 Yusuke Fukuda
Yusuke Fukuda Yihang Li
Yihang Li Rosalind A. Segal
Rosalind A. Segal- 1Department of Neurobiology, Harvard Medical School, Boston, MA, United States
- 2Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA, United States
Chemotherapeutic agents cause many short and long term toxic side effects to peripheral nervous system (PNS) that drastically alter quality of life. Chemotherapy-induced peripheral neuropathy (CIPN) is a common and enduring disorder caused by several anti-neoplastic agents. CIPN typically presents with neuropathic pain, numbness of distal extremities, and/or oversensitivity to thermal or mechanical stimuli. This adverse side effect often requires a reduction in chemotherapy dosage or even discontinuation of treatment. Currently there are no effective treatment options for CIPN. While the underlying mechanisms for CIPN are not understood, current data identify a “dying back” axon degeneration of distal nerve endings as the major pathology in this disorder. Therefore, mechanistic understanding of axon degeneration will provide insights into the pathway and molecular players responsible for CIPN. Here, we review recent findings that expand our understanding of the pathogenesis of CIPN and discuss pathways that may be shared with the axonal degeneration that occurs during developmental axon pruning and during injury-induced Wallerian degeneration. These mechanistic insights provide new avenues for development of therapies to prevent or treat CIPN.
Introduction
Cancer therapies result in multiple toxic side effects that limit the doses used and cause long-lasting damage to patients. Chemotherapy-induced peripheral neuropathy (CIPN) is a severe and long lasting side effect caused by diverse anticancer agents that damage sensory and/or motor nerves. Symptoms of CIPN include numbness, pain, burning, tingling, heat/cold hyperalgesia, and mechanical allodynia, as well as reduced motor function. CIPN commonly presents with a “Glove-and-Stocking” distribution with the most distal portions of the limb exhibiting the greatest deficits (Brewer et al., 2016). CIPN occurs in 30–70% of patients treated with specific categories of anticancer agents (Seretny et al., 2014). Symptoms usually begin after multiple doses of the chemotherapeutic agents, and progress as treatment continues. After the treatments cease, they can resolve in a short time period, or persist as a long-lasting sequela of cancer therapy.
Clinical assessment of CIPN is usually based on patient-derived questionnaires and physician-based grading scales such as the common toxicity criteria (CTC) scale. These evaluation methods suffer from subjectivity and inconsistency (Cavaletti et al., 2010; Brewer et al., 2016). More objective evaluation methods have also been developed, such as quantitative sensory testing (QST), which measures the detection threshold for both mechanical and thermal sensory inputs. Nerve conduction studies (NCS), which assess both sensory and motor nerve action potential can also be used in patient assessment (Brewer et al., 2016), as decreased amplitude of sensory action potential are commonly observed in CIPN patients (Chaudhry et al., 1994; Park et al., 2013). Skin biopsy and quantitative assessment of intraepidermal nerve fiber (IENF) density provides a sensitive, objective and quantitative measurement of the small nerve fiber neuropathy commonly seen in CIPN (Periquet et al., 1999; Kroigard et al., 2014). However, these more objective measurements are not easily deployed in clinical assessments.
Chemotherapeutic Drugs that Cause CIPN
Multiple anticancer drugs cause peripheral neuropathies; the most common are the taxanes, vinca alkaloids, platinum-based drugs, and protease inhibitors (Brewer et al., 2016; Table 1). Taxane agents (e.g., paclitaxel and docetaxel) exert their antimitotic effect by binding to polymerized tubulin within microtubules, and thereby preventing microtubule depolymerization (Jordan and Wilson, 2004). Vinca alkaloids (e.g., vincristine and vinblastine), another class of agents targeting microtubules, promote microtubule depolymerization and thereby disrupt mitotic spindles and cause cell cycle arrest (Jordan and Wilson, 2004; Kavallaris, 2010). As highly polarized dorsal root ganglion (DRG) sensory neurons require proper microtubule dynamics for axonal transport of mRNAs, proteins, mitochondria and other organelles, it is perhaps not surprising that these microtubule binding agents can cause degeneration of peripheral nerve fibers and symptoms of CIPN (Authier et al., 2000; Ja'afer et al., 2006; Gornstein and Schwarz, 2014; Geisler et al., 2016). As these two drugs with opposite effects on microtubule stability both can cause CIPN, it appears likely that diverse perturbations in microtubule dynamics may play a critical role instigating CIPN.
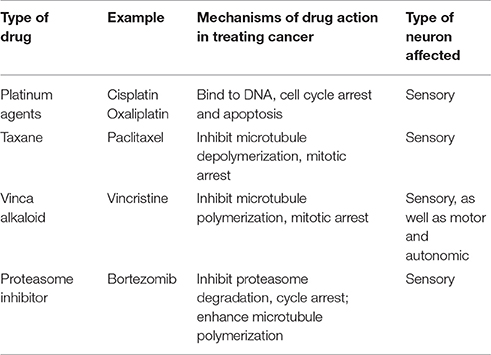
Table 1. Chemotherapeutic drugs implicated in CIPN.
The platinum-based chemotherapeutics (e.g., cisplatin and oxaliplatin) exert their antineoplastic activity by forming interstrand DNA adduct (Suchankova et al., 2012), leading to cell cycle arrest (Johnstone et al., 2014). Patients treated with platinum agents exhibit predominantly sensory neuropathy (Addington and Freimer, 2016). It is not known whether this sensory neuropathy is due to damage of nuclear or mitochondrial DNA in DRG neurons or other cells, or reflects other actions of platinum-based compounds.
Proteasome inhibitors represent a novel class of anticancer drugs that result in protein accumulation and apoptosis (Adams, 2004). Bortezomib, an inhibitor of the 20S subunit of the proteasome, was the first proteasome inhibitor approved for clinical treatment of multiple myeloma (Curran and McKeage, 2009). Bortezomib induces a peripheral sensory neuropathy. As bortezomib exerts a microtubule stabilizing activity similar to paclitaxel in addition to proteasome inhibition (Poruchynsky et al., 2008), it is not clear whether the neuropathic effect reflects proteasome inhibition or microtubule changes.
While other agents have been reported to cause neuropathy in some patients, the above chemotherapeutic agents represent the major drug categories currently responsible for CIPN. These agents have different mechanisms of action, and so it is not known whether they cause CIPN by a common pathway. Given the poor understanding of the disorder, it is not surprising that there are currently no effective treatments of CIPN, and that multiple clinical trials have had disappointing results. Among recent trials, duloxetine, an antidepressant therapy that inhibits serotonin and norepinephrine reuptake, was shown to have a small, but significant beneficial effect on CIPN (Smith et al., 2013). However, the other 14 out of 15 therapies tested failed to show beneficial effects on CIPN patients (Majithia et al., 2016). Thus, there is a clear need for greater understanding of CIPN to enable development and testing of new therapies. This review summarizes the up-to-date understanding of CIPN, discusses the known mechanisms of general axon degeneration and how this understanding provides insight into for future research into the pathogenesis of CIPN.
Pre-clinical Models for Studying CIPN
Animal Models
To understand the biology of CIPN, a variety of preclinical models have been developed. Chemotherapy drugs are typically given to rodents (rats or mice) through intraperitoneal or intravenous injection, followed by behavioral, electrophysiological, or morphological analysis. These models enable a rigorous assessment of the neuropathological features consistently observed following chemotherapeutic treatment. A recent review (Hoke and Ray, 2014) provides a thorough overview of the current animal models used for CIPN study.
A “dying back” axon degeneration is a prototypical pathological feature of CIPN in patients, and this can be assessed both in patients and in rodent models by skin biopsy and measurement of IENF density (Authier et al., 2000). Both the number of innervating nerve fibers and the neuronal subtypes perturbed by chemotherapeutic agents can be readily and quantitatively assessed in animal models. Additional studies of sensory and motor nerve conduction including sensory and compound muscle action potentials and latency of evoked response can be carried out to assess CIPN in animal models. Several groups have reported decreased sciatic nerve fiber diameter in rats treated with various dosages of paclitaxel (Authier et al., 2000; Persohn et al., 2005; Arrieta et al., 2011), as well as decreased peripheral nerve conduction (Persohn et al., 2005); however motor function is not altered (Authier et al., 2000). Peripheral nerve fiber degeneration has also been reported in rats/mice treated with vincristine (Ja'afer et al., 2006; Geisler et al., 2016), bortezomib (Cavaletti et al., 2007; Carozzi et al., 2010; Meregalli et al., 2010) and cisplatin (Carozzi et al., 2010; Arrieta et al., 2011). Although sensory neurons represent the most common target of CIPN, damage to motor and autonomic neurons have also been reported (Mora et al., 2016).
Behavioral studies in rodent models of CIPN assess mechanical allodynia, mechanical hyperalgesia, and thermal hypo- and hyperalgesia following exposure to chemotherapeutic drugs. Mechanical allodynia, the over sensitization to touch, is assessed by applying a series of monofilaments, the Von Frey filaments, to the hind paw of the animal and the threshold of pressure applied that causes the animals to withdraw their paw is measured. To measure changes in responses to temperature, a hot or cold stimulus is applied by tail immersion or radiant heat, and the response as assessed by tail flick or paw withdrawal is quantified. It is not yet known which of these behaviors best reflects the clinical condition. Thermal hyperalgesia is commonly observed in rodent models of CIPN (Authier et al., 2000; Cata et al., 2008; Zheng et al., 2012), while mechanical hyperalgesia (Authier et al., 2000) and thermal allodynia (Arrieta et al., 2011) are also reported. Unfortunately, the behaviors observed are inconsistent among different research groups, which is likely due to the fact that current rodent models are not standardized for animal strain, age, sex, or drug dosing schedule (Hoke and Ray, 2014). The diverse methods of initiating and assessing CIPN in rodent models make it difficult to compare data across laboratories.
Non-mammalian models have also been reported, but are less commonly studied. Lisse and colleagues reported on a Zebrafish model, in which DRG axon degeneration and impaired twitching response were observed after a 4-day treatment with paclitaxel (Lisse et al., 2016). Several groups have also established Drosophila models to study the underlying mechanisms of CIPN. To establish these models, adult flies or larvae are fed with food that contains chemotherapeutic drugs. Drosophila larvae fed paclitaxel exhibited axonal swellings and axon loss in sensory neurons without alterations at the neuromuscular junction (Bhattacharya et al., 2012). In this Drosophila model, axon degeneration occurs without loss of the neuronal cell bodies. Interestingly, overexpression of NMNAT, a protective protein during axon injury, prevented this paclitaxel-induced axon degeneration. Cisplatin fed adult Drosophila exhibited enhanced neuronal apoptosis in the brain and displayed defective climbing behavior (Podratz et al., 2011b, 2013). These results are consistent with findings from rodent models and from people, suggesting that these non-mammalian systems can provide valuable models to study CIPN.
Tissue Culture Models
In addition to animal models, tissue culture preparations of rat and mouse DRG sensory neurons are used to study CIPN at a mechanistic level (Malgrange et al., 1994; Yang et al., 2009; Guo et al., 2017). While most studies have relied on embryonic tissue, a recent study by Gornstein et al. developed a DRG culture from adult mice to study paclitaxel-induced neuropathy (Gornstein and Schwarz, 2017). Recent methods for turning human adult cells into induced pluripotent stem cells (iPSCs) and reprogramming these cells to generate peripheral sensory neurons have enabled studies of CIPN on cultured human cells (Chambers et al., 2012; Wainger et al., 2015). A recent study analyzed the effects on different chemotherapy agents on neurite outgrowth using commercially available iPSC-derived neurons coupled with high content imaging analysis. Both paclitaxel and vincristine-treated neurons showed decreased neurite outgrowth without increased cell death, while cisplatin treatment induced cell death (Wheeler et al., 2015). Moreover, knocking down TUBB2A, a gene encoding a tubulin isoform that has previously been identified as the locus for a single nucleotide polymorphism associated with enhanced risk of CIPN (Leandro-Garcia et al., 2012), increased the sensitivity of iPSC-derived neurons to paclitaxel treatment (Wheeler et al., 2015). This result supports the hypothesis that disruption of microtubule dynamics may be one mechanism contributing to CIPN. These findings also suggest that human iPSC-induced neurons can provide a reliable and powerful in vitro human model for mechanistic and therapeutic studies of CIPN.
Pathology of CIPN
The mechanism(s) whereby chemotherapies cause CIPN are not yet understood, nor is it clear whether distinct agents converge on a shared pathway to induce symptoms. While effects of chemotherapeutic agents on neurons, glial cells, and skin cells have each been suggested to initiate the neurological symptoms, studies in patients and in animal models implicate axonal degeneration as a common process in CIPN pathology. Specifically, chemotherapeutic agents either directly or indirectly trigger a “dying back” axon degeneration that proceeds in a distal-to-proximal manner. Potential mechanisms for initiating axon degeneration include defects in axon transport, altered mitochondrial function, or altered Ca+2 homeostasis (Figure 1).
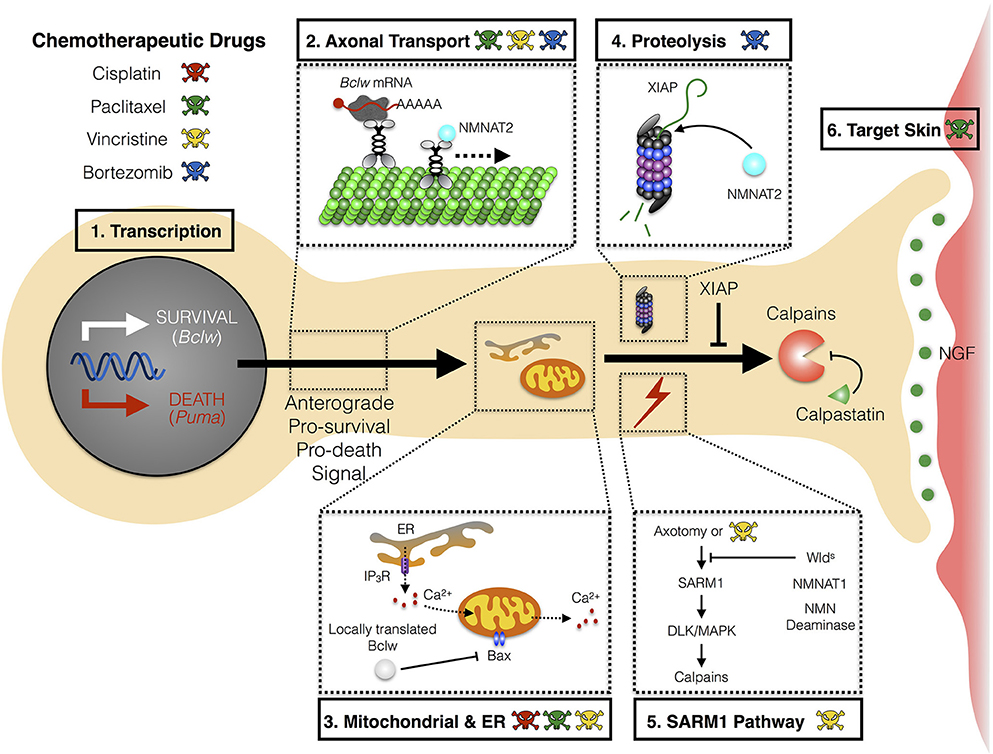
Figure 1. Mechanism of axon degeneration and sites of action of chemotherapeutic drugs. Axon degeneration is determined by factors that can either enhance death and/or inhibit survival pathways. Key mechanisms that underlie this pathway are transcriptional programs, microtubule-dependent transport, local translation to modulate mitochondrial function, and proteolysis. Chemotherapeutic agents (illustrated by colored skull and cross bones) that are currently known to modulate these key mechanisms are depicted (see text for detail). (1) During developmental axon pruning, target-derived neurotrophins instruct transcription of key pro-survival genes (Bclw). Under neurotrophic deprivation, the function of pro-survival genes is overcome by induction of pro-death genes (Puma). (2) Key transcripts of neurotrophin-dependent retrograde response genes (Bclw) are transported in an anterograde manner by kinesin motor proteins toward distal axons. Cell soma-derived factors (NMNAT2) are also transported to axons to replenish axonal pools of proteins with fast turnover rate. (3) Local translation of Bclw and LaminB2 (not shown) modulates mitochondrial function to inhibit the caspase cascade. Mitochondrial and ER integrity is also critical for maintaining Ca2+ homeostasis to prevent activation of Ca2+-dependent calpains. (4) Neurotrophic deprivation leads to degradation of XIAP, a key inhibitor of the caspase cascade. NMNAT2, a survival factor with a short half-life, is possibly degraded through the proteasome in axons. (5) Vincristine and axotomy activates SARM1 and DLK/MAPK signaling, leading to activation of calpains, the ultimate convergence point for executing axon degeneration. Expression of Wlds, axonal NMNAT1, or NMN deaminase provides protection against vincristine or injury-induced degeneration. (6) Non neuron-autonomous effects from other cell types can potentially sensitize axons to degeneration during CIPN. Paclitaxel-induced neurotoxicity can mediate inflammatory response and epithelial damage to perturb skin homeostasis. Bclw, Bcl-2-like protein 2; Puma, p53 upregulated modulator of apoptosis; NMNAT, nicotinamide mononucleotide adenylyltransferase; ER, endoplasmic reticulum; IP3R, inositol 1,4,5-triphosphate receptor; BAX, Bcl-2-like protein 4; XIAP, X-linked inhibitor of apoptosis protein; SARM1, sterile α-motif-containing and armadillo-motif-containing protein; DLK, dual leucine zipper kinase; MAPK, mitogen-activated protein kinase; Wlds, Wallerian degeneration slow; NMN, nicotinamide mononucleotide; CIPN, chemotherapy-induced peripheral neuropathy.
CIPN and Axon Transport
Paclitaxel, vincristine, and bortezomib all affect microtubule dynamics by inhibiting the processes of tubulin depolymerization and polymerization (Figure 1). Paclitaxel causes retraction bulbs at the tips of sensory nerve axons, indicating axon degeneration (Gornstein and Schwarz, 2017). Epothilone B, a chemotherapy drug currently in clinical trials, is a structurally distinct compound that binds to the same site on tubulin as paclitaxel. Interestingly, DRG cultures treated with epothilone B showed a similar axon tip retraction as cultures treated with paclitaxel (Gornstein and Schwarz, 2017). This further indicates that altered microtubule dynamics contributes to taxane-induced axonopathy. As both anterograde and retrograde axonal transport rely on microtubule integrity and dynamics, CIPN drugs targeting microtubule are likely to result in altered axonal transport of essential cellular components. LaPointe and colleagues used an in vitro vesicle motility assay and discovered that vincristine and paclitaxel inhibit anterograde axonal transport in axoplasm isolated from squid giant axons, and that vincristine inhibits retrograde transport as well (LaPointe et al., 2013). Additional studies in simple systems affirm that paclitaxel can reduce axonal transport (Theiss and Meller, 2000; Shemesh and Spira, 2010). In support of the possibility that impaired transport causes CIPN, a recent study demonstrated reductions in the levels of several axonal mRNAs in the distal nerves of mice treated with paclitaxel (Bobylev et al., 2015). In contrast, time lapse studies detected little change in mitochondria and late endosome/lysosome transport along microtubules following paclitaxel treatment (Gornstein and Schwarz, 2017). Thus, it is possible that paclitaxel causes defective mRNA transport, rather than a general disruption of microtubule-based motility.
Mitotoxicity in CIPN
Mitochondrial dysfunction is commonly observed in CIPN (Figure 1). Abnormal mitochondrial morphology including swelling, vacuolation, enlargement, and loss of cristae structure have been observed in peripheral nerve axons (Flatters and Bennett, 2006; Xiao et al., 2012; Zheng et al., 2012; Bobylev et al., 2017), but not in the surrounding Schwann cells (Xiao et al., 2012) in several CIPN animal models. Potential causes of altered mitochondria include paclitaxel-induced depletion of mRNAs encoding mitochondrial fission/fusion machinery in distal axons (Bobylev et al., 2015). Mitochondria dynamics may be altered in cisplatin-treated animals due to decreased level of mitochondrial fusion protein MFN2 in distal axon segments (Bobylev et al., 2017). Moreover, increased mitochondrial fragmentation, as well as decreased mitochondrial fission/fusion dynamics and motility, occur rapidly in vincristine-treated DRG cultures and precede axon degeneration (Berbusse et al., 2016). Platinum agents can also affect mitochondrial function through damaging mitochondrial DNA (mtDNA), and thereby impeding mtDNA replication and transcription (Podratz et al., 2011a). Thus, mitochondrial dysfunction may represent a common mechanism for axon degeneration in CIPN (Figure 1).
Ca2+ Homeostasis and Cation Channels
Ca2+ homeostasis is critical for neuronal and axonal health. Mitochondria and the endoplasmic reticulum (ER) both function as intracellular stores of Ca2+, and paclitaxel treatment alters Ca2+ homeostasis, potentially by inducing Ca2+ release from the mitochondria (Kidd et al., 2002; Figure 1). An alternative mechanism for alterations in intracellular Ca2+ dynamics was presented by Boehmerle and colleagues, who reported that paclitaxel-induced Ca2+ oscillation reflects altered function of inositol 1,4,5-trisphosphate receptors (IP3R) in the ER (Boehmerle et al., 2006; Figure 1). Indeed, chronic paclitaxel exposure leads to impaired phosphoinositide-mediated Ca2+ signaling in both neuroblastoma cell and DRG cultures (Boehmerle et al., 2007). Increased intracellular Ca2+ can activate the potent protease calpain, which directly triggers axon degeneration (Wang et al., 2012). Moreover, Ca2+ reducing drugs alleviate mechanical allodynia and hyperalgesia in paclitaxel and vincristine rat models (Siau and Bennett, 2006).
Chemotherapeutic agents may alter cation channels more generally. Recent studies showed that cisplatin, paclitaxel or bortezomib treatment result in increased expression of the non-selective cation channels, TRPV1 and TRPA1, in cultured DRG neurons (Ta et al., 2010; Hara et al., 2013; Quartu et al., 2014). As TRP channels are critical for pain signaling, alterations in these cation channels may be an important component of CIPN. Therefore, approaches that target Ca2+ homeostasis and/or cation channels provide potential future therapies.
Central Sensitization and CNS Glial Activation
While abundant data indicate that peripheral nerves and epidermal innervation are affected in CIPN, changes in the central nervous system (CNS) may also contribute to the disorder. Central sensitization may occur as a direct or indirect consequence of chemotherapeutic agents. Low concentrations of chemotherapeutic drugs, oxaliplatin, and paclitaxel, can be detected in the CNS after systemic dosing that produce hyperalgesia in rats (Huang et al., 2016). Paclitaxel can directly sensitize spinal neurons to TRPV1-mediated capsaicin response (Li et al., 2015), while oxaliplatin can increase expression and release of chemokine C-X3-C motif ligand 1 from spinal cord neurons (Huang et al., 2016). Thus, chemotherapeutic agents can possibly act directly on spinal neurons to mediate central sensitization.
The release of chemokines and cytokines from spinal neurons may further potentiate neuropathic pain by activating microglia and astrocytes. Paclitaxel can induce activation of astrocytes, and in some cases microglia, in the dorsal horn of the spinal cord (Zhang et al., 2012; Ruiz-Medina et al., 2013). Consistent with these effects, drugs that affect astrocytes and microglia, including cannabinoid agonists and minocycline, have been reported to alleviate mechanical allodynia in rats (Boyette-Davis et al., 2011; Burgos et al., 2012). Interestingly, some chemotherapeutics can also activate astrocytes and microglia in regions of the brain associated with chronic pain and nociceptive processing, as paclitaxel increases expression of GFAP in anterior cingulate cortex concurrent with behavioral evidence of thermal hyperalgesia (Masocha, 2015). Collectively, these evidences demonstrate a potential contribution of CNS sensitization and glial activation to the neuropathic pain that occurs in CIPN.
Mechanistic Insights into CIPN and Axon Degeneration
The recent failures of multiple clinical trials for CIPN (Majithia et al., 2016) underscore the need to explore and address underlying molecular mechanisms more broadly. Since all the relevant chemotherapeutic agents, either directly or indirectly, trigger a “dying back” axon degeneration, a greater understanding of the biology of axon degeneration process per-se can identify major players and signaling pathways that can be targeted. Axon degeneration can be provoked by multiple stimuli in addition to chemotherapeutic agents. Axon pruning during development and axotomy also entail axon degeneration. Here we will discuss what is currently known about the cellular pathways for axon degeneration in these contexts, to provide insights for addressing the molecular mechanisms of axon degeneration during CIPN.
Developmental Axon Pruning
Axon degeneration constitutes a necessary and healthy end result of pruning that allows plasticity of neuronal circuitry during development. The process of axon pruning has been studied in diverse species including mice, Drosophila, C. elegans, and zebrafish (Kage et al., 2005; Hayashi et al., 2009; Poulain and Chien, 2013; Yu and Schuldiner, 2014; Riccomagno and Kolodkin, 2015). Early in development, excess innervation can be observed in both the CNS and PNS. Subsequently, refinement of neuronal connectivity through axon pruning enables the establishment of the robust mature circuitry. Multiple signaling processes critical for axon pruning and refinement have been identified in studies of both central and peripheral neurons.
Repulsive axon guidance cues such as the semaphorins bind to pruning receptors and initiate local axon degeneration in the CNS. For example, in the hippocampus, stereotyped pruning of infrapyramidal bundle (IPB) is initiated by semaphorin3F expressed in interneurons in the stratum oriens, and is achieved by signaling complexes containing neuropilin-2 and plexin-A3 (Bagri et al., 2003). A similar role for semaphorin3F in axon pruning has been observed in corticospinal tract from layer 5 of the visual cortex (Low et al., 2008). In Drosophila, mushroom body (MB) γ neurons provide another system where stereotyped axon pruning occurs during development. Pruning of dorsal and medial axonal branches during metamorphosis requires signaling through a TGF-β receptor complex in MB γ neurons, and is initiated by myoglianin secreted from nearby glial cells (Awasaki et al., 2011). Although TGF-β signaling is essential for axon fragmentation, it is not sufficient. Thus, glial cells initiate the process of axon fragmentation in these cells but other factors are required to fully execute axon pruning. Similarly, chemotherapeutic agents acting directly on nearby glial cells may sensitize neurons subsequently undergo axon degeneration. Although paclitaxel has been shown to induce inflammatory responses and promote epithelial damage prior to the induction of axon degeneration (Lisse et al., 2016; Zhang et al., 2016), this model of sensitization has not yet been explored with other chemotherapeutic drugs.
Much of our understanding of developmental axon pruning is derived from studies of neurotrophins, nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF) and neurotrophins 3 and 4 (NT3 and NT4), which are released in a limited amount by target tissues and promote axon and cell body survival (Harrington and Ginty, 2013). The role of neurotrophins in regulating axonal survival and degeneration is most widely studied in the PNS, particularly sympathetic and sensory neurons. Target-derived neurotrophins bind and activate tropomyosin receptor kinase (Trk) receptors in the innervating axon terminals. Endocytosis of the activated receptors results in signaling endosomes that are retrogradely trafficked along the axons. The signaling endosomes initiate instructive programs that promote both axonal and cell survival (Tasdemir-Yilmaz and Segal, 2016). Consequently, neurons that fail to receive neurotrophins lose their innervation, and may undergo apoptosis. This selection process of both axonal and cell degeneration is further enhanced by active signaling to eliminate axon processes. In superior cervical ganglion (SCG) sympathetic neurons, active signaling through the p75 neurotrophin receptor (p75NTR) can promote axon degeneration or even apoptosis (Singh et al., 2008). These examples provide evidence of overlap between cellular and axonal survival and death processes, and suggest that cumulative responses to pro-survival and pro-death signals may also play a role in the axon degeneration observed in CIPN.
Regulation of Axonal Survival during Development: Coordination of Transcription, Transport, Translation, and Proteolysis
Regulation occurs at multiple levels to determine whether axons will survive or degenerate, including nuclear transcription, axonal translation and protein activity (Tasdemir-Yilmaz and Segal, 2016). In vitro culture systems using compartmented culture platforms have proven to be valuable tools that recapitulate the spatially and fluidically isolated cell body and distal axon compartment observed in vivo, enabling mechanistic understanding of the multiple regulatory steps involved in determining and implementing axonal survival or degeneration.
In these compartmented systems, stimulation of axon terminals with target-derived neurotrophins promotes axon outgrowth and prevents both axon degeneration and cell body apoptosis. In contrast, neurotrophin stimulation of cell bodies prevents apoptosis, but does not prevent axon degeneration. Therefore, some of the retrograde response genes that are selectively transcribed in response to neurotrophin stimulation of axons and not by stimulation of cell bodies are likely to encode components needed for axonal survival. These transcriptional changes can be closely linked to events in the axon, as many newly transcribed mRNAs are transported to the distal axons, where they are translated into protein. For example, the Bcl2 family member, Bclw (aka Bcl2l2) is a retrograde response gene, and newly transcribed Bclw mRNA is transported to the axons, where it is locally translated and the resultant protein promotes axonal survival (Pazyra-Murphy et al., 2009; Courchesne et al., 2011; Cosker et al., 2013; Figure 1).
Recently, the RNA binding protein SFPQ was shown to bind multiple axonal transcripts including Bclw and LaminB2 mRNAs (Cosker et al., 2016). While LaminB2 is predominantly a nuclear membrane protein, locally translated LaminB2 associates with axonal mitochondria and adjusts mitochondrial function (Yoon et al., 2012). SFPQ is needed for transport and axonal localization of Bclw, LaminB2, and others, and so functions to promote axonal survival and prevent axon degeneration (Cosker et al., 2016; Thomas-Jinu et al., 2017). Together these studies demonstrate that regulated transcription, axonal transport, and local translation work in concert to determine and implement axon survival pathways. Currently, there is no evidence for chemotherapeutic agents affecting transcription of pro-survival genes or translation of axonal mRNAs. However, as this pathway is critical in axon survival, such studies represent an important direction for future research.
In addition to the neurotrophin-mediated axon survival pathways above, key transcriptional programs have recently been uncovered that actively promote axon degeneration during trophic deprivation. Simon et al. demonstrated that loss of neurotrophic support induces transcription of Puma, which encodes a pro-apoptotic BH3-only family protein Puma, through the DLK/MAPK signaling pathway (Simon et al., 2016; Figure 1). Maor-Nof et al. provided further mechanistic insight into this transcriptional program, demonstrating that the phosphatase Dusp16 functions to put a brake on the degenerative response by inhibiting Puma transcription (Maor-Nof et al., 2016). At present it is not clear whether the Puma protein subsequently functions in the cell bodies or in axons to trigger axon degeneration. In either case, it will be important to assess whether this pro-degenerative component contributes to the biology of CIPN.
A final step in executing axon degeneration involves proteolytic cleavage of axonal proteins by caspases and calpains. Initiator caspase-9 and the effector caspases-3 and -6 have all been implicated in axon degenerative cascades (Nikolaev et al., 2009; Schoenmann et al., 2010; Simon et al., 2012; Cusack et al., 2013; Unsain et al., 2013). Caspase-mediated degeneration is usually held in check by inhibitor of apoptosis protein (IAP) (Figure 1) (Potts et al., 2003; Cusack et al., 2013; Unsain et al., 2013). Calpains are Ca2+-activated proteases that execute axonal degeneration by proteolysis of multiple cellular proteins. Calpastatin inhibits calpain activation during developmental pruning and so limits the extent of axon degeneration (Yang et al., 2013). Similarly, calpain-mediated proteolysis has been implicated as a convergent pathway that contributes to axon degeneration in CIPN (Figure 1). Mice treated with paclitaxel that are administered calpain inhibitors show reduced signs of axonal degeneration in sensory neurons and improved clinical measures of neuropathy (Wang et al., 2004) One of the targets of calpain during paclitaxel-induced peripheral neuropathy is neuronal calcium sensor-1 (NCS-1). NCS-1 binds to IP3R to enhance intracellular calcium signaling. Paclitaxel disrupts IP3R-mediated intracellular calcium signaling via NCS-1 degradation (Boehmerle et al., 2007). In the future, it will be important to identify other molecular downstream targets of calpain relevant to peripheral neuropathy. Nonetheless, these studies demonstrate calpain as a promising therapeutic target for CIPN.
Wallerian Degeneration
Axon degeneration due to traumatic injury represents another useful model for mechanistic studies of axon degeneration or survival. Following axotomy, an initial latent phase is followed by fragmentation of the cytoskeleton, destruction of organelles, and ultimately disintegration of axons distal to the severed site (Gerdts et al., 2016). This process of axon degeneration is referred to as Wallerian degeneration (WD). Much of the mechanistic understanding of WD derives from a spontaneous genetic mutation, Wallerian degeneration slow (Wlds), which delays the process of WD. This mutation encodes a chimeric fusion protein of the N-terminal fragment of E4 ubiquitin ligase Ube4b and the enzyme nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1; Conforti et al., 2000). The axonal protective effect of the mutant Wlds depends on the NMNAT1 portion of the fusion protein, which synthesizes nicotinamide adenine dinucleotide (NAD+) from nicotinamide mononucleotide (NMN; Araki et al., 2004). This protective role of Wlds is conserved evolutionarily across diverse species (MacDonald et al., 2006; Martin et al., 2010).
Building on the exciting discovery that the Wlds mutation slows the process of axon degeneration, multiple studies have addressed the roles of NMNAT enzymes in axonal survival. There are three mammalian NMNATs. Loss of function mutations in NMNAT2, but not the other NMNATs triggers Wallerian-like degeneration in undamaged axons (Gilley and Coleman, 2010; Gilley et al., 2013). As NMNAT2 is selectively expressed in axons and has a very short half-life, NMNAT activity in axons requires continued anterograde transport of this enzyme from the cell soma (Figure 1). Slowing the turnover of NMNAT2 can significantly delay injury-induced WD (Xiong et al., 2012; Babetto et al., 2013; Milde et al., 2013a,b). These studies on NMNAT2 highlight the essential role of axonal transport and proteasomal degradation in maintaining homeostatic protein content in axons. Disruption of either pathway can reduce any levels of critical axonal protein levels below a necessary threshold and so induce degeneration cascade. Therefore, chemotherapy-induced changes in axonal transport, translation and proteasomal degradation represent important avenues of investigation in future studies of CIPN.
Studies of axotomy have also identified SARM1, a scaffolding molecule, as a critical regulator of axonal degeneration. Loss of function of SARM1 significantly delays axon degeneration after injury, indicating that SARM1 functions as a pro-degeneration factor in WD (Osterloh et al., 2012; Gerdts et al., 2013). Depletion of SARM1 prevents the axon degeneration phenotype seen with NMNAT2 loss of function, indicating that these two components act in the same signaling cascade, with SARM1 downstream of NMNAT2 (Gilley et al., 2015). To understand the functions of SARM1 and NMNAT2, several investigations have analyzed the importance of NAD+ vs. NMN and ATP, the product and the substrates of NMNAT2 enzymatic activity, respectively. It is not yet clear whether loss of NMNAT2 triggers axon degeneration as a result of NAD+ deficiency, or accumulation of NMN, or other outcomes. Whether SARM1 alters NAD+ or NMN to induce axon degeneration is an area of active investigation (Di Stefano et al., 2015, 2017; Gerdts et al., 2015; Sasaki et al., 2016; Essuman et al., 2017). One important consequence of SARM1 activation or of NMNAT2 loss is activation of DLK/MAPK signaling, which ultimately triggers calpain-mediated axon degeneration (Yang et al., 2013, 2015; Figure 1).
Calpain and many additional components implicated in axonal responses to injury are linked to CIPN. Initial evidence of these similarities came from studies demonstrating that Wlds mutant neurons display resistance to vincristine-induced neuropathy (Wang et al., 2001). More recently, SARM1 knockout mice were shown to be resistant to vincristine in an in vivo model (Geisler et al., 2016). Essuman et al. demonstrated that SARM1 functions as a NAD+-depleting enzyme and that the intrinsic NADase activity of SARM1 is required for vincristine-induced axon degeneration (Essuman et al., 2017). Additional evidence that the SARM1 pathway plays a critical role in CIPN includes data that expression of axonal NMNAT1 and constitutive expression of the NMN deaminase that consumes NMN, both protect against vincristine-induced peripheral neuropathy (Sasaki et al., 2009; Di Stefano et al., 2017). Therefore, inhibition of SARM1 enzymatic activity might be a useful therapeutic strategy in CIPN.
Human Variants
In addition to these models of development and axotomy, a final approach for identifying molecular cascades implicated in CIPN is to discover human variations that alter susceptibility to CIPN. The incidence of CIPN varies significantly from person to person. Increased age, diabetes, and previous neuropathy are known risk factors for CIPN. However, it is likely that genetic variations are also important for susceptibility. Several genome wide association studies (GWAS) have identified gene variants associated with increased vulnerability to CIPN. Single nucleotide polymorphisms of FGD4 and EPHA5 were significantly associated with susceptibility to paclitaxel-induced peripheral neuropathy in both European and African ancestral groups (Baldwin et al., 2012). FGD4 (FYVE, RhoGEF, and PH Domain Containing 4) is a Rho GTP/GDP exchange protein regulating actin cytoskeleton dynamics (Obaishi et al., 1998). Mutations of FGD4 in humans lead to autosomal recessive demyelinating Charcot-Marie-Tooth neuropathy (type 4H; De Sandre-Giovannoli et al., 2005; Delague et al., 2007; Stendel et al., 2007). The EPHA family is a group of tyrosine kinase receptors that bind to the Ephrin ligands. Eph/Ephrin signaling has been implicated in regulating axon outgrowth and guidance during development (Dickson, 2002; Egea and Klein, 2007). A recent study performed next generation sequencing on patients who developed significant neuropathy after paclitaxel, and identified variations of three related receptors, EPHA5/6/8, that correlated with susceptibility to paclitaxel-induced neuropathy (Apellaniz-Ruiz et al., 2017). Other CIPN associated genes identified by GWAS studies also play critical roles in neurite outgrowth and nerve development including FZD3 (Baldwin et al., 2012), TUBB2A (Leandro-Garcia et al., 2012), VAC14 (Hertz et al., 2016). These candidates may affect CIPN by directly affecting the cytoskeleton organization (TUBB2A, FGD4), by regulating intracellular trafficking (VAC14) or may function as signaling molecules regulating axon growth (EPHA, FZD3). Future verifications are required to confirm the involvement of these genes in CIPN pathology, and how these factors might contribute to the disorder.
Concluding Remarks
As summarized in this review, multiple chemotherapeutic agents that cause CIPN are directed against different molecular targets. Preclinical studies have generated several hypotheses to explain the pathogenesis of CIPN by these agents, including defective axon transport, mitotoxicity, and altered Ca2+ homeostasis. While each agent may initially affect the nervous system in a distinctive manner, the pathologies observed in CIPN converge to cause an axon degenerative process. Mechanistic studies of the phenotypically similar axon degeneration processes during developmental axon pruning or during injury-induced WD are likely to shed new light on the molecular basis of CIPN. Indeed, several signaling molecules on the SARM1, DLK/MAPK, and NMNAT pathways that protect axons from WD can also impact CIPN. Concurrently, human GWAS studies have identified pathways implicated in neurite outgrowth and/or axon maintenance that may be relevant for axon degeneration in CIPN. These findings suggest that axon degeneration is central to CIPN pathology. Enhanced understanding of the pathologic process of axon degeneration will be essential for developing an arsenal of effective therapies of CIPN.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Segal lab for helpful discussions. Work in the Segal lab on CIPN has been funded by the Barr-Weaver Award, and NCI R01CA205255.
References
Adams, J. (2004). The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer 4, 349–360. doi: 10.1038/nrc1361
Addington, J., and Freimer, M. (2016). Chemotherapy-induced peripheral neuropathy: an update on the current understanding [version 1; referees: 2 approved]. F1000Res. 5:1466. doi: 10.12688/f1000research.8053.1
Apellaniz-Ruiz, M., Tejero, H., Inglada-Perez, L., Sanchez-Barroso, L., Gutierrez-Gutierrez, G., Calvo, I., et al. (2017). Targeted sequencing reveals low-frequency variants in epha genes as markers of paclitaxel-induced peripheral neuropathy. Clin. Cancer Res. 23, 1227–1235. doi: 10.1158/1078-0432.CCR-16-0694
Araki, T., Sasaki, Y., and Milbrandt, J. (2004). Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010–1013. doi: 10.1126/science.1098014
Arrieta, O., Hernandez-Pedro, N., Fernandez-Gonzalez-Aragon, M. C., Saavedra-Perez, D., Campos-Parra, A. D., Rios-Trejo, M. A., et al. (2011). Retinoic acid reduces chemotherapy-induced neuropathy in an animal model and patients with lung cancer. Neurology 77, 987–995. doi: 10.1212/WNL.0b013e31822e045c
Authier, N., Gillet, J. P., Fialip, J., Eschalier, A., and Coudore, F. (2000). Description of a short-term Taxol-induced nociceptive neuropathy in rats. Brain Res. 887, 239–249. doi: 10.1016/S0006-8993(00)02910-3
Awasaki, T., Huang, Y., O'Connor, M. B., and Lee, T. (2011). Glia instruct developmental neuronal remodeling through TGF-beta signaling. Nat. Neurosci. 14, 821–823. doi: 10.1038/nn.2833
Babetto, E., Beirowski, B., Russler, E. V., Milbrandt, J., and DiAntonio, A. (2013). The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep. 3, 1422–1429. doi: 10.1016/j.celrep.2013.04.013
Bagri, A., Cheng, H. J., Yaron, A., Pleasure, S. J., and Tessier-Lavigne, M. (2003). Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell 113, 285–299. doi: 10.1016/S0092-8674(03)00267-8
Baldwin, R. M., Owzar, K., Zembutsu, H., Chhibber, A., Kubo, M., Jiang, C., et al. (2012). A genome-wide association study identifies novel loci for paclitaxel-induced sensory peripheral neuropathy in CALGB 40101. Clin. Cancer Res. 18, 5099–5109. doi: 10.1158/1078-0432.CCR-12-1590
Berbusse, G. W., Woods, L. C., Vohra, B. P., and Naylor, K. (2016). Mitochondrial dynamics decrease prior to axon degeneration induced by vincristine and are partially rescued by overexpressed cytNmnat1. Front. Cell. Neurosci. 10:179. doi: 10.3389/fncel.2016.00179
Bhattacharya, M. R., Gerdts, J., Naylor, S. A., Royse, E. X., Ebstein, S. Y., Sasaki, Y., et al. (2012). A model of toxic neuropathy in Drosophila reveals a role for MORN4 in promoting axonal degeneration. J. Neurosci. 32, 5054–5061. doi: 10.1523/JNEUROSCI.4951-11.2012
Bobylev, I., Joshi, A. R., Barham, M., Neiss, W. F., and Lehmann, H. C. (2017). Depletion of mitofusin-2 causes mitochondrial damage in cisplatin-induced neuropathy. Mol. Neurobiol. doi: 10.1007/s12035-016-0364-7. [Epub ahead of print].
Bobylev, I., Joshi, A. R., Barham, M., Ritter, C., Neiss, W. F., Hoke, A., et al. (2015). Paclitaxel inhibits mRNA transport in axons. Neurobiol. Dis. 82, 321–331. doi: 10.1016/j.nbd.2015.07.006
Boehmerle, W., Splittgerber, U., Lazarus, M. B., McKenzie, K. M., Johnston, D. G., Austin, D. J., et al. (2006). Paclitaxel induces calcium oscillations via an inositol 1,4,5-trisphosphate receptor and neuronal calcium sensor 1-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 103, 18356–18361. doi: 10.1073/pnas.0607240103
Boehmerle, W., Zhang, K., Sivula, M., Heidrich, F. M., Lee, Y., Jordt, S. E., et al. (2007). Chronic exposure to paclitaxel diminishes phosphoinositide signaling by calpain-mediated neuronal calcium sensor-1 degradation. Proc. Natl. Acad. Sci. U.S.A. 104, 11103–11108. doi: 10.1073/pnas.0701546104
Boyette-Davis, J., Xin, W., Zhang, H., and Dougherty, P. M. (2011). Intraepidermal nerve fiber loss corresponds to the development of taxol-induced hyperalgesia and can be prevented by treatment with minocycline. Pain 152, 308–313. doi: 10.1016/j.pain.2010.10.030
Brewer, J. R., Morrison, G., Dolan, M. E., and Fleming, G. F. (2016). Chemotherapy-induced peripheral neuropathy: current status and progress. Gynecol. Oncol. 140, 176–183. doi: 10.1016/j.ygyno.2015.11.011
Burgos, E., Gomez-Nicola, D., Pascual, D., Martin, M. I., Nieto-Sampedro, M., and Goicoechea, C. (2012). Cannabinoid agonist WIN 55,212-2 prevents the development of paclitaxel-induced peripheral neuropathy in rats. Possible involvement of spinal glial cells. Eur. J. Pharmacol. 682, 62–72. doi: 10.1016/j.ejphar.2012.02.008
Carozzi, V. A., Canta, A., Oggioni, N., Sala, B., Chiorazzi, A., Meregalli, C., et al. (2010). Neurophysiological and neuropathological characterization of new murine models of chemotherapy-induced chronic peripheral neuropathies. Exp. Neurol. 226, 301–309. doi: 10.1016/j.expneurol.2010.09.004
Cata, J. P., Weng, H. R., and Dougherty, P. M. (2008). Behavioral and electrophysiological studies in rats with cisplatin-induced chemoneuropathy. Brain Res. 1230, 91–98. doi: 10.1016/j.brainres.2008.07.022
Cavaletti, G., Frigeni, B., Lanzani, F., Mattavelli, L., Susani, E., Alberti, P., et al. (2010). Chemotherapy-Induced Peripheral Neurotoxicity assessment: a critical revision of the currently available tools. Eur. J. Cancer 46, 479–494. doi: 10.1016/j.ejca.2009.12.008
Cavaletti, G., Gilardini, A., Canta, A., Rigamonti, L., Rodriguez-Menendez, V., Ceresa, C., et al. (2007). Bortezomib-induced peripheral neurotoxicity: a neurophysiological and pathological study in the rat. Exp. Neurol. 204, 317–325. doi: 10.1016/j.expneurol.2006.11.010
Chambers, S. M., Qi, Y., Mica, Y., Lee, G., Zhang, X. J., Niu, L., et al. (2012). Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat. Biotechnol. 30, 715–720. doi: 10.1038/nbt.2249
Chaudhry, V., Rowinsky, E. K., Sartorius, S. E., Donehower, R. C., and Cornblath, D. R. (1994). Peripheral neuropathy from taxol and cisplatin combination chemotherapy: clinical and electrophysiological studies. Ann. Neurol. 35, 304–311. doi: 10.1002/ana.410350310
Conforti, L., Tarlton, A., Mack, T. G., Mi, W., Buckmaster, E. A., Wagner, D., et al. (2000). A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc. Natl. Acad. Sci. U.S.A. 97, 11377–11382. doi: 10.1073/pnas.97.21.11377
Cosker, K. E., Fenstermacher, S. J., Pazyra-Murphy, M. F., Elliott, H. L., and Segal, R. A. (2016). The RNA-binding protein SFPQ orchestrates an RNA regulon to promote axon viability. Nat. Neurosci. 19, 690–696. doi: 10.1038/nn.4280
Cosker, K. E., Pazyra-Murphy, M. F., Fenstermacher, S. J., and Segal, R. A. (2013). Target-derived neurotrophins coordinate transcription and transport of bclw to prevent axonal degeneration. J. Neurosci. 33, 5195–5207. doi: 10.1523/JNEUROSCI.3862-12.2013
Courchesne, S. L., Karch, C., Pazyra-Murphy, M. F., and Segal, R. A. (2011). Sensory neuropathy attributable to loss of Bcl-w. J. Neurosci. 31, 1624–1634. doi: 10.1523/JNEUROSCI.3347-10.2011
Curran, M. P., and McKeage, K. (2009). Bortezomib: a review of its use in patients with multiple myeloma. Drugs 69, 859–888. doi: 10.2165/00003495-200969070-00006
Cusack, C. L., Swahari, V., Hampton Henley, W., Michael Ramsey, J., and Deshmukh, M. (2013). Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat. Commun. 4, 1876. doi: 10.1038/ncomms2910
De Sandre-Giovannoli, A., Delague, V., Hamadouche, T., Chaouch, M., Krahn, M., Boccaccio, I., et al. (2005). Homozygosity mapping of autosomal recessive demyelinating Charcot-Marie-Tooth neuropathy (CMT4H) to a novel locus on chromosome 12p11.21-q13.11. J. Med. Genet. 42, 260–265. doi: 10.1136/jmg.2004.024364
Delague, V., Jacquier, A., Hamadouche, T., Poitelon, Y., Baudot, C., Boccaccio, I., et al. (2007). Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot-Marie-Tooth type 4H. Am. J. Hum. Genet. 81, 1–16. doi: 10.1086/518428
Di Stefano, M., Loreto, A., Orsomando, G., Mori, V., Zamporlini, F., Hulse, R. P., et al. (2017). NMN deamidase delays wallerian degeneration and rescues axonal defects caused by NMNAT2 deficiency in vivo. Curr. Biol. 27, 784–794. doi: 10.1016/j.cub.2017.01.070
Di Stefano, M., Nascimento-Ferreira, I., Orsomando, G., Mori, V., Gilley, J., Brown, R., et al. (2015). A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Diff. 22, 731–742. doi: 10.1038/cdd.2014.164
Dickson, B. J. (2002). Molecular mechanisms of axon guidance. Science 298, 1959–1964. doi: 10.1126/science.1072165
Egea, J., and Klein, R. (2007). Bidirectional Eph-ephrin signaling during axon guidance. Trends Cell Biol. 17, 230–238. doi: 10.1016/j.tcb.2007.03.004
Essuman, K., Summers, D. W., Sasaki, Y., Mao, X., DiAntonio, A., and Milbrandt, J. (2017). The SARM1 toll/interleukin-1 receptor domain possesses intrinsic NAD+ cleavage activity that promotes pathological axonal degeneration. Neuron 93, 1334.e5–1343.e5. doi: 10.1016/j.neuron.2017.02.022
Flatters, S. J., and Bennett, G. J. (2006). Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain 122, 245–257. doi: 10.1016/j.pain.2006.01.037
Geisler, S., Doan, R. A., Strickland, A., Huang, X., Milbrandt, J., and DiAntonio, A. (2016). Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 139, 3092–3108. doi: 10.1093/brain/aww251
Gerdts, J., Brace, E. J., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2015). SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 348, 453–457. doi: 10.1126/science.1258366
Gerdts, J., Summers, D. W., Milbrandt, J., and DiAntonio, A. (2016). Axon self-destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron 89, 449–460. doi: 10.1016/j.neuron.2015.12.023
Gerdts, J., Summers, D. W., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2013). Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J. Neurosci. 33, 13569–13580. doi: 10.1523/JNEUROSCI.1197-13.2013
Gilley, J., Adalbert, R., Yu, G., and Coleman, M. P. (2013). Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J. Neurosci. 33, 13410–13424. doi: 10.1523/JNEUROSCI.1534-13.2013
Gilley, J., and Coleman, M. P. (2010). Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 8:e1000300. doi: 10.1371/journal.pbio.1000300
Gilley, J., Orsomando, G., Nascimento-Ferreira, I., and Coleman, M. P. (2015). Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. 10, 1974–1981. doi: 10.1016/j.celrep.2015.02.060
Gornstein, E. L., and Schwarz, T. L. (2017). Neurotoxic mechanisms of paclitaxel are local to the distal axon and independent of transport defects. Exp. Neurol. 288, 153–166. doi: 10.1016/j.expneurol.2016.11.015
Gornstein, E., and Schwarz, T. L. (2014). The paradox of paclitaxel neurotoxicity: mechanisms and unanswered questions. Neuropharmacology 76(Pt A), 175–183. doi: 10.1016/j.neuropharm.2013.08.016
Guo, L., Hamre, J. 3rd, Eldridge, S., Behrsing, H. P., Cutuli, F. M., Mussio, J., et al. (2017). Multiparametric image analysis of rat dorsal root ganglion cultures to evaluate peripheral neuropathy-inducing chemotherapeutics. Toxicol. Sci. 156, 275–288. doi: 10.1093/toxsci/kfw254
Hara, T., Chiba, T., Abe, K., Makabe, A., Ikeno, S., Kawakami, K., et al. (2013). Effect of paclitaxel on transient receptor potential vanilloid 1 in rat dorsal root ganglion. Pain 154, 882–889. doi: 10.1016/j.pain.2013.02.023
Harrington, A. W., and Ginty, D. D. (2013). Long-distance retrograde neurotrophic factor signalling in neurons. Nat. Rev. Neurosci. 14, 177–187. doi: 10.1038/nrn3253
Hayashi, Y., Hirotsu, T., Iwata, R., Kage-Nakadai, E., Kunitomo, H., Ishihara, T., et al. (2009). A trophic role for Wnt-Ror kinase signaling during developmental pruning in Caenorhabditis elegans. Nat. Neurosci. 12, 981–987. doi: 10.1038/nn.2347
Hertz, D. L., Owzar, K., Lessans, S., Wing, C., Jiang, C., Kelly, W. K., et al. (2016). Pharmacogenetic discovery in CALGB (Alliance) 90401 and mechanistic validation of a VAC14 Polymorphism that increases risk of docetaxel-induced neuropathy. Clin. Cancer Res. 22, 4890–4900. doi: 10.1158/1078-0432.CCR-15-2823
Hoke, A., and Ray, M. (2014). Rodent models of chemotherapy-induced peripheral neuropathy. ILAR J. 54, 273–281. doi: 10.1093/ilar/ilt053
Huang, Z. Z., Li, D., Ou-Yang, H. D., Liu, C. C., Liu, X. G., Ma, C., et al. (2016). Cerebrospinal fluid oxaliplatin contributes to the acute pain induced by systemic administration of oxaliplatin. Anesthesiology 124, 1109–1121. doi: 10.1097/ALN.0000000000001084
Ja'afer, F. M., Hamdan, F. B., and Mohammed, F. H. (2006). Vincristine-induced neuropathy in rat: electrophysiological and histological study. Exp. Brain Res. 173, 334–345. doi: 10.1007/s00221-006-0499-2
Johnstone, T. C., Park, G. Y., and Lippard, S. J. (2014). Understanding and improving platinum anticancer drugs–phenanthriplatin. Anticancer Res. 34, 471–476.
Jordan, M. A., and Wilson, L. (2004). Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–265. doi: 10.1038/nrc1317
Kage, E., Hayashi, Y., Takeuchi, H., Hirotsu, T., Kunitomo, H., Inoue, T., et al. (2005). MBR-1, a novel helix-turn-helix transcription factor, is required for pruning excessive neurites in Caenorhabditis elegans. Curr. Biology: CB 15, 1554–1559. doi: 10.1016/j.cub.2005.07.057
Kavallaris, M. (2010). Microtubules and resistance to tubulin-binding agents. Nature reviews Cancer 10, 194–204. doi: 10.1038/nrc2803
Kidd, J. F., Pilkington, M. F., Schell, M. J., Fogarty, K. E., Skepper, J. N., Taylor, C. W., et al. (2002). Paclitaxel affects cytosolic calcium signals by opening the mitochondrial permeability transition pore. J. Biol. Chem. 277, 6504–6510. doi: 10.1074/jbc.M106802200
Kroigard, T., Schroder, H. D., Qvortrup, C., Eckhoff, L., Pfeiffer, P., Gaist, D., et al. (2014). Characterization and diagnostic evaluation of chronic polyneuropathies induced by oxaliplatin and docetaxel comparing skin biopsy to quantitative sensory testing and nerve conduction studies. Eur. J. Neurol. 21, 623–629. doi: 10.1111/ene.12353
LaPointe, N. E., Morfini, G., Brady, S. T., Feinstein, S. C., Wilson, L., and Jordan, M. A. (2013). Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology 37, 231–239. doi: 10.1016/j.neuro.2013.05.008
Leandro-Garcia, L. J., Leskela, S., Jara, C., Green, H., Avall-Lundqvist, E., Wheeler, H. E., et al. (2012). Regulatory polymorphisms in beta-tubulin IIa are associated with paclitaxel-induced peripheral neuropathy. Clin. Cancer Res. 18, 4441–4448. doi: 10.1158/1078-0432.CCR-12-1221
Li, Y., Adamek, P., Zhang, H., Tatsui, C. E., Rhines, L. D., Mrozkova, P., et al. (2015). The cancer chemotherapeutic paclitaxel increases human and rodent sensory neuron responses to TRPV1 by activation of TLR4. J. Neurosci. 35, 13487–13500. doi: 10.1523/JNEUROSCI.1956-15.2015
Lisse, T. S., Middleton, L. J., Pellegrini, A. D., Martin, P. B., Spaulding, E. L., Lopes, O., et al. (2016). Paclitaxel-induced epithelial damage and ectopic MMP-13 expression promotes neurotoxicity in zebrafish. Proc. Natl. Acad. Sci. U.S.A. 113, E2189–2198. doi: 10.1073/pnas.1525096113
Low, L. K., Liu, X. B., Faulkner, R. L., Coble, J., and Cheng, H. J. (2008). Plexin signaling selectively regulates the stereotyped pruning of corticospinal axons from visual cortex. Proc. Natl. Acad. Sci. U.S.A. 105, 8136–8141. doi: 10.1073/pnas.0803849105
MacDonald, J. M., Beach, M. G., Porpiglia, E., Sheehan, A. E., Watts, R. J., and Freeman, M. R. (2006). The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 50, 869–881. doi: 10.1016/j.neuron.2006.04.028
Majithia, N., Temkin, S. M., Ruddy, K. J., Beutler, A. S., Hershman, D. L., and Loprinzi, C. L. (2016). National Cancer Institute-supported chemotherapy-induced peripheral neuropathy trials: outcomes and lessons. Support. Care Cancer 24, 1439–1447. doi: 10.1007/s00520-015-3063-4
Malgrange, B., Delree, P., Rigo, J. M., Baron, H., and Moonen, G. (1994). Image analysis of neuritic regeneration by adult rat dorsal root ganglion neurons in culture: quantification of the neurotoxicity of anticancer agents and of its prevention by nerve growth factor or basic fibroblast growth factor but not brain-derived neurotrophic factor or neurotrophin-3. J. Neurosci. Methods 53, 111–122. doi: 10.1016/0165-0270(94)90151-1
Maor-Nof, M., Romi, E., Sar Shalom, H., Ulisse, V., Raanan, C., Nof, A., et al. (2016). Axonal degeneration is regulated by a transcriptional program that coordinates expression of pro- and anti-degenerative factors. Neuron 92, 991–1006. doi: 10.1016/j.neuron.2016.10.061
Martin, S. M., O'Brien, G. S., Portera-Cailliau, C., and Sagasti, A. (2010). Wallerian degeneration of zebrafish trigeminal axons in the skin is required for regeneration and developmental pruning. Development 137, 3985–3994. doi: 10.1242/dev.053611
Masocha, W. (2015). Astrocyte activation in the anterior cingulate cortex and altered glutamatergic gene expression during paclitaxel-induced neuropathic pain in mice. PeerJ 3:e1350. doi: 10.7717/peerj.1350
Meregalli, C., Canta, A., Carozzi, V. A., Chiorazzi, A., Oggioni, N., Gilardini, A., et al. (2010). Bortezomib-induced painful neuropathy in rats: a behavioral, neurophysiological and pathological study in rats. Eur. J. Pain 14, 343–350. doi: 10.1016/j.ejpain.2009.07.001
Milde, S., Fox, A. N., Freeman, M. R., and Coleman, M. P. (2013a). Deletions within its subcellular targeting domain enhance the axon protective capacity of Nmnat2 in vivo. Sci. Rep. 3:2567. doi: 10.1038/srep02567
Milde, S., Gilley, J., and Coleman, M. P. (2013b). Subcellular localization determines the stability and axon protective capacity of axon survival factor Nmnat2. PLoS Biol. 11:e1001539. doi: 10.1371/journal.pbio.1001539
Mora, E., Smith, E. M., Donohoe, C., and Hertz, D. L. (2016). Vincristine-induced peripheral neuropathy in pediatric cancer patients. Am. J. Cancer Res. 6, 2416–2430.
Nikolaev, A., McLaughlin, T., O'Leary, D. D., and Tessier-Lavigne, M. (2009). APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457, 981–989. doi: 10.1038/nature07767
Obaishi, H., Nakanishi, H., Mandai, K., Satoh, K., Satoh, A., Takahashi, K., et al. (1998). Frabin, a novel FGD1-related actin filament-binding protein capable of changing cell shape and activating c-Jun N-terminal kinase. J. Biol. Chem. 273, 18697–18700. doi: 10.1074/jbc.273.30.18697
Osterloh, J. M., Yang, J., Rooney, T. M., Fox, A. N., Adalbert, R., Powell, E. H., et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. doi: 10.1126/science.1223899
Park, S. B., Goldstein, D., Krishnan, A. V., Lin, C. S., Friedlander, M. L., Cassidy, J., et al. (2013). Chemotherapy-induced peripheral neurotoxicity: a critical analysis. CA Cancer J. Clin. 63, 419–437. doi: 10.3322/caac.21204
Pazyra-Murphy, M. F., Hans, A., Courchesne, S. L., Karch, C., Cosker, K. E., Heerssen, H. M., et al. (2009). A retrograde neuronal survival response: target-derived neurotrophins regulate MEF2D and bcl-w. J. Neurosci. 29, 6700–6709. doi: 10.1523/JNEUROSCI.0233-09.2009
Periquet, M. I., Novak, V., Collins, M. P., Nagaraja, H. N., Erdem, S., Nash, S. M., et al. (1999). Painful sensory neuropathy: prospective evaluation using skin biopsy. Neurology 53, 1641–1647. doi: 10.1212/WNL.53.8.1641
Persohn, E., Canta, A., Schoepfer, S., Traebert, M., Mueller, L., Gilardini, A., et al. (2005). Morphological and morphometric analysis of paclitaxel and docetaxel-induced peripheral neuropathy in rats. Eur. J. Cancer 41, 1460–1466. doi: 10.1016/j.ejca.2005.04.006
Podratz, J. L., Knight, A. M., Ta, L. E., Staff, N. P., Gass, J. M., Genelin, K., et al. (2011a). Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol. Dis. 41, 661–668. doi: 10.1016/j.nbd.2010.11.017
Podratz, J. L., Staff, N. P., Boesche, J. B., Giorno, N. J., Hainy, M. E., Herring, S. A., et al. (2013). An automated climbing apparatus to measure chemotherapy-induced neurotoxicity in Drosophila melanogaster. Fly 7, 187–192. doi: 10.4161/fly.24789
Podratz, J. L., Staff, N. P., Froemel, D., Wallner, A., Wabnig, F., Bieber, A. J., et al. (2011b). Drosophila melanogaster: a new model to study cisplatin-induced neurotoxicity. Neurobiol. Dis. 43, 330–337. doi: 10.1016/j.nbd.2011.03.022
Poruchynsky, M. S., Sackett, D. L., Robey, R. W., Ward, Y., Annunziata, C., and Fojo, T. (2008). Proteasome inhibitors increase tubulin polymerization and stabilization in tissue culture cells: a possible mechanism contributing to peripheral neuropathy and cellular toxicity following proteasome inhibition. Cell Cycle 7, 940–949. doi: 10.4161/cc.7.7.5625
Potts, P. R., Singh, S., Knezek, M., Thompson, C. B., and Deshmukh, M. (2003). Critical function of endogenous XIAP in regulating caspase activation during sympathetic neuronal apoptosis. J. Cell Biol. 163, 789–799. doi: 10.1083/jcb.200307130
Poulain, F. E., and Chien, C. B. (2013). Proteoglycan-mediated axon degeneration corrects pretarget topographic sorting errors. Neuron 78, 49–56. doi: 10.1016/j.neuron.2013.02.005
Quartu, M., Carozzi, V. A., Dorsey, S. G., Serra, M. P., Poddighe, L., Picci, C., et al. (2014). Bortezomib treatment produces nocifensive behavior and changes in the expression of TRPV1, CGRP, and substance P in the rat DRG, spinal cord, and sciatic nerve. Biomed Res. Int. 2014:180428. doi: 10.1155/2014/180428
Riccomagno, M. M., and Kolodkin, A. L. (2015). Sculpting neural circuits by axon and dendrite pruning. Annu. Rev. Cell Dev. Biol. 31, 779–805. doi: 10.1146/annurev-cellbio-100913-013038
Ruiz-Medina, J., Baulies, A., Bura, S. A., and Valverde, O. (2013). Paclitaxel-induced neuropathic pain is age dependent and devolves on glial response. Eur. J. Pain 17, 75–85. doi: 10.1002/j.1532-2149.2012.00172.x
Sasaki, Y., Nakagawa, T., Mao, X., DiAntonio, A., and Milbrandt, J. (2016). NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD+ depletion. eLife 5:e19749. doi: 10.7554/eLife.19749
Sasaki, Y., Vohra, B. P., Baloh, R. H., and Milbrandt, J. (2009). Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J. Neurosci. 29, 6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009
Schoenmann, Z., Assa-Kunik, E., Tiomny, S., Minis, A., Haklai-Topper, L., Arama, E., et al. (2010). Axonal degeneration is regulated by the apoptotic machinery or a NAD+-sensitive pathway in insects and mammals. J. Neurosci. 30, 6375–6386. doi: 10.1523/JNEUROSCI.0922-10.2010
Seretny, M., Currie, G. L., Sena, E. S., Ramnarine, S., Grant, R., MacLeod, M. R., et al. (2014). Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: a systematic review and meta-analysis. Pain 155, 2461–2470. doi: 10.1016/j.pain.2014.09.020
Shemesh, O. A., and Spira, M. E. (2010). Paclitaxel induces axonal microtubules polar reconfiguration and impaired organelle transport: implications for the pathogenesis of paclitaxel-induced polyneuropathy. Acta Neuropathol. 119, 235–248. doi: 10.1007/s00401-009-0586-0
Siau, C., and Bennett, G. J. (2006). Dysregulation of cellular calcium homeostasis in chemotherapy-evoked painful peripheral neuropathy. Anesth. Analg. 102, 1485–1490. doi: 10.1213/01.ane.0000204318.35194.ed
Simon, D. J., Pitts, J., Hertz, N. T., Yang, J., Yamagishi, Y., Olsen, O., et al. (2016). Axon degeneration gated by retrograde activation of somatic pro-apoptotic signaling. Cell 164, 1031–1045. doi: 10.1016/j.cell.2016.01.032
Simon, D. J., Weimer, R. M., McLaughlin, T., Kallop, D., Stanger, K., Yang, J., et al. (2012). A caspase cascade regulating developmental axon degeneration. J. Neurosci. 32, 17540–17553. doi: 10.1523/JNEUROSCI.3012-12.2012
Singh, K. K., Park, K. J., Hong, E. J., Kramer, B. M., Greenberg, M. E., Kaplan, D. R., et al. (2008). Developmental axon pruning mediated by BDNF-p75NTR-dependent axon degeneration. Nat. Neurosci. 11, 649–658. doi: 10.1038/nn.2114
Smith, E. M., Pang, H., Cirrincione, C., Fleishman, S., Paskett, E. D., Ahles, T., et al. (2013). Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA 309, 1359–1367. doi: 10.1001/jama.2013.2813
Stendel, C., Roos, A., Deconinck, T., Pereira, J., Castagner, F., Niemann, A., et al. (2007). Peripheral nerve demyelination caused by a mutant Rho GTPase guanine nucleotide exchange factor, frabin/FGD4. Am. J. Hum. Genet. 81, 158–164. doi: 10.1086/518770
Suchankova, T., Kubicek, K., Kasparkova, J., Brabec, V., and Kozelka, J. (2012). Platinum-DNA interstrand crosslinks: molecular determinants of bending and unwinding of the double helix. J. Inorg. Biochem. 108, 69–79. doi: 10.1016/j.jinorgbio.2011.09.025
Ta, L. E., Bieber, A. J., Carlton, S. M., Loprinzi, C. L., Low, P. A., and Windebank, A. J. (2010). Transient Receptor Potential Vanilloid 1 is essential for cisplatin-induced heat hyperalgesia in mice. Mol. Pain 6:15. doi: 10.1186/1744-8069-6-15
Tasdemir-Yilmaz, O. E., and Segal, R. A. (2016). There and back again: coordinated transcription, translation and transport in axonal survival and regeneration. Curr. Opin. Neurobiol. 39, 62–68. doi: 10.1016/j.conb.2016.04.006
Theiss, C., and Meller, K. (2000). Taxol impairs anterograde axonal transport of microinjected horseradish peroxidase in dorsal root ganglia neurons in vitro. Cell Tissue Res. 299, 213–224. doi: 10.1007/s004410050019
Thomas-Jinu, S., Gordon, P. M., Fielding, T., Taylor, R., Smith, B. N., Snowden, V., et al. (2017). Non-nuclear pool of splicing factor SFPQ regulates axonal transcripts required for normal motor development. Neuron 94, 322.e5–336.e5. doi: 10.1016/j.neuron.2017.03.026
Unsain, N., Higgins, J. M., Parker, K. N., Johnstone, A. D., and Barker, P. A. (2013). XIAP regulates caspase activity in degenerating axons. Cell Rep. 4, 751–763. doi: 10.1016/j.celrep.2013.07.015
Wainger, B. J., Buttermore, E. D., Oliveira, J. T., Mellin, C., Lee, S., Saber, W. A., et al. (2015). Modeling pain in vitro using nociceptor neurons reprogrammed from fibroblasts. Nat. Neurosci. 18, 17–24. doi: 10.1038/nn.3886
Wang, J. T., Medress, Z. A., and Barres, B. A. (2012). Axon degeneration: molecular mechanisms of a self-destruction pathway. J. Cell Biol. 196, 7–18. doi: 10.1083/jcb.201108111
Wang, M. S., Davis, A. A., Culver, D. G., Wang, Q., Powers, J. C., and Glass, J. D. (2004). Calpain inhibition protects against Taxol-induced sensory neuropathy. Brain 127, 671–679. doi: 10.1093/brain/awh078
Wang, M.-S., Wu, Y., Culver, D. G., and Glass, J. D. (2001). The gene for slow Wallerian degeneration (Wlds) is also protective against vincristine neuropathy. Neurobiol. Dis. 8, 155–161. doi: 10.1006/nbdi.2000.0334
Wheeler, H. E., Wing, C., Delaney, S. M., Komatsu, M., and Dolan, M. E. (2015). Modeling chemotherapeutic neurotoxicity with human induced pluripotent stem cell-derived neuronal cells. PLoS ONE 10:e0118020. doi: 10.1371/journal.pone.0118020
Xiao, W. H., Zheng, H., and Bennett, G. J. (2012). Characterization of oxaliplatin-induced chronic painful peripheral neuropathy in the rat and comparison with the neuropathy induced by paclitaxel. Neuroscience 203, 194–206. doi: 10.1016/j.neuroscience.2011.12.023
Xiong, X., Hao, Y., Sun, K., Li, J., Li, X., Mishra, B., et al. (2012). The Highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol. 10:e1001440. doi: 10.1371/journal.pbio.1001440
Yang, I. H., Siddique, R., Hosmane, S., Thakor, N., and Hoke, A. (2009). Compartmentalized microfluidic culture platform to study mechanism of paclitaxel-induced axonal degeneration. Exp. Neurol. 218, 124–128. doi: 10.1016/j.expneurol.2009.04.017
Yang, J., Weimer, R. M., Kallop, D., Olsen, O., Wu, Z., Renier, N., et al. (2013). Regulation of axon degeneration after injury and in development by the endogenous calpain inhibitor calpastatin. Neuron 80, 1175–1189. doi: 10.1016/j.neuron.2013.08.034
Yang, J., Wu, Z., Renier, N., Simon, D. J., Uryu, K., Park, D. S., et al. (2015). Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 160, 161–176. doi: 10.1016/j.cell.2014.11.053
Yoon, B. C., Jung, H., Dwivedy, A., O'Hare, C. M., Zivraj, K. H., and Holt, C. E. (2012). Local translation of extranuclear lamin B promotes axon maintenance. Cell 148, 752–764. doi: 10.1016/j.cell.2011.11.064
Yu, F., and Schuldiner, O. (2014). Axon and dendrite pruning in Drosophila. Curr. Opin. Neurobiol. 27, 192–198. doi: 10.1016/j.conb.2014.04.005
Zhang, H., Li, Y., de Carvalho-Barbosa, M., Kavelaars, A., Heijnen, C. J., Albrecht, P. J., et al. (2016). Dorsal root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy-induced peripheral neuropathy. J. Pain 17, 775–786. doi: 10.1016/j.jpain.2016.02.011
Zhang, H., Yoon, S. Y., and Dougherty, P. M. (2012). Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of Paclitaxel-induced painful neuropathy. J. Pain 13, 293–303. doi: 10.1016/j.jpain.2011.12.002
Keywords: axon, chemotherapy, CIPN, degeneration, DRG, neuropathy, sensory neuron, Wallerian
Citation: Fukuda Y, Li Y and Segal RA (2017) A Mechanistic Understanding of Axon Degeneration in Chemotherapy-Induced Peripheral Neuropathy. Front. Neurosci. 11:481. doi: 10.3389/fnins.2017.00481
Received: 23 May 2017; Accepted: 14 August 2017;
Published: 31 August 2017.
Edited by:
Robert W. Burgess, The Jackson Laboratory, United StatesReviewed by:
Sandra Rieger, Mount Desert Island Biological Laboratory, United StatesChristian Gonzalez-Billault, Universidad de Chile, Chile
Copyright © 2017 Fukuda, Li and Segal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosalind A. Segal, Um9zYWxpbmRfc2VnYWxAZGZjaS5oYXJ2YXJkLmVkdQ==
†These authors have contributed equally to this work.