 Josh Allen
Josh Allen Raquel Romay-Tallon
Raquel Romay-Tallon Kyle J. Brymer
Kyle J. Brymer Hector J. Caruncho
Hector J. Caruncho Lisa E. Kalynchuk
Lisa E. Kalynchuk- 1Division of Medical Sciences, University of Victoria, Victoria, BC, Canada
- 2Department of Psychology, University of Saskatchewan, Saskatoon, SK, Canada
Human and animal studies suggest an intriguing link between mitochondrial diseases and depression. Although depression has historically been linked to alterations in monoaminergic pharmacology and adult hippocampal neurogenesis, new data increasingly implicate broader forms of dampened plasticity, including plasticity within the cell. Mitochondria are the cellular powerhouse of eukaryotic cells, and they also regulate brain function through oxidative stress and apoptosis. In this paper, we make the case that mitochondrial dysfunction could play an important role in the pathophysiology of depression. Alterations in mitochondrial functions such as oxidative phosphorylation (OXPHOS) and membrane polarity, which increase oxidative stress and apoptosis, may precede the development of depressive symptoms. However, the data in relation to antidepressant drug effects are contradictory: some studies reveal they have no effect on mitochondrial function or even potentiate dysfunction, whereas other studies show more beneficial effects. Overall, the data suggest an intriguing link between mitochondrial function and depression that warrants further investigation. Mitochondria could be targeted in the development of novel antidepressant drugs, and specific forms of mitochondrial dysfunction could be identified as biomarkers to personalize treatment and aid in early diagnosis by differentiating between disorders with overlapping symptoms.
Mitochondria
Mitochondria are the main energy factories of eukaryotic cells. The brain is particularly dependent on mitochondrial activity due to both its high levels of energy use and its inability to store large amounts of energy reserves in the form of glycogen. As a result of the their roles in energy production, mitochondria also generate reactive oxygen species (ROS) that may have a toxic effects in cells. In addition, mitochondria also play a prominent role in the regulation of apoptotic cell death (for examples, see Davidson and Hardison, 1984; Herrmann and Neupert, 2000; Calabrese et al., 2001; Chan, 2006; Chipuk et al., 2006; Fattal et al., 2006; McBride et al., 2006; Youle and van der Bliek, 2012; Tobe, 2013; Bansal and Kuhad, 2016).
The focus of this review is the link between mitochondrial dysfunction and major depression. Depression has historically been considered a disorder of altered pharmacology and altered hippocampal neurogenesis. However, recent evidence has opened the door to an expanded notion of the neurobiology of depression, such that a reduction in ATP levels, enhancement of oxidative stress, and acceleration of apoptosis are now considered to be important events (reviewed in Rezin et al., 2008a). In this review, we summarize some of the latest knowledge on mitochondrial dysregulation in major depression (depicted in Figure 1) and also discuss how mitochondrial dysfunction could instigate downstream changes in extracellular matrix proteins such as reelin, neuronal nitric oxide (nNOS), oxidative stress, and inflammation, and finally adult hippocampal neurogenesis. Uncovering how all these factors influence one another could lead to new vistas in the development of novel therapeutics for the treatment of this problematic disorder.
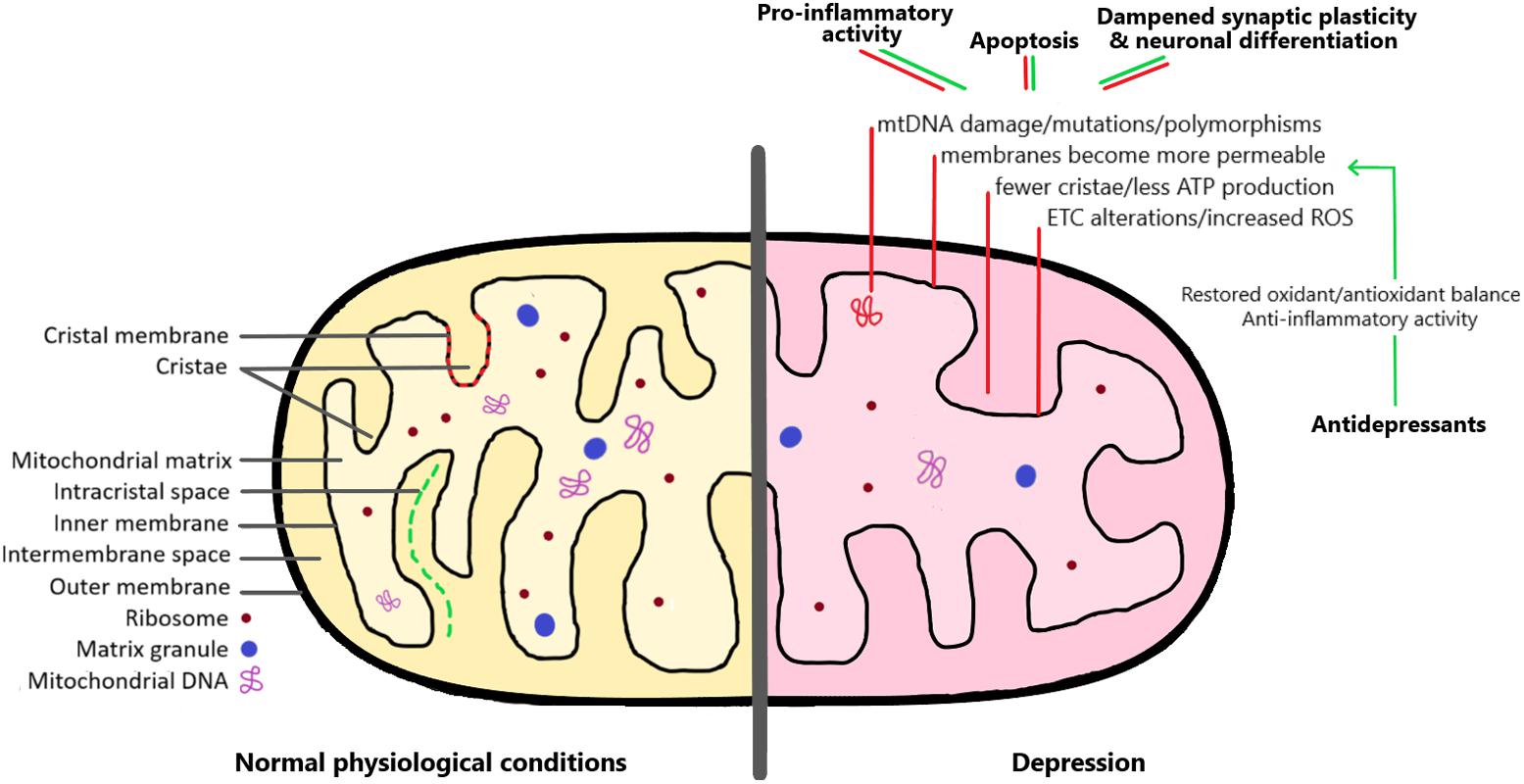
FIGURE 1. The mitochondrion under normal physiological conditions and in the depression brain. As detailed in the right side of the image, there are a series of mitochondrial alterations that have been observed both in depressed patients and in animal models of depression (red lines). These include changes affecting mitochondrial DNA, membrane permeability, and increased formation of reactive oxygen species (ROS). As a consequence, these alterations lead to pro-inflammatory activity, increased apoptosis, and dampened synaptic plasticity and neuronal differentiation. Interestingly, antidepressant medication can restore the mitochondrial oxidant/antioxidant balance, and therefore help to rescue the negative effects of mitochondrial dysregulation (green lines). See the text for more detailed explanations.
Hypotheses About the Neurobiological Basis of Depression
Depression is a common neuropsychiatric disorder, affecting up to 20% of the population (Kessler et al., 1994). The presence and severity of symptoms vary among individuals, and can include low mood and anhedonia, decreased energy, altered appetite and weight, irritability, sleep disturbances, and cognitive deficits (Nemeroff, 1998). Patients with depression have a higher rate of other physical illnesses (i.e., comorbidities with cardiovascular disorders, stroke, etc.), decreased social functioning, and a high mortality rate (Nemeroff, 1998). The complexity of this disorder is further compounded by the fact that it often co-occurs with other psychiatric conditions. For example, about 50% of depression patients also suffer from anxiety disorders (Kessler et al., 1996), which can indicate a more severe form of the disease, with delayed recovery, increased risk of relapse, greater disability, and increased suicide attempts (Hirschfeld, 2001). It is generally thought that a combination of environmental and genetic factors influences the development of depression (Nestler et al., 2002; Kalia, 2005). However, despite extensive clinical and preclinical research efforts, there is still a fundamental lack of understanding about the specific biological changes that give rise to depressive symptoms.
For more than 50 years, the dominant theory for the pathogenesis of depression was the monoamine hypothesis (Schildkraut, 1965), which arose from observations that antidepressant drugs work by inhibiting the reuptake of monoamines such as serotonin and norepinephrine. However, this theory has largely fallen out of favor due to a number of discrepancies, such as the fact that the therapeutic effects of antidepressants take weeks to develop even though monoamine levels are elevated within hours of administration, and the fact that only about 40% of patients respond satisfactorily to treatment (Trivedi et al., 2006). More recent theories about the neurobiological basis of depression have focused on the neurogenesis theory, which posits that stress-induced decreases in hippocampal neurogenesis could be a causal factor in depression (Jacobs et al., 2000). This hypothesis is supported by observations of decreased hippocampal volume in patients with depression (MacQueen et al., 2003; Campbell et al., 2004), decreased cell proliferation and survival in preclinical animal models of depression (Brummelte and Galea, 2010; Schoenfeld and Cameron, 2015), and increased cell proliferation and survival after antidepressant treatment (Santarelli et al., 2003; Fenton et al., 2015). Our laboratory has contributed to this literature using a well-validated preclinical model of depression in which rats or mice are subjected to daily injections of the stress hormone corticosterone (CORT) for several weeks, followed by behavioral testing and then tissue collection for further analyses (Gregus et al., 2005; Johnson et al., 2006; Sterner and Kalynchuk, 2010). Using this approach, we showed that the time course for the onset of depression-like behavior in rats is paralleled by dampened hippocampal neurogenesis and neuronal maturation (Lussier et al., 2013). Importantly, this work also implicated the extracellular matrix protein reelin in the pathogenesis of depression (see Caruncho et al., 2016). There is evidence that reelin can regulate adult hippocampal neurogenesis and dendritic spine plasticity (Pujadas et al., 2010), and our data show that rats subjected to repeated CORT injections have dampened reelin expression selectively in the proliferative subgranular zone of the dentate gyrus (Lussier et al., 2009) and also that antidepressant treatment normalizes depression-like behavior, hippocampal neurogenesis, and hippocampal reelin expression in tandem (Fenton et al., 2015). The reelin link is relevant to mitochondrial dysfunction because reelin in the periphery interacts with the immune system and a loss of reelin can magnify markers of inflammation that influence mitochondria (Green-Johnson et al., 1995). This issue is discussed in more detail in the section below on reelin and inflammation.
It is important to note that the neurogenesis hypothesis of depression is somewhat controversial because depression-like symptoms can occur even when cell proliferation is not decreased and the behavioral actions of antidepressants do not always coincide with increases in the number of hippocampal neurons (Surget et al., 2008; Bessa et al., 2009; David et al., 2009). Instead, some data suggest that depression and the therapeutic actions of antidepressants may be more related to alterations in dendritic complexity and neuronal remodeling than cell number per se (Bessa et al., 2009; Lussier et al., 2013). The neurogenesis hypothesis of depression may therefore be better described as the neuroplasticity hypothesis of depression – a broadening of concept that would include plasticity within the cell, such as mitochondrial activity.
Because the brain has high aerobic activity, requiring about 20 times more energy than the rest of the body by weight (Kety, 1950), it is highly vulnerable to conditions stemming from impaired energy production. A resting cortical neuron consumes 4.7 billion ATP molecules every second (Zhu et al., 2012). Evidence from post-mortem (Kato et al., 1997; Konradi et al., 2004; Iwamoto et al., 2005; Munakata et al., 2005), imaging (Frey et al., 2007), genetic (Kato et al., 1997; Kato and Kato, 2000; Iwamoto et al., 2005; Benes et al., 2006), and cellular (Cataldo et al., 2010) studies is plentiful in showing the involvement of mitochondrial dysfunction in bipolar disorder and schizophrenia. Studies are also emerging providing evidence that mitochondria-mediated mechanisms are related to depressive symptoms (reviewed in Castren, 2005; Tobe, 2013; Shimamoto and Rappeneau, 2017; Petschner et al., 2018), that mitochondrial mutations are witnessed in individuals diagnosed with depression (Munakata et al., 2007; Ben-Shachar and Karry, 2008) and that the two diseases are often comorbid (Koene et al., 2009; Morava et al., 2010).
Mitochondria could play a role in the dampened plasticity associated with depression. Depression is associated with abnormalities in intracellular second messenger signal transduction cascades resulting from 5HT and NE receptor activation (Perez et al., 2000; Popoli et al., 2000) and dysregulated and desensitized monoamine receptors (Hamon and Blier, 2013). These observations can be related to mitochondrial dysfunction because ATP is needed for the activation of downstream signaling following the binding of neurotransmitters to receptors (Moretti et al., 2003). ATP is also necessary to attend to the energy demands of vesicle transport and neurotransmitter release (reviewed in Vos et al., 2010; and more recently in Devine and Kittler, 2018). Furthermore, patients with mitochondrial diseases or mitochondrial DNA (mtDNA) mutations and polymorphisms often present symptoms characteristic of mood disorders (Suomalainen et al., 1992; Onishi et al., 1997; Kato et al., 2001; Fattal et al., 2006; Morava et al., 2010). Higher rates of mitochondrial biogenesis are needed for neuronal differentiation (Calingasan et al., 2008) and therefore, dysfunctional mitochondria could result in impaired neuroplasticity in depressed patients.
Genetics
There have been many observations of links between mtDNA and depression. As mentioned above, depression is a heterogeneous disorder, with several different symptom profiles, and genetic background contributes to its development (Lesch, 2004). Prevalence rates for depression are as high as 54% in patients with mitochondrial diseases (Fattal et al., 2007). However, not all patients who have the same mitochondrial gene mutations develop depressive symptoms, indicating a genetic and non-genetic interplay of factors (Koene et al., 2009). Mitochondrial disorders may result from mutations in nuclear DNA or mtDNA, with the amount of mtDNA mutations possibly correlating with disease severity (Chinnery et al., 2000). In one interesting study, long-PCR revealed that 68% of patients with depression have mtDNA deletions, compared to 36% of control subjects (Gardner et al., 2003). Similarly, in leukocytes of depressed patients, mtDNA copy number variates were significantly lower than in control subjects, and mtDNA oxidative damage was increased (Chang et al., 2015). Interestingly, oxidized mtDNA activates pro-inflammatory cytokines (Adzic et al., 2016) and increased inflammation is known to play a role in the development of depressive symptoms (e.g., Brymer et al., 2018; Wang et al., 2018). Variations in mtDNA have also been shown to cause cognitive impairments in mice (Sharpley et al., 2012) and in humans (Inczedy-Farkas et al., 2014; Petschner et al., 2018), and cognitive deficits are a common symptom associated with depression.
Beyond these general linkages between mtDNA and depression, recent research has implicated a number of specific mitochondrial genes in depression. This topic has been reviewed in detail recently (Petschner et al., 2018), so only a few examples will be mentioned here. A recent systematic assessment using mitochondrial PCR array profiling identified 16 genes that were differentially expressed in the dorsolateral prefrontal cortex of post-mortem brains from depressed patients compared to control subjects (Wang and Dwivedi, 2017). The identified genes are ones known to govern oxidative stress and neuronal ATP levels, suggesting for the first time that mitochondrial genes might be altered in tissue samples from human patients. Similarly, the mitochondrial ATP6V1B2 gene has been implicated in depression, possibly through its effects on neurotransmission and receptor-mediated endocytosis (Petschner et al., 2018). Less direct evidence comes from observations that mice with mutations of the POLG gene, which encodes a subunit of mtDNA polymerase, exhibit depression-like symptoms (Kasahara et al., 2006), and that polymorphisms of genes that code for mitochondrial enzymes, such as MTHFD1L, are associated with negative rumination, which is a precursor to depression (Eszlari et al., 2016). This polymorphism is also associated with high levels of homocysteine, which has been related to hippocampal volume and depression (Moustafa et al., 2014).
These examples point to an intriguing link between mtDNA or gene expression and depression, though more work needs to be done in this area, particularly to identify gene alterations in tissue from human patients.
Proteomic Studies
There have been many studies indicating the involvement of the oxidative phosphorylation (OXPHOS) pathway in depression. Proteomic studies conducted on post-mortem brains from depressed patients suggest that about 21% of dysregulated proteins are also commonly dysregulated in patients with schizophrenia and bipolar disorder (Saia-Cereda et al., 2017; Villa et al., 2017). In a mutant mouse model of depression, a dysregulated OXPHOS pathway was seen in the hippocampus (Zubenko et al., 2014), which is a key region of dampened plasticity in both human depression and rodent models (Sheline et al., 2003; Frodl et al., 2006; Sterner and Kalynchuk, 2010). In addition, proteomic studies using different animal models of depression have revealed alterations in specific proteins involved in OXPHOS and also confirmed the effect of antidepressant treatment in the expression of these proteins (reviewed in Carboni, 2015).
In depressed patients, most of the differentially expressed proteins are involved in cellular assembly, organization, function, and maintenance, as well as cardiovascular system development and function, but they are mainly related to deregulation of energy metabolism pathways (Martins-de-Souza et al., 2012). Twenty different subunits of the OXPHOS complex were increased in post-mortem brains from depressed patients (Martins-de-Souza et al., 2012), whereas the opposite effect has been seen in brains from patients with schizophrenia (Martins-de-Souza et al., 2011). A proteomics approach also revealed that the SSRI fluoxetine upregulated and downregulated 23 and 60 cytosolic mitochondrial-related proteins, respectively (Filipović et al., 2017). In addition, 60 non-synaptic mitochondrial-related proteins were upregulated whereas three were downregulated. These effects were largely confirmed in a subsequent study (Głombik et al., 2017). When looking at samples from the dorsolateral prefrontal cortex, which shows reduced activation and volume in patients with depression (Drevets et al., 1998; Halari et al., 2009), a shotgun label-free approach revealed that 32% of differentially expressed proteins associated with depression were involved in metabolic/energy pathways (Martins-de-Souza et al., 2012). Two other proteomic studies showed that several proteins involved in energy metabolism, such as carbonic anhydrase and aldolase C, were increased in the frontal cortex (Johnston-Wilson et al., 2000) and anterior cingulate cortex (Beasley et al., 2006) of depressed patients. These results are consistent with PET findings of a reduction in cerebral glucose metabolism in the brains of depressed patients (Baxter et al., 1989), which was reversed by 6 weeks of treatment with the SSRI paroxetine (Kennedy et al., 2001). Similarly, PET studies also revealed that depressed patients had reduced blood flow and bioenergetic metabolism in the prefrontal cortex (Drevets et al., 1997; Mayberg et al., 1999; Moretti et al., 2003), cingulate gyrus, and basal ganglia (Videbech, 2000).
Psychiatric disorders almost always have overlapping symptoms, which might reflect common mechanisms when compared against controls. An interesting study addressing this issue showed that patients with major depression that included psychosis had more differentially expressed proteins associated with energy metabolism, whereas patients with depression without psychosis had changes in proteins associated with cell growth and maintenance, although 53.7% of the altered proteins overlapped (Martins-de-Souza et al., 2012). Subtle differences in proteome fingerprints may become useful biomarkers that could be used to stratify patients with different symptoms profiles and to formulate effective personalized treatment plans. Proteomic studies support the view that mitochondrial dysfunction is one of many important factors involved in depression, and may identify novel pathogenic mechanisms of psychiatric disorders.
That alterations in mitochondria bioenergetics pathways contributed to the pathophysiology of depression also raise the possibility of developing mitochondrial biomarkers that can illustrate a better therapeutic approach to the treatment of depression. However, this field is still undeveloped and additional studies are needed characterizing specific mitochondrial dysfunctions in depression in relation to therapeutic response to antidepressants, or to evaluate the possibility of identifying mitochondrial drug targets that could be used to develop novel antidepressant drugs (Klinedinst and Regenold, 2015).
Decreased ATP Production
The production of ATP through OXPHOS is a key method by which mitochondria provide energy to the cell. Several lines of research have confirmed that depression is associated with lower than normal levels of ATP production. For example, brain levels of ATP are generally lower in the brains of depressed patients compared to control subjects (Moretti et al., 2003; Martins-de-Souza et al., 2012). This may be related to the dampened neuronal plasticity and impaired hippocampal neurogenesis thought to be operative in depression (Caruncho et al., 2016), as neurogenesis is a metabolically demanding process. Other researchers have found changes in ATP in depression in areas outside the brain. Gardner et al. (2003) found that mitochondrial ATP production rates and mitochondrial enzyme ratios in electron transport chain (ETC) complexes I–IV were significantly decreased in the muscles of patients with depression compared to controls. A correlation was found between altered biochemistry and depression rating scale scores further evidencing the relationship between mitochondrial dysfunction and psychopathology. In peripheral blood mononuclear cells, ATP turnover-related respiration was lowered in depressed patients compared to age-matched controls, as well as routine and uncoupled respiration and coupling efficiency (Karabatsiakis et al., 2014). Moreover, ATP-binding cassette transporters, which utilize the energy of ATP, are also altered in depressed patients and single nucleotide polymorphisms in the gene that codes for these transporters may be indicators of severity of the disorder and patient responsiveness to antidepressants (Lin et al., 2011).
Decreased ATP production has also been observed in preclinical animal models of depression. Female rats displaying anhedonia (i.e., decreased preference for sucrose) after 40 days of mild stress also had decreased hippocampal NA+, K+-ATPase activity (Gamaro et al., 2003). Fluoxetine reversed the effects of stress on enzymatic activity suggesting that NA+, K+-ATPase activity may be involved in the depression-like phenotype. In another study, fluoxetine restored sucrose preference, and normalized ATP synthesis rate and mitochondrial respiratory control in the raphe nucleus after 18 days of chronic unpredictable stress (Wen et al., 2014). Another study using the chronic mild stress paradigm revealed that mice with decreased sucrose preference and increased immobility in the tail suspension test (i.e., learned helplessness) also showed damaged mitochondrial ultrastructure, impaired respiration rates, and altered membrane potentials in the hippocampus, hypothalamus, and the cortex (Gong et al., 2011).
Oxidative Stress
Mitochondria are the primary source of ROS, which under normal conditions play important roles in cell signaling and homeostasis. ROS are produced in the OXPHOS pathway; however, in normal physiological conditions, mitochondria create protective factors that can neutralize harmful free radicals (Petschner et al., 2018). For example, there is a mitochondrial matrix thiol system that has an important role in antioxidant protection (Murphy, 2012). In the ETC, complexes donate electrons to oxygen producing radicals like superoxide and peroxidases, and high levels of such radicals and oxidative stress cause damage to lipids, enhance DNA breaks, and oxidize nuclear and mtDNA (Tobe, 2013; Czarny et al., 2015). Lower levels of ROS also play a role in normal cellular functioning, such as differentiation of cells, tissue regeneration, redox biology, and promoting adaptation to environmental changes (Vakifahmetoglu-Norberg et al., 2017). The production of highly reactive free radicals is increased when premature leakage of electrons to oxygen occurs in the ETC, increasing oxidative stress. Superoxide is a precursor for ROS, and complex I and III are mainly responsible for its production (Vakifahmetoglu-Norberg et al., 2017). Oxidative stress could be the cause or consequence of damage to mitochondria and mtDNA (Xie et al., 2017). Martins-de-Souza et al. (2012) speculated that a reduction in ATP could be due to oxidative stress and that the increased levels of subunits of OXPHOS complexes were compensatory. In fact, ATP reduction and its relation to oxidative stress have been linked not only to depression (detailed below), but also to psychotic disorders (see Chouinard et al., 2017), autism (Rose et al., 2014), anxiety (Kumar and Chanana, 2017), Alzheimer’s disease (recently reviewed in Tramutola et al., 2017), and Huntington disease (Quintanilla et al., 2017).
Several papers have reported links between oxidative stress and depression. Ben-Shachar and Karry (2008) reported an increase in oxidative damage and alterations in ETC complex I in the prefrontal cortex of depressed patients. Other researchers noted decreased levels of antioxidants and antioxidant enzymes in depression and related these changes to deficits in cognition (Anderson, 2018). In an immobilization stress preclinical model of depression, in which animals were restrained for 6 h a day, levels of the cellular antioxidant glutathione were reduced by 36.7% after 21 days, while lipid peroxidation increased (Madrigal et al., 2001). The authors speculated that lipid peroxidation could cause mitochondrial dysfunction by damaging membranes and causing excitotoxicity, which could be potentiated by increased production of reactive molecules (Braughler and Hall, 1989) or decreased antioxidant levels. In the olfactory bulbectomy model of depression, glutathione levels were also decreased whereas ROS superoxide, nitric oxide (NO), and lipid hydroperoxide levels were increased in mice (Holzmann et al., 2015) and rats (Almeida et al., 2017).
As previously mentioned, the effect of 21 days of environmental stress (i.e., restraint) on mitochondrial dysfunction in rats was investigated (Madrigal et al., 2001). The authors reported that mitochondrial activity of ETC complexes I–III were significantly decreased after just 7 days of restraint stress (6 h per day); however, there was no difference in complex IV and no stress-induced decreases in oxygen consumption throughout the 21-day period. They speculated that mitochondrial dysfunction was a result of overproduction of NO, as an accumulation of NO metabolites was found in the brain tissue. Similarly, 40 days of chronic variable stress decreased sucrose preference as well as inhibited ETC complexes I, II, and IV in the cerebral cortex and cerebellum of rats (Rezin et al., 2008b). Interestingly, increased expression of proteins related to mitochondrial import and transport in the OXPHOS pathway was also seen in a preclinical mouse model of anxiety, a disorder that is highly comorbid with depression (Filiou et al., 2011). Moreover, in this model, the expression of enzymes involved in catalyzing glycolysis pathway reactions was also dysregulated.
The mechanisms by which environmental stress negatively impacts the brain are still not fully understood. However, there is evidence that free-radicals such as NO cause rapid damage to certain cell macromolecules that are involved in the ETC system, which in turn will decrease production of ATP and may be implicated in cytotoxic effects in the central nervous system (Cleeter et al., 1994; Lizasoain et al., 1996). Madrigal et al. (2001) did not find changes in ATP levels, which provides further evidence that a threshold of ETC complex dysfunction may have to be reached before the capability of mitochondria to maintain homeostasis diminishes (Davey and Clark, 1996).
Antidepressant treatment improves oxidative stress parameters in patients with depression. For example, a higher serum total oxidant status and a lower serum total antioxidant capacity in depressed patients were normalized after 42 days of antidepressant treatment (Cumurcu et al., 2009). Similar findings have been reported in animal models of depression. Venlafaxine increased expression of antioxidant mitochondrial genes in the mouse brain, which reduced levels of hydrogen peroxide and peroxynitrite (Goemaere and Knoops, 2012; Tamási et al., 2014). Furthermore, in the chronic mild stress paradigm, lamotrigine, aripiprazole, and escitalopram all normalized glutathione and glutathione peroxidase activity in rat cortical regions (Eren et al., 2007a). Lipid peroxidation in the cortex and plasma was increased by chronic mild stress, but also reversed by the same three treatments. A similar study revealed that venlafaxine can reverse chronic mild stress-induced decreases in glutathione peroxidase activity and vitamin C, and increases in lipid peroxidation and NO in the rat cortex (Eren et al., 2007b). Moreover, unpredictable stress in mice resulted in increased open field test exploration along with decreased liver glutathione, superoxide dismutase, and total antioxidant capability, which was reversed by the traditional Chinese medicine, Shudihuang, in a dose-dependent manner (Zhang et al., 2009).
Reelin, Oxidative Stress, Inflammation, and Depression
A further link between ROS and depression has been suggested by recent work focused on the extracellular matrix protein reelin. Reelin has been linked to depression in preclinical models of depression: A decline in reelin expression in the hippocampal subgranular zone is associated with the emergence of depression-like behavior, and heterozygous reeler mice (HRM) with 50% of the normal levels of reelin are highly susceptible to the depressogenic effects of stress hormones (Lussier et al., 2011, 2013). Interestingly, a subpopulation of reelin containing cells also coexpress nNOS, and the percentage of neurons coexpressing both markers is specifically decreased in the subgranular zone and molecular layer of the dentate gyrus in HRM (Romay-Tallon et al., 2010).
As reelin secretion by neurons in the subgranular zone may be involved in regulating the maturation of adult hippocampal newborn neurons (Lussier et al., 2009), and as deficits in adult hippocampal neurogenesis have been proposed to be a key event underlying the development of a depressive phenotype (detailed above in section “Hypotheses About the Neurobiological Basis of Depression”), we recently examined the effects of repeated CORT injections on coexpression of reelin and nNOS across hippocampal subregions in brains from HRM and wildtype mice. We found that repeated CORT (administered at a dose that induces depression-like behavior in HRM; Lussier et al., 2011) creates an imbalance between reelin and nNOS expression in the proliferative subgranular zone of the dentate gyrus, with CORT inducing a decrease in colocalization of reelin and nNOS in wildtype mice but a significant increase in colocalization of these markers in HRM. We interpreted these results as being indicative of profound excitotoxicity in dentate gyrus neurons after chronic exposure to stress hormones to a degree that produces depression-like behavior (Romay-Tallon et al., 2010, 2015).
Nitric oxide and other ROS inhibit mitochondrial 2-oxoglutarate dehydrogenase giving rise to increased levels of glutamate, which eventually leads to glutamate excitotoxicity and cell death (Weidinger et al., 2017). The reelin–nNOS connection should receive more experimental attention, as a number of reports indicate that alterations in reelin expression within the dentate gyrus may result in deficient maturation of newborn granule neurons and dampened hippocampal plasticity, and may represent a key event in the pathophysiology of depression (reviewed in Caruncho et al., 2016).
There is also a key link with inflammation to consider in the context of these experiments. Many studies support the idea that inflammatory processes are involved in depression, and in fact targeting of inflammatory cytokines to reduce depression symptoms is a very active area of research (recently reviewed by Shariq et al., 2018). Studies have shown that pro-inflammatory cytokines alter ETC complexes and complex associated enzymes (Samavati et al., 2008), and that they activate pro-apoptotic proteins and the caspase cascade (Bansal and Kuhad, 2016). In mice, the injection of lipopolysaccharide, which induces strong immune responses and secretion of pro-inflammatory cytokines, significantly increased depression-like behavior in the sucrose preference and forced swim tests, and decreased ATP levels and mitochondrial membrane potential in the hippocampus (Chen et al., 2017).
Alterations in several components of the immune system, and in inflammatory markers, have also been observed in animals with low or null reelin expression (Green-Johnson et al., 1995). We have reported that these animals not only are quite susceptible to the depressogenic effects of repeated CORT (Lussier et al., 2011), but also show alterations in the clustering of specific membrane proteins in lymphocytes (Rivera-Baltanás et al., 2010) which prompted us to investigate membrane protein clustering in lymphocytes in depression patients, and to propose that the pattern of clustering of specific proteins along the plasma membrane of lymphocytes could be a putative biomarker of depression, and perhaps underlie some of the inflammatory events observed in depression patients (Rivera-Baltanas et al., 2012, 2014, 2015). In fact, alterations in oxidative stress in lymphocytes have been clearly demonstrated in depression (Szuster-Ciesielska et al., 2008; Czarny et al., 2018). Following up this line of thought, we have recently demonstrated that peripheral injections of the anti-inflammatory drug etanercept (which is unable to cross the blood–brain–barrier) not only rescues the depression-like behavior induced by repeated CORT but also normalizes the neurochemical phenotype of reelin expressing cells in the hippocampal dentate gyrus. We speculated that both peripheral and secondary central actions may be operative in the antidepressant effects of etanercept injections (Brymer et al., 2018). It seems clear that additional studies would be required to determine the connection between reelin, oxidative stress, and inflammation in depression, not only to determine how these factors may be an important component of the pathophysiology of depression, but also to evaluate them as possible targets to develop novel antidepressant drugs.
Apoptosis
Mitochondria have a clear role in cell metabolism, and evidence suggests that mitochondrial morphology also affects metabolic enzymes through fusion and fission (Chen et al., 2005). The formation and morphology of cristae on the inner membrane, which regulate mitochondrial metabolism, may require fusion machinery as a loss of such machinery results in decreased metabolism (McBride et al., 2006). Abnormal cell structure and function could result in alterations in synaptic signaling and neural circuits and vice versa (Kaidanovich-Beilin et al., 2012). Excessive glutamatergic activation of NMDA receptors was shown to increase ROS levels and alter mitochondrial membrane polarity, which led to elevated apoptosis rates in cardiomyocytes, possibly as a result of increased calcium ion influx (Gao et al., 2007). Mitochondria are present at synapses and responsive to synaptic stimulation (McBride et al., 2006). As the hippocampus is highly vulnerable to the depressogenic effects of chronic stress, it is likely that hippocampal mitochondria behave abnormally in depressed patients. For example, the clustering of mitochondria in dendritic spines in response to neural activity may be altered (Li et al., 2004). Glucocorticoid receptors (GRs) are also highly prevalent in the hippocampus. These receptors are activated when stress hormone levels are high, such as during periods of chronic stress. GRs coordinate OXPHOS enzyme biosynthesis (Simoes et al., 2012) and regulate mitochondrial gene transcription such as cytochrome oxidase 1 and 3, the activity of which correlates with levels of ATP (Adzic et al., 2013). In the hippocampus, chronic stress altered the phosphorylation of mitochondrial GRs, whereas in the prefrontal cortex, chronic stress significantly increased mitochondrial GR levels (Adzic et al., 2013). In the gut, stress increased serum CORT levels, which activated GR recruitment to instigate decreased ETC complex I activity, hyper-fission, and accumulation of ROS inducing apoptosis (De et al., 2017). This is the intrinsic pathway of apoptosis, which is affected by oxidative stress, elevated Ca2+ levels, and damaged DNA (Green and Kroemer, 1998; Kroemer et al., 2007).
Mitochondria are also involved in apoptosis through the extrinsic pathway, in which the death-inducing signaling complex is formed, leading to activation of caspase-8 and then downstream caspases that target substrates leading to programmed cell death (Vakifahmetoglu-Norberg et al., 2017). Other proteins such as K-Ras or BH3 interacting domain death agonist can also induce cell death when activated by caspases by translocating to mitochondria, where they trigger the release of the executioner caspases (Bivona et al., 2006; Bansal and Kuhad, 2016).
It is important to note that the effect of stress on mitochondrial function may depend on the nature of the stressor or period of chronicity. Although chronic stress and high levels of circulating stress hormones are a clear risk factor for depression (Gibbons and McHugh, 1962; Holsboer, 2001; Parker et al., 2003), low levels of stress hormones can be beneficial. For example, the effects of the stress hormone CORT on depression-like behavior in rodent models depend on the dose and time period of administration: higher doses and longer periods of administration produce robust increases in depression-like behavior but low doses or high doses given for short periods do not (Johnson et al., 2006; Lussier et al., 2013). This is consistent with the effects of stress hormones on mitochondria. Glucocorticoids can translocate to mitochondria, where they inhibit the release of cytochrome c and decrease apoptosis (Du et al., 2009). However, this is dependent on the level of glucocorticoids present in the tissue. Du et al. (2009) revealed that low doses of CORT were neuroprotective through regulation of mitochondria, but high doses were neurotoxic. Similarly, inhibiting mitochondrial protein synthesis completely impairs neuronal differentiation, but inhibiting ATP synthetase alone does not affect neurogenesis (Vayssiere et al., 1992). It would be of interest to map the dose-dependent effects of glucocorticoids on markers of mitochondrial function along with depression-like behavior to further confirm these relationships.
The Effect of Antidepressants on Mitochondria
There has been quite a bit of work done to try to understand the effect of antidepressant drugs on mitochondrial function. Most antidepressants work by increasing synaptic levels of serotonin and/or norepinephrine, and adverse side effects are commonly reported. Much of the research done to examine links between antidepressant drugs and mitochondrial function have used the SSRI fluoxetine, which may either inhibit or trigger mitochondrial apoptosis and alter activity of the ETC, depending on the cell type (de Oliveira, 2016). In the rat liver, fluoxetine administered in vitro inhibited state 3 of mitochondrial respiration for α-ketoglutarate and succinate oxidation, stimulated state 4 for succinate, and decreased the respiratory control ratio for both oxidizable substrates (Souza et al., 1994). The same effects were found in a later study on the rat brain, with fluoxetine decreasing the rate of ATP synthesis (Curti et al., 1999), and a study on the pig brain, showing that fluoxetine can inhibit mitochondrial function (Hroudová and Fišar, 2012). These results indicate that high doses of fluoxetine have negative effects on mitochondria. Fluoxetine crosses mitochondrial membranes with ease, and it is possible that fluoxetine could interfere with membrane-bound proteins causing pro-apoptotic events (de Oliveira, 2016).
In vivo studies reveal a slightly more complex scenario, in that fluoxetine has both beneficial and detrimental effects on mitochondria when given systemically. After a single injection of fluoxetine (25 mg/kg), activity of the Krebs cycle enzyme citrate synthase was increased in the striatum, but not in the prefrontal cortex or hippocampus, and the striatal increase was no longer evident after 28 days of treatment (Agostinho et al., 2011a). Fluoxetine at the same dose also increased activity of ETC complex I in the hippocampus after one injection, but not in the prefrontal cortex or striatum (Agostinho et al., 2011b). However, after 28 days of daily injections, complex IV activity was decreased in the hippocampus. In another study, 21 days of low dose fluoxetine injections (5 mg/kg) increased expression of cytochrome oxidase 1 and cytochrome oxidase 3 mRNA in the prefrontal cortex in female rats, but not male rats, and decreased cytochrome oxidase 1 and cytochrome oxidase 3 mRNA in the hippocampus of male rats but not female rats (Adzic et al., 2013). These results suggest sex and region specific effects of systemic fluoxetine on mitochondrial function.
There has been some work done to examine the effect of other antidepressants. For example, chronic treatment with the tricyclic antidepressant imipramine as well as electroconvulsive shocks increased levels of cytochrome b mRNA in the rat cortex but not in the hippocampus, cerebellum or liver (Huang et al., 1997). Cytochrome b mRNA translates a protein that is involved in ETC complex III functioning. In addition, the SNRI venlafaxine actually had detrimental effects on complex IV of the ETC, although it increased expression of anti-apoptotic and antioxidant mitochondrial genes (Tamási et al., 2014). Finally, fluoxetine and desipramine enhanced cytochrome oxidase and glutamate dehydrogenase in presynaptic mitochondria located in the rat hippocampus (Villa et al., 2017). These data highlight the importance of antidepressants at a subcellular level and suggest that mitochondrial energy metabolism could be a mechanism of antidepressant drug action.
Gender Differences in Depression and Mitochondria
Women are more than twice as likely to suffer from depression than men, but it is not yet clear why this occurs and whether or not it has a biological basis. There is evidence that gender differences might arise due to decreased levels of circulating estrogens (Bloch et al., 2003), which is reinforced by observations that ovariectomy increases depression-like behavior in mice subjected to a chronic unpredictable stress paradigm (Lagunas et al., 2010). Furthermore, administering estradiol alleviates depression-like symptoms in ovariectomized rats (Rachman et al., 1998) and may accelerate antidepressant effects in humans (Rasgon et al., 2007). The evidence linking mitochondria to estradiol and depression is sparse, but emerging. Some studies have indicated a protective role of estradiol in mitochondria, showing that it can inhibit the passage of ROS into mitochondria as well as preventing mitochondrial collapse and increasing the rate of ATP synthesis (Wang et al., 2003; Shimamoto and Rappeneau, 2017). Mitochondria are known to express estrogen and GRs in lung tissue, suggesting that mitochondria are responsive to fluctuating levels of stress hormones and estradiol (Walf and Frye, 2006). It seems that mitochondrial estrogen and GRs in lung tissue are involved in the biosynthesis of OXPHOS enzymes, which will affect other mitochondrial functions such as apoptosis and ROS production (Simoes et al., 2012). It would be quite interesting to follow these studies with an investigation of brain mitochondria and estrogen receptors to determine whether sex steroid hormones in the brain might be involved in the gender differences seen in the prevalence of depression.
Conclusion
The specific biological mechanisms underlying major depression have yet to be elucidated. This review highlights the potential importance of mitochondrial function in depression. This is an area that has received relatively little experimental attention, but the data that have been published to date are promising and should be pursued. Although one must be cautious in extrapolating findings from preclinical animal models to the human condition, there is evidence that chronic stress-induced inhibition of ETC complexes in the inner membrane of mitochondria is a contributing factor in the pathophysiology of depression. Dysfunctional mitochondria decrease the pool of available ATP, which could have detrimental effects on signal transduction pathways, dampening activity in neuronal circuits, and interfering with mitochondrial fusion and fission. This negative cascade would ultimately increase oxidative stress, inflammatory responses, and pro-apoptotic events, some of which are known to be involved in the pathogenesis of depression. Viewed this way, it seems logical that reversing the early stages of mitochondrial dysfunction could provide a novel target for therapeutic intervention.
Author Contributions
All authors contributed to the writing of this manuscript. JA wrote the first draft. RR-T, KB, HC, and LK edited the draft. JA constructed the figure. LK finalized the manuscript.
Funding
This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) to LK and HC. KB was supported by an NSERC Doctoral Canada Graduate Scholarship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adzic, M., Brkic, Z., Bulajic, S., Mitic, M., and Radojcic, M. B. (2016). Antidepressant action on mitochondrial dysfunction in psychiatric disorders. Drug Dev. Res. 77, 400–406. doi: 10.1002/ddr.21332
Adzic, M., Lukic, I., Mitic, M., Djordjevic, J., Elakovic, I., Djordjevic, A., et al. (2013). Brain region- and sex-specific modulation of mitochondrial glucocorticoid receptor phosphorylation in fluoxetine treated stressed rats: effects on energy metabolism. Psychoneuroendocrinology 38, 2914–2924. doi: 10.1016/j.psyneuen.2013.07.019
Agostinho, F. R., Réus, G. Z., Stringari, R. B., Ribeiro, K. F., Ferraro, A. K., Benedet, J., et al. (2011a). Treatment with olanzapine, fluoxetine and olanzapine/fluoxetine alters citrate synthase activity in rat brain. Neurosci. Lett. 487, 278–281. doi: 10.1016/j.neulet.2010.10.037
Agostinho, F. R., Réus, G. Z., Stringari, R. B., Ribeiro, K. F., Ferreira, G. K., Jeremias, I. C., et al. (2011b). Olanzapine plus fluoxetine treatment alters mitochondrial respiratory chain activity in the rat brain. Acta Neuropsychiatr. 23, 282–291. doi: 10.1111/j.1601-5215.2011.00569.x
Almeida, R. F. D., Ganzella, M., Machado, D. G., Loureiro, S. O., Leffa, D., Quincozes-Santos, A., et al. (2017). Olfactory bulbectomy in mice triggers transient and long-lasting behavioral impairments and biochemical hippocampal disturbances. Prog. Neuropsychopharmacol. Biol. Psychiatry 76, 1–11. doi: 10.1016/j.pnpbp.2017.02.013
Anderson, G. (2018). Linking the biological underpinnings of depression: role of mitochondria interactions with melatonin, inflammation, sirtuins, tryptophan catabolites, DNA repair and oxidative and nitrosative stress, with consequences for classification and cognition. Prog. Neuropsychopharmacol. Biol. Psychiatry 80, 255–266. doi: 10.1016/j.pnpbp.2017.04.022
Bansal, Y., and Kuhad, A. (2016). Mitochondrial dysfunction in depression. Curr. Neuropharmacol. 14, 610–618. doi: 10.2174/1570159X14666160229114755
Baxter, L. R. Jr., Schwartz, J. M., Phelps, M. E., Mazziotta, J. C., Guze, B. H., Selin, C. E., et al. (1989). Reduction of prefrontal cortex glucose metabolism common to three types of depression. Arch. Gen. Psychiatry 46, 243–250. doi: 10.1001/archpsyc.1989.01810030049007
Beasley, C. L., Pennington, K., Behan, A., Wait, R., Dunn, M. J., and Cotter, D. (2006). Proteomic analysis of the anterior cingulate cortex in the major psychiatric disorders: evidence for disease-associated changes. Proteomics 6, 3414–3425. doi: 10.1002/pmic.200500069
Benes, F. M., Matzilevich, D., Burke, R. E., and Walsh, J. (2006). The expression of proapoptosis genes is increased in bipolar disorder, but not in schizophrenia. Mol. Psychiatry 11, 241–251. doi: 10.1038/sj.mp.4001758
Ben-Shachar, D., and Karry, R. (2008). Neuroanatomical pattern of mitochondrial complex I pathology varies between schizophrenia, bipolar disorder and major depression. PLoS One 3:e3676. doi: 10.1371/journal.pone.0003676
Bessa, J. M., Ferreira, D., Melo, I., Marques, F., Cerqueira, J. J., Palha, J. A., et al. (2009). The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol. Psychiatry 14, 764–773, 739. doi: 10.1038/mp.2008.119
Bivona, T. G., Quatela, S. E., Bodemann, B. O., Ahearn, I. M., Soskis, M. J., Mor, A., et al. (2006). PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 21, 481–493. doi: 10.1016/j.molcel.2006.01.012
Bloch, M., Daly, R. C., and Rubinow, D. R. (2003). Endocrine factors in the etiology of postpartum depression. Compr. Psychiatry 44, 234–246. doi: 10.1016/s0010-440x(03)00034-8
Braughler, J. M., and Hall, E. D. (1989). Central nervous system trauma and stroke. I. Biochemical considerations for oxygen radical formation and lipid peroxidation. Free Radic. Biol. Med. 6, 289–301. doi: 10.1016/0891-5849(89)90056-7
Brummelte, S., and Galea, L. A. (2010). Chronic high corticosterone reduces neurogenesis in the dentate gyrus of adult male and female rats. Neuroscience 168, 680–690. doi: 10.1016/j.neuroscience.2010.04.023
Brymer, K. J., Fenton, E. Y., Kalynchuk, L. E., and Caruncho, H. J. (2018). Peripheral etanercept administration normalizes behavior, hippocampal neurogenesis, and hippocampal reelin and GABAA receptor expression in a preclinical model of depression. Front. Pharmacol. 9:121. doi: 10.3389/fphar.2018.00121
Calabrese, V., Scapagnini, G., Giuffrida Stella, A. M., Bates, T. E., and Clark, J. B. (2001). Mitochondrial involvement in brain function and dysfunction: relevance to aging, neurodegenerative disorders and longevity. Neurochem. Res. 26, 739–764. doi: 10.1023/A:1010955807739
Calingasan, N. Y., Ho, D. J., Wille, E. J., Campagna, M. V., Ruan, J., Dumont, M., et al. (2008). Influence of mitochondrial enzyme deficiency on adult neurogenesis in mouse models of neurodegenerative diseases. Neuroscience 153, 986–996. doi: 10.1016/j.neuroscience.2008.02.071
Campbell, S., Marriott, M., Nahmias, C., and MacQueen, G. M. (2004). Lower hippocampal volume in patients suffering from depression: a meta-analysis. Am. J. Psychiatry 161, 598–607. doi: 10.1176/appi.ajp.161.4.598
Carboni, L. (2015). The contribution of proteomic studies in humans, animal models, and after antidepressant treatments to investigate the molecular neurobiology of major depression. Proteomics Clin. Appl. 9, 889–898. doi: 10.1002/prca.201400139
Caruncho, H. J., Brymer, K., Romay-Tallon, R., Mitchell, M. A., Rivera-Baltanas, T., Botterill, J., et al. (2016). Reelin-related disturbances in depression: implications for translational studies. Front. Cell. Neurosci. 10:48. doi: 10.3389/fncel.2016.00048
Cataldo, A. M., McPhie, D. L., Lange, N. T., Punzell, S., Elmiligy, S., Ye, N. Z., et al. (2010). Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am. J. Pathol. 177, 575–585. doi: 10.2353/ajpath.2010.081068
Chan, D. C. (2006). Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 22, 79–99. doi: 10.1146/annurev.cellbio.22.010305.104638
Chang, C. C., Jou, S. H., Lin, T. T., Lai, T. J., and Liu, C. S. (2015). Mitochondria DNA change and oxidative damage in clinically stable patients with major depressive disorder. PLoS One 10:e0125855. doi: 10.1371/journal.pone.0125855
Chen, H., Chomyn, A., and Chan, D. C. (2005). Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 280, 26185–26192. doi: 10.1074/jbc.M503062200
Chen, W. J., Du, J. K., Hu, X., Yu, Q., Li, D. X., Wang, C. N., et al. (2017). Protective effects of resveratrol on mitochondrial function in the hippocampus improves inflammation-induced depressive-like behavior. Physiol. Behav. 182, 54–61. doi: 10.1016/j.physbeh.2017.09.024
Chinnery, P. F., Elliott, C., Green, G. R., Rees, A., Coulthard, A., Turnbull, D. M., et al. (2000). The spectrum of hearing loss due to mitochondrial DNA defects. Brain 123, 82–92. doi: 10.1093/brain/123.1.82
Chipuk, J. E., Bouchier-Hayes, L., and Green, D. R. (2006). Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 13, 1396–1402. doi: 10.1038/sj.cdd.4401963
Chouinard, V.-A., Kim, S.-Y., Valeri, L., Yuksel, C., Ryan, K. P., Chouinard, G., et al. (2017). Brain bioenergetics and redox state measured by 31P magnetic resonance spectroscopy in unaffected siblings of patients with psychotic disorders. Schizophr. Res. 187, 11–16. doi: 10.1016/j.schres.2017.02.024
Cleeter, M. W., Cooper, J. M., Darley-Usmar, V. M., Moncada, S., and Schapira, A. H. (1994). Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 345, 50–54. doi: 10.1016/0014-5793(94)00424-2
Cumurcu, B. E., Ozyurt, H., Etikan, I., Demir, S., and Karlidag, R. (2009). Total antioxidant capacity and total oxidant status in patients with major depression: impact of antidepressant treatment. Psychiatry Clin. Neurosci. 63, 639–645. doi: 10.1111/j.1440-1819.2009.02004.x
Curti, C., Mingatto, F. E., Polizello, A. C., Galastri, L. O., Uyemura, S. A., and Santos, A. C. (1999). Fluoxetine interacts with the lipid bilayer of the inner membrane in isolated rat brain mitochondria, inhibiting electron transport and F1F0-ATPase activity. Mol. Cell. Biochem. 199, 103–109. doi: 10.1023/A:1006912010550
Czarny, P., Kwiatkowski, D., Kacperska, D., Kawczynska, D., Talarowska, M., Orzechowska, A., et al. (2015). Elevated level of DNA damage and impaired repair of oxidative DNA damage in patients with recurrent depressive disorder. Med. Sci. Monit. 21, 412–418. doi: 10.12659/msm.892317
Czarny, P., Wigner, P., Galecki, P., and Sliwinski, T. (2018). The interplay between inflammation, oxidative stress, DNA damage, DNA repair and mitochondrial dysfunction in depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 80, 309–321. doi: 10.1016/j.pnpbp.2017.06.036
Davey, G. P., and Clark, J. B. (1996). Threshold effects and control of oxidative phosphorylation in nonsynaptic rat brain mitochondria. J. Neurochem. 66, 1617–1624. doi: 10.1046/j.1471-4159.1996.66041617.x
David, D. J., Samuels, B. A., Rainer, Q., Wang, J.-W., Marsteller, D., Mendez, I., et al. (2009). Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 62, 479–493. doi: 10.1016/j.neuron.2009.04.017
Davidson, C. S., and Hardison, W. G. M. (1984). Molecular Biology of the Cell. By Bruce Alberts, Dennis Bray, Julian Lewis, Martin Raff, Keith Roberts, and James D. Watson, xxxix + 1146 pp. New York: Garland, 1983. $39.95. Hepatology 4, 348–348. doi: 10.1002/hep.1840040230
De, R., Mazumder, S., Sarkar, S., Debsharma, S., Siddiqui, A. A., Saha, S. J., et al. (2017). Acute mental stress induces mitochondrial bioenergetic crisis and hyper-fission along with aberrant mitophagy in the gut mucosa in rodent model of stress-related mucosal disease. Free Radic. Biol. Med. 113, 424–438. doi: 10.1016/j.freeradbiomed.2017.10.009
de Oliveira, M. R. (2016). Fluoxetine and the mitochondria: a review of the toxicological aspects. Toxicol. Lett. 258, 185–191. doi: 10.1016/j.toxlet.2016.07.001
Devine, M. J., and Kittler, J. T. (2018). Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 19, 63–80. doi: 10.1038/nm.2017.170
Drevets, W. C., Ongur, D., and Price, J. L. (1998). Neuroimaging abnormalities in the subgenual prefrontal cortex: implications for the pathophysiology of familial mood disorders. Mol. Psychiatry 3, 220–226, 190–191. doi: 10.1038/sj.mp.4000380
Drevets, W. C., Price, J. L., Simpson, J. R. Jr., Todd, R. D., Reich, T., Vannier, M., et al. (1997). Subgenual prefrontal cortex abnormalities in mood disorders. Nature 386, 824–827. doi: 10.1038/386824a0
Du, J., Wang, Y., Hunter, R., Wei, Y., Blumenthal, R., Falke, C., et al. (2009). Dynamic regulation of mitochondrial function by glucocorticoids. Proc. Natl. Acad. Sci. U.S.A. 106, 3543–3548. doi: 10.1073/pnas.0812671106
Eren, I., Naziroglu, M., and Demirdas, A. (2007a). Protective effects of lamotrigine, aripiprazole and escitalopram on depression-induced oxidative stress in rat brain. Neurochem. Res. 32, 1188–1195. doi: 10.1007/s11064-007-9289-x
Eren, I., Naziroglu, M., Demirdas, A., Celik, O., Uguz, A. C., Altunbasak, A., et al. (2007b). Venlafaxine modulates depression-induced oxidative stress in brain and medulla of rat. Neurochem. Res. 32, 497–505. doi: 10.1007/s11064-006-9258-9
Eszlari, N., Kovacs, D., Petschner, P., Pap, D., Gonda, X., Elliott, R., et al. (2016). Distinct effects of folate pathway genes MTHFR and MTHFD1L on ruminative response style: a potential risk mechanism for depression. Transl. Psychiatry 6:e745. doi: 10.1038/tp.2016.19
Fattal, O., Budur, K., Vaughan, A. J., and Franco, K. (2006). Review of the Literature on Major Mental Disorders in Adult Patients With Mitochondrial Diseases. Psychosomatics 47, 1–7. doi: 10.1176/appi.psy.47.1.1
Fattal, O., Link, J., Quinn, K., Cohen, B. H., and Franco, K. (2007). Psychiatric comorbidity in 36 adults with mitochondrial cytopathies. CNS Spectr. 12, 429–438. doi: 10.1017/S1092852900015303
Fenton, E. Y., Fournier, N. M., Lussier, A. L., Romay-Tallon, R., Caruncho, H. J., and Kalynchuk, L. E. (2015). Imipramine protects against the deleterious effects of chronic corticosterone on depression-like behavior, hippocampal reelin expression, and neuronal maturation. Prog. Neuro Psychopharmacol. Biol. Psychiatry 60, 52–59. doi: 10.1016/j.pnpbp.2015.02.001
Filiou, M. D., Zhang, Y., Teplytska, L., Reckow, S., Gormanns, P., Maccarrone, G., et al. (2011). Proteomics and metabolomics analysis of a trait anxiety mouse model reveals divergent mitochondrial pathways. Biol. Psychiatry 70, 1074–1082. doi: 10.1016/j.biopsych.2011.06.009
Filipović, D., Costina, V., Perić, I., Stanisavljević, A., and Findeisen, P. (2017). Chronic fluoxetine treatment directs energy metabolism towards the citric acid cycle and oxidative phosphorylation in rat hippocampal nonsynaptic mitochondria. Brain Res. 1659, 41–54. doi: 10.1016/j.brainres.2017.01.025
Frey, B. N., Stanley, J. A., Nery, F. G., Monkul, E. S., Nicoletti, M. A., Chen, H. H., et al. (2007). Abnormal cellular energy and phospholipid metabolism in the left dorsolateral prefrontal cortex of medication-free individuals with bipolar disorder: an in vivo 1H MRS study. Bipolar Disord. 9, 119–127. doi: 10.1111/j.1399-5618.2007.00454.x
Frodl, T., Schaub, A., Banac, S., Charypar, M., Jäger, M., Kümmler, P., et al. (2006). Reduced hippocampal volume correlates with executive dysfunctioning in major depression. J. Psychiatry Neurosci. 31, 316–325.
Gamaro, G. D., Streck, E. L., Matte, C., Prediger, M. E., Wyse, A. T., and Dalmaz, C. (2003). Reduction of hippocampal Na+, K+-ATPase activity in rats subjected to an experimental model of depression. Neurochem. Res. 28, 1339–1344. doi: 10.1023/A:1024988113978
Gao, X., Xu, X., Pang, J., Zhang, C., Ding, J. M., Peng, X., et al. (2007). NMDA receptor activation induces mitochondrial dysfunction, oxidative stress and apoptosis in cultured neonatal rat cardiomyocytes. Physiol. Res. 56, 559–569.
Gardner, A., Johansson, A., Wibom, R., Nennesmo, I., von Döbeln, U., Hagenfeldt, L., et al. (2003). Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J. Affect. Disord. 76, 55–68. doi: 10.1016/S0165-0327(02)00067-8
Gibbons, J. L., and McHugh, P. R. (1962). Plasma cortisol in depressive illness. J. Psychiatr. Res. 1, 162–171. doi: 10.1016/0022-3956(62)90006-7
Głombik, K., Stachowicz, A., Trojan, E., Olszanecki, R., Ślusarczyk, J., Suski, M., et al. (2017). Evaluation of the effectiveness of chronic antidepressant drug treatments in the hippocampal mitochondria – A proteomic study in an animal model of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 78, 51–60. doi: 10.1016/j.pnpbp.2017.05.014
Goemaere, J., and Knoops, B. (2012). Peroxiredoxin distribution in the mouse brain with emphasis on neuronal populations affected in neurodegenerative disorders. J. Comp. Neurol. 520, 258–280. doi: 10.1002/cne.22689
Gong, Y., Chai, Y., Ding, J. H., Sun, X. L., and Hu, G. (2011). Chronic mild stress damages mitochondrial ultrastructure and function in mouse brain. Neurosci. Lett. 488, 76–80. doi: 10.1016/j.neulet.2010.11.006
Green, D., and Kroemer, G. (1998). The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol. 8, 267–271.
Green-Johnson, J. M., Zalcman, S., Vriend, C. Y., Nance, D. M., and Greenberg, H. (1995). Suppressed T cell and macrophage function in the “reeler” (rl/rl) mutant, a murine strain with elevated cerebellar norepinephrine concentration. Brain Behav. Immun. 9, 47–60. doi: 10.1006/brbi.1995.1005
Gregus, A., Wintink, A. J., Davis, A. C., and Kalynchuk, L. E. (2005). Effect of repeated corticosterone injections and restraint stress on anxiety and depression-like behavior in male rats. Behav. Brain Res. 156, 105–114. doi: 10.1016/j.bbr.2004.05.013
Halari, R., Simic, M., Pariante, C. M., Papadopoulos, A., Cleare, A., Brammer, M., et al. (2009). Reduced activation in lateral prefrontal cortex and anterior cingulate during attention and cognitive control functions in medication-naive adolescents with depression compared to controls. J. Child Psychol. Psychiatry 50, 307–316. doi: 10.1111/j.1469-7610.2008.01972.x
Hamon, M., and Blier, P. (2013). Monoamine neurocircuitry in depression and strategies for new treatments. Prog. Neuropsychopharmacol. Biol. Psychiatry 45, 54–63. doi: 10.1016/j.pnpbp.2013.04.009
Herrmann, J. M., and Neupert, W. (2000). Protein transport into mitochondria. Curr. Opin. Microbiol. 3, 210–214. doi: 10.1016/S1369-5274(00)00077-1
Hirschfeld, R. M. (2001). When to hospitalize patients at risk for suicide. Ann. N. Y. Acad. Sci. 932, 188–196; discussion 196–199. doi: 10.1111/j.1749-6632.2001.tb05806.x
Holsboer, F. (2001). Stress, hypercortisolism and corticosteroid receptors in depression: implications for therapy. J. Affect. Disord. 62, 77–91. doi: 10.1016/S0165-0327(00)00352-9
Holzmann, I., da Silva, L. M., Correa da Silva, J. A., Steimbach, V. M., and de Souza, M. M. (2015). Antidepressant-like effect of quercetin in bulbectomized mice and involvement of the antioxidant defenses, and the glutamatergic and oxidonitrergic pathways. Pharmacol. Biochem. Behav. 136, 55–63. doi: 10.1016/j.pbb.2015.07.003
Hroudová, J., and Fišar, Z. (2012). In vitro inhibition of mitochondrial respiratory rate by antidepressants. Toxicol. Lett. 213, 345–352. doi: 10.1016/j.toxlet.2012.07.017
Huang, N.-Y., Strakhova, M., Layer, R. T., and Skolnick, P. (1997). Chronic antidepressant treatments increase cytochrome b mRNA levels in mouse cerebral cortex. J. Mol. Neurosci. 9, 167–176. doi: 10.1007/BF02800499
Inczedy-Farkas, G., Trampush, J. W., Perczel Forintos, D., Beech, D., Andrejkovics, M., Varga, Z., et al. (2014). Mitochondrial DNA mutations and cognition: a case-series report. Arch. Clin. Neuropsychol. 29, 315–321. doi: 10.1093/arclin/acu016
Iwamoto, K., Bundo, M., and Kato, T. (2005). Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum. Mol. Genet. 14, 241–253. doi: 10.1093/hmg/ddi022
Jacobs, B. L., van Praag, H., and Gage, F. H. (2000). Depression and the birth and death of brain cells: the turnover of neurons in the hippocampus might help to explain the onset of and recovery from clinical depression. Am. Sci. 88, 340–345. doi: 10.1511/2000.29.775
Johnson, S. A., Fournier, N. M., and Kalynchuk, L. E. (2006). Effect of different doses of corticosterone on depression-like behavior and HPA axis responses to a novel stressor. Behav. Brain Res. 168, 280–288. doi: 10.1016/j.bbr.2005.11.019
Johnston-Wilson, N. L., Sims, C. D., Hofmann, J. P., Anderson, L., Shore, A. D., Torrey, E. F., et al. (2000). Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley Neuropathology Consortium. Mol. Psychiatry 5, 142–149. doi: 10.1038/sj.mp.4000696
Kaidanovich-Beilin, O., Cha, D. S., and McIntyre, R. S. (2012). Crosstalk between metabolic and neuropsychiatric disorders. F1000 Biol. Rep. 4:14. doi: 10.3410/B4-14
Kalia, M. (2005). Neurobiological basis of depression: an update. Metabolism 54, 24–27. doi: 10.1016/j.metabol.2005.01.009
Karabatsiakis, A., Bock, C., Salinas-Manrique, J., Kolassa, S., Calzia, E., Dietrich, D. E., et al. (2014). Mitochondrial respiration in peripheral blood mononuclear cells correlates with depressive subsymptoms and severity of major depression. Transl. Psychiatry 4:e397. doi: 10.1038/tp.2014.44
Kasahara, T., Kubota, M., Miyauchi, T., Noda, Y., Mouri, A., Nabeshima, T., et al. (2006). Mice with neuron-specific accumulation of mitochondrial DNA mutations show mood disorder-like phenotypes. Mol. Psychiatry 11, 577–593, 523. doi: 10.1038/sj.mp.4001824
Kato, T., and Kato, N. (2000). Mitochondrial dysfunction in bipolar disorder. Bipolar Disord. 2, 180–190. doi: 10.1034/j.1399-5618.2000.020305.x
Kato, T., Kunugi, H., Nanko, S., and Kato, N. (2001). Mitochondrial DNA polymorphisms in bipolar disorder. J. Affect. Disord. 62, 151–164. doi: 10.1016/S0165-0327(99)00173-1
Kato, T., Stine, O. C., McMahon, F. J., and Crowe, R. R. (1997). Increased levels of a mitochondrial DNA deletion in the brain of patients with bipolar disorder. Biol. Psychiatry 42, 871–875. doi: 10.1016/s0006-3223(97)00012-7
Kennedy, S. H., Evans, K. R., Kruger, S., Mayberg, H. S., Meyer, J. H., McCann, S., et al. (2001). Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am. J. Psychiatry 158, 899–905. doi: 10.1176/appi.ajp.158.6.899
Kessler, R. C., McGonagle, K. A., Zhao, S., Nelson, C. B., Hughes, M., Eshleman, S., et al. (1994). Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch. Gen. Psychiatry 51, 8–19. doi: 10.1001/archpsyc.1994.03950010008002
Kessler, R. C., Nelson, C. B., McGonagle, K. A., Liu, J., Swartz, M., and Blazer, D. G. (1996). Comorbidity of DSM-III-R major depressive disorder in the general population: results from the US National Comorbidity Survey. Br. J. Psychiatry Suppl. 30, 17–30.
Kety, S. S. (1950). Blood flow and metabolism of the human brain in health and disease. Trans. Stud. Coll. Physicians Phila. 18, 103–108.
Klinedinst, N. J., and Regenold, W. T. (2015). A mitochondrial bioenergetic basis of depression. J. Bioenerg. Biomembr. 47, 155–171. doi: 10.1007/s10863-014-9584-6
Koene, S., Kozicz, T. L., Rodenburg, R. J., Verhaak, C. M., de Vries, M. C., Wortmann, S., et al. (2009). Major depression in adolescent children consecutively diagnosed with mitochondrial disorder. J. Affect. Disord. 114, 327–332. doi: 10.1016/j.jad.2008.06.023
Konradi, C., Eaton, M., MacDonald, M. L., Walsh, J., Benes, F. M., and Heckers, S. (2004). Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch. Gen. Psychiatry 61, 300–308. doi: 10.1001/archpsyc.61.3.300
Kroemer, G., Galluzzi, L., and Brenner, C. (2007). Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163. doi: 10.1152/physrev.00013.2006
Kumar, A., and Chanana, P. (2017). Role of nitric oxide in stress-induced anxiety: from pathophysiology to therapeutic target. Vitam. Horm. 103, 147–167. doi: 10.1016/bs.vh.2016.09.004
Lagunas, N., Calmarza-Font, I., Diz-Chaves, Y., and Garcia-Segura, L. M. (2010). Long-term ovariectomy enhances anxiety and depressive-like behaviors in mice submitted to chronic unpredictable stress. Horm. Behav. 58, 786–791. doi: 10.1016/j.yhbeh.2010.07.014
Lesch, K. P. (2004). Gene-environment interaction and the genetics of depression. J. Psychiatry Neurosci. 29, 174–184.
Li, Z., Okamoto, K.-I., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Lin, K. M., Chiu, Y. F., Tsai, I. J., Chen, C. H., Shen, W. W., Liu, S. C., et al. (2011). ABCB1 gene polymorphisms are associated with the severity of major depressive disorder and its response to escitalopram treatment. Pharmacogenet. Genomics 21, 163–170. doi: 10.1097/FPC.0b013e32833db216
Lizasoain, I., Moro, M. A., Knowles, R. G., Darley-Usmar, V., and Moncada, S. (1996). Nitric oxide and peroxynitrite exert distinct effects on mitochondrial respiration which are differentially blocked by glutathione or glucose. Biochem. J. 314, 877–880. doi: 10.1042/bj3140877
Lussier, A. L., Caruncho, H. J., and Kalynchuk, L. E. (2009). Repeated exposure to corticosterone, but not restraint, decreases the number of reelin-positive cells in the adult rat hippocampus. Neurosci. Lett. 460, 170–174. doi: 10.1016/j.neulet.2009.05.050
Lussier, A. L., Lebedeva, K., Fenton, E. Y., Guskjolen, A., Caruncho, H. J., and Kalynchuk, L. E. (2013). The progressive development of depression-like behavior in corticosterone-treated rats is paralleled by slowed granule cell maturation and decreased reelin expression in the adult dentate gyrus. Neuropharmacology 71, 174–183. doi: 10.1016/j.neuropharm.2013.04.012
Lussier, A. L., Romay-Tallón, R., Kalynchuk, L. E., and Caruncho, H. J. (2011). Reelin as a putative vulnerability factor for depression: examining the depressogenic effects of repeated corticosterone in heterozygous reeler mice. Neuropharmacology 60, 1064–1074. doi: 10.1016/j.neuropharm.2010.09.007
MacQueen, G. M., Campbell, S., McEwen, B. S., Macdonald, K., Amano, S., Joffe, R. T., et al. (2003). Course of illness, hippocampal function, and hippocampal volume in major depression. Proc. Natl. Acad. Sci. U.S.A. 100, 1387–1392. doi: 10.1073/pnas.0337481100
Madrigal, J. L., Olivenza, R., Moro, M. A., Lizasoain, I., Lorenzo, P., Rodrigo, J., et al. (2001). Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology 24, 420–429. doi: 10.1016/s0893-133x(00)00208-6
Martins-de-Souza, D., Guest, P. C., Harris, L. W., Vanattou-Saifoudine, N., Webster, M. J., Rahmoune, H., et al. (2012). Identification of proteomic signatures associated with depression and psychotic depression in post-mortem brains from major depression patients. Transl. Psychiatry 2:e87. doi: 10.1038/tp.2012.13
Martins-de-Souza, D., Harris, L. W., Guest, P. C., and Bahn, S. (2011). The role of energy metabolism dysfunction and oxidative stress in schizophrenia revealed by proteomics. Antioxid. Redox Signal. 15, 2067–2079. doi: 10.1089/ars.2010.3459
Mayberg, H. S., Liotti, M., Brannan, S. K., McGinnis, S., Mahurin, R. K., Jerabek, P. A., et al. (1999). Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am. J. Psychiatry 156, 675–682. doi: 10.1176/ajp.156.5.675
McBride, H. M., Neuspiel, M., and Wasiak, S. (2006). Mitochondria: more than just a powerhouse. Curr. Biol. 16, R551–R560. doi: 10.1016/j.cub.2006.06.054
Morava, E., Gardeitchik, T., Kozicz, T., de Boer, L., Koene, S., de Vries, M. C., et al. (2010). Depressive behaviour in children diagnosed with a mitochondrial disorder. Mitochondrion 10, 528–533. doi: 10.1016/j.mito.2010.05.011
Moretti, A., Gorini, A., and Villa, R. F. (2003). Affective disorders, antidepressant drugs and brain metabolism. Mol. Psychiatry 8, 773–785. doi: 10.1038/sj.mp.4001353
Moustafa, A. A., Hewedi, D. H., Eissa, A. M., Frydecka, D., and Misiak, B. (2014). Homocysteine levels in schizophrenia and affective disorders-focus on cognition. Front. Behav. Neurosci. 8:343. doi: 10.3389/fnbeh.2014.00343
Munakata, K., Fujii, K., Nanko, S., Kunugi, H., and Kato, T. (2007). Sequence and functional analyses of mtDNA in a maternally inherited family with bipolar disorder and depression. Mutat. Res. 617, 119–124. doi: 10.1016/j.mrfmmm.2007.01.006
Munakata, K., Iwamoto, K., Bundo, M., and Kato, T. (2005). Mitochondrial DNA 3243A< G mutation and increased expression of LARS2 gene in the brains of patients with bipolar disorder and schizophrenia. Biol. Psychiatry 57, 525–532. doi: 10.1016/j.biopsych.2004.11.041
Murphy, M. P. (2012). Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 16, 476–495. doi: 10.1089/ars.2011.4289
Nemeroff, C. B. (1998). The neurobiology of depression. Sci. Am. 278, 42–49. doi: 10.1038/scientificamerican0698-42
Nestler, E. J., Barrot, M., DiLeone, R. J., Eisch, A. J., Gold, S. J., and Monteggia, L. M. (2002). Neurobiology of depression. Neuron 34, 13–25. doi: 10.1016/S0896-6273(02)00653-0
Onishi, H., Kawanishi, C., Iwasawa, T., Osaka, H., Hanihara, T., Inoue, K., et al. (1997). Depressive disorder due to mitochondrial transfer RNALeu(UUR) mutation. Biol. Psychiatry 41, 1137–1139. doi: 10.1016/S0006-3223(97)00005-X
Parker, K. J., Schatzberg, A. F., and Lyons, D. M. (2003). Neuroendocrine aspects of hypercortisolism in major depression. Horm. Behav. 43, 60–66. doi: 10.1016/S0018-506X(02)00016-8
Perez, J., Tardito, D., Mori, S., Racagni, G., Smeraldi, E., and Zanardi, R. (2000). Abnormalities of cAMP signaling in affective disorders: implications for pathophysiology and treatment. Bipolar Disord. 2, 27–36. doi: 10.1034/j.1399-5618.2000.020104.x
Petschner, P., Gonda, X., Baksa, D., Eszlari, N., Trivaks, M., Juhasz, G., et al. (2018). Genes linking mitochondrial function, cognitive impairment and depression are associated with endophenotypes serving precision medicine. Neuroscience 370, 207–217. doi: 10.1016/j.neuroscience.2017.09.049
Popoli, M., Brunello, N., Perez, J., and Racagni, G. (2000). Second messenger-regulated protein kinases in the brain: their functional role and the action of antidepressant drugs. J. Neurochem. 74, 21–33. doi: 10.1046/j.1471-4159.2000.0740021.x
Pujadas, L., Gruart, A., Bosch, C., Delgado, L., Teixeira, C. M., Rossi, D., et al. (2010). Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J. Neurosci. 30, 4636–4649. doi: 10.1523/JNEUROSCI.5284-09.2010
Quintanilla, R. A., Tapia, C., and Perez, M. J. (2017). Possible role of mitochondrial permeability transition pore in the pathogenesis of Huntington disease. Biochem. Biophys. Res. Commun. 483, 1078–1083. doi: 10.1016/j.bbrc.2016.09.054
Rachman, I. M., Unnerstall, J. R., Pfaff, D. W., and Cohen, R. S. (1998). Estrogen alters behavior and forebrain c-fos expression in ovariectomized rats subjected to the forced swim test. Proc. Natl. Acad. Sci. U.S.A. 95, 13941–13946. doi: 10.1073/pnas.95.23.13941
Rasgon, N. L., Dunkin, J., Fairbanks, L., Altshuler, L. L., Troung, C., Elman, S., et al. (2007). Estrogen and response to sertraline in postmenopausal women with major depressive disorder: a pilot study. J. Psychiatr. Res. 41, 338–343. doi: 10.1016/j.jpsychires.2006.03.009
Rezin, G. T., Amboni, G., Zugno, A. I., Quevedo, J., and Streck, E. L. (2008a). Mitochondrial dysfunction and psychiatric disorders. Neurochem. Res. 34, 1021–1029. doi: 10.1007/s11064-008-9865-8
Rezin, G. T., Cardoso, M. R., Gonçalves, C. L., Scaini, G., Fraga, D. B., Riegel, R. E., et al. (2008b). Inhibition of mitochondrial respiratory chain in brain of rats subjected to an experimental model of depression. Neurochem. Int. 53, 395–400. doi: 10.1016/j.neuint.2008.09.012
Rivera-Baltanas, T., Agis-Balboa, R. C., Romay-Tallón, R., Kalynchuk, L. E., Olivares, J. M., and Caruncho, H. J. (2015). Serotonin transporter clustering in blood lymphocytes predicts the outcome on anhedonia scores in naïve depressive patients treated with antidepressant medication. Ann. Gen. Psychiatry 14:45. doi: 10.1186/s12991-015-0085-8
Rivera-Baltanas, T., Olivares, J. M., Calado-Otero, M., Kalynchuk, L. E., Martinez- Villamarin, J. R., and Caruncho, H. J. (2012). Serotonin transporter clustering in blood lymphocytes as a putative biomarker of therapeutic efficacy in major depressive disorder. J. Affect. Disord. 137, 46–55. doi: 10.1016/j.jad.2011.12.041
Rivera-Baltanas, T., Olivares, J. M., Martinez-Villamarin, J. R., Fenton, E. Y., Kalynchuk, L. E., and Caruncho, H. J. (2014). Serotonin 2A receptor clustering in peripheral lymphocytes is altered in major depression and may be a biomarker of therapeutic efficacy. J. Affect. Disord. 163, 47–55. doi: 10.1016/j.jad.2014.03.011
Rivera-Baltanás, T., Romay-Tallón, R., Dopeso-Reyes, I. G., and Caruncho, H. J. (2010). Serotonin transporter clustering in blood lymphocytes of reeler mice. Cardiovasc. Psychiatry Neurol. 2010:396282. doi: 10.1155/2010/396282
Romay-Tallon, R., Dopeso-Reyes, I. G., Lussier, A. L., Kalynchuk, L. E., and Caruncho, H. J. (2010). The coexpression of reelin and neuronal nitric oxide synthase in a subpopulation of dentate gyrus neurons is downregulated in heterozygous reeler mice. Neural Plast. 2010:130429. doi: 10.1155/2010/130429
Romay-Tallon, R., Rivera-Baltanas, T., Kalynchuk, L. E., and Caruncho, H. J. (2015). Differential effects of corticosterone on the colocalization of reelin and neuronal nitric oxide synthase in the adult hippocampus in wild type and heterozygous reeler mice. Brain Res. 1594, 274–283. doi: 10.1016/j.brainres.2014.10.050
Rose, S., Frye, R. E., Slattery, J., Wynne, R., Tippett, M., Pavliv, O., et al. (2014). Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS One 9:e85436. doi: 10.1371/journal.pone.0085436
Saia-Cereda, V. M., Cassoli, J. S., Martins-de-Souza, D., and Nascimento, J. M. (2017). Psychiatric disorders biochemical pathways unraveled by human brain proteomics. Eur. Arch. Psychiatry Clin. Neurosci. 267, 3–17. doi: 10.1007/s00406-016-0709-2
Samavati, L., Lee, I., Mathes, I., Lottspeich, F., and Huttemann, M. (2008). Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J. Biol. Chem. 283, 21134–21144. doi: 10.1074/jbc.M801954200
Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., et al. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301, 805–809. doi: 10.1126/science.1083328
Schildkraut, J. J. (1965). The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am. J. Psychiatry 122, 509–522. doi: 10.1176/ajp.122.5.509
Schoenfeld, T. J., and Cameron, H. A. (2015). Adult neurogenesis and mental illness. Neuropsychopharmacology 40, 113–128. doi: 10.1038/npp.2014.230
Shariq, A. S., Brietzke, E., Rosenblat, J. D., Barendra, V., Pan, Z., and McIntyre, R. S. (2018). Targeting cytokines in reduction of depressive symptoms: a comprehensive review. Prog. Neuropsychopharmacol. Biol. Psychiatry 83, 86–91. doi: 10.1016/j.pnpbp.2018.01.003
Sharpley, M. S., Marciniak, C., Eckel-Mahan, K., McManus, M., Crimi, M., Waymire, K., et al. (2012). Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 151, 333–343. doi: 10.1016/j.cell.2012.09.004
Sheline, Y. I., Gado, M. H., and Kraemer, H. C. (2003). Untreated depression and hippocampal volume loss. Am. J. Psychiatry 160, 1516–1518. doi: 10.1176/appi.ajp.160.8.1516
Shimamoto, A., and Rappeneau, V. (2017). Sex-dependent mental illnesses and mitochondria. Schizophr. Res. 187, 38–46. doi: 10.1016/j.schres.2017.02.025
Simoes, D. C., Psarra, A. M., Mauad, T., Pantou, I., Roussos, C., Sekeris, C. E., et al. (2012). Glucocorticoid and estrogen receptors are reduced in mitochondria of lung epithelial cells in asthma. PLoS One 7:e39183. doi: 10.1371/journal.pone.0039183
Souza, M. E. J., Polizello, A. C. M., Uyemura, S. A., Castro-Silva, O., and Curti, C. (1994). Effect of fluoxetine on rat liver mitochondria. Biochem. Pharmacol. 48, 535–541. doi: 10.1016/0006-2952(94)90283-6
Sterner, E. Y., and Kalynchuk, L. E. (2010). Behavioral and neurobiological consequences of prolonged glucocorticoid exposure in rats: relevance to depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 777–790. doi: 10.1016/j.pnpbp.2010.03.005
Suomalainen, A., Majander, A., Haltia, M., Somer, H., Lönnqvist, J., Savontaus, M. L., et al. (1992). Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. J. Clin. Invest. 90, 61–66. doi: 10.1172/JCI115856
Surget, A., Saxe, M., Leman, S., Ibarguen-Vargas, Y., Chalon, S., Griebel, G., et al. (2008). Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol. Psychiatry 64, 293–301. doi: 10.1016/j.biopsych.2008.02.022
Szuster-Ciesielska, A., Slotwinska, M., Stachura, A., Marmurowska-Michalowska, H., Dubas-Slemp, H., Bojarska-Junak, A., et al. (2008). Accelerated apoptosis of blood leucocytes and oxidative stress in blood of patients with major depression. Prog. Neruophychopharmacol. Biol. Psychiatry 32, 686–694. doi: 10.1016/j.pnpbp.2007.11.012
Tamási, V., Petschner, P., Adori, C., Kirilly, E., Ando, R. D., Tothfalusi, L., et al. (2014). Transcriptional evidence for the role of chronic venlafaxine treatment in neurotrophic signaling and neuroplasticity including also glutatmatergic- and insulin-mediated neuronal processes. PLoS One 9:e113662. doi: 10.1371/journal.pone.0113662
Tobe, E. H. (2013). Mitochondrial dysfunction, oxidative stress, and major depressive disorder. Neuropsychiatr. Dis. Treat. 9, 567–573. doi: 10.2147/ndt.s44282
Tramutola, A., Lanzillotta, C., Perluigi, M., and Butterfield, D. A. (2017). Oxidative stress, protein modification and Alzheimer disease. Brain Res. Bull. 133, 88–96. doi: 10.1016/j.brainresbull.2016.06.005
Trivedi, M. H., Rush, A. J., Wisniewski, S. R., Nierenberg, A. A., Warden, D., Ritz, L., et al. (2006). Evaluation of outcomes with citalopram for depression using measurement-based care in STAR∗D: implications for clinical practice. Am. J. Psychiatry 163, 28–40. doi: 10.1176/appi.ajp.163.1.28
Vakifahmetoglu-Norberg, H., Ouchida, A. T., and Norberg, E. (2017). The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 482, 426–431. doi: 10.1016/j.bbrc.2016.11.088
Vayssiere, J. L., Cordeau-Lossouarn, L., Larcher, J. C., Basseville, M., Gros, F., and Croizat, B. (1992). Participation of the mitochondrial genome in the differentiation of neuroblastoma cells. In Vitro Cell. Dev. Biol. 28A, 763–772. doi: 10.1007/BF02631065
Videbech, P. (2000). PET measurements of brain glucose metabolism and blood flow in major depressive disorder: a critical review. Acta Psychiatr. Scand. 101, 11–20. doi: 10.1034/j.1600-0447.2000.101001011.x
Villa, R. F., Ferrari, F., Bagini, L., Gorini, A., Brunello, N., and Tascedda, F. (2017). Mitochondrial energy metabolism of rat hippocampus after treatment with the antidepressants desipramine and fluoxetine. Neuropharmacology 121, 30–38. doi: 10.1016/j.neuropharm.2017.04.025
Vos, M., Lauwers, E., and Verstreken, P. (2010). Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic Neurosci. 2:139. doi: 10.3389/fnsyn.2010.00139
Walf, A. A., and Frye, C. A. (2006). A review and update of mechanisms of estrogen in the hippocampus and amygdala for anxiety and depression behavior. Neuropsychopharmacology 31, 1097–1111. doi: 10.1038/sj.npp.1301067
Wang, J., Hodes, G. E., Zhang, H., Zhang, S., Zhao, W., and Golden, S. A. (2018). Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat. Commun. 9:477. doi: 10.1038/s41467-017-02794-5
Wang, Q., and Dwivedi, Y. (2017). Transcriptional profiling of mitochondria associated genes in prefrontal cortex of subjects with major depressive disorder. World J. Biol. Psychiatry 18, 592–603. doi: 10.1080/15622975.2016.1197423
Wang, X., Simpkins, J. W., Dykens, J. A., and Cammarata, P. R. (2003). Oxidative damage to human lens epithelial cells in culture: estrogen protection of mitochondrial potential, ATP, and cell viability. Invest. Ophthalmol. Vis. Sci. 44, 2067–2075. doi: 10.1167/iovs.02-0841
Weidinger, A., Moldzio, R., Törö, G., Mkrtchyan, G., Dumitrescu, S., Trofimova, L., et al. (2017). 94 - glutamate excitotoxicity induced by nitric oxide mediated dysfunction of the mitochondrial 2-oxoglutarate dehydrogenase complex. Free Radic. Biol. Med. 112:74. doi: 10.1016/j.freeradbiomed.2017.10.107
Wen, L., Jin, Y., Li, L., Sun, S., Cheng, S., Zhang, S., et al. (2014). Exercise prevents raphe nucleus mitochondrial overactivity in a rat depression model. Physiol. Behav. 132, 57–65. doi: 10.1016/j.physbeh.2014.04.050
Xie, X., Chen, Y., Ma, L., Shen, Q., Huang, L., Zhao, B., et al. (2017). Major depressive disorder mediates accelerated aging in rats subjected to chronic mild stress. Behav. Brain Res. 329, 96–103. doi: 10.1016/j.bbr.2017.04.022
Youle, R. J., and van der Bliek, A. M. (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. doi: 10.1126/science.1219855
Zhang, D., Wen, X. S., Wang, X. Y., Shi, M., and Zhao, Y. (2009). Antidepressant effect of Shudihuang on mice exposed to unpredictable chronic mild stress. J. Ethnopharmacol. 123, 55–60. doi: 10.1016/j.jep.2009.02.029
Zhu, X. H., Qiao, H., Du, F., Xiong, Q., Liu, X., Zhang, X., et al. (2012). Quantitative imaging of energy expenditure in human brain. Neuroimage 60, 2107–2117. doi: 10.1016/j.neuroimage.2012.02.013
Zubenko, G. S., Hughes, H. B. III, Jordan, R. M., Lyons-Weiler, J., and Cohen, B. M. (2014). Differential hippocampal gene expression and pathway analysis in an etiology-based mouse model of major depressive disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 165B, 457–466. doi: 10.1002/ajmg.b.32257
Keywords: depression, behavior, reelin, mitochondria, oxidative phosphorylation, antidepressants
Citation: Allen J, Romay-Tallon R, Brymer KJ, Caruncho HJ and Kalynchuk LE (2018) Mitochondria and Mood: Mitochondrial Dysfunction as a Key Player in the Manifestation of Depression. Front. Neurosci. 12:386. doi: 10.3389/fnins.2018.00386
Received: 27 February 2018; Accepted: 22 May 2018;
Published: 06 June 2018.
Edited by:
Victor Tapias, Cornell University, United StatesReviewed by:
Carmen Sandi, École Polytechnique Fédérale de Lausanne, SwitzerlandSandeep Kumar Barodia, The University of Alabama at Birmingham, United States
Copyright © 2018 Allen, Romay-Tallon, Brymer, Caruncho and Kalynchuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa E. Kalynchuk, lkalynchuk@uvic.ca