 Qiu Han1,2†Yong-An Sun1,3†Yu Zong4Chun Chen1,5Hui-Fu Wang4
Qiu Han1,2†Yong-An Sun1,3†Yu Zong4Chun Chen1,5Hui-Fu Wang4 Lan Tan1* and Alzheimer's Disease Neuroimaging Initiative
Lan Tan1* and Alzheimer's Disease Neuroimaging Initiative- 1Department of Neurology, Qingdao Clinical Medical School, Qingdao Municipal Hospital, Nanjing Medical University, Qingdao, China
- 2Department of Neurology, The Affiliated Huaian Hosipital of Xuzhou Medical University, Huai'an, China
- 3Department of Neurology, First Affiliated Hospital of Kangda School, Nanjing Medical University, Lianyungang, China
- 4Department of Neurology, School of Medicine, Qingdao Municipal Hospital, Qingdao University, Qingdao, China
- 5Department of Neurology, Hongze Huai'an District People's Hospital, Huai'an, China
The Plexin-A 4 (PLXNA4) gene, has recently been identified in genome wide association studies (GWAS), as a novel genetic player associated with Alzheimer's disease (AD). Additionally, PLXNA4 genetic variations were also found to increase AD risk by tau pathology in vitro. However, the potential roles of PLXNA4 variants in the amyloid-β (Aβ) pathology, were not evaluated. Five targeted loci capturing the top common variations in PLXNA4, were extracted using tagger methods. Multiple linear regression models were used to explore whether these variations can affect the cerebrospinal fluid (CSF) (Aβ1−42, T-tau, and P-tau) phenotypes in the Alzheimer's disease Neuroimaging Initiative (ADNI) dataset. We detected that two loci (rs6467431, rs67468325) were significantly associated with CSF Aβ1−42 levels in the hybrid population (rs6467431: P = 0.01376, rs67468325: P = 0.006536) and the significance remained after false discovery rate (FDR) correction (rs6467431: Pc = 0.03441, rs67468325: Pc = 0.03268). In the subgroup analysis, we further confirmed the association of rs6467431 in the cognitively normal (CN) subgroup (P = 0.01904, Pc = 0.04761). Furthermore, rs6467431-A carriers and rs67468325-G carriers showed higher CSF Aβ1−42 levels than non-carriers. Nevertheless, we did not detect any significant relationships between the levels of T-tau, P-tau and these PLXNA4 loci. Our findings provided preliminary evidence that PLXNA4 variants can confer AD risk through modulating the Aβ deposition.
Introduction
Globally, an estimated 47 million people had dementia in 2015 and this figure is projected to triple by 2050 (Livingston et al., 2017). AD is the leading cause of dementia and is one of the biggest health-care challenges of the twenty first century (Scheltens et al., 2016). The earliest recognizable pathological event in AD is brain amyloid-β aggregation (Bateman et al., 2012). As the initial event and the most important factor, it is responsible for a battery of downstream abnormalities in AD (Cummings et al., 2007; Mormino et al., 2009; Jack et al., 2010, 2013). Novel research criteria for AD in the preclinical phase, highlight the occurrence of amyloid pathology in the first stage of the disease (Sperling et al., 2011; Dubois et al., 2014). However, the etiological mechanisms underlying the neuropathological changes remain unclear (Reitz and Mayeux, 2014). Family studies revealed that complex genetic mechanisms along with environmental factors, contribute to disease risk (Jiang et al., 2013; Yu et al., 2014). Indeed, the estimated heritability for late-onset Alzheimer's disease (LOAD) is between 60 and 80% (Gatz et al., 2006), suggesting that genetic determinants are involved in most of the pathophysiological pathways in AD. Therefore, genetics might play an important role in the underlying pathogenesis of AD.
Plexin-A 4, a member of the Plexin-A family, which is located on chromosome 7q32.3, is a receptor for secreted semaphorin class 3 (SEMA3A) and class 6 (SEMA6) proteins, which mediate the effects of multiple semaphorins, including controlling diverse aspects of the nervous system development and plasticity, ranging from axon guidance and neuron migration to synaptic organization (Sun et al., 2013; Andermatt et al., 2014; Kong et al., 2016). Previous studies have shown that Plexin-A 4 (encoded by PLXNA4) is activated by Sema3A and can mediate growth cone collapse and axon repulsion, as well as dendrite morphogenesis in different neuronal populations during development (Suto et al., 2005; Yaron et al., 2005; Tran et al., 2009; Pasterkamp, 2012), both in cells and in vivo. Accumulation of SEMA3A was previously detected in susceptible areas of the hippocampal neurons during AD progression and colocalized with phosphorylated tau (Good et al., 2004). Phosphorylated CRMP2 protein, an intracellular signaling molecule for the semaphorin-plexin signaling pathway, has been observed in neurofibrillary tangles in the brains of autopsied AD patients (Cole et al., 2004).
A genome-wide significant association study (GWAS) on humans has identified that several single nucleotide polymorphisms (SNPs) in PLXNA4 can increase the risk of AD (Jun et al., 2014). However, the concrete mechanisms PLXNA4 contribute to AD risk, remain elusive. Jun et al. found that tau phosphorylation mediated by PLXNA4 is an independent upstream event contributing to AD-related neurofibrillary tangles in neurons in vitro (Jun et al., 2014). Additionally, Kang et al. demonstrated that Plexin A4 (PLXNA4) is a novel, high affinity receptor for CLU in the adult CNS (Kang et al., 2016). Their data demonstrated that the level of PLXNA4 significantly impacts CSF levels of CLU in vivo in mice and that PLXNA4 is genetically associated with CLU levels in the CSF of humans (Kang et al., 2016). Nevertheless, induction of intracellular signal pathways mediated by PLXNA4 in the adult brain in general, or particularly by CLU-PLXNA4 interactions, remain unexplored (Kang et al., 2016). Cogent evidence reveals that the physiological interplay of CLU with Aβ has an important influence on AD pathogenesis (Yu and Tan, 2012). We therefore hypothesized that PLXNA4 would also modulate Aβ accumulation to modify AD risk, compared to tau pathology.
By demonstrating that these genetic risk factors of AD also influence CSF traits, important verification of the roles for these loci will be supported and will indicate the mechanisms by which they might act. To test this hypothesis, we aimed to explore the involvement of PLXNA4 variants in the underlying pathogenesis of AD, by investigating the effects of PLXNA4 polymorphisms on the brain amyloid burden through CSF phenotypes in the ADNI dataset.
Materials and Methods
ADNI Database
The ADNI is a large, multicentered, ongoing, longitudinal neuroimaging study, started in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, nonprofit organizations, and private pharmaceutical companies (Mueller et al., 2005). As an initial goal, 800 subjects were recruited, but the ADNI has since been followed by ADNI-GO and ADNI-2. Thus far, over 1,500 adults (aged between 55 and 90) have been recruited in the three protocols, including people with AD, people with early or late MCI and cognitively normal older individuals. This study was upheld by the institutional review boards of centers involved and written informed consent was acquired from all participants or from authorized representatives, after a comprehensive description of the ADNI was provided, in accordance with the 1975 Declaration of Helsinki (Carrillo et al., 2012). The present study was authorized by the institutional review boards of Qingdao Municipal Hospital, Memory, and Aging Center at the University of California, and the ADNI. Informed consent was acquired from all participants or from authorized representatives. Additionally, we conformed to the approved guidelines when performing these methods.
Participants
We included all participants from the ADNI database (http://adni.loni.usc.edu), in this study. Based on the National Institute of Neurological and Communication Disorders/Alzheimer's Disease and Related Disorders Association criteria for probable AD (NINCDS/ADRDA: probable AD), AD patients were enrolled if they had a Mini Mental State Examination (MMSE) score of between 20 and 26, a CDRSB score of between 1.0 and 9.0 and a global Clinical Dementia Rating (CDR) score of between 0.5 and 1.0. Amnestic MCI cases had a MMSE score of between 24 and 30 and a CDR score of at least 0.5, while cognitively normal individuals had a CDR score of 0. Moreover, individuals were excluded if they had a history of brain lesions or trauma, psychoactive medication use (including chronic anxiolytics, antidepressants, sedative hypnotics, or neuroleptics), or serious neurological diseases other than possible AD. Finally, the present study included 812 subjects, including 48 AD, 483 MCI, and 281 CN individuals at baseline. We downloaded the basic data of individuals in our study from the ADNI website in 2015.
Genotyping and SNP Selection
We used Bead Studio 3.2 software and a recent Genome Studio v2009.1 (Illumina) to produce SNP genotypes from bead intensity data (Saykin et al., 2010). In this study, we extracted the PLXNA4 genotypes in the ADNI PLINK data format and conducted the quality control procedures using PLINK software. We filtered criteria as follows: minimum minor allele frequencies (MAF) >0.01, Hardy-Weinberg equilibrium test P > 0.001, minimum call rates >90%. We preferentially chose eight SNPs (rs277470, rs277472, rs277484, rs277476, rs12539196, rs75460865, rs13232207, rs10273901) reported to be significantly associated with AD for analysis (Jun et al., 2014). Furthermore, five promising tag SNPs (rs78036292, rs6467431, rs67468325, rs1863015, rs156676), capturing the top common variations in PLXNA4 were chosen with tagger methods in the Haploview 4.2 platform. A total of 13 SNPs were initially screened and eight reported SNPs were excluded due to their absence in the ADNI. Ultimately, the remaining five loci were selected as the target SNPs (Table 2) in our study.
CSF Biomarkers
Firstly, CSF specimens were collected and transported to the ADNI Biomarker Core laboratory at the University of Pennsylvania Medical Center, within dry ice and a preparation of aliquots (0.5 ml) was prepared from the collected specimens after thawing (1 h) at room temperature and moderate mixing, then preserved in barcode–labeled polypropylene vials in −80°C. The CSF biomarkers, such as Aβ1−42, Phosphorylated tau181p and Total-tau, were detected via the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX) using Innogenetics (INNO-BIA AlzBio3; Ghent, Belgium; for research use-only reagents) immunoassay kit–based reagents. Quality control procedures and further analysis details are displayed on the website (http://adni.loni.ucla.edu). This study was a cross-sectional evaluation at baseline with regard to CSF measures. Finally, a total of 627 cases (206 CN, 377 MCI, and 44 AD) with a baseline CSF and corresponding genetic data, were included in the CSF analysis from the ADNI database.
Statistical Analyses
Differences in continuous variables were compared with the one-way analysis of variance (ANOVA) and categorical variables were examined with a chi-square test. Possible correlations between various biomarkers and the PLXNA4 genotypes were tested with a multiple linear regression model which controlled gender, education, age, and the APOE4 status. FDR, a statistical method developed by Hochberg and Benjamini (Hochberg and Benjamini, 1990), was used to adjust multiple hypothesis testing instead of a Bonferroni correction, which was inappropriate due to the non-independence of tests (Biffi et al., 2010). P < 0.05 was defined as a significant difference according to the FDR correction. Firstly, we screened significant CSF-related phenotypes associated with PLXNA4 loci in all individuals. We then stratified subjects into three groups (CN, MCI, and AD) to explore the influences of the PLXNA4 variants in these phenotypes at deferent clinical stages separately. All statistical analyses were conducted by PLINK 1.07 (http://pngu.mgh.harvard.edu/wpurcell/plink/) and R 3.12 (http://www.r-project.org/).
Results
Baseline Characteristics of Included Subjects
The baseline information of individuals included in the study are summarized in Table 1. Finally, 48 AD patients (18 women, 75.51 ± 9.23 years), 483 MCI (201 women, 72.28 ± 7.45 years), and 281 cognitively normal individuals (145 women, 74.51 ± 5.56 years) were enrolled in our study (Table 1). Compared to the MCI and CN group, patients in the AD group had the highest frequency of the ε4 allele within the APOE gene as well as the lowest cognitive function based on the scores of the five neuropsychological scales (ADAS11, MMSE, ADAS13, RAVLT, FAQ), lower CSF Aβ1−42 levels, higher CSF T-tau and P-tau levels, and the most severe atrophy in the hippocampus.
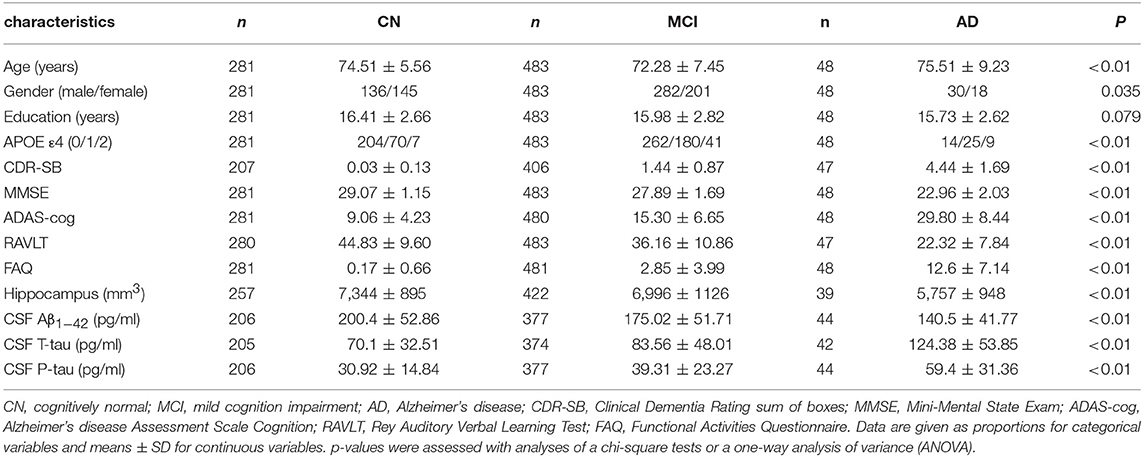
Table 1. The characteristics of the ADNI subjects at baseline.
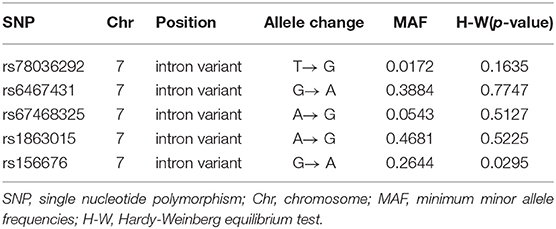
Table 2. The characteristics of targeted SNP.
Genotypes of PLXNA4 Effect on CSF Markers
At the baseline, we found that two loci (rs6467431, rs67468325) showed marked associations with the levels of CSF Aβ1−42, in the hybrid group (rs6467431: P = 0.01376, rs67468325: P = 0.006536; Figure 1) and the difference reached the significance level in the FDR test (rs6467431: Pc = 0.03441; rs67468325: Pc = 0.03268; Figure 1). For rs6467431, minor A allele carriers had higher CSF Aβ1−42 levels than non-carriers (AG>AA>GG). For rs67468325, minor G allele carriers had higher CSF Aβ1−42 levels in a dose-dependent manner (GG>AG>AA). The minor allele carriers had higher CSF Aβ1−42 levels, suggesting that the minor A allele for rs6467431 and G allele for rs67468325, were correlated with less amyloid loads. To determine this result, we then conducted a subgroup analyses and only confirmed the association between the levels of Aβ and rs6467431 in the CN subgroup (P = 0.01904, Pc = 0.04761; Figure 1). In the CN subgroup, rs6467431-A carriers had higher CSF Aβ1−42 levels than G allele homozygotes subjects (AG>AA>GG). Although rs67468325 showed a trend to associate with less amyloid loads in the MCI subgroup (P = 0.0443), it failed in the FDR test (Pc = 0.2215). Nevertheless, we did not find any remarkable relationships between the levels of T-tau, P-tau and these PLXNA4 loci in any individuals and any subgroups.
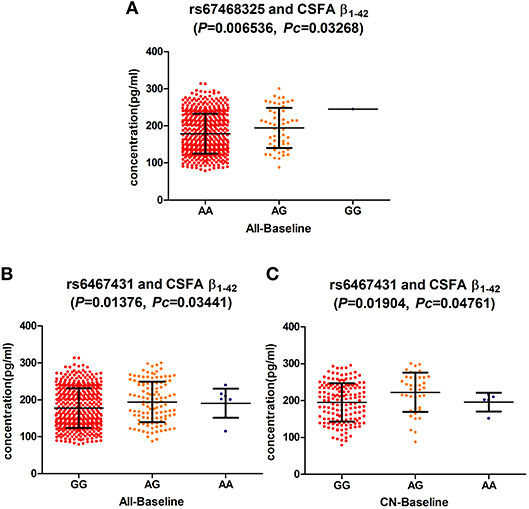
Figure 1. (A) The significant association of PLXNA4 rs67468325 with Cerebrospinal fluid (CSF) Aβ1-42 levels in the hybrid population. (B) The significant association of PLXNA4 rs6467431 with Cerebrospinal fluid (CSF) Aβ1-42 levels in the hybrid population. (C) The significant association of PLXNA4 rs6467431 with Cerebrospinal fluid (CSF) Aβ1-42 levels in the CN group.
Discussion
To our knowledge, this is the first study to explore the roles of PLXNA4 SNPs in AD pathogenesis using CSF methods. Our main findings indicated that two PLXNA4 variants modified the risk for AD through Aβ pathology. Genotypes at the PLXNA4 loci (rs6467431, rs67468325) were related to the trajectory of the Aβ deposition, assuming minor allele effects and these results were significant independent of the APOE status. Nevertheless, PLXNA4 genotypes did not affect CSF tau levels.
Jun et al. conducted a two-stage family-based AD GWAS, using a novel method which incorporates the entire family structure and reduces diagnostic misclassification in the association test and renders the result less prone to type I errors, even for rare variants (Choi et al., 2011; Jun et al., 2014). The analysis of genotyped SNPs (n = 341, 492 post quality control) in the Framingham Heart Study (FHS) dataset, using the normalized liability scores for AD, indicated little genomic inflation (λ = 1.01) with strong evidence of an association in three regions of the genome (Jun et al., 2014). A genome-wide significant association was identified with a PLXNA4 locus (rs277470; meta-analysis p-value [meta-P] = 4.1 × 10−8; Jun et al., 2014). Moreover, these PLXNA4 loci (rs277470, rs277472, rs277476, and rs277484) were also significant (P < 10−9 for each) and in complete linkage disequilibrium (LD) (Jun et al., 2014). The most significant SNPs (rs10273901 in Caucasians [meta-P] = 3.9 × 10−5, rs75460865 in African Americans [meta-P] = 8.0 × 10−4, rs13232207 in Japanese P = 1.2 × 10−4) in each population group, were further examined with the meta-analyzed results in the Alzheimer's Disease Genetics Consortium (ADGC) datasets (Jun et al., 2014). The results support the association of PLXNA4 SNPs with AD in these ethnic samples in the ADGC datasets. It was regrettable that we could not extract these PLXNA4 SNPs due to the absence of these variants in the ADNI database. We therefore extracted 5 targeted PLXNA4 loci to explore their relations with CSF markers in the ADNI dataset using tagger methods. Our CSF-related Phenotypes analysis in the ADNI dataset showed that PLXNA4 (rs6467431, rs67468325) were strongly associated with CSF Aβ1−42 levels in all subjects. We then validated the association between the levels of CSF Aβ1−42 and rs6467431 in the CN subgroup, suggesting that rs6467431 may act in the preclinical phase. MAF for rs6467431 almost 40%, a common variant in PLXNA4rs6467431 mainly correlated with Aβ deposition and minor A allele carriers had higher CSF Aβ1−42 levels than G allele homozygotes subjects in the CN subgroup (AG>AA>GG). All the above results showed good agreement with previous reports that amyloid pathology often occurs in older CN individuals, at the early stage of the disease (Aizenstein et al., 2008; Jack et al., 2010, 2013; Sperling et al., 2011). Furthermore, rs6467431-A carriers and rs67468325-G carriers had higher CSF Aβ1−42 levels than non-carriers, suggesting that the minor A and G allele play protective roles in AD, respectively.
Thus far, few investigations have provided biological evidence of the influence of PLXNA4 on AD. A vitro study revealed that PLXNA4 plays a role in AD pathogenesis through isoform-specific influences on tau phosphorylation (Jun et al., 2014). Intriguingly, no evidence supports that PLXNA4 genotypes affect CSF tau levels, in our study. There are several reasons that might be responsible for this discrepancy. Firstly, a lack of power arose from the limited effect size of the individual variations. Secondly, abnormal levels of tau appear later than the occurrence of the Aβ deposition (Jack et al., 2010, 2013; Buchhave et al., 2012) and we could not observe longitudinal changes of CSF tau due to cross-sectional measurements. Furthermore, as experimental research in vitro, Jun's findings could not completely reflect the effects of PLXNA4 on tau phosphorylation under physiological circumstances. In physiological conditions, the targeted genetic variants can perform a direct effect on phenotypes, but also mediate an influence through the interaction with other genes, or via a downstream functional change. Future studies should ascertain whether these SNPs are authentic regulatory variants (and if so, how it may impact protein expression) or if they are simply flagging a region comprised of another functional variant (Roussotte et al., 2015).
Genetic studies with endophenotypes, not only support enough power to identify novel relations with smaller sample sizes than case–control studies, but can also assist in understanding biological mechanisms of disease (Deming et al., 2017). However, some caveats in this study should be noted. Firstly, our sample was of limited size especially in the subgroup. Secondly, our participants were limited to Caucasians, to prevent genetic stratification across ethnicities and our findings can therefore not be generalized to other ethnicities. Thirdly, we could not obtain longitudinal changes of CSF biomarkers due to the cross-sectional nature of the present study. Furthermore, these PLXNA4 loci were restricted because of a lack of exact replications. These results should therefore be treated with caution and future replication studies with larger sample sizes, different ethnicities, and longer follow-up are imperative to validate these findings.
Conclusions
In summary, we detected that two PLXNA4 loci (rs6467431, rs67468325) correlated with amyloid loads, suggesting that PLXNA4 may participate in pathogenesis of AD through Aβ pathology. These findings further support that PLXNA4 genotypes modulate the alterations of Aβ deposition to modify the susceptibility of AD. Although these variants mainly impact subjects at the baseline, it could still provide clues to the underlying relationship between PLXNA4 and AD. Further studies are warranted to unravel the detailed mechanisms underlying the impacts of PLXNA4 on AD.
Author Contributions
LT designed the study Y-AS and QH analyzed the data QH drafted the manuscript LT and QH revised the manuscript QH, H-FW, CC, Y-AS, YZ, and LT interpreted the findings of study. All authors approved the final version for publication.
Alzheimer's Disease Neuroimaging Initiative
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Funding
This work was also supported by grants from the National Key R&D Program of China (2016YFC1305803), Qingdao Key Health Discipline Development Fund, Qingdao Outstanding Health Professional Development Fund, and Shandong Provincial Collaborative Innovation Center for Neurodegenerative Disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). The ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research provide funds to support the ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study was coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
References
Aizenstein, H. J., Nebes, R. D., Saxton, J. A., Price, J. C., Mathis, C. A., Tsopelas, N. D., et al. (2008). Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol. 65, 1509–1517. doi: 10.1001/archneur.65.11.1509
Andermatt, I., Wilson, N. H., Bergmann, T., Mauti, O., Gesemann, M., Sockanathan, S., et al. (2014). Semaphorin 6B acts as a receptor in post-crossing commissural axon guidance. Development 141, 3709–3720. doi: 10.1242/dev.112185
Bateman, R. J., Xiong, C., Benzinger, T. L., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Biffi, A., Anderson, C. D., Desikan, R. S., Sabuncu, M., Cortellini, L., Schmansky, N., et al. (2010). Genetic variation and neuroimaging measures in Alzheimer disease. Arch. Neurol. 67, 677–685. doi: 10.1001/archneurol.2010.108
Buchhave, P., Minthon, L., Zetterberg, H., Wallin, A. K., Blennow, K., and Hansson, O. (2012). Cerebrospinal fluid levels of beta-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 69, 98–106. doi: 10.1001/archgenpsychiatry.2011.155
Carrillo, M. C., Bain, L. J., Frisoni, G. B., and Weiner, M. W. (2012). Worldwide Alzheimer's disease neuroimaging initiative. Alzheimers. Dement. 8, 337–342. doi: 10.1016/j.jalz.2012.04.007
Choi, S. H., Liu, C., Dupuis, J., Logue, M. W., and Jun, G. (2011). Using linkage analysis of large pedigrees to guide association analyses. BMC Proc. 5(Suppl. 9):S79. doi: 10.1186/1753-6561-5-S9-S79
Cole, A. R., Knebel, A., Morrice, N. A., Robertson, L. A., Irving, A. J., Connolly, C. N., et al. (2004). GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J. Biol. Chem. 279, 50176–50180. doi: 10.1074/jbc.C400412200
Cummings, J. L., Doody, R., and Clark, C. (2007). Disease-modifying therapies for Alzheimer disease: challenges to early intervention. Neurology 69, 1622–1634. doi: 10.1212/01.wnl.0000295996.54210.69
Deming, Y., Li, Z., Kapoor, M., Harari, O., Del-Aguila, J. L., Black, K., et al. (2017). Genome-wide association study identifies four novel loci associated with Alzheimer's endophenotypes and disease modifiers. Acta Neuropathol. 133, 839–856. doi: 10.1007/s00401-017-1685-y
Dubois, B., Feldman, H. H., Jacova, C., Hampel, H., Molinuevo, J. L., Blennow, K., et al. (2014). Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. 13, 614–629. doi: 10.1016/S1474-4422(14)70090-0
Gatz, M., Reynolds, C. A., Fratiglioni, L., Johansson, B., Mortimer, J. A., Berg, S., et al. (2006). Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174. doi: 10.1001/archpsyc.63.2.168
Good, P. F., Alapat, D., Hsu, A., Chu, C., Perl, D., Wen, X., et al. (2004). A role for semaphorin 3A signaling in the degeneration of hippocampal neurons during Alzheimer's disease. J. Neurochem. 91, 716–736. doi: 10.1111/j.1471-4159.2004.02766.x
Hochberg, Y., and Benjamini, Y. (1990). More powerful procedures for multiple significance testing. Stat. Med. 9, 811–818. doi: 10.1002/sim.4780090710
Jack, C. R. Jr., Knopman, D. S., Jagust, W. J., Petersen, R. C., Weiner, M. W., Aisen, P. S., et al. (2013). Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. doi: 10.1016/S1474-4422(12)70291-0
Jack, C. R. Jr., Knopman, D. S., Jagust, W. J., Shaw, L. M., Aisen, P. S., Weiner, M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 9, 119–128. doi: 10.1016/S1474-4422(09)70299-6
Jiang, T., Yu, J. T., Tian, Y., and Tan, L. (2013). Epidemiology and etiology of Alzheimer's disease: from genetic to non-genetic factors. Curr. Alzheimer Res. 10, 852–867. doi: 10.2174/15672050113109990155
Jun, G., Asai, H., Zeldich, E., Drapeau, E., Chen, C., Chung, J., et al. (2014). PLXNA4 is associated with Alzheimer disease and modulates tau phosphorylation. Ann. Neurol. 76, 379–392. doi: 10.1002/ana.24219
Kang, S. S., Kurti, A., Wojtas, A., Baker, K. E., Liu, C. C., Kanekiyo, T., et al. (2016). Identification of plexin A4 as a novel clusterin receptor links two Alzheimer's disease risk genes. Hum. Mol. Genet. 25, 3467–3475. doi: 10.1093/hmg/ddw188
Kong, Y., Janssen, B. J., Malinauskas, T., Vangoor, V. R., Coles, C. H., Kaufmann, R., et al. (2016). Structural basis for plexin activation and regulation. Neuron 91, 548–560. doi: 10.1016/j.neuron.2016.06.018
Livingston, G., Sommerlad, A., Orgeta, V., Costafreda, S. G., Huntley, J., Ames, D., et al. (2017). Dementia prevention, intervention, and care. Lancet 390, 2673–2734. doi: 10.1016/S0140-6736(17)31363-6
Mormino, E. C., Kluth, J. T., Madison, C. M., Rabinovici, G. D., Baker, S. L., Miller, B. L., et al. (2009). Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain 132, 1310–1323. doi: 10.1093/brain/awn320
Mueller, S. G., Weiner, M. W., Thal, L. J., Petersen, R. C., Jack, C., Jagust, W., et al. (2005). The Alzheimer's disease neuroimaging initiative. Neuroimaging Clin N Am 15, 869–877, xi-xii. doi: 10.1016/j.nic.2005.09.008
Pasterkamp, R. J. (2012). Getting neural circuits into shape with semaphorins. Nat. Rev. Neurosci. 13, 605–618. doi: 10.1038/nrn3302
Reitz, C., and Mayeux, R. (2014). Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 88, 640–651. doi: 10.1016/j.bcp.2013.12.024
Roussotte, F. F., Gutman, B. A., Hibar, D. P., Madsen, S. K., Narr, K. L., and Thompson, P. M. (2015). Carriers of a common variant in the dopamine transporter gene have greater dementia risk, cognitive decline, and faster ventricular expansion. Alzheimers. Dement. 11, 1153–1162. doi: 10.1016/j.jalz.2014.10.011
Saykin, A. J., Shen, L., Foroud, T. M., Potkin, S. G., Swaminathan, S., Kim, S., et al. (2010). Alzheimer's Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: Genetics core aims, progress, and plans. Alzheimers. Dement. 6, 265–273. doi: 10.1016/j.jalz.2010.03.013
Scheltens, P., Blennow, K., Breteler, M. M., De Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer's disease. Lancet 388, 505–517. doi: 10.1016/S0140-6736(15)01124-1
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers. Dement. 7, 280–292. doi: 10.1016/j.jalz.2011.03.003
Sun, L. O., Jiang, Z., Rivlin-Etzion, M., Hand, R., Brady, C. M., Matsuoka, R. L., et al. (2013). On and off retinal circuit assembly by divergent molecular mechanisms. Science 342:1241974. doi: 10.1126/science.1241974
Suto, F., Ito, K., Uemura, M., Shimizu, M., Shinkawa, Y., Sanbo, M., et al. (2005). Plexin-a4 mediates axon-repulsive activities of both secreted and transmembrane semaphorins and plays roles in nerve fiber guidance. J. Neurosci. 25, 3628–3637. doi: 10.1523/JNEUROSCI.4480-04.2005
Tran, T. S., Rubio, M. E., Clem, R. L., Johnson, D., Case, L., Tessier-Lavigne, M., et al. (2009). Secreted semaphorins control spine distribution and morphogenesis in the postnatal CNS. Nature 462, 1065–1069. doi: 10.1038/nature08628
Yaron, A., Huang, P. H., Cheng, H. J., and Tessier-Lavigne, M. (2005). Differential requirement for Plexin-A3 and -A4 in mediating responses of sensory and sympathetic neurons to distinct class 3 Semaphorins. Neuron 45, 513–523. doi: 10.1016/j.neuron.2005.01.013
Yu, J. T., and Tan, L. (2012). The role of clusterin in Alzheimer's disease: pathways, pathogenesis, and therapy. Mol. Neurobiol. 45, 314–326. doi: 10.1007/s12035-012-8237-1
Keywords: Plexin-A 4 (PLXNA4), variant, Alzheimer's disease (AD), amyloid-β (Aβ), association
Citation: Han Q, Sun Y-A, Zong Y, Chen C, Wang H-F, Tan L and Alzheimer's Disease Neuroimaging Initiative (2018) Common Variants in PLXNA4 and Correlation to CSF-related Phenotypes in Alzheimer's Disease. Front. Neurosci. 12:946. doi: 10.3389/fnins.2018.00946
Received: 06 September 2018; Accepted: 29 November 2018;
Published: 18 December 2018.
Edited by:
Qinghua Jiang, Harbin Institute of Technology, ChinaReviewed by:
Keshen Li, Jinan University, ChinaJun Zhang, Texas Tech University Health Sciences Center, United States
Copyright © 2018 Han, Sun, Zong, Chen, Wang, Tan and Alzheimer's Disease Neuroimaging Initiative. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lan Tan, ZHIudGFubGFuQDE2My5jb20=
†These authors have contributed equally to this work