 Geoffrey Canet1,2,3
Geoffrey Canet1,2,3 Nathalie Chevallier1,2,3Charleine Zussy1,2,3
Nathalie Chevallier1,2,3Charleine Zussy1,2,3 Catherine Desrumaux1,2,3
Catherine Desrumaux1,2,3 Laurent Givalois1,2,3*
Laurent Givalois1,2,3*- 1Molecular Mechanisms in Neurodegenerative Dementia Laboratory, INSERM, U1198, Team Environmental Impact in Alzheimer’s Disease and Related Disorders (EiAlz), Montpellier, France
- 2University of Montpellier, Montpellier, France
- 3EPHE, Paris, France
Alzheimer’s disease (AD) is the principal neurodegenerative pathology in the world displaying negative impacts on both the health and social ability of patients and inducing considerable economic costs. In the case of sporadic forms of AD (more than 95% of patients), even if mechanisms are unknown, some risk factors were identified. The principal risk is aging, but there is growing evidence that lifetime events like chronic stress or stress-related disorders may increase the probability to develop AD. This mini-review reinforces the rationale to consider major depressive disorder (MDD) as an important risk factor to develop AD and points the central role played by the hypothalamic-pituitary-adrenal (HPA) axis, glucocorticoids (GC) and their receptors (GR) in the etiology of MDD and AD. Several strategies directly targeting GR were tested to neutralize the HPA axis dysregulation and GC overproduction. Given the ubiquitous expression of GR, antagonists have many undesired side effects, limiting their therapeutic potential. However, a new class of molecules was developed, highly selective and acting as modulators. They present the advantage to selectively abrogate pathogenic GR-dependent processes, while retaining beneficial aspects of GR signaling. In fact, these “selective GR modulators” induce a receptor conformation that allows activation of only a subset of downstream signaling pathways, explaining their capacity to combine agonistic and antagonistic properties. Thus, targeting GR with selective modulators, alone or in association with current strategies, becomes particularly attractive and relevant to develop novel preventive and/or therapeutic strategies to tackle disorders associated with a dysregulation of the HPA axis.
General Aspects
Alzheimer’s disease is the principal neurodegenerative pathology in the world. This pathology is characterized by a progressive impairment of cognitive functions associated with synaptic and neuronal loss, the presence in the brain of senile plaques and NFT. Plaques are composed of insoluble extracellular aggregates consisting principally of Aβ peptides, while NFT result from intracellular hyper- and abnormal phosphorylation of the microtubule-stabilizing protein Tau (Selkoe, 2001; Mattson, 2004).
There are several forms of AD. Familial forms with known mutations of specific genes, representing less than 5% of AD cases, and sporadic forms representing more than 95% of patients, with unknown mechanisms, but identified risk factors. The principal risk factor for sporadic AD is aging. The risk doubles every 5 years after age 65, and prevalence reaches 50% over the age of 85. There is also growing evidence that lifetime events like chronic stress or stress-related disorders may increase the probability to develop AD (Heininger, 2000; Blennow et al., 2006; Querfurth and Laferla, 2010). This view is particularly supported by the fact that in AD patients, psychological symptoms and cognitive deficits are associated with an early dysregulation of the HPA axis, which is highly involved in stress responses (de Kloet et al., 2005; Figure 1). In AD, HPA axis dysregulation is associated with elevated levels of GC (cortisol in human and corticosterone in rodent) in plasma and cerebrospinal fluid (Hartmann et al., 1997; Swanwick et al., 1998; Csernansky et al., 2006; Hoogendijk et al., 2006).
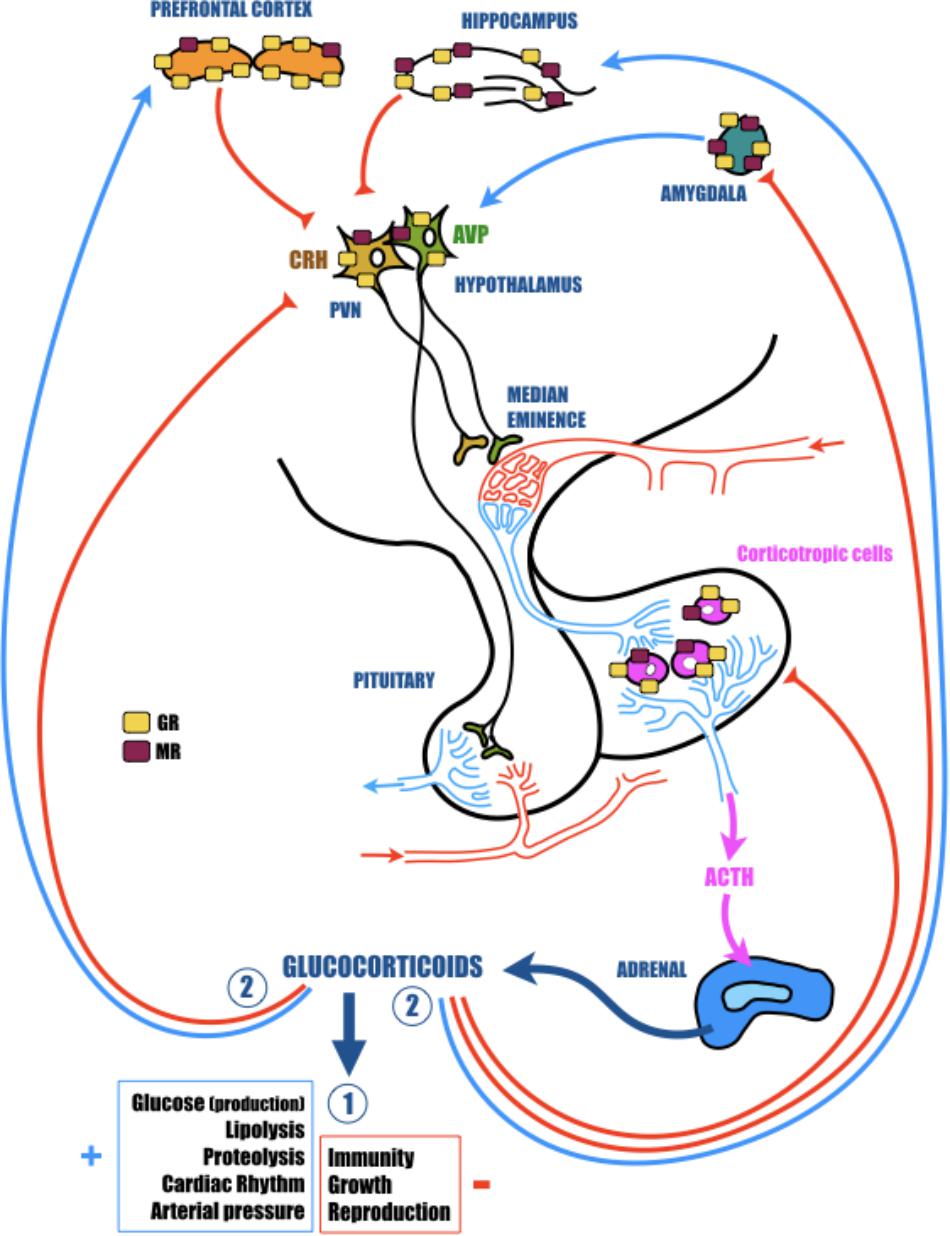
FIGURE 1. The hypothalamic-pituitary-adrenal (HPA) axis plays a vital role in adaptation of the organism to homeostatic challenge. The activation of the HPA axis by stress leads to rapid secretion of corticotropin releasing hormone (CRH) and arginine vasopressin (AVP) from the hypothalamic paraventricular nucleus (PVN). In turn, CRH and AVP activate the secretion of adrenocorticotropic hormone (ACTH) from the anterior pituitary, which finally stimulates the release of glucocorticoids (GC; cortisol in primates and corticosterone in rodents) from the adrenal glands. CRH is principally synthesized in the parvocellular portion of the PVN where it is co-localized with AVP. AVP acts in synergy with CRH and potentiates the ability of CRH to induce ACTH secretion. AVP is largely synthesized in the other portion of the PVN, the magnocellular region. Here, this hormone is preponderantly conveyed to the posterior pituitary, where it is stocked until release into the general circulation to participate in several actions particularly related to water control. GC readily cross the blood-brain barrier and bind to low-affinity glucocorticoid receptors (GR) and high-affinity mineralocorticoid receptors (MR). These nuclear receptors are indispensable for regular cellular activity and crucial for many central nervous system functions, including learning and memory. While MR are essentially localized in the hippocampus, GR are more ubiquitous and are particularly found in the hypothalamus and in the pituitary, but also in several structures of the limbic system (prefrontal cortex, hippocampus, and amygdala), which are highly involved in cognitive and psychological functions. All of these cerebral regions are important components of the neural circuitry mediating HPA axis activity. Indeed, while hippocampus and prefrontal cortex exert a tonic inhibition, amygdala stimulates hypothalamic neurons. In fact, though structural plasticity in hippocampus and prefrontal cortex might mediate cognitive impairment caused by severe stress, changes in amygdala are more likely to contribute to the affective aspect of stress disorders. In response to homeostatic challenge, GC stimulate in a first time (Circle 1) all physiological functions necessary to induce an adapted response to the stress (cardiovascular and metabolic) and inhibit all functions non-immediately necessary (immunity, reproduction, and growth). In a second time (Circle 2), to avoid a runaway of the HPA axis, GC exert a negative feedback at all levels of the axis (limbic, hypothalamic, and pituitary).
Glucorticoids are steroid hormones that freely cross the blood–brain barrier and bind to high-affinity (Kd = 0.5 nM) MR and low-affinity (Kd = 5 nM) GR (Reul and de Kloet, 1985). Globally, MR are necessary for regular cellular activity, but are also the receptors for aldosterone (involved in specific cells to the enzymatic degradation of GC). By contrast, GR are involved in stress responses, exert a negative feedback on the HPA axis activity and are essential for many CNS functions, including learning and memory (Roozendaal, 2000). While MR are essentially localized in the hippocampus, GR are more ubiquitous. They are particularly found in several structures of the limbic system (prefrontal cortex, hippocampus, and amygdala), which are especially involved in psychological and cognitive functions, but also are important elements of the neural circuitry mediating HPA axis activity (Jankord and Herman, 2008; Figure 1). Therefore, while structural plasticity in the hippocampus and prefrontal cortex may mediate cognitive impairment induced by severe stress, modifications in amygdala are more likely to contribute to the affective aspect of stress disorders (Vyas et al., 2004). Furthermore, because GC act synergistically with excitatory amino acids (like glutamate), disruptions of the HPA axis, GC overexposure or a modification of GR functioning could be extremely toxic, particularly in the limbic structures (hippocampus, prefrontal cortex, or amygdala) and thus contribute to the cognitive decline associated with stress-related disorders (McEwen, 2008). For instance, it was established a long time ago that chronic stress and subsequent GC over-secretion severely impact the structure, function and plasticity of synapses in the hippocampus. Repeated stress causes atrophy of dendrites in the CA3 region, and both acute and chronic stress suppress neurogenesis in dentate gyrus neurons (McEwen and Sapolsky, 1995; Galea et al., 1997; McEwen, 1999; Vyas et al., 2002). This loss of synaptic plasticity is in the heart of AD (Selkoe, 2002), and could be in part responsible for the cognitive decline observed in patients, and thus making a link between stress, stress-related disorders, GC, and AD.
As previously established by Ownby et al. (2006) and recently reviewed by Ishijima et al. (2018), epidemiological studies demonstrated that MDD (the stress-related disorder by excellence) may be considered as an important risk factor to develop AD (Ownby et al., 2006; Ishijima et al., 2018). Early life MDD (more than 25 years before the diagnosis of dementia) is correlated with a belated development of AD (Robert et al., 2003), and has systematically been associated with a more than twofold increase in dementia risk (Byers and Yaffe, 2011). In fact, the risk to develop AD increases with every new affective episode associated to mood disorders and especially, the level of dementia tended to increase by 13% with every episode of MDD (Kessing and Andersen, 2004). Additionally, numerous proofs suggest that late-life MDD also increases the risk to develop dementia (Baldwin et al., 2006; Thomas and O’Brien, 2008). It appears that MDD accelerates age-related cognitive decline (Gualtieri and Johnson, 2008) and promotes the conversion of mild cognitive impairments into AD (Modrego and Ferrández, 2004; Houde et al., 2008). Finally, MDD may occur in 30–40% of the AD patients (Assal and Cummings, 2002; Starkstein et al., 2005) and affects the clinical evolution of AD (Shim and Yang, 2006). Senile plaques and NFT are more marked in the hippocampus of AD patients with comorbid MDD as compared with AD patients without depression (Rapp et al., 2008).
However, even though mechanisms of the switch from MDD to AD remain unclear, some findings suggest that one of the links between these two disorders could be a dysregulation of the HPA axis activity, associated with impaired GC signaling (Ownby et al., 2006; Caraci et al., 2010; Notarianni, 2013; Givalois, 2014; Herbert and Lucassen, 2016). Thus in the present review, we will examine this evidence, focusing on the HPA axis dysregulation in AD and MDD, and on the new molecules targeting selectively GR for the treatment of both MDD and AD.
HPA Axis Dysregulation
The links between AD, HPA axis, stress and GC come from observations in humans, but also from different animal models. In humans, there is considerable evidence involving HPA axis dysfunction in AD patients. This dysregulation is reflected not only by elevated levels of circulating cortisol, but also by the failure to show cortisol suppression following a DEX challenge, suggesting the inability for the HPA axis to maintain homeostasis (Martignoni et al., 1990; Hartmann et al., 1997; Weiner et al., 1997; Swanwick et al., 1998; Csernansky et al., 2006; Elgh et al., 2006; Hoogendijk et al., 2006; Popp et al., 2009). Chronic stress, such as mourning or sleep deprivation, in addition to cause memory impairments, increases the susceptibility to develop AD (Mejía et al., 2003; Wilson et al., 2005, 2006). In AD patients treated with prednisone (a synthetic GC used for its anti-inflammatory properties), behavioral decline was increased when compared with the placebo-treated patients (Aisen et al., 2000). Besides, de Quervain et al. (2004) evidenced a haplotype in the gene of an enzyme involved in the activation of GC (11β-hydroxysteroid dehydrogenase – 11β-HSD) that increases by six the risk to develop AD (de Quervain et al., 2004).
In different Tg animal models of AD, chronic stress improved plaque pathology, accelerated the inception of cognitive deficits, triggered APP misprocessing, reduced Aβ clearance, increased Aβ levels, stimulated Tau hyperphosphorylation and its neuronal accumulation (Dong et al., 2004; Green et al., 2006; Jeong et al., 2006; Huang et al., 2011; Rothman et al., 2012). The presence of a GRE in the promoter regions of the APP and BACE1 genes (Lahiri, 2004; Sambamurti et al., 2004) may explain the impact of chronic stress and the role of GC in APP misprocessing and induction of the Aβ pathway. Regarding Tau, some data suggest that the modification of Tau system could be an indirect consequence of chronic stress, due to stress-induced Aβ increase (Tomidokoro et al., 2001; Tu et al., 2014). However it seems that chronic stress, or GC-excess could directly impact Tau phosphorylation (Yan et al., 2010), mainly through the over-activation of GSK-3β and Cdk5 enzymes (Papadopoulou et al., 2015; Dey et al., 2017; Yi et al., 2017). In addition, another factor linking chronic stress and Tau has also been identified, the corticotropin-releasing factor receptor (CRF1), since this receptor appears to be directly involved in the progression of Tau pathology (Rissman et al., 2007; Caroll et al., 2011).
In an acute model of AD, injection of an oligomeric solution of an Aβ fragment (oAβ25-35) in cerebral ventricles (icv) induces a wide pattern of central modifications reminiscent of the human pathophysiology (Zussy et al., 2011, 2013; Pineau et al., 2016). This Aβ fragment is found early in AD patients and originates from proteolysis of parent amyloid proteins (Kaneko et al., 2001; Kubo et al., 2002; Gruden et al., 2007). It also induces a strong and long-lasting activation of the HPA axis, which is associated with a modification of the expression and functioning of GR (Brureau et al., 2013; Pineau et al., 2016).
Interestingly, this deregulation of the HPA axis associated with AD, is also the most prevalent and well-documented neuroendocrine abnormality in stress-related disorders and especially in MDD (Holsboer and Barden, 1996). This pathology appears like a prodromal stage and an important element of AD, but could also be a trigger for developing AD (Herbert and Lucassen, 2016; Ishijima et al., 2018). In fact, in addition to be a risk factor in AD (Heininger, 2000; Blennow et al., 2006; Querfurth and Laferla, 2010), chronic exposure to stress and stressful life events also seem to lead to the development of MDD (Pariante, 2003; Charney and Manji, 2004; Czéh and Lucassen, 2007; Pittenger and Duman, 2008; Herbert and Lucassen, 2016). Taken together these data demonstrate a central role of HPA axis dysregulation and high levels of GC both in MDD and AD etiology and suggest the possibility that GR might be suitable targets both for antidepressant and antidementia drugs.
GR, a Potential Therapeutic Target
Based on the above-mentioned observations in humans and in animal models, suggesting a deregulation of GR functioning, several strategies targeting directly GR were tested in AD and MDD, and seem to have an important therapeutic potential (Bachmann et al., 2003; Caraci et al., 2010; Figure 2). However, given the ubiquitous expression of these receptors (Sapolsky et al., 2000), antagonists could have many undesired side effects and should be used with caution.
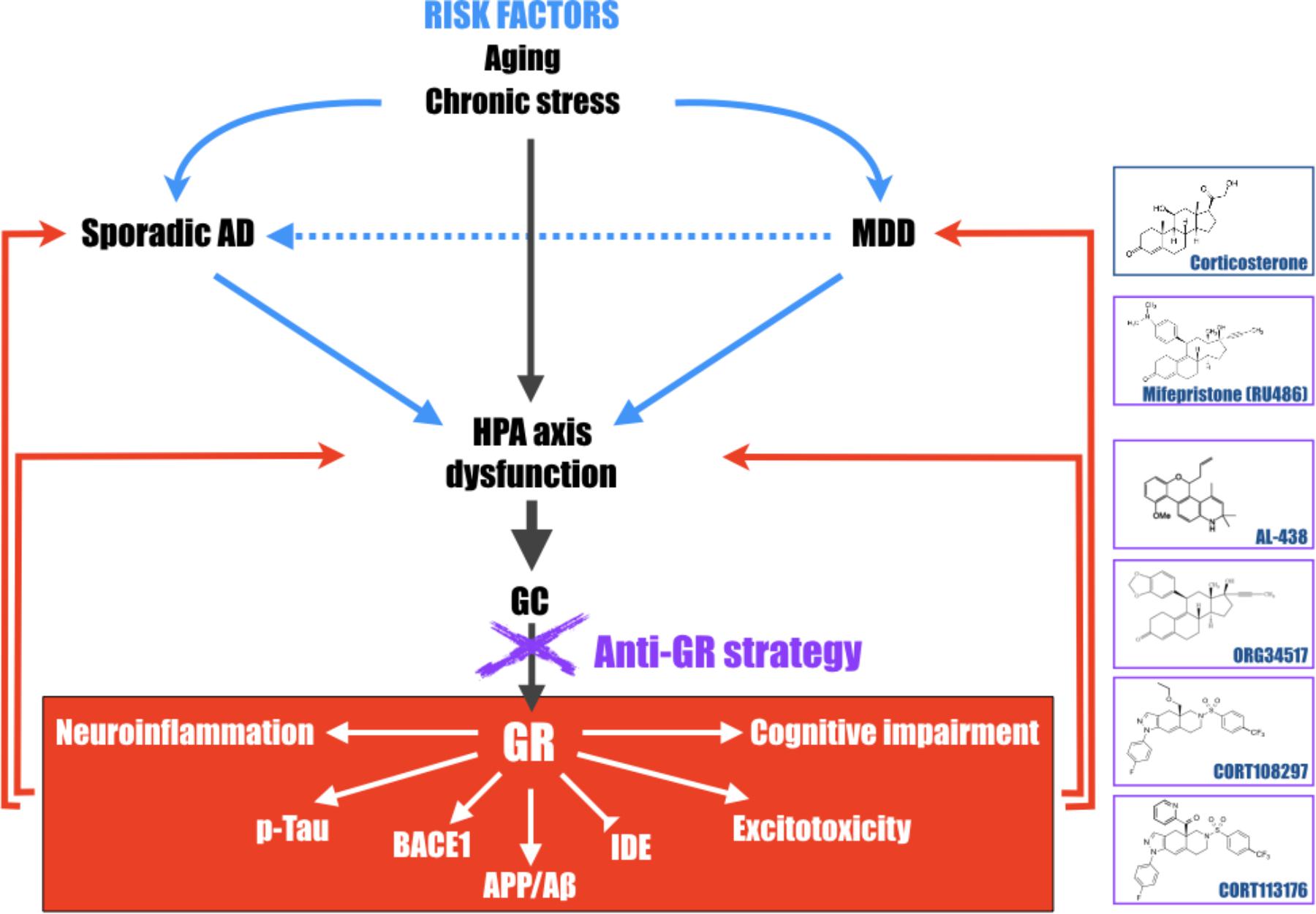
FIGURE 2. Schematic representation of the central role of the hypothalamic-pituitary-adrenal (HPA) axis dysfunction, GC and GR in the development of major depressive disorder (MDD) and Alzheimer’s disease (AD). Long-term exposure to stress or stress-related disorders (like MDD) contributes to cognitive impairment, Aβ accumulation, Tau hyperphosphorylation, excitotoxicity, and neuroinflammation processes, leading to development of AD. This schematic representation highlights that pathologies associated with a dysregulation of the HPA axis must be considered, with aging, as important risk factors to develop AD and evidence that therapies aiming at reducing high GC levels upstream, become particularly attractive and relevant in the treatment of the elderly, MDD or early AD patients. Chemical structures of molecules described in this review; corticosterone, mifepristone, AL-438 (Coghlan et al., 2003), ORG34517 (Bachmann et al., 2003), CORT108297, and CORT113176 (Pineau et al., 2016).
In MDD, but also in psychotic depression and bipolar disorder, preclinical and clinical studies were realized with the prototypical GR non-selective antagonist mifepristone (RU486) (Figure 2) and showed that this molecule seemed to be effective in inducing a rapid improvement of psychotic and depressive symptoms, while being well tolerated by patients (Belanoff et al., 2001, 2002b; Young et al., 2004; Oomen et al., 2007; Schatzberg and Lindley, 2008; Blasey et al., 2009; Wulsin et al., 2010; Howland, 2013; Block et al., 2017).
In AD, first studies with mifepristone provided very hopeful results. A chronic treatment in 3 × Tg-AD mice reversed cognitive deficits, clearly reduced Aβ levels, as well as phosphorylation and accumulation of Tau (Baglietto-Vargas et al., 2013). In Tg2576 mice, mifepristone rescued early episodic memory and synaptic plasticity deficits (Lanté et al., 2015). In the oAβ25-35 model, mifepristone restored basal circulating CORT levels, reversed synaptic deficits and apoptosis in the hippocampus. However, this non-selective antagonist only partially reversed cognitive and Aβ clearance deficits, hippocampal APP misprocessing, and neuroinflammatory processes, suggesting limits in its efficacy (Pineau et al., 2016). This limitation was also observed in humans since mifepristone, even if it slows the progression of cognitive decline in AD patients (Belanoff et al., 2002a), increases morning levels of blood GC (Pomara et al., 2006), suggesting potential side effects and thus a limited therapeutic usefulness.
Thus, several potent and selective GR ligand series were recently developed and seem to have an interesting therapeutic potential. The first non-steroidal selective GR molecules come from anti-inflammatory studies. Indeed, GC are generally prescribed in inflammatory diseases, however, chronic treatment with steroids leads to various undesired effects, most likely due to the wild range of genes targeted by GR and not directly involved in inflammatory processes. The objective of Abbott Laboratories (Abbott Park, IL, United States) was to create a series of molecules capable to have anti-inflammatory properties without deleterious side effects (Coghlan et al., 2003). The Abbott-Ligand 438 (AL-438) (Figure 2) was obtained by modifying a synthetic progestin scaffold resulting in the discovery of a series of high affinity, selective ligands for GR (Elmore et al., 2001). In comparison with prednisone, the steroidal anti-inflammatory molecule of reference, AL-438 shares the same affinity for MR as well as high affinity for GR as prednisone. Even if, in MR-dependent reporter gene assays, AL-438 showed low antagonist properties, whereas prednisone is a full agonist at nanomolar concentrations. In vivo, AL-438 preserved full anti-inflammatory efficacy and potency equivalent to steroids while side effects were especially reduced (Coghlan et al., 2003). Unfortunately, this compound and its derivatives were never tested, to our knowledge, in AD or MDD studies.
More recently, a new molecule was tested in an experimental model of ethanol dependence showing HPA axis and GR impairments (Reynolds et al., 2015). The ORG 34517 (Figure 2) is a 11,21-Bisphenyl-19-norpregnane steroid originally discovered by Organon (Oss, Netherlands). This compound, highly selective for GR (Peeters et al., 2008), has insignificant affinity for human PR, since it possesses a nearly 500-fold greater affinity for human GR (Gebhard et al., 1994). In addition, ORG 34517 is unable to occupy MR after an acute subcutaneous injection (Bachmann et al., 2003). At this time, this selective GR antagonist was envisaged as a promising therapeutic alternative in MDD (Bachmann et al., 2003). However, to our knowledge, this compound was never tested in MDD or in AD.
Another series of selective non-steroidal molecules (1H-pyrazolo[3,4-g]hexahydro-isoquinoline sulfonamides) come from Corcept Therapeutics (Menlo Park, CA, United States). These GR ligands and in particular CORT108297 and CORT113176 (Figure 2) present excellent affinity only for GR, and none for the other nuclear hormone receptors PR, AR, MR, and ER (Clark et al., 2008; Beaudry et al., 2014; Hunt et al., 2015; Pineau et al., 2016). Several cell-based assays were developed to assess their functionality. DEX, the GR agonist of reference, increases the activity and expression of TAT in liver cells. In HepG2 human cells or in human hepatocytes, both CORT108297 and CORT113176 act as full antagonists since they are able to prevent the DEX-induced increase in TAT activity and to induce non-measurable agonist activity in the absence of DEX. However, when tested in a similar assay in rat hepatocytes, both molecules display incomplete antagonism and partial agonist activity (Beaudry et al., 2014; Pineau et al., 2016). An additional cell-based functional assay was developed in the A549 cell line to investigate the effect of these two compounds on IL-1β-induced IL-6 production. Both ligands demonstrated partial agonist effects, and also acted as partial antagonists when tested in presence of DEX (Pineau et al., 2016). Thus, their particular modulator properties make this family of molecules really interesting in AD or MDD. In fact, these ligands have the capacity to more selectively abrogate pathogenic GR-dependent progressions in the brain, while retaining positive aspects of GR signaling. Onno Meijer’s team showed that CORT108297 clear antagonist effects on the brain were accompanied by a lack of negative-feedback inhibition of the HPA axis, which suggests “the possibility of antagonizing a number of GR effects without affecting systemic basal GC levels” (Zalachoras et al., 2013). In fact, it appears that this family of molecules acts as “selective GR modulators” rather than pure antagonists. They induce a receptor conformation that permits activation of only a subset of downstream signaling pathways, explaining their capacity to combine agonistic and antagonistic properties (Zalachoras et al., 2013; Meijer et al., 2018).
In a 3 × Tg mice model of AD, CORT108297 reduces APP-C-terminal fragment (C83) and Tau hyperphosphorylation via reductions in p25 levels (Baglietto-Vargas et al., 2013). Furthermore, in the acute model, we recently showed that treatments with CORT108297 and CORT113176 reverse the hippocampal amyloidogenic pathway induced by the icv injection of oAβ25-35 through the inhibition of the principal enzyme involved in Aβ synthesis (BACE1) and the increase of one enzyme mainly involved in the elimination of Aβ (IDE). In addition, selective GR modulators reestablish hippocampal levels of synaptic markers, reverse hippocampal apoptotic processes and neuroinflammation, re-establish basal plasma levels of GC and in fine cognitive functions (Pineau et al., 2016).
In rodents, CORT108297 treatment decreases immobility in the FST suggesting potential antidepressant properties (Solomon et al., 2014). By contrast, treatment with another member of this family, CORT118335, which is a GR modulator but also a MR antagonist, did not affect immobility in the FST (Nguyen et al., 2018), suggesting a differential specificity and efficacy of each molecule.
Thus, the difference of efficacy between all of these compounds could be due to the difference of selectivity and affinity for GR (Coghlan et al., 2003; Clark et al., 2008; Peeters et al., 2008; Beaudry et al., 2014; Hunt et al., 2015; Pineau et al., 2016), but also to the intrinsic properties of GR and their ability to differentially recruit nuclear receptor coregulators after ligands binding (Coghlan et al., 2003; Zalachoras et al., 2013; Atucha et al., 2015; Meijer et al., 2018). These coregulators are transcriptionally active proteins, which mediate the transcriptional properties of nuclear receptors. They have tissue-, ligand-, and cell-specific expression patterns, and display gene- and receptor-specific interactions (Meijer et al., 2000; Lachize et al., 2009; Zalachoras et al., 2013; Meijer et al., 2018). Recently, Onno Meijer’s team, established that each GR compound induced a specific profile of interaction with these coregulators. They suggested, as previously envisaged by Coghlan et al. (2003) that these specific profiles could explain the difference of functionality and efficacy of these particular GR ligands and their capacity to combine antagonistic and agonistic properties (Atucha et al., 2015; Meijer et al., 2018). Accordingly, as recently suggested by Meijer et al. (2018), a better knowledge of the specific molecular interaction profiles of each GR compound, combined with the regional distribution of each coregulator in the brain, could assist in dissecting the molecular signaling pathways underlying pathologies associated with high levels of GC. This strategy will participate to create new avenues of investigation on GC and GR, and to exploit these avenues to develop novel preventive and/or therapeutic strategies to tackle disorders (neurodegenerative or not), associated with a dysregulation of the HPA axis.
GR activity can also be indirectly modulated by side regulations which could be additional potential targets. It opens the door to multiple approaches to target the GR pathway. Recently, it was demonstrated that inhibiting the adenosine A2A receptor, which is upregulated in the forebrain of AD patients, reverses memory deficits through HPA axis feedback and corticosterone circadian levels reestablishment (Batalha et al., 2013). Authors also evidenced that A2A receptor is a major regulator of GR function since its inhibition reduces GR hippocampal levels, and acts on GR nuclear translocation and GR-dependent transcriptional regulation (Batalha et al., 2016). Interestingly, some studies showed an anti-depressive effect of A2A receptor antagonists in MDD models (López-Cruz et al., 2018; Padilla et al., 2018). A2A receptor is an example among others. Indeed, annexin A1 is a GC-induced molecule that is known to replicate many of the described anti-inflammatory effects of GC (Yang et al., 2013). Even if there is no study about the role of annexin A1 in MDD, emerging evidence suggest a role of this protein in the clearance and the degradation of Aβ peptides, and in the neuroprotective role of microglia (McArthur et al., 2010; Ries et al., 2016).
Conclusion
All these findings in favor of the “GC theory” reinforce the hypothesis that long-term exposure to stress or stress-related disorders (like MDD or Cushing’s syndrome for instance), contributes to cognitive impairment, Aβ accumulation, Tau hyperphosphorylation, excitotoxicity, and neuroinflammation processes, leading to later development of AD. They also evidence that pathologies associated with a dysregulation of the HPA axis must be considered as important risk factors for AD (Figure 2). Therefore, therapies aiming at reducing high GC levels upstream, in the elderly or in early AD patients, could be envisaged. This review also evidences that modulator molecules targeting selectively GR could abrogate pathogenic GR-dependent processes induced by a dysregulation of the HPA axis and retain beneficial and primordial aspects of GR signaling. Thus, this class of compounds, alone or in association with current treatments against MDD (anti-depressant compounds) or AD (anti-NMDA and anti-cholinesterase molecules), becomes particularly attractive and relevant candidates in the treatment of stress-related disorders or neurodegenerative diseases, and particularly AD.
Author Contributions
GC, NC, CZ, CD, and LG equally contributed to the definition of the scope and to writing of the manuscript.
Funding
LG was supported by “France Alzheimer” and “Fédération pour la Recherche sur le Cerveau” (Grant AAP SM2016#1512). CD and CZ were supported by the “Agence Nationale de la Recherche” (ANR) under the program “Investissements d’Avenir” (ANR-11-LABEX-0021-LipSTIC). GC was supported by a Ph.D. fellowship from the University of Montpellier, France (CBS2 Ph.D. program).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
Aβ, amyloid-β peptide, AD, Alzheimer’s disease, APP, amyloid precursor protein, AR, androgen receptors, BACE1, β-APP cleaving enzyme type 1, Cdk5, cyclin dependent kinase 5, CNS, central nervous system, DEX, dexamethasone, ER, estrogen receptors, FST, forced swim test, GC, glucocorticoids, GR, glucocorticoid receptors, GRE, glucocorticoid response element, GSK-3β, glycogen synthase kinase 3β, HPA axis, hypothalamic-pituitary-adrenal axis, Icv, intracerebroventricular, IDE, insulin-degrading enzyme, MDD, major depressive disorder, MR, mineralocorticoid receptors, NFT, neurofibrillary tangles, oAβ25-35, oligomers of Aβ fragment [25-35], PR, progesterone receptors, TAT, tyrosine aminotransferase activity assay, Tg, transgenic.
References
Aisen, P. S., Davis, K. L., Berg, J. D., Schafer, K., Campbell, K., Thomas, R. G., et al. (2000). A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s disease cooperative study. Neurology 54, 588–593. doi: 10.1212/WNL.54.3.588
Assal, F., and Cummings, J. L. (2002). Neuropsychiatric symptoms in the dementia. Curr. Opin. Neurol. 15, 445–450. doi: 10.1097/00019052-200208000-00007
Atucha, E., Zalachoras, I., van den Heuvel, J. K., van Weert, L. T., Melchers, D., Mol, I. M., et al. (2015). A mixed glucocorticoid/mineralocorticoid selective modulator with dominant antagonism in the male rat brain. Endocrinology 156, 4105–4114. doi: 10.1210/en.2015-1390
Bachmann, C. G., Linthorst, A. C., Holsboer, F., and Reul, J. M. H. (2003). Effect of chronic administration of selective glucocorticoid receptor antagonists on the rat hypothalamic-pituitary-adrenocortical axis. Neuropsychopharmacology 28, 1056–1067. doi: 10.1038/sj.npp.1300158
Baglietto-Vargas, D., Medeiros, R., Martinez-Coria, H., LaFerla, F. M., and Green, K. N. (2013). Mifepristone alters amyloid precursor protein processing to preclude amyloid beta and also reduces tau pathology. Biol. Psychiatry 74, 357–366. doi: 10.1016/j.biopsych.2012.12.003
Baldwin, R. C., Gallagley, A., Gourlay, M., Jackson, A., and Burns, A. (2006). Prognosis of late life depression: a three-year cohort study of outcome and potential predictors. Int. J. Geriatr. Psychiatry 21, 57–63. doi: 10.1002/gps.1424
Batalha, V. L., Ferreira, D. G., Coelho, J. E., Valadas, J. S., Gomes, R., Temido-Ferreira, M., et al. (2016). The caffeine-binding adenosine A2A receptor induces age-like HPA-axis dysfunction by targeting glucocorticoid receptor function. Sci. Rep. 6:31493. doi: 10.1038/srep31493
Batalha, V. L., Pego, J. M., Fontinha, B. M., Costenla, A. R., Valadas, J. S., Baqi, Y., et al. (2013). Adenosine A(2A) receptor blockade reverts hippocampal stress-induced deficits and restores corticosterone circadian oscillation. Mol. Psychiatry 18, 320–331. doi: 10.1038/mp.2012.8
Beaudry, J., Dunford, E. C., Teich, T., Zaharieva, D., Hunt, H., Belanoff, J. K., et al. (2014). Effects of selective and non-selective glucocorticoid receptor II antagonists on rapid-onset diabetes in young rats. PLoS One 9:e91248. doi: 10.1371/journal.pone.0091248
Belanoff, J. K., Flores, B. H., Kalezhan, M., Sund, B., and Schatzberg, A. F. (2001). Rapid reversal of psychotic depression using mifepristone. J. Clin. Psychopharmacol. 21, 516–521. doi: 10.1097/00004714-200110000-00009
Belanoff, J. K., Jurik, J., Schatzberg, L. D., DeBattista, C., and Schatzberg, A. F. (2002a). Slowing the progression of cognitive decline in Alzheimer’s disease using mifepristone. J. Mol. Neurosci. 19, 201–206.
Belanoff, J. K., Rothschild, A. J., Cassidy, F., DeBattista, C., Baulieu, E. E., Schold, C., et al. (2002b). An open label trial of C-1073 (mifepristone) for psychotic major depression. Biol. Psychiatry 52, 386–392.
Blasey, C. M., Debattista, C., Roe, R., Block, T., and Belanoff, J. K. (2009). A multisite trial of mifepristone for the treatment of psychotic depression: a site-by-treatment interaction. Contemp. Clin. Trials 30, 284–288. doi: 10.1016/j.cct.2009.03.001
Blennow, K., de Leon, M. J., and Zetterberg, H. (2006). Alzheimer’s disease. Lancet 368, 387–403. doi: 10.1016/S0140-6736(06)69113-7
Block, T., Petrides, G., Kushner, H., Kalin, N., Belanoff, J., and Schatzberg, A. (2017). Mifepristone plasma level and glucocorticoid receptor antagonism associated with response in patients with psychotic depression. J. Clin. Psychopharmacol. 37, 505–511. doi: 10.1097/JCP.0000000000000744
Brureau, A., Zussy, C., Delair, B., Ogier, C., Ixart, G., Maurice, T., et al. (2013). Deregulation of HPA axis functions in an Alzheimer’s disease rat model. Neurobiol. Aging 34, 1426–1439. doi: 10.1016/j.neurobiolaging.2012.11.015
Byers, A. L., and Yaffe, K. (2011). Depression and risk of developing dementia. Front. Neurosci. 7:323–331. doi: 10.1038/nrneurol.2011.60
Caraci, F., Copani, A., Nicoletti, F., and Drago, F. (2010). Depression and Alzheimer’s disease: neurobiological links and common pharmacological targets. Eur. J. Pharmacol. 626, 64–71. doi: 10.1016/j.ejphar.2009.10.022
Caroll, J. C., Iba, M., Bangasser, D. A., Valentino, R. J., James, M. J., Brunden, K. R., et al. (2011). Chronic stress exacerbates tau pathology, neurodegeneration, and cognitive performance through a corticotropin-releasing factor receptor-dependent mechanism in a transgenic mouse model of tauopathy. J. Neurosci. 31, 14436–14449. doi: 10.1523/JNEUROSCI.3836-11.2011
Charney, D. S., and Manji, H. K. (2004). Life stress, genes, and depression: multiple pathways lead to increased risk and new opportunities for intervention. Sci. STKE 225:re5.
Clark, R. D., Ray, N. C., Williams, K., Blaney, P., Ward, S., Crackett, P. H., et al. (2008). 1H-Pyrazolo[3,4-g]hexahydro-isoquinolines as selective glucocorticoid receptor antagonists with high functional activity. Bioorg. Med. Chem. Lett. 18, 1312–1317. doi: 10.1016/j.bmcl.2008.01.027
Coghlan, M. J., Jacobson, P. B., Lane, B., Nakane, M., Lin, C. W., Elmore, S. W., et al. (2003). A novel antiinflammatory maintains glucocorticoid efficacy with reduced side effects. Mol. Endocrinol. 17, 860–869. doi: 10.1210/me.2002-0355
Csernansky, J. G., Dong, H., Fagan, A. M., Wang, L., Xiong, C., Holtzman, D. M., et al. (2006). Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am. J. Psychiatry 163, 2164–2169. doi: 10.1176/ajp.2006.163.12.2164
Czéh, B., and Lucassen, P. J. (2007). What causes the hippocampal volume decrease in depression? Are neurogenesis, glial changes and apoptosis implicated? Eur. Arch. Psychiatry Clin. Neurosci. 257, 250–260. doi: 10.1007/s00406-007-0728-0
de Kloet, E. R., Joëls, M., and Holsboer, F. (2005). Stress and the brain: from adaptation to disease. Nat. Rev. Neurosci. 6, 463–475. doi: 10.1038/nrn1683
de Quervain, D. J., Poirier, R., Wollmer, M. A., Grimaldi, L. M., Tsolaki, M., Streffer, J. R., et al. (2004). Glucocorticoid-related genetic susceptibility for Alzheimer’s disease. Hum. Mol. Genet. 13, 47–52. doi: 10.1093/hmg/ddg361
Dey, A., Hao, S., Wosiski-Kuhn, M., and Stranahan, A. M. (2017). Glucocorticoid-mediated activation of GSK3β promotes tau phosphorylation and impairs memory in type 2 diabetes. Neurobiol. Aging 57, 75–83. doi: 10.1016/j.neurobiolaging.2017.05.010
Dong, H., Goico, B., Martin, M., Csernansky, C. A., Bertchume, A., and Csernansky, J. G. (2004). Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience 127, 601–609. doi: 10.1016/j.neuroscience.2004.05.040
Elgh, E., Lindqvist Astot, A., Fagerlund, M., Eriksson, S., Olsson, T., and Nasman, B. (2006). Cognitive dysfunction, hippocampal atrophy and glucocorticoid feedback in Alzheimer’s disease. Biol. Psychiatry 59, 155–161. doi: 10.1016/j.biopsych.2005.06.017
Elmore, S. W., Coghlan, M. J., Anderson, D. D., Pratt, J. K., Green, B. E., Wang, A. X., et al. (2001). Nonsteroidal selective glucocorticoid modulators: the effect of C-5 alkyl substitution on the transcriptional activation/repression profile of 2,5-dihydro-10-methoxy-2,2,4-trimethyl-1H-[1]benzopyrano[3,4-f]quinolines. J. Med. Chem. 44, 4481–4491. doi: 10.1021/jm010367u
Galea, L. A. M., McEwen, B. S., Tanapat, P., Deak, T., Spencer, R. L., and Dhabhar, F. S. (1997). Sex differences in dendritic atrophy of CA3 pyramidal neurons in response to chronic restraint stress. Neuroscience 81, 689–697. doi: 10.1016/S0306-4522(97)00233-9
Gebhard, R., van der Voort, H., Schuts, W., and Schoonen, W. (1994). 11,21-Bisphenyl-19-norpregnane derivatives are selective antiglucocorticoids. Bioorg. Med. Chem. Lett. 7, 2229–2234. doi: 10.1016/S0960-894X(97)00397-1
Givalois, L. (2014). The glucocorticoid receptors regulation in Alzheimer’s disease. Neurobiol. Aging 35, e17–e18.
Green, K., Billings, L., Roozendaal, B., McGaugh, J., and LaFerla, F. M. (2006). Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 26, 9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006
Gruden, M., Davidova, T., Malisauskas, M., Sewell, R., Voskresenskaya, N., Wilhelm, K., et al. (2007). Differential neuroimmune markers to the onset of Alzheimer’s disease neurodegeneration and dementia: autoantibodies to Aβ(25–35) oligomers, S100b and neurotransmitters. J. Neuroimmunol. 186, 181–192. doi: 10.1016/j.jneuroim.2007.03.023
Gualtieri, C. T., and Johnson, L. G. (2008). Age-related cognitive decline in patients with mood disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 962–967. doi: 10.1016/j.pnpbp.2007.12.030
Hartmann, A., Veldhuis, J. D., Deuschle, M., Standhardt, H., and Heuser, I. (1997). Twenty-four hour cortisol release profiles in patients with Alzheimer’s and Parkinson’s disease compared to normal controls: ultradian secretory pulsatility and diurnal variation. Neurobiol. Aging 18, 285–289. doi: 10.1016/S0197-4580(97)80309-0
Heininger, K. (2000). A unifying hypothesis of Alzheimer’s disease. III. Risk factors. Hum. Psychopharmacol. Clin. Exp. 15, 1–70. doi: 10.1002/(SICI)1099-1077(200001)15:1<1::AID-HUP153>3.0.CO;2-1
Herbert, J., and Lucassen, P. J. (2016). Depression as a risk factor for Alzheimer’s disease: genes, steroid, cytokines and neurogenesis – What do we need to know? Front. Neuroendocrinol. 41, 153–171. doi: 10.1016/j.yfrne.2015.12.001
Holsboer, F., and Barden, N. (1996). Antidepressants and hypothalamic-pituitary-adrenocortical regulation. Endocr. Rev. 17, 187–205. doi: 10.1210/edrv-17-2-187
Hoogendijk, W. J., Meynen, G., Endert, E., Hofman, M. A., and Swaab, D. F. (2006). Increased cerebrospinal fluid cortisol level in Alzheimer’s disease is not related to depression. Neurobiol. Aging 27, 780.e1–780.e2.
Houde, M., Bergman, H., Whitehead, V., and Chertkow, H. (2008). A predictive depression pattern inmild cognitive impairment. Int. J. Geriatr. Psychopharmacol. 23, 1028–1033. doi: 10.1002/gps.2028
Howland, R. H. (2013). Mifepristone as a therapeutic agent in psychiatry. J. Psychosoc. Nurs. Ment. Health Serv. 51, 11–14. doi: 10.3928/02793695-20130513-01
Huang, H. J., Liang, K. C., Ke, H. C., Chang, Y. Y., and Hsieh-Li, H. M. (2011). Long-term social isolation exacerbates the impairment of spatial working memory in APP/PS1 transgenic mice. Brain Res. 1371, 150–160. doi: 10.1016/j.brainres.2010.11.043
Hunt, H., Belanoff, J. K., Golding, E., Gourdet, B., Phillips, T., Swift, D., et al. (2015). 1H-Pyrazolo[3,4-g]hexahydro-isoquinolines as potent GR antagonists with reduced hERG inhibition and an improved pharmacokinetic profile. Bioorg. Med. Chem. Lett. 25, 5720–5725. doi: 10.1016/j.bmcl.2015.10.097
Ishijima, S., Baba, H., Maeshima, H., Shimano, T., Inoue, M., Suzuki, T., et al. (2018). Glucocorticoid may influence amyloid β metabolism in patients with depression. Psychiatry Res. 259, 191–196. doi: 10.1016/j.psychres.2017.10.008
Jankord, R., and Herman, J. P. (2008). Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann. N. Y. Acad. Sci. 1148, 64–73. doi: 10.1196/annals.1410.012
Jeong, Y. H., Park, C. H., Yoo, J., Shin, K. Y., Ahn, S. M., Kim, H. S., et al. (2006). Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV717I-CT100 transgenic mice, an Alzheimer’s disease model. FASEB J. 20, 729–731. doi: 10.1096/fj.05-4265fje
Kaneko, I., Morimoto, K., and Kubo, T. (2001). Drastic neuronal loss in vivo by β-amyloid racemized at Ser26 residue: conversion of non-toxic [D-Ser26] β-amyloid 1-40 to toxic and proteinase-resistant fragments. Neuroscience 104, 1003–1011. doi: 10.1016/S0306-4522(01)00155-5
Kessing, L. V., and Andersen, P. K. (2004). Does the risk of developing dementia increase with the number of episodes in patients with depressive disorder and in patients with bipolar disorder? J. Neurol. Neurosurg. Psychiatry 75, 1662–1666. doi: 10.1136/jnnp.2003.031773
Kubo, T., Nishimura, S., Kumagae, Y., and Kaneko, I. (2002). In vivo conversion of racemized β-amyloid ([D-Ser26]Aβ1-40) to truncated and toxic fragments ([D-Ser26]Aβ25-35/40) and fragment presence in the brains of Alzheimer’s patients. J. Neurosci. Res. 70, 474–478. doi: 10.1002/jnr.10391
Lachize, S., Apostolakis, E. M., van der Laan, S., Tijssen, A. M., Xu, J., de Kloet, E. R., et al. (2009). Steroid receptor coactivator-1 is necessary for regulation of corticotropin-releasing hormone by chronic stress and glucocorticoids. Proc. Natl. Acad. Sci. U.S.A. 106, 8038–8042. doi: 10.1073/pnas.0812062106
Lahiri, D. K. (2004). Functional characterization of amyloid beta precursor protein regulatory elements: rationale for the identification of genetic polymorphism. Ann. N. Y. Acad. Sci. 1030, 282–288. doi: 10.1196/annals.1329.035
Lanté, F., Chafai, M., Raymond, E. F., Pereira, A. R., Mouska, X., Kootar, S., et al. (2015). Subchronic glucocorticoid receptor inhibition rescues early episodic memory and synaptic plasticity deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 40, 1772–1781. doi: 10.1038/npp.2015.25
López-Cruz, L., Salamone, J. D., and Correa, M. (2018). Caffeine and selective adenosine receptor antagonists as new therapeutic tools for the motivational symptoms of depression. Front. Pharmacol. 9:526. doi: 10.3389/fphar.2018.00526
Martignoni, E., Petraglia, F., Costa, A., Bono, G., Genazzani, A. R., and Nappi, G. (1990). Dementia of the Alzheimer type and hypothalamus-pituitary-adrenocortical axis: changes in cerebrospinal fluid corticotropin releasing factor and plasma cortisol levels. Acta Neurol. Scand. 81, 452–456. doi: 10.1111/j.1600-0404.1990.tb00994.x
Mattson, M. P. (2004). Pathways towards and away from Alzheimer’s disease. Nature 430, 631–639. doi: 10.1038/nature02621
McArthur, S., Cristante, E., Paterno, M., Christian, H., Roncaroli, F., Gillies, G. E., et al. (2010). Annexin A1: a central player in the anti-inflammatory and neuroprotective role of microglia. J. Immunol. 185, 6317–6328. doi: 10.4049/jimmunol.1001095
McEwen, B. S. (1999). Stress and hippocampal plasticity. Annu. Rev. Neurosci. 22, 105–122. doi: 10.1146/annurev.neuro.22.1.105
McEwen, B. S. (2008). Central effects of stress hormones in health and disease: understanding the protective and damaging effects of stress and stress mediators. Eur. J. Pharmacol. 583, 174–185. doi: 10.1016/j.ejphar.2007.11.071
McEwen, B. S., and Sapolsky, R. M. (1995). Stress and cognitive function. Curr. Opin. Neurobiol. 5, 205–216. doi: 10.1016/0959-4388(95)80028-X
Meijer, O. C., Koorneef, L. L., and Kroon, J. (2018). Glucocorticoid receptor modulators. Ann. Endocrinol. 79, 107–111. doi: 10.1016/j.ando.2018.03.004
Meijer, O. C., Steenbergen, P. J., and de Kloet, E. R. (2000). Differential expression and regional distribution of steroid receptor coactivators SRC-1 and SRC-2 in brain and pituitary. Endocrinology 141, 2192–2199. doi: 10.1210/endo.141.6.7489
Mejía, S., Giraldo, M., Pineda, D., Ardila, A., and Lopera, F. (2003). Nongenetic factors as modifiers of the age of onset of familial Alzheimer’s disease. Int. Psychogeriatr. 15, 337–349. doi: 10.1017/S1041610203009591
Modrego, P. J., and Ferrández, J. (2004). Depression in patients with mild cognitive impairment increases the risk of developing dementia of Alzheimer type: a prospective cohort study. Arch. Neurol. 61, 1290–1293. doi: 10.1001/archneur.61.8.1290
Nguyen, E. T., Caldwell, J. L., Streicher, J., Ghisays, V., Balmer, N. J., Estrada, C. M., et al. (2018). Differential effects of imipramine and CORT118335 (Glucocorticoid receptor modulator/mineralocorticoid receptor antagonist) on brain-endocrine stress responses and depression-like behavior in female rats. Behav. Brain Res. 336, 99–110. doi: 10.1016/j.bbr.2017.08.045
Notarianni, E. (2013). Hypercortisolemia and glucocorticoid receptor-signaling insufficiency in Alzheimer’s disease initiation and development. Curr. Alzheimer Res. 10, 714–731. doi: 10.2174/15672050113109990137
Oomen, C. A., Mayer, J. L., de Kloet, E. R., Joëls, M., and Lucassen, P. J. (2007). Brief treatment with the glucocorticoid receptor antagonist mifepristone normalizes the reduction in neurogenesis after chronic stress. Eur. J. Neurosci. 26, 3395–3401. doi: 10.1111/j.1460-9568.2007.05972.x
Ownby, R. L., Crocco, E., Acevedo, A., John, V., and Loewenstein, D. (2006). Depression and risk for Alzheimer disease: systematic review, meta-analysis, and metaregression analysis. Arch. Gen. Psychiatry 63, 530–538. doi: 10.1001/archpsyc.63.5.530
Padilla, K. M., Quintanar-Setephano, A., López-Vallejo, F., Berumen, L. C., Miledi, R., and García-Alcocer, G. (2018). Behavioral changes induced through adenosine A2A receptor ligands in a rat depression model induced by olfactory bulbectomy. Brain Behav. 8:e00952. doi: 10.1002/brb3.952
Papadopoulou, A., Siamastras, T., Delgado-Morales, R., Amin, N. D., Shukla, V., Zheng, Y. L., et al. (2015). Acute and chronic stress differentially regulate cyclin-dependent kinase 5 in mouse brain: implications to glucocorticoid actions and major depression. Transl. Psychiatry 5:e578. doi: 10.1038/tp.2015.72
Pariante, C. M. (2003). Depression, stress and the adrenal axis. J. Neuroendocrinol. 15, 811–812. doi: 10.1046/j.1365-2826.2003.01058.x
Peeters, B. W., Ruigt, G. S. F., Craighead, M., and Kitchener, P. (2008). Differential effects of the new glucocorticoid receptor antagonist ORG 34517 and RU486 (mifepristone) on glucocorticoid receptor nuclear translocation in the AtT20cell line. Ann. N. Y. Acad. Sci. 1148, 536–541. doi: 10.1196/annals.1410.072
Pineau, F., Canet, G., Desrumaux, C., Hunt, H., Chevallier, N., Ollivier, M., et al. (2016). New selective glucocorticoid receptor modulators reverse amyloid-β peptide-induced hippocampus toxicity. Neurobiol. Aging 45, 109–122. doi: 10.1016/j.neurobiolaging.2016.05.018
Pittenger, C., and Duman, R. S. (2008). Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33, 88–109. doi: 10.1038/sj.npp.1301574
Pomara, N., Hernando, R. T., de la Pena, C. B., Sidtis, J. J., Cooper, T. B., and Ferris, S. (2006). The effect of mifepristone (RU486) on plasma cortisol in Alzheimer’s disease. Neurochem. Res. 31, 585–588. doi: 10.1007/s11064-006-9055-5
Popp, J., Schaper, K., Kölsch, H., Cvetanovska, G., Rommel, F., Klingmüller, D., et al. (2009). CSF cortisol in Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 30, 498–500. doi: 10.1016/j.neurobiolaging.2007.07.007
Querfurth, H. X., and Laferla, F. M. (2010). Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Rapp, M. A., Schnaider-Beeri, M., Purohit, D. P., Perl, D. P., Haroutunian, V., and Sano, M. (2008). Increased neurofibrillary tangles in patients with Alzheimer disease with comorbid depression. Am. J. Geriatr. Psychiatry 16, 168–174. doi: 10.1097/JGP.0b013e31816029ec
Reul, J. M., and de Kloet, E. R. (1985). Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 117, 2505–2511. doi: 10.1210/endo-117-6-2505
Reynolds, A. R., Saunders, M. A., Brewton, H. W., Winchester, S. R., Elgumati, I. S., and Prendergast, M. A. (2015). Acute oral administration of the novel, competitive and selective glucocorticoid receptor antagonist ORG 34517 reduces the severity of ethanol withdrawal and related hypothalamic-pituitary-adrenal axis activation. Drug Alcohol. Depend. 154, 100–104. doi: 10.1016/j.drugalcdep.2015.06.018
Ries, M., Loiola, R., Shah, U. N., Gentleman, S. M., Solito, E., and Sastre, M. (2016). The anti-inflammatory Annexin A1 induces the clearance and degradation of the amyloid-β peptide. J. Neuroinflammation 13:234. doi: 10.1186/s12974-016-0692-6
Rissman, R. A., Lee, K. F., Vale, W., and Sawchenko, P. E. (2007). Corticotropin-releasing factor receptors differentially regulate stress-induced Tau phosphorylation. Neurobiol. Dis. 27, 6552–6562.
Robert, P. H., Schuck, S., Dubois, B., Lepine, J. P., Gallarda, T., Olie, J. P., et al. (2003). Validation of the short cognitive battery (B2C). Value in screening for Alzheimer’s disease and depressive disorders in psychiatric practice. Encephale 29, 266–272.
Roozendaal, B. (2000). Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology 25, 213–238. doi: 10.1016/S0306-4530(99)00058-X
Rothman, S. M., Herdener, N., Camandola, S., Texel, S. J., Mughal, M. R., Cong, W. N., et al. (2012). 3xTgAD mice exhibit altered behavior and elevated Aβ after chronic mild social stress. Neurobiol. Aging 33, 830.e1–830.e12. doi: 10.1016/j.neurobiolaging.2011.07.005
Sambamurti, K., Kinsey, R., Maloney, B., Ge, Y.-W., and Lahiri, D. K. (2004). Gene structure and organization of the human beta-secretase (BACE) promoter. FASEB J. 18, 1034–1036. doi: 10.1096/fj.03-1378fje
Sapolsky, R. M., Romero, L. M., and Munck, A. U. (2000). How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endoc. Rev. 21, 55–89.
Schatzberg, A. F., and Lindley, S. (2008). Glucocorticoid antagonists in neuropsychotic disorders. Eur. J. Pharmacol. 583, 358–364. doi: 10.1016/j.ejphar.2008.01.001
Selkoe, D. J. (2001). Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766. doi: 10.1152/physrev.2001.81.2.741
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1007/978-981-10-7757-9_11
Shim, Y. S., and Yang, D. W. (2006). Depression as prognostic factor: 6 months follow-up in a geriatric institution. Arch. Gerontol. Geriatr. 43, 277–283. doi: 10.1016/j.archger.2005.11.002
Solomon, M. B., Wulsin, A. C., Rice, T., Wick, D., Myers, B., McKlveen, J., et al. (2014). The selective glucocorticoid receptor antagonist CORT 108297 decreases neuroendocrine stress responses and immobility in the forced swim test. Horm. Behav. 65, 363–371. doi: 10.1016/j.yhbeh.2014.02.002
Starkstein, S. E., Jorge, R., Mizrahi, R., and Robinson, R. G. (2005). The construct of minor and major depression in Alzheimer’s disease. Am. J. Psychol. 162, 2086–2093. doi: 10.1176/appi.ajp.162.11.2086
Swanwick, G., Kirby, M., Bruce, I., Buggy, F., Coen, R., Coakley, D., et al. (1998). Hypothalamic-pituitary-adrenal axis dysfunction in Alzheimer’s disease: lack of association between longitudinal and cross-sectional findings. Am. J. Psychiatry 155, 286–289.
Thomas, A. J., and O’Brien, J. T. (2008). Depression and cognition in older adults. Curr. Opin. Psychiatry 21, 8–13. doi: 10.1097/YCO.0b013e3282f2139b
Tomidokoro, Y., Ishiguro, K., Harigaya, Y., Matsubara, E., Ikeda, M., Park, J. M., et al. (2001). Abeta amyloidosis induces the initial stage of tau accumulation in APP(Sw) mice. Neurosci. Lett. 299, 169–172. doi: 10.1016/S0304-3940(00)01767-5
Tu, S., Okamoto, S. I., Lipton, S. A., and Xu, H. (2014). Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 9, 1–48. doi: 10.1186/1750-1326-9-48
Vyas, A., Mitra, R., Shankaranarayana, B. S., and Chattarji, S. (2002). Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J. Neurosci. 22, 6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002
Vyas, A., Pillai, A. G., and Chattarji, S. (2004). Recovery after chronic stress fails to reverse amygdaloid neuronal hypertrophy and enhanced anxiety-like behavior. Neuroscience 128, 667–673. doi: 10.1016/j.neuroscience.2004.07.013
Weiner, M. F., Vobach, S., Olsson, K., Svetlik, D., and Risser, R. C. (1997). Cortisol secretion and Alzheimer’s disease progression. Biol. Psychiatry 42, 1030–1038. doi: 10.1016/S0006-3223(97)00165-0
Wilson, R. S., Arnold, S. E., Schneider, J. A., Kelly, J. F., Tang, Y., and Bennett, D. A. (2006). Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology 27, 143–153. doi: 10.1159/000095761
Wilson, R. S., Barnes, L. L., Bennett, D. A., Li, Y., Bienias, J. L., Mendes de Leon, C. F., et al. (2005). Proneness to psychological distress and risk of Alzheimer disease in a biracial community. Neurology 64, 380–382. doi: 10.1212/01.WNL.0000149525.53525.E7
Wulsin, A. C., Herman, J. P., and Solomon, M. B. (2010). Mifepristone decreases depression-like behavior and modulates neuroendocrine and central hypothalamic-pituitary-adrenocortical axis responsiveness to stress. Psychoneuroendocrinology 35, 1100–1112. doi: 10.1016/j.psyneuen.2010.01.011
Yan, J., Sun, X. B., Wang, H. Q., Zhao, H., Zhao, X. Y., Xu, Y. X., et al. (2010). Chronic restraint stress alters the expression and distribution of phosphorylated tau and MAP2 in cortex and hippocampus of rat brain. Brain Res. 1347, 132–141. doi: 10.1016/j.brainres.2010.05.074
Yang, Y. H., Morand, E., and Leech, M. (2013). Annexin A1: potential for glucocorticoid sparing in RA. Nat. Rev. Rheumatol. 9, 595–603. doi: 10.1038/nrrheum.2013.126
Yi, J. H., Brown, C., Whitehead, G., Piers, T., Lee, Y. S., Perez, C. M., et al. (2017). Glucocorticoids activate a synapse weakening pathway culminating in tau phosphorylation in the hippocampus. Pharmacol. Res. 121, 42–51. doi: 10.1016/j.phrs.2017.04.015
Young, A. H., Gallagher, P., Watson, S., Del-Estal, D., Owen, B. M., and Ferrier, I. N. (2004). Improvements in neurocognitive function and mood following adjunctive treatment with mifepristone (RU-486) in bipolar disorder. Neuropsychopharmacology 29, 1538–1545. doi: 10.1038/sj.npp.1300471
Zalachoras, I., Houtman, R., Atucha, E., Devos, R., Tijssen, A. M., Hu, P., et al. (2013). Differential targeting of brain stress circuits with a selective glucocorticoid receptor modulator. Proc. Natl. Acad. Sci. U.S.A. 110, 7910–7915. doi: 10.1073/pnas.1219411110
Zussy, C., Brureau, A., Delair, B., Marchal, S., Keller, E., Naert, G., et al. (2011). Time-course and regional analyses of the physiopathological changes induced after cerebral injection of an amyloid-β fragment in rats. Am. J. Pathol. 179, 315–334. doi: 10.1016/j.ajpath.2011.03.021
Keywords: Alzheimer’s disease, depression, risk factor, HPA axis, glucocorticoids, selective GR modulators
Citation: Canet G, Chevallier N, Zussy C, Desrumaux C and Givalois L (2018) Central Role of Glucocorticoid Receptors in Alzheimer’s Disease and Depression. Front. Neurosci. 12:739. doi: 10.3389/fnins.2018.00739
Received: 11 June 2018; Accepted: 25 September 2018;
Published: 16 October 2018.
Edited by:
Jean-Michel Verdier, Université de Sciences Lettres de Paris, FranceReviewed by:
David Blum, INSERM U1172 Centre de Recherche Jean Pierre Aubert, FranceEmmanuel Planel, Laval University, Canada
Copyright © 2018 Canet, Chevallier, Zussy, Desrumaux and Givalois. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurent Givalois, bGF1cmVudC5naXZhbG9pc0B1bW9udHBlbGxpZXIuZnI=