and Kettenmann, 2007; Gadani et al., 2015; McMahon et al., 2015). For example, CB2 receptor agonists dramatically attenuate iNOS induction and ROS generation in LPS-activated microglia (Ribeiro et al., 2013). The incitement of inflammation signals microglia to migrate, proliferate, synthesize and release pro-inflammatory mediators that maintain neuron activation. In keeping with the fact that an intrathecal injection of activated microglia induces both thermal hyperalgesia and tactile allodynia (Tsuda et al., 2003; Narita et al., 2006), while resting microglia or activated astrocytes are without effect (Narita et al., 2006). Blockades of p38 MAPK (Tanga et al., 2005; Ji et al., 2009), CX3CR-1 (Milligan et al., 2004; Verge et al., 2004; Sun et al., 2007) or P2X4R (Tsuda et al., 2003) have been shown to alleviate chronic pain in rodents. Other chemokines, such as RANTES, IP-10, and SDF1 are also implicated in enhanced microglia migration, infiltration, phagocytosis; they are therefore contributors to microglia-induced neuropathic pain (White et al., 2007).
Salter and Beggs have made several discoveries linking neuronal hypersensitivity to overactive microglia (Beggs and Salter, 2016). Firstly, they were able to show that nerve injury activates microglia and causes them to express P2X4R. They demonstrated that the IRF-8/IRF-5 transcriptional cascade clearly regulates the expression of P2X4R gene. Additionally, they showed that external stimulation (CCL2 and LPS) leads to the translocation of P2X4R protein from lysosome to cell surface (Tsuda and Inoue, 2016). A targeted silencing of P2X4R suppressed injury-induced tactile allodynia, while an intraspinal administration of P2X4R-expressing glia had the opposite effect (Tsuda et al., 2003). They also identified spinal dorsal horn neurons as a source of ATP (Masuda et al., 2016), and that ATP-stimulated microglia release brain derived neurotrophic factor (BDNF). BDNF limits GABAergic inhibitory signals sent to afferent nociceptor neurons (Torsney and MacDermott, 2005); this enhances their activity by uncoupling the transmembrane anion gradient (Coull et al., 2005). A disruption of chloride transport changed spinal lamina I neurons' phenotype, causing them to (i) increase nociceptive responsiveness, (ii) relay innocuous mechanical inputs, and (iii) generate spontaneous bursts of activity; respectively, accounting for (a) hyperalgesia, (b) mechanical allodynia, and (c) spontaneous pain (Keller et al., 2007). The microglia-to-neuron P2X4R -BDNF-KCC2 axis was also found to drive opioid-induced thermal hyperalgesia. Interestingly, Salter and Beggs found that this hyperalgesia mechanistically differs from opioid-induced tolerance (Beggs and Salter, 2016). Microglia-derived BDNF also emerged in their research as a negative regulator of reward in opioid-dependent states (Taylor et al., 2016), while the Panx1-mediated release of microglial ATP controls morphine withdrawal without affecting opiate-induced analgesia (Burma et al., 2017).
Peripheral tissue injury was found to increase the intensity, spatial distribution, and persistence of Iba-1+ microglial activity within the spinal dorsal horn, resulting in a long-lasting priming of withdrawal reflex sensitivity and microglial responsiveness (Beggs et al., 2012). MCP-1+ neurons drive the bone marrow-derived microglial infiltration of injured spinal cords. These neurons also facilitate glial activation and drive mechanical allodynia (Zhang et al., 2007). CCL21, a microglial activator, is also found to be upregulated in the cell bodies of spinothalamic tract neurons following nerve injury. This molecule is transported rostrally to the thalamus, where it activates microglia and drives neuron hyperexcitability (Zhao et al., 2007b). Spinal microglia consequently remain activated for more than 3 months following nerve injury in rodents, thereby maintaining chronic pain (Echeverry et al., 2017). Newly-generated microglia also appear to be coded with an inherent “memory” of previous injuries, contributing to long-lasting neuropathic pain (Yao et al., 2016).
Following a spinal cord injury, microglial ERK kinases are activated, prompting the intraspinal release of PGE2, which results in spinal cord dorsal horn neuron hyperresponsiveness (Zhao et al., 2007a). Following the spinal release of IL-1β and IL-6, fractalkine triggers pain by activating microglia expressing CX3CR1 (Milligan et al., 2004, 2005). IL-1β upregulates both neuronal NMDAR phosphorylation and expression. These effects enhance NMDAR conductivity (Zhang et al., 2008) and calcium influx (Viviani et al., 2003), increasing neuronal excitability and synaptic strength (Beattie et al., 2002; Stellwagen and Malenka, 2006). Microglial NO and PGE2 can also increase the excitability of pain-projecting neurons (Besson, 1999). Overall, a modulation of microglial polarization was shown to alleviate neuropathic pain (Chang et al., 2010; Piotrowska et al., 2016; Xu et al., 2018). Intriguingly, microglia only induce mechanical pain hypersensitivity in male rodents (Mapplebeck et al., 2018), as T-lymphocytes appear to be responsible for this mechanism in females (Sorge et al., 2015).
Microglia, an Emerging Target in DPN
Given that db/db mice and T2D patients show increased levels of activated microglia (Arroba and Valverde, 2017), and that the pharmacological inhibition of spinal resident microglia reverses painful neuropathy (Wodarski et al., 2009; Sun et al., 2015; Lin, 2017; Zhang et al., 2018), we take the overall view that microglia may drive DPN (Figure 1). Activated microglia levels have been found to correlate with thalamus hyperresponsiveness (Fischer and Waxman, 2010), and the brain thalami of DPN patients demonstrate increases in blood flow (Paulson et al., 2007), spontaneous neuronal activity (Fischer et al., 2009), receptive field size enhancement (Fischer et al., 2009), and alterations in neuronal connectivity (Cauda et al., 2010). Additionally, activated microglia drive DPN in STZ-treated rats (Wodarski et al., 2009; Talbot et al., 2010); this process can successfully be reversed by gabapentin (Wodarski et al., 2009) or minocycline treatment (Talbot et al., 2010).
Additionally, diabetes-induced hyperglycemia enhances microglial NADPH oxidase and iNOS activation (Figure 3), promoting the production of ROS (Quan et al., 2007, 2011) and peroxynitrite (Li et al., 2005). As a result of the iNOS-NO-NRS axis, activated microglia are a major source of free radicals in the spinal cords of animals with DPN (Li et al., 2005; Candelario-Jalil et al., 2007). Glial-released NO inhibits neuronal cytochrome oxidase, blocking mitochondrial respiration, which in turn depletes the production of ATP (Brown and Bal-Price, 2003). Moreover, ROS and RNS deplete endothelial cell NO levels, contributing to the generation of microangiopathy (section Microangiopathy). A reduction in neuronal blood supply leads to hypoxia and mitochondrial dysfunction; this results in cellular energy deficits, and ultimately, neuron death. Finally, elevated ROS levels also impair axonal transport (Larsen and Sidenius, 1989) and the capacity of the nerve to repair itself (Longo et al., 1986), further worsening nerve health in DPN patients.
Microglia exposed to high glucose levels show increases in mRNA expression, and secrete TNFα and MCP-1, leading to neuronal activation (Quan et al., 2011). The spinal microglia's activation of P2Y12 and P2Y13 receptors triggers the production of IL-1β and IL-6 in a rat model of DPN (Liu et al., 2017; Zhou et al., 2018). The activation of microglial RAGE (Thornalley, 1998) leads to the release of chemokines CCL3, CCL5, and CXCL12, which activate microglia (Bianchi et al., 2011). The resulting activated microglia actively phagocyte neuronal myelin, thereby promoting DPN (Mosley and Cuzner, 1996).
Microangiopathy
Microangiopathy is characterized by the shrinking and weakening of small blood vessels. This pathology leads to a reduction in blood flow, protein leakage, and bleeding. In diabetic states, endothelial cells take up excessively large quantities of circulating glucose, raising the production of AGE (section Advanced Glycation End Products). These AGEs enhance the proliferation of endothelial cells, which leads to a thickening of basal membranes and progressive vessel occlusion, which are associated with reduced neuronal blood flow (Tuck et al., 1984; Cameron et al., 1991) and ischemia. In turn, the ischemia reduces neuronal oxygen and nutrient supplies, triggering nerve fiber loss (Yagihashi et al., 2007). Microangiopathy in neuron-irrigating vessels also impairs their capacity to regenerate and repair (Kennedy and Zochodne, 2005). A thickening of basement membranes and endothelial cell swelling thereby positively correlates with reduced nerve fiber myelin densities (Yagihashi et al., 2007). The occlusion of neuronal capillaries promotes axonal degeneration and a dysfunctional loss of sensory perception, as typically observed in the later stages of DPN.
Multifocal nerve lesions and alterations in endoneurial capillaries indicate a role for circulatory factors in the symmetrical form of DPN (Dyck et al., 1986; King et al., 1989). Peripheral nerve fiber loss in DPN is well-associated with the increased migration and infiltration of inflammatory cytokines released by T-lymphocytes and macrophages (Said et al., 2003; Said, 2007). These cytokines increase vessel damage and promote microangiopathy (Said, 2007). Considering that microglia and macrophages share similar roles and inflammatory characteristics, it is probable that microglia may also promote microangiopathy within the CNS (Figure 3).
Conclusion and Future Therapeutic Directions
Multiple molecular mechanisms generate and amplify hyperglycemia-induced neuropathic pain. This collaboration involves voltage-gated ion channels, ligand-gated channels, cytokine receptors, direct myelin damage (Edwards et al., 2008; Vincent et al., 2008), neuronal depolarization (Abbadie et al., 2009), conduction impairment (Kramer et al., 2004), a loss of interneuron inhibitory input (Wood, 2008), Aβ-fiber sprouting (Yasuda et al., 2003), and neuronal death (Inoue, 2006). This multicentric view accounts for why hyperglycemia-induced DPN remains highly refractory to treatment paradigms. Given such potential heterogeneity amongst patients, personalizing of DPN management may prove useful. This will necessitate improved diagnostic methods and personalized medicine tailored to the specific pathology in question.
While design, safety and drug efficacy often vary between rodent and clinical models of disease, calcium channel α2-delta ligands (pregabalin, gabapentin), antidepressants (TCA and SSRI), and opioid-like drugs as well as topical agents including capsaicin, lidocaine, or botulinum toxin A can help alleviate patients' DPN (Finnerup et al., 2015). The long-term efficacies of these approaches have yet to be demonstrated. Overall, the high plasticity of microglial phenotypic and transcriptomic changes persists even after glycemic control, inciting and maintaining neuron sensitization (Milligan et al., 2008). Therefore, the microglia, which potentially act upstream of pain neurons, should rightly be considered as a crucial therapeutic target in the treatment of diabetic pain neuropathy. Future therapies may therefore involve targeting specific receptors and signaling cascades which engage such deleterious neuro-immune crosstalk.
Author Contributions
TR, RC, AC, and ST conceived the study and wrote the manuscript while SCT, J-CW, MA, MB, TC, and JD contributed to its design.
Funding
This project has been made possible by the Canada Research Chair program (#950-231859) and the Brain Canada Foundation (ST) through the Canada Brain Research Fund, with the financial support of Health Canada and the Azrieli Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbadie, C., Bhangoo, S., De Koninck, Y., Malcangio, M., Melik-Parsadaniantz, S., and White, F. A. (2009). Chemokines and pain mechanisms. Brain Res. Rev. 60, 125–134. doi: 10.1016/j.brainresrev.2008.12.002
Arroba, A. I., and Valverde, A. M. (2017). Modulation of microglia in the retina: new insights into diabetic retinopathy. Acta Diabetol. 54, 527–533. doi: 10.1007/s00592-017-0984-z
Aschner, P., Gagliardino, J. J., Ilkova, H. M., Lavalle-Gonzalez, F. J., Ramachandran, A., Kaddaha, G., et al. (2018). No improvement in glycemic control or rates of diabetes-related complications for people with type 2 diabetes—results from 10 years of the international diabetes management practices study (IDMPS). Am. Diabetes Assoc. 67 (Suppl. 1):1583-P. doi: 10.2337/db18-1583-P
Balamurugan, A. N., Naziruddin, B., Lockridge, A., Tiwari, M., Loganathan, G., Takita, M., et al. (2014). Islet product characteristics and factors related to successful human islet transplantation from the Collaborative Islet Transplant Registry (CITR) 1999-2010. Am. J. Transl. 14, 2595–2606. doi: 10.1111/ajt.12872
Balbi, M. E., Tonin, F. S., Mendes, A. M., Borba, H. H., Wiens, A., Fernandez-Llimos, F., et al. (2018). Antioxidant effects of vitamins in type 2 diabetes: a meta-analysis of randomized controlled trials. Diabetol. Metab. Syndr. 10:18. doi: 10.1186/s13098-018-0318-5
Bautista, D. M., Jordt, S. E., Nikai, T., Tsuruda, P. R., Read, A. J., Poblete, J., et al. (2006). TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124, 1269–1282. doi: 10.1016/j.cell.2006.02.023
Baynes, J. W., and Thorpe, S. R. (1999). Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 48, 1–9. doi: 10.2337/diabetes.48.1.1
Beattie, E. C., Stellwagen, D., Morishita, W., Bresnahan, J. C., Ha, B. K., Von Zastrow, M., et al. (2002). Control of synaptic strength by glial TNFalpha. Science 295, 2282–2285. doi: 10.1126/science.1067859
Beggs, S., Currie, G., Salter, M. W., Fitzgerald, M., and Walker, S. M. (2012). Priming of adult pain responses by neonatal pain experience: maintenance by central neuroimmune activity. Brain 135, 404–417. doi: 10.1093/brain/awr288
Beggs, S., and Salter, M. W. (2016). SnapShot: microglia in disease. Cell 165, 1294–1294.e1. doi: 10.1016/j.cell.2016.05.036
Bertolotto, F., and Massone, A. (2012). Combination of alpha lipoic acid and superoxide dismutase leads to physiological and symptomatic improvements in diabetic neuropathy. Drugs R D 12, 29–34. doi: 10.2165/11599200-000000000-00000
Besson, J. M. (1999). The neurobiology of pain. Lancet 353, 1610–1615. doi: 10.1016/S0140-6736(99)01313-6
Bianchi, R., Kastrisianaki, E., Giambanco, I., and Donato, R. (2011). S100B protein stimulates microglia migration via RAGE-dependent up-regulation of chemokine expression and release. J. Biol. Chem. 286, 7214–7226. doi: 10.1074/jbc.M110.169342
Biggs, J. E., Lu, V. B., Stebbing, M. J., Balasubramanyan, S., and Smith, P. A. (2010). Is BDNF sufficient for information transfer between microglia and dorsal horn neurons during the onset of central sensitization? Mol. Pain 6:44. doi: 10.1186/1744-8069-6-44
Billings, L. K., and Florez, J. C. (2010). The genetics of type 2 diabetes: what have we learned from GWAS? Ann. N. Y. Acad. Sci. 1212, 59–77. doi: 10.1111/j.1749-6632.2010.05838.x
Binshtok, A. M., Wang, H., Zimmermann, K., Amaya, F., Vardeh, D., Shi, L., et al. (2008). Nociceptors are interleukin-1beta sensors. J. Neurosci. 28, 14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008
Brown, G. C., and Bal-Price, A. (2003). Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol. Neurobiol. 27, 325–355. doi: 10.1385/MN:27:3:325
Brownlee, M. (2001). Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820. doi: 10.1038/414813a
Brownlee, M. (2005). The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54, 1615–1625. doi: 10.2337/diabetes.54.6.1615
Bruck, W., and Friede, R. L. (1990). L-fucosidase treatment blocks myelin phagocytosis by macrophages in vitro. J. Neuroimmunol. 27, 217–227. doi: 10.1016/0165-5728(90)90072-U
Burma, N. E., Bonin, R. P., Leduc-Pessah, H., Baimel, C., Cairncross, Z. F., Mousseau, M., et al. (2017). Blocking microglial pannexin-1 channels alleviates morphine withdrawal in rodents. Nat. Med. 23, 355–360. doi: 10.1038/nm.4281
Cameron, N. E., Cotter, M. A., and Low, P. A. (1991). Nerve blood flow in early experimental diabetes in rats: relation to conduction deficits. Am. J. Physiol. 261, E1–E8. doi: 10.1152/ajpendo.1991.261.1.E1
Cameron, N. E., Eaton, S. E., Cotter, M. A., and Tesfaye, S. (2001). Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia 44, 1973–1988. doi: 10.1007/s001250100001
Candelario-Jalil, E., de Oliveira, A. C., Graf, S., Bhatia, H. S., Hull, M., Munoz, E., et al. (2007). Resveratrol potently reduces prostaglandin E2 production and free radical formation in lipopolysaccharide-activated primary rat microglia. J. Neuroinflammation 4:25. doi: 10.1186/1742-2094-4-25
Cauda, F., D'Agata, F., Sacco, K., Duca, S., Cocito, D., Paolasso, I., et al. (2010). Altered resting state attentional networks in diabetic neuropathic pain. J. Neurol. Neurosurg. Psychiatry 81, 806–811. doi: 10.1136/jnnp.2009.188631
Cevikbas, F., Kempkes, C., Buhl, T., Mess, C., Buddenkotte, J., and Steinhoff, M. (2014). “Role of interleukin-31 and oncostatin M in itch and neuroimmune communication,” in Itch: Mechanisms and Treatment, eds E. Carstens and T. Akiyama (Boca Raton, FL: CRC Press/Taylor & Francis), 237–256.
Chang, K. H., de Pablo, Y., Lee, H. P., Lee, H. G., Smith, M. A., and Shah, K. (2010). Cdk5 is a major regulator of p38 cascade: relevance to neurotoxicity in Alzheimer's disease. J. Neurochem. 113, 1221–1229. doi: 10.1111/j.1471-4159.2010.06687.x
Chiu, I. M., Barrett, L. B., Williams, E. K., Strochlic, D. E., Lee, S., Weyer, A. D., et al. (2014). Transcriptional profiling at whole population and single cell levels reveals somatosensory neuron molecular diversity. Elife 3:e04660. doi: 10.7554/eLife.04660
Chiu, I. M., Heesters, B. A., Ghasemlou, N., Von Hehn, C. A., Zhao, F., Tran, J., et al. (2013). Bacteria activate sensory neurons that modulate pain and inflammation. Nature 501, 52–57. doi: 10.1038/nature12479
Clement, K., Vaisse, C., Lahlou, N., Cabrol, S., Pelloux, V., Cassuto, D., et al. (1998). A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 392, 398–401. doi: 10.1038/32911
Costigan, M., Mannion, R. J., Kendall, G., Lewis, S. E., Campagna, J. A., Coggeshall, R. E., et al. (1998). Heat shock protein 27: developmental regulation and expression after peripheral nerve injury. J. Neurosci. 18, 5891–5900. doi: 10.1523/JNEUROSCI.18-15-05891.1998
Coull, J. A., Beggs, S., Boudreau, D., Boivin, D., Tsuda, M., Inoue, K., et al. (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021. doi: 10.1038/nature04223
Craner, M. J., Klein, J. P., Renganathan, M., Black, J. A., and Waxman, S. G. (2002). Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann. Neurol. 52, 786–792. doi: 10.1002/ana.10364
Creange, A., Boerio, D., and Lefaucheur, J. P. (2006). Inflammatory demyelinating neuropathies: classification, evolution and prognosis. J. Soc. Biol. 200, 301–306. doi: 10.1051/jbio:2006035
Crofford, O. B. (1995). Diabetes control and complications. Annu. Rev. Med. 46, 267–279. doi: 10.1146/annurev.med.46.1.267
Daousi, C., MacFarlane, I. A., Woodward, A., Nurmikko, T. J., Bundred, P. E., and Benbow, S. J. (2004). Chronic painful peripheral neuropathy in an urban community: a controlled comparison of people with and without diabetes. Diabet. Med. 21, 976–982. doi: 10.1111/j.1464-5491.2004.01271.x
Das Evcimen, N., and King, G. L. (2007). The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol. Res. 55, 498–510. doi: 10.1016/j.phrs.2007.04.016
Daulhac, L., Mallet, C., Courteix, C., Etienne, M., Duroux, E., Privat, A. M., et al. (2006). Diabetes-induced mechanical hyperalgesia involves spinal mitogen-activated protein kinase activation in neurons and microglia via N-methyl-D-aspartate-dependent mechanisms. Mol. Pharmacol. 70, 1246–1254. doi: 10.1124/mol.106.025478
Davis, J. B., Gray, J., Gunthorpe, M. J., Hatcher, J. P., Davey, P. T., Overend, P., et al. (2000). Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405, 183–187. doi: 10.1038/35012076
De Brito Gariepy, H., Talbot, S., Senecal, J., and Couture, R. (2013). Brain kinin B(1) receptor contributes to the onset of stereotypic nocifensive behavior in rat. Behav. Brain Res. 241, 17–26. doi: 10.1016/j.bbr.2012.11.032
Di Stasi, A. M., Mallozzi, C., Macchia, G., Maura, G., Petrucci, T. C., and Minetti, M. (2002). Peroxynitrite affects exocytosis and SNARE complex formation and induces tyrosine nitration of synaptic proteins. J. Neurochem. 82, 420–429. doi: 10.1046/j.1471-4159.2002.00980.x
Downing, J. E., and Miyan, J. A. (2000). Neural immunoregulation: emerging roles for nerves in immune homeostasis and disease. Immunol. Today 21, 281–289. doi: 10.1016/S0167-5699(00)01635-2
Du, X. L., Edelstein, D., Rossetti, L., Fantus, I. G., Goldberg, H., Ziyadeh, F., et al. (2000). Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. U.S.A. 97, 12222–12226. doi: 10.1073/pnas.97.22.12222
Duran-Jimenez, B., Dobler, D., Moffatt, S., Rabbani, N., Streuli, C. H., Thornalley, P. J., et al. (2009). Advanced glycation end products in extracellular matrix proteins contribute to the failure of sensory nerve regeneration in diabetes. Diabetes 58, 2893–2903. doi: 10.2337/db09-0320
Dyck, P. J., Lais, A., Karnes, J. L., O'Brien, P., and Rizza, R. (1986). Fiber loss is primary and multifocal in sural nerves in diabetic polyneuropathy. Ann. Neurol. 19, 425–439. doi: 10.1002/ana.410190503
Echeverry, S., Shi, X. Q., Yang, M., Huang, H., Wu, Y., Lorenzo, L. E., et al. (2017). Spinal microglia are required for long-term maintenance of neuropathic pain. Pain 158, 1792–1801. doi: 10.1097/j.pain.0000000000000982
Edwards, J. L., Vincent, A. M., Cheng, H. T., and Feldman, E. L. (2008). Diabetic neuropathy: mechanisms to management. Pharmacol. Ther. 120, 1–34. doi: 10.1016/j.pharmthera.2008.05.005
Eriksson, N. P., Persson, J. K., Svensson, M., Arvidsson, J., Molander, C., and Aldskogius, H. (1993). A quantitative analysis of the microglial cell reaction in central primary sensory projection territories following peripheral nerve injury in the adult rat. Exp. Brain Res. 96, 19–27. doi: 10.1007/BF00230435
Fernyhough, P., and Calcutt, N. A. (2010). Abnormal calcium homeostasis in peripheral neuropathies. Cell Calcium 47, 130–139. doi: 10.1016/j.ceca.2009.11.008
Figueroa-Romero, C., Sadidi, M., and Feldman, E. L. (2008). Mechanisms of disease: the oxidative stress theory of diabetic neuropathy. Rev. Endocr. Metab. Disord. 9, 301–314. doi: 10.1007/s11154-008-9104-2
Finnerup, N. B., Attal, N., Haroutounian, S., McNicol, E., Baron, R., Dworkin, R. H., et al. (2015). Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 14, 162–173. doi: 10.1016/S1474-4422(14)70251-0
Fischer, T. Z., Tan, A. M., and Waxman, S. G. (2009). Thalamic neuron hyperexcitability and enlarged receptive fields in the STZ model of diabetic pain. Brain Res. 1268, 154–161. doi: 10.1016/j.brainres.2009.02.063
Fischer, T. Z., and Waxman, S. G. (2010). Neuropathic pain in diabetes–evidence for a central mechanism. Nat. Rev. Neurol. 6, 462–466. doi: 10.1038/nrneurol.2010.90
Foster, S. L., Seehus, C. R., Woolf, C. J., and Talbot, S. (2017). Sense and immunity: context-dependent neuro-immune interplay. Front. Immunol. 8:1463. doi: 10.3389/fimmu.2017.01463
Foster, S. L., Talbot, S., and Woolf, C. J. (2015). CNS injury: IL-33 sounds the alarm. Immunity 42, 403–405. doi: 10.1016/j.immuni.2015.02.019
Gadani, S. P., Walsh, J. T., Smirnov, I., Zheng, J., and Kipnis, J. (2015). The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron 85, 703–709. doi: 10.1016/j.neuron.2015.01.013
Gao, Y. J., and Ji, R. R. (2010). Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol. Ther. 126, 56–68. doi: 10.1016/j.pharmthera.2010.01.002
Gerbi, A., Maixent, J. M., Barbey, O., Jamme, I., Pierlovisi, M., Coste, T., et al. (1998). Alterations of Na,K-ATPase isoenzymes in the rat diabetic neuropathy: protective effect of dietary supplementation with n-3 fatty acids. J. Neurochem. 71, 732–740. doi: 10.1046/j.1471-4159.1998.71020732.x
Gispen, W. H., and Hamers, F. P. (1994). Calcium and neuronal dysfunction in peripheral nervous system. Ann. N. Y. Acad. Sci. 747, 419–430. doi: 10.1111/j.1749-6632.1994.tb44426.x
Gosselin, R. D., Suter, M. R., Ji, R. R., and Decosterd, I. (2010). Glial cells and chronic pain. Neuroscientist 16, 519–531. doi: 10.1177/1073858409360822
Green, D. R., and Reed, J. C. (1998). Mitochondria and apoptosis. Science 281, 1309–1312. doi: 10.1126/science.281.5381.1309
Groop, L., and Lyssenko, V. (2008). Genes and type 2 diabetes mellitus. Curr. Diabetes Rep. 8, 192–197. doi: 10.1007/s11892-008-0033-y
Guastella, V., and Mick, G. (2009). Strategies for the diagnosis and treatment of neuropathic pain secondary to diabetic peripheral sensory polyneuropathy. Diabetes Metab. 35, 12–19. doi: 10.1016/j.diabet.2008.09.003
Haddad, Y., and Couture, R. (2016). Interplay between the kinin B1 receptor and inducible nitric oxide synthase in insulin resistance. Br. J. Pharmacol. 173, 1988–2000. doi: 10.1111/bph.13491
Haigh, R. C., and Blake, D. R. (2001). Understanding pain. Clin. Med. 1, 44–48. doi: 10.7861/clinmedicine.1-1-44
Hammond, T. R., Robinton, D., and Stevens, B. (2018). Microglia and the brain: complementary partners in development and disease. Annu. Rev. Cell Dev. Biol. 34, 523–544. doi: 10.1146/annurev-cellbio-100616-060509
Haslbeck, K. M., Schleicher, E., Bierhaus, A., Nawroth, P., Haslbeck, M., Neundorfer, B., et al. (2005). The AGE/RAGE/NF-(kappa)B pathway may contribute to the pathogenesis of polyneuropathy in impaired glucose tolerance (IGT). Exp. Clin. Endocrinol. Diabetes 113, 288–291. doi: 10.1055/s-2005-865600
HD iPSC Consortium. (2017). Developmental alterations in Huntington's disease neural cells and pharmacological rescue in cells and mice. Nat. Neurosci. 20, 648–660. doi: 10.1038/nn.4532
Hefti, F. F., Rosenthal, A., Walicke, P. A., Wyatt, S., Vergara, G., Shelton, D. L., et al. (2006). Novel class of pain drugs based on antagonism of NGF. Trends Pharmacol. Sci. 27, 85–91. doi: 10.1016/j.tips.2005.12.001
Hickey, W. F., and Kimura, H. (1988). Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science 239, 290–292. doi: 10.1126/science.3276004
Hirai, A., Yasuda, H., Joko, M., Maeda, T., and Kikkawa, R. (2000). Evaluation of diabetic neuropathy through the quantitation of cutaneous nerves. J. Neurol. Sci. 172, 55–62. doi: 10.1016/S0022-510X(99)00290-7
Hong, S., Morrow, T. J., Paulson, P. E., Isom, L. L., and Wiley, J. W. (2004). Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. Biol. Chem. 279, 29341–29350. doi: 10.1074/jbc.M404167200
Hotta, N., Kawamori, R., Fukuda, M., and Shigeta, Y. and Aldose Reductase Inhibitor-Diabetes Complications Trial Study Group. (2012). Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on progression of diabetic neuropathy and other microvascular complications: multivariate epidemiological analysis based on patient background factors and severity of diabetic neuropathy. Diabetes Med. 29, 1529–1533. doi: 10.1111/j.1464-5491.2012.03684.x
Hotta, N., Sakamoto, N., Shigeta, Y., Kikkawa, R., and Goto, Y. (1996). Clinical investigation of epalrestat, an aldose reductase inhibitor, on diabetic neuropathy in Japan: multicenter study. Diabetic Neuropathy Study Group in Japan. J. Diabetes Complications 10, 168–172. doi: 10.1016/1056-8727(96)00113-4
Howe, J. F., Loeser, J. D., and Calvin, W. H. (1977). Mechanosensitivity of dorsal root ganglia and chronically injured axons: a physiological basis for the radicular pain of nerve root compression. Pain 3, 25–41. doi: 10.1016/0304-3959(77)90033-1
Hutchinson, M. R., Zhang, Y., Brown, K., Coats, B. D., Shridhar, M., Sholar, P. W., et al. (2008). Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4). Eur. J. Neurosci. 28, 20–29. doi: 10.1111/j.1460-9568.2008.06321.x
Inoguchi, T., Li, P., Umeda, F., Yu, H. Y., Kakimoto, M., Imamura, M., et al. (2000). High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49, 1939–1945. doi: 10.2337/diabetes.49.11.1939
Inoue, K. (2006). The function of microglia through purinergic receptors: neuropathic pain and cytokine release. Pharmacol. Ther. 109, 210–226. doi: 10.1016/j.pharmthera.2005.07.001
Inoue, K. (2008). Purinergic systems in microglia. Cell Mol. Life Sci. 65, 3074–3080. doi: 10.1007/s00018-008-8210-3
Ishii, D. N. (1995). Implication of insulin-like growth factors in the pathogenesis of diabetic neuropathy. Brain Res. Rev. 20, 47–67. doi: 10.1016/0165-0173(94)00005-A
Ji, R. R., Befort, K., Brenner, G. J., and Woolf, C. J. (2002a). ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J. Neurosci. 22, 478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002
Ji, R. R., Gereau, R. W. IV, Malcangio, M., and Strichartz, G. R. (2009). MAP kinase and pain. Brain Res. Rev. 60, 135–148. doi: 10.1016/j.brainresrev.2008.12.011
Ji, R. R., Samad, T. A., Jin, S. X., Schmoll, R., and Woolf, C. J. (2002b). p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36, 57–68. doi: 10.1016/S0896-6273(02)00908-X
Ji, R. R., and Suter, M. R. (2007). p38 MAPK, microglial signaling, and neuropathic pain. Mol. Pain 3:33. doi: 10.1186/1744-8069-3-33
Jin, S. X., Zhuang, Z. Y., Woolf, C. J., and Ji, R. R. (2003). p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J. Neurosci. 23, 4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003
Julius, D. (2013). TRP channels and pain. Annu. Rev. Cell Dev. Biol. 29, 355–384. doi: 10.1146/annurev-cellbio-101011-155833
Kawasaki, Y., Xu, Z. Z., Wang, X., Park, J. Y., Zhuang, Z. Y., Tan, P. H., et al. (2008). Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat. Med. 14, 331–336. doi: 10.1038/nm1723
Keller, A. F., Beggs, S., Salter, M. W., and De Koninck, Y. (2007). Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Mol. Pain 3:27. doi: 10.1186/1744-8069-3-27
Kennedy, J. M., and Zochodne, D. W. (2005). Impaired peripheral nerve regeneration in diabetes mellitus. J. Peripher. Nerv. Syst. 10, 144–157. doi: 10.1111/j.1085-9489.2005.0010205.x
Kerr, B. J., Souslova, V., McMahon, S. B., and Wood, J. N. (2001). A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport 12, 3077–3080. doi: 10.1097/00001756-200110080-00019
Khoutorsky, A., and Price, T. J. (2017). Translational control mechanisms in persistent pain. Trends Neurosci. 41, 100–114. doi: 10.1016/j.tins.2017.11.006
Kim, D., Kim, M. A., Cho, I. H., Kim, M. S., Lee, S., Jo, E. K., et al. (2007). A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J. Biol. Chem. 282, 14975–14983. doi: 10.1074/jbc.M607277200
King, G. L., and Loeken, M. R. (2004). Hyperglycemia-induced oxidative stress in diabetic complications. Histochem. Cell Biol. 122, 333–338. doi: 10.1007/s00418-004-0678-9
King, R. H. (2001). The role of glycation in the pathogenesis of diabetic polyneuropathy. Mol. Pathol. 54, 400–408. doi: 10.1136/mp.54.6.400
King, R. H., Llewelyn, J. G., Thomas, P. K., Gilbey, S. G., and Watkins, P. J. (1989). Diabetic neuropathy: abnormalities of Schwann cell and perineurial basal laminae. Implications for diabetic vasculopathy. Neuropathol. Appl. Neurobiol. 15, 339–355. doi: 10.1111/j.1365-2990.1989.tb01234.x
Knip, M., and Siljander, H. (2008). Autoimmune mechanisms in type 1 diabetes. Autoimmun. Rev. 7, 550–557. doi: 10.1016/j.autrev.2008.04.008
Kostyuk, E., Voitenko, N., Kruglikov, I., Shmigol, A., Shishkin, V., Efimov, A., et al. (2001). Diabetes-induced changes in calcium homeostasis and the effects of calcium channel blockers in rat and mice nociceptive neurons. Diabetologia 44, 1302–1309. doi: 10.1007/s001250100642
Kramer, H. H., Schmelz, M., Birklein, F., and Bickel, A. (2004). Electrically stimulated axon reflexes are diminished in diabetic small fiber neuropathies. Diabetes 53, 769–774. doi: 10.2337/diabetes.53.3.769
Kumar, V., Abbas, A. K., Fausto, N., Robbins, S. L., and Cotran, R. S. (2005). Robbins and Cotran Pathologic Basis of Disease. Philadelphia, PA: Elsevier Saunders.
Larsen, J. R., and Sidenius, P. (1989). Slow axonal transport of structural polypeptides in rat, early changes in streptozocin diabetes, and effect of insulin treatment. J. Neurochem. 52, 390–401. doi: 10.1111/j.1471-4159.1989.tb09134.x
Lassmann, H., Schmied, M., Vass, K., and Hickey, W. F. (1993). Bone marrow derived elements and resident microglia in brain inflammation. Glia 7, 19–24. doi: 10.1002/glia.440070106
Lawson, L. J., Perry, V. H., and Gordon, S. (1992). Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48, 405–415. doi: 10.1016/0306-4522(92)90500-2
Leinninger, G. M., Backus, C., Sastry, A. M., Yi, Y. B., Wang, C. W., and Feldman, E. L. (2006). Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiol. Dis. 23, 11–22. doi: 10.1016/j.nbd.2006.01.017
Li, J., Baud, O., Vartanian, T., Volpe, J. J., and Rosenberg, P. A. (2005). Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc. Natl. Acad. Sci. U.S.A. 102, 9936–9941. doi: 10.1073/pnas.0502552102
Li, Y., and Kim, J. (2017). Distinct roles of neuronal and microglial CB2 cannabinoid receptors in the mouse hippocampus. Neuroscience 363, 11–25. doi: 10.1016/j.neuroscience.2017.08.053
Lin, W.-W. (2017). Trehalose-6, 6′-dibehenate inhibits lipopolysaccharide-induced NF-κB activation via PLC-γ1-PKC-ERK signaling and promotes M2 microglial polarization. Am. Assoc. Immnol. 198 (Suppl. 1), 129.14.
Liu, B., Tai, Y., Achanta, S., Kaelberer, M. M., Caceres, A. I., Shao, X., et al. (2016). IL-33/ST2 signaling excites sensory neurons and mediates itch response in a mouse model of poison ivy contact allergy. Proc. Natl. Acad. Sci. U.S.A. 113, E7572–E7579. doi: 10.1073/pnas.1606608113
Liu, X. S., Fan, B., Szalad, A., Jia, L., Wang, L., Wang, X., et al. (2017). MicroRNA-146a mimics reduce the peripheral neuropathy in type 2 diabetic mice. Diabetes 66, 3111–3121. doi: 10.2337/db16-1182
Longo, F. M., Powell, H. C., Lebeau, J., Gerrero, M. R., Heckman, H., and Myers, R. R. (1986). Delayed nerve regeneration in streptozotocin diabetic rats. Muscle Nerve 9, 385–393. doi: 10.1002/mus.880090502
Ludbrook, V. J., Hicks, K. J., Hanrott, K. E., Patel, J. S., Binks, M. H., Wyres, M. R., et al. (2016). Investigation of selective JAK1 inhibitor GSK2586184 for the treatment of psoriasis in a randomized placebo-controlled phase IIa study. Br. J. Dermatol. 174, 985–995. doi: 10.1111/bjd.14399
Lyssenko, V. (2008). The transcription factor 7-like 2 gene and increased risk of type 2 diabetes: an update. Curr. Opin. Clin. Nutr. Metab. Care 11, 385–392. doi: 10.1097/MCO.0b013e328304d970
Malin, S. A., Molliver, D. C., Koerber, H. R., Cornuet, P., Frye, R., Albers, K. M., et al. (2006). Glial cell line-derived neurotrophic factor family members sensitize nociceptors in vitro and produce thermal hyperalgesia in vivo. J. Neurosci. 26, 8588–8599. doi: 10.1523/JNEUROSCI.1726-06.2006
Mapplebeck, J. C. S., Dalgarno, R., Tu, Y., Moriarty, O., Beggs, S., Kwok, C. H. T., et al. (2018). Microglial P2X4R-evoked pain hypersensitivity is sexually dimorphic in rats. Pain 159, 1752–1763. doi: 10.1097/j.pain.0000000000001265
Marchand, F., Perretti, M., and McMahon, S. B. (2005). Role of the immune system in chronic pain. Nat. Rev. Neurosci. 6, 521–532. doi: 10.1038/nrn1700
Marieb, E. N., Kollett, L. S., and Zao, P. Z. (2009). Human Anatomy & Physiology Laboratory Manual. San Francisco, CA: Pearson/Benjamin Cummings.
Masuda, T., Ozono, Y., Mikuriya, S., Kohro, Y., Tozaki-Saitoh, H., Iwatsuki, K., et al. (2016). Dorsal horn neurons release extracellular ATP in a VNUT-dependent manner that underlies neuropathic pain. Nat. Commun. 7:12529. doi: 10.1038/ncomms12529
Mathieu, C., Gysemans, C., Giulietti, A., and Bouillon, R. (2005). Vitamin D and diabetes. Diabetologia 48, 1247–1257. doi: 10.1007/s00125-005-1802-7
Matsui, T., Svensson, C. I., Hirata, Y., Mizobata, K., Hua, X. Y., and Yaksh, T. L. (2010). Release of prostaglandin E(2) and nitric oxide from spinal microglia is dependent on activation of p38 mitogen-activated protein kinase. Anesth. Analg. 111, 554–560. doi: 10.1213/ANE.0b013e3181e3a2a2
McMahon, S. B., Cafferty, W. B., and Marchand, F. (2005). Immune and glial cell factors as pain mediators and modulators. Exp. Neurol. 192, 444–462. doi: 10.1016/j.expneurol.2004.11.001
McMahon, S. B., La Russa, F., and Bennett, D. L. (2015). Crosstalk between the nociceptive and immune systems in host defence and disease. Nat. Rev. Neurosci. 16, 389–402. doi: 10.1038/nrn3946
Metz, T. O., Alderson, N. L., Thorpe, S. R., and Baynes, J. W. (2003). Pyridoxamine, an inhibitor of advanced glycation and lipoxidation reactions: a novel therapy for treatment of diabetic complications. Arch. Biochem. Biophys. 419, 41–49. doi: 10.1016/j.abb.2003.08.021
Midaoui, A. E., Talbot, S., Lahjouji, K., Dias, J. P., Fantus, I. G., and Couture, R. (2015). Effects of alpha-lipoic acid on oxidative stress and kinin receptor expression in obese zucker diabetic fatty rats. J. Diabetes Metab. 6, 1–7. doi: 10.4172/2155-6156.1000556
Miki, K., Fukuoka, T., Tokunaga, A., and Noguchi, K. (1998). Calcitonin gene-related peptide increase in the rat spinal dorsal horn and dorsal column nucleus following peripheral nerve injury: up-regulation in a subpopulation of primary afferent sensory neurons. Neuroscience 82, 1243–1252. doi: 10.1016/S0306-4522(97)00258-3
Milligan, E., Zapata, V., Schoeniger, D., Chacur, M., Green, P., Poole, S., et al. (2005). An initial investigation of spinal mechanisms underlying pain enhancement induced by fractalkine, a neuronally released chemokine. Eur. J. Neurosci. 22, 2775–2782. doi: 10.1111/j.1460-9568.2005.04470.x
Milligan, E. D., Sloane, E. M., and Watkins, L. R. (2008). Glia in pathological pain: a role for fractalkine. J. Neuroimmunol. 198, 113–120. doi: 10.1016/j.jneuroim.2008.04.011
Milligan, E. D., and Watkins, L. R. (2009). Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 10, 23–36. doi: 10.1038/nrn2533
Milligan, E. D., Zapata, V., Chacur, M., Schoeniger, D., Biedenkapp, J., O'Connor, K. A., et al. (2004). Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur. J. Neurosci. 20, 2294–2302. doi: 10.1111/j.1460-9568.2004.03709.x
Misawa, S., Kuwabara, S., Kanai, K., Tamura, N., Hiraga, A., Nakata, M., et al. (2005). Axonal potassium conductance and glycemic control in human diabetic nerves. Clin. Neurophysiol. 116, 1181–1187. doi: 10.1016/j.clinph.2004.12.019
Misawa, S., Sakurai, K., Shibuya, K., Isose, S., Kanai, K., Ogino, J., et al. (2009). Neuropathic pain is associated with increased nodal persistent Na(+) currents in human diabetic neuropathy. J. Peripher. Nerv. Syst. 14, 279–284. doi: 10.1111/j.1529-8027.2009.00239.x
Molines, L., Darmon, P., and Raccah, D. (2010). Charcot's foot: newest findings on its pathophysiology, diagnosis and treatment. Diabetes Metab. 36, 251–255. doi: 10.1016/j.diabet.2010.04.002
Monnier, V. M. (2003). Intervention against the Maillard reaction in vivo. Arch. Biochem. Biophys. 419, 1–15. doi: 10.1016/j.abb.2003.08.014
Mori, T., Miyamoto, T., Yoshida, H., Asakawa, M., Kawasumi, M., Kobayashi, T., et al. (2011). IL-1beta and TNFalpha-initiated IL-6-STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int. Immunol. 23, 701–712. doi: 10.1093/intimm/dxr077
Mosley, K., and Cuzner, M. L. (1996). Receptor-mediated phagocytosis of myelin by macrophages and microglia: effect of opsonization and receptor blocking agents. Neurochem. Res. 21, 481–487. doi: 10.1007/BF02527713
Narita, M., Yoshida, T., Nakajima, M., Miyatake, M., Takagi, T., Yajima, Y., et al. (2006). Direct evidence for spinal cord microglia in the development of a neuropathic pain-like state in mice. J. Neurochem. 97, 1337–1348. doi: 10.1111/j.1471-4159.2006.03808.x
Nassar, M. A., Stirling, L. C., Forlani, G., Baker, M. D., Matthews, E. A., Dickenson, A. H., et al. (2004). Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc. Natl. Acad. Sci. U.S.A. 101, 12706–12711. doi: 10.1073/pnas.0404915101
Nestor, J., Arinuma, Y., Huerta, T. S., Kowal, C., Nasiri, E., Kello, N., et al. (2018). Lupus antibodies induce behavioral changes mediated by microglia and blocked by ACE inhibitors. J. Exp. Med. 215, 2554–2566. doi: 10.1084/jem.20180776
Nishikawa, T., Edelstein, D., Du, X. L., Yamagishi, S., Matsumura, T., Kaneda, Y., et al. (2000). Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404, 787–790. doi: 10.1038/35008121
Noda, M., Kariura, Y., Pannasch, U., Nishikawa, K., Wang, L., Seike, T., et al. (2007). Neuroprotective role of bradykinin because of the attenuation of pro-inflammatory cytokine release from activated microglia. J. Neurochem. 101, 397–410. doi: 10.1111/j.1471-4159.2006.04339.x
Noguchi, H. (2009). Pancreatic islet transplantation. World J. Gastrointest. Surg. 1, 16–20. doi: 10.4240/wjgs.v1.i1.16
Noguchi, H. (2010). Pancreatic stem/progenitor cells for the treatment of diabetes. Rev. Diabet. Stud. 7, 105–111. doi: 10.1900/RDS.2010.7.105
Oates, P. J. (2002). Polyol pathway and diabetic peripheral neuropathy. Int. Rev. Neurobiol. 50, 325–392. doi: 10.1016/S0074-7742(02)50082-9
Oates, P. J. (2008). Aldose reductase, still a compelling target for diabetic neuropathy. Curr. Drug Targets 9, 14–36. doi: 10.2174/138945008783431781
Obrosova, I. G. (2009). Diabetic painful and insensate neuropathy: pathogenesis and potential treatments. Neurotherapeutics 6, 638–647. doi: 10.1016/j.nurt.2009.07.004
Oetjen, L. K., Mack, M. R., Feng, J., Whelan, T. M., Niu, H., Guo, C. J., et al. (2017). Sensory neurons Co-opt classical immune signaling pathways to mediate chronic itch. Cell 171, 217–228 e13. doi: 10.1016/j.cell.2017.08.006
Oka, M., and Kato, N. (2001). Aldose reductase inhibitors. J. Enzym. Inhib. 16, 465–473. doi: 10.1080/14756360127568
Opree, A., and Kress, M. (2000). Involvement of the proinflammatory cytokines tumor necrosis factor-alpha, IL-1 beta, and IL-6 but not IL-8 in the development of heat hyperalgesia: effects on heat-evoked calcitonin gene-related peptide release from rat skin. J. Neurosci. 20, 6289–6293. doi: 10.1523/JNEUROSCI.20-16-06289.2000
Orman, D., Vardi, N., Ates, B., Taslidere, E., and Elbe, H. (2015). Aminoguanidine mitigates apoptosis, testicular seminiferous tubules damage, and oxidative stress in streptozotocin-induced diabetic rats. Tissue Cell. 47, 284–290. doi: 10.1016/j.tice.2015.03.006
Oyama, T., Miyasita, Y., Watanabe, H., and Shirai, K. (2006). The role of polyol pathway in high glucose-induced endothelial cell damages. Diabetes Res. Clin. Pract. 73, 227–234. doi: 10.1016/j.diabres.2006.02.010
Oyenihi, A. B., Ayeleso, A. O., Mukwevho, E., and Masola, B. (2015). Antioxidant strategies in the management of diabetic neuropathy. Biomed. Res. Int. 2015:515042. doi: 10.1155/2015/515042
Pabreja, K., Dua, K., Sharma, S., Padi, S. S., and Kulkarni, S. K. (2011). Minocycline attenuates the development of diabetic neuropathic pain: possible anti-inflammatory and anti-oxidant mechanisms. Eur. J. Pharmacol. 661, 15–21. doi: 10.1016/j.ejphar.2011.04.014
Paulson, P. E., Wiley, J. W., and Morrow, T. J. (2007). Concurrent activation of the somatosensory forebrain and deactivation of periaqueductal gray associated with diabetes-induced neuropathic pain. Exp. Neurol. 208, 305–313. doi: 10.1016/j.expneurol.2007.09.001
Pereira, P. J., Machado, G. D., Danesi, G. M., Canevese, F. F., Reddy, V. B., Pereira, T. C., et al. (2015). GRPR/PI3Kgamma: partners in central transmission of itch. J. Neurosci. 35, 16272–16281. doi: 10.1523/JNEUROSCI.2310-15.2015
Petrenko, A. B., Yamakura, T., Baba, H., and Shimoji, K. (2003). The role of N-methyl-D-aspartate (NMDA) receptors in pain: a review. Anesth. Analg. 97, 1108–1116. doi: 10.1213/01.ANE.0000081061.12235.55
Piotrowska, A., Kwiatkowski, K., Rojewska, E., Makuch, W., and Mika, J. (2016). Maraviroc reduces neuropathic pain through polarization of microglia and astroglia - Evidence from in vivo and in vitro studies. Neuropharmacology 108, 207–219. doi: 10.1016/j.neuropharm.2016.04.024
Pocock, J. M., and Kettenmann, H. (2007). Neurotransmitter receptors on microglia. Trends Neurosci. 30, 527–535. doi: 10.1016/j.tins.2007.07.007
Pop-Busui, R., Sima, A., and Stevens, M. (2006). Diabetic neuropathy and oxidative stress. Diabetes Metab. Res. Rev. 22, 257–273. doi: 10.1002/dmrr.625
Popiolek-Barczyk, K., and Mika, J. (2016). Targeting the microglial signaling pathways: new insights in the modulation of neuropathic pain. Curr. Med. Chem. 23, 2908–2928. doi: 10.2174/0929867323666160607120124
Prabodha, L. B. L., Sirisena, N. D., and Dissanayake, V. H. W. (2018). Susceptible and prognostic genetic factors associated with diabetic peripheral neuropathy: a comprehensive literature review. Int. J. Endocrinol. 2018:8641942. doi: 10.1155/2018/8641942
Quan, X., Lim, S. O., and Jung, G. (2011). Reactive oxygen species downregulate catalase expression via methylation of a CpG Island in the Oct-1 promoter. FEBS Lett. 585, 3436–3441. doi: 10.1016/j.febslet.2011.09.035
Quan, Y., Du, J., and Wang, X. (2007). High glucose stimulates GRO secretion from rat microglia via ROS, PKC, and NF-kappaB pathways. J. Neurosci. Res. 85, 3150–3159. doi: 10.1002/jnr.21421
Raccah, D. (1998). Physiopathology of diabetic neuropathies. Functional exploration of peripheral involvement. Diabetes Metab. 24 (Suppl. 3), 73–78.
Ribeiro, R., Wen, J., Li, S., and Zhang, Y. (2013). Involvement of ERK1/2, cPLA2 and NF-kappaB in microglia suppression by cannabinoid receptor agonists and antagonists. Prostaglandins Other Lipid Mediat. 100–101, 1–14. doi: 10.1016/j.prostaglandins.2012.11.003
Riddell, D. R., and Owen, J. S. (1999). Nitric oxide and platelet aggregation. Vitam. Horm. 57, 25–48. doi: 10.1016/S0083-6729(08)60639-1
Riol-Blanco, L., Ordovas-Montanes, J., Perro, M., Naval, E., Thiriot, A., Alvarez, D., et al. (2014). Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 510, 157–161. doi: 10.1038/nature13199
Romero-Sandoval, E. A., Horvath, R. J., and DeLeo, J. A. (2008). Neuroimmune interactions and pain: focus on glial-modulating targets. Curr. Opin. Investig. Drugs 9, 726–734.
Rosas-Ballina, M., Olofsson, P. S., Ochani, M., Valdes-Ferrer, S. I., Levine, Y. A., Reardon, C., et al. (2011). Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334, 98–101. doi: 10.1126/science.1209985
Rother, K. I. (2007). Diabetes treatment–bridging the divide. N. Engl. J. Med. 356, 1499–1501. doi: 10.1056/NEJMp078030
Said, G. (2007). Diabetic neuropathy–a review. Nat. Clin. Pract. Neurol. 3, 331–340. doi: 10.1038/ncpneuro0504
Said, G., Lacroix, C., Lozeron, P., Ropert, A., Plante, V., and Adams, D. (2003). Inflammatory vasculopathy in multifocal diabetic neuropathy. Brain 126, 376–385. doi: 10.1093/brain/awg029
Salter, M. W., and Stevens, B. (2017). Microglia emerge as central players in brain disease. Nat. Med. 23, 1018–1027. doi: 10.1038/nm.4397
Samad, T. A., Moore, K. A., Sapirstein, A., Billet, S., Allchorne, A., Poole, S., et al. (2001). Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 410, 471–475. doi: 10.1038/35068566
Samad, T. A., Sapirstein, A., and Woolf, C. J. (2002). Prostanoids and pain: unraveling mechanisms and revealing therapeutic targets. Trends Mol. Med. 8, 390–396. doi: 10.1016/S1471-4914(02)02383-3
Schemmel, K. E., Padiyara, R. S., and D'Souza, J. J. (2009). Aldose reductase inhibitors in the treatment of diabetic peripheral neuropathy: a review. J. Diabetes Complications 24, 354–360. doi: 10.1016/j.jdiacomp.2009.07.005
Scholz, J., and Woolf, C. J. (2002). Can we conquer pain? Nat. Neurosci. 5 (Suppl.), 1062–1067. doi: 10.1038/nn942
Scholz, J., and Woolf, C. J. (2007). The neuropathic pain triad: neurons, immune cells and glia. Nat. Neurosci. 10, 1361–1368. doi: 10.1038/nn1992
Shi, Q., Chowdhury, S., Ma, R., Le, K. X., Hong, S., Caldarone, B. J., et al. (2017). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 9:eaaf6295. doi: 10.1126/scitranslmed.aaf6295
Shim, W. S., Tak, M. H., Lee, M. H., Kim, M., Kim, M., Koo, J. Y., et al. (2007). TRPV1 mediates histamine-induced itching via the activation of phospholipase A2 and 12-lipoxygenase. J. Neurosci. 27, 2331–2337. doi: 10.1523/JNEUROSCI.4643-06.2007
Singh Grewal, A., Bhardwaj, S., Pandita, D., Lather, V., and Sekhon, B. S. (2016). Updates on aldose reductase inhibitors for management of diabetic complications and non-diabetic diseases. Mini. Rev. Med. Chem. 16, 120–162. doi: 10.2174/1389557515666150909143737
Singh, V. P., Bali, A., Singh, N., and Jaggi, A. S. (2014). Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 18, 1–14. doi: 10.4196/kjpp.2014.18.1.1
Sorge, R. E., Mapplebeck, J. C., Rosen, S., Beggs, S., Taves, S., Alexander, J. K., et al. (2015). Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 18, 1081–1083. doi: 10.1038/nn.4053
Spruce, M. C., Potter, J., and Coppini, D. V. (2003). The pathogenesis and management of painful diabetic neuropathy: a review. Diabet. Med. 20, 88–98. doi: 10.1046/j.1464-5491.2003.00852.x
Stellwagen, D., and Malenka, R. C. (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440, 1054–1059. doi: 10.1038/nature04671
Sugimoto, K., Yasujima, M., and Yagihashi, S. (2008). Role of advanced glycation end products in diabetic neuropathy. Curr. Pharm. Des. 14, 953–961. doi: 10.2174/138161208784139774
Sun, J. S., Yang, Y. J., Zhang, Y. Z., Huang, W., Li, Z. S., and Zhang, Y. (2015). Minocycline attenuates pain by inhibiting spinal microglia activation in diabetic rats. Mol. Med. Rep. 12, 2677–2682. doi: 10.3892/mmr.2015.3735
Sun, S., Cao, H., Han, M., Li, T. T., Pan, H. L., Zhao, Z. Q., et al. (2007). New evidence for the involvement of spinal fractalkine receptor in pain facilitation and spinal glial activation in rat model of monoarthritis. Pain 129, 64–75. doi: 10.1016/j.pain.2006.09.035
Svensson, C. I., Fitzsimmons, B., Azizi, S., Powell, H. C., Hua, X. Y., and Yaksh, T. L. (2005). Spinal p38beta isoform mediates tissue injury-induced hyperalgesia and spinal sensitization. J. Neurochem. 92, 1508–1520. doi: 10.1111/j.1471-4159.2004.02996.x
Sweitzer, S. M., White, K. A., Dutta, C., and DeLeo, J. A. (2002). The differential role of spinal MHC class II and cellular adhesion molecules in peripheral inflammatory versus neuropathic pain in rodents. J. Neuroimmunol. 125, 82–93. doi: 10.1016/S0165-5728(02)00036-X
Talbot, S., Abdulnour, R. E., Burkett, P. R., Lee, S., Cronin, S. J., Pascal, M. A., et al. (2015). Silencing nociceptor neurons reduces allergic airway inflammation. Neuron 87, 341–354. doi: 10.1016/j.neuron.2015.06.007
Talbot, S., Chahmi, E., Dias, J. P., and Couture, R. (2010). Key role for spinal dorsal horn microglial kinin B1 receptor in early diabetic pain neuropathy. J. Neuroinflamm. 7:36. doi: 10.1186/1742-2094-7-36
Talbot, S., De Brito Gariepy, H., Saint-Denis, J., and Couture, R. (2012). Activation of kinin B1 receptor evokes hyperthermia through a vagal sensory mechanism in the rat. J. Neuroinflamm. 9:214. doi: 10.1186/1742-2094-9-214
Talbot, S., Dias, J. P., El Midaoui, A., and Couture, R. (2016a). Beneficial effects of kinin B1 receptor antagonism on plasma fatty acid alterations and obesity in Zucker diabetic fatty rats. Can. J. Physiol. Pharmacol. 94, 752–757. doi: 10.1139/cjpp-2016-0063
Talbot, S., Foster, S. L., and Woolf, C. J. (2016b). Neuroimmunity: physiology and pathology. Annu. Rev. Immunol. 34, 421–447. doi: 10.1146/annurev-immunol-041015-055340
Tanga, F. Y., Nutile-McMenemy, N., and DeLeo, J. A. (2005). The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc. Natl. Acad. Sci. U.S.A. 102, 5856–5861. doi: 10.1073/pnas.0501634102
Tanga, F. Y., Raghavendra, V., and DeLeo, J. A. (2004). Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem. Int. 45, 397–407. doi: 10.1016/j.neuint.2003.06.002
Taylor, A. M., Castonguay, A., Ghogha, A., Vayssiere, P., Pradhan, A. A., Xue, L., et al. (2016). Neuroimmune regulation of GABAergic neurons within the ventral tegmental area during withdrawal from chronic morphine. Neuropsychopharmacology 41, 949–959. doi: 10.1038/npp.2015.221
Taylor, A. M., Castonguay, A., Taylor, A. J., Murphy, N. P., Ghogha, A., Cook, C., et al. (2015). Microglia disrupt mesolimbic reward circuitry in chronic pain. J. Neurosci. 35, 8442–8450. doi: 10.1523/JNEUROSCI.4036-14.2015
Tender, G. C., Li, Y. Y., and Cui, J. G. (2010). Brain-derived neurotrophic factor redistribution in the dorsal root ganglia correlates with neuropathic pain inhibition after resiniferatoxin treatment. Spine J. 10, 715–720. doi: 10.1016/j.spinee.2010.03.029
Tesfaye, S., and Kempler, P. (2005). Painful diabetic neuropathy. Diabetologia 48, 805–807. doi: 10.1007/s00125-005-1721-7
Thacker, M. A., Clark, A. K., Bishop, T., Grist, J., Yip, P. K., Moon, L. D., et al. (2009). CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur. J. Pain 13, 263–272. doi: 10.1016/j.ejpain.2008.04.017
Thamotharampillai, K., Chan, A. K., Bennetts, B., Craig, M. E., Cusumano, J., Silink, M., et al. (2006). Decline in neurophysiological function after 7 years in an adolescent diabetic cohort and the role of aldose reductase gene polymorphisms. Diabetes Care 29, 2053–2057. doi: 10.2337/dc06-0678
Thornalley, P. J. (1998). Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs. Cell Mol. Biol. 44, 1013–1023.
Tomlinson, D. R., Fernyhough, P., and Diemel, L. T. (1997). Role of neurotrophins in diabetic neuropathy and treatment with nerve growth factors. Diabetes 46 (Suppl. 2), S43–S49. doi: 10.2337/diab.46.2.S43
Torsney, C., and MacDermott, A. B. (2005). Neuroscience: a painful factor. Nature 438, 923–925. doi: 10.1038/438923a
Tsuda, M., and Inoue, K. (2016). Neuron-microglia interaction by purinergic signaling in neuropathic pain following neurodegeneration. Neuropharmacology 104, 76–81. doi: 10.1016/j.neuropharm.2015.08.042
Tsuda, M., Inoue, K., and Salter, M. W. (2005). Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 28, 101–107. doi: 10.1016/j.tins.2004.12.002
Tsuda, M., Mizokoshi, A., Shigemoto-Mogami, Y., Koizumi, S., and Inoue, K. (2004). Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia 45, 89–95. doi: 10.1002/glia.10308
Tsuda, M., Shigemoto-Mogami, Y., Koizumi, S., Mizokoshi, A., Kohsaka, S., Salter, M. W., et al. (2003). P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424, 778–783. doi: 10.1038/nature01786
Tuck, R. R., Schmelzer, J. D., and Low, P. A. (1984). Endoneurial blood flow and oxygen tension in the sciatic nerves of rats with experimental diabetic neuropathy. Brain 107(Pt 3), 935–950.
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
Vague, P., Dufayet, D., Lamotte, M. F., Mouchot, C., and Raccah, D. (1997). Genetic factors, Na K ATPase activity and neuropathy in diabetics. Bull. Acad. Natl. Med. 181, 1811–1821; discussion 1821–1823.
Vardeh, D., Wang, D., Costigan, M., Lazarus, M., Saper, C. B., Woolf, C. J., et al. (2009). COX2 in CNS neural cells mediates mechanical inflammatory pain hypersensitivity in mice. J. Clin. Invest. 119, 287–294. doi: 10.1172/JCI37098
Vareniuk, I., Pavlov, I. A., and Obrosova, I. G. (2008). Inducible nitric oxide synthase gene deficiency counteracts multiple manifestations of peripheral neuropathy in a streptozotocin-induced mouse model of diabetes. Diabetologia 51, 2126–2133. doi: 10.1007/s00125-008-1136-3
Veiga-Fernandes, H., and Mucida, D. (2016). Neuro-immune interactions at barrier surfaces. Cell 165, 801–811. doi: 10.1016/j.cell.2016.04.041
Verge, G. M., Milligan, E. D., Maier, S. F., Watkins, L. R., Naeve, G. S., and Foster, A. C. (2004). Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur. J. Neurosci. 20, 1150–1160. doi: 10.1111/j.1460-9568.2004.03593.x
Vincent, A. M., Edwards, J. L., Sadidi, M., and Feldman, E. L. (2008). The antioxidant response as a drug target in diabetic neuropathy. Curr. Drug Targets 9, 94–100. doi: 10.2174/138945008783431754
Virtanen, S. M., Saukkonen, T., Savilahti, E., Ylonen, K., Rasanen, L., Aro, A., et al. (1994). Diet, cow's milk protein antibodies and the risk of IDDM in Finnish children. Childhood diabetes in finland study group. Diabetologia 37, 381–387. doi: 10.1007/BF00408475
Viviani, B., Bartesaghi, S., Gardoni, F., Vezzani, A., Behrens, M. M., Bartfai, T., et al. (2003). Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 23, 8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003
von Hehn, C. A., Baron, R., and C., Woolf, J. J. N. (2012). Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 73, 638–652. doi: 10.1016/j.neuron.2012.02.008
Wabitsch, M., Funcke, J. B., von Schnurbein, J., Denzer, F., Lahr, G., Mazen, I., et al. (2015). Severe early-onset obesity due to bioinactive leptin caused by a p.N103K mutation in the leptin gene. J. Clin. Endocrinol. Metab. 100, 3227–3230. doi: 10.1210/jc.2015-2263
Wagner, R., and Myers, R. R. (1996). Endoneurial injection of TNF-alpha produces neuropathic pain behaviors. Neuroreport 7, 2897–2901. doi: 10.1097/00001756-199611250-00018
Wall, P. D., and Gutnick, M. (1974). Ongoing activity in peripheral nerves: the physiology and pharmacology of impulses originating from a neuroma. Exp. Neurol. 43, 580–593. doi: 10.1016/0014-4886(74)90197-6
Watkins, L. R., Milligan, E. D., and Maier, S. F. (2001a). Glial activation: a driving force for pathological pain. Trends Neurosci. 24, 450–455. doi: 10.1016/S0166-2236(00)01854-3
Watkins, L. R., Milligan, E. D., and Maier, S. F. (2001b). Spinal cord glia: new players in pain. Pain 93, 201–205. doi: 10.1016/S0304-3959(01)00359-1
White, F. A., Jung, H., and Miller, R. J. (2007). Chemokines and the pathophysiology of neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 104, 20151–20158. doi: 10.1073/pnas.0709250104
Wilson, S. R., The, L., Batia, L. M., Beattie, K., Katibah, G. E., McClain, S. P., et al. (2013). The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 155, 285–295. doi: 10.1016/j.cell.2013.08.057
Wodarski, R., Clark, A. K., Grist, J., Marchand, F., and Malcangio, M. (2009). Gabapentin reverses microglial activation in the spinal cord of streptozotocin-induced diabetic rats. Eur. J. Pain 13, 807–811. doi: 10.1016/j.ejpain.2008.09.010
Wood, P. B. (2008). Role of central dopamine in pain and analgesia. Expert Rev. Neurother. 8, 781–797. doi: 10.1586/14737175.8.5.781
Woolf, C. J. (2004). Dissecting out mechanisms responsible for peripheral neuropathic pain: implications for diagnosis and therapy. Life Sci. 74, 2605–2610. doi: 10.1016/j.lfs.2004.01.003
Woolf, C. J. (2007). Central sensitization. Anesthesiology 106, 864–867. doi: 10.1097/01.anes.0000264769.87038.55
Woolf, C. J., Shortland, P., and Coggeshall, R. E. (1992). Peripheral nerve injury triggers central sprouting of myelinated afferents. Nature 355, 75–78. doi: 10.1038/355075a0
Xia, P., Inoguchi, T., Kern, T. S., Engerman, R. L., Oates, P. J., and King, G. L. (1994). Characterization of the mechanism for the chronic activation of diacylglycerol-protein kinase C pathway in diabetes and hypergalactosemia. Diabetes 43, 1122–1129. doi: 10.2337/diab.43.9.1122
Xu, J., Zhang, L., Xie, M., Li, Y., Huang, P., Saunders, T. L., et al. (2018). Role of complement in a rat model of paclitaxel-induced peripheral neuropathy. J. Immunol. 200, 4094–4101. doi: 10.4049/jimmunol.1701716
Yagihashi, S., Yamagishi, S., and Wada, R. (2007). Pathology and pathogenetic mechanisms of diabetic neuropathy: correlation with clinical signs and symptoms. Diabetes Res. Clin. Pract. 77 (Suppl. 1), S184–S189. doi: 10.1016/j.diabres.2007.01.054
Yao, Y., Echeverry, S., Shi, X. Q., Yang, M., Yang, Q. Z., Wang, G. Y., et al. (2016). Dynamics of spinal microglia repopulation following an acute depletion. Sci. Rep. 6:22839. doi: 10.1038/srep22839
Yasuda, H., Terada, M., Maeda, K., Kogawa, S., Sanada, M., Haneda, M., et al. (2003). Diabetic neuropathy and nerve regeneration. Prog. Neurobiol. 69, 229–285. doi: 10.1016/S0301-0082(03)00034-0
Zhang, B., Yu, Y., Aori, G., Wang, Q., Kong, D., Yang, W., et al. (2018). Tanshinone IIA attenuates diabetic peripheral neuropathic pain in experimental rats via inhibiting inflammation. J. Evidence Based Complementary Alter. Med. 8:2789847. doi: 10.1155/2018/2789847
Zhang, J., and De Koninck, Y. (2006). Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J. Neurochem. 97, 772–783. doi: 10.1111/j.1471-4159.2006.03746.x
Zhang, J., Shi, X. Q., Echeverry, S., Mogil, J. S., De Koninck, Y., and Rivest, S. (2007). Expression of CCR2 in both resident and bonemarrow-derived microglia plays a critical role in neuropathic pain. J. Neurosci. 27, 12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007
Zhang, N., Inan, S., Cowan, A., Sun, R., Wang, J. M., Rogers, T. J., et al. (2005). A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc. Natl. Acad. Sci. U.S.A. 102, 4536–4541. doi: 10.1073/pnas.0406030102
Zhang, R. X., Li, A., Liu, B., Wang, L., Ren, K., Zhang, H., et al. (2008). IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain 135, 232–239. doi: 10.1016/j.pain.2007.05.023
Zhao, P., Waxman, S. G., and Hains, B. C. (2007a). Extracellular signal-regulated kinase-regulated microglia-neuron signaling by prostaglandin E2 contributes to pain after spinal cord injury. J. Neurosci. 27, 2357–2368. doi: 10.1523/JNEUROSCI.0138-07.2007
Zhao, P., Waxman, S. G., and Hains, B. C. (2007b). Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J. Neurosci. 27, 8893–8902. doi: 10.1523/JNEUROSCI.2209-07.2007
Zhou, R., Xu, T., Liu, X., Chen, Y., Kong, D., Tian, H., et al. (2018). Activation of spinal dorsal horn P2Y13 receptors can promote the expression of IL-1beta and IL-6 in rats with diabetic neuropathic pain. J. Pain Res. 11, 615–628. doi: 10.2147/JPR.S154437
Zhou, Z., Peng, X., Hagshenas, J., Insolera, R., Fink, D. J., and Mata, M. (2010). A novel cell-cell signaling by microglial transmembrane TNFalpha with implications for neuropathic pain. Pain 151, 296–306. doi: 10.1016/j.pain.2010.06.017
Zhu, W., Xu, P., Cuascut, F. X., Hall, A. K., and Oxford, G. S. (2007). Activin acutely sensitizes dorsal root ganglion neurons and induces hyperalgesia via PKC-mediated potentiation of transient receptor potential vanilloid I. J. Neurosci. 27, 13770–13780. doi: 10.1523/JNEUROSCI.3822-07.2007
Ziegler, D. (2008). Painful diabetic neuropathy: treatment and future aspects. Diabetes Metab. Res. Rev. 24 (Suppl. 1), S52–S57. doi: 10.1002/dmrr.817
Zimmermann, M. (2001). Pathobiology of neuropathic pain. Eur. J. Pharmacol. 429, 23–37. doi: 10.1016/S0014-2999(01)01303-6
Zochodne, D. W., and Levy, D. (2005). Nitric oxide in damage, disease and repair of the peripheral nervous system. Cell Mol. Biol. 51, 255–267. doi: 10.1170/T626
Keywords: pain, diabetes, neuropathy, neurons, microglia, oxidative stress, hyperglycemia
Citation: Rajchgot T, Thomas SC, Wang J-C, Ahmadi M, Balood M, Crosson T, Dias JP, Couture R, Claing A and Talbot S (2019) Neurons and Microglia; A Sickly-Sweet Duo in Diabetic Pain Neuropathy. Front. Neurosci. 13:25. doi: 10.3389/fnins.2019.00025
Received: 21 October 2018; Accepted: 11 January 2019;
Published: 31 January 2019.
Edited by:
Avital Schurr, University of Louisville, United StatesReviewed by:
Manuel Guzmán, Complutense University of Madrid, SpainBrenda Bartnik-Olson, Loma Linda University, United States
Copyright © 2019 Rajchgot, Thomas, Wang, Ahmadi, Balood, Crosson, Dias, Couture, Claing and Talbot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sébastien Talbot, U2ViYXN0aWVuLnRhbGJvdEB1bW9udHJlYWwuY2E=