 Víctor Fernández-Dueñas
Víctor Fernández-Dueñas Andrea Pérez-Arévalo1,2
Andrea Pérez-Arévalo1,2 Xavier Altafaj
Xavier Altafaj Sergi Ferré
Sergi Ferré Francisco Ciruela
Francisco Ciruela
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Neurosci. , 22 November 2017
Sec. Neurodegeneration
Volume 11 - 2017 | https://doi.org/10.3389/fnins.2017.00652
This article is part of the Research Topic Adenosine Receptors and Neurodegeneration View all 8 articles
Parkinson's disease (PD) is a progressive, neurodegenerative disorder that affects ~1% of individuals over the age of 60, which turns to 5% in subjects up to 85 years (de Lau and Breteler, 2006). On the other hand, a form of PD, called early-onset PD (EOPD), arises at an earlier age (<45; Bonifati et al., 2005; Ylikotila et al., 2015). EOPD patients generally display a slower progression of the disease and present a better response to dopaminergic treatments; however, they may finally develop a full PD symptomatology (i.e., bradykinesia, resting tremor, muscular rigidity and postural instability, drug-induced dyskinesia; Olgiati et al., 2016). The etiology of both PD and EOPD is still not completely elucidated. Thus, although genetic studies have provided some information about the main genes involved, epidemiological data showed that behavioral and environmental factors play a key role in the pathogenesis and progression of PD (Puschmann, 2013; Ascherio and Schwarzschild, 2016). Importantly, the contribution of genetic causes in EOPD has been extensively studied. For instance, mutations in PD-associated genes, such as PRKN (PARK2; MIM number 600116), PINK1 (PARK5; MIM number 605909), and DJ-1 (PARK7; MIM number 602533), have often been associated to autosomal-recessive forms of EOPD (Lücking et al., 2000; Bonifati et al., 2005; Olgiati et al., 2016). Recently, an Iranian research group described a new autosomal-recessive mutation in two siblings (30 and 34 years old) with consanguine parents, which was associated to EOPD (Jaberi et al., 2016). Interestingly, while both brothers did not present alterations in the main PD-related genes (i.e., PRKN, PINK1, and DJ-1), a homozygous missense mutation (c.835G > A) in the adenosine A1 receptor (A1R) gene (ADORA1) was found (Jaberi et al., 2016). This nucleotide point mutation in ADORA1 involves the substitution of a highly conserved amino acid (p.Gly279Ser) within the transmembrane 7 (TM7) domain, but the functional consequences remain unknown. In contrast, it was recently determined that mutations affecting ADORA1 gene and more particularly the missense matution ADORA1 (p.G279S), are not a common risk factor for PD in the European population, arguing against ADORA1 as a candidate gene in PD (Blauwendraat et al., 2017). Altogether, these opposing data indicate that additional work must be done toward the elucidation of the potential contribution of ADORA1 mutations in PD pathogenesis, and the contribution of genetic and environmental factors.
A1R has a widespread distribution in the brain, with the highest levels detected in the cortex, hippocampus, and cerebellum (Sebastião and Ribeiro, 2009). In addition, A1R is markedly expressed in the basal ganglia. Thus, A1R can be found in the major striatal neuronal population, the GABAergic medium-sized spiny neurons (MSNs; Ferré et al., 1996), together with the expression in the cortico-thalamic glutamatergic afferent fibers. These fibers, together with the dopaminergic projections from the substantia nigra pars compacta control the striatal circuitry that are critical in the control of the motor function (Sebastião and Ribeiro, 2009). The selective death of dopaminergic fibers is the primary cause and a hallmark of PD; however, the dysregulation of cortico-thalamic glutamatergic signaling is also involved in the progression of the disease (Fredholm et al., 2005; Gomes et al., 2011). Under physiological conditions, GABAergic MSNs are continuously activated by cortico-thalamic glutamatergic terminals, but a complex array of presynaptic receptors, which include A1R, adenosine A2A receptor (A2AR), cannabinoid CB1 receptor (CB1R) and dopamine D2 and D4 receptors (D2R and D4R, respectively) modulate this tonic stimulation (Ciruela et al., 2006a; González et al., 2012; Mathur and Lovinger, 2012; Ferreira et al., 2015; Bonaventura et al., 2017). Potentially, the dysregulation of these presynaptic modulatory receptors can lead to abnormal glutamate release in the synaptic cleft, which may over activate postsynaptic glutamate receptors, trigger excitotoxicity and, ultimately, lead to neurodegenerative processes affecting brain circuits involved in the control of motor function (Gomes et al., 2011).
Interestingly, A1R colocalizes and interacts with A2AR at the presynaptic membrane of cortico-thalamic glutamatergic terminals, forming functional receptor heteromers in the striatum (Figure 1; Ciruela et al., 2006a). Importantly, the striatal A1R/A2AR heteromer plays a pivotal role controlling glutamate release, thus acting as an adenosine concentration-dependent switch (Ciruela et al., 2006b; Figure 1). Hence, low to moderate extracellular adenosine concentrations (homeostatic basal levels) mostly stimulate A1R, since it displays higher affinity for adenosine compared to A2AR, and a net inhibition of glutamate release is achieved (Figure 1). Conversely, moderate to high concentrations of striatal adenosine, which should theoretically trigger, in theory, both A1R and A2AR activation, ultimately lead to a predominant A2AR activation. In such way, A2AR may block heteromeric A1R through a receptor-receptor allosteric trans-inhibition, thus leading to a predominant facilitation of glutamate release (Figure 1; Ciruela et al., 2006b). At this point, the question consists of whether the ADORA1 (p.G279S) mutation abolishes A1R function and whether this alteration depends on its heteromerization with A2AR receptor, specifically disrupting the function of the adenosine concentration-dependent switch. In the absence of experimental data, we can speculate that the mutation can be affecting the A1R/A2AR heteromer, resulting in a potential alteration of the fine-tuning modulation of striatal glutamatergic neurotransmission. Indeed, in such scenario, we can hypothesize that moderate concentrations of striatal adenosine would facilitate glutamate release and reduce the excitotoxicity threshold (Figure 1).
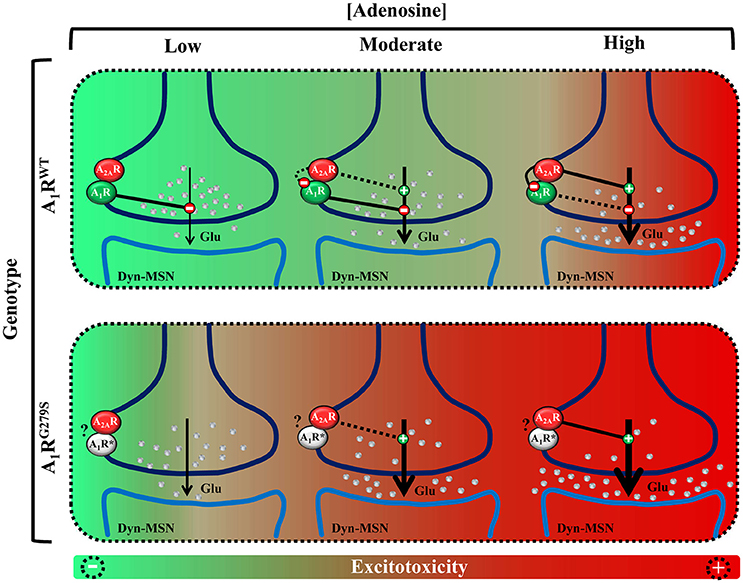
Figure 1. Schematic representation of the potential impact of A1R mutation in the fine-tuning modulation of striatal glutamatergic neurotransmission. (Up) Model of glutamate release control by the A1R/A2AR heteromer adenosine concentration-dependent switch. Low to moderate concentrations of adenosine activate predominantly A1R, inhibiting glutamate release. Moderate to high concentrations of adenosine also activate A2AR which, by means of the A1R–A2AR intramembrane interaction, antagonizes A1R function, therefore facilitating glutamate release. (Bottom) Model of ADORA1(p.G279S) mutation pathogenic impact (A1RG279S or A1R*) in the striatal glutamatergic neurotransmission. The proposed A1R mutant loss-of-function would implicate a dysregulation of the adenosinergic presynaptic control of striatal glutamate release, which may ultimately lead to a higher risk of inducing excitotoxicity and neurodegeneration.
The dysregulation of this presynaptic module may lead to uncontrolled glutamate release which, in addition, might be potentiated by low dopamine innervation, which would not act upon inhibitory presynaptic D2R and D4R. Consequently, managing the disturbance of the adenosine switch mechanism regulating glutamatergic striatal innervation (caused either by a direct ADORA1 mutation or mutations affecting A1R/A2AR heteromers function), may help to restore the normal functioning of the basal ganglia. In this sense, the A1R/A2AR heteromer could be considered as a potential therapeutic target for EOPD. Alternatively, the glutamatergic component of these forms of EOPD would represent an initial or master pathogenic event to dopamine denervation, as proposed in Hungtinton's disease pathophysiology (Gomes et al., 2011). In such way, the predominant role of an aberrant glutamatergic signaling could explain at the molecular level the high effectiveness of PD dopamine-based therapies, either in terms of higher or long-lasting efficacy. Nevertheless, in order to restore physiological neurotransmission it would be necessary to focus not exclusively on dopamine availability, but also in the control of glutamate release which is partially modulated by the A1R/A2AR oligomer (i.e., A1R activation and A2AR inhibition). In this sense, the use of A2AR antagonists has been assessed for the treatment of PD (Vallano et al., 2011). Regarding the potential use of A1R-based therapies, there is a major hurdle related to the A1R ubiquitous expression pattern that might lead to deleterious side-effects. In order to bypass these limitations, novel approaches based on (i) local A1R activation or (ii) pharmacological increase of the adenosine tone (below the threshold of A2AR activation) using adenosine transporters blockers and/or metabolizing enzymes, are expected to reach an effective treatment for EOPD.
Overall, the discovery of a novel mutation in ADORA1 presumably leading to EOPD supports the potential beneficial use of a multimodal approach for the pharmacological treatment of this neurodegenerative condition. This approach, based on the combination of pharmacological therapies (i.e., dopaminergic compounds and drugs targeting the A1R/A2AR oligomer) could be potentially extended to all forms of PD.
VF-D, XA, SF: wrote the paper; AP-A: conceived the idea; FC: conceived the idea and wrote the paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by MINECO/ISCIII (SAF2014-55700-P, PIE14/00034, and PS16/00851), IWT (SBO-140028), and Fundació la Marató de TV3 (Grant 20152031 and Grant 20140210). FC, XA, AP-A, and VF-D belong to the “Neuropharmacology and Pain” accredited research group (Generalitat de Catalunya, 2014 SGR 1251), and by the intramural funds of the National Institute on Drug Abuse to SF.
Ascherio, A., and Schwarzschild, M. A. (2016). The epidemiology of Parkinson's disease: risk factors and prevention. Lancet Neurol. 15, 1257–1272. doi: 10.1016/S1474-4422(16)30230-7
Blauwendraat, C., Nalls, M. A., Federoff, M., Pletnikova, O., Ding, J., Letson, C., et al. (2017). ADORA1 mutations are not a common cause of Parkinson's disease and dementia with Lewy bodies. Mov. Dis. 32, 298–299. doi: 10.1002/mds.26886
Bonaventura, J., Quiroz, C., Cai, N.-S., Rubinstein, M., Tanda, G., and Ferré, S. (2017). Key role of the dopamine D4 receptor in the modulation of corticostriatal glutamatergic neurotransmission. Sci. Adv. 3:e1601631. doi: 10.1126/sciadv.1601631
Bonifati, V., Rohé, C. F., Breedveld, G. J., Fabrizio, E., De Mari, M., Tassorelli, C., et al. (2005). Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology 65, 87–95. doi: 10.1212/01.wnl.0000167546.39375.82
Ciruela, F., Casadó, V., Rodrigues, R. J., Luján, R., Burgueño, J., Canals, M., et al. (2006a). Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J. Neurosci. 26, 2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006
Ciruela, F., Ferré, S., Casadó, V., Cortés, A., Cunha, R. A., Lluis, C., et al. (2006b). Heterodimeric adenosine receptors: a device to regulate neurotransmitter release. Cell. Mol. Life Sci. 63, 2427–2431. doi: 10.1007/s00018-006-6216-2
de Lau, L. M. L., and Breteler, M. M. B. (2006). Epidemiology of Parkinson's disease. Lancet Neurol. 5, 525–535. doi: 10.1016/S1474-4422(06)70471-9
Ferré, S., O'Connor, W. T., Svenningsson, P., Bjorklund, L., Lindberg, J., Tinner, B., et al. (1996). Dopamine D1 receptor-mediated facilitation of GABAergic neurotransmission in the rat strioentopenduncular pathway and its modulation by adenosine A1 receptor-mediated mechanisms. Eur. J. Neurosci. 8, 1545–1553. doi: 10.1111/j.1460-9568.1996.tb01617.x
Ferreira, S. G., Gonçalves, F. Q., Marques, J. M., Tomé, Â. R., Rodrigues, R. J., Nunes-Correia, I., et al. (2015). Presynaptic adenosine A2A receptors dampen cannabinoid CB1 receptor-mediated inhibition of corticostriatal glutamatergic transmission. Br. J. Pharmacol. 172, 1074–1086. doi: 10.1111/bph.12970
Fredholm, B. B., Chen, J. F., Cunha, R. A., Svenningsson, P., and Vaugeois, J. M. (2005). Adenosine and brain function. Int. Rev. Neurobiol. 63, 191–270. doi: 10.1016/S0074-7742(05)63007-3
Gomes, C. V., Kaster, M. P., Tomé, A. R., Agostinho, P. M., and Cunha, R. A. (2011). Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim. Biophys. Acta 1808, 1380–1399. doi: 10.1016/j.bbamem.2010.12.001
González, S., Moreno-Delgado, D., Moreno, E., Pérez-Capote, K., Franco, R., Mallol, J., et al. (2012). Circadian-related heteromerization of adrenergic and dopamine D4 receptors modulates melatonin synthesis and release in the pineal gland. PLoS Biol. 10:e1001347. doi: 10.1371/journal.pbio.1001347
Jaberi, E., Rohani, M., Shahidi, G. A., Nafissi, S., Arefian, E., Soleimani, M., et al. (2016). Mutation in ADORA1 identified as likely cause of early-onset parkinsonism and cognitive dysfunction. Mov. Dis. 31, 1004–1011. doi: 10.1002/mds.26627
Lücking, C. B., Dürr, A., Bonifati, V., Vaughan, J., De Michele, G., Gasser, T., et al. (2000). Association between early-onset Parkinson's disease and mutations in the parkin gene. N. Engl. J. Med. 342, 1560–1567. doi: 10.1056/NEJM200005253422103
Mathur, B. N., and Lovinger, D. M. (2012). Endocannabinoid–dopamine interactions in striatal synaptic plasticity. Front. Pharmacol. 3:66. doi: 10.3389/fphar.2012.00066
Olgiati, S., Quadri, M., Fang, M., Rood, J. P. M. A., Saute, J. A., Chien, H. F., et al. (2016). DNAJC6 mutations associated with early-onset Parkinson's disease. Ann. Neurol. 79, 244–256. doi: 10.1002/ana.24553
Puschmann, A. (2013). Monogenic Parkinson's disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat. Disord. 19, 407–415. doi: 10.1016/j.parkreldis.2013.01.020
Sebastião, A. M., and Ribeiro, J. A. (2009). Adenosine receptors and the central nervous system. Handb. Exp. Pharmacol. 193, 471–534. doi: 10.1007/978-3-540-89615-9_16
Vallano, A., Fernandez-Duenas, V., Pedros, C., Arnau, J. M., and Ciruela, F. (2011). An update on adenosine A2A receptors as drug target in Parkinson's disease. CNS Neurol. Disord. Drug Targets 10, 659–669. doi: 10.2174/187152711797247803
Keywords: early-onset Parkinson's disease, adenosine A1 receptor, oligomer
Citation: Fernández-Dueñas V, Pérez-Arévalo A, Altafaj X, Ferré S and Ciruela F (2017) Adenosine A1-A2A Receptor Heteromer as a Possible Target for Early-Onset Parkinson's Disease. Front. Neurosci. 11:652. doi: 10.3389/fnins.2017.00652
Received: 04 October 2017; Accepted: 09 November 2017;
Published: 22 November 2017.
Edited by:
Manuella P. Kaster, Universidade Federal de Santa Catarina, BrazilReviewed by:
Maria José Diógenes, Faculdade de Medicina da Universidade de Lisboa, PortugalCopyright © 2017 Fernández-Dueñas, Pérez-Arévalo, Altafaj, Ferré and Ciruela. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francisco Ciruela, ZmNpcnVlbGFAdWIuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.