 Alberto Piperno
Alberto Piperno Massimo Alessio
Massimo Alessio
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurosci., 04 December 2018
Sec. Neurodegeneration
Volume 12 - 2018 | https://doi.org/10.3389/fnins.2018.00903
This article is part of the Research TopicIron and NeurodegenerationView all 30 articles
Aceruloplasminemia is an ultra-rare hereditary disorder caused by defective production of ceruloplasmin. Its phenotype is characterized by iron-restricted erythropoiesis and tissue iron overload, diabetes, and progressive retinal and neurological degeneration. Ceruloplasmin is a ferroxidase that plays a critical role in iron homeostasis through the oxidation and mobilization of iron from stores and subsequent incorporation of ferric iron into transferrin (Tf), which becomes available for cellular uptake via the Tf receptor. In addition, ceruloplasmin has antioxidant properties preventing the production of deleterious reactive oxygen species via the Fenton reaction. Some recent findings suggest that aceruloplasminemia phenotypes can be more heterogeneous than previously believed, varying within a wide range. Within this large heterogeneity, microcytosis with or without anemia, low serum iron and high serum ferritin, and diabetes are the early hallmarks of the disease, while neurological manifestations appear 10–20 years later. The usual therapeutic approach is based on iron chelators that are efficacious in reducing systemic iron overload. However, they have demonstrated poor efficacy in counteracting the progression of neurologic manifestations, and also often aggravate anemia, thereby requiring drug discontinuation. Open questions remain regarding the mechanisms leading to neurological manifestation and development of diabetes, and iron chelation therapy (ICT) efficacy. Recent studies in animal models of aceruloplasminemia support the possibility of new therapeutic approaches by parenteral ceruloplasmin administration. In this review we describe the state of the art of aceruloplasminemia with particular attention on the pathogenic mechanisms of the disease and therapeutic approaches, both current and perspective.
Aceruloplasminemia (ACP) was firstly described in 1987 as an autosomal recessive disease caused by an inactivating mutation of the ceruloplasmin gene (CP) (Miyajima et al., 1987). The rarity of the disease (estimate prevalence about 1:2,000,000) is a major limit for a comprehensive definition of phenotype, genotype-phenotype associations, therapeutic efficiency, and development of disease markers and drugs. Indeed, the typical manifestations make ACP a unique iron overload disease being: (i) The only one among the Neurodegeneration with Brain Iron Overload disorders, to whom ACP belongs, manifesting systemic iron overload; (ii) The unique systemic iron overload disease characterized by neuropathy as major cause of morbidity and microcytic anemia with low serum iron and transferrin (Tf) saturation as a common manifestation. This phenotype is related to the function of ceruloplasmin (Cp) in iron homeostasis at both systemic and brain levels. The lack of Cp induces cellular iron retention and progressive overload on the one hand, and low cellular iron release leading to iron-restricted erythropoiesis and iron deficiency anemia, on the other hand. However, the physiopathology of organ damage, as well as the pathways and efficiency of chelators in brain iron removal, are highly controversial issues which remain to be fully elucidated.
Recent studies showed that ACP phenotypes are more heterogeneous than previously believed and that some manifestations may precede neurological manifestation even by decades (Pelucchi et al., 2018). Thus, this could allow early diagnosis, hopefully preventing the development of neurological derangements, which are the most severe complication of this disease. Neurological symptoms usually appear in the fifth decade of life and vary within a wide spectrum that includes cerebellar ataxia, involuntary movements, parkinsonism, mood and behavior disturbances, and cognitive impairment (McNeill et al., 2008; Kono, 2012). As magnetic resonance imaging (MRI) is increasingly used in neurological diagnostics, brain iron accumulation can be found in a few patients with neuro-psychiatric symptoms addressing diagnosis to ACP. However, ACP patients without brain iron overload or patients with brain iron overload without or with only very mild neurological manifestations even after 50 years of age have been reported (Skidmore et al., 2008; Kassubek et al., 2017; Pelucchi et al., 2018), suggesting that genetic and/or acquired factors may partially modify neurologic phenotype.
Retinal manifestations are reported in over 75% of Japanese patients (Miyajima, 1993; Kono, 2012), but these were less frequent in Italian patients (Pelucchi et al., 2018). However, accurate descriptions of retinopathy are available in only two patients (He et al., 2007) ranging from retinal discoloration to macular degeneration, indicating that further studies are needed to better characterize ACP-dependent retinopathy.
Diabetes mellitus is considered an early manifestation of ACP, being reported as the first symptom in 68.5% of patients at a median age of 38.5 years in a recent overview. Diabetes was insulin dependent in 60% of patients, undefined in 30%, and less than 10% orally and/or dietary treated (Vroegindeweij et al., 2015). Little information is available on family susceptibility, concomitant overweight, and beta-cells function, which would be useful in understanding diabetes pathogenesis in ACP.
Often not included in the classical triad of ACP (diabetes, retinopathy, and neuropathy), anemia and/or microcytosis are common manifestations being reported in 80% of ACP patients in Japan (Miyajima, 1993) and recorded as the earliest manifestations of disease in 86% of Italian patients, preceding diagnosis even by decades (Pelucchi et al., 2018). However, in a retrospective review of reported cases, anemia was recorded as the first symptom only in 24% of patients (Vroegindeweij et al., 2015). The high frequency of mild anemia and/or microcytosis in the general population, which are likely to be related to iron deficiency or thalassemia traits, can lead to the underestimation of these signs as early ACP-related manifestations. Thus, despite ACP rarity, Cp measurement should be included in the diagnostic work-up of anemia. The presence of low serum iron and Tf saturation with normal/high serum ferritin may serve to further enhance disease suspicion.
Ceruloplasmin is part of the multicopper ferroxidase family, a group of tissue specific proteins (Cp, hephaestin, and zyklopen) that facilitate cellular iron efflux in conjunction with the membrane ferrous iron exporter ferroportin (Fpn), by oxidizing ferrous iron to the ferric state (Vashchenko and MacGillivray, 2013). Cp is recognized as a serum protein secreted by the liver, but it has also been found as glycosylphosphatidylinositol (GPI)-linked protein in astrocytes and leptomeningeal cells in the central nervous system (CNS), but also in macrophages, hepatocytes and many other tissues (Musci et al., 2014). Cp-KO mice revealed an impairment in hepatocyte and reticuloendothelial iron efflux (Harris et al., 1999), and showed that Cp-GPI is relevant for brain iron metabolism in regulating iron efflux from astrocytes (Jeong and David, 2003). A molecular connection between Cp and Fpn has been established with the finding that ferroxidase activity is required to stabilize Fpn at the cell surface in cells expressing Cp-GPI (De Domenico et al., 2007). Thus, Cp can be considered as a second determinant of Fpn stability after hepcidin. These findings suggest that differently from most disorders of iron overload that reflect changes in the absolute amount of iron, ACP results, at least in early stages, from iron imbalance caused by impairment in the rate of iron efflux from storage sites. Serum hepcidin is decreased in ACP patients, possibly related to the low Tf saturation, suggesting that suppressed hepcidin synthesis might contribute to the development of iron overload (Kaneko et al., 2010). While it is clear that microcytosis, anemia and low serum iron depend on reduced iron availability due to cellular iron sequestration, the development of diabetes and neurodegeneration in ACP are not evidently explained at cellular and molecular levels. It has been suggested that both diabetes and neurodegeneration are secondary to iron accumulation and toxicity (Kono, 2012), however, no clear evidence of intracellular iron deposition in the endocrine pancreas and in neurons has been reported in neither ACP patients nor mice models of ACP (Kato et al., 1997; Jeong and David, 2006; Chen et al., 2018). These findings suggest that neurons and islet beta-cells distress might be related to an elevated sensitivity/vulnerability to iron toxicity or to the iron deficient status induced by iron retention in the surrounding cells (Figure 1).
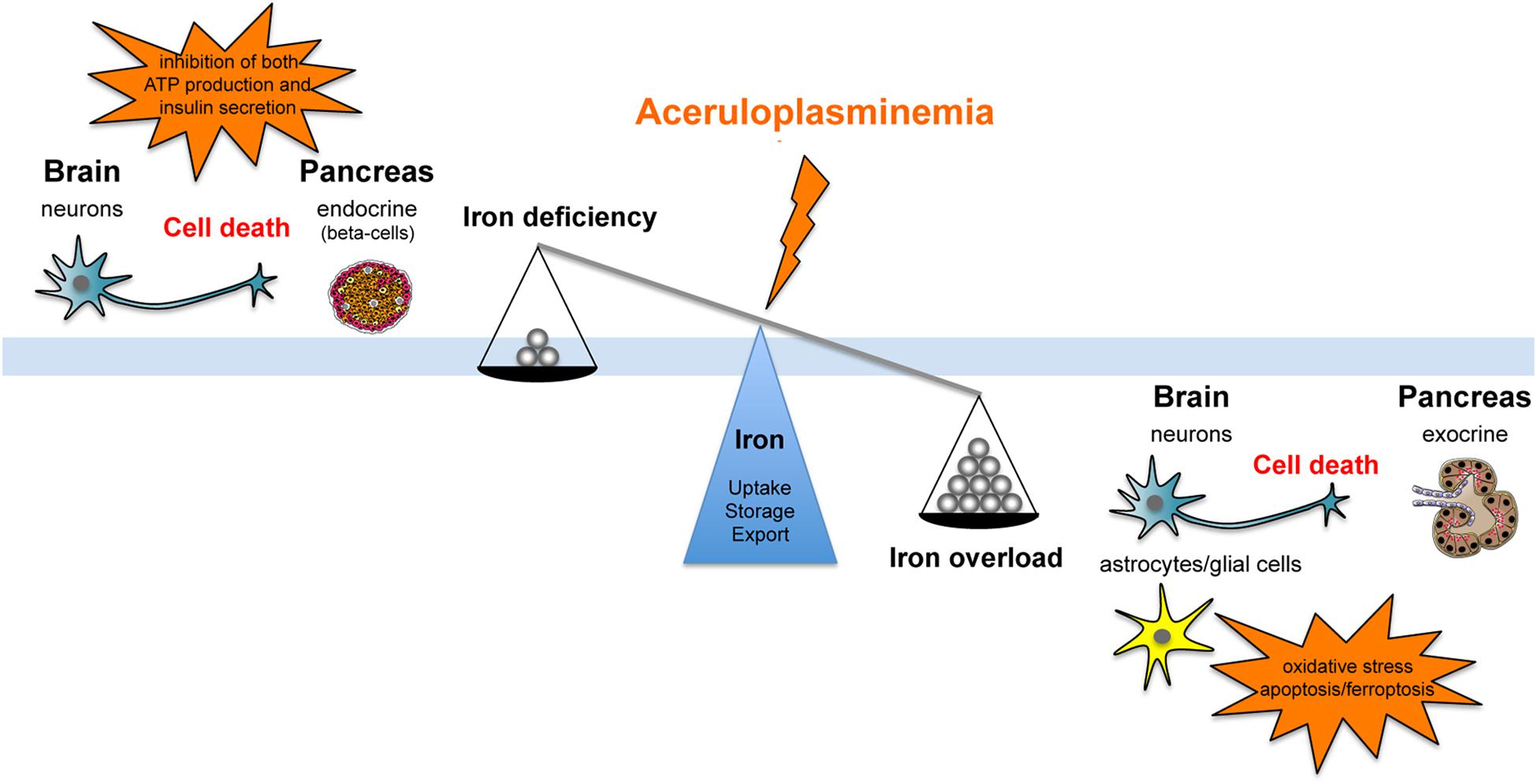
FIGURE 1. Schematic representation of the effect that aceruloplasminemia can promote on iron balance either fostering iron overload or iron deficiency, inducing neurons and islet beta-cells distress due to elevated sensitivity/vulnerability to iron toxicity or to iron deficient status consequent to metal retention in the surrounding cells.
Iron enters the CNS via the brain barrier systems, namely the blood brain barrier (BBB) and the blood-cerebrospinal fluid barrier (BCSFB), which separate the blood from the interstitial- and cerebrospinal-fluid (CSF), respectively. The mechanism of iron uptake/export in cells depends on the iron transport molecules they express. Transferrin-bound-iron, internalized by transferrin receptor 1 (TfR1) in brain capillary endothelial cells (BCECs), can cross the BBB by: (1) transcytosis and direct release as ferric iron in the brain parenchyma, or (2) iron release from endosomes to cytosol via divalent metal ion transporter 1 (DMT1), and exported via Fpn assisted by the GPI-Cp expressed on perivascular astrocytes (McCarthy and Kosman, 2015; Simpson et al., 2015). The absence of iron accumulation in BCECs in both ACP patients and murine models indicate transcytosis and direct iron release as the more plausible mechanism in ACP (Miyajima, 1993; Zanardi et al., 2018). The BCSFB comprises choroid plexus epithelial cells (CPEpiCs) that are in contact with the CSF on one side, and with fenestrated endothelial cells on the other. In CPEpiCs iron is internalized via TfR1 and then exported to the CSF by the action of Fpn and Cp, which are both expressed by CPEpiCs (Rouault et al., 2009; Zheng and Monnot, 2012). Large iron deposition in CPEpiCs precedes accumulation in other brain regions in Cp-KO mice (Zanardi et al., 2018) suggesting an early deficit in iron handling by these cells in ACP. However, the pathophysiology of choroid plexus damage in ACP patients is largely undefined.
Neurons acquire iron prevalently from Tf and TfR1 while oligodendrocytes and astrocytes do not express TfR1, which suggests they import iron using other mechanisms (Zhang et al., 2006; Belaidi and Bush, 2016). Astrocytes may acquire iron released by BCECs being in close contact with them via endfeets that highly express DMT1 (McCarthy and Kosman, 2015). However, DMT1 uptake requires ferric iron to be reduced, but the mechanism of iron reduction by astrocytes is still unclear. Oligodendrocytes and astrocytes are rich in ferritin, suggesting that they function as iron storage site for the whole brain (Finazzi and Arosio, 2014). Regardless of import mechanisms, iron efflux from all brain cells involve Fpn, and depends on the extracellular ferroxidase activity fostered primarily by both the membrane Cp-GPI and the soluble Cp circulating in the interstitial fluid and CSF (Patel and David, 1997; Olivieri et al., 2011). Therefore, in ACP, the absence of Cp-ferroxidase activity promotes Fpn internalization and degradation favoring intracellular iron accumulation (Kono et al., 2010; Persichini et al., 2012).
Brain iron accumulation, particularly in the basal ganglia and cerebellum, is reported by MRI in a large number of ACP patients (Grisoli et al., 2005; Kono, 2012). Nevertheless, the few autopsy studies performed indicated that iron deposition was more evident in glial and astrocytes than in neurons, in particular in the perivascular regions of brain areas where neuronal loss is prevalent (Kawanami et al., 1996; Kaneko et al., 2002, 2012; Oide et al., 2006). Thus, it has been hypothesized that apoptosis/ferroptosis of iron-engulfed astrocytes/glial cells might release large amounts of toxic ferrous iron promoting oxidative neuronal damage, as inferred by large lipids peroxidation observed in the brain of ACP patients (Miyajima et al., 1998, 2001a; Kohno et al., 2000; Yoshida et al., 2000). Neuronal damage could be aggravated by the loss of glial-derived growth factors necessary for neuronal survival (Jeong and David, 2006). It is also conceivable that neuronal injury might depend on iron deficiency as the consequence of impaired mobilization of the accumulated iron in glial cells (Jeong and David, 2006; Kono, 2013).
In the liver of ACP patients, iron accumulation prevails in hepatocytes, a surprising finding considering the role of Cp in regulating cellular iron efflux via Fpn. (Morita et al., 1995; Kawanami et al., 1996; Hellman et al., 2000; Bosio et al., 2002; Loreal et al., 2002; Finkenstedt et al., 2010; Rusticeanu et al., 2014). Indeed, liver iron distribution differs in patients with classical Fpn disease and ACP, being prevalent in reticuloendothelial cells in the former and in hepatocytes in the latter, indicating the need to expand our knowledge on the relative function of Fpn and Cp and their interactions in different tissues. Despite hepatocellular iron overload, cirrhosis is uncommon in ACP, however, some patients developed moderate/severe fibrosis in the presence of heavy hepatic iron overload (Pelucchi et al., 2018). This suggests that in ACP patients the risk of liver fibrosis depends on the iron burden, a similar scenario to hereditary hemochromatosis (HH) (Wood et al., 2008), and that liver iron overload in ACP patients generally does not reach the threshold associated with liver damage risk.
Clinically, diabetes may precede neurological derangements even by decades, but the mechanism of endocrine pancreatic damage in ACP is unclear. Iron accumulation in the exocrine pancreas has been commonly reported in mouse models characterized by systemic iron loading (hypotransferrinemic mice, bone morphogenetic protein 6, hemojuvelin-, and hepcidin-deficient strains) (Chen et al., 2018). About 20% of HH patients show diabetes, all of them being heavy iron overloaded and the majority having liver fibrosis/cirrhosis (McClain et al., 2006). In HH-patients iron deposition is present in the exocrine pancreas and, varied from case to case, in beta-cells together with loss of endocrine granules (Rahier et al., 1987). In mouse models of HFE-HH, iron accumulates specifically in the endocrine pancreas resulting in increased beta-cell death (Cooksey et al., 2004). The total pancreatic- and per islet-insulin content were low in Hfe-KO mice (Huang et al., 2011), suggesting an iron-related beta-cell oxidative stress and decreased insulin secretory capacity secondary to beta-cell apoptosis and desensitization of glucose-induced insulin secretion (Backe et al., 2016). However, in other mouse models of hemochromatosis, the endocrine pancreas was unaffected. Hepcidin-KO mice developed chronic pancreatitis owing to exocrine iron overload (Lunova et al., 2017), and, similarly, a murine model of Fpn mutant resistant to hepcidin binding showed exocrine pancreatic failure and iron overload (Altamura et al., 2014). Iron deposition and functional deficit restricted to the exocrine pancreas has also been reported in the double Cp/Hephaestin-KO mice model that did not display structural or functional (insulin secretion) deficiency of pancreatic islets (Chen et al., 2018).
Few studies describe pancreatic alteration in ACP patients mainly reporting generic iron accumulation without histological description of the cells and structures involved (exocrine vs. endocrine) (Daimon et al., 1995; Kawanami et al., 1996). Three reports described the absence or mild iron accumulation in the islets compared to the exocrine pancreas (Morita et al., 1995; Kato et al., 1997; Miyajima et al., 2001b). In a single ACP patient, an autopsy study showed marked reduction of insulin-containing cells without iron accumulation and degeneration/loss of islet cells, despite massive iron deposition in the exocrine pancreas (Kato et al., 1997). The patient’s long type-2 diabetes history may suggest beta-cell exhaustion as a possible explanation for such findings. Nevertheless, the pathogenesis of diabetes in ACP and the role of iron remain elusive, also considering that diabetes can occur early when systemic iron overload is still minor (Hatanaka et al., 2003; Muroi et al., 2006; Pelucchi et al., 2018).
Current treatment in ACP (Table 1) is primarily focused on reducing iron overload using iron chelators (deferasirox, deferoxamine, and deferiprone). In the majority of patients, iron chelation therapy (ICT) was effective in reducing systemic iron overload, but there is no information on whether it can improve glucose metabolism derangement and retinopathy. ICT was less or not effective on neurological symptoms [reviewed in (Kono, 2013; Dusek et al., 2016)]. Thus, there is the need to find treatments efficient in rescuing/preventing neurological manifestations, taking advantage of the window between the appearance of the first manifestations and neurological symptoms (Vroegindeweij et al., 2015; Pelucchi et al., 2018). The variable efficacy of ICT on neurological symptoms may depend on the ability of different chelators to cross the BBB, the timeliness of the treatment, the large heterogeneity of therapeutic protocols, and the unclear kinetics of iron chelators in the brain in vivo [reviewed in (Dusek et al., 2016)]. In the large majority, therapeutic efficacy was evaluated in a short time, and the few data available in long-term follow-up studies showed progression of neurological derangements (Pelucchi et al., 2018). ICT seems more promising if started before the onset of neurological symptoms, although the majority of the cases that remained asymptomatic have not yet reached the risk age [reviewed in (Dusek et al., 2016)]. Also, ICT must be often discontinued because of the aggravation of functional iron deficiency anemia, limiting the long-term therapy required to mobilize iron from the brain (Loreal et al., 2002; Mariani et al., 2004; Fasano et al., 2008; Finkenstedt et al., 2010). Due to the antioxidant properties of zinc sulfate and minocycline, the inhibitory effect on iron absorption of zinc sulfate, and chelating properties of minocycline iron, these have been proposed as alternatives to ICT when the latter must be discontinued, but the results, although promising, are limited to only two patients (Kuhn et al., 2007; Hayashida et al., 2016). To prevent tissue damage, antioxidants like vitamin E and C have been used along with ICT (Kuhn et al., 2007; Pelucchi et al., 2018).
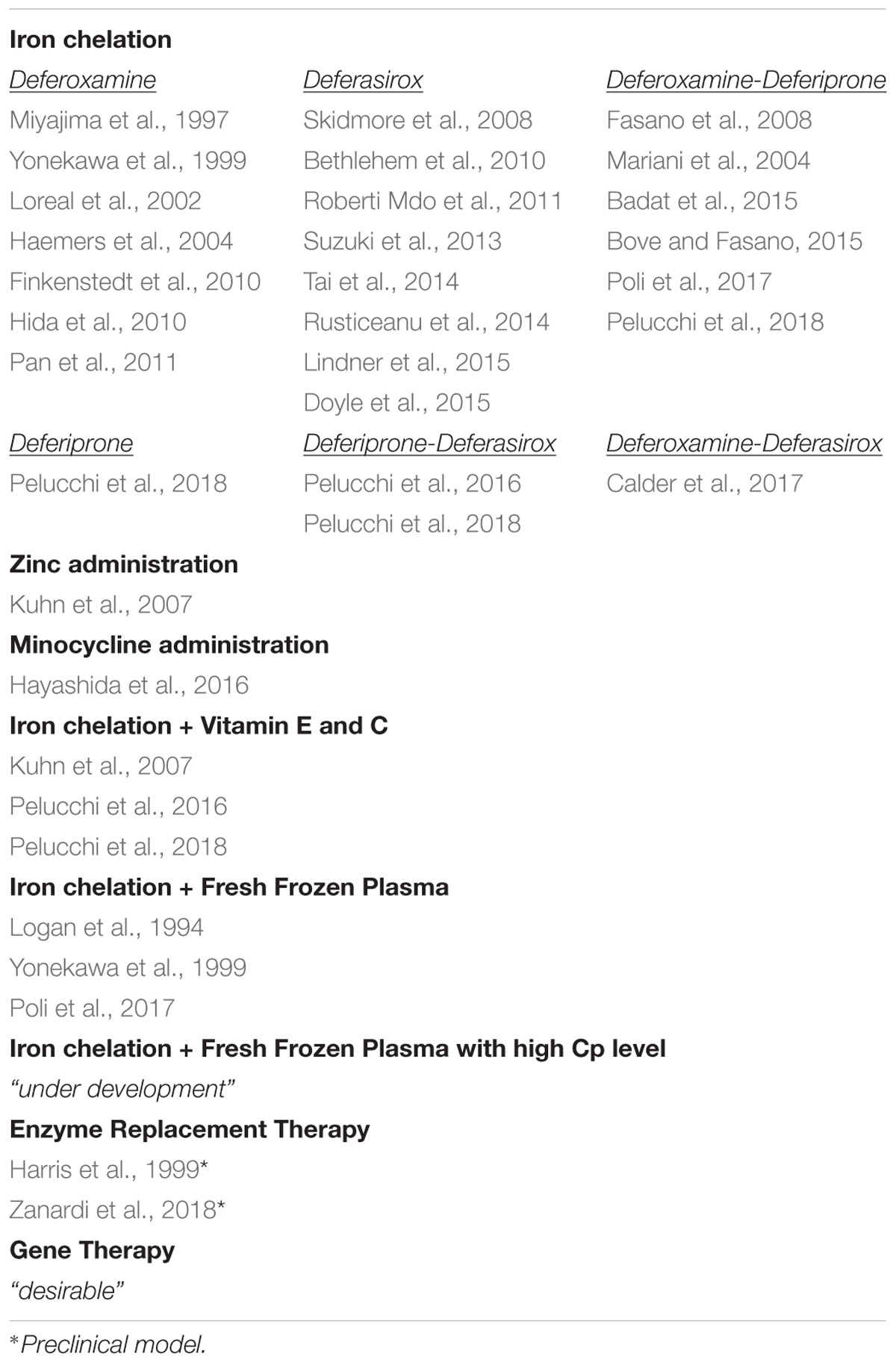
TABLE 1. Therapeutic approaches for aceruloplasminemia.
In some cases, ICT was combined with fresh-frozen plasma (FFP) administration. Due to the half-life of Cp (5.5 days) (Hellman and Gitlin, 2002), FFP administration could partially/temporarily restore circulating Cp. In two cases, the combined therapy improved neurological symptoms, visibly reducing brain iron deposition (Yonekawa et al., 1999; Poli et al., 2017). This implies that circulating Cp can enter the CNS and be functionally effective. Since physiological serum Cp concentration ranges between 21 and 54 mg/dL (Gibbs and Walshe, 1979; Gaasch et al., 2007), the selection of high-Cp-content FFP transfusions might be more effective, thereby advancing this therapeutic approach. A post-transfusion Cp level of 8–10 mg/dL, comparable to that of ACP heterozygotes, could be enough to rescue iron homeostasis as inferred by the absence of clinical symptoms in the vast majority of heterozygous subjects (Miyajima, 1993; Kono, 2013). Nevertheless, the risks associated with long-term repeated transfusion could limit this approach.
A neuroprotective effect of ceruloplasmin administration was reported in various pathological models (Ayton et al., 2013, 2014; Tuo et al., 2017; Zanardi et al., 2018). Indeed, it has been demonstrated that intraperitoneally administered Cp was able to enter the Cp-KO brain crossing the barrier systems (Ayton et al., 2013; Zanardi et al., 2018). The potential of the Cp-enzyme replacement therapy (ERT) in reducing neurological manifestation was recently demonstrated in the preclinical model of ACP (Zanardi et al., 2018). The ferroxidase activity was restored in Cp-KO brain, inducing reduction in iron deposition, rescue of neuronal loss, and amelioration of motor incoordination (Zanardi et al., 2018). The mechanism by which administered Cp crosses the brain barrier systems in Cp-KO, but not in wild-type mice, is not known. However, the iron accumulation found in the choroid plexus of Cp-KO mice suggested that Cp enters the brain likely through BCSFB impairment (Zanardi et al., 2018). A similar impairment would also explain the effectiveness of the FFP therapy in ACP patients. Cp administration also mobilizes iron and temporarily restores iron homeostasis in Cp-KO mice (Harris et al., 1999; Zanardi et al., 2018). Thus, ERT seems to be efficacious on both systemic and neurological symptoms, and may be a potential therapeutic opportunity in humans.
As for many monogenic rare diseases, the ideal ultimate cure consists in gene therapy approaches to correct the defect, but further studies and several years will be necessary before its clinical application.
In conclusion, current therapies in ACP are effective in reducing systemic iron overload but have little or no effect on neurological symptoms. Their capability in improving pancreatic and retinal damage is unclear, and might have deleterious side effects that limit long-term therapy. FFP administration can be a suitable source of functional Cp, and although cost-benefit effects need to be further evaluated, can represent a step toward the development of more efficient therapies in ACP.
AP and MA contributed the same to conception and writing of the work.
Associazione per lo Studio dell’Emocromatosi e della Malattie da Sovraccarico di Ferro-ONLUS will pay for the review. The Association was funded in Monza in 1996 and is part of the European Federation of Associations of Patients with Hemochromatosis (EFAPH). This no-profit Italian Association is involved in supporting information to patients, general practitioners, specialized physician, institutions, research in the field of iron overload disorders.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Associazione per lo Studio dell’Emocromatosi e delle Malattie da Sovraccarico di Ferro (+Fe – ONLUS) for supporting our work. We also thank Dr. Serena Melgrati, Imperial College London, for English language editing of the manuscript.
Altamura, S., Kessler, R., Grone, H. J., Gretz, N., Hentze, M. W., Galy, B., et al. (2014). Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab. 20, 359–367. doi: 10.1016/j.cmet.2014.07.007
Ayton, S., Lei, P., Adlard, P. A., Volitakis, I., Cherny, R. A., Bush, A. I., et al. (2014). Iron accumulation confers neurotoxicity to a vulnerable population of nigral neurons: implications for Parkinson’s disease. Mol. Neurodegener. 9:27. doi: 10.1186/1750-1326-9-27
Ayton, S., Lei, P., Duce, J. A., Wong, B. X., Sedjahtera, A., Adlard, P. A., et al. (2013). Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann. Neurol. 73, 554–559. doi: 10.1002/ana.23817
Backe, M. B., Moen, I. W., Ellervik, C., Hansen, J. B., and Mandrup-Poulsen, T. (2016). Iron regulation of pancreatic beta-cell functions and oxidative stress. Annu. Rev. Nutr. 36, 241–273. doi: 10.1146/annurev-nutr-071715-050939
Badat, M., Kaya, B., and Telfer, P. (2015). Combination-therapy with concurrent deferoxamine and deferiprone is effective in treating resistant cardiac iron-loading in aceruloplasminaemia. Br. J. Haematol. 171, 430–432. doi: 10.1111/bjh.13401
Belaidi, A. A., and Bush, A. I. (2016). Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J. Neurochem. 139(Suppl. 1), 179–197. doi: 10.1111/jnc.13425
Bethlehem, C., van Harten, B., and Hoogendoorn, M. (2010). Central nervous system involvement in a rare genetic iron overload disorder. Neth. J. Med. 68, 316–318.
Bosio, S., De Gobbi, M., Roetto, A., Zecchina, G., Leonardo, E., Rizzetto, M., et al. (2002). Anemia and iron overload due to compound heterozygosity for novel ceruloplasmin mutations. Blood 100, 2246–2248. doi: 10.1182/blood-2002-02-0584
Bove, F., and Fasano, A. (2015). Iron chelation therapy to prevent the manifestations of aceruloplasminemia. Neurology 85, 1085–1086. doi: 10.1212/WNL.0000000000001956
Calder, G. L., Lee, M. H., Sachithanandan, N., Bell, S., Zeimer, H., and MacIsaac, R. J. (2017). Aceruloplasminaemia: a disorder of diabetes and neurodegeneration. Intern. Med. J. 47, 115–118. doi: 10.1111/imj.13309
Chen, M., Zheng, J., Liu, G., Xu, E., Wang, J., Fuqua, B. K., et al. (2018). Ceruloplasmin and hephaestin jointly protect the exocrine pancreas against oxidative damage by facilitating iron efflux. Redox. Biol. 17, 432–439. doi: 10.1016/j.redox.2018.05.013
Cooksey, R. C., Jouihan, H. A., Ajioka, R. S., Hazel, M. W., Jones, D. L., Kushner, J. P., et al. (2004). Oxidative stress, beta-cell apoptosis, and decreased insulin secretory capacity in mouse models of hemochromatosis. Endocrinology 145, 5305–5312. doi: 10.1210/en.2004-0392
Daimon, M., Kato, T., Kawanami, T., Tominaga, M., Igarashi, M., Yamatani, K., et al. (1995). A nonsense mutation of the ceruloplasmin gene in hereditary ceruloplasmin deficiency with diabetes mellitus. Biochem. Biophys. Res. Commun. 217, 89–95. doi: 10.1006/bbrc.1995.2749
De Domenico, I., Ward, D. M., Di Patti, M. C., Jeong, S. Y., David, S., Musci, G., et al. (2007). Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 26, 2823–2831. doi: 10.1038/sj.emboj.7601735
Doyle, A., Rusli, F., and Bhathal, P. (2015). Aceruloplasminaemia: a rare but important cause of iron overload. BMJ Case Rep. 2015:bcr-2014-207541. doi: 10.1136/bcr-2014-207541
Dusek, P., Schneider, S. A., and Aaseth, J. (2016). Iron chelation in the treatment of neurodegenerative diseases. J. Trace Elem. Med. Biol. 38, 81–92. doi: 10.1016/j.jtemb.2016.03.010
Fasano, A., Colosimo, C., Miyajima, H., Tonali, P. A., Re, T. J., and Bentivoglio, A. R. (2008). Aceruloplasminemia: a novel mutation in a family with marked phenotypic variability. Mov. Disord. 23, 751–755. doi: 10.1002/mds.21938
Finazzi, D., and Arosio, P. (2014). Biology of ferritin in mammals: an update on iron storage, oxidative damage and neurodegeneration. Arch. Toxicol. 88, 1787–1802. doi: 10.1007/s00204-014-1329-0
Finkenstedt, A., Wolf, E., Hofner, E., Gasser, B. I., Bosch, S., Bakry, R., et al. (2010). Hepatic but not brain iron is rapidly chelated by deferasirox in aceruloplasminemia due to a novel gene mutation. J. Hepatol. 53, 1101–1107. doi: 10.1016/j.jhep.2010.04.039
Gaasch, J. A., Lockman, P. R., Geldenhuys, W. J., Allen, D. D., and Van Der Schyf, C. J. (2007). Brain iron toxicity: differential responses of astrocytes, neurons, and endothelial cells. Neurochem. Res. 32, 1196–1208. doi: 10.1007/s11064-007-9290-4
Gibbs, K., and Walshe, J. M. (1979). A study of the caeruloplasmin concentrations found in 75 patients with Wilson’s disease, their kinships and various control groups. Q. J. Med. 48, 447–463.
Grisoli, M., Piperno, A., Chiapparini, L., Mariani, R., and Savoiardo, M. (2005). MR imaging of cerebral cortical involvement in aceruloplasminemia. AJNR Am. J. Neuroradiol. 26, 657–661.
Haemers, I., Kono, S., Goldman, S., Gitlin, J. D., and Pandolfo, M. (2004). Clinical, molecular, and PET study of a case of aceruloplasminaemia presenting with focal cranial dyskinesia. J. Neurol. Neurosurg. Psychiatry 75, 334–337. doi: 10.1136/jnnp.2003.017434
Harris, Z. L., Durley, A. P., Man, T. K., and Gitlin, J. D. (1999). Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc. Natl. Acad. Sci. U.S.A. 96, 10812–10817. doi: 10.1073/pnas.96.19.10812
Hatanaka, Y., Okano, T., Oda, K., Yamamoto, K., and Yoshida, K. (2003). Aceruloplasminemia with juvenile-onset diabetes mellitus caused by exon skipping in the ceruloplasmin gene. Intern. Med. 42, 599–604. doi: 10.2169/internalmedicine.42.599
Hayashida, M., Hashioka, S., Miki, H., Nagahama, M., Wake, R., Miyaoka, T., et al. (2016). Aceruloplasminemia with psychomotor excitement and neurological sign was improved by minocycline (case report). Medicine 95:e3594. doi: 10.1097/MD.0000000000003594
He, X., Hahn, P., Iacovelli, J., Wong, R., King, C., Bhisitkul, R., et al. (2007). Iron homeostasis and toxicity in retinal degeneration. Prog. Retin. Eye Res. 26, 649–673. doi: 10.1016/j.preteyeres.2007.07.004
Hellman, N. E., and Gitlin, J. D. (2002). Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 22, 439–458. doi: 10.1146/annurev.nutr.22.012502.114457
Hellman, N. E., Schaefer, M., Gehrke, S., Stegen, P., Hoffman, W. J., Gitlin, J. D., et al. (2000). Hepatic iron overload in aceruloplasminaemia. Gut 47, 858–860. doi: 10.1136/gut.47.6.858
Hida, A., Kowa, H., Iwata, A., Tanaka, M., Kwak, S., and Tsuji, S. (2010). Aceruloplasminemia in a Japanese woman with a novel mutation of CP gene: clinical presentations and analysis of genetic and molecular pathogenesis. J. Neurol. Sci. 298, 136–139. doi: 10.1016/j.jns.2010.08.019
Huang, J., Jones, D., Luo, B., Sanderson, M., Soto, J., Abel, E. D., et al. (2011). Iron overload and diabetes risk: a shift from glucose to Fatty Acid oxidation and increased hepatic glucose production in a mouse model of hereditary hemochromatosis. Diabetes Metab. Res. Rev. 60, 80–87. doi: 10.2337/db10-0593
Jeong, S. Y., and David, S. (2003). Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 278, 27144–27148. doi: 10.1074/jbc.M301988200
Jeong, S. Y., and David, S. (2006). Age-related changes in iron homeostasis and cell death in the cerebellum of ceruloplasmin-deficient mice. J. Neurosci. 26, 9810–9819. doi: 10.1523/JNEUROSCI.2922-06.2006
Kaneko, K., Hineno, A., Yoshida, K., Ohara, S., Morita, H., and Ikeda, S. (2012). Extensive brain pathology in a patient with aceruloplasminemia with a prolonged duration of illness. Hum. Pathol. 43, 451–456. doi: 10.1016/j.humpath.2011.05.016
Kaneko, K., Yoshida, K., Arima, K., Ohara, S., Miyajima, H., Kato, T., et al. (2002). Astrocytic deformity and globular structures are characteristic of the brains of patients with aceruloplasminemia. J. Neuropathol. Exp. Neurol. 61, 1069–1077. doi: 10.1093/jnen/61.12.1069
Kaneko, Y., Miyajima, H., Piperno, A., Tomosugi, N., Hayashi, H., Morotomi, N., et al. (2010). Measurement of serum hepcidin-25 levels as a potential test for diagnosing hemochromatosis and related disorders. J. Gastroenterol. 45, 1163–1171. doi: 10.1007/s00535-010-0259-8
Kassubek, R., Uttner, I., Schonfeldt-Lecuona, C., Kassubek, J., and Connemann, B. J. (2017). Extending the aceruloplasminemia phenotype: NBIA on imaging and acanthocytosis, yet only minor neurological findings. J. Neurol. Sci. 376, 151–152. doi: 10.1016/j.jns.2017.03.019
Kato, T., Daimon, M., Kawanami, T., Ikezawa, Y., Sasaki, H., and Maeda, K. (1997). Islet changes in hereditary ceruloplasmin deficiency. Hum. Pathol. 28, 499–502. doi: 10.1016/S0046-8177(97)90041-1
Kawanami, T., Kato, T., Daimon, M., Tominaga, M., Sasaki, H., Maeda, K., et al. (1996). Hereditary caeruloplasmin deficiency: clinicopathological study of a patient. J. Neurol. Neurosurg. Psychiatry 61, 506–509. doi: 10.1136/jnnp.61.5.506
Kohno, S., Miyajima, H., Takahashi, Y., Suzuki, H., and Hishida, A. (2000). Defective electron transfer in complexes I and IV in patients with aceruloplasminemia. J. Neurol. Sci. 182, 57–60. doi: 10.1016/S0022-510X(00)00452-4
Kono, S. (2012). Aceruloplasminemia. Curr. Drug Targets 13, 1190–1199. doi: 10.2174/138945012802002320
Kono, S. (2013). Aceruloplasminemia: an update. Int. Rev. Neurobiol. 110, 125–151. doi: 10.1016/B978-0-12-410502-7.00007-7
Kono, S., Yoshida, K., Tomosugi, N., Terada, T., Hamaya, Y., Kanaoka, S., et al. (2010). Biological effects of mutant ceruloplasmin on hepcidin-mediated internalization of ferroportin. Biochim. Biophys. Acta 1802, 968–975. doi: 10.1016/j.bbadis.2010.07.011
Kuhn, J., Bewermeyer, H., Miyajima, H., Takahashi, Y., Kuhn, K. F., and Hoogenraad, T. U. (2007). Treatment of symptomatic heterozygous aceruloplasminemia with oral zinc sulphate. Brain Dev. 29, 450–453. doi: 10.1016/j.braindev.2007.01.001
Lindner, U., Schuppan, D., Schleithoff, L., Habeck, J.-O., Grodde, T., Kirchhof, K., et al. (2015). Aceruloplasminaemia: a family with a novel mutation and long-term therapy with deferasirox. Horm. Metab. Res. 47, 303–308. doi: 10.1055/s-0034-1383650
Logan, J. I., Harveyson, K. B., Wisdom, G. B., Hughes, A. E., and Archbold, G. P. R. (1994). Hereditary caeruloplasmin deficiency, dementia and diabetes mellitus. QJM 87, 663–670. doi: 10.1093/oxfordjournals.qjmed.a068881
Loreal, O., Turlin, B., Pigeon, C., Moisan, A., Ropert, M., Morice, P., et al. (2002). Aceruloplasminemia: new clinical, pathophysiological and therapeutic insights. J. Hepatol. 36, 851–856. doi: 10.1016/S0168-8278(02)00042-9
Lunova, M., Schwarz, P., Nuraldeen, R., Levada, K., Kuscuoglu, D., Stutzle, M., et al. (2017). Hepcidin knockout mice spontaneously develop chronic pancreatitis owing to cytoplasmic iron overload in acinar cells. J. Pathol. 241, 104–114. doi: 10.1002/path.4822
Mariani, R., Arosio, C., Pelucchi, S., Grisoli, M., Piga, A., Trombini, P., et al. (2004). Iron chelation therapy in aceruloplasminaemia: study of a patient with a novel missense mutation. Gut 53, 756–758. doi: 10.1136/gut.2003.030429
McCarthy, R. C., and Kosman, D. J. (2015). Mechanisms and regulation of iron trafficking across the capillary endothelial cells of the blood-brain barrier. Front. Mol. Neurosci. 8:31. doi: 10.3389/fnmol.2015.00031
McClain, D. A., Abraham, D., Rogers, J., Brady, R., Gault, P., Ajioka, R., et al. (2006). High prevalence of abnormal glucose homeostasis secondary to decreased insulin secretion in individuals with hereditary haemochromatosis. Diabetologia 49, 1661–1669. doi: 10.1007/s00125-006-0200-0
McNeill, A., Pandolfo, M., Kuhn, J., Shang, H., and Miyajima, H. (2008). The neurological presentation of ceruloplasmin gene mutations. Eur. Neurol. 60, 200–205. doi: 10.1159/000148691
Miyajima, H. (1993). “Aceruloplasminemia,” in GeneReviews(R), eds R. A. Pagon, M. P. Adam, H. H. Ardinger, S. E. Wallace, A. Amemiya, L. J. H. Bean, et al. (Seattle, WA: University of Washington).
Miyajima, H., Adachi, J., Kohno, S., Takahashi, Y., Ueno, Y., and Naito, T. (2001a). Increased oxysterols associated with iron accumulation in the brains and visceral organs of acaeruloplasminaemia patients. QJM 94, 417–422.
Miyajima, H., Fujimoto, M., Kohno, S., Kaneko, E., and Gitlin, J. D. (1998). CSF abnormalities in patients with aceruloplasminemia. Neurology 51, 1188–1190. doi: 10.1212/WNL.51.4.1188
Miyajima, H., Kono, S., Takahashi, Y., Sugimoto, M., Sakamoto, M., and Sakai, N. (2001b). Cerebellar ataxia associated with heteroallelic ceruloplasmin gene mutation. Neurology 57, 2205–2210.
Miyajima, H., Nishimura, Y., Mizoguchi, K., Sakamoto, M., Shimizu, T., and Honda, N. (1987). Familial apoceruloplasmin deficiency associated with blepharospasm and retinal degeneration. Neurology 37, 761–767. doi: 10.1212/WNL.37.5.761
Miyajima, H., Takahashi, Y., Kamata, T., Shimizu, H., Sakai, N., and Gitlin, J. D. (1997). Use of desferrioxamine in the treatment of aceruloplasminemia. Ann. Neurol. 41, 404–407. doi: 10.1002/ana.410410318
Morita, H., Ikeda, S., Yamamoto, K., Morita, S., Yoshida, K., Nomoto, S., et al. (1995). Hereditary ceruloplasmin deficiency with hemosiderosis: a clinicopathological study of a Japanese family. Ann. Neurol. 37, 646–656. doi: 10.1002/ana.410370515
Muroi, R., Yagyu, H., Kobayashi, H., Nagata, M., Sato, N., Ideno, J., et al. (2006). Early onset insulin-dependent diabetes mellitus as an initial manifestation of aceruloplasminaemia. Diabet. Med. 23, 1136–1139. doi: 10.1111/j.1464-5491.2006.01883.x
Musci, G., Polticelli, F., and Bonaccorsi Di Patti, M. C. (2014). Ceruloplasmin-ferroportin system of iron traffic in vertebrates. World J. Biol. Chem. 5, 204–215. doi: 10.4331/wjbc.v5.i2.204
Oide, T., Yoshida, K., Kaneko, K., Ohta, M., and Arima, K. (2006). Iron overload and antioxidative role of perivascular astrocytes in aceruloplasminemia. Neuropathol. Appl. Neurobiol. 32, 170–176. doi: 10.1111/j.1365-2990.2006.00710.x
Olivieri, S., Conti, A., Iannaccone, S., Cannistraci, C. V., Campanella, A., Barbariga, M., et al. (2011). Ceruloplasmin oxidation, a feature of Parkinson’s disease CSF, inhibits ferroxidase activity and promotes cellular iron retention. J. Neurosci. 31, 18568–18577. doi: 10.1523/JNEUROSCI.3768-11.2011
Patel, B. N., and David, S. (1997). A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J. Biol. Chem. 272, 20185–20190. doi: 10.1074/jbc.272.32.20185
Pan, P.-L., Tang, H.-H., Chen, Q., Song, W., and Shang, H.-F. (2011). Desferrioxamine treatment of aceruloplasminemia: long-term follow-up. Mov. Disord. 26, 2142–2144. doi: 10.1002/mds.23797
Pelucchi, S., Mariani, R., Ravasi, G., Pelloni, I., Marano, M., Tremolizzo, L., et al. (2018). Phenotypic heterogeneity in seven Italian cases of aceruloplasminemia. Parkinsonism. Relat. Disord. 51, 36–42. doi: 10.1016/j.parkreldis.2018.02.036
Pelucchi, S., Pelloni, I., Arosio, C., Mariani, R., and Piperno, A. (2016). Does aceruloplasminemia modulate iron phenotype in thalassemia intermedia? Blood Cells Mol. Dis. 57, 112–114. doi: 10.1016/j.bcmd.2015.12.011
Persichini, T., De Francesco, G., Capone, C., Cutone, A., Di Patti, M. C., Colasanti, M., et al. (2012). Reactive oxygen species are involved in ferroportin degradation induced by ceruloplasmin mutant Arg701Trp. Neurochem. Int. 60, 360–364. doi: 10.1016/j.neuint.2012.01.010
Poli, L., Alberici, A., Buzzi, P., Marchina, E., Lanari, A., Arosio, C., et al. (2017). Is aceruloplasminemia treatable? Combining iron chelation and fresh-frozen plasma treatment. Neurol. Sci. 38, 357–360. doi: 10.1007/s10072-016-2756-x
Rahier, J., Loozen, S., Goebbels, R. M., and Abrahem, M. (1987). The haemochromatotic human pancreas: a quantitative immunohistochemical and ultrastructural study. Diabetologia 30, 5–12. doi: 10.1007/BF01788899
Roberti Mdo, R., Borges Filho, H. M., Goncalves, C. H., and Lima, F. L. (2011). Aceruloplasminemia: a rare disease – diagnosis and treatment of two cases. Rev. Bras. Hematol. Hemoter. 33, 389–392. doi: 10.5581/1516-8484.20110104
Rouault, T. A., Zhang, D. L., and Jeong, S. Y. (2009). Brain iron homeostasis, the choroid plexus, and localization of iron transport proteins. Metab. Brain Dis. 24, 673–684. doi: 10.1007/s11011-009-9169-y
Rusticeanu, M., Zimmer, V., Schleithoff, L., Wonney, K., Viera, J., Zimmer, A., et al. (2014). Novel ceruloplasmin mutation causing aceruloplasminemia with hepatic iron overload and diabetes without neurological symptoms. Clin. Genet. 85, 300–301. doi: 10.1111/cge.12145
Simpson, I. A., Ponnuru, P., Klinger, M. E., Myers, R. L., Devraj, K., Coe, C. L., et al. (2015). A novel model for brain iron uptake: introducing the concept of regulation. J. Cereb. Blood Flow Metab. 35, 48–57. doi: 10.1038/jcbfm.2014.168
Skidmore, F. M., Drago, V., Foster, P., Schmalfuss, I. M., Heilman, K. M., and Streiff, R. R. (2008). Aceruloplasminaemia with progressive atrophy without brain iron overload: treatment with oral chelation. J. Neurol. Neurosurg. Psychiatry 79, 467–470. doi: 10.1136/jnnp.2007.120568
Suzuki, Y., Yoshida, K., Aburakawa, Y., Kuroda, K., Kimura, T., Terada, T., et al. (2013). Effectiveness of oral iron chelator treatment with deferasirox in an aceruloplasminemia patient with a novel ceruloplasmin gene mutation. Intern. Med. 52, 1527–1530. doi: 10.2169/internalmedicine.52.0102
Tai, M., Matsuhashi, N., Ichii, O., Suzuki, T., Ejiri, Y., Kono, S., et al. (2014). Case of presymptomatic aceruloplasminemia treated with deferasirox. Hepatol. Res. 44, 1253–1258. doi: 10.1111/hepr.12292
Tuo, Q. Z., Lei, P., Jackman, K. A., Li, X. L., Xiong, H., Li, X. L., et al. (2017). Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 22, 1520–1530. doi: 10.1038/mp.2017.171
Vashchenko, G., and MacGillivray, R. T. (2013). Multi-copper oxidases and human iron metabolism. Nutrients 5, 2289–2313. doi: 10.3390/nu5072289
Vroegindeweij, L. H., Van Der Beek, E. H., Boon, A. J., Hoogendoorn, M., Kievit, J. A., Wilson, J. H., et al. (2015). Aceruloplasminemia presents as Type 1 diabetes in non-obese adults: a detailed case series. Diabet. Med. 32, 993–1000. doi: 10.1111/dme.12712
Wood, M. J., Powell, L. W., and Ramm, G. A. (2008). Environmental and genetic modifiers of the progression to fibrosis and cirrhosis in hemochromatosis. Blood 111, 4456–4462. doi: 10.1182/blood-2007-11-122374
Yonekawa, M., Okabe, T., Asamoto, Y., and Ohta, M. (1999). A case of hereditary ceruloplasmin deficiency with iron deposition in the brain associated with chorea, dementia, diabetes mellitus and retinal pigmentation: administration of fresh-frozen human plasma. Eur. Neurol. 42, 157–162. doi: 10.1159/000008091
Yoshida, K., Kaneko, K., Miyajima, H., Tokuda, T., Nakamura, A., Kato, M., et al. (2000). Increased lipid peroxidation in the brains of aceruloplasminemia patients. J. Neurol. Sci. 175, 91–95. doi: 10.1016/S0022-510X(00)00295-1
Zanardi, A., Conti, A., Cremonesi, M., D’adamo, P., Gilberti, E., Apostoli, P., et al. (2018). Ceruloplasmin replacement therapy ameliorates neurological symptoms in a preclinical model of aceruloplasminemia. EMBO Mol. Med. 10, 91–106. doi: 10.15252/emmm.201708361
Zhang, X., Surguladze, N., Slagle-Webb, B., Cozzi, A., and Connor, J. R. (2006). Cellular iron status influences the functional relationship between microglia and oligodendrocytes. Glia 54, 795–804. doi: 10.1002/glia.20416
Keywords: aceruloplasminemia, neurodegeneration, iron overload, ferroxidase, diabetes, ceruloplasmin, iron chelation, plasma
Citation: Piperno A and Alessio M (2018) Aceruloplasminemia: Waiting for an Efficient Therapy. Front. Neurosci. 12:903. doi: 10.3389/fnins.2018.00903
Received: 26 September 2018; Accepted: 19 November 2018;
Published: 04 December 2018.
Edited by:
Isabella Zanella, Università degli Studi di Brescia, ItalyReviewed by:
Scott Ayton, Florey Institute of Neuroscience and Mental Health, AustraliaCopyright © 2018 Piperno and Alessio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alberto Piperno, YWxiZXJ0by5waXBlcm5vQHVuaW1pYi5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.